

Abstract
Mitochondrial dysfunction has repeatedly been reported associated with type 2 diabetes mellitus (T2DM) and metabolic syndrome (MS), as have mitochondrial DNA (mtDNA) tRNA and duplication mutations and mtDNA haplogroup lineages. We identified 19 Taiwanese T2DM and MS pedigrees from Taiwan, with putative matrilineal transmission, one of which harbored the pathogenic mtDNA tRNALeu(UUR) nucleotide (nt) 3243A > G mutation on the N9a3 haplogroup background. We then recruited three independent Taiwanese cohorts, two from Taipei (N = 498, mean age 52 and N = 1002, mean age 44) and one from a non-urban environment (N = 501, mean age 57). All three cohorts were assessed for an array of metabolic parameters, their mtDNA haplogroups determined, and the haplogroups correlated with T2DM/MS phenotypes. Logistic regression analysis revealed that mtDNA haplogroups D5, F4, and N9a conferred T2DM protection, while haplogroups F4 and N9a were risk factors for hypertension (HTN), and F4 was a risk factor for obesity (OB). Additionally, the 5263C > T (ND2 A165V) variant commonly associated with F4 was associated with hypertension (HTN). Cybrids were prepared with macro-haplogroup N (defined by variants m.ND3 10398A (114T) and m.ATP6 8701A (59T)) haplogroups B4 and F1 mtDNAs and from macro-haplogroup M (variants m.ND3 10398G (114A) and m.ATP6 8701G (59A)) haplogroup M9 mtDNAs. Additionally, haplogroup B4 and F1 cybrids were prepared with and without the mtDNA variant in ND1 3394T > C (Y30H) reported to be associated with T2DM. Assay of mitochondria complex I in these cybrids revealed that macro-haplogroup N cybrids had lower activity than M cybrids, that haplogroup F cybrids had lower activity than B4 cybrids, and that the ND1 3394T > C (Y30H) variant reduced complex I on both the B4 and F1 background but with very different cumulative effects. These data support the hypothesis that functional mtDNA variants may contribute to the risk of developing T2DM and MS.
Keywords: Mitochondria, mtDNA, Haplogroup, Diabetes, Obesity, Cybrids
1. Introduction
Considerable evidence has accumulated implicating mitochondrial dysfunction in T2DM and MS [1,2]. Studies of patients with T2DM have revealed that mitochondrial function and gene expression are generally down-regulated and insulin-resistant offspring of T2DM patients have impaired mitochondrial energetics, as assessed by 31P-MR spectroscopy [3]. T2DM patients consistently show a down-regulation in the expression of nuclear DNA (nDNA) encoded mitochondrial genes, in association with reduced peroxisome-proliferation-activated receptor γ (PPARγ)-coactivator 1 (PGC-1) activity [4,5]. Mitochondrial reactive oxygen species (ROS) production may be relevant to T2DM risk since C57BL6/J mice which lack the mitochondrial antioxidant enzyme nicotinamide nucleotide transhydrogenase are prone to age-related diabetes and this can be reversed by introduction of a mitochondrial-targeted catalase transgene [6,7].
mtDNA variation is implicated in human T2DM, since the probability that the mother was the affected parent increases to a ratio of 3:1 for patients with a mean age-of-onset of 46 years [8,9]. Clinically relevant mtDNA variation falls into three categories: pathogenic mutations that result in maternally inherited disease; ancient functional polymorphisms associated with specific mtDNA lineages, known as haplogroups; and the accumulation of mutations during development or in post-mitotic tissues [10].
Pathogenic mtDNA mutations have already been linked to T2DM and MS. The mtDNA tRNALeu(UUR) nt 3243A > G gene mutation [11] is a common cause of T2DM, accounting for between 0.5% and 2.8% of all cases throughout Eurasia [12]. The 3243A > G mutation is remarkably variable in its clinical manifestations due to shifts in the heteroplasmic percentage of mutant mtDNAs. When present at 10–30% mutant in blood, it is frequently associated with T2DM [13,14], at ~50–80% mutant with neuromuscular disease, and at ~90–100% mutant with lethal pediatric disease. This clinical variability correlates with the differential action of clusters of nDNA transcription factors, which create discrete gene expression profiles that correspond to the clinical phenotypes [15]. Maternally-transmitted duplications have also been linked to T2DM [16,17], and a mtDNA tRNAIle np 4291T > C mutation has been associated with a large MS pedigree involving hypertension, hypercholesterolemia, hypomagnesemia (renal ductal convoluted tubule defect), migraine, hearing loss, hypertrophic cardiomyopathy, mitochondrial myopathy, and reduced mitochondrial ATP production [18].
Ancient mtDNA haplogroups have been founded by functional mtDNA mutations, which were adaptive in the context in which they arose and therefore regionally enriched by selection. This created groups of related regional haplotypes, a haplogroup. However, these same variants can become maladaptive in another context. Because the mtDNA is exclusively maternally inherited, all the accumulated functional variants within a mtDNA remain linked to produce a cumulative physiological phenotype. Moreover, a number of functional mtDNA variants have arisen multiple independent times on different mtDNA lineages. Therefore, haplogroups generally provide the best way to represent the collective action of linked mtDNA variants.
Some functional mtDNA variants are very ancient and distinguish large groups of haplogroups, known as macro-haplogroups. All Asian mtDNAs fall into two macro-haplogroups, M and N, with N defined by two foundational variants, m.ND3 10398A (114T) and m.ATP6 8701A (59T) and macro-haplogroup M defined by m.ND3 10398G (114A) and m.ATP6 8701G (59A) [10,19,20]. Many additional variants have arisen in M and N macro-haplogroup mtDNAs to found the various Asian haplogroups. Other mtDNA variants such as the ND1 nt 3394T > C (Y30H) variant have arisen multiple independent times on different haplogroups [21].
2. Genetic analysis of East Asian mtDNA variants with T2DM and MS
The first report of Asian mtDNA haplogroups associated with T2DM and MS involved Japanese and Koreans subjects and revealed that haplogroup N9a was at decreased risk of T2DM while haplogroups D4/D5 and F were at increased risk [22]. In Japanese women haplogroups N9a, G1, and D5 have been reported as protective of MS [23] while in Japanese males N9a was found to be protective of myocardial infarctions [24]. Also in the Japanese, M8a was found to be at increased risk of T2DM and B4c with predisposition to OB [25]. By contrast, in the Chinese haplogroup N9a has been found to be at increased risk of T2DM [26] and diabetic retinopathy [27]. Among Han Chinese M9 was found to be at increased risk of T2DM [28] as was M8a [27]. In a Taiwanese sample, haplogroups B4a1a and E2b1 were found to be significantly associated with T2DM, while haplogroup D4 was protective [29]. Among the Uyghur of China, haplogroups H and D4 were reported to be at increased risk for T2DM [30].
The common ND3 nt 10398A 114T variant has been associated with T2DM in the Han Chinese [28], and in India [31]. The ND1 nt 3394C (30H) variant has been found to be at increased T2DM risk in the Han Chinese [28], and the mtDNA control region hypervariable nt 16189C variant has been associated with T2DM in multiple Eurasian populations [28,32].
2.1. Population and pedigree samples
To assess the role of mtDNA variation in the development of T2DM and MS in Asians, we have conducted a 15-year study on the mtDNA variation in Taiwan. Taiwan was chosen because repeated migrations to Taiwan have resulted in the Taiwanese encompassing virtually all of the major Asian mtDNA lineages, presumably dispersed across the array of Asian nDNA genetic alleles. While originally collected for different clinical reasons, most of the Taiwanese subjects were evaluated for the following continuous clinical and metabolic traits: Before meal glucose (ante cibum, AC, mg/dL), after meal glucose (post cibum, PC mg/dL), Body Mass Index (BMI, kg/m2), Waist Circumference (Waist, cm), Systolic Blood Pressure (SBP, mm Hg), Diastolic Blood Pressure (DBP, mm Hg), High Density Lipoprotein (HDL, mg/dL), Low Density Lipoprotein (LDL, mg/dL), Triglycerides (TG, mg/dL), and Total Cholesterol (TCHO, mg/dL).
2.2. A T2DM pedigree with the tRNALeu(UUR) nt 3243G mutation on the N9a background
We first analyzed the mtDNA variation of 27 Taipei T2DM pedigrees, 19 of which were suggestive of maternally inheritance. These 19 pedigrees were screened for the tRNALeu(UUR) nt 3243A > G mutation and one pedigree was found to harbor this mutation (Fig. 1). In this pedigree, individual II5 had very high AC levels. Analyzing the blood cell DNA from individual II3, revealed the 3243G mutation at a heteroplasmy level of ~88%, which is surprisingly high given her relatively mild phenotype. However, on sequencing her mtDNA, we discovered that her mtDNA lineage was Asian haplogroup N9a3, which has been reported as protective of T2DM [22]. Her mtDNA also harbored the non-T2DM associated 16189T variant [28,32]. Hence, the N9a3 & 16189T mtDNA background may have ameliorated the toxicity of the high heteroplasmic 3243G mutation.
Fig. 1.
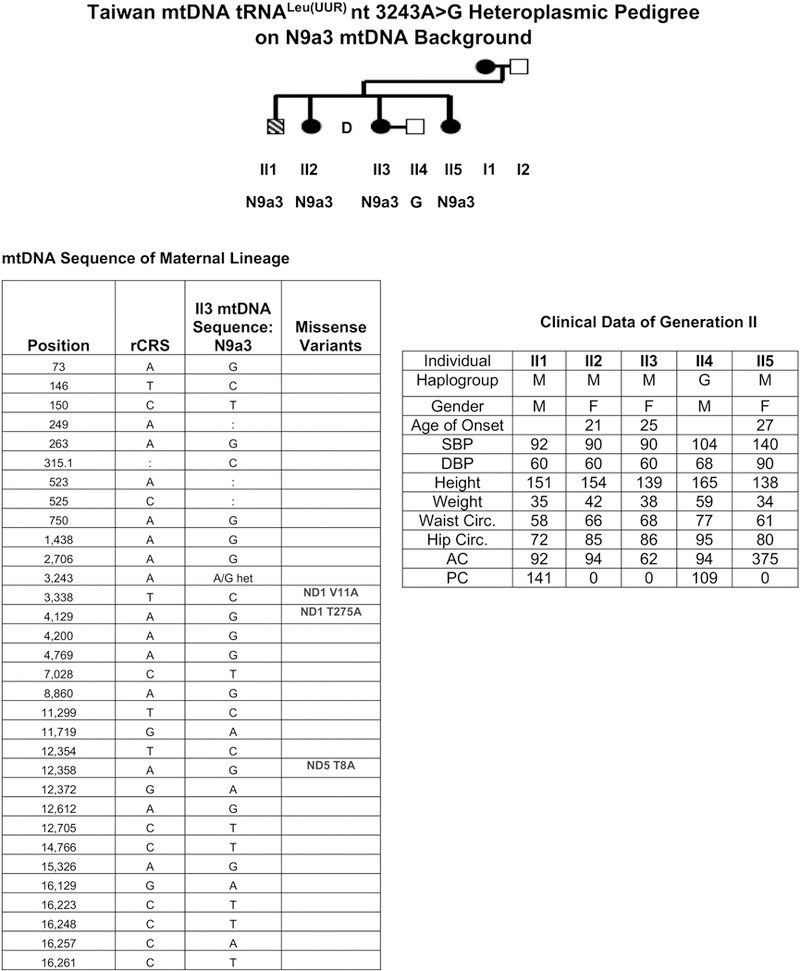
Taiwanese T2DM pedigree #14 harboring the heteroplasmic tRNALeu(UUR) nt 3243G on the haplogroup N9a3 mtDNA background. This N9a3 mtDNA sequence encompasses three complex I missense variants: ND1 nt 3338C (V11A), ND1 nt 4129G (T275A), and ND5 nt 12,358 A (T8A). The clinical characteristics of the generation II show individual II5 as having T2DM.
Based on the sequences of mtDNA control region hypervariable segment I (HVS1) the remaining 18 putative maternal pedigrees were found to have an assortment of mtDNA haplogroups: A (n = 1), B4 (3), D or G (3), F (1), F1 (4), F4 (1), G (1), M9 (1), and Y (1). The pedigrees and HSV1 sequences are provided in Fig. S1.
2.3. Asian mtDNA haplogroup versus T2DM and MS parameter associations
The potential role of N9a3 in modulating the 3243G mutation pathogenicity prompted us to compare the frequency of Taiwanese mtDNA haplogroups with the phenotypic parameters associated with T2DM and MS using a case-control study design. Accordingly, we collected three Taiwanese cohorts and characterized their mtDNA haplogroup status.
Cohort 1 encompassed 498 Taiwanese collected from Taipei, Taiwan with a mean age of 52 years. These patients had originally been ascertained for T2DM, HTN, and OB. Cohort 2 encompassed 501 Taiwanese from non-urban Taiwan with a mean age of 57 years and assessed for T2DM. Cohort 3 encompassed 1002 Taiwanese collected from Taipei with a mean age of 44 years and evaluated for OB.
We then evaluated the clinical phenotypes of the three Taiwanese case-control cohorts. Summary statistics of the cohorts are presented stratified by cohort and gender for T2DM (Table S1), HTN (Table S2), and OB (Table S3). In addition to the rural cohort 2 being overall significantly older (mean2 = 57 years old) than the two Taipei cohorts (mean1 = 52, mean3 = 44 years old), age differences were observed between T2DM and controls within each cohort. Among the T2DM patients, in cohorts 1 and 2 the patients were older than controls, whereas in cohort 3 the T2DM patients were younger than controls (Table S1). Among the HTN patients, the patients were older than the controls (Table S2), while among the OB patients, the controls were older than the patients (Table S3).
Among the T2DM patients (Table S1) defined by elevated AC in the three cohorts, PC was also elevated where tested in cohort 1. Cohort 1 T2DM patients also had slightly lower BMI that controls, cohort 3 patients had higher BMIs, and cohort 2 patient and control BMIs were similar. SBP was generally higher in T2DM patients than controls and DBP was elevated in several contexts. LDL levels were elevated in cohort 1 T2DM patients but reduced in cohort 2 patients. TG was elevated in the patients of all three cohorts (Table S1).
Among the HTN patients (Table S2) defined by elevated SBP and DBP in all three cohorts, PC was elevated in cohort 1 and LDL was elevated in cohort 3.
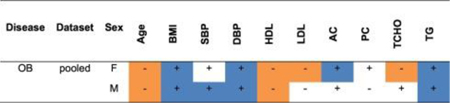
Among the OB patients (Table S3) defined by elevated BMI in the three cohorts, AC was elevated in cohort 3 but not cohorts 1 and 2, TCHO was reduced in cohort 1 but not cohorts 2 & 3, and TG was elevated in cohort 3 but not cohort 1 & 2 (Table S3). Hence, there was considerable phenotypic heterogeneity among the three cohorts, even though they were from the same island population.
The female:male ratio for T2DM, HTN, OB, American Heart Association definition of Metabolic Syndrome (AHA-MS), and International Diabetes Foundation definition of Metabolic Syndrome (IDF-MS) subjects was roughly 1 in T2DM, but differed significantly in OB, AHA-MS, and IDF-MS (> 1), and in HTN (< 1) (Table S4). Additionally, age distributions between the three Taiwan cohorts, stratified by gender, were different (Table S5), with cohort 3 being the youngest and cohort 2 the oldest. The female:male ratio was roughly 1 in all three cohorts (Table S6). Thus, the most likely difference between cohorts 1 and 3 versus 2 was environment (urban versus rural) and between cohorts 1 and 2 versus 3 was age.
2.4. Haplogroups and nucleotide variants in the Taiwanese cohorts
The mtDNA haplogroups were determined for cohorts 1 and 2 using a combination of HVSI sequencing and screening for the informative sites shown in Fig. 2. Cohort 3 was genotyped for D5, F4, and N9a haplogroups.
Fig. 2.
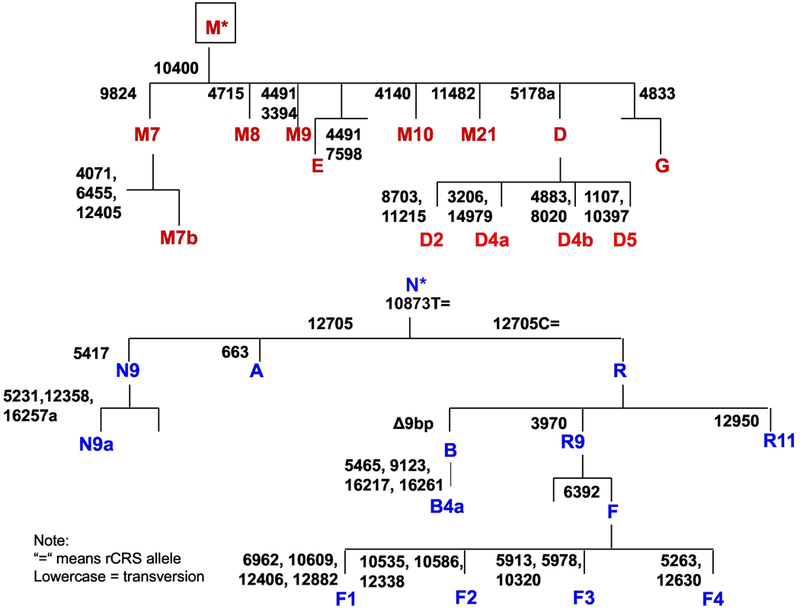
Asian mtDNA variants that define the haplogroups of macro-haplogroups M and N.
Table S7 presents the detailed haplogroup breakdown of the three cohorts (32 identified haplogroups), and Fig. 3 combines these into the 15 major haplogroups (Table S8). Haplogroup B was found to be the most prevalent Asian mtDNA lineage in both urban and rural Taiwanese. But there were also marked differences between urban cohort 1 and rural cohort 2 for the F, M* and M9 haplogroups (Figs. 3, S2 and S3).
Fig. 3.
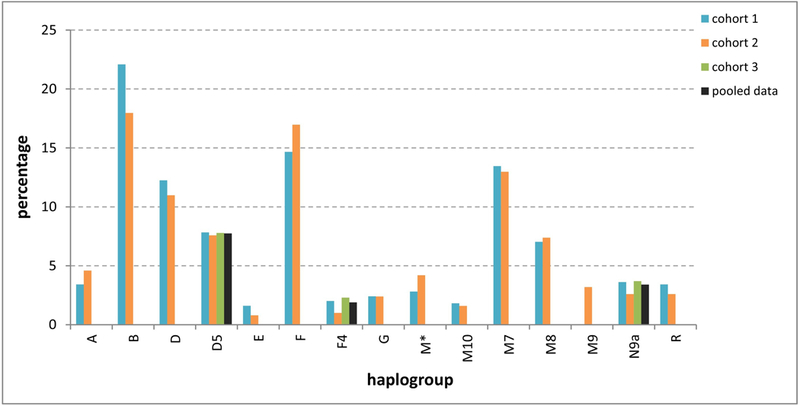
Distribution of 15 mitochondrial haplogroups in Taiwan (in cohort 3 only haplogroups D5, F4, and N9a were typed).
Initially, the mtDNA haplogroups were determined for cohort 1 and cohort 2 (Fig. 3) in preparation for correlating mtDNA variants with the risk of T2DM and assorted MS clinical manifestations. However, about this time a report was published that haplogroup N9a was protective of T2DM in Japanese and Koreans, that haplogroups F and D5 were at increased risk of T2DM in Japanese, and that D4/D5 was at increased risk of T2DM in Koreans. Since the 3243G pedigree was associated with N9a3, this report shifted our focus to determining if the Japanese and Korean N9a, D, and F haplogroup associations with T2DM also held for the Taiwanese. Accordingly, we recruited our third Taiwanese cohort from Taipei and typed these samples for the mtDNA haplogroups D, F, and N9a (Fig. 2, confirmation Table S9). We then analyzed our three Taiwanese cohorts for T2DM, HTN, and OB and compared the association of these phenotypes with the incidence of mtDNA haplogroups D5, F4, and N9a.
The combined data for cohorts 1, 2, and 3 was stratified by ascertainment phenotype and gender resulting in: Table 1 for T2DM, Table 2 for HTN, and Table 3 for OB. Stratification of the complete dataset for T2DM based on elevated AC and PC levels revealed associations with elevated SBP and TG, but reduced LDL in both genders (Table 1). Stratification of the dataset for HTN based on elevation of SBP and DBP gave associations with elevated AC and PD and TG in both genders (Table 2). Stratification of the dataset for OB based on elevated BMI gave associations with elevated DBP and TG and reduced HDL (Table 3).
Table 1.
Descriptive statistics for continuous Metabolic Syndrome traits grouped by gender (pooled data). Traits were compared between Type-2 Diabetes Mellitus (T2DM) patients and control individuals by using the Student’s t-test, assuming unequal variances, α = 0.05.
| Gender | Trait | Controls | T2DM | t-Score | p-Value | ||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean | Std Dev | n | Mean | Std Dev | ||||
| F | Age | 914 | 48.86 | 13.53 | 218 | 53.85 | 15.34 | 4.40 | <0.0001 |
| BMI | 905 | 28.17 | 7.26 | 216 | 29.03 | 7.36 | 1.55 | 0.1200 | |
| SBP | 841 | 127.15 | 18.32 | 182 | 135.75 | 20.10 | 5.31 | <0.0001 | |
| DBP | 841 | 80.37 | 10.79 | 182 | 80.22 | 10.36 | −0.17 | 0.8647 | |
| HDL | 750 | 53.64 | 12.08 | 162 | 47.93 | 12.12 | −5.43 | <0.0001 | |
| LDL | 622 | 125.93 | 35.47 | 151 | 117.58 | 36.29 | −2.54 | 0.0115 | |
| AC | 916 | 93.22 | 10.27 | 217 | 167.67 | 56.07 | 19.48 | <0.0001 | |
| PC | 81 | 125.12 | 29.21 | 53 | 235.35 | 75.32 | 10.16 | <0.0001 | |
| TCHO | 912 | 201.29 | 37.89 | 210 | 203.66 | 43.48 | 0.72 | 0.4680 | |
| TG | 914 | 119.33 | 73.68 | 216 | 180.05 | 125.83 | 6.82 | <0.0001 | |
| M | Age | 669 | 47.20 | 15.38 | 178 | 54.35 | 13.89 | 5.95 | <0.0001 |
| BMI | 666 | 28.20 | 7.45 | 177 | 27.51 | 7.88 | −1.05 | 0.2938 | |
| SBP | 632 | 130.99 | 17.67 | 161 | 138.06 | 19.96 | 4.12 | <0.0001 | |
| DBP | 632 | 84.62 | 11.77 | 161 | 82.59 | 11.36 | −2.00 | 0.0456 | |
| HDL | 578 | 44.71 | 10.43 | 131 | 41.77 | 22.49 | −1.45 | 0.1468 | |
| LDL | 501 | 125.69 | 33.85 | 126 | 112.16 | 35.99 | −3.81 | 0.0002 | |
| AC | 671 | 92.50 | 11.06 | 178 | 166.89 | 57.64 | 17.13 | <0.0001 | |
| PC | 77 | 118.71 | 28.77 | 42 | 228.50 | 77.65 | 8.83 | <0.0001 | |
| TCHO | 670 | 194.78 | 35.14 | 169 | 187.68 | 44.47 | −1.92 | 0.0550 | |
| TG | 669 | 148.92 | 109.84 | 177 | 211.52 | 297.35 | 2.75 | 0.0065 | |
Summary of statistically significant mean differences between T2DM patients and controls for continuous traits (blue or orange cells).
+ sign: mean higher in patients;
– sign: mean lower in patients.
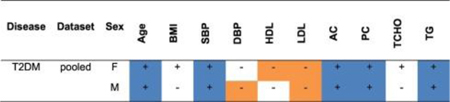
Table 2.
Descriptive statistics for continuous Metabolic Syndrome traits grouped by gender (pooled data). Traits were compared between hypertensive (HTN) patients and control individuals by using the Student’s t-test, assuming unequal variances, α = 0.05.
| Gender | Trait | Controls | HTN | t-Score | p-Value | ||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean | Std Dev | n | Mean | Std Dev | ||||
| F | Age | 741 | 49.08 | 13.58 | 297 | 54.27 | 13.49 | 5.59 | <0.0001 |
| BMI | 739 | 27.50 | 7.02 | 298 | 28.16 | 6.16 | 1.49 | 0.1347 | |
| SBP | 741 | 119.91 | 11.25 | 282 | 151.71 | 15.35 | 31.69 | <0.0001 | |
| DBP | 741 | 76.30 | 7.82 | 282 | 90.96 | 9.99 | 22.17 | <0.0001 | |
| HDL | 595 | 53.46 | 12.57 | 240 | 51.12 | 11.17 | −2.62 | 0.0088 | |
| LDL | 502 | 125.22 | 32.93 | 195 | 132.44 | 39.38 | 2.27 | 0.0239 | |
| AC | 740 | 103.70 | 36.22 | 298 | 112.78 | 44.25 | 3.14 | 0.0018 | |
| PC | 46 | 141.32 | 58.68 | 19 | 238.42 | 95.95 | 4.10 | 0.0004 | |
| TCHO | 736 | 201.84 | 36.45 | 293 | 208.85 | 41.86 | 2.50 | 0.0124 | |
| TG | 740 | 124.48 | 89.62 | 297 | 141.47 | 84.50 | 2.87 | 0.0042 | |
| M | Age | 482 | 48.72 | 15.18 | 323 | 49.52 | 15.32 | 0.73 | 0.4649 |
| BMI | 482 | 26.53 | 6.22 | 324 | 29.29 | 8.01 | 5.22 | <0.0001 | |
| SBP | 482 | 121.74 | 10.55 | 312 | 149.04 | 15.37 | 27.45 | <0.0001 | |
| DBP | 482 | 77.96 | 7.48 | 312 | 93.86 | 10.42 | 23.32 | <0.0001 | |
| HDL | 416 | 44.78 | 15.61 | 263 | 43.80 | 9.58 | −1.01 | 0.3098 | |
| LDL | 370 | 122.41 | 33.22 | 227 | 127.73 | 34.91 | 1.84 | 0.0663 | |
| AC | 482 | 104.34 | 36.80 | 325 | 111.42 | 47.02 | 2.28 | 0.0228 | |
| PC | 72 | 145.55 | 70.60 | 19 | 204.63 | 96.53 | 2.49 | 0.0200 | |
| TCHO | 477 | 192.75 | 36.57 | 322 | 196.52 | 37.51 | 1.40 | 0.1598 | |
| TG | 481 | 152.16 | 183.241 | 324 | 175.50 | 154.408 | 1.94 | 0.0516 | |
Summary of statistically significant mean differences between HTN patients and controls for continuous traits (blue or orange cells).
+ sign: mean higher in patients
— sign: mean lower in patients.
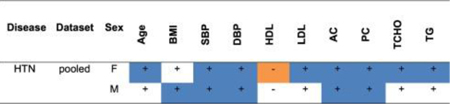
Table 3.
Descriptive statistics for continuous Metabolic Syndrome traits grouped by gender (pooled data). Traits were compared between and Obese (OB) and control individuals by using the Student’s t-test, assuming unequal variances, α = 0.05.
| Gender | Trait | Controls | OB | t-Score | p-Value | ||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean | Std Dev | n | Mean | Std Dev | ||||
| F | Age | 760 | 54.80 | 11.14 | 359 | 39.00 | 13.48 | −19.30 | 0.0000 |
| BMI | 761 | 24.27 | 2.88 | 360 | 36.93 | 6.21 | 36.83 | <0.0001 | |
| SBP | 726 | 128.14 | 19.91 | 295 | 130.07 | 16.24 | 1.60 | 0.1094 | |
| DBP | 726 | 79.50 | 10.32 | 295 | 82.49 | 11.35 | 3.91 | 0.0001 | |
| HDL | 670 | 54.06 | 12.50 | 237 | 48.48 | 10.64 | −6.61 | <0.0001 | |
| LDL | 615 | 126.48 | 35.69 | 153 | 114.89 | 34.91 | −3.65 | 0.0003 | |
| AC | 760 | 105.76 | 38.84 | 360 | 111.06 | 40.06 | 2.08 | 0.0372 | |
| PC | 72 | 163.38 | 74.15 | 62 | 174.91 | 76.58 | 0.88 | 0.3795 | |
| TCHO | 752 | 205.39 | 39.43 | 358 | 193.86 | 36.66 | −4.77 | <0.0001 | |
| TG | 759 | 119.04 | 79.90 | 359 | 155.91 | 102.55 | 6.00 | <0.0001 | |
| M | Age | 626 | 52.12 | 14.07 | 215 | 38.34 | 14.21 | −12.29 | 0.0000 |
| BMI | 628 | 24.57 | 2.81 | 216 | 38.19 | 7.80 | 25.09 | <0.0001 | |
| SBP | 604 | 130.73 | 18.06 | 188 | 137.86 | 18.35 | 4.66 | <0.0001 | |
| DBP | 604 | 82.82 | 10.83 | 188 | 88.42 | 13.13 | 5.31 | <0.0001 | |
| HDL | 548 | 45.30 | 14.43 | 158 | 40.08 | 8.54 | −5.69 | <0.0001 | |
| LDL | 514 | 123.96 | 33.88 | 110 | 118.12 | 37.20 | −1.51 | 0.1312 | |
| AC | 627 | 107.06 | 38.29 | 216 | 111.15 | 49.39 | 1.10 | 0.2686 | |
| PC | 95 | 158.54 | 79.21 | 24 | 153.16 | 45.32 | −0.43 | 0.6637 | |
| TCHO | 618 | 192.82 | 36.87 | 215 | 194.36 | 37.66 | 0.51 | 0.6041 | |
| TG | 624 | 141.16 | 102.20 | 216 | 223.98 | 277.26 | 4.29 | <0.0001 | |
Summary of statistically significant mean differences between OB patients and controls for continuous traits (blue or orange cells).
+ sign: mean higher in patients
– sign: mean lower in patients.
The mtDNA haplogroups of patient and control samples were parsed into 5 haplogroup categories [D5, F4, N9a, macro-M (D, E, G, M*, M7, M8, M9, M10), and macro-N (A, B, F, R)] and haplogroups D5, F4, and N9a were correlated with the three major phenotypes [T2DM (Table 4), HTN (Table 5) and OB (Table 6)] in the context of macro-M and macro-N.
Table 4.
Haplogroup distribution in controls and patients with T2DM (cohorts 1, 2, and 3).
| Parsed haplogroup | Haplogroup | Controls (total %) | T2DM (total %) | Total (total %) |
|---|---|---|---|---|
| D5 | D5 | 119 (10.76) | 32 (2.89) | 151 (13.65) |
| F4 | F4 | 33 (2.98) | 5 (0.45) | 38 (3.44) |
| N9a | N9a | 58 (5.24) | 10 (0.90) | 68 (6.15) |
| Macro-M | D | 73 (6.60) | 41 (3.71) | 114 (10.31) |
| E | 8 (0.72) | 4 (0.36) | 12 (1.08) | |
| G | 12 (1.08) | 12 (1.08) | 24 (2.17) | |
| M* | 23 (2.08) | 12 (1.08) | 35 (3.16) | |
| M10 | 13 (1.18) | 4 (0.36) | 17 (1.54) | |
| M7 | 81 (7.32) | 51 (4.61) | 132 (11.93) | |
| M8 | 46 (4.16) | 26 (2.35) | 72 (6.51) | |
| M9 | 12 (1.08) | 4 (0.36) | 16 (1.45) | |
| Macro-N | A | 28 (2.53) | 12 (1.08) | 40 (3.62) |
| B | 125 (11.30) | 74 (6.69) | 199 (17.99) | |
| F | 116 (10.49) | 42 (3.80) | 158 (14.29) | |
| R | 19 (1.72) | 11 (0.99) | 30 (2.71) | |
| Total | 766 (69.26) | 340 (30.74) | 1106 | |
Table 5.
Haplogroup distribution in controls and patients with HTN (cohorts 1, 2, and 3).
| Parsed haplogroup | Haplogroup | Controls (total %) | HTN (total %) | Total (total %) |
|---|---|---|---|---|
| D5 | D5 | 94 (9.68) | 47 (4.84) | 141 (14.52) |
| F4 | F4 | 14 (1.44) | 18 (1.85) | 32 (3.30) |
| N9a | N9a | 33 (3.40) | 27 (2.78) | 60 (6.18) |
| Macro-M | D | 56 (5.77) | 43 (4.43) | 99 (10.20) |
| E | 8 (0.82) | 3 (0.31) | 11 (1.13) | |
| G | 11 (1.13) | 7 (0.72) | 18 (1.85) | |
| M* | 25 (2.57) | 8 (0.82) | 33 (3.40) | |
| M10 | 9 (0.93) | 6 (0.62) | 15 (1.54) | |
| M7 | 72 (7.42) | 43 (4.43) | 115 (11.84) | |
| M8 | 42 (4.33) | 20 (2.06) | 62 (6.39) | |
| M9 | 11 (1.13) | 5 (0.51) | 16 (1.65) | |
| Macro-N | A | 25 (2.57) | 12 (1.24) | 37 (3.81) |
| B | 115 (11.84) | 55 (5.66) | 170 (17.51) | |
| F | 94 (9.68) | 44 (4.53) | 138 (14.21) | |
| R | 18 (1.85) | 6 (0.62) | 24 (2.47) | |
| Total | 627 (64.57) | 344 (35.43) | 971 | |
Table 6.
Haplogroup distribution in controls and patients with OB (cohorts 1, 2, and 3).
| Parsed haplogroup | Haplogroup | Controls (total %) | OB patients (total %) | Total (total %) |
|---|---|---|---|---|
| D5 | D5 | 106 (9.74) | 42 (3.86) | 148 (13.60) |
| F4 | F4 | 20 (1.84) | 18 (1.65) | 38 (3.49) |
| N9a | N9a | 48 (4.41) | 19 (1.75) | 67 (6.16) |
| Macro-M | D | 89 (8.18) | 26 (2.39) | 115 (10.57) |
| E | 11 (1.01) | 0 (0.00) | 11 (1.01) | |
| G | 19 (1.75) | 4 (0.37) | 23 (2.11) | |
| M* | 29 (2.67) | 6 (0.55) | 35 (3.22) | |
| M10 | 11 (1.01) | 6 (0.55) | 17 (1.56) | |
| M7 | 98 (9.01) | 32 (2.94) | 130 (11.95) | |
| M8 | 49 (4.50) | 22 (2.02) | 71 (6.53) | |
| M9 | 11 (1.01) | 5 (0.46) | 16 (1.47) | |
| Macro-N | A | 32 (2.94) | 8 (0.74) | 40 (3.68) |
| B | 158 (14.52) | 37 (3.40) | 195 (17.92) | |
| F | 121 (11.12) | 33 (3.03) | 154 (14.15) | |
| R | 22 (2.02) | 6 (0.55) | 28 (2.57) | |
| Total | 824 (75.74) | 264 (24.26) | 1088 | |
2.5. Modeling of haplogroup versus T2DM and MS phenotypes
Multivariate logistic regression with adjustment for age and gender showed that mitochondrial haplogroups had significant effects on T2DM, HTN, and OB and related MS clinical phenotypes (Tables 7–10).
Table 7.
MtDNA haplogroups are associated with Type 2 Diabetes Mellitus (T2DM), Hypertension (HTN), and Obesity (OB). Five mitochondrial haplogroups (HG), age, and gender were used as effects.
| Variable | Effects | Model p- value |
R2 | AICc | Lack of Fit p- value |
n | Effect | Effect p- value# |
FDR |
|---|---|---|---|---|---|---|---|---|---|
| T2DM | Age, HG, gender |
<0.0001 | 0.0402 | 1317.24 | 0.5473 | 1100 | Age | <0.0001 | 0.00001 |
| HG | <0.0001 | 0.00012 | |||||||
| Gender | 0.4912 | 0.49121 | |||||||
| HTN | Age, HG, gender |
<0.0001 | 0.0359 | 1229.28 | 0.0278 | 969 | Age | <0.0001 | 0.00000 |
| HG | 0.0026 | 0.00394 | |||||||
| Gender | 0.0075 | 0.00753 | |||||||
| OB | Age, HG, gender |
<0.0001 | 0.1335 | 1057.55 | 0.4180 | 1085 | Age | <0.0001 | 0.00000 |
| HG | 0.0562 | 0.08437 | |||||||
| Gender | 0.1444 | 0.14439 | |||||||
R2: The proportion of the total uncertainty that is attributed to the model fit. Because certainty in the predicted probabilities is rare for logistic models, R2 tends to be small.
AICc: Corrected Akaike Information Criterion that assesses model fit based on –2LogLikelihood.
Lack of Fit: Chi-square statistics on the negative log-likelihood error due to lack of fit, error in a saturated model (pure error), and the total error in the fitted model. High p-values denote that the lack of fit Chi-square is not significant and support the conclusion that there is little to be gained by introducing additional variables.
FDR: Benjamini-Hochberg false discovery rate.
Age levels: advanced ≥60, 30 ≤medium < 60, young < 30.
HG levels: D5, F4, N9a, macro-M (D, E, G, M*, M7, M8, M9, M10), and macro-N (A, B, F, R).
Likelihood-Ratio test p-value.
Table 10.
Significant odds ratios for the different levels of haplogroup, age, and gender levels in the linear model analysis of MS clinical phenotypes (high versus low values).
| Variable | Effect | Level 1 | Level 2 | Odds ratio | p-Valuea | FDRb | Lower 95% CIa | Upper 95% CIa |
|---|---|---|---|---|---|---|---|---|
| AC | Age | Advanced | Medium | 1.55 | 0.0037 | 0.01690 | 1.15 | 2.08 |
| Advanced | Young | 4.21 | 0.0012 | 0.01560 | 1.76 | 10.04 | ||
| Medium | Young | 2.72 | 0.0229 | 0.04962 | 1.15 | 6.44 | ||
| HG | D5 | Macro-M | 0.56 | 0.0176 | 0.04576 | 0.34 | 0.90 | |
| N9a | Macro-M | 0.28 | 0.0039 | 0.01690 | 0.12 | 0.66 | ||
| N9a | Macro-N | 0.34 | 0.0157 | 0.04576 | 0.14 | 0.82 | ||
| BMI | Age | Medium | Advanced | 2.37 | <0.0001 | 0.00043 | 1.64 | 3.41 |
| Young | Advanced | 36.07 | <0.0001 | 0.00043 | 17.92 | 72.59 | ||
| Young | Medium | 15.23 | <0.0001 | 0.00043 | 7.96 | 29.14 | ||
| HG | F4 | D5 | 2.34 | 0.0343 | 0.07432 | 1.06 | 5.13 | |
| F4 | N9a | 2.52 | 0.0445 | 0.08264 | 1.02 | 6.19 | ||
| F4 | Macro-M | 2.55 | 0.0109 | 0.02834 | 1.24 | 5.25 | ||
| F4 | Macro-N | 3.10 | 0.0023 | 0.00748 | 1.50 | 6.41 | ||
| DBP | Age | Medium | Advanced | 1.39 | 0.0334 | 0.00520 | 1.03 | 1.88 |
| HG | F4 | Macro-M | 2.48 | 0.0185 | 0.04008 | 1.16 | 5.28 | |
| F4 | Macro-N | 2.78 | 0.0083 | 0.02158 | 1.30 | 5.93 | ||
| D5 | Macro-M | 1.79 | 0.0050 | 0.01625 | 1.19 | 2.70 | ||
| D5 | Macro-N | 2.01 | 0.0009 | 0.00585 | 1.33 | 3.03 | ||
| N9a | Macro-N | 1.87 | 0.0364 | 0.05915 | 1.04 | 3.37 | ||
| Gender | Male | Female | 1.65 | 0.0004 | 0.00520 | 1.25 | 2.18 | |
| SBP | Age | Advanced | Medium | 2.72 | <0.0001 | 0.00130 | 2.05 | 3.62 |
| HG | F4 | Macro-M | 2.91 | 0.0088 | 0.03987 | 1.31 | 6.49 | |
| F4 | Macro-N | 2.90 | 0.0092 | 0.03987 | 1.30 | 6.46 | ||
| TCHO | Age | Advanced | Young | 3.86 | <0.0001 | 0.00043 | 2.06 | 7.23 |
| Medium | Young | 3.90 | <0.0001 | 0.00043 | 2.11 | 7.18 | ||
| HG | D5 | Macro-M | 1.49 | 0.0435 | 0.12766 | 1.01 | 2.19 | |
| D5 | Macro-N | 1.47 | 0.0491 | 0.12766 | 1.00 | 2.17 | ||
| Gender | Female | Male | 1.69 | <0.0001 | 0.00043 | 1.32 | 2.17 | |
Age levels: advanced ≥ 60, 30 ≤ medium < 60, young < 30.
HG levels: D5, F4, N9a, macro-M (D, E, G, M*, M7, M8, M9, M10), and macro-N (A, B, F, R).
Wald test.
Benjamini-Hochberg false discovery rate.
Subjects with mitochondrial haplogroups D5, F4, and N9a had a significantly reduced risk for T2DM (OR: 0.30–0.60, CI: 0.11–0.96) (Table 8). Even after adjustment for TCHO, SBP, DBP, and BMI, the regression analysis demonstrated that haplogroups D5, F4, and N9a were protective factors against T2DM for East Asian subjects (OR: 0.21–0.47, CI: 0.06–0.94) (Tables S10 and S11). Haplogroups D5 and N9a were also protective factors for increased levels of AC (OR: 0.28–0.56, CI: 0.12–0.90) (Table 10).
Table 8.
Significant odds ratios of haplogroup, age, and gender levels in the linear model analysis of T2DM, HTN, and OB (patients versus controls).
| Variable | Effect | Level 1 | Level 2 | Odds ratio | p-Valuea | FDRb | Lower 95% CIa | Upper 95% CIa |
|---|---|---|---|---|---|---|---|---|
| T2DM | Age | Advanced | Medium | 1.72 | 0.0001 | 0.00130 | 1.31 | 2.26 |
| Advanced | Young | 3.74 | 0.0002 | 0.00130 | 1.85 | 7.57 | ||
| Medium | Young | 2.18 | 0.0284 | 0.04615 | 1.08 | 4.37 | ||
| HG | D5 | Macro-M | 0.49 | 0.0018 | 0.00585 | 0.32 | 0.77 | |
| D5 | Macro-N | 0.60 | 0.0279 | 0.04615 | 0.39 | 0.95 | ||
| F4 | Macro-M | 0.30 | 0.0138 | 0.02990 | 0.11 | 0.78 | ||
| F4 | Macro-N | 0.36 | 0.0410 | 0.05922 | 0.14 | 0.96 | ||
| N9a | Macro-M | 0.32 | 0.0018 | 0.00585 | 0.16 | 0.66 | ||
| N9a | Macro-N | 0.40 | 0.0111 | 0.02886 | 0.20 | 0.81 | ||
| HTN | Age | Advanced | Medium | 2.11 | <0.0001 | 0.00130 | 1.59 | 2.80 |
| Advanced | Young | 2.27 | 0.0302 | 0.05609 | 1.08 | 4.78 | ||
| HG | F4 | D5 | 3.03 | 0.0063 | 0.01976 | 1.37 | 6.70 | |
| F4 | Macro-M | 2.83 | 0.0064 | 0.01976 | 1.34 | 5.98 | ||
| F4 | Macro-N | 3.71 | 0.0006 | 0.00390 | 1.75 | 7.86 | ||
| N9a | Macro-N | 2.08 | 0.0117 | 0.02535 | 1.18 | 3.68 | ||
| Gender | Male | Female | 1.45 | 0.0076 | 0.01976 | 1.10 | 1.90 | |
| OB | Age | Medium | Advanced | 2.39 | <0.0001 | 0.00043 | 1.66 | 3.44 |
| Young | Advanced | 36.12 | <0.0001 | 0.00043 | 17.94 | 72.71 | ||
| Young | Medium | 15.12 | <0.0001 | 0.00043 | 7.90 | 28.92 | ||
| HG | F4 | D5 | 2.33 | 0.0346 | 0.07497 | 1.06 | 5.12 | |
| F4 | N9a | 2.51 | 0.0448 | 0.08320 | 1.02 | 6.18 | ||
| F4 | Macro-M | 2.51 | 0.0124 | 0.03224 | 1.22 | 5.16 | ||
| F4 | Macro-N | 3.09 | 0.0024 | 0.00780 | 1.49 | 6.40 | ||
Age levels: advanced ≥ 60, 30 ≤ medium < 60, young < 30.
HG levels: D5, F4, N9a, macro-M (D, E, G, M*, M7, M8, M9, M10), and macro-N (A, B, F, R).
Wald test.
Benjamini-Hochberg false discovery rate.
Analogous modeling with adjustment for age and gender showed that subjects in the mitochondrial haplogroups F4 and N9a were at increased risk for HTN (OR: 2.08–3.71, CI: 1.18–7.86) (Table 8). Similarly to the T2DM case, even after adjustment for AC, TCHO, and BMI, the analysis showed that both F4 and N9a were risk factors for HTN (OR: 2.18–3.86, CI: 1.23–8.27) (Tables S10 and S11). Also, haplogroup F4 was a significant risk factor for increased levels of DBP and SBP (OR: 2.48–2.91, CI: 1.16–6.49) (Table 10) and haplogroup D5 was a risk factor for DBP (OR: 2.01, CI:1.19–3.03) (Table 10).
Similar analysis with adjustment for age and gender revealed that haplogroup F4 was a risk factor for OB (OR: 2.33–3.09, CI: 1.02–6.40) (Table 8). After adjustment for additional MS phenotypes such as TCHO, SBP, DBP and AC, the analysis confirmed that F4 was a risk factor for OB (OR: 2.83, CI: 1.22–6.55) (Tables S10 and S11). Hap- logroup F4 was also a significant risk factor for the OB-related trait BMI (OR: 2.34–3.10, CI: 1.02–6.41) (Table 10).
To determine if age was a major determining variable, we stratified the patients and controls by age and compared the haplogroup associations with those of the aggregate populations. Age stratification lowered the sample size and thus significance without changing the overall haplogroup associations. Hence, we chose to report the results from the aggregate data.
2.6. Analysis of D5, F4, and N9a nucleotide variation and T2DM and MS phenotypes
The major mtDNA variants associated with haplogroups D5, F4, and N9a are: for D5 m.ND2 nt 5301A > G, I278V; for F4 m.ND5 nt 5263C > T, synonymous; and for N9a m.ND5 nt 12358A > G, T8A (Table S9). The distribution of these variants across the Asian mtDNA haplogroups is presented in Fig. S4. While variant m.ND2 nt 5301A > G, I278V is confined to haplogroup D mtDNAs and m.ND5 nt 5263C > T to F mtDNAs, variant m.ND5 nt 12358A > G, T8A is dispersed throughout the Asian mtDNA haplogroups (Fig. S4, Table S12).
Consistent with the association of the m.ND5 nt 5263C > T with haplogroup F mtDNAs, we found this variant was significantly associated with HTN (OR: 2.30–2.53, CI: 1.02–5.32). However, none of these single nucleotide variants associated significantly with T2DM or OB (Tables S13 and S14).
2.7. Conclusions from case-control studies
Our modeling studies on the relationship between mtDNA haplogroups D5, F4, and N9a for T2DM, HTN and OB phenotypes have shown that all three haplogroups are at reduced risk for T2DM, and haplogroups D5 and N9a are protective factors for elevated levels of AC. Haplogroup F4 imparts increased risk for HTN with high levels of DBP and SBP and OB through increased BMI. We also found that using individual variants in association studies, even when the variant is enriched in a particular haplogroup, it is not as informative as using the mtDNA haplogroup, which encompasses all of the linked mtDNA functional variants.
The lack of correspondence between haplogroup associations and nucleotide variant associations is likely the result of two factors. The first is the total linkage disequilibrium of the mtDNA creating an integrated physiological unit. The second is that the same functional mtDNA variants have arisen and been selected multiple times on different mtDNA haplogroup backgrounds with differential effects.
3. Biochemical analysis of mtDNA haplogroups associated with Asian T2DM and MS phenotypes
The association of haplogroups D4/D5, F and N9a mtDNAs in Japanese and Koreans [22] was followed up by the generation of cybrids by the Korean investigators harboring haplogroups D5, F and N9a mtDNAs on the 143B(TK–) osteosarcoma nuclear background [33]. Biochemical analysis did not detect any significant mitochondrial physiological differences between the different haplogroup-baring cybrids. However, microarray analysis of these cybrids revealed that the D5 and F cybrids had similar gene expression profiles while N9a cybrids had an increased expression of OXPHOS genes relative to the D5 and F cybrids [33]. This suggests that cybrids harboring N9a mtDNA may have a higher mitochondrial function than D5 and F cybrids and this is protective for T2DM.
However, an independent Chinese study found that haplogroup N9a was at increased risk of T2DM [26]. This study was associated with a detailed analysis of 143B(TK–) cybrids harboring N9a versus D4, G3, and Y mtDNAs. This study confirmed that the N9a cybrids have higher levels of nDNA coded OXPHOS gene transcripts. However, physiological and molecular analysis concluded that this was the result of retrograding signaling in response to reduced N9a mitochondrial function. The N9a cybrids were found to have a 30% lower mtDNA copy number along with reduced mtDNA transcripts, complex I and IV specific activities, respiration rate, ATP levels, NAD + /NADH ratio and the long form of OpaI, the latter being associated with increased mitochondrial fragmentation and ROS production. These mitochondrial effects were associated with the activation of the signal transduction pathways for Wnt, ERK1/2, and p38-MAPK [26]. This study suggests that N9a has reduced mitochondrial function and thus is prone to T2DM.
Other studies of 143B(TK–) cybrids harboring Asian mtDNA haplogroups revealed that cybrids that harbored mtDNAs with the linked macro-haplogroup N ND3 nt 10398A and ATP6 nt 8701A variants have reduced mitochondrial pH, increased mitochondrial Ca++, and increased cytosolic Ca++ after histamine treatment [34]. The macro-haplogroup 10398A variant has been associated with predisposition to T2DM [28,31] as well as Alzheimer Disease, Parkinson Disease, bipolar disorder, and cancer [34]. The low mitochondrial pH and inability to buffer excess cytosolic Ca++ implies that these haplogroup N markers impart reduced mitochondrial coupling and energetic efficiency. Since the 10398A variant defines macro-haplogroup N and thus divides Asian mtDNAs in half, it encompasses sufficient mtDNAs to permit associations in its own right. Cybrids harboring haplogroup D4a were found to have a high mitochondrial Ca++ retention, relative to cybrids with haplogroup G mtDNAs [35].
3.1. Identification of Asian mtDNAs from macro-haplogroup N and M mtDNAs & ND1 3394T > C variants
Taiwanese haplogroup B4a1a has been found to be at increased risk of T2DM, while haplogroup D4 was protective [29]. Also, the mtDNA ND1 nt 3394T > C (Y30H) polymorphism has been found to be associated with increased risk of Asian T2DM [27]. Since cybrid studies have examined the relative mitochondrial physiology of the macro-haplogroup N and haplogroup N9a mtDNAs, we used 143B(TK–) cybrids to study the mitochondrial biochemistry of macro-haplogroups N versus M; macro-haplogroup N haplogroups B4a and F1; and variant m.ND1 nt 3394T > C (Y30H).
To prepare cybrids to test for the biochemical effects of haplogroups and individual missense mutations requires screening a population for individuals with the desired mtDNA haplotypes. Blood platelets are then used to transfer the mtDNAs into cultured cells. Finding individuals with macro-haplogroup M and N mtDNAs and with haplogroups B4 and F mtDNAs is relatively easy. However, to find individual mtDNAs with the same haplogroup that differed by a single nucleotide variant, such as m.ND1 nt 339T > C Y30H, requires screening large populations of individuals.
To accomplish this, we recruited an additional Chinese cohort in Southern California, which encompassed 390 individuals (Table S15). After appropriate informed consent, these individuals were evaluated for blood glucose level, systolic and diastolic blood pressure, oral temperature, weight (kg), height (cm), body mass index (BMI), waist circumference (cm), hip circumference (cm), and waist/hip ratio (Table S15). A blood sample was also collected, and then screened for the mtDNA haplogroups and for the m.ND1 nt 3394 T versus C variants. We were able to identify individuals from macro-haplogroup N harboring haplogroups B4a and F1 with either the 3394 T or C variant and to identify macro-haplogroup M individuals harboring the 3394 C allele, haplogroup M9 [21].
3.2. Cybrid preparation and complex I assays
We prepared 143B(TK–) cybrids harboring macro-haplogroup N haplogroups B4 and F versus M mtDNAs with and without the 3394T versus C variants. We chose to assay for complex I activity since the nt 10398A variant causes the complex I ND3 114T missense mutation and the nt 3394C variant causes the complex I ND1 30H missense mutation.
We fused platelets from the subjects to 143B(TK–) ρo cells, the resulting cybrids being selected in bromodeoxyuridine and in uridine free media [36]. Cybrid fusions were performed for two subjects with the 3394C B4c mtDNA, two subjects with the 3394T B4c mtDNA, one with the 3394C F1 mtDNA, and two with the 3394T F1 mtDNA. Cybrids with the desired genotype were then expanded and the mitochondria assayed using our ultrasensitive complex I assay [21].
3.3. Complex I activity is strongly affected by both haplogroup and individual nucleotide variant
These studies revealed that macro-haplogroup N mtDNAs have generally lower complex I activities than macro-haplogroup M mtDNAs, consistent with the presence of the ND3 nt 10398A amino acid 114T variant in the haplogroup N mtDNAs. Within macro-haplogroup N, haplogroup F mtDNAs had lower complex I activities than B4 mtDNAs. Moreover, mtDNAs with the m.ND1 3394C codon 30H variant had lower complex I activities on both the F1 and B4c backgrounds relative to those haplogroups with the m.ND1 3394T codon 30T variant (Fig. 4). However, the difference between F1 and B4c with the common 3394T variant was greater than the difference for either the F1 or B4c with the 3394 T versus C variant. Moreover, the complex I activity of F1 cybrids with the 3394T variant was the same as the activity of the B4c cybrids with the 3394C variant and substantially less than the activity of the B4c cybrids with the 3394T allele. Finally, macro-haplogroups M haplogroup M9 cybrids had the highest complex I activity, even when harboring the 3394C variant, with only the B4c cybrids with the 3394T allele having a comparable complex I activity (Fig. 4) [21].
Fig. 4.
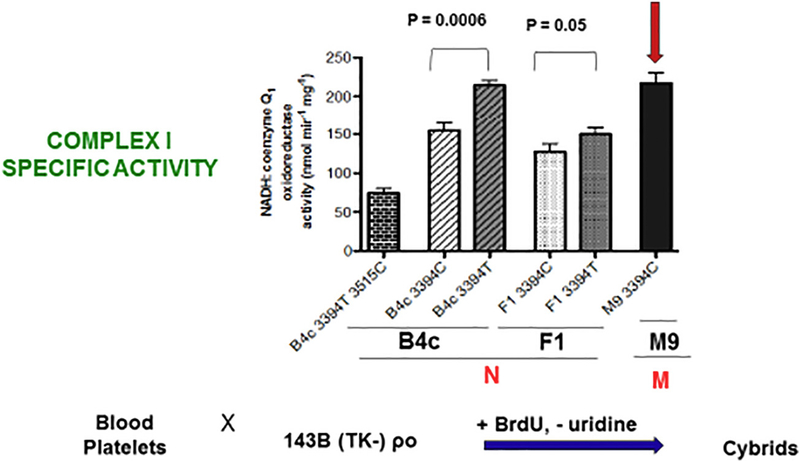
Generation of mtDNA haplogroup cybrids and determination of their complex I specific activity. NADH: coenzyme q1 oxidoreductase activity of mitochondria isolated from B4c, F1, and M9 haplogroup cybrids. The number of independent determinations for each sample is: B4c-T 3515C (n = 6); B4c-C (n = 19); B4c-T (n = 21); F1-C (n = 5); F1-T (n = 5); M9-C (n = 11). On the B4a haplogroup background, the nucleotide 3515C (L70P) missense mutation was associated with a complex I specific activity reduction of 65% (p < 0.0001). On the B4c haplogroup background, the 3394C variant was associated with a 28% reduction in complex I activity relative to the 3394T variant (p = 0.0006). On the F1 background, the 3394C variant was associated with a 15% reduction in complex I activity relative to the 3394T variant (p = 0.047).(Reprinted from [21]).
Overall, macro-haplogroup N derived haplogroups with the m.ND3 nt 10398A 114T variant have a lower complex I activity than macro-haplogroup M mtDNAs with the m.ND3 nt 10398G 114A variant. The complex I activity of the cells with m.ND1 3394C 30H codon also have lower complex I activity than cells with the m.ND1 3394T 30Y variant. Generally, these lower complex I levels correlate with increased risk for metabolic phenotypes such as HTN and OB.
Individuals with the F1 haplogroup had the lowest complex I and this correlates with their predisposition to OB and HTN. Furthermore, the low complex I activity of F1 seems consistent with the study of Japanese and Koreans that found that F and D5 mtDNAs are at increased risk of T2DM [22]. However, the low complex I activity of F1 is discordant with our observation that haplogroups D5 and F4 are protective of T2DM, and that of an independent Taiwanese study that observed that haplogroup D4 is protective of T2DM [29]. Finally, the high B4 complex I activity would seem to be discordant with the report that Taiwanese haplogroup B4a1a was found to be at increased risk of T2DM [29].
If lower complex I activity is a factor in diabetes risk, then the lower complex I activity of the macro-haplogroup N mtDNA is consistent with the lower mitochondrial function reported for haplogroup N9a cybrids found in the Chinese study and the associated report of increased T2DM risk of N9a mtDNAs [26]. However, it is at variance with ours and other reports that haplogroup N9a is protective of T2DM [22]. Perhaps, the risk of T2DM is not a direct consequence of complex I activity but of an associated physiological process such as mitochondrial ROS production, Ca++ regulation, or the modulation nuclear gene expression by mitochondrial metabolic intermediates [15,26].
4. Discussion
The current study provides additional support for the hypothesis that mitochondrial dysfunction is an important contributor to the etiology of T2DM, HTN, and OB. However, it also revealed the multifactorial nature of the mitochondrial involvement.
4.1. The tRNALeu(UUR) nt 3243A > G mutation in Asian diabetes
The tRNALeu(UUR) nt 3243A > G mutation was found in one of the 19 maternal T2DM and MS pedigrees from Taipei proving that mtDNA mutations can cause T2DM. Surprisingly, however, the heteroplasmy level in the proband’s blood was 88%, which is significantly higher than the 20–30% heteroplasmy commonly required to produce T2DM [14,37]. In studies of Europeans, 88% 3243G is in the range of the heteroplasmy level associated with predisposition to neuromuscular disease, the respective phenotypes correlating with distinctive nuclear gene transcription profiles [15]. This anomaly, might be explained by the discovery that the background mtDNA is Asian haplogroup N9a, since both our Taiwanese data and of the Japanese-Korean study [22] determined N9a is protective of T2DM. Perhaps the N9a3 background significantly ameliorated the pathophysiology of the 3243G mutation, requiring a higher heteroplasmy level to produce the T2DM phenotype.
4.2. mtDNA association summaries and their relationship to mitochondrial complex I
Consistent with the increased risk of the mtDNAs harboring the macro-haplogroups N ND3 10398A 114T variant versus the macro-haplogroup M 10398G 114A variant for an array of metabolic and degenerative diseases, we have found that macro-haplogroup N mtDNAs have an overall lower complex I activity than macro-haplogroup M mtDNAs. Moreover, within macro-haplogroup N, we found marked additional differences in complex I activities for different haplogroups with haplogroup F having a significantly lower complex I activity than haplogroup B. Addition of the ND1 nt 3394C (30H) variant further reduced the complex I activity on each haplogroup background.
It is important to keep in mind that complex I is only one of the four central mitochondrial OXPHOS complexes encoded by the mtDNA. For example, macro-haplogroup N mtDNAs also differ from macro-haplogroup M mtDNA in the ATP6 8701G > A (A59T) variant. Alterations in the OXPHOS complexes can affect an array of cellular functions including energy production, ROS production, nuclear gene expression profiles, Ca2+ regulation, and intermediate metabolism. The complex array of interacting biochemical variants within a single mtDNA haplotype provides insight into the reason why different studies on the role of individual mtDNA variants in complex diseases appear to be inconsistent.
Our current association studies have shown that F4 is at increased risk for OB. This is consistent with the very low complex I activity associated with haplogroup F. Similarly, haplogroups F4 and N9a are at increased risk of HTN and these haplogroups would share the lower complex I activity associated with the macro-haplogroup N ND3 10398A 114T variant. Hence, there is some association between complex I activity, OB and HTN.
Our association studies of Taiwanese support the report of the previous Japanese and Korean study that N9a is protective of T2DM [22], but both studies are at variance with a Chinese finding that haplogroup N9a is at increased risk of T2DM [26]. Our results that D5 and F4 haplogroups are protective of T2DM in the Taiwanese is also at variance with observations in the Japanese and Koreans [22], but is consistent with an independent Taiwanese study reporting that haplogroup D4 was protective of T2DM [29]. This inconsistency is additionally interesting since haplogroup F has the lowest complex I activity. So, if T2DM risk was related to complex I activity then the Taiwanese data would be at variance with the biochemical data while the Japanese and Korean data would not.
To address the reason for these inconsistencies we compared the haplogroup frequencies reported for the Japanese and Korean samples [22] and our samples. This revealed that while the distribution of N9a mtDNAs between patients and controls was similar in the Japanese-Korean versus the Taiwanese samples, the distribution of the haplogroup D5 and F mtDNAs was markedly different between the two studies (Fig. S5). Hence, the different effects of haplogroups D5 and F4 on T2DM between the two studies may be differences in the haplogroup distributions of the various populations.
That mtDNA haplogroups are variably associated with altered T2DM risk is a result that has been observed for a plethora of studies on mtDNA variation in T2DM and MS among Europeans. In various studies, haplogroups U [38,39] and J/T [40–42] have been proposed to be risk factors for T2DM. However, other studies have failed to demonstrate these mtDNA haplogroup associations. One major source of variability in these associations might be variation in the nuclear genetic background. In a carefully controlled study of Israeli Jewish diabetes patients, subhaplogroup J1 was found to be 2.4 fold underrepresented in T2DM patients whose parents were not diabetic versus patients whose parents were diabetic. The conclusion was that sub-haplogroup J1 increases the risk of T2DM in association with additional nDNA genetic factors [41,43]. A further study of Israeli Jews revealed that the risk for the clinical sequelae associated with T2DM were influenced by different mtDNA haplogroups with haplogroup H being associated with retinopathy, H3 with neuropathy, U3 with nephropathy, and V with renal failure, [44].
Beyond T2DM and its sequelae, European haplogroup T has been associated with increased risk for coronary artery disease [45], and T [46] and K with ischemic stroke [47]. So in Europeans, mitochondrial genetic background, presumably including both nDNA and the mtDNA variants, may act in concert to determine the genetic risk for T2DM and MS.
While such relative effects seem anomalous from the perspective of classical nDNA genetics, in which the “mutant” is generally a null allele, and its severity defines the directionality of the clinical effect, this is not the case for the mitochondria. Mitochondrial alterations are of necessity subtle, and, because of the central nature of the mitochondrial physiology, they have broad consequences on an array of cellular functions.
It has been shown that mice lacking the nDNA-coded mitochondrial nicotinamide nucleotide transhydrogenase, which generates the NADPH from NADH required for detoxifying mitochondrial hydrogen peroxide, are prone to T2DM, and this predisposition can be ameliorated by introduction to catalase into the mitochondrion matrix to hydrolyze the hydrogen peroxide [7]. Hence, an important factor in the etiology of T2DM may be mitochondrial ROS (hydrogen peroxide) toxicity. Subtle changes in complex I are known to alter mitochondrial ROS production and all of the genes encoding the mitochondrial antioxidant genes are nuclear. Hence, variation in both nDNA and mtDNA genes will regulate ROS production and risk of T2DM. Therefore, the N9a T2DM phenotypes of Japanese, Korean, and Taiwanese patients might differ from the Chinese patients due to the influence of mitochondrial ROS production which is modulated not only by complex I activity but also by the variable expression of nDNA antioxidant genes.
This type of complex regulation has been well documented in another mtDNA disease associated with complex I defects, Leber Hereditary Optic Neuropathy (LHON). LHON is caused by mild homoplasmic mtDNA complex I missense mutations. Individuals, homoplasmic for the causal mutation, show high variability in their predilection to go blind, such that a substantial proportion of the homoplasmic mutant individuals never lose their sight, with males being four times more prone to blindness than females [48,49]. The first parameter found to modulate the expressivity of the LHON mtDNA complex I missense mutations was the mtDNA background, specifically mtDNA haplogroup J in Europeans [50–53] and the ND1 nt 3394T > C Y30H variant [21]. Another mitigating factor is mtDNA copy number. Individuals that harbor a LHON mtDNA complex I mutation that go blind have a higher mtDNA copy number than individuals without the complex I mutation. However, individuals that harbor an LHON complex I gene mutation but do not go blind have even higher mtDNA copy number than individuals with the LHON mutation that do go blind. Thus partial complex I defects can be compensated by simply making more mitochondria, but individuals differ in their capacities to exploit this compensatory mechanism [54]. The third factor that affects the penetrance of LHON mtDNA mutations is the environment. Individuals homoplasmic for LHON complex I mutations that smoke or are heavy drinkers are more likely to go blind [49,55]. nDNA genetic variants is a fourth factor affecting LHON penetrance [56]. The final factor affecting the expressivity of LHON mutations is gender which acts through modulation of mitochondrial ROS production. A mouse model of LHON has revealed that a major component of the pathophysiology of LHON mutations is chronically increased mitochondrial ROS production. Women, which are less prone to go blind, may be protected by their regular production of estrogen. Twenty % of the estrogen receptor β (ERβ) is located in the mitochondrial matrix, and when ERβ is activated by estradiol the specific activity of the mitochondrial antioxidant enzyme, Mn superoxide dismutase, doubles [57]. In cell culture models of LHON addition of the combination of phytoestrogens that activate ERp [genistein + daidzein + equol (G + D + E)], increase mitochondrial respiration back to normal and up-regulate critical mitochondrial biogenesis genes [58]. If both LHON and T2DM are the result of chronic mitochondrial ROS production, then the same factors that modulate the penetrance of LHON might also affect the penetrance of the complex I haplogroup variants associated with T2DM.
4.3. Conclusions
Our 15-year study of Taiwanese on the role of mtDNA variation on risk for T2DM and MS has provided direct evidence that mtDNA variation is an important factor in T2DM/MS etiology. This was demonstrated by both the identification of the 3234A > G mutation in one pedigree and by the overall association between mtDNA haplogroups D5, F4, and N9a and altered risk of T2DM, HTN and OB. However, our study also demonstrated the importance of environmental factors, gender, age, and nDNA and mtDNA generic background factors in determining the clinical manifestations of T2DM and MS.
5. Methods and procedures
5.1. Population studies
5.1.1. Cohort population samples
This study recruited three separate Taiwanese cohorts, with informed consent, for which the mtDNA haplogroups were correlated with T2DM, HTN, and OB (Fig. S3).
Cohort 1 (or initial dataset) was collected through the endocrine unit of the National Taiwan University Hospital (NTUH). The patients were collected from patient populations undergoing weight-loss surgery (obesity, OB), treatment for hypertension (HTN), and treatment for diabetes mellitus (Type 2 diabetes, T2DM). Finally a control group was included which presented with normal glucose tolerance during health check-up.
Cohort 2 (or replication dataset) was collected from the Yunlin branch of NTUH. These patients were being seen for treatment of diabetes and the controls were individuals found to have normal glucose tolerance during community screening.
Cohort 3 (the second Taipei population) was again collected from NTUH. These patients were undergoing treatment for obesity and the controls were again individuals found to have normal glucose tolerance during health check-up.
Cohort 4 (Southern California Chinese) was collected in Southern California. With informed consent 390 Southern California Chinese were recruited, blood samples collected and information on metabolic status collected (Table S15). These samples were screened for macro-haplogroup N and M markers and for haplogroup markers that identified haplogroups B4a and F1, within which we were able to identify individuals the ND1 3394T versus 3394C variants.
5.1.2. Haplogroups variables
The first and second cohorts involved correlating the various clinical parameters with all of the major Asian mtDNA haplogroups, a total of 32. The first cohort study pointed to haplogroups D5, F4, and N9a as having the greatest potential effect on T2DM and MS clinical manifestations. Accordingly, the cohort 3 was only genotyped for these three haplogroups and the combined cohorts 1–3 were analyzed for these three haplogroups to minimize the effects of multiple testing noise.
5.1.3. Clinical variables and variable coding
mtDNA haplogroups were then tested against 15 clinical variables (six binary and nine continuous). The binary traits included: Obesity (OB), Type 2 Diabetes Mellitus (T2DM), Hypertension (HTN), Metabolic Syndrome as defined by the American Heart Association (AHA-MS), Metabolic Syndrome as defined by the International Diabetes Federation (IDF-MS), and Gender.
The continuous traits included: Body Mass Index (BMI) (kg/m2), Fasting Plasma Glucose (AC) (mg/dL), Triglycerides (TG) (mg/dL), Total Cholesterol (TCHO) (mg/dL), Systolic Blood Pressure (SBP) (mm Hg), Diastolic Blood Pressure (DBP) (mm Hg), High Density Lipoprotein (HDL) (mg/dL), post cibum (after meal) glucose (PC) (mg/ dL), and age (years).
The values of each variable were used as they were for descriptive statistics, or the variables were transformed to binary ones with values 0 or 1 for association and modeling analyses.
When continuous variables were reduced to binary variables, the following discriminating values were employed: OB was defined as BMI ≥ 30 kg/m2; Diabetes was defined as fasting glucose (AC) > 126 mg/dL and post cibum glucose (PC) ≥ 200 mg/dL (when available); Hypertension was defined as SBP/DBP ≥ 140/90 mm Hg; the IDF (2005) criteria (IDF-MS) included: waist circumference ≥ 90 cm (male) or ≥ 80 cm (female) or BMI > 30 kg/m2 plus at least two of the following criteria TG ≥ 150 mg/dL, HDL < 40 mg/dL (male) or < 50 mg/dL (female), SBP/DBP ≥ 130/ 85 mm Hg, AC ≥ 100 mg/dL; the AHA/NHLBI criteria (AHA-MS) included at least three of the following five criteria: waist circumference ≥ 90 cm (male) or ≥ 80 cm (female) or BMI ≥ 30 kg/m2, TG ≥ 150 mg/dL, HDL < 40 mg/dL (male) or < 50 mg/dL (female), SBP/DBP ≥ 130/85 mm Hg, and AC ≥ 100 mg/dL.
5.1.4. Clinical variables used in haplogroup/SNP association studies
Continuous variables were analyzed either as continuous or as binary after transformation by using the thresholds below: BMI ≥ 30 kg/m2 → 1, otherwise 0; TCHO ≥ 200 mg/dL → 1, otherwise 0; DBP ≥ 85 mm Hg → 1, otherwise 0; SBP ≥ 130mmHg → 1, otherwise 0; TG ≥ 150 mg/dL → 1, otherwise 0; HDL for Males < 40 mg/dL or Females < 50 mg/dL → 1, otherwise 0; T2DM, OB, HTN, and HTN as defined above.
When stratified for gender, cohort, or age variables were coded as: Gender strata: 1 = Male, 0 = Female; Cohort strata: 1 = cohort 1, 2 = cohort 2, 3 = cohort 3; Age strata: young = < 30, medium = 30 ≤ x < 60, advanced = ≥60 years.
Recursive partitioning analysis was performed for defining the age strata. In this method we recursively partitioned the data according to the relationship between the MS variables (T2DM, HTN, and obesity) and age, gender and haplogroup values, creating a tree of partitions per cohort as well as pooled data. The method resulted in a set of groupings of age values that best predicted the used MS variable. We employed this method in JMP with the default significance maximization criterion.
5.2. mtDNA sequencing
Total DNA was extracted from blood samples for probands of 27 pedigrees and mtDNA fragments were PCR-amplified using mtDNA-specific primers (Table S15). The hypervariable segment I (HVS I) of the control region was amplified with primers htmL15591/htmH112, haplogroup-specific sites within the coding region of mtDNA were amplified using primers to generate targeted amplicons. For complete mtDNA sequencing mtDNA was amplified in eight overlapping fragments ranging from 1643 to 2851 bp in length. Primer pairs for 8 fragments amplification were as follows: hmtL569/hmtH2941, hmtL2797/hmtH5193, hmtL5061/hmtH7497, hmtL7336/hmtH9819, hmtL9611/hmtH12111, hmtL11727/hmtH14559, hmtL14130/ hmtH112 and hmtL15591/hmtH626 (Table S14). All sequencing was performed using ABI Big Dye Terminator V3.1 Cycle Sequencing Kits (ABI) on ABI Prism 3130 Genetic Analyzer. Trace files were analyzed using the Sequencher Software and were compared with the revised Cambridge Reference Sequence (rCRS).
5.3. mtDNA haplogroup analysis
The mtDNAs were haplogrouped by a combination of control region hypervariable region sequencing and testing for haplogroup informative restriction site and sequence polymorphisms. The restriction sites analyzed included gain of HaeIII 663, gain of HhaI 4833, gain of Tsp 5417, gain of Hinfl 9820, loss of BsaW1 4715, loss of Alul 5176, loss of Taq 10181 and loss of Hinfl 14976. Nucleotide variants evaluated included 10400, 7598, 4140, 11482, [4071, 6455, 7853], 11215, 10397, 10873, 12705, [16223, 16257, 16261], 3970, 11061, 6392, [6962, 10609, 12406, 12882], [10535, 10586, 12338], [5913, 5973, 10320, 11065], [5263, 12630] and 8281–8289. Primers to encompass variant sites are listed in Suppl. Table S16. The use of these variants to define the haplogroups within the Asian macro-haplogroups M and N are shown in Fig. 2.
5.4. Statistical analysis - modeling
We used logistic regression analysis to describe the nature of the association between MS phenotypes and mtDNA haplogroups.
Logistic Regression analysis of binary dependent variables was performed for T2DM, HTN, and OB, as well as AC, SBP, DBP, BMI, and TCHO in their nominal format.
To avoid random associations due to high number of multiple testing, we first pooled samples in haplogroups D, F, M7, and R, resulting into 15 major haplogroups. Also, because the number of samples in haplogroups M21, N*, M28, and Y was very small, these samples were excluded from analysis. After having established that haplogroups D5, F4, and N9a are the only ones with significant effects on MS phenotypes and following the human mtDNA phylogeny, we grouped haplogroups D, E, G, M*, M7-M10 in macro-M haplogroup (n = 424, 38.09%) and haplogroups A, B, F, and R in macro-N haplogroup (n = 428, 38.45%). Therefore, as independent variables we used 5 haplogroups (D5, F4, N9a, macro-M, and macro-N) adjusted for age and gender, and MS clinical phenotypes.
Chi-square test was used to test the hypothesis that all regression parameters are zero. A p-value > χ2 denotes the probability for obtaining a greater χ2 value by chance alone if the specified model fits no better than the model that includes only intercepts. R2 is the uncertainty coefficient, which ranges from 0 to 1. A logistic model rarely has a high R2. Wald or likelihood ratio tests were used to test whether each individual coefficient equals to 0. The likelihood ratio test refitted the model after deleting each explanatory parameter. Parameters were coded using effect coding. Odds ratios were estimated for all levels of each parameter. The corrected Akaike Information Criterion and lack of fit chi-square statistics were used to evaluate model fit. The False Discovery Rate p-value for each model effect was calculated using the Benjamini-Hochberg [59] technique. This technique adjusts the p-values to control the false discovery rate for multiple testing.
Because this is a retrospective design (case-control study), the odds ratio is a useful measure of association, and in our study it indicated that the odds for a metabolic phenotype is x times higher or lower for the tested effect. If the confidence intervals include the value of 1, the odds ratio is not considered significantly different from 1. Wide confidence bounds indicated that the odds ratio estimate had low precision. SAS and JMP software were used for statistical analyses.
5.5. Preparation mtDNA cybrids
Cybrids were prepared by fusing subject blood platelets to human mtDNA-deficient (ρo) osteosarcoma 143B (TK –) cells. Cybrids were selected in DMEM with 10% (vol/vol) FCS containing 30 μg/mL BrdU in the absence of uridine [36].
5.6. Mitochondrial complex I (NADH: coenzyme Q1 oxidoreductase) activity
Mitochondria were isolated by differential centrifugation and the pellets were resuspended in iso-osmotic solution and frozen. The mitochondria were permeabilized by incubation in assay buffer with alamethicin (40 μg/mL). Complex I was assayed using 25 μg of mitochondria, coenzyme q1, and NADH. NADH oxidation was monitored at 340 nm using a dual-beam spectrophotometer, with or without rotenone, and rotenone-sensitivity calculated. The complex I assays of the B4c cybrids involved two independent subjects for each allele, encompassing eight and nine separate mitochondrial isolations from B4c 3394T and B4c 3394C clones, respectively. The B4c 3394T 3515C cybrid clone complex I was assayed six times. The complex I activity of the F1a cybrid clones involved three separate mitochondrial isolations from a cybrid clone of the F1a 3394C subject, two mitochondrial isolations from a cybrid clone of one of the F1a 3394T subjects and one mitochondrial isolation from the cybrid clone of the other F1a 3394C subject. The results of 10 complex I assays were averaged for the three cybrid clones. The mtDNA/nDNA ratios of all clones assayed for complex I activity were within 20% of the mean of the mtDNA/nDNA ratio of all clones and did not correlate with enzyme activities.
Supplementary Material
Table 9.
MtDNA haplogroups are associated with MS clinical phenotypes. Five mitochondrial haplogroups (HG), age, and gender were used as effects.
| Variable | Effects | Model p- value |
R2 | AICc | Lack of Fit p- value |
n | Effect | Effect p- value# |
FDR |
|---|---|---|---|---|---|---|---|---|---|
| AC | Age, HG, gender |
<0.0001 | 0.0336 | 1157.52 | 0.0777 | 1099 | Age | 0.0001 | 0.00035 |
| HG | 0.0013 | 0.00197 | |||||||
| Gender | 0.9825 | 0.98254 | |||||||
| BMI | Age, HG, gender |
<0.0001 | 0.1335 | 1055.54 | 0.4095 | 1085 | Age | <0.0001 | 0.00000 |
| HG | 0.0565 | 0.08476 | |||||||
| Gender | 0.16645 | 0.16447 | |||||||
| DBP | Age, HG, gender |
<0.0001 | 0.0356 | 1167.01 | 0.0112 | 940 | Age | 0.0444 | 0.4445 |
| HG | 0.0012 | 0.00173 | |||||||
| Gender | 0.0004 | 0.00123 | |||||||
| SBP | Age, HG, gender |
<0.0001 | 0.0438 | 1261.39 | 0.1223 | 940 | Age | <0.0001 | 0.00000 |
| HG | 0.0789 | 0.11836 | |||||||
| Gender | 0.1471 | 0.14713 | |||||||
| TCHO | HG, Age, gender | <0.0001 | 0.0297 | 1457.93 | <0.0001 | 1081 | Age | <0.0001 | 0.00002 |
| HG | 0.1643 | 0.16428 | |||||||
| Gender | <0.0001 | 0.00005 | |||||||
R2: The proportion of the total uncertainty that is attributed to the model fit. Because certainty in the predicted probabilities is rare for logistic models, R2 tends to be small.
AlCc: Corrected Akaike Information Criterion that assesses model fit based on – 2LogLikelihood.
Lack of Fit: Chi-square statistics on the negative log-likelihood error due to lack of fit, error in a saturated model (pure error), and the total error in the fitted model. High p-values denote that the lack of fit Chi-square is not significant, and support the conclusion that there is little to be gained by introducing additional variables.
FDR: Benjamini-Hochberg false discovery rate.
Age levels: advanced ≥ 60, 30 ≤ medium < 60, young < 30.
HG levels: D5, F4, N9a, macro-M (D, E, G, M*, M7, M8, M9, M10), and macro-N (A, B, F, R).
Likelihood-Ratio test p-value.
Acknowledgements
DCW would like to acknowledge the excellent efforts of Dr. Mariella Simon, Ms. Julia Platt, and Dr. Shiqin Xu in the recruitment of the Southern California Han Chinese cohort and Kierstin Keller and Ashwini Ramachandran for assistance with data organization. This work was supported by NIH grants NS021328, DK73691; Doris Duke Foundation grant 2005057; and SP Accure Labs Pvt. Ltd. grant awarded to DC Wallace; National Science Council of Taiwan grant 89-B-FA01–1-4 to L.-M. Chuang; and NIH UCI Clinical and Translational Science (ICTS) grant UL1 RR031985 (currently UL1 TR001414) which supported the collection of the Southern California Han Chinese sample.
Footnotes
This article is part of a Special Issue entitled 20th European Bioenergetics Conference, edited by László Zimányi and László Tretter.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbabio.2018.07.002.
Transparency document
The Transparency document associated with this article can be found, in online version.
References
- [1].Kwak SH, Park KS, Lee KU, Lee HK, Mitochondrial metabolism and diabetes, J. Diabetes Investig. 1 (2010) 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wallace DC, A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine, Annu. Rev. Genet. 39 (2005) 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI, Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes, N. Engl. J. Med. 350 (2004) 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC, PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes, Nat. Genet. 34 (2003) 267–273. [DOI] [PubMed] [Google Scholar]
- [5].Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ, Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 8466–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS, Extension of murine life span by overexpression of catalase targeted to mitochondria, Science 308 (2005) 1909–1911. [DOI] [PubMed] [Google Scholar]
- [7].Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, Petersen KF, Samuel VT, Shulman GI, Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance, Cell Metab. 12 (2010) 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dorner G, Mohnike A, Steindel E, On possible genetic and epigenetic modes of diabetes transmission, Endokrinologie 66 (1975) 225–227. [PubMed] [Google Scholar]
- [9].Dorner G, Plagemann A, Reinagel H, Familial diabetes aggregation in type I diabetics: gestational diabetes an apparent risk factor for increased diabetes susceptibility in the offspring, Exp. Clin. Endocrinol. 89 (1987) 84–90. [DOI] [PubMed] [Google Scholar]
- [10].Wallace DC, Mitochondrial DNA variation in human radiation and disease, Cell 163 (2015) 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Goto Y, Nonaka I, Horai S, A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies, Nature 348 (1990) 651–653. [DOI] [PubMed] [Google Scholar]
- [12].Suzuki S, Oka Y, Kadowaki T, Kanatsuka A, Kuzuya T, Kobayashi M, Sanke T, Seino Y, Nanjo K, The Research Committee for Specific Types of Diabetes Mellitus with Gene Mutations of the Japan Diabetes Society, Clinical features of diabetes mellitus with the mitochondrial DNA 3243 (A-G) mutation in Japanese: maternal inheritance and mitochondria-related complications, Diabetes Res. Clin. Pract. 59 (2003) 207–217. [DOI] [PubMed] [Google Scholar]
- [13].van den Ouweland JM, Lemkes HHP, Ruitenbeek W, Sandkjujl LA, deVijlder MF, Struyvenberg PAA, van de Kamp JJP, Maassen JA, Mutation in mitochondrial tRNALeu(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness, Nat. Genet. 1 (1992) 368–371. [DOI] [PubMed] [Google Scholar]
- [14].van den Ouweland JM, Lemkes HH, Gerbitz KD, Maassen JA, Maternally inherited diabetes and deafness (MIDD): a distinct subtype of diabetes associated with a mitochondrial tRNALeu(UUR) gene point mutation, Muscle Nerve 3 (1995) S124–S130. [DOI] [PubMed] [Google Scholar]
- [15].Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O’Hearn S, Levy S, Potluri P, Lvova M, Davila A, Lin CS, Perin JC, Rappaport EF, Hakonarson IA Trounce, V. Procaccio, D.C. Wallace, Progressive increase in mtDNA 3243A > G heteroplasmy causes abrupt transcriptional reprogramming, Proc. Natl. Acad. Sci. U. S. A. 111 (2014) E4033–E4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ballinger SW, Shoffner JM, Hedaya EV, Trounce I, Polak MA, Koontz DA, Wallace DC, Maternally transmitted diabetes and deafness associated with a 10.4kb mitochondrial DNA deletion, Nat. Genet. 1 (1992) 11–15. [DOI] [PubMed] [Google Scholar]
- [17].Ballinger SW, Shoffner JM, Gebhart S, Koontz DA, Wallace DC, Mitochondrial diabetes revisited, Nat. Genet. 7 (1994) 458–459. [DOI] [PubMed] [Google Scholar]
- [18].Wilson FH, Hariri A, Farhi A, Zhao H, Petersen KF, Toka HR, Nelson-Williams C, Raja KM, Kashgarian M, Shulman GI, Scheinman SJ, Lifton RP, A cluster of metabolic defects caused by mutation in a mitochondrial tRNA, Science 306 (2004) 1190–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mishmar D, Ruiz-Pesini EE, Golik P, Macaulay V, Clark AG, Hosseini S, Brandon M, Easley K, Chen E, Brown MD, Sukernik RI, Olckers A, Wallace DC, Natural selection shaped regional mtDNA variation in humans, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC, Effects of purifying and adaptive selection on regional variation in human mtDNA, Science 303 (2004) 223–226. [DOI] [PubMed] [Google Scholar]
- [21].Ji F, Sharpley MS, Derbeneva O, Alves LS, Qian P, Wang Y, Chalkia D, Lvova M, Xu J, Yao W, Simon M, Platt J, Xu S, Angelin A, Davila A, Huang T, Wang PH, Chuang LM, Moore LG, Qian G, Wallace DC, Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fuku N, Park KS, Yamada Y, Nishigaki Y, Cho YM, Matsuo H, Segawa T, Watanabe S, Kato K, Yokoi K, Nozawa Y, Lee HK, Tanaka M, Mitochondrial haplogroup N9a confers resistance against type 2 diabetes in Asians, Am. J. Hum. Genet. 80 (2007) 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tanaka M, Fuku N, Nishigaki Y, Matsuo H, Segawa T, Watanabe S, Kato K, Yokoi K, Ito M, Nozawa Y, Yamada Y, Women with mitochondrial haplogroup N9a are protected against metabolic syndrome, Diabetes 56 (2007) 518–521. [DOI] [PubMed] [Google Scholar]
- [24].Nishigaki Y, Yamada Y, Fuku N, Matsuo H, Segawa T, Watanabe S, Kato K, Yokoi K, Yamaguchi S, Nozawa Y, Tanaka M, Mitochondrial haplogroup N9b is protective against myocardial infarction in Japanese males, Hum. Genet. 120 (2007) 827–836. [DOI] [PubMed] [Google Scholar]
- [25].Guo LJ, Oshida Y, Fuku N, Takeyasu T, Fujita Y, Kurata M, Sato Y, Ito M, Tanaka M, Mitochondrial genome polymorphisms associated with type-2 diabetes or obesity, Mitochondrion 5 (2005) 15–33. [DOI] [PubMed] [Google Scholar]
- [26].Fang H, Hu N, Zhao Q, Wang B, Zhou H, Fu Q, Shen L, Chen X, Shen F, Lyu J, mtDNA haplogroup N9a increases the risk of T2DM by altering mitochondrial function and intracellular mitochondrial signals, Diabetes (2018), 10.2337/db17-0974 (ePub ahead of print). [DOI] [PubMed] [Google Scholar]
- [27].Niu Q, Zhang W, Wang H, Guan X, Lu J, Li W, Effects of mitochondrial haplogroup N9a on type 2 diabetes mellitus and its associated complications, Exp. Ther. Med. 10 (2015) 1918–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liao WQ, Pang Y, Yu CA, Wen JY, Zhang YG, Li XH, Novel mutations of mitochondrial DNA associated with type 2 diabetes in Chinese Han population, Tohoku J. Exp. Med. 215 (2008) 377–384. [DOI] [PubMed] [Google Scholar]
- [29].Loo JH, Trejaut JA, Yen JC, Chen ZS, Ng WM, Huang CY, Hsu KN, Hung KH, Hsiao Y, Wei YH, Lin M, Mitochondrial DNA association study of type diabetes with or without ischemic stroke in Taiwan, BMC Res. Notes 7 (2014) 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jiang W, Li R, Zhang Y, Wang P, Wu T, Lin J, Yu J, Gu M, Mitochondrial DNA mutations associated with type 2 diabetes mellitus in Chinese Uyghur population, Sci. Rep. 7 (2017) 16989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sharma V, Sharma I, Singh VP, Verma S, Pandita A, Singh V, Rai E, Sharma S, mtDNA G10398A variation provides risk to type 2 diabetes in population group from the Jammu region of India, Meta Gene 2 (2014) 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Poulton J, Luan J, Macaulay V, Hennings S, Mitchell J, Wareham NJ, Type 2 diabetes is associated with a common mitochondrial variant: evidence from a population-based case-control study, Hum. Mol. Genet. 11 (2002) 1581–1583. [DOI] [PubMed] [Google Scholar]
- [33].Hwang S, Kwak SH, Bhak J, Kang HS, Lee YR, Koo BK, Park KS, Lee HK, Cho YM, Gene expression pattern in transmitochondrial cytoplasmic hybrid cells harboring type 2 diabetes-associated mitochondrial DNA haplogroups, PLoS One 6 (2011) e22116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kazuno AA, Munakata K, Nagai T, Shimozono S, Tanaka M, Yoneda M, Kato N, Miyawaki A, Kato T, Identification of mitochondrial DNA polymorphisms that alter mitochondrial matrix pH and intracellular calcium dynamics, PLoS Genet. 2 (2006) e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kazuno AA, Munakata K, Tanaka M, Kato N, Kato T, Relationships between mitochondrial DNA subhaplogroups and intracellular calcium dynamics, Mitochondrion 8 (2008) 164–169. [DOI] [PubMed] [Google Scholar]
- [36].Chomyn A, Platelet-mediated transformation of human mitochondrial DNA-less cells, Methods Enzymol. 264 (1996) 334–339. [DOI] [PubMed] [Google Scholar]
- [37].van den Ouweland JM, Maechler P, Wollheim CB, Attardi G, Maassen JA, Functional and morphological abnormalities of mitochondria harbouring the tRNA(Leu)(UUR) mutation in mitochondrial DNA derived from patients with maternally inherited diabetes and deafness (MIDD) and progressive kidney disease, Diabetologia 42 (1999) 485–492. [DOI] [PubMed] [Google Scholar]
- [38].Martikainen MH, Ronnemaa T, Majamaa K, Prevalence of mitochondrial diabetes in southwestern Finland: a molecular epidemiological study, Acta Diabetol. 50 (2013) 737–741. [DOI] [PubMed] [Google Scholar]
- [39].Martikainen MH, Ronnemaa T, Majamaa K, Association of mitochondrial DNA haplogroups and vascular complications of diabetes mellitus: a population-based study, Diab. Vasc. Dis. Res. 12 (2015) 302–304. [DOI] [PubMed] [Google Scholar]
- [40].Crispim D, Canani LH, Gross JL, Tschiedel B, Souto KE, Roisenberg I, The European-specific mitochondrial cluster J/T could confer an increased risk of insulin-resistance and type 2 diabetes: an analysis of the m.4216T > C and m.4917A > G variants, Ann. Hum. Genet. 70 (2006) 488–495. [DOI] [PubMed] [Google Scholar]
- [41].Feder J, Ovadia O, Blech I, Cohen J, Wainstein J, Harman-Boehm I, Glaser B, Mishmar D, Parental diabetes status reveals association of mitochondrial DNA haplogroup J1 with type 2 diabetes, BMC Med. Genet. 10 (2009) 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mohlke KL, Jackson AU, Scott LJ, Peck EC, Suh YD, Chines PS, Watanabe RM, Buchanan TA, Conneely KN, Erdos MR, Narisu N, Enloe S, Valle TT, Tuomilehto J, Bergman RN, Boehnke M, Collins FS, Mitochondrial polymorphisms and susceptibility to type 2 diabetes-related traits in Finns, Hum. Genet. 118 (2005) 245–254. [DOI] [PubMed] [Google Scholar]
- [43].Gershoni M, Levin L, Ovadia O, Toiw Y, Shani N, Dadon S, Barzilai N, Bergman A, Atzmon G, Wainstein J, Tsur A, Nijtmans L, Glaser B, Mishmar D, Disrupting mitochondrial-nuclear coevolution affects OXPHOS complex I integrity and impacts human health, Genome Biol. Evol. 6 (2014) 2665–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Achilli A, Olivieri A, Pala M, Hooshiar Kashani B., Carossa V, Perego UA, Gandini F, Santoro A, Battaglia V, Grugni V, Lancioni H, Sirolla C, Bonfigli AR, Cormio A, Boemi M, Testa I, Semino O, Ceriello A, Spazzafumo L, Gadaleta MN, Marra M, Testa R, Franceschi C, Torroni A, Mitochondrial DNA backgrounds might modulate diabetes complications rather than T2DM as a whole, PLoS One 6 (2011) e21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kofler B, Mueller EE, Eder W, Stanger O, Maier R, Weger M, Haas A, Winker R, Schmut O, Paulweber B, Iglseder B, Renner W, Wiesbauer M, Aigner I, Santic D, Zimmermann FA, Mayr JA, Sperl W, Mitochondrial DNA haplogroup T is associated with coronary artery disease and diabetic retinopathy: a case control study, BMC Med. Genet. 10 (2009) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nardelli C, Labruna G, Liguori R, Mazzaccara C, Ferrigno M, Capobianco V, Pezzuti M, Castaldo G, Farinaro E, Contaldo F, Buono P, Sacchetti L, Pasanisi F, Haplogroup T is an obesity risk factor: mitochondrial DNA haplotyping in a morbid obese population from southern Italy, Biomed. Res. Int. 2013 (2013) 631082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chinnery PF, Elliott HR, Syed A, Rothwell PM, Mitochondrial DNAhaplogroups and risk of transient ischaemic attack and ischaemic stroke: a genetic association study, Lancet Neurol. 9 (2010) 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, Nikoskelainen EK, Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy, Science 242 (1988) 1427–1430. [DOI] [PubMed] [Google Scholar]
- [49].Sadun AA, La Morgia C, Carelli V, Leber’s hereditary optic neuropathy, Curr. Treat. Options Neurol. 13 (2011) 109–117. [DOI] [PubMed] [Google Scholar]
- [50].Brown MD, Starikovskaya E, Derbeneva O, Hosseini S, Allen JC, Mikhailovskaya IE, Sukernik RI, Wallace DC, The role of mtDNA background in disease expression: a new primary LHON mutation associated with Western Eurasian haplogroup J, Hum. Genet. 110 (2002) 130–138. [DOI] [PubMed] [Google Scholar]
- [51].Brown MD, Sun F, Wallace DC, Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage, Am. J. Hum. Genet. 60 (1997) 381–387. [PMC free article] [PubMed] [Google Scholar]
- [52].Brown MD, Torroni A, Reckord CL, Wallace DC, Phylogenetic analysis of Leber’s hereditary optic neuropathy mitochondrial DNA’s indicates multiple independent occurrences of the common mutations, Hum. Mutat. 6 (1995) 311–325. [DOI] [PubMed] [Google Scholar]
- [53].Torroni A, Petrozzi M, D’Urbano L, Sellitto D, Zeviani M, Carrara F, Carducci C, Leuzzi V, Carelli V, Barboni P, De Negri A, Scozzari R, Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484, Am. J. Hum. Genet. 60 (1997) 1107–1121. [PMC free article] [PubMed] [Google Scholar]
- [54].Giordano C, Iommarini L, Giordano L, Maresca A, Pisano A, Valentino ML, Caporali L, Liguori R, Deceglie S, Roberti M, Fanelli F, Fracasso F, Ross-Cisneros FN, D’Adamo P, Hudson G, Pyle A, Yu-Wai-Man P, Chinnery PF, Zeviani M, Salomao SR, Berezovsky A, Belfort R Jr., Ventura DF, Moraes M, Moraes Filho M, Barboni P, Sadun F, De Negri A, Sadun AA, Tancredi A, Mancini M, Amati G. d, Loguercio Polosa P, Cantatore P, Carelli V, Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy, Brain 137 (2014) 335–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Amaral-Fernandes MS, Marcondes AM, Miranda PM, Maciel-Guerra AT, Sartorato EL, Mutations for Leber hereditary optic neuropathy in patients with alcohol and tobacco optic neuropathy, Mol. Vis. 17 (2011) 3175–3179. [PMC free article] [PubMed] [Google Scholar]
- [56].Shankar SP, Fingert JH, Carelli V, Valentino ML, King TM, Daiger SP, Salomao SR, Berezovsky A, Belfort R Jr., T.A. Braun, V.C. Sheffield, A.A. Sadun, E.M. Stone, Evidence for a novel X-linked modifier locus for Leber hereditary optic neuropathy, Ophthalmic Genet. 29 (2008) 17–24. [DOI] [PubMed] [Google Scholar]
- [57].Pedram A, Razandi M, Wallace DC, Levin ER, Functional estrogen receptors in the mitochondria of breast cancer cells, Mol. Biol. Cell 17 (2006) 2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Pisano A, Preziuso C, Iommarini L, Perli E, Grazioli P, Campese AF, Maresca A, Montopoli M, Masuelli L, Sadun AA, d’Amati G, Carelli V, Ghelli A, Giordano C, Targeting estrogen receptor beta as preventive therapeutic strategy for Leber’s hereditary optic neuropathy, Hum. Mol. Genet. 24 (2015) 6921–6931. [DOI] [PubMed] [Google Scholar]
- [59].Benjamini Y, Hochberg Y, Controlling the false discovery rate: a practical and powerful approach to multiple testing, J. R. Stat. Soc. B 57 (1995) 289–300. Available online at http://www.jstor.org/stable/2346101. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.