

Abstract
Interleukin (IL)‐18, formerly known as interferon (IFN)‐γ‐inducing factor, is a crucial mediator of host defence and inflammation. Control of IL‐18 bioactivity by its endogenous antagonist IL‐18 binding protein (IL‐18BP) is a major objective of immunoregulation. IL‐18BP is strongly up‐regulated by IFN‐γ, thereby establishing a negative feedback mechanism detectable in cell culture and in vivo. Here we sought to investigate in D.L. Dexter (DLD) colon carcinoma cells molecular mechanisms of IL‐18BP induction under the influence of IFN‐γ. Mutational analysis revealed that a proximal γ‐activated sequence (GAS) at the IL‐18BP promoter is of pivotal importance for expression by IFN‐γ‐activated cells. Use of siRNA underscored the essential role of the signal transducer and activator of transcription (STAT)‐1 in this process. Indeed, electrophoretic mobility shift assay and chromatin immunoprecipitation analysis proved STAT1 binding to this particular GAS site. Maximal expression of IL‐18BP was dependent on de novo protein synthesis but unaffected by silencing of interferon regulatory factor‐1. Altogether, data presented herein indicate that direct action of STAT1 on the IL‐18BP promoter at the proximal GAS element is key to IL‐18BP expression by IFN‐γ‐stimulated DLD‐1 colon carcinoma cells.
Keywords: interferon‐γ, IL‐18 binding protein, STAT1
Introduction
Interleukin (IL)‐18 is a pro‐inflammatory member of the IL‐1 cytokine family that is constitutively expressed primarily in monocytes/macrophages, neutrophils and epithelial cells [1, 2, 3]. This cellular pattern of expression suggests a key role of IL‐18 in the cytokine network orchestrating innate immune activation and successive acquired immunity. The function of IL‐18 in immunity is mediated by its capability to rapidly initiate an inflammatory cytokine cascade [4, 5] and to act as potent co‐stimulus for production of interferon (IFN)‐γ[1, 2, 3]. In concurrence with its pro‐inflammatory nature, IL‐18 production has been linked to severity of disease in models of inflammation such as experimental colitis [6] and arthritis [7]. Accordingly, up‐regulation of IL‐18 is detectable in inflammatory syndromes in patients [3].
Due to inherent pathogenicity in association with constitutive expression in cells of the innate immune system, control of IL‐18 bioactivity is a major objective of immunoregulation. Two principal checkpoints are in place. One is pro‐IL‐18 maturation by caspase‐1 [3]. The second modulatory principle is suppression of IL‐18 function by its natural antagonist, IL‐18 binding protein (IL‐18BP) [8, 9, 10]. IL‐18BP is a circulating decoy receptor able to bind to and inactivate IL‐18 biological activity. Of the four isoenzymes of human IL‐18BP, IL‐18BPa and IL‐18BPc neutralize IL‐18 with high efficacy. IL‐18BPa is by far the most abundant splice variant in human cDNA libraries [8, 11] and thus is concentrated on and referred to as IL‐18BP throughout the study. IL‐18BP is constitutively expressed in the spleen [8] thereby establishing a physiological buffer directed against unwanted basal IL‐18 bioactivity that may arise from dying cells. Notably, IL‐18BP is up‐regulated by immunoactivation as seen in sepsis [12] and under conditions of chronic inflammation such as Crohn’s disease [13], psoriasis [14] and Wegener’s granulomatosis [15]. The significance of the IL‐18/IL‐18BP system is furthermore highlighted by the existence of viral forms of IL‐18BP that are secreted by infected cells and likely contribute to the pathophysiology of infection. For example, expression of vIL‐18BP may mediate the phenomenon of absent inflammation in the context of infection by the molluscum contagiosum virus [16]. We and others have previously identified IFN‐γ as chief mediator of IL‐18BP induction. Interestingly, induction of IL‐18BP appears to especially apply to non‐leucocytic cells [17, 18, 19, 20] and could be demonstrated in the ex vivo setting of colonic organ cultures [18]. Notably, this regulatory path operates in vivo[21] and may curb IL‐18 biological activity as part of a negative feedback loop [10]. In the present study, we set out to investigate molecular mechanisms that direct expression of IL‐18BP in human DLD‐1 colon carcinoma cells under the influence of IFN‐γ.
Materials and methods
Reagents
IFN‐γ was from Peprotech (Frankfurt, Germany), IL‐1 receptor antagonist (IL‐1Ra) from R&D Systems (Wiesbaden, Germany), IL‐1β from Invitrogen (Solingen, Germany) and Cycloheximide (CHX) from Sigma (Taufkirchen, Germany). Tumour necrosis factor (TNF)‐α was kindly provided by the Knoll AG (Ludwigshafen, Germany).
Cultivation of DLD‐1 colon carcinoma cells, freshly isolated peripheral blood mononuclear cells (PBMC) and monocytic THP‐1 cells
DLD‐1 cells (Center of Applied Microbiology and Research, Salisbury, UK) were maintained in Dulbecco’s modified eagle medium (DMEM) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin and 10% heat‐inactivated foetal bovine serum (FCS; GIBCO‐BRL, Eggenstein, Germany). Unless otherwise indicated, cells were incubated under the specified experimental conditions on polystyrene plates (Greiner, Frickenhausen, Germany) in the aforementioned culture medium. The study protocol and consent documents for isolation of PBMC from healthy donors were approved by the ‘Ethik Kommission’ of the Klinikum der Johann Wolfgang Goethe‐Universität. Informed consent was obtained from volunteers. Healthy donors abstained from using drugs during 2 weeks before the study. PBMC were freshly isolated using Histopaque®‐1077 (Sigma). PBMC were resuspended in Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin and 1% heat‐ inactivated human serum (Invitrogen, Karlsruhe, Germany) and seeded at 3 × 106 cells/ml in round‐bottom polypropylene tubes (Greiner, Frickenhausen, Germany). Monocytic THP‐1 cells were obtained from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Cells were maintained and stimulated in RPMI 1640 supplemented with 10 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin and 10% heat‐inactivated FCS (GIBCO‐BRL) using polystyrene flasks and plates (Greiner). All incubations of cell cultures were performed at 37°C and 5% CO2.
Detection of IL‐18BP, interferon regulatory factor (IRF)‐1 and signal transducer and activator of transcription‐1 (STAT1) by immunoblot analysis
For detection of secreted IL‐18BP (cultivation of cells without FCS), cell‐free supernatants were trichloroacetic acid (TCA) precipitated, as previously described [18]. To detect total STAT1, activated phosphorylated pSTAT1, and IRF‐1, nuclear extracts of DLD‐1 cells were isolated as previously described [22]. For detection of total nuclear STAT1, blots were stripped and reprobed. Antibodies: IL‐18BPa, goat polyclonal antibody (R&D Systems); total STAT1, rabbit polyclonal antibody (Santa Cruz Biotechnology, Heidelberg, Germany); pSTAT1‐Y701, rabbit polyclonal antibody (Cell Signaling, Frankfurt, Germany); IRF1, rabbit polyclonal antibody (Santa Cruz Biotechnology) and β‐actin, mouse monoclonal antibody (Sigma).
Luciferase reporter assays
An IL‐18BP promoter fragment was cloned into pGL3‐Basic (Promega, Mannheim, Germany) and entitled pGL3‐BPwt. Position of cloned DNA, AF110798: nt128‐nt794 (667 nt long, spanning until –657 bp relative to the transcriptional start site at nt785 of AF110798). The following primers were used for cloning of pGL3‐BPwt: forward, 5′‐ TCTTACGCGTGTCTTGGAGCTCCTAGAGGGA‐3′ reverse, 5′‐TCGCAGATCTGCTTCTGGGAAGGGCAG‐3′. Site directed mutagenesis was performed by using the QuikChange site‐directed mutagenesis kit (Stratagene, Amsterdam, Netherlands) in order to generate promoter fragments that show a dysfunctional proximal γ‐activated sequence (GAS) (pGL3‐BPmt/prox, located at –25 bp to –33 bp), a dysfunctional putative distal GAS site (pGL3‐BPmt/dist, located at –625 bp to –633 bp) and a double mutation of both GAS sites (pGL3‐BPmt/dist/prox). The following primers were used for that purpose: pGL3‐BPmt/dist, forward: 5′‐TAGAGGGAAGCGATTGGAAAGGAAGGCTCT‐3′; pGL3‐BPmt/prox, forward: 5′‐GGCAGTGCTGATATCGAAGGATTGCTC‐3′. The identity of the mutants was confirmed by sequencing. For each transfection experiment (on six‐well polystyrene plates) 1 μg of the indicated plasmids were transfected using Lipofectamine2000 (Invitrogen). For control of transfection efficiency 0.1 μg pRL‐TK (Promega) coding for Renilla luciferase were cotransfected. After 4 hrs of transfection, culture medium was exchanged and DLD‐1 cells were rested for 20 hrs. Thereafter, cells were either kept as unstimulated control or stimulated with IFN‐γ (20 ng/ml). After another 6‐hr stimulation period, cells were harvested and fold induction of luciferase activity by IFN‐γ was determined by using the dual reporter gene system (Promega) with control conditions (unstimulated cells transfected with the same respective promoter fragment) set to 1.
Electrophoretic mobility shift assay (EMSA analysis)
Preparation of nuclear extracts was performed as previously described [22]. A double‐stranded oligonucleotide reflecting the specific proximal GAS site at the IL‐18BP promoter was used for EMSA analysis: EMSA/GAS oligonucleotide (–18 bp to –40 bp relative to the IL‐18BP transcriptional start site [20]): 5′‐CAGTGCTTTCCCAGAAGGATTGC‐3′. In addition, two oligonucleotides with mutations at the specific proximal GAS site were used to further characterize the retarded complex and are entitled: GASprox‐5′mut: 5′‐CAGTGCTGATATCGAAGGATTGC‐3′; GASprox‐3′mut: 5′‐CAGTGCTTTCCCATCCGGATTGC‐3′. Complementary oligonucleotides were labelled by T4 polynucleotide kinase (MBI Fermentas, St. Leon‐Rot, Germany) using [γ‐32P]ATP (6000 Ci/mmol, Amersham Bioscience, Braunschweig, Germany). Binding reactions were performed for 45 min. on ice with either 5 μg (Fig. 2C, left panel) or 1 μg of protein (Fig. 2C, right panel) in 20 μl of buffer containing 4% Ficoll, 20 mM n‐2‐hydroxyethylpiperazine‐n′‐2‐ethane sulfonic acid (HEPES) (pH 7.9), 50 mM KCl, 1 mM ethylenediaminetetraacetic acid, 1 mM DTT, 1 mM phenylmethanesulfonylfluoride (PMSF), 0.25 mg/ml bovine serum albumin, 1.25 μg of poly(dI‐dC) and 50.000 cpm of 32P‐labelled oligonucleotide. To characterize retarded complexes, nuclear proteins were pre‐incubated (30 min., RT) with two different rabbit polyclonal anti‐STAT1 antibodies (Santa Cruz Biotechnology and Upstate Biotechnology, Hamburg, Germany).
Figure 2.
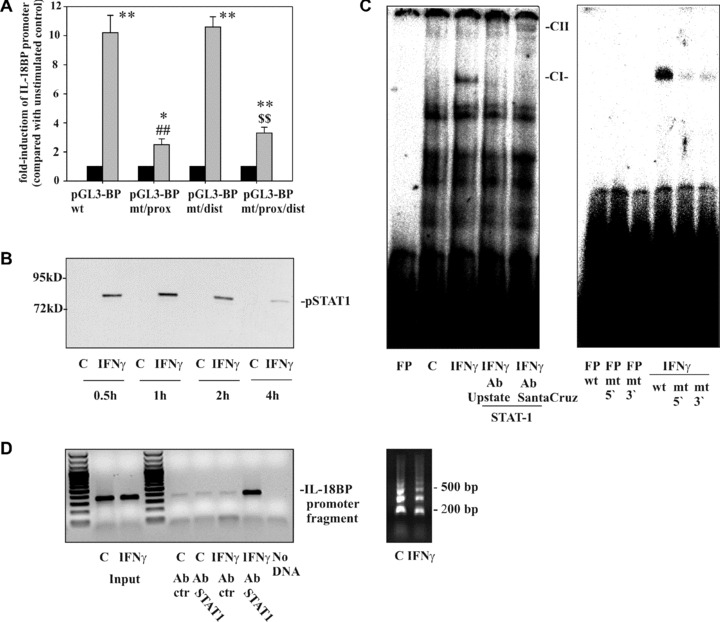
The role of the STAT1/GAS axis in IL‐18BP promoter activation as detected in DLD‐1 cells. (A) DLD‐1 cells were transfected with the indicated IL‐18BP promoter constructs (pGL3‐BPwt, pGL3‐BPmt/prox, pGL3‐BPmt/dist, pGL3‐BPmt/prox/dist) together with pRL‐TK (Promega) as described in the ‘Materials and methods’ section. After 24 hrs, cells were kept as unstimulated control or stimulated with IFN‐γ (20 ng/ml). After another 6 hrs, cells were harvested and luciferase assays were performed. Data are expressed as mean fold‐luciferase induction ± S.D. (compared to the unstimulated control transfected with the same plasmid) obtained from three independently performed experiments. Black bars: unstimulated control cells, gray bars: IFN‐γ (20 ng/ml). *, P < 0.05 and **, P < 0.01 compared with the respective unstimulated control; ##, P < 0.01 compared with pGL3‐BPwt under the influence of IFN‐γ; $$, P < 0.01 compared with pGL3‐BPmt/dist under the influence of IFN‐γ. (B) DLD‐1 cells were kept as unstimulated control or stimulated with IFN‐γ (10 ng/ml) for the indicated times. Nuclear pSTAT1 was evaluated by immunoblot analysis. One representative of three independently performed experiments is shown. (C) Cells were kept as unstimulated control or stimulated with IFN‐γ (10 ng/ml). After 1 hr, nuclear lysates were prepared and protein binding to oligonucleotides reflecting the proximal GAS site (–18 bp/–40 bp) was assessed by electrophoretic mobility shift assay analysis. Where indicated, anti‐STAT1 antibodies were added to characterize the IFN‐γ‐inducible retarded complex denoted CI. FP, free probe; CII, supershifted complex. One representative of six independently performed experiments is shown. Right panel: protein binding to wild‐type oligonucleotides was compared with binding to the mutated oligonucleotides GASprox‐5′mut and GASprox‐3′mut, respectively. One representative of three independently performed experiments is shown. (D) DLD‐1 cells were kept as unstimulated control or stimulated with IFN‐γ (10 ng/ml). After 1 hr, cells were harvested followed by ChIP analyses as described in the ‘Materials and methods’ section. One representative of six independently performed experiments is shown (left panel). Right panel: sheared chromatin of the experiment shown in the left panel.
Chromatin immunoprecipitation (ChIP analysis)
After fixation (RT, 10 min., 1% formaldehyde) and addition of glycine‐buffer, cells were harvested in phosphate buffered saline (PBS)/PMSF (1 mM) followed by incubation in lysis‐buffer, supplemented with PMSF (1mM) and proteinase inhibitor cocktail (Enzymatic Shearing Kit, Active Motif, Rixensart, Belgium). After 30 min. on ice, douncing and centrifugation (15,000 ×g, 4°C, 10 min.), nuclei were resuspendend in digestion buffer. Genomic DNA was digested by adding enzymatic shearing‐cocktail solution (200 U/ml). After addition of 20 μl 0.5 M EDTA, 10 min. on ice and centrifugation, 1/10 of the digested genomic DNA was removed for control of shearing efficiency and PCR‐input, respectively. Nine out of 10 of the digested DNA‐protein‐complexes were immunoprecipitated either with 3 μg of IgG control or STAT1 antibody (Santa Cruz). After washing in PBS/RIPA‐buffer (10 mM Tris‐HCl, pH 8.0, 140 mM NaCl, 1% triton‐X‐100, 0.1% SDS), DNA‐protein complexes were eluted from the beads twice by 200 μl of 1% SDS (in TE‐buffer) and 200 μl of 0.67% SDS (in TE‐buffer) at 65°C (10 min.). After RNase (4 hrs, 65°C) and proteinase K (2 h, 42°C) digestion, sheared genomic DNA was precipitated. To amplify the IL‐18BP promoter fragment containing the GAS site under investigation (–25 bp to –33 bp relative to the IL‐18BP transcriptional start site [20]), the following primers were used: forward, 5′‐GCTGAAGGTTTCTGGCCA‐3′; reverse, 5′‐TAGCCTGGAGCTCTT TCA‐3′. PCR conditions: 95°C for 10 min. (1 cycle); 95°C for 30 sec., 54°C for 30 sec. and 72°C for 1 min. for 40 cycles, and a final extension phase at 72°C for 7 min. Identity of amplicons (360 bp) was confirmed by sequencing (AbiPrism 310 Genetic Analyzer, Applied Biosystems, Darmstadt, Germany).
Analysis of IL‐18BP by quantitative realtime PCR
Realtime PCR for IL‐18BP and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was performed as previously described [22]. Primers and probe for IL‐18BPa were designed using Primer Express (Applied Biosystems) according to AF110798: forward, 5′‐ACCTCCCAGGCCGACTG‐3′; reverse, 5′‐CCTTGCACAGCTGCGTACC‐3′; probe 5′‐CACCAGCCGGGAACGTGGGA‐3′. Amplification of genomic DNA was avoided by selecting an amplicon that crosses an exon/intron boundary. For GAPDH pre‐developed assay reagents were used (4310884E; Applied Biosystems). Quantitative realtime PCR was performed on AbiPrism 7700 Sequence Detector (Applied Biosystems): One initial step at 50°C for 2 min. and 95°C for 2 min. was followed by 40 cycles at 95°C for 15 sec. and 60°C for 1 min. Detection, calculation of threshold cycles (Ct values) and data analysis were performed by Sequence Detector software. mRNA was quantified by use of cloned cDNA standards for IL‐18BP and GAPDH. Data for IL‐18BP were normalized to those of GAPDH.
Suppression of STAT1 and IRF1 expression by siRNA
For experiments, DLD‐1 cells were transfected with 100 nM of either STAT1 siRNA (SI02662324, Qiagen, Hilden, Germany), IRF1 siRNA (5′‐AGACCAGAGCAGGAACAAGTTdtdt‐3′, MWG), or control siRNA (Silencer®Negative Control siRNA, #4611, Ambion) using Lipofectamine2000 (Invitrogen). All cultures without siRNA or control siRNA were mock transfected. After 48 hrs, cells were further stimulated with IFN‐γ and harvested thereafter for further analysis. Transfection of siRNA (control; STAT1 or IRF1 targeting) by the experimental protocol used herein did not mediate activation of IRF3 in DLD‐1 cells indicating that the procedure was not associated with cellular activation induced by RNA sensing mechanisms of innate immunity (data not shown).
Determination of IL‐8 release by enzyme‐linked immunosorbent assay (ELISA)
IL‐8 in cell‐free supernatants was determined by ELISA according to the manufacturer’s instructions (BD Biosciences, Heidelberg, Germany).
Statistical analysis
Data are shown as means ± S.D. and are presented as ng/ml, as fold induction compared with unstimulated control, or as percent of IFN‐γ alone. Raw data were analysed by one‐way ANOVA with post hoc Bonferroni correction (GraphPad 4.0).
Results
Optimal induction of IL‐18BP in DLD‐1 cells is dependent on protein translation
In previous work, we identified IFN‐γ as a potent stimulus for production of IL‐18BP [17, 18, 19], particularly in non‐leucocytic cells (Table 1). The crucial role of signal transduction pathways that are activated by IFN‐γ is underscored by the observation that IL‐18BP secretion cannot be initiated by the combination IL‐1β/TNF‐α (Fig. 1A), although those cytokines are particularly efficient in activating DLD‐1 cells, e.g. for IL‐8 secretion (Fig. 1D). To clarify whether full induction of IL‐18BP mRNA requires de novo protein synthesis, cells were activated in the presence of the translational inhibitor CHX. Figure 1B demonstrates that protein synthesis is mandatory for optimal mRNA induction, even though significant up‐regulation of IL‐18BP can be achieved upon blockage of translation. Notably, under those conditions, induction of IRF1, a crucial component of the IFN‐γ response [23], was suppressed in cells exposed to CHX (Fig. 1B, inset). Recently, it has been proposed that endogenous IL‐1α contributes to immunoregulatory actions of IFN‐γ, including up‐regulation of IL‐18BP in epithelial‐like cells [21]. To identify a potential function of endogenously produced IL‐1, effects of IL‐1Ra were assessed. Figure 1C demonstrates that pre‐incubation with IL‐1Ra, even at 5 μg/ml, was unable to modulate IL‐18BP expression in response to IFN‐γ. As expected, IL‐1Ra completely suppressed IL‐1β‐mediated IL‐8 in this same set of experiments (Fig. 1D). Those data suggest that it is very unlikely that endogenously produced IL‐1α contributes to IFN‐γ‐induced IL‐18BP expression in DLD‐1 cells.
Table 1.
IL‐18BP induction primarily detected in non‐leucocytic cells
| IFNγ | |
|---|---|
| PBMC (n= 5) | 2.06 ± 0.63 |
| THP‐1 (n= 3) | 2.52 ± 0.48 |
| DLD‐1 (n= 3) | 16.54 ± 3.77 ** |
PBMC, THP‐1 cells and DLD‐1 cells were either kept as unstimulated control or were stimulated with IFN‐γ (PBMC, THP‐1: 20 ng/ml; DLD‐1: 10 ng/ml). After 12 hrs, IL‐18BP mRNA was evaluated by realtime PCR. IL‐18BP mRNA was normalized to that of GAPDH. Data are expressed as fold induction compared with unstimulated control ± S.D., **P < 0.01 compared to unstimulated control.
Figure 1.
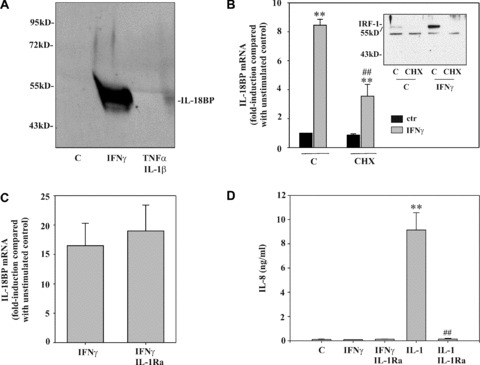
Optimal induction of IL‐18BP in DLD‐1 cells requires protein translation. (A) Cells were kept as unstimulated control, stimulated with IFN‐γ (10 ng/ml) or with IL‐1β/TNF‐α (both 20 ng/ml) for 24 hrs. TCA‐precipitated supernatants were analysed by immunoblot analysis. One representative of three independently performed experiments is shown. (B) Cells were kept as unstimulated control or stimulated with CHX (10 μg/ml), IFN‐γ (10 ng/ml), or CHX (10 μg/ml)/IFN‐γ (10 ng/ml). CHX was always added 2 hrs before IFN‐γ. After 8 hrs, IL‐18BP mRNA was evaluated by realtime PCR. IL‐18BP mRNA was normalized to that of GAPDH. Data are expressed as fold induction compared with unstimulated control ± S.D. (n= 3). **, P < 0.01 compared with unstimulated control or with cycloheximide alone; ##, P < 0.01 compared with IFN‐γ alone. Inset: DLD‐1 cells were kept as unstimulated control or stimulated as described above. After 1 hr, nuclear IRF1 was evaluated by immunoblot analysis. (C) DLD‐1 cells were kept as unstimulated control or stimulated for 12 hrs with IFN‐γ (10 ng/ml) or with IFN‐γ (10 ng/ml)/IL‐1Ra (5 μg/ml). IL‐1Ra was always added 5 hrs before IFN‐γ. IL‐18BP mRNA was evaluated by realtime PCR. IL‐18BP mRNA was normalized to that of GAPDH. Data are expressed as fold induction compared with unstimulated control ± S.D. (n= 3). (D) Supernatants generated by the experiments shown in (C) were evaluated for IL‐8 levels by ELISA. Additional conditions: IL‐1β (2 ng/ml) alone and IL‐1β (2 ng/ml)/IL‐1Ra (5 μg/ml). Data are shown as mean ± S.D. (n= 3). **, P < 0.01 compared with unstimulated control; ##, P < 0.01 compared with IL‐1β.
The role of the STAT1/GAS axis in IL‐18BP promoter activation by DLD‐1 cells
The transcriptional start site of IL‐18BP was previously identified [20]. Notably, a proximal GAS site imposes within the IL‐18BP promoter that is directly located adjacent to the transcriptional start site (–25 bp to –33 bp relative to the transcriptional start site at nt785 of AF110798). In addition, a second distal GAS site that applies to the sequence requirements for putative GAS elements [24, 25] is located at –625 bp to –633 bp. A third potential STAT1 binding site included in the promoter fragment used herein is an IRF‐E/ISRE‐like site located at –54 bp to –69 bp. However, this site is unlikely relevant in the context of the current study as it binds ISGF3, a heterotrimeric transcription factor composed of STAT1/STAT2/IRF9. Notably, STAT2 is activated by type I IFNs but not by IFN‐γ[24].
Luciferase reporter assays were performed in order to analyse mechanisms that direct IL‐18BP promoter activation under the influence of IFN‐γ. Figure 2A demonstrates strong induction of the IL‐18BP wild‐type promoter (pGL3‐BPwt) in response to IFN‐γ. Promoter activation was significantly reduced in the context of a mutated proximal GAS site (pGL3‐BPmt/prox). In contrast, a dysfunctional distal GAS site (pGL3‐BPmt/dist) left reporter gene induction unaffected. The double mutation at the proximal and the distal GAS site (pGL3‐BPmt/prox/dist) was not associated with further suppression of reporter gene activity as compared to the single mutation at the proximal site (pGL3‐BPmt/prox). Those results clearly indicate that the integrity of the proximal GAS site at the IL‐18BP promoter is crucial for gene activation in response to IFN‐γ.
In accordance with previous data [22], robust activation of STAT1 was obvious in DLD‐1 cells under the influence of IFN‐γ (Fig. 2B). EMSA analysis was performed in order to identify in vitro protein/DNA interactions at the proximal GAS site of the IL‐18BP promoter (Fig. 2C). For that purpose, double‐stranded DNA probes representing this specific site were exposed to nuclear extracts generated from unstimulated or IFN‐γ‐treated cells. A single retarded complex (CI) was inducible by IFN‐γ. No such complex was detectable if extracts of unstimulated cells were used. Formation of this particular complex, but not of the constitutive complexes, was abolished by addition of two different STAT1 antibodies suggesting an interaction between the labelled probe representing this specific GAS site and pSTAT1 homodimers induced by IFN‐γ. Notably, a super‐shifted complex (CII) became visible in case of co‐incubation with the antibody obtained from Santa Cruz (Fig. 2C, left panel, 5 μg of protein). Figure 2C (right panel, 1 μg of protein) demonstrates impaired formation of the otherwise retarded protein complex if oligonucleotides were used in EMSA analysis that contain a mutation in the area reflecting the specific proximal GAS site under investigation. For each EMSA experiment, corresponding pSTAT1 immunoblot analysis was performed and validated activation of STAT1 signalling by IFN‐γ (data not shown). We conclude that pSTAT1, present in nuclei of IFN‐γ‐activated DLD‐1 cells, is able to bind in vitro to oligonucleotides reflecting the specific nucleotide context at the proximal GAS site of the IL‐18BP promoter.
Based on those observations, we sought to assess by ChIP analysis whether STAT1 is physically bound to this specific proximal GAS site in intact DLD‐1 cells under the influence of IFN‐γ. In Fig. 2D (left panel), we demonstrate that a promoter fragment containing the proximal GAS site and being targeted by the ChIP‐PCR is only amplified in those samples derived from sheared‐chromatin that had been obtained from IFN‐γ‐stimulated cells and subsequently underwent STAT1 immunoprecipitation. No such amplicons were generated by PCR if either sheared chromatin from unstimulated cells was used or if immunoprecipitation was performed with IgG control. These observations demonstrate that activated STAT1 in fact physically binds to the proximal GAS site of the IL‐18BP promoter in DLD‐1 cells exposed to IFN‐γ. Figure 2D (right panel) shows sheared chromatin of the experiment shown in the left panel.
Modulation of STAT1 by siRNA impairs IFN‐γ‐induced IL‐18BP in DLD‐1 cells
By using the siRNA approach, we set out to investigate the functional significance of STAT1 for IL‐18BP expression. In addition, the role of IRF1 was investigated in these same experiments. Figure 3A demonstrates efficient inhibition of STAT1 and IRF1 activation in DLD‐1 cells that had been transfected with siRNA targeting STAT1 or IRF1 mRNA, respectively. In agreement with the role of STAT1 in IRF1 induction [23], we also observed a reduction of IRF1 expression in cells transfected with STAT1 siRNA. Whereas reduction of IRF1 protein by IRF1 siRNA did not affect IL‐18BP expression, significant modulation was evident under the influence of siRNA targeting STAT1. However, although STAT1 siRNA was able to down‐regulate STAT1 activation on average by 81.5% (based on levels of nuclear pSTAT1), IL‐18BP expression was reduced by 48.9% only (Fig. 3B).
Figure 3.
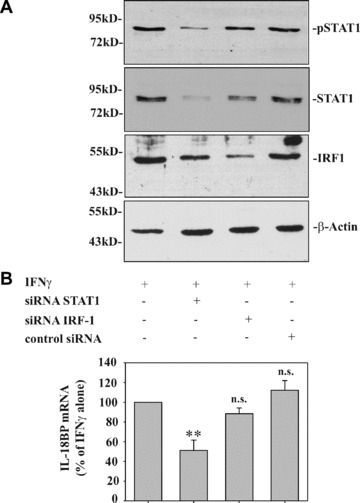
Modulation of STAT1 by siRNA impairs IFN‐γ‐induced IL‐18BP in DLD‐1 cells. (A) DLD‐1 cells were either mock transfected or transfected with the indicated siRNA. After 2d, cells were stimulated with IFN‐γ (10 ng/ml) for 1 hr. Nuclear pSTAT1, total STAT1 and IRF1 content was assessed by immunoblot analysis. One representative of three independently performed experiments is shown. (B) IL‐18BP expression by cells treated as described in (A) but stimulated for 8 hrs was evaluated by realtime PCR. IL‐18BP mRNA was normalized to that of GAPDH. Data are expressed as% of mock‐transfected IFN‐γ‐stimulated cells ± S.D.; for mock transfection, STAT1 siRNA, control siRNA: n = 6; for IRF1 siRNA: n = 3. **, P < 0.01 compared to mock‐transfected IFN‐γ stimulated cells.
Discussion
In the present study, mechanisms underlying IFN‐γ‐induced IL‐18BP expression in the human colon carcinoma cell line DLD‐1 were investigated. DLD‐1 colon carcinoma cells are regarded as a prototypic and well‐characterized cell culture model for studying gene regulation in colon epithelial cells under the influence of IFN‐γ[18, 22, 26, 27]. Moreover, it is well described that IL‐18 bioac‐ tivity likely contributes to colonic pathophysiology with regard to inflammation and carcinogenesis [3, 13, 28, 29]. Here, we report that STAT1, activated by IFN‐γ, physically binds to a GAS element located proximal at the IL‐18BP promoter as detected by ChIP analysis. Mutational analysis of luciferase reporter constructs further substantiated the relevance of this particular regulatory site.
In fact, activation of GAS elements by STAT1 homodimers is key to IFN‐γ biological activity and mediates induction of numerous genes [24]. Notably, significant induction of IL‐18BP in DLD‐1 cells was achieved in the absence of translation, though optimal gene activation required protein synthesis. Those observations were complemented by use of the siRNA technology and altogether strongly suggest a direct and pivotal role of STAT1 in IL‐18BP regulation as observed in DLD‐1 cells. Notably, we could recently demonstrate that STAT3 activation has no influence on IL‐18BP expression [22].
Data presented herein reveal cell type specificity concerning the usage of the key GAS element at the proximal IL‐18BP promoter. In fact, a previous study likewise identified this same proximal GAS site as crucial for IFN‐γ‐induced IL‐18BP in HepG2 cells. However, in those cells, instead of STAT1, a complex consisting of IRF1 and C/EBPβ is binding to the specific GAS site under investigation [20]. Whereas C/EBPβ is constitutively expressed in most cell types [30], IRF1 biological activity is dependent on de novo protein synthesis and up‐regulated by IFN‐γ[23]. Consequently, induction of IL‐18BP in response to IFN‐γ is fully dependent on translation in HepG2 cells [20]. In contrast, a mandatory role for IRF1 in the up‐regulation of IL‐18BP by DLD‐1 cells appears unlikely in light of significant IL‐18BP induction under blockage of translation and of results obtained in the context of IRF1 silencing. Furthermore, only a single IFN‐γ‐induced retarded complex became apparent in EMSA analysis and was identified as being at least partially composed of STAT1. Differences between DLD‐1 and HepG2 cells may relate to a divergent repertoire of transcription factors. Interestingly, compared to DLD‐1 cells, HepG2 cells express higher levels of C/EBPβ (data not shown). Therefore, it is tempting to speculate that, once IRF1 is induced by IFN‐γ, STAT1 homodimers and IRF1:C/EBPβ heterodimers compete for binding to this particular GAS element at the proximal IL‐18BP promoter.
Data presented in the current study also indicate that mechanisms besides binding of STAT1 to the proximal GAS site affect the extent of IL‐18BP promoter activation. Not only was most efficient induction of IL‐18BP dependent on protein synthesis, mutational analysis in luciferase‐reporter assays and siRNA experiments targeting STAT1 further suggest that additional mechanisms contribute to IFN‐γ‐induced IL‐18BP gene activation. Those likely STAT1‐independent actions of IFN‐γ are currently under investigation. Among signalling pathways that possibly synergize with STAT1 are those culminating in NF‐κB activation [31, 32]. In fact it has been demonstrated that endogenously produced IL‐1α and subsequent NF‐κB activation must be regarded a cofactor for IL‐18BP production in Wistar Institute Susan Hayflick (WISH) cells and HaCaT keratinocytes [21]. However, a basal level of NF‐κB activity is characteristic for DLD‐1 cells [22, 33, 34] and may be related to the inability of IL‐1Ra to modulate IFN‐γ‐induced IL‐18BP in this cell type.
Taken together, data presented herein demonstrate a direct action of STAT1 on the proximal GAS site of the IL‐18BP promoter that is crucial for gene induction as detected in DLD‐1 cells. Furthermore, present and previous observations support a concept of cell type specific, promiscuous usage of GAS elements by either STAT1 homodimes or IRF1:C/EBPβ heterodimers at a given IFN‐γ‐inducible promoter.
Acknowledgements
The skilful technical assistance of Silke Kusch and Stephan Hartmann (ChIP protocol) is gratefully acknowledged. This work was supported by the Deutsche Forschungsgemeinschaft DFG MU 1284/3‐2 (to H.M.).
References
- 1. Boraschi D, Dinarello CA. IL‐18 in autoimmunity: review. Eur Cytokine Netw. 2006; 17: 224–52. [PubMed] [Google Scholar]
- 2. Arend WP, Palmer G, Gabay C. IL‐1, IL‐18, and IL‐33 families of cytokines. Immunol Rev. 2008; 223: 20–38. [DOI] [PubMed] [Google Scholar]
- 3. Mühl H, Pfeilschifter J. Interleukin‐18 bioactivity: a novel target for immunopharmacological anti‐inflammatory intervention. Eur J Pharmacol. 2004; 500: 63–71. [DOI] [PubMed] [Google Scholar]
- 4. Puren AJ, Fantuzzi G, Gu Y, et al . Interleukin‐18 (IFNgamma‐inducing factor) induces IL‐8 and IL‐1beta via TNFalpha production from non‐CD14+ human blood mononuclear cells. J Clin Invest. 1998; 101: 711–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Netea MG, Kullberg BJ, Verschueren I, et al . Interleukin‐18 induces production of proinflammatory cytokines in mice: no intermediate role for the cytokines of the tumor necrosis factor family and interleukin‐1beta. Eur J Immunol. 2000; 30: 3057–60. [DOI] [PubMed] [Google Scholar]
- 6. Siegmund B, Fantuzzi G, Rieder F, et al . Neutralization of interleukin‐18 reduces severity in murine colitis and intestinal IFN‐gamma and TNF‐alpha production. Am J Physiol Regul Integr Comp Physiol. 2001; 281: R1264–R73. [DOI] [PubMed] [Google Scholar]
- 7. Canetti CA, Leung BP, Culshaw S, et al . IL‐18 enhances collagen‐induced arthritis by recruiting neutrophils via TNF‐alpha and leukotriene B4. J Immunol. 2003; 171: 1009–15. [DOI] [PubMed] [Google Scholar]
- 8. Novick D, Kim SH, Fantuzzi G, et al . Interleukin‐18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999; 10: 127–36. [DOI] [PubMed] [Google Scholar]
- 9. Aizawa Y, Akita K, Taniai M, et al . Cloning and expression of interleukin‐18 binding protein. FEBS Lett. 1999; 445: 338–42. [DOI] [PubMed] [Google Scholar]
- 10. Mühl H, Pfeilschifter J. Anti‐inflammatory properties of pro‐inflammatory interferon‐gamma. Int Immunopharmacol. 2003; 3: 1247–55. [DOI] [PubMed] [Google Scholar]
- 11. Kim SH, Eisenstein M, Reznikov L, et al. Structural requirements of six naturally occurring isoforms of the IL‐18 binding protein to inhibit IL‐18. Proc Natl Sci USA. 2000; 97: 1190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Novick D, Schwartsburd B, Pinkus R, et al . A novel IL‐18BP ELISA shows elevated serum IL‐18BP in sepsis and extensive decrease of free IL‐18. Cytokine. 2001; 14: 334–42. [DOI] [PubMed] [Google Scholar]
- 13. Corbaz A, ten Hove T, Herren S, et al . IL‐18 binding protein expression by endothelial cells and macrophages is upregulated during active Crohn’s disease. J Immunol. 2002; 168: 3608–16. [DOI] [PubMed] [Google Scholar]
- 14. Ohta Y, Hamada Y, Katsuoka K. Expression of IL‐18 in psoriasis. Arch Dermatol Res. 2001; 293: 334–42. [DOI] [PubMed] [Google Scholar]
- 15. Novick D, Elbitt D, Dinarello CA, et al . Interleukin‐18 binding protein in the sera of patients with Wegener’s granulomatosis. J Clin Immunol. 2009; 29: 38–45. [DOI] [PubMed] [Google Scholar]
- 16. Smith VP, Bryant NA, Alcami A. Ectromelia, vaccinia and cowpox viruses encode secreted Interleukin‐18 binding proteins. J Gen Virol. 2000; 81: 1223–30. [DOI] [PubMed] [Google Scholar]
- 17. Mühl H, Kämpfer H, Bosmann M, et al . Interferon‐gamma mediates gene expression of IL‐18 binding protein in nonleukocytic cells. Biochem Biophys Res Commun. 2000; 267: 960–3. [DOI] [PubMed] [Google Scholar]
- 18. Paulukat J, Bosmann M, Nold M, et al . Expression and release of IL‐18 binding protein in response to IFN‐gamma. J Immunol. 2001; 167: 7038–43. [DOI] [PubMed] [Google Scholar]
- 19. Möller B, Paulukat J, Nold M, et al . Interferon‐gamma induces expression of interleukin‐18 binding protein in fibroblast‐like synoviocytes. Rheumatology. 2003; 42: 442–5. [DOI] [PubMed] [Google Scholar]
- 20. Hurgin V, Novick D, Rubinstein M. The promoter of IL‐18 binding protein: activation by an IFN‐gamma ‐induced complex of IFN regulatory factor 1 and CCAAT/enhancer binding protein beta. Proc Natl Acad Sci USA. 2002; 99: 16957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hurgin V, Novick D, Werman A, et al . Antiviral and immunoregulatory activities of IFN‐gamma depend on constitutively expressed IL‐1alpha. Proc Natl Acad Sci USA. 2007; 104: 5044–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ziesché E, Bachmann M, Kleinert H, et al . The interleukin‐22/STAT3 pathway potentiates expression of inducible nitric‐oxide synthase in human colon carcinoma cells. J Biol Chem. 2007; 282: 16006–15. [DOI] [PubMed] [Google Scholar]
- 23. Taniguchi T, Ogasawara K, Takaoka A, et al . IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001; 19: 623–55. [DOI] [PubMed] [Google Scholar]
- 24. Platanias LC. Mechanisms of type‐I‐ and type‐II‐interferon‐mediated signaling. Nat Rev Immunol. 2005; 5: 375–86. [DOI] [PubMed] [Google Scholar]
- 25. Schroder K, Hertzog PJ, Ravasi T, et al . Interferon‐gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004; 75: 163–89. [DOI] [PubMed] [Google Scholar]
- 26. Jin Y, Heck DE, DeGeorge G, et al . 5‐Fluorouracil suppresses nitric oxide biosynthesis in colon carcinoma cells. Cancer Res. 1996; 56: 1978–82. [PubMed] [Google Scholar]
- 27. Pautz A, Linker K, Hubrich T, et al . The polypyrimidine tract‐binding protein (PTB) is involved in the post‐transcriptional regulation of human inducible nitric oxide synthase expression. J Biol Chem. 2006; 281: 32294–302. [DOI] [PubMed] [Google Scholar]
- 28. Leach ST, Messina I, Lemberg DA, et al . Local and systemic interleukin‐18 and interleukin‐18‐binding protein in children with inflammatory bowel disease. Inflamm Bowel Dis. 2008; 14: 68–74. [DOI] [PubMed] [Google Scholar]
- 29. Pagès F, Berger A, Henglein B, et al . Modulation of interleukin‐18 expression in human colon carcinoma: consequences for tumor immune surveillance. Int J Cancer. 1999; 84: 326–30. [DOI] [PubMed] [Google Scholar]
- 30. Kalvakolanu DV, Roy SK. CCAAT/enhancer binding proteins and interferon signaling pathways. J Interferon Cytokine Res. 2005; 25: 757–69. [DOI] [PubMed] [Google Scholar]
- 31. Robinson CM, Hale PT, Carlin JM. NF‐kappa B activation contributes to indoleamine dioxygenase transcriptional synergy induced by IFN‐gamma and tumor necrosis factor‐alpha. Cytokine. 2006; 35: 53–61. [DOI] [PubMed] [Google Scholar]
- 32. Tamassia N, Calzetti F, Ear T, et al . Molecular mechanisms underlying the synergistic induction of CXCL10 by LPS and IFN‐gamma in human neutrophils. Eur J Immunol. 2007; 37: 2627–34. [DOI] [PubMed] [Google Scholar]
- 33. Kleinert H, Wallerath T, Fritz G, et al . Cytokine induction of NO synthase II in human DLD‐1 cells: roles of the JAK‐STAT, AP‐1 and NF‐kappaB‐signaling pathways. Br J Pharmacol. 1998; 125: 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hellmuth M, Wetzler C, Nold M, et al . Expression of interleukin‐8, heme oxygenase‐1, and vascular endothelial growth factor in DLD‐1 colon carcinoma cells exposed to pyrrolidine dithiocarbamate. Carcinogenesis. 2002; 23: 1273–9. [DOI] [PubMed] [Google Scholar]