

Our study describes the first in vivo transposon sequencing (Tn-seq) analysis of B. pertussis and identifies genes predicted to be essential for in vivo growth in a murine model of intranasal infection, generating key resources for future investigations into B. pertussis pathogenesis and vaccine design.
KEYWORDS: Bordetella, Bordetella pertussis, Tn-seq, gene essentiality, in vivo, metabolism
ABSTRACT
Bordetella pertussis is the causative agent of whooping cough, a serious respiratory illness affecting children and adults, associated with prolonged cough and potential mortality. Whooping cough has reemerged in recent years, emphasizing a need for increased knowledge of basic mechanisms of B. pertussis growth and pathogenicity. While previous studies have provided insight into in vitro gene essentiality of this organism, very little is known about in vivo gene essentiality, a critical gap in knowledge, since B. pertussis has no previously identified environmental reservoir and is isolated from human respiratory tract samples. We hypothesize that the metabolic capabilities of B. pertussis are especially tailored to the respiratory tract and that many of the genes involved in B. pertussis metabolism would be required to establish infection in vivo. In this study, we generated a diverse library of transposon mutants and then used it to probe gene essentiality in vivo in a murine model of infection. Using the CON-ARTIST pipeline, 117 genes were identified as conditionally essential at 1 day postinfection, and 169 genes were identified as conditionally essential at 3 days postinfection. Most of the identified genes were associated with metabolism, and we utilized two existing genome-scale metabolic network reconstructions to probe the effects of individual essential genes on biomass synthesis. This analysis suggested a critical role for glucose metabolism and lipooligosaccharide biosynthesis in vivo. This is the first genome-wide evaluation of in vivo gene essentiality in B. pertussis and provides tools for future exploration.
IMPORTANCE Our study describes the first in vivo transposon sequencing (Tn-seq) analysis of B. pertussis and identifies genes predicted to be essential for in vivo growth in a murine model of intranasal infection, generating key resources for future investigations into B. pertussis pathogenesis and vaccine design.
INTRODUCTION
Whooping cough, caused by the Gram-negative bacterium Bordetella pertussis, is a serious respiratory illness in children and adults that can be fatal (1). Despite widespread vaccination, whooping cough has reemerged in recent years, reinvigorating research into B. pertussis pathogenesis and vaccine design (2). Humans are the only natural host for B. pertussis, but animal models, including baboons and mice (3–6), have been developed to facilitate study. Compared with other Bordetellae, the B. pertussis genome is smaller with more pseudogenes (9.4% of the genome) and substantial rearrangements mediated by insertion elements (7). This restricted genetic content is thought to be a result of adaptation to the human host (7); however, the genetic elements and changes that facilitate host specificity have not been identified.
Some insight into B. pertussis-host interaction has been gained by single gene mutations and transposon mutagenesis in vitro (8–10). Select mutants with altered virulence phenotypes in vitro have subsequently been evaluated for the effect of mutation on B. pertussis survival in vivo, identifying virulence genes important for infection (11, 12). More recently, high-throughput transposon sequencing (Tn-seq) has been used to identify genes essential for in vitro growth of B. pertussis (13). Unlike traditional transposon mutagenesis, which is based on the isolation of individual mutant clones in a transposon library, Tn-seq is based on sequencing of a pool of clones as a community. Because all members of the population are competing for the same limited nutrients, metabolism-associated genes are most often identified by Tn-seq, and Fyson et al. utilized Tn-seq to validate a computational model of B. pertussis metabolism in vitro (13).
Because metabolism-associated genes are central to host-pathogen specificity, we used Tn-seq to probe in vivo gene essentiality in B. pertussis. We generated a transposon library with high insertion coverage and measured growth in vitro and in vivo using an intranasal model of murine infection. These studies reveal novel observations about B. pertussis metabolism, gene regulation, and metabolism-associated virulence factors in vitro and within the host environment.
RESULTS
Construction and validation of Tn library.
The B. pertussis library was constructed through introduction of the pSAM-Km transposon (14) into B. pertussis strain UT25-lux (as described in Text S1 in the supplemental material) by conjugation with an Escherichia coli diaminopimelic acid (DAP) auxotrophic donor strain (15). The library contains approximately 3.0 × 106 clones, determined by enumeration of CFU of the library before harvest from BG plates. High-throughput sequencing on the Illumina Hi-Seq platform revealed that 73.6% of the possible insertion sites (based on the TA insertion site sequence for mariner transposons) and 88.5% of genes were occupied by insertions, which is representative of a highly complex library. Figure 1 shows the distribution of insertions throughout the genome (16). The high insertion coverage occurred despite high GC content of the genome (7, 17).
FIG 1.

B. pertussis transposon library. A map of insertions (red) and their distribution across the genome was generated by CGView (16). Coding regions of genes (coding sequences [CDS]) are shown in blue and separated by strand. Locations of tRNA and rRNA are shown in purple and green, respectively. GC content is represented in black.
Supplemental methods. Download Text S1, DOCX file, 0.04 MB (38.6KB, docx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
High- and low-density libraries can be distinguished by the separation between peaks on a histogram plotting the percentage of potential insertion sites occupied per gene (Fig. 2A); a more-dense library is characterized by a distinct and greater magnitude difference between peaks (18). The greatest number of genes with a low percentage of TA site occupation is considered the essential gene peak, and the genes with the highest percentage of TA site occupation form the second, nonessential peak. As demonstrated in Fig. 2A, there is a clear separation between the two peaks, which is consistent with a well-saturated library.
FIG 2.

Analysis of in vitro gene essentiality using TRANSIT and ARTIST. (A) Histogram showing the TA site insertion saturation per gene. (B) A Venn diagram of genes identified as essential by ARTIST (red) and TRANSIT (blue). A total of 609 genes were identified by both analysis pipelines (purple).
Tn-seq data (from samples grown in vitro on BG agar) were analyzed using two analysis programs, TRANSIT (19) and ARTIST (20), that both utilize a hidden Markov model (HMM). In Tn-seq analysis, HMMs identify the most probable essentiality state of a region by assessing a sequence of TA sites and incorporating information from neighboring TA sites in order to improve the accuracy of the essentiality determination (21). These programs classify genes as essential based on the essentiality classification of regions within a gene. Classification of a gene as essential presumes that gene loss is associated with an impairment of growth that can be inferred by the number of transposon insertions present in that gene. Using these two HMM-based methods, we generated a list of 609 genes (Fig. 2B; see also Table S2 in the supplemental material) that were classified as essential by both programs and are required for efficient in vitro growth (Text S1).
Genes identified as essential by conservative analysis. In this conservative method, only genes with no TA site hits in the first 95% of gene were considered essential. Based on five input samples grown on BG media, the analysis identified 400 essential genes (including tRNAs and rRNAs). Download Table S1, XLSX file, 0.01 MB (12.6KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Full list of genes identified by ARTIST and TRANSIT. This list includes lists of genes identified individually by ARTIST and TRANSIT as well as the list of 609 genes identified by both programs. It also includes domain essential genes identified by ARTIST. Download Table S2, XLSX file, 0.05 MB (52.5KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We further analyzed the set of genes classified as essential by both TRANSIT and ARTIST, presuming that genes within this combined set were more likely than either set individually to represent genes that are truly essential in vitro. Identification of multiple essential genes within the same metabolic pathway would suggest the importance of that pathway for B. pertussis metabolism and viability. To organize the data, we manually added KEGG pathway classifications to 499 essential genes, which for many genes resulted in more than one annotation per gene (Table S3). The remaining 110 essential genes lacked an identified pathway classification. Mapping to KEGG pathways revealed information about essential metabolic pathways under our growth conditions (Table S3 and Fig. S1). The most represented categories were biosynthesis of secondary metabolites, biosynthesis of antibiotics, ribosome, microbial metabolism in diverse environments, biosynthesis of amino acids, and metabolism of cofactors and vitamins.
Representation of in vitro (thick black lines) and in vivo (red lines) essential genes within the network of bacterial metabolic processes. Colored boxes indicate the approximate locations of selected metabolic reactions associated with a related end product or substrate, including lipooligosaccharide synthesis (green), pentose phosphate (violet), glycolysis and gluconeogenesis (blue), TCA cycle (orange), amino acid synthesis (dark gray), vitamin and cofactor synthesis (yellow), and lipid metabolism (light gray). The figure was created using the KEGG Mapper function on the KEGG database. Download FIG S1, TIF file, 2.5 MB (2.5MB, tif) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
KEGG pathway assignments for in vitro essential genes. Download Table S3, XLSX file, 0.03 MB (35.3KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Multiple genes within the gluconeogenesis and pentose phosphate pathways were classified as essential (Fig. 3), supporting prior findings that these pathways are present and functional in B. pertussis (7). Originally, the tricarboxylic acid (TCA) cycle in B. pertussis was considered nonfunctional despite the observation that the genome contains all required genes (7, 22). Recently, the enzymatic activities thought to be absent (citrate synthase, aconitase, and isocitrate dehydrogenase) in B. pertussis were shown to be present (23). In support of these findings, our data indicated that genes encoding citrate synthase and isocitrate dehydrogenase are essential in vitro in our screen, as well as the majority (all but two) of the remaining genes assigned to reactions within the tricarboxylic acid cycle (Fig. 3). The two nonessential genes (BP2014 and BP2021) are annotated as aconitate hydratases (responsible for interconversion of citrate and isocitrate), and these genes may be functionally redundant. Together, these data suggest not only a functional TCA cycle but that the TCA cycle is critical for growth under our in vitro growth conditions on BG agar.
FIG 3.

Carbon metabolism in B. pertussis. Pathways with all or many genes identified as essential for in vitro growth are shown in green. Genes involved in the interconversion of glucose-1P and fructose-6P were classified as conditionally essential in vivo, and this reaction is shown in red.
Unexpectedly, the Bvg two-component system (bvgAS) was computationally classified as essential for in vitro growth by our analysis by TRANSIT and ARTIST. However, bvgAS did tolerate a small number of insertions (three insertions in bvgS and one insertion in bvgA, all at distinct TA sites) and is not identified as essential by stringent analysis (24). BvgAS has been characterized in many strains of Bordetellae, including strains of B. pertussis, and these strains are able to tolerate mutations within the bvgAS genes, suggesting that it is not truly essential for in vitro growth. To confirm that bvgAS could be deleted in the strain used for this study, we generated B. pertussis UT25 ΔbvgAS. This strain grows under the conditions that the library was generated (static growth on BG agar at 37°C), suggesting that bvgAS are not true essential genes. One explanation is that lacking BvgAS was disadvantageous for growth under our conditions. We tested the hypothesis by comparing the growth of B. pertussis UT25 ΔbvgAS to the growth of wild-type B. pertussis UT25 under both modulating (active BvgAS) and nonmodulating (inactive BvgAS) conditions. B. pertussis UT25 exhibited a growth defect when BvgAS was inactive, either genetically or through chemical modulation with MgSO4 (Fig. 4), supporting the hypothesis that lacking bvgAS was disadvantageous for growth. It is important to note that these growth studies were performed in Stainer-Scholte medium, a standard, defined liquid growth medium for B. pertussis, rather than on BG agar, which was used to generate the library. Demonstration of a BvgAS-dependent growth defect provides a potential explanation for identification of bvgAS as essential in our screen, but other explanations for this observation exist, highlighting the limitations of Tn-seq analysis for determining gene essentiality.
FIG 4.
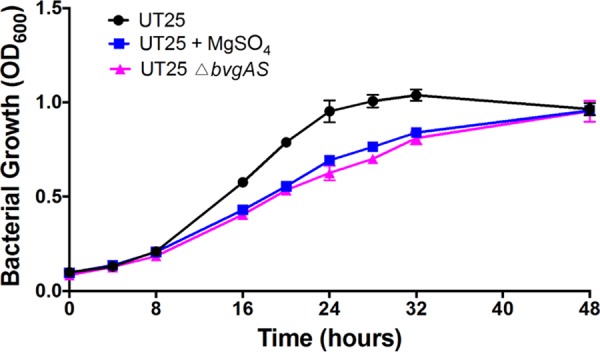
Growth curve of BvgAS-active and BvgAS-inactive B. pertussis UT25. Wild-type (black) and △bvgAS B. pertussis UT25 (pink) were grown in SSM as described in Materials and Methods. To modulate wild-type B. pertussis UT25 to a BvgAS-inactive state, 40 mM MgSO4 was added to all passages in SSM (blue). Bacterial growth was determined by the optical density at 600 nm (OD600).
Conditional essentiality in vivo.
To determine which genes contribute to infection, CD1 mice were intranasally infected with approximately 5 × 106 CFU of the library cultivated on BG agar. The mice were euthanized at either day 1 or day 3 postinfection, and B. pertussis conditional gene essentiality was determined at each of these time points. The average bacterial burden in combined lung and trachea samples at day 1 was 7.4 × 105 CFU/organ, and at day 3, it was 1.2 × 107 CFU/organ (Fig. 5), consistent with published work using this strain of mouse and a similar inoculum (25). The entire organ homogenate was plated on BG agar, and then bacterial colonies were harvested after 3 days of growth, genomic DNA was isolated, and samples were prepared for high-throughput sequencing by Illumina HiSeq platform.
FIG 5.

Bacterial burden in organs of B. pertussis-infected mice. The lungs and trachea of infected mice were removed and homogenized at the indicated time point, and the homogenates were diluted and plated, and the bacteria were enumerated to determine CFU/organ. Each point represents the total bacteria enumerated from the combined lung and trachea from one animal (n = 11 at day 1 and n = 12 at day 3). The means ± standard deviations (error bars) are shown.
We used ARTIST (20) for in vitro to in vivo comparisons, which utilizes simulation-based normalization followed by initial analysis by Mann-Whitney U tests and further refinement using a hidden Markov model. Genes that are classified as essential for in vitro growth are excluded from in vivo analysis, as clones with insertions in these genes are absent from our library. The ARTIST pipeline includes analysis to determine whether a bottleneck is present under the experimental conditions that could influence essentiality classifications despite normalization. When the bottleneck simulation was run on our in vitro data using parameters from our in vivo experimental conditions, the false-positive rates calculated for each of the three input samples were 2.9, 5.3, and 4.0% with standard deviations of 0.08, 0.12, and 0.08%, respectively. This result indicates an estimated false-positive rate of about 4% for each individual in vivo sample. In order to reduce the chance that we would falsely designate a gene as essential, we utilized the following consensus method. After ARTIST computationally classified each individual in vivo sample by comparison to the respective in vitro sample, we examined the ARTIST-derived gene essentiality for each of the 11 or 12 replicates of each gene at each time point (Table S4). If a replicate was classified as essential, the gene associated with that replicate would earn a point. The sum of the replicate points was the consensus score. We designated a gene as essential based on the consensus score. As anticipated, increasing the stringency, by requiring a greater consensus score in order to designate a gene as essential, resulted in designation of a smaller number of genes as conditionally essential (Fig. S2). We designated a gene as conditionally essential if >50% of the replicates (a score of 6) for that gene were computationally classified as essential as previously described (20). Thus, genes designated as essential by this consensus method reduce false-positive essentiality designations attributable to the effect of the bottleneck on computational gene classification (20).
Number of genes classified as essential decreases as stringency in the consensus score increases. Download FIG S2, TIF file, 0.4 MB (405.1KB, tif) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Table used for consensus determination for in vivo essential genes. For each sequencing sample (derived from one infected animal), ARTIST classified each gene or intergenic region as essential (designated as 1) or nonessential (designated as 0). The consensus score was calculated by taking the sum of this essentiality classifications; for example, a gene that was identified as essential in seven animals would have a consensus score of 7. Download Table S4, XLSX file, 0.6 MB (580.3KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We chose to perform these analyses at both day 1 and day 3 postinfection to determine whether gene essentiality differed as infection progressed. Three days postinfection is at or near the peak of bacterial burden (25). We hypothesized that the number of essential genes would increase at day 3 compared to day 1 as the bacterial population continued to be exposed to selection pressure. We identified 117 genes as conditionally essential at day 1 postinfection and 169 genes as conditionally essential at day 3 postinfection, with 94 genes shared between the two time points (Fig. 6 and Table S5). When all the genes identified at either day 1 or day 3 postinfection are included, 192 genes are identified as conditionally essential (Table S5).
FIG 6.
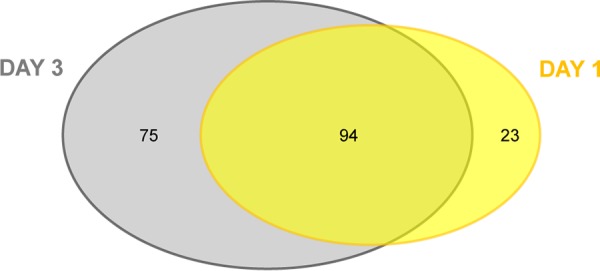
B. pertussis genes that are conditionally essential in vivo. CON-ARTIST was used for this analysis; 117 essential genes identified at 1 day postinfection are shown in yellow, and 169 genes identified as essential at 3 days postinfection are shown in gray.
Genes identified by ARTIST as essential on day 1 and day 3 postinfection. Genes had a consensus score greater than or equal to 6 for day 1 or 7 for day 3, which corresponds to being classified as essential in greater than 50% of the animals. Annotations for genes identified as essential on day 1 and day 3 postinfection are also included. Annotations were based on KEGG pathway assignments. Download Table S5, XLSX file, 0.03 MB (31KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
B. pertussis in vivo metabolism.
Very few prototypical virulence factors were identified, and this was anticipated based on the methodology of Tn-seq. Clones in the library with mutations of genes encoding secreted factors (for example, cyaA encoding adenylate cyclase toxin and ptxA encoding pertussis toxin) are complemented by other library clones within the population and are not identified in this screen. B. pertussis carries genes encoding an arsenal of adhesins, including Fim2, Fim3, and filamentous hemagglutinin that are included in the acellular vaccine, and not identifying these factors is expected, as their functions may be redundant in our model of infection. A recent study utilized RNA-seq to characterize the BvgAS regulon, including describing metabolic genes under BvgAS control (26). BvgAS is well characterized for its role in regulation of B. pertussis virulence, and we hypothesized that some metabolic genes identified as essential in our in vivo model would be under BvgAS control because of their importance in adaptation to the host nutritional environment. Because BvgAS-activated genes and Bvg-repressed genes have been shown to be expressed during B. pertussis infection in murine models (27), we compared our in vivo essential list with known BvgAS-activated and BvgAS-repressed genes (26). Out of the 192 genes identified at day 1 or day 3 postinfection, 7 were positively regulated by BvgAS, and 22 were negatively regulated by BvgAS (Table 1). Most of these 29 genes are associated with metabolism, suggesting a role for BvgAS in coordination of B. pertussis metabolism in vivo.
TABLE 1.
BvgAS regulation of conditionally essential genes in vivo
| Gene category and locus tag/gene name |
Annotation |
|---|---|
| BvgAS-activated genes | |
| BP0790/ilvH | Acetolactate synthase 3 regulatory subunit |
| BP1127 | |
| BP2930/cyoD | Cytochrome 0 ubiquinol oxidase |
| BP2932/cyoB | Ubiquinol oxidase subunit I |
| BP2934 | Two-component system response regulator |
| BP2935 | Two-component system, histidine kinase |
| BP3494/brkA | Serum resistance protein |
| BvgAS-repressed genes | |
| BP0250 | Hypothetical protein |
| BP0618/lpdA_1 | Dihydrolipoamide dehydrogenase |
| BP0619 | Branched-chain amino acid ABC transporter ATP-binding protein |
| BP0670 | Acyl-CoA dehydrogenase |
| BP0671 | Hypothetical protein |
| BP0682 | Putative exported protein |
| BP0683 | 4,5-Dihydroxyphthalate decarboxylase |
| BP0684A | 2Fe-2S ferredoxin |
| BP0808 | Hypothetical protein |
| BP1631/wcbA | Capsular polysaccharide export protein |
| BP1818/dadA_1 | d-Amino acid dehydrogenase small subunit |
| BP2368/prpC | 2-Methylcitrate synthase |
| BP2644 | Glycerol-3-phosphate dehydrogenase |
| BP3228/minD | Septum site-determining protein |
| BP3575 | Hypothetical protein |
| BP3671 | Glycosyltransferase family protein |
| BP3672 | Hypothetical protein |
| BP3679 | TetR family transcriptional regulator |
| BP3740/colII | Cytochrome c oxidase subunit II |
| BP3743/ctaD | Cytochrome c oxidase polypeptide I |
| BP3744 | Cytochrome c oxidase subunit II |
| BP3831 | Amino acid ABC transporter substrate-binding protein |
The majority of genes required for infection had metabolic functions. Out of the total 192 genes designated as conditionally essential in vivo, 117 had one or more KEGG pathway classifications (Table S5 and Fig. S1). Genes associated with transport, biosynthesis of secondary metabolites, and biosynthesis of antibiotics were enriched in our analysis (Table S5 and Fig. S1). Twenty-eight genes annotated as transporters were conditionally essential in vivo (Table S5). The specificity of the majority of these transporters is not yet known. We identified BP3494, encoding the BrkA autotransporter, as conditionally essential in vivo. B. pertussis strains lacking brkA are more sensitive to serum in vitro and are less virulent in mice (28–30), and our results are consistent with these previous observations. Another autotransporter, the vaccine component pertactin, was not identified, possibly due to the redundancy of autotransporter function. Genes within the bhu operon (BP0344-BP0346), encoding a heme iron acquisition system (31), were essential at day 3 but not day 1, consistent with previous literature that suggested that heme becomes more available to B. pertussis later in infection perhaps through B. pertussis-mediated host damage (32).
Genes encoding a quinol oxidase (cyoABCD) were conditionally essential in vivo. Similarly, these genes were identified in an in vivo transposon mutagenesis screen in P. aeruginosa (14). Identification of this complex as important in two very different models of infection with two distinct pathogens may highlight a role of the Cyo terminal oxidase in aerobic respiration in vivo, potentially in adaptation to lower oxygen concentrations within the host (33).
In silico analysis of Tn-seq data.
Most of the genes identified in our in vivo analysis were involved in metabolic functions, based on current annotation and homology, and we used computational tools to obtain a more complete understanding of each gene’s role in B. pertussis growth. Genome-scale metabolic network reconstructions (GENREs) are computational representations of all the information known about the metabolism of an organism, generated from literature and from genomic, proteomic, and transcriptomic data sets. By mathematically representing the known metabolic genes, enzymes, metabolites, and reactions involved in carrying out the chemical processes in a network, it is possible to relate the genotype and phenotype. Through the use of constraint-based analyses (34, 35), computational predictions of the organism’s metabolic capabilities, including the impact of gene knockouts and of varied medium conditions on growth, allow generation of novel hypotheses.
Two genome-scale metabolic network reconstructions have been recently published for B. pertussis (13, 36), and these models were used as the basis for our calculations. For any model, input nutrients must be defined for simulating biomass production, and we chose components and stoichiometry based on existing defined medium formulations. Biomass production was simulated using each model with an array of nutrient inputs, including unrestricted nutrient availability, two different published formulations of SSM (36, 37), and sputum media (SCFM) (38). BG medium is not defined and cannot be used as an input for modeling biomass production. SSM medium formulations are defined, but they were not originally designed with the goal of simulating the in vivo environment and likely are not representative of nutrient availability within the host. SCFM was developed as a chemically defined medium to approximate nutrients available in the respiratory tracts of humans with cystic fibrosis (CF). Although the nutrient composition of CF sputum is different than that of patients affected by pertussis, SCFM is a suitable medium for application to the GENRE and may better represent growth conditions for B. pertussis in the respiratory tract.
The Fyson et al. GENRE (13) and the Branco dos Santos et al. GENRE (36) each generated biomass when grown in silico on SCFM media and when provided with unrestricted nutrient availability. Under the SCFM condition, both models indicated lower biomass production yields relative to unrestricted nutrient availability for uptake and export, as would be expected when comparing a defined media where some metabolites are limited or not provided. The Branco dos Santos et al. model indicated generation of biomass with both formulations of SSM as the input media, but the Fyson et al. model, which was tested previously with nutrients provided by charcoal agar, suggested that biomass could not be synthesized on SSM. When single gene deletions were simulated under the SCFM conditions, the Fyson et al. model predicted that 332 of the model’s 796 genes are essential for growth and the Branco dos Santos et al. model predicted that 241 of the model’s 770 genes are essential for growth. The model-predicted essential genes lists were each compared to genes found to be experimentally essential in vivo in our Tn-seq analyses (Table S6).
Results of in silico analysis using two published GENREs. Download Table S6, XLSX file, 0.03 MB (29.2KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We used the two GENREs to probe the function of Tn-seq-designated in vivo essential genes associated with B. pertussis metabolism using SCFM as the input medium. Based on Tn-seq analysis, two genes encoding products involved in glucose metabolism were essential despite the fact that B. pertussis does not utilize glucose for growth. At both 1 day and 3 days postinfection, BP3141 and BP3142, encoding a phosphoglucomutase and a glucose-6-phosphate isomerase, respectively, were conditionally essential. These enzymes facilitate the conversion of glucose to fructose-6-phosphate, which can then enter either the glycolysis pathway or the pentose phosphate pathway (Fig. 3). Since glycolysis is not functional in B. pertussis, we hypothesized that these enzymes were likely shuttling glucose into the pentose phosphate pathway.
When the genes were deleted in silico, model-simulated biomass production was inhibited due to the inability to synthesize lipooligosaccharide (LOS) (Table S6). When genes involved in the pentose phosphate pathway were deleted in silico, model-simulated biomass production was similarly inhibited, but outputs derived from other metabolic pathways in addition to LOS biosynthesis were affected as well (Table S6). This result suggested that BP3141 and BP3142 were likely contributing to LOS biosynthesis independently of the known LOS biosynthesis precursors generated by the pentose phosphate pathway. In agreement with this finding, West et al. reported that a strain lacking the homolog to BP3141 in the related organism Bordetella bronchiseptica was less able to survive in a murine model of respiratory infection and exhibited an altered lipopolysaccharide (LPS) profile with no O antigen and a truncated core oligosaccharide (39). Glucose is a component of the outer core Bordetella LOS, and we next hypothesized that BP3141 and BP3142 are required for generation of glucose-1-phosphate (glucose-1P) as a precursor for LOS outer core synthesis. To address this hypothesis, we investigated whether the genes required for the addition of glucose to the outer core were similarly required for in vivo fitness. A UDP-glucose:LOS-beta-1,4-glucosyltransferase adds the first glucose residue to the HepI moiety of LOS. The gene encoding this activity (BP2329) has been identified within a novel lipopolysaccharide core biosynthesis gene cluster in B. pertussis (40). BP2329 is classified by our Tn-seq analysis as conditionally essential in vivo, as well as the two other genes within this locus that also result in a truncated LOS (BP2328 and BP2330) (40). Mutation of the remaining gene within this operon, BP2331, did not result in a truncated LOS molecule (40) and was not identified as conditionally essential in vivo in our analyses. Other genes annotated as having a role in LOS biosynthesis that were assigned as conditionally essential in vivo by Tn-seq were BP2325 and BP0388. BP2325 encodes a putative UDP-glucose:heptosyl-alpha-1,3-glucosyltransferase which adds N-acetylglucosamine (a derivative of glucose) to the HepII moiety. BP0388 is one of the few genes in the major LOS pathway that was not essential in vitro, and it encodes phosphoheptose isomerase, which incorporates sedoheptulose-7P into the LOS molecule. Cumulatively, these results suggest a vital role of biosynthesis of full-length LOS in our in vivo model and demonstrate how the GENREs aid in interpreting Tn-seq data.
Correlation of gene essentiality and single nucleotide polymorphism frequency in human isolates of B. pertussis.
By definition, mutations or variants in essential genes of a genome are more likely than variants in nonessential genes to be associated with a growth or fitness defect. We hypothesized that this concept would be applicable to B. pertussis within its natural human host, and we tested whether data on variation in individual genes from human isolates correlated with our gene essentiality data determined by Tn-seq. For this comparison, we utilized the densities of single nucleotide variants (SNVs) within genes from B. pertussis isolates as described previously (41). SNV densities were calculated based on a collection of 343 strains isolated between 1920 and 2010 (41). The essential gene list derived from our data contained the 609 genes that were designated essential by both TRANSIT and ARTIST analysis (Table S2), and the nonessential list included the remaining 3015 genes encoded in the Tohama genome. Based on analysis using an unpaired t test with Welch’s correction, there were significantly fewer (P < 0.0001) SNVs, both silent and nonsilent, in genes that are essential in vitro (mean total SNV density of 0.0009588) versus genes that are not essential in vitro (mean total SNV density of 0.001391).
Additionally, we hypothesized that genes that are essential in vivo would also be associated with fewer SNVs in this data set than genes identified in vivo as nonessential. Genes that are essential in vitro are unable to be assessed in vivo, and therefore, these genes were excluded from this analysis. For in vivo essential genes identified by our screen, we used 192 genes classified as conditionally essential at either day 1 or day 3 postinfection (Table S5) and compared SNV density in those genes to SNV density in genes that are not essential in vivo (2,823 genes) using an unpaired t test with Welch’s correction. There was no significant difference (P = 0.0735) in the total SNV density between essential genes (mean total SNV density of 0.001227) and nonessential genes (mean total SNV density of 0.001402). Nonsilent SNVs are more likely to affect function of a gene product and are therefore more similar to transposon insertion. In contrast to the total SNV density comparison, there were fewer nonsilent SNVs in in vivo essential genes (mean SNV density of 0.0006634) compared to nonessential genes (mean SNV density of 0.0008771) (P = 0.0023). Altogether, these findings suggest that Tn-seq-derived gene essentiality presented in this study may correlate with gene variation identified in clinical isolates, an interesting finding given the differences between the method of Tn-seq and the nature of variation in the natural host.
DISCUSSION
This study describes the first in vivo Tn-seq analysis of B. pertussis, thus identifying genes predicted to be necessary for wild-type levels of both in vitro and in vivo growth. These data have been incorporated into two published metabolic models. One is a method for exploring meaningful connections between essential genes, and the other is aimed at improving understanding of essential gene function. We also incorporated data from SNVs of clinical isolates, thus integrating our in vivo essentiality predictions with data from human B. pertussis infections.
While many previous studies have investigated B. pertussis metabolism in order to optimize growth in vitro, the resulting medium formulations likely do not reflect nutrient availability in vivo. Because the evolution of B. pertussis has involved both massive gene loss and a narrowing of its host range, we reasoned that its metabolic activities might be especially tailored to the human respiratory tract and that as a result, many of the genes involved might be critical for establishing infection in vivo. Additionally, the syntheses of some known virulence factors depend on intermediates generated by central or secondary metabolic pathways, for example, lipopolysaccharides, exopolysaccharides, siderophores, and quorum-sensing molecules (42). In this sense, any gene that contributes to the fitness and survival of the pathogen may be considered a virulence gene (43). Here, we use Tn-seq and metabolic modeling to establish links between a known virulence factor, LOS, and its metabolic building blocks.
B. pertussis does not grow on typical carbohydrates as the sole carbon source due to an incomplete glycolysis pathway; three required genes (glucokinase, phosphofructokinase, and fructose-1,6-bis-phosphotase) are absent from the genome (7). However, the pathway for gluconeogenesis is apparently fully functional. Our data show that this pathway is essential, as transposon insertions in the genes encoding enzymes that convert pyruvate to fructose-6P were underrepresented in in vitro growth conditions. Our in vivo analysis also revealed that the genes interconverting fructose-6P and glucose-1P, BP3141 and BP3142, were both essential at day 1 and at day 3 postinfection. As glycolysis is not functional, we hypothesize that the selective advantage conferred by this pathway is in the utilization of glucose to produce full-length lipo-oligosaccharide, as glucose-1P is a component of the LOS outer core. Consistent with this finding is the observation that a mutation in the homolog to BP3141 in B. bronchiseptica resulted in an altered LPS profile, with no O antigen produced, a truncated core oligosaccharide, and decreased survival in a murine model of respiratory infection (39). This further supports the importance of the BP3142 phosphoglucomutase in Bordetellae and suggests that the role of this enzyme in B. pertussis pathogenesis may be in LOS biosynthesis during infection. Interpretation of these data was aided by in silico analysis using GENREs and highlights the utility of these programs in interpreting Tn-seq data sets. Our identification in this study of additional genes involved in the production of mature, full-length LOS provides compelling evidence that this pathway is crucial for B. pertussis survival in the murine respiratory tract.
An unusual finding was the identification of the Bvg two-component system as contributing to wild-type levels of in vitro growth under our culture conditions. Mutations in bvgAS have been constructed in multiple B. pertussis strains, including a B. pertussis UT25 ΔbvgAS strain generated in this study. This strain was able to be generated and grown under the conditions used for the screen, which is validation that these genes are not truly “essential.” However, we found that a B. pertussis UT25 ΔbvgAS strain exhibited a growth defect in comparison to a wild-type parental strain, and this growth defect is replicated by chemical modulation to a Bvg inactive state with MgSO4. We speculate that this growth defect was likely further exacerbated under competitive growth conditions in a pool of clones possessing wild-type bvgAS alleles. Other potential explanations for this finding include the potential for functionally or structurally guarded genes that do not permit transposon insertion in Bvg such as extensive binding proteins or a three-dimensional structure of the gene (44). Although the finding with bvgAS serves as evidence that some of the genes identified in our screen, more likely through the hidden Markov model analyses than the more stringent analyses, are not true essential genes, these genes contribute to efficient growth under our conditions and therefore are important for exploration.
We asked whether essential genes identified by Tn-seq predict which genes in B. pertussis are subject to variation and tested this question by comparing in vivo Tn-seq-based gene essentiality to SNVs identified in 343 strains of B. pertussis collected between 1920 and 2010 (41). We found that in vitro essential genes had a lower total and nonsilent SNV density than in vitro nonessential genes. Genes that were conditionally essential in vivo also had a lower nonsilent SNV density than genes that were not essential in vivo. These data suggest that genes identified in the in vitro and in vivo studies as essential are associated with gene variation identified in clinical isolates from human infection.
There are caveats to this suggestion that the comparison of in vivo essential genes to SNV data supports a connection of the Tn-seq essentiality assignments to human infection. The screen was performed in an intranasal murine infection model, which differs from human infection with B. pertussis and does not replicate the clinical features of pertussis. The method of inoculation and burden of infection may result in different selective pressure due to anatomy, nutrient availability, and different inflammatory responses among other aspects. The early time points of the Tn-seq in vivo studies likely include different selective pressures than those to which human isolates are subjected in the first several weeks of human infection prior to cultures being obtained. Nonetheless, we expect that some of the selective pressures and growth requirements in humans and mice over these differing times are comparable, particularly in the case of metabolic genes included in the Tn-seq data set. In fact, the abundance of metabolic genes favors the association between Tn-seq and SNV analysis, as virulence factors vary more than nonvirulence factors (45).
In addition to host- and infection-related differences between Tn-seq data and SNV data, there are fundamental methodologic differences between these data. In Tn-seq, a complex library of clones containing unique insertions located across the genome is created simultaneously in a single isolate and then subjected to pressure. The forward introduction of variation is therefore presumed to be equal across the genome except for experimental biases introduced by the transposon’s (e.g., TA site preference) ability to insert into structurally guarded genes (e.g., protein binding regions or H-NS regions [44]) and the in vitro conditions under which the library was generated. It is essentially also comprehensive, if saturating Tn mutagenesis is performed. SNVs, on the other hand, accumulate over time, both randomly and in response to host selection, their introduction is far from comprehensive, and their detection is limited by the degree of sampling of the genome, allelic frequency, and number of samples interrogated (46, 47). The effects of SNVs, with the exception of nonsense and frameshift mutations, on gene function will be impossible to predict in the vast majority of cases. Transposon insertion into a gene, on the other hand, will usually disrupt gene function.
Presumably, increased time of the Tn-seq pool within the host will subject it to prolonged and/or additional pressures, which may alter the spectrum of genes identified as essential in vivo. Balancing this, prolonged residence in the host may result in fewer bacteria recovered, resulting in loss of diversity in the Tn-seq output pool, due to random loss of subpopulations stochastically. Robust sequencing and analysis require sufficient numbers of bacteria in order to maintain statistical and analytical power. With a smaller number of bacteria recovered, the number of replicates would have to be increased substantially for statistical power. Therefore, the time point of 3 days, which is at or near the peak of bacterial burden (25) was chosen. Bacteria recovered from the host at day 1 postinfection have been subjected to limited host pressure, and output pool analysis at this earlier time point could logically be expected to show fewer essential genes than at day 3 postinfection. Furthermore, analysis of day 1 or day 3 output pools is expected to be more indicative of early growth requirements in the host and/or of selective pressures from innate, rather than adaptive, immune mechanisms. Future studies will be required to assess selective pressures experienced at later time points and may be challenging due to decreased bacterial recovery.
In this study, we generated a diverse library of transposon mutants and used this library to probe gene essentiality in vivo in a murine model of intranasal infection. This screen generated a large data set that will be a valuable resource for future investigations into B. pertussis pathogenesis to support the design of improved vaccines. It also enabled novel observations about metabolism and gene regulation in vitro and in vivo. Integration of these data with other published reports and available metabolic models provided a more comprehensive understanding of metabolism and gene essentiality in this pathogen that has undergone genome condensation and reduction of its host range during its evolution. Understanding B. pertussis metabolism and gene essentiality in vivo may also be used to direct the design of growth media that more accurately reflect in vivo growth and that may therefore enable identification of new vaccine antigen candidates.
MATERIALS AND METHODS
Additional methods are included in Text S1 in the supplemental material.
Ethics statement.
All animal work was approved by the University of Virginia Institutional Animal Care and Use Committee protocol 4004.
Construction of library.
B. pertussis strain UT25-lux (Text S1) was grown as indicated on BG and then passaged into SSM for growth for 20 to 24 h at 35.5°C shaking. The culture was diluted to an optical density at 600 nm (OD600) of 0.08 and grown for 18 to 20 h to an OD600 of 0.7 to 0.8. E. coli strain RHO3 (15) was grown shaking at 35.5°C for 16 to 18 h in the presence of DAP. Cultures of B. pertussis and E. coli were washed to remove antibiotics, combined in SSM (50 to 100 μl), and incubated statically on BG+MgSO4+DAP for 18 to 24 h at 37°C. The mixed mating was then harvested, washed once, and plated on BG+kanamycin in the absence of DAP. Colonies were visible after 3 days of growth at 37°C. Transposon insertion was verified by positive PCR for the kanamycin cassette and negative PCR for the pSAM backbone, suggesting integration of the transposon into chromosomal DNA and loss of the pSAM-Km plasmid. Southern blot analysis indicated that most clones had a single insertion site.
In vitro and in vivo screening of library.
For preparation of bacterial inocula, 1 × 108 CFU of library was plated on four large plates, grown for 3 days on BG+Kan, collected, washed, and replated on fresh BG for 2 days. The colonies were harvested by swabbing, washed, and diluted to obtain the desired dose. Four-week-old CD1 mice (Charles River) were infected intranasally with 20 μl of PBS containing approximately 5 × 106 CFU. Genomic DNA from the remainder of the inoculum was prepared and used as the input sample for comparative analysis and the BG-grown sample for in vitro analysis. Mice were euthanized at day 1 (n = 11) or day 3 (n = 12), and lungs and trachea were extracted, weighed, pooled, and homogenized. Organ homogenates were serially diluted and plated to determine bacterial load; the remainder of the homogenized sample was plated on BG, grown for 2 days, and collected for preparation of genomic DNA. Genomic DNA was prepared following the manufacturer’s instructions (Wizard genomic DNA purification kit; Promega).
Accession number(s).
The mapped reads are available at the Sequence Read Archive (SRA) under BioProject accession number PRJNA542053 and SRA accession number SRP197242.
Oligonucleotide sequences. Download Table S7, XLSX file, 0.01 MB (10.1KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ACKNOWLEDGMENTS
This work was funded in whole or in part by federal funds from the National Institutes of Allergy and Infectious Diseases, National Institutes of Health, under contracts HHSN272201200005C-416476 to J.C.E. and 5R01AI18000-33 to E.L.H. L.A.G. received a Hartwell Foundation Postdoctoral Fellowship.
REFERENCES
- 1.Jakinovich A, Sood SK. 2014. Pertussis: still a cause of death, seven decades into vaccination. Curr Opin Pediatr 26:597–604. doi: 10.1097/MOP.0000000000000139. [DOI] [PubMed] [Google Scholar]
- 2.Warfel JM, Edwards KM. 2015. Pertussis vaccines and the challenge of inducing durable immunity. Curr Opin Immunol 35:48–54. doi: 10.1016/j.coi.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Warfel JM, Beren J, Kelly VK, Lee G, Merkel TJ. 2012. Nonhuman primate model of pertussis. Infect Immun 80:1530–1536. doi: 10.1128/IAI.06310-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warfel JM, Beren J, Merkel TJ. 2012. Airborne transmission of Bordetella pertussis. J Infect Dis 206:902–906. doi: 10.1093/infdis/jis443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elahi S, Holmstrom J, Gerdts V. 2007. The benefits of using diverse animal models for studying pertussis. Trends Microbiol 15:462–468. doi: 10.1016/j.tim.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Mills KHG, Gerdts V. 2014. Mouse and pig models for studies of natural and vaccine-induced immunity to Bordetella pertussis. J Infect Dis 209Suppl 1:S16–S19. doi: 10.1093/infdis/jit488. [DOI] [PubMed] [Google Scholar]
- 7.Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MTG, Churcher CM, Bentley SD, Mungall KL, Cerdeño-Tárraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O’Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, Whitehead S, Barrell BG, Maskell DJ. 2003. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet 35:32–40. doi: 10.1038/ng1227. [DOI] [PubMed] [Google Scholar]
- 8.Merkel TJ, Stibitz S. 1995. Identification of a locus required for the regulation of bvg-repressed genes in Bordetella pertussis. J Bacteriol 177:2727–2736. doi: 10.1128/jb.177.10.2727-2736.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knapp S, Mekalanos JJ. 1988. Two trans-acting regulatory genes (vir and mod) control antigenic modulation in Bordetella pertussis. J Bacteriol 170:5059–5066. doi: 10.1128/jb.170.11.5059-5066.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiss AA, Melton AR, Walker KE, Andraos-Selim C, Meidl JJ. 1989. Use of the promoter fusion transposon Tn5 lac to identify mutations in Bordetella pertussis vir-regulated genes. Infect Immun 57:2674–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodwin MS, Weiss AA. 1990. Adenylate cyclase toxin is critical for colonization and pertussis toxin is critical for lethal infection by Bordetella pertussis in infant mice. Infect Immun 58:3445–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marr N, Oliver DC, Laurent V, Poolman J, Denoël P, Fernandez RC. 2008. Protective activity of the Bordetella pertussis BrkA autotransporter in the murine lung colonization model. Vaccine 26:4306–4311. doi: 10.1016/j.vaccine.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 13.Fyson N, King J, Belcher T, Preston A, Colijn C. 2017. A curated genome-scale metabolic model of Bordetella pertussis metabolism. PLoS Comput Biol 13:e1005639. doi: 10.1371/journal.pcbi.1005639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skurnik D, Roux D, Aschard H, Cattoir V, Yoder-Himes D, Lory S, Pier GB. 2013. A comprehensive analysis of in vitro and in vivo genetic fitness of Pseudomonas aeruginosa using high-throughput sequencing of transposon libraries. PLoS Pathog 9:e1003582. doi: 10.1371/journal.ppat.1003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.López CM, Rholl DA, Trunck LA, Schweizer HP. 2009. Versatile dual-technology system for markerless allele replacement in Burkholderia pseudomallei. Appl Environ Microbiol 75:6496–6503. doi: 10.1128/AEM.01669-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stothard P, Wishart DS. 2005. Circular genome visualization and exploration using CGView. Bioinformatics 21:537–539. doi: 10.1093/bioinformatics/bti054. [DOI] [PubMed] [Google Scholar]
- 17.Stibitz S, Garletts TL. 1992. Derivation of a physical map of the chromosome of Bordetella pertussis Tohama I. J Bacteriol 174:7770–7777. doi: 10.1128/jb.174.23.7770-7777.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chao MC, Abel S, Davis BM, Waldor MK. 2016. The design and analysis of transposon insertion sequencing experiments. Nat Rev Microbiol 14:119–128. doi: 10.1038/nrmicro.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeJesus MA, Ambadipudi C, Baker R, Sassetti C, Ioerger TR. 2015. TRANSIT–a software tool for Himar1 TnSeq analysis. PLoS Comput Biol 11:e1004401. doi: 10.1371/journal.pcbi.1004401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pritchard JR, Chao MC, Abel S, Davis BM, Baranowski C, Zhang YJ, Rubin EJ, Waldor MK. 2014. ARTIST: high-resolution genome-wide assessment of fitness using transposon-insertion sequencing. PLoS Genet 10:e1004782. doi: 10.1371/journal.pgen.1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeJesus MA, Ioerger TR. 2013. A hidden Markov model for identifying essential and growth-defect regions in bacterial genomes from transposon insertion sequencing data. BMC Bioinformatics 14:303. doi: 10.1186/1471-2105-14-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thalen M, van den IJssel J, Jiskoot W, Zomer B, Roholl P, de Gooijer C, Beuvery C, Tramper J. 1999. Rational medium design for Bordetella pertussis: basic metabolism. J Biotechnol 75:147–159. doi: 10.1016/S0168-1656(99)00155-8. [DOI] [PubMed] [Google Scholar]
- 23.Izac M, Garnier D, Speck D, Lindley ND. 2015. A functional tricarboxylic acid cycle operates during growth of Bordetella pertussis on amino acid mixtures as sole carbon substrates. PLoS One 10:e0145251. doi: 10.1371/journal.pone.0145251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brutinel ED, Gralnick JA. 2012. Anomalies of the anaerobic tricarboxylic acid cycle in Shewanella oneidensis revealed by Tn-seq. Mol Microbiol 86:273–283. doi: 10.1111/j.1365-2958.2012.08196.x. [DOI] [PubMed] [Google Scholar]
- 25.Alonso S, Pethe K, Mielcarek N, Raze D, Locht C. 2001. Role of ADP-ribosyltransferase activity of pertussis toxin in toxin-adhesin redundancy with filamentous hemagglutinin during Bordetella pertussis infection. Infect Immun 69:6038–6043. doi: 10.1128/IAI.69.10.6038-6043.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moon K, Bonocora RP, Kim DD, Chen Q, Wade JT, Stibitz S, Hinton DM. 2017. The BvgAS regulon of Bordetella pertussis. mBio 8:e01526-17. doi: 10.1128/mBio.01526-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Beek LF, de Gouw D, Eleveld MJ, Bootsma HJ, de Jonge MI, Mooi FR, Zomer A, Diavatopoulos DA. 2018. Adaptation of Bordetella pertussis to the respiratory tract. J Infect Dis 217:1987–1996. doi: 10.1093/infdis/jiy125. [DOI] [PubMed] [Google Scholar]
- 28.Fernandez RC, Weiss AA. 1994. Cloning and sequencing of a Bordetella pertussis serum resistance locus. Infect Immun 62:4727–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elder KD, Harvill ET. 2004. Strain-dependent role of BrkA during Bordetella pertussis infection of the murine respiratory tract. Infect Immun 72:5919–5924. doi: 10.1128/IAI.72.10.5919-5924.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pishko EJ, Betting DJ, Hutter CS, Harvill ET. 2003. Bordetella pertussis acquires resistance to complement-mediated killing in vivo. Infect Immun 71:4936–4942. doi: 10.1128/iai.71.9.4936-4942.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vanderpool CK, Armstrong SK. 2001. The Bordetella bhu locus is required for heme iron utilization. J Bacteriol 183:4278–4287. doi: 10.1128/JB.183.14.4278-4287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brickman TJ, Hanawa T, Anderson MT, Suhadolc RJ, Armstrong SK. 2008. Differential expression of Bordetella pertussis iron transport system genes during infection. Mol Microbiol 70:3–14. doi: 10.1111/j.1365-2958.2008.06333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lunak ZR, Noel KD. 2015. A quinol oxidase, encoded by cyoABCD, is utilized to adapt to lower O2 concentrations in Rhizobium etli CFN42. Microbiology 161:203–212. doi: 10.1099/mic.0.083386-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oberhardt MA, Palsson BØ, Papin JA. 2009. Applications of genome-scale metabolic reconstructions. Mol Syst Biol 5:320. doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunphy LJ, Papin JA. 2018. Biomedical applications of genome-scale metabolic network reconstructions of human pathogens. Curr Opin Biotechnol 51:70–79. doi: 10.1016/j.copbio.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Branco dos Santos F, Olivier BG, Boele J, Smessaert V, De Rop P, Krumpochova P, Klau GW, Giera M, Dehottay P, Teusink B, Goffin P. 2017. Probing the genome-scale metabolic landscape of Bordetella pertussis, the causative agent of whooping cough. Appl Environ Microbiol 83:e01528-17. doi: 10.1128/AEM.01528-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stainer DW, Scholte MJ. 1970. A simple chemically defined medium for the production of phase I Bordetella pertussis. J Gen Microbiol 63:211–220. doi: 10.1099/00221287-63-2-211. [DOI] [PubMed] [Google Scholar]
- 38.Palmer KL, Aye LM, Whiteley M. 2007. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J Bacteriol 189:8079–8087. doi: 10.1128/JB.01138-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.West NP, Jungnitz H, Fitter JT, McArthur JD, Guzmán CA, Walker MJ. 2000. Role of phosphoglucomutase of Bordetella bronchiseptica in lipopolysaccharide biosynthesis and virulence. Infect Immun 68:4673–4680. doi: 10.1128/iai.68.8.4673-4680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geurtsen J, Dzieciatkowska M, Steeghs L, Hamstra H-J, Boleij J, Broen K, Akkerman G, el Hassan H, Li J, Richards JC, Tommassen J, van der Ley P. 2009. Identification of a novel lipopolysaccharide core biosynthesis gene cluster in Bordetella pertussis, and influence of core structure and lipid A glucosamine substitution on endotoxic activity. Infect Immun 77:2602–2611. doi: 10.1128/IAI.00033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bart MJ, van Gent M, van der Heide HGJ, Boekhorst J, Hermans P, Parkhill J, Mooi FR. 2010. Comparative genomics of prevaccination and modern Bordetella pertussis strains. BMC Genomics 11:627. doi: 10.1186/1471-2164-11-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartell JA, Blazier AS, Yen P, Thøgersen JC, Jelsbak L, Goldberg JB, Papin JA. 2017. Reconstruction of the metabolic network of Pseudomonas aeruginosa to interrogate virulence factor synthesis. Nat Commun 8:14631. doi: 10.1038/ncomms14631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Lorenzo V. 2015. Pseudomonas aeruginosa: the making of a pathogen. Environ Microbiol 17:1–3. doi: 10.1111/1462-2920.12620. [DOI] [PubMed] [Google Scholar]
- 44.Kimura S, Hubbard TP, Davis BM, Waldor MK. 2016. The nucleoid binding protein H-NS biases genome-wide transposon insertion landscapes. mBio 7:e01351-16. doi: 10.1128/mBio.01351-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sealey KL, Harris SR, Fry NK, Hurst LD, Gorringe AR, Parkhill J, Preston A. 2015. Genomic analysis of isolates from the United Kingdom 2012 pertussis outbreak reveals that vaccine antigen genes are unusually fast evolving. J Infect Dis 212:294–301. doi: 10.1093/infdis/jiu665. [DOI] [PubMed] [Google Scholar]
- 46.Leaché AD, Oaks JR. 2017. The utility of single nucleotide polymorphism (SNP) data in phylogenetics. Annu Rev Ecol Evol Syst 48:69–84. doi: 10.1146/annurev-ecolsys-110316-022645. [DOI] [Google Scholar]
- 47.Carlton VEH, Ireland JS, Useche F, Faham M. 2006. Functional single nucleotide polymorphism-based association studies. Hum Genomics 2:391–402. doi: 10.1186/1479-7364-2-6-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. Download Text S1, DOCX file, 0.04 MB (38.6KB, docx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes identified as essential by conservative analysis. In this conservative method, only genes with no TA site hits in the first 95% of gene were considered essential. Based on five input samples grown on BG media, the analysis identified 400 essential genes (including tRNAs and rRNAs). Download Table S1, XLSX file, 0.01 MB (12.6KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Full list of genes identified by ARTIST and TRANSIT. This list includes lists of genes identified individually by ARTIST and TRANSIT as well as the list of 609 genes identified by both programs. It also includes domain essential genes identified by ARTIST. Download Table S2, XLSX file, 0.05 MB (52.5KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Representation of in vitro (thick black lines) and in vivo (red lines) essential genes within the network of bacterial metabolic processes. Colored boxes indicate the approximate locations of selected metabolic reactions associated with a related end product or substrate, including lipooligosaccharide synthesis (green), pentose phosphate (violet), glycolysis and gluconeogenesis (blue), TCA cycle (orange), amino acid synthesis (dark gray), vitamin and cofactor synthesis (yellow), and lipid metabolism (light gray). The figure was created using the KEGG Mapper function on the KEGG database. Download FIG S1, TIF file, 2.5 MB (2.5MB, tif) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
KEGG pathway assignments for in vitro essential genes. Download Table S3, XLSX file, 0.03 MB (35.3KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Number of genes classified as essential decreases as stringency in the consensus score increases. Download FIG S2, TIF file, 0.4 MB (405.1KB, tif) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Table used for consensus determination for in vivo essential genes. For each sequencing sample (derived from one infected animal), ARTIST classified each gene or intergenic region as essential (designated as 1) or nonessential (designated as 0). The consensus score was calculated by taking the sum of this essentiality classifications; for example, a gene that was identified as essential in seven animals would have a consensus score of 7. Download Table S4, XLSX file, 0.6 MB (580.3KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes identified by ARTIST as essential on day 1 and day 3 postinfection. Genes had a consensus score greater than or equal to 6 for day 1 or 7 for day 3, which corresponds to being classified as essential in greater than 50% of the animals. Annotations for genes identified as essential on day 1 and day 3 postinfection are also included. Annotations were based on KEGG pathway assignments. Download Table S5, XLSX file, 0.03 MB (31KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Results of in silico analysis using two published GENREs. Download Table S6, XLSX file, 0.03 MB (29.2KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Oligonucleotide sequences. Download Table S7, XLSX file, 0.01 MB (10.1KB, xlsx) .
Copyright © 2019 Gonyar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.