

Abstract
Current treatments for Mycobacterium tuberculosis infections require long and complicated regimens that can lead to patient non-compliance, increasing incidences of antibiotic-resistant strains, and lack of efficacy against latent stages of disease. Thus, new therapeutics are needed to improve tuberculosis standard of care. One strategy is to target protein homeostasis pathways by inhibiting molecular chaperones such as GroEL/ES (HSP60/10) chaperonin systems. M. tuberculosis has two GroEL homologs: GroEL1 is not essential but is important for cytokine-dependent granuloma formation, while GroEL2 is essential for survival and likely functions as the canonical housekeeping chaperonin for folding proteins. Another strategy is to target the protein tyrosine phosphatase B (PtpB) virulence factor that M. tuberculosis secretes into host cells to help evade immune responses. In the present study, we have identified a series of GroEL/ES inhibitors that inhibit M. tuberculosis growth in liquid culture and biochemical function of PtpB in vitro. With further optimization, such dual-targeting GroEL/ES and PtpB inhibitors could be effective against all stages of tuberculosis – actively replicating bacteria, bacteria evading host cell immune responses, and granuloma formation in latent disease – which would be a significant advance to augment current therapeutics that primarily target actively replicating bacteria.
Keywords: GroEL, GroES, HSP60, HSP10, molecular chaperone, chaperonin, proteostasis, phosphatases, small molecule inhibitors, Mycobacterium tuberculosis, antibiotics, polypharmacology
Graphical Abstract
Mycobacterium tuberculosis, the causative agent of tuberculosis, infects about 1 in 4 people worldwide. In 2017, over 10 million active cases of tuberculosis infection were reported, with 1.6 million deaths attributed to this deadly disease.1 M. tuberculosis is a facultative intracellular bacterium that is transmitted as an airborne particulate generated when people with active tuberculosis cough or sneeze. When inhaled, the bacteria traverse the respiratory tract to alveoli where they infect phagocytic cells, namely macrophages.2–4 The bacteria replicate within the macrophage and induce cytokines that initiate an inflammatory response in the lungs. Macrophages and lymphocytes migrate to the site of infection and form granulomas, which lead to asymptomatic latent disease where pools of bacteria can reside in dormant states for years. However, a weakened immune system can lead to re-activation of dormant M. tuberculosis from the granulomas, allowing bacteria to once again actively replicate, which can be fatal if left untreated.
First-line antibiotics to treat tuberculosis include isoniazid, rifampicin, ethambutol, and pyrazinamide.1, 3, 5 Unfortunately, these antibiotics are most effective against actively-replicating bacteria, which is problematic since M. tuberculosis replicates slowly (18–54 h doubling times) and can persist in granulomas in a more metabolically inactive state.6 To increase treatment efficacy, combination therapy is typically administered for at least six-to-nine months. This extensive regimen often leads to patient non-compliance, which contributes to the development of antibiotic resistant strains. To help combat drug resistance, two new drugs have recently been approved – bedaquiline and delamanid.7, 8 Unfortunately, resistance is also arising to these two drugs.9–11 Thus, new drug candidates are needed that act on previously unexploited targets and pathways not predisposed to resistance, and that are additionally effective against the asymptomatic latent phase of infections.
A new paradigm in antibiotic research is to exploit protein homeostasis pathways, such as targeting molecular chaperones.12–24 A network of molecular chaperones and proteases collectively functions to maintain protein homeostasis by assisting proteins to fold to their native, functional states, or ensuring their proper degradation.25–31 In particular, all organisms contain at least one homolog of the 60 kDa class of molecular chaperone (HSP60) that is essential under all conditions. Thus, targeting HSP60 chaperonin systems with small molecule inhibitors should be an effective antibacterial strategy. Escherichia coli GroEL is the prototypical member of the HSP60 chaperonin family, which has been studied extensively. GroEL is a homo-tetradecameric protein that forms two, seven-subunit rings that stack back-to-back with one another. Through a series of events driven by ATP binding and hydrolysis, unfolded substrate polypeptides are bound within the central cavity of a GroEL ring and are encapsulated by the GroES co-chaperonin lid, allowing protein folding within the sequestered chamber. We refer readers to these other studies for a more detailed understanding of the structure and function of GroEL/ES chaperonin systems.32, 33
M. tuberculosis has two GroEL homologs, GroEL1 and GroEL2, which interestingly diverge in their sequences and share 61% amino acid identity with each other.34–40 GroEL2 is the putative housekeeping molecular chaperone for folding proteins as it is essential for M. tuberculosis survival and contains the GGM repeat motif found in the canonical chaperonins of other organisms.35 Thus, targeting GroEL2 with small molecule inhibitors should be an effective strategy to kill actively replicating M. tuberculosis. While GroEL1 is not essential, it is important in regulating cytokine-dependent granuloma formation.35, 38 When infected with a strain of M. tuberculosis deficient in GroEL1 (Δcpn60.1), both mice and guinea pigs produced equal numbers of bacteria as the WT-GroEL1 strain, but the mutant strain failed to produce granulomatous inflammation.35 Thus, targeting GroEL1 with small molecule inhibitors might be effective at preventing the formation of granulomas that contribute to latent disease, or targeting bacteria that are already present in established granulomas. These enticing findings suggest that a small molecule that could concurrently inhibit both GroEL1 and GroEL2 in M. tuberculosis might be effective at treating active and latent stages of tuberculosis, which would be superior to current tuberculosis therapeutics.
Towards our goal of exploiting HSP60/10 and GroEL/ES chaperonin systems as an antibiotic strategy, we previously reported a high-throughput screen for small molecule inhibitors of the E. coli GroEL/ES chaperonin system.41 This initial study identified compound 1 as one of our most potent GroEL/ES inhibitors (Figure 1A). Our subsequent study found 1 lacked any appreciable antibacterial efficacy against a panel of Gram-positive and Gram-negative bacteria known as the ESKAPE pathogens – an acronym that stands for Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species.42 However, our third study identified isostructural benzoxazole analogs (2–14, Figure 1B) that exhibit antibiotic effects against Trypanosoma brucei parasites, the causative agents of African sleeping sickness. 43We refer to these as “pseudosymmetric full-molecules” as they have two sulfonamide end-capping groups with R1 substructures being identical on both the right and left-hand sides of the molecule. Furthermore, our fourth study identified 2-chlorothiophene-based analogs (15, 16R-34R, and 16L-34L, Figure 1C) that exhibited antibiotic effects against Gram-positive bacteria, in particular Staphylococcus aureus and methicillin-resistant S. aureus (MRSA).44 We refer to these as “asymmetric full-molecules” as they have two sulfonamide end-capping groups, but the substructures differ between the Right- and Left-hand sides of the molecule (i.e. R- and L-series full-molecules). Since the M. tuberculosis GroEL1 and GroEL2 homologs share 52% and 59% sequence identity with E. coli GroEL, respectively, we reasoned that they may also exhibit antibiotic effects against M. tuberculosis. Thus, in the present study, we evaluated the previously developed compound 1 analogs for their ability to inhibit M. tuberculosis growth in liquid media (see discussion below). Because we were unsure whether or not these compounds required both of the sulfonamide end-capping groups to maintain potent inhibition, we developed a new series that we term “half-molecules”, as they contain only one sulfonamide end-capping group on either the Right- or Left-hand sides of the molecule (R- and L-series half-molecules, Figure 1D). We also wanted to determine whether simplifying inhibitors in this manner might reduce the cytotoxicity of this series to human cells, while still maintaining antibiotic efficacy.
Figure 1.

Progression of compound 1 analog development and evaluation through our recent studies.41–44 In each panel, the R1-R3 groups represent a variety of alkyl, aryl, and ortho-, meta-, and para-substituted phenyl substructures (refer to the Tables S1–S6 in the Supporting Information for specific R-substructures). Each successive study (panels A-C) included testing of compounds developed from the previous studies in order to more thoroughly characterize SAR. Panel D presents compounds newly developed in the current study, which we term “half-molecules” as they contain only one sulfonamide end-capping group on either the Right- or Left-hand sides of the molecules (R- and L-series half-molecules). We have evaluated these, as well as all the previously-developed “full-molecules” (i.e. two sulfonamide end-capping groups as shown in panels A-C), for their ability to inhibit the proliferation of M. tuberculosis, as well as the protein tyrosine phosphatase B (PtpB) virulence factor that they secrete into macrophages to evade host cell immune responses.
Another possible strategy to treat tuberculosis infections is by targeting a virulence factor that M. tuberculosis secretes into the cytoplasm of host macrophages, protein tyrosine phosphatase B (PtpB). By secreting PtpB into the cytosol of host macrophages, M. tuberculosis disrupts host cell immune responses by blocking ERK1/2 and p38 mediated IL-6 production and promoting host cell survival by interfering with Akt signaling.45 Deletion of PtpB has been shown to block intracellular survival of M. tuberculosis in IFN-γ activated macrophages and reduce the bacterial load in a guinea pig model.45, 46 Several studies have shown the possibility of selectively inhibiting M. tuberculosis PtpB over human phosphatases, which can result from structural differences such as PtpB containing alpha-helices that can occlude the active site, yet are mobile enough to allow substrate and competitive inhibitors to bind.45, 47–54 Furthermore, previous studies have demonstrated the feasibility of inhibiting M. tuberculosis PtpB with small molecules and reducing bacterial loads in macrophages.45, 47, 48
In their 2007 study, Grundner et al. reported a crystal structure of PtpB in complex with the selective inhibitor (oxalylamino-methylene)-thiophene sulfonamide (OMTS – Figure 2A).50 In that structure, two OMTS molecules were found bound in the crystal structure, with the sulfonamides of each interacting with a number of ordered water molecules and binding site residues (e.g. Arg59, Arg63, His94, Glu129, and Arg136). Examining the crystal structure, we were intrigued that the sulfonamides of each OMTS molecule reside ~11–12 Åapart from one another (Figure 2B). Since the two sulfonamides of our compound 1 analogs are also ~11–12 Å apart (albeit in a head-to-head configuration, whereas the OMTS sulfonamides are head-to-tail), we envisioned that our analog series might also be capable of inhibiting M. tuberculosis PtpB in a conformation that would bridge across the two OMTS binding sites, making similar polar interactions as OMTS, while positioning the sulfonamide end-capping groups upwards to what would be the phosphotyrosine active site catalytic residues and adjacent pocket. This raises the enticing possibility that this class of molecules could inhibit the two M. tuberculosis GroEL homologs to target actively replicating mycobacteria and dormant mycobacteria in granulomas, as well as inhibit PtpB to target intracellular M. tuberculosis that are evading host cell immune responses. Thus, this class of inhibitors could be effective against all stages of M. tuberculosis infection. The present study was designed to explore such dual-targeting GroEL/ES and PtpB inhibitors in vitro and in cell culture, with future studies envisioned to build from the established structure-activity relationships (SAR) to further optimize lead candidates for evaluating in animal infection models.
Figure 2.

A. Grundner et al. previously reported that the OMTS compound was a potent and selective inhibitor of M. tuberculosis PtpB.50 B. Two OMTS molecules were found occupying the active site of M. tuberculosis PtpB (PDB ID 2OZ5). On examination, the sulfonamides of each OMTS molecule reside ~11–12 Å apart, similar to our compound 1 analogs. Thus, we envisioned compound 1 analogs might be able to bind in a manner that bridges the distal and proximal parts of the active site. In such a conformation, the sulfonamides could interact with the binding site similarly to those of OMTS, with the R-substructures pointing upwards to fill the binding cavity and engaging the catalytic residues located in the proximal part of the active site.
Extending from our previous studies that explored the bis-sulfonamide full-molecule series (compounds 1–34 – see Tables S1–S4 in the Supporting Information), we synthesized a library of half-molecule analogs that had the R3-substituted sulfonamide end caps on either the Right (R-series) or Left (L-series) sides of the 2-phenylbenzoxazole core (Scheme 1).43, 44 Representative synthetic protocols for the sulfonamide coupling and methoxy-to-hydroxy deprotection reactions are presented in the Supporting Information along with complete compound characterizations (1H-NMR, MS, and RPHPLC). These syntheses resulted in the development of 58 new half-molecules – 29 each of the R- and L-series analogs. As we previously found that aryl-sulfonamides were the most potent GroEL/ES inhibitors, we developed analogs bearing substituted phenyl-sulfonamides. Furthermore, we biased the analogs to contain a variety of halide substituents and substitution patterns as our previous antibacterial study indicated that halide-bearing compounds were typically more effective at inhibiting bacterial proliferation than compounds with other substituents.44
Scheme 1a.

a Reagents and conditions: (a) R3-SO2Cl, pyridine, CH2Cl2, rt; (b) BBr3, CH2Cl2, rt.
We next employed a series of established biochemical assays to evaluate the inhibitory effects of the new half-molecules against the GroEL/ES chaperonin system.41–44, 55 For these assays, we used E. coli GroEL/ES as a surrogate, as obtaining functional GroEL oligomeric rings from M. tuberculosis has so far proven difficult.12, 36, 37, 39 However, we anticipate a high probability that inhibitors would bind to and inhibit the GroEL/ES chaperonin systems in M. tuberculosis since E. coli GroEL shares 52% identity with the GroEL1 homolog, and 59% identity with GroEL2. We employed two chaperonin-mediated folding assays using malate dehydrogenase (MDH) and rhodanese (Rho) as the unfolded reporter enzymes. We have described these assays elsewhere, and have included detailed protocols of each in the Supporting Information.41–44, 55 Briefly, these are coupled assays where we monitor the enzymatic activity of two reporter enzymes (MDH and Rho), which are denatured and then get efficiently refolded by GroEL/ES in the absence of inhibition. We further counter-screened compounds for their ability to inhibit native MDH and Rho to identify false-positives that inhibit the enzymatic reporter reactions of the refolding assays. IC50 results for the testing of compounds in these assays are shown in Tables S5A–S6A in the Supporting Information. The pseudosymmetric and asymmetric full-molecules had been evaluated in these assays in our previous studies, and thus we refer readers to those publications for a detailed account of their results.43, 44
As visualized in the correlation plot in Figure 3A, compounds were nearly equipotent at inhibiting in both of the GroEL/ES-mediated folding assays (Spearman correlation coefficient is 0.923,p < 0.0001). As SAR for the full-molecule series has been more thoroughly discussed for these assays in our previous studies,43, 44 we will primarily present comparisons between the full vs. half-molecule scaffolds herein. In this context, the full-molecules were dramatically more potent than the half-molecules, indicating that the presence of both aryl-sulfonamide substructures are required for potent inhibition. While some compounds were found to inhibit in either the native MDH or Rho reporter activity counter-screens, only one previously reported compound (28R) inhibited in both native assays, and only weakly (Figure 3B).43, 44 As the IC50 values for all the compounds in the native MDH and Rho reporter counter-screens were much higher than in the corresponding refolding assays, these results support that compounds were on target for inhibiting the GroEL/ES-mediated folding cycle.
Figure 3.

Correlation plots of IC50 and EC50 values for compounds tested in the respective GroEL/ES-dMDH and dRho refolding assays (A), native MDH and Rho reporter counter-screens (B), and M. tuberculosis proliferation assay (C). Each data point represents results for an individual compound (plotted from results presented in Tables S1–S6 in the Supporting Information). Full molecules (i.e. compounds 1–34) are represented by blue circles, and half-molecules (i.e. compounds 35–51) are represented by yellow squares. A & B. Compounds inhibit nearly equipotently in the GroEL/ES-dMDH and -dRho refolding assays (Spearman correlation coefficient is 0.923, p < 0.0001), but are weak or inactive in the native MDH or Rho enzymatic reporter counter-screens, supporting on-target effects for inhibiting chaperonin-mediated substrate refolding. As indicated in panel A, we consider compounds with IC50 values plotted in the grey zones to be inactive (i.e. greater than the maximum concentrations tested), >30 μM to be weak inhibitors, 10–30 μM moderate inhibitors, 1–10 μM potent inhibitors, and <1 μM very potent and acting near stoichiometrically since the concentration of GroEL subunits is 700 nM. C. Some GroEL/ES inhibitors exhibit weak-to-moderate inhibition of M. tuberculosis proliferation, but the correlation with inhibiting GroEL/ES refolding functions in vitro is weak (Spearman correlation coefficient is 0.439, p = 0.0011). Compounds showing <50% inhibition of M. tuberculosis proliferation when tested at a single 200 μM concentration were excluded from this analysis.
We next evaluated how effective our GroEL/ES inhibitors would be at inhibiting the growth of M. tuberculosis in liquid culture, as inhibiting the essential GroEL2 homolog should be bactericidal. We tested the ability of each compound to inhibit growth at a fixed concentration of 200 μM after 5 days (see the Supporting Information for the assay protocol).56 Compounds exhibiting >50% inhibition were then tested as serial dilutions in order to determine EC50 values, defined as the concentration at which growth was inhibited by 50%. Percent inhibition and EC50 results are presented in Tables S1–S6 in the Supporting Information. Intriguingly, while the half-molecules were twice as likely to exhibit >50% inhibition at the single 200 μM test concentration (28/58, or 48%) compared to the full-molecules (22/89, or 25%), the only analogs with EC50s <100 μM were the full molecules. This latter finding was not entirely surprising since the full-molecules were much more potent GroEL/ES inhibitors than the half-molecules. While this coarse SAR helps support that inhibitors are on target for GroEL/ES in mycobacteria, we note only a weak correlation between M. tuberculosis EC50 results compared to IC50 results for the GroEL/ES-dMDH refolding assay (Figure 3C – Spearman correlation coefficient is 0.439, p = 0.0011). More thorough experiments need to be conducted in the future to conclusively determine the mechanism of action in cells. While the most potent compounds exhibited only moderate EC50s in the 20–30 μM range, these were encouraging results since this was the first exploratory study we have conducted to see whether or not GroEL/ES inhibitors would have antibiotic efficacy against M. tuberculosis. Thus, the current results have provided valuable insights to guide future pharmacological optimization studies.
A potential caveat to targeting GroEL/ES chaperonin systems as an antibiotic strategy is that human cells contain homologous machinery in their mitochondria. Human mitochondrial HSP60 shares 48% sequence identity with E. coli GroEL (similar to the M. tuberculosis GroEL1 and GroEL2 homologs), which raises the possibility of potential HSP60-dependent cytotoxicity to human cells. However, as we continue to study this and other chaperonin inhibitor scaffolds, we become less and less concerned with potential cytotoxicities associated with targeting human HSP60/10. In many instances, compounds that we have found to inhibit HSP60/10 in vitro exhibit low-to-no cytotoxicity when tested in our liver and kidney cell viability assays.42, 43, 55 Furthermore, we have recently identified numerous known drugs and natural products that are able to inhibit HSP60/10 in vitro.43, 55, 57 For example, while we found that suramin can inhibit human HSP60/10 in vitro, it has been safely used for over 100 years as a first-line therapeutic for treating African sleeping sickness, which is caused by Trypanosoma brucei parasites. Nonetheless, we continue to counter-screen compounds in our standard HSP60/10-dMDH refolding assay as it does prove useful for helping to understand inhibitor mechanisms of action at the protein level. This assay is analogous to the GroEL/ES-dMDH assay so that IC50 results can be directly compared between the two chaperonin systems. A detailed protocol is presented in the Supporting Information, with results for the half-molecules presented in Tables S5A–S6A. For an account of results for the pseudosymmetric and asymmetric full-molecules, we refer readers to our previous studies.43, 44
As we observed with GroEL/ES, the full-molecules were more potent at inhibiting HSP60/10 than the half-molecules (Figure 4A). While some compounds were nearly equipotent between the two chaperonin systems, many exhibited selectivity towards E. coli GroEL/ES. We previously noted this SAR for the pseudosymmetric (1–15) and R-series asymmetric full molecules (16R-34R), demonstrating that it is possible to tune this scaffold to selectively target GroEL/ES.43, 44 However, as noted above, this may be inconsequential for cytotoxicity purposes as the mitochondrial membrane is highly impermeable to penetration by small molecules, and thus inhibitors may never reach HSP60/10 in the mitochondrial matrix.
Figure 4.

Correlation plots of IC50 and CC50 values for compounds tested in the respective GroEL/ES-dMDH and HSP60/10-dMDH refolding assays (A), and liver/kidney cell viability assays (B & C). A. While many compounds inhibit human HSP60/10 nearly as well as E. coli GroEL/ES (Spearman correlation coefficient for all molecules is 0.740, p < 0.0001), we previously reported being able to tune for selectivity with the pseudosymmetric and asymmetric R-series full molecules.43, 44 B. Cytotoxicities of compounds are comparable between human THLE-3 liver and HEK 293 kidney cells (Spearman correlation coefficient is 0.857, p < 0.0001). C. HSP60/10 inhibitors exhibit weak-to-moderate cytotoxicity to human THLE-3 liver cells, with a low correlation evident between the two assays (Spearman correlation coefficient is 0.244, p = 0.0027).
As a moderate-throughput first-pass indicator of cellular toxicity, we typically employ two Alamar Blue-based cell viability assays, where compounds are incubated with human liver (THLE-3) and kidney (HEK 293) cells over a 48 h time period. A detailed protocol for these assays is presented in the Supporting Information, with cell viability results (cytotoxicity CC50 values) presented in Tables S5B–S6B. We again refer readers to our previous studies for cell viability CC50 results for the pseudosymmetric and asymmetric full-molecules (compounds 1–34).43, 44 Similar to results from the biochemical assays, the full-molecules were more potent inhibitors (i.e. more cytotoxic) than the half-molecules were in the two cell viability assays (Figure 4B). However, we note that there is a poor correlation when comparing IC50 and CC50 results from the HSP60/10-dMDH and cell viability assays (Figure 4C – Spearman correlation coefficient is 0.244, p = 0.0027 when comparing with liver cells), which could indicate off-target effects not related to inhibiting the HSP60/10 chaperonin system, and/or potential cell or mitochondrial permeability differences between compounds. These possibilities highlight that results from the cell viability assays are the critical indicators of potential cytotoxic effects that may be encountered in vivo.
While the above results support the feasibility of identifying GroEL/ES inhibitors that can kill actively replicating M. tuberculosis, we were particularly intrigued by the possibility of this series also being able to target the protein tyrosine phosphatase B (PtpB) virulence factor that intracellular M. tuberculosis secrete into macrophages to evade host cell immune responses. To investigate this possibility, we obtained a His-tagged version of M. tuberculosis PtpB, which was previously developed by Grundner et al., to generate recombinantly-expressed and purified enzyme.49, 50 Following previously reported procedures by Zhou et al., which monitored for phosphatase activity using paranitrophenyl phosphate (pNPP), we evaluated all compounds in dose-response format to obtain IC50 values.45, 47, 49, 50 As a preliminary indication of selectively targeting M. tuberculosis PtpB, we counter-screened against three human phosphatases, PTPN1 (PTP1B), PTPN2 (TCPTP), and PTPN5 (STEP), using analogous procedures. Detailed protocols are presented in the Supporting Information for these assays, with IC50 results for all compounds presented in Tables S1–S6.
While both the full- and half-molecules were able to inhibit M. tuberculosis PtpB phosphatase activity, as noted above for the other assays, the full-molecules were generally more potent than the half-molecules (Figure 5A). An interesting discovery was that, while the full-molecules all inhibit M. tuberculosis PtpB activity with increasing compound concentrations, most half-molecules exhibit a biphasic response first activating, then inhibiting phosphatase activity (Figure 5B). This could be the result of two half-molecules binding to PtpB in a manner like that previously observed for OMTS.50 Thus, binding of the first molecule at the distal part of the active site may open the alpha-helical lid, allowing pNPP to more effectively occupy the proximal part next to the catalytic residues. Then at higher concentrations, the second half-molecule could competitively displace the pNPP, thereby showing inhibition. It should be noted, though, that this may be an artifact of this particular assay protocol using pNPP, as binding to either site would theoretically compete with a phosphorylated peptide. Thus, IC50 values could be more potent in a physiological context than what we have reported (presuming binding affinities were high enough), although future studies would need to confirm this.
Figure 5.

A. Correlation plot of IC50 values for compounds tested in the GroEL/ES-dMDH refolding and M. tuberculosis PtpB phosphatase assays (Spearman correlation coefficient is 0.756, p < 0.0001). B. Dose-response curves for analogs 2h-m, R51-m, and L51-m evaluated in the M. tuberculosis PtpB phosphatase assay. While the full-molecules all inhibit M. tuberculosis PtpB phosphatase activity with increasing compounds concentrations, most half-molecules exhibit a bi-phasic response where compounds activate then inhibit phosphatase activity, to varying extents, as shown for analogs R51-m and L51-m. Data point errors represent standard deviations from 6–8 replicates.
While we were excited to see so many GroEL/ES inhibitors were also able to potently inhibit PtpB, we were intrigued that such a high correlation was observed between the two data sets (Spearman correlation coefficient is 0.756, p < 0.0001) as there is no a priori reason why the binding sites of GroEL and PtpB would be similar. However, we note that the SAR is relatively flat for the full-molecules for inhibiting PtpB (i.e. most are within the 1–10 μM range), which could be consistent with the bissulfonamide core predominantly contributing to binding affinity and the end-capping groups not fully occupying the remainder of the active site. This is important since, on the whole, this scaffold class exhibits only slight selectivity for M. tuberculosis PtpB over the three human phosphatases (Figure 6). Thus, with extra space that compounds could more fully occupy in the active site, there should be room to optimize inhibitor potency and selectivity profiles against M. tuberculosis PtpB. This also boosts confidence that there should be ample chemical space to simultaneously optimize inhibition of GroEL/ES and PtpB function to target M. tuberculosis in both active and latent stages of infection. Such optimization studies will be the focus of future investigations.
Figure 6.

Correlation plots of IC50 values for compounds tested against the M. tuberculosis PtpB phosphatase compared to the human PTPN1 (PTP1B, panel A), PTPN2 (TCPTP, panel B), and PTPN5 (STEP, panel C). While some individual compounds are able to selectively inhibit the M. tuberculosis PtpB over the human phosphatases, on the whole, this scaffold class only exhibits a slight selectivity for the bacterial phosphatase – Spearman correlation coefficients are 0.828 (p < 0.0001) for PTPN1, 0.755 (p < 0.0001) for PTPN2, and 0.786 (p < 0.0001) for PTPN5.
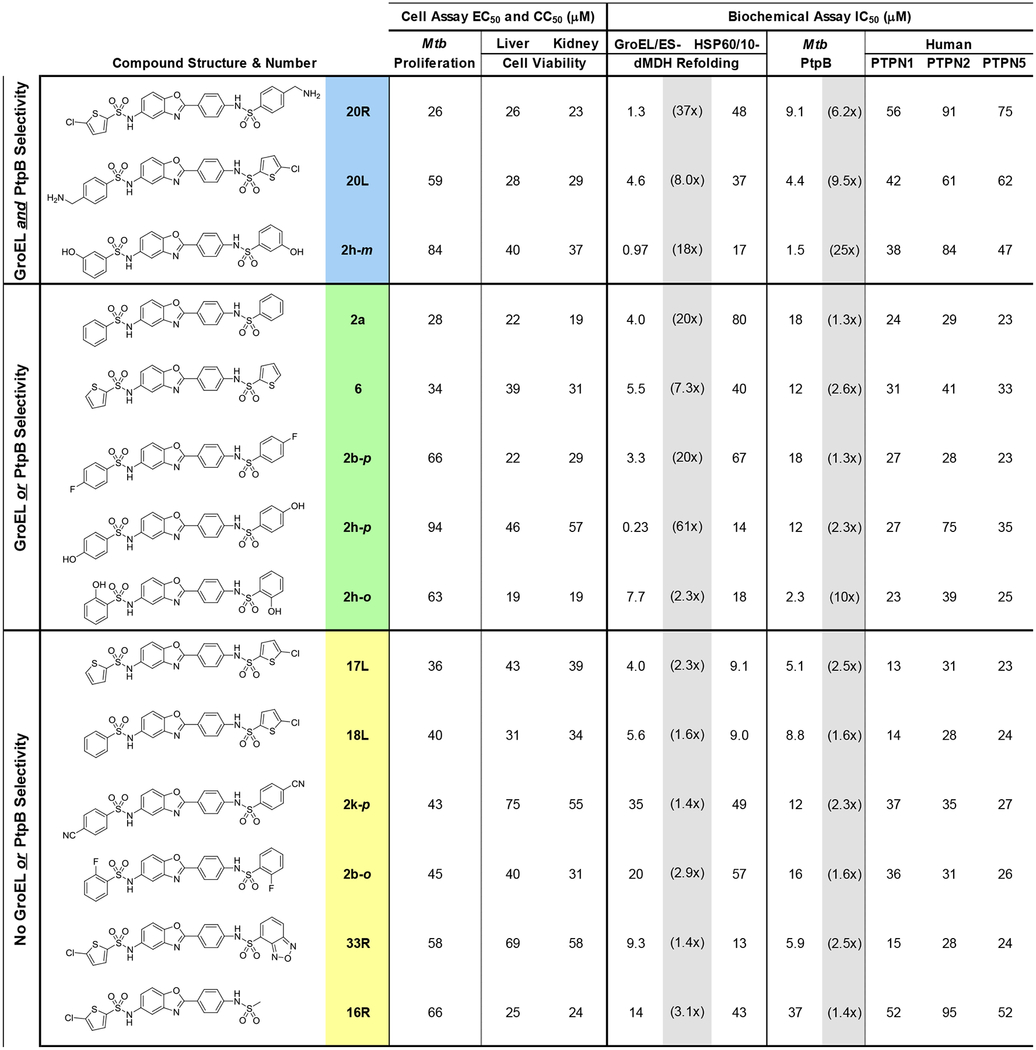
In summary, in the present study, we evaluated whether or not a series of GroEL/ES inhibitors, based on the compound 1 scaffold, would be effective at inhibiting the growth of M. tuberculosis. Furthermore, we envisioned that these could be dual-targeting molecules capable of inhibiting the M. tuberculosis GroEL/ES chaperonin systems as well as the PtpB virulence factor that they secrete into macrophages. This study identified 14 GroEL/ES inhibitors that exhibited EC50 values <100 μM against M. tuberculosis growth in liquid culture (Table 1). We have categorized these into three groups, depending on their selectivity profiles for inhibiting GroEL/ES over human HSP60/10, as well as for inhibiting M. tuberculosis PtpB over the panel of three human phosphatases. Compounds within the blue category exhibit selectivity for both GroEL/ES and PtpB; compounds within the green category exhibit selectivity for either GroEL/ES or PtpB, but not both; and compounds in the yellow category are not inherently selective. From our continued studies, though, we note that even if compounds can inhibit human HSP60/10 in vitro, many exhibit only low or no cytotoxicity to human cells. Furthermore, as PtpB is highly adaptable to compounds that bind within the active site, there should still be ample chemical space to optimize inhibitors for binding affinity, efficacy, and selectivity.
Table 1.
Biochemical and cell-based assay results for lead analogs exhibiting M. tuberculosis proliferation EC50 values <100 μM. Fold-selectivities for inhibiting GroEL/ES over human HSP60/10, and M. tuberculosis PtpB over the three human phosphatases, are shown in brackets in the shaded columns. Compounds colored blue exhibit >5x selectivities for both GroEL and PtpB; compounds colored green exhibit >5x selectivities for either GroEL or PtpB (but not both); and compounds colored yellow exhibit between 1–5x selectivities for GroEL or PtpB.
|
Two of the lead compounds we identified were 20R and 20L, which had primary amines either on the right or left-hand sides of the scaffold. These were unique compounds within this study as they were the only analogs to contain primary amines that would be charged under physiological conditions. Thus, future studies will explore new analogs that contain amines on either the right or left-hand sulfonamides, while varying groups on the other side. The 2h-o/m/p analogs were also unique lead molecules – the only hydroxylated full-molecule analogs – to which a similar SAR strategy will be applied. In either case, additional areas of exploration to optimize inhibitors are varying the sulfonamide linkers and employing scaffolds other than the 2-phenylbenzoxazole core. By doing so, we are optimistic that we will be able to enhance the potency and selectivity of lead inhibitors for M. tuberculosis GroEL/ES chaperonin systems and PtpB, and increase their selectivity indices for inhibiting M. tuberculosis proliferation over cytotoxicity to human cells. Once we have developed more viable lead candidates, we will pursue additional studies looking at the effects of inhibiting M. tuberculosis PtpB in macrophage models, as well as antibiotic efficacy in an in vivo infection model. With an antibiotic strategy that concomitantly inhibits the two M. tuberculosis GroEL homologs and PtpB, these polypharmacologic agents are predicted to target actively replicating bacteria, intracellular bacteria that are evading host cell immune responses, and dormant bacteria that have formed granulomas. Thus, we are hopeful that this could be an effective strategy for treating all stages of tuberculosis.
Supplementary Material
Acknowledgments:
Research reported in this publication was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under Award Number R01GM120350. QQH and YP additionally acknowledge support by NIH grants 5R01GM111639 and 5R01GM115844. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was also supported by startup funds from the IU School of Medicine (SMJ) and the University of Arizona (EC). Technical assistance from Megha Gupta, Junitta Guzman, and Aaron Korkegian (IDRI, Seattle, WA) is also greatly appreciated. The human HSP60 expression plasmid (lacking the 26 amino acid N-terminal mitochondrial signal peptide) was generously donated by Dr. Abdussalam Azem from Tel Aviv University, Faculty of Life Sciences, Department of Biochemistry, Israel. We thank Dr. Zhong-Yin Zhang (Purdue University, West Lafayette, IN, USA) for providing the M. tuberculosis PtpB expression plasmid, with permission from Dr. Christoph Grundner (Center for Infectious Disease Research, Seattle, WA, USA), who originally developed the plasmid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information: Supporting information associated with this article can be found in the online version, which includes tabulation of all biochemical IC50, bacterial proliferation EC50, and human cell viability CC50 results; log(IC50), log(EC50), and log(CC50) results with standard deviations; synthetic protocols and characterization data for test compounds (HPLC purity, MS, and 1H-NMR); experimental protocols for protein synthesis and purification, and all biochemical and cell-based assays.
References
- 1.World Health Organization. Global Tuberculosis Report 2018; Geneva, Switzerland: World Health Organization, 2018. [Google Scholar]
- 2.Flynn JL; Chan J Infect. Immun 2001, 69, 4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pai M; Behr MA; Dowdy D; Dheda K; Divangahi M; Boehme CC; Ginsberg A; Swaminathan S; Spigelman M; Getahun H; Menzies D; Raviglione M Nat. Rev. Dis. Primers 2016, 2, 16076. [DOI] [PubMed] [Google Scholar]
- 4.Smith I Clin. Microbiol. Rev 2003, 16, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. Treatment of Tuberculosis: Guidelines.; Geneva, Switzerland: World Health Organization, 2010. [Google Scholar]
- 6.Gill WP; Harik NS; Whiddon MR; Liao RP; Mittler JE; Sherman DR Nat. Med 2009, 15, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Ambrosio L; Centis R; Tiberi S; Tadolini M; Dalcolmo M; Rendon A; Esposito S; Migliori GB J. Thorac. Dis 2017, 9, 2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Migliori GB; Pontali E; Sotgiu G; Centis R; D’Ambrosio L; Tiberi S; Tadolini M; Esposito S Int. J. Mol. Sci 2017, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen TVA; Anthony RM; Banuls AL; Nguyen TVA; Vu DH; Alffenaar JC Clin. Infect. Dis 2018, 66, 1625. [DOI] [PubMed] [Google Scholar]
- 10.Fujiwara M; Kawasaki M; Hariguchi N; Liu Y; Matsumoto M Tuberculosis (Edinb) 2018, 108, 186. [DOI] [PubMed] [Google Scholar]
- 11.Stinson K; Kurepina N; Venter A; Fujiwara M; Kawasaki M; Timm J; Shashkina E; Kreiswirth BN; Liu Y; Matsumoto M; Geiter L Antimicrob. Agents Chemother 2016, 60, 3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lupoli TJ; Vaubourgeix J; Burns-Huang K; Gold B ACS Infect. Dis 2018, 4, 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar A; Balbach J Sci. Rep 2017, 7, 42141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiappori F; Fumian M; Milanesi L; Merelli I Plos One 2015, 10, e0124563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arita-Morioka K; Yamanaka K; Mizunoe Y; Ogura T; Sugimoto S Antimicrob. Agents Chemother 2015, 59, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sass P; Josten M; Famulla K; Schiffer G; Sahl HG; Hamoen L; Brotz-Oesterhelt H Proc. Natl. Acad. Sci. U.S.A 2011, 108, 17474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans CG; Chang L; Gestwicki JE J. Med. Chem 2010, 53, 4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piper PW; Millson SH Open Biol. 2012, 2, 120138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liebscher M; Jahreis G; Lucke C; Grabley S; Raina S; Schiene-Fischer C. J. Biol. Chem 2007, 282, 4437. [DOI] [PubMed] [Google Scholar]
- 20.Otvos L Jr.; O I; Rogers ME; Consolvo PJ; Condie BA; Lovas S; Bulet P; Blaszczyk-Thurin M Biochemistry 2000, 39, 14150. [DOI] [PubMed] [Google Scholar]
- 21.Otvos L Jr.; Wade JD; Lin F; Condie BA; Hanrieder J; Hoffmann RJ Med. Chem 2005, 48, 5349. [DOI] [PubMed] [Google Scholar]
- 22.Kragol G; Hoffmann R; Chattergoon MA; Lovas S; Cudic M; Bulet P; Condie BA; Rosengren KJ; Montaner LJ; Otvos L Jr. Eur. J. Biochem 2002, 269, 4226. [DOI] [PubMed] [Google Scholar]
- 23.Kragol G; Lovas S; Varadi G; Condie BA; Hoffmann R; Otvos L Jr. Biochemistry 2001, 40, 3016. [DOI] [PubMed] [Google Scholar]
- 24.Mohammadi-Ostad-Kalayeh S; Hrupins V; Helmsen S; Ahlbrecht C; Stahl F; Scheper T; Preller M; Surup F; Stadler M; Kirschning A; Zeilinger C Bioorg. Med. Chem 2017, 25, 6345. [DOI] [PubMed] [Google Scholar]
- 25.Chapman E; Farr GW; Usaite R; Furtak K; Fenton WA; Chaudhuri TK; Hondorp ER; Matthews RG; Wolf SG; Yates JR; Pypaert M; Horwich AL Proc. Natl. Acad. Sci. U.S.A 2006, 103, 15800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartl FU; Bracher A; Hayer-Hartl M Nature 2011, 475, 324. [DOI] [PubMed] [Google Scholar]
- 27.Hartl FU; Hayer-Hartl M Science 2002, 295, 1852. [DOI] [PubMed] [Google Scholar]
- 28.Stefani M; Dobson CM J. Mol. Med. (Berl.) 2003, 81, 678. [DOI] [PubMed] [Google Scholar]
- 29.Maisonneuve E; Ezraty B; Dukan S J. Bacter 2008, 190, 6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carmichael J; Chatellier J; Woolfson A; Milstein C; Fersht AR; Rubinsztein DC Proc. Natl. Acad. Sci. U.S.A 2000, 97, 9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bao YP; Cook LJ; O’Donovan D; Uyama E; Rubinsztein DC J. Biol. Chem 2002, 277, 12263. [DOI] [PubMed] [Google Scholar]
- 32.Horwich AL; Fenton WA; Chapman E; Farr GW Annu. Rev. Cell. Dev. Biol 2007, 23, 115. [DOI] [PubMed] [Google Scholar]
- 33.Horwich AL; Farr GW; Fenton WA Chem. Rev 2006, 106, 1917. [DOI] [PubMed] [Google Scholar]
- 34.Kong TH; Coates ARM; Butcher PD; Hickman CJ; Shinnick TM Proc. Natl. Acad. Sci. U.S.A 1993, 90, 2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu YM; Henderson B; Lund PA; Tormay P; Ahmed MT; Gurcha SS; Besra GS; Coates ARM Infect. Immun 2008, 76, 1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qamra R; Mande SC; Coates AR; Henderson B Tuberculosis (Edinb) 2005, 85, 385. [DOI] [PubMed] [Google Scholar]
- 37.Qamra R; Srinivas V; Mande SC J. Mol. Biol 2004, 342, 605. [DOI] [PubMed] [Google Scholar]
- 38.Hu YM; Coates ARM; Liu A; Lund PA; Henderson B Tuberculosis 2013, 93, 442. [DOI] [PubMed] [Google Scholar]
- 39.Fan M; Rao T; Zacco E; Ahmed MT; Shukla A; Ojha A; Freeke J; Robinson CV; Benesch JL; Lund PA Mol. Microbiol 2012, 85, 934. [DOI] [PubMed] [Google Scholar]
- 40.Henderson B; Lund PA; Coates AR Tuberculosis (Edinb) 2010, 90, 119. [DOI] [PubMed] [Google Scholar]
- 41.Johnson SM; Sharif O; Mak PA; Wang HT; Engels IH; Brinker A; Schultz PG; Horwich AL; Chapman E Bioorg. Med. Chem. Lett 2014, 24, 786. [DOI] [PubMed] [Google Scholar]
- 42.Abdeen S; Salim N; Mammadova N; Summers CM; Frankson R; Ambrose AJ; Anderson GG; Schultz PG; Horwich AL; Chapman E; Johnson SM Bioorg. Med. Chem. Lett 2016, 26, 3127. [DOI] [PubMed] [Google Scholar]
- 43.Abdeen S; Salim N; Mammadova N; Summers CM; Goldsmith-Pestana K; McMahon-Pratt D; Schultz PG; Horwich AL; Chapman E; Johnson SM Bioorg. Med. Chem. Lett 2016, 26, 5247. [DOI] [PubMed] [Google Scholar]
- 44.Abdeen S; Kunkle T; Salim N; Ray AM; Mammadova N; Summers C; Stevens M; Ambrose AJ; Park Y; Schultz PG; Horwich AL; Hoang QQ; Chapman E; Johnson SM J. Med. Chem 2018, 61, 7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou B; He Y; Zhang X; Xu J; Luo Y; Wang Y; Franzblau SG; Yang Z; Chan RJ; Liu Y; Zheng J; Zhang ZY Proc. Natl. Acad. Sci. U.S.A 2010, 107, 4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh R; Rao V; Shakila H; Gupta R; Khera A; Dhar N; Singh A; Koul A; Singh Y; Naseema M; Narayanan PR; Paramasivan CN; Ramanathan VD; Tyagi AK Mol. Microbiol 2003, 50, 751. [DOI] [PubMed] [Google Scholar]
- 47.Chen L; Zhou B; Zhang S; Wu L; Wang Y; Franzblau SG; Zhang ZY ACS Med. Chem. Lett 2010, 1, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beresford NJ; Mulhearn D; Szczepankiewicz B; Liu G; Johnson ME; Fordham-Skelton A; Abad-Zapatero C; Cavet JS; Tabernero L J. Antimicrob. Chemother 2009, 63, 928. [DOI] [PubMed] [Google Scholar]
- 49.Grundner C; Ng HL; Alber T Structure 2005, 13, 1625. [DOI] [PubMed] [Google Scholar]
- 50.Grundner C; Perrin D; Hooft van Huijsduijnen R; Swinnen D; Gonzalez J; Gee CL; Wells TN; Alber T Structure 2007, 15, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiaradia LD; Martins PG; Cordeiro MN; Guido RV; Ecco G; Andricopulo AD; Yunes RA; Vernal J; Nunes RJ; Terenzi H J. Med. Chem 2012, 55, 390. [DOI] [PubMed] [Google Scholar]
- 52.Mascarello A; Mori M; Chiaradia-Delatorre LD; Menegatti AC; Delle Monache F; Ferrari F; Yunes RA; Nunes RJ; Terenzi H; Botta B; Botta M Plos One 2013, 8, e77081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mascarello A; Chiaradia-Delatorre LD; Mori M; Terenzi H; Botta B Curr. Pharm. Design 2016, 22, 1561. [DOI] [PubMed] [Google Scholar]
- 54.Mascarello A; Orbem Menegatti AC; Calcaterra A; Martins PGA; Chiaradia-Delatorre LD; D’Acquarica I; Ferrari F; Pau V; Sanna A; De Logu A; Botta M; Botta B; Terenzi H; Mori M Eur. J. Med. Chem 2018, 144, 277. [DOI] [PubMed] [Google Scholar]
- 55.Kunkle T; Abdeen S; Salim N; Ray AM; Stevens M; Ambrose AJ; Victorino J; Park Y; Hoang QQ; Chapman E; Johnson SM J. Med. Chem 2018, 61, 10651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ollinger J; Bailey MA; Moraski GC; Casey A; Florio S; Alling T; Miller MJ; Parish T Plos One 2013, 8, e60531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stevens M; Abdeen S; Salim N; Ray AM; Washburn A; Chitre S; Sivinski J; Park Y; Hoang QQ; Chapman E; Johnson SM Bioorganic & medicinal chemistry letters 2019, 29, 1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.