

Abstract
Background
Currently the World Health Organization only recommend fluoroquinolones for people with presumed drug‐sensitive tuberculosis (TB) who cannot take standard first‐line drugs. However, use of fluoroquinolones could shorten the length of treatment and improve other outcomes in these people. This review summarises the effects of fluoroquinolones in first‐line regimens in people with presumed drug‐sensitive TB.
Objectives
To assess fluoroquinolones as substitute or additional components in antituberculous drug regimens for drug‐sensitive TB.
Search methods
We searched the Cochrane Infectious Diseases Group Specialized Register; CENTRAL (The Cochrane Library 2013, Issue 1); MEDLINE; EMBASE; LILACS; Science Citation Index; Databases of Russian Publications; and metaRegister of Controlled Trials up to 6 March 2013.
Selection criteria
Randomized controlled trials (RCTs) of antituberculous regimens based on rifampicin and pyrazinamide and containing fluoroquinolones in people with presumed drug‐sensitive pulmonary TB.
Data collection and analysis
Two authors independently applied inclusion criteria, assessed the risk of bias in the trials, and extracted data. We used the risk ratio (RR) for dichotomous data and the fixed‐effect model when it was appropriate to combine data and no heterogeneity was present. We assessed the quality of evidence using the GRADE approach.
Main results
We identified five RCTs (1330 participants) that met the inclusion criteria. None of the included trials examined regimens of less than six months duration.
Fluoroquinolones added to standard regimens
A single trial (174 participants) added levofloxacin to the standard first‐line regimen. Relapse and treatment failure were not reported. For death, sputum conversion, and adverse events we are uncertain if there is an effect (one trial, 174 participants, very low quality evidence for all three outcomes).
Fluoroquinolones substituted for ethambutol in standard regimens
Three trials (723 participants) substituted ethambutol with moxifloxacin, gatifloxacin, and ofloxacin into the standard first‐line regimen. For relapse, we are uncertain if there is an effect (one trial, 170 participants, very low quality evidence). No trials reported on treatment failure. For death, sputum culture conversion at eight weeks, or serious adverse events we do not know if there was an effect (three trials, 723 participants, very low quality evidence for all three outcomes).
Fluoroquinolones substituted for isoniazid in standard regimens
A single trial (433 participants) substituted moxifloxacin for isoniazid. Treatment failure and relapse were not reported. For death, sputum culture conversion, or serious adverse events the substitution may have little or no difference (one trial, 433 participants, low quality evidence for all three outcomes).
Fluoroquinolines in four month regimens
Six trials are currently in progress testing shorter regimens with fluoroquinolones.
Authors' conclusions
Ofloxacin, levofloxacin, moxifloxacin, and gatifloxacin have been tested in RCTs of standard first‐line regimens based on rifampicin and pyrazinamide for treating drug‐sensitive TB. There is insufficient evidence to be clear whether addition or substitution of fluoroquinolones for ethambutol or isoniazid in the first‐line regimen reduces death or relapse, or increases culture conversion at eight weeks. Much larger trials with fluoroquinolones in short course regimens of four months are currently in progress.
6 June 2018
No update planned
Other
No update is currently planned; this is not currently a priority topic for update
Keywords: Humans; Drug Substitution; Antitubercular Agents; Antitubercular Agents/therapeutic use; Ciprofloxacin; Ciprofloxacin/therapeutic use; Fluoroquinolones; Fluoroquinolones/therapeutic use; Levofloxacin; Ofloxacin; Ofloxacin/therapeutic use; Randomized Controlled Trials as Topic; Tuberculosis, Multidrug‐Resistant; Tuberculosis, Multidrug‐Resistant/drug therapy; Tuberculosis, Pulmonary; Tuberculosis, Pulmonary/drug therapy
Plain language summary
Substituting or adding fluoroquinolones to established first‐line antituberculous drug regimens gives no additional benefit or risks
Tuberculosis is an infectious disease caused by Mycobacterium tuberculosis bacteria. Over two billion people worldwide are believed to be latently infected with TB and approximately 10% of these people will develop active TB later in life. The World Health Organization currently only recommend treatment with fluoroquinolones for patients who cannot take standard first‐line drugs. In this review, we examined the effect of including fluoroquinolones in first‐line treatment regimens on people with presumed drug‐sensitive tuberculosis.
We examined the research published up to 6 March 2013 and we identified five randomised controlled trials (1330 people) that met the inclusion criteria. The trials were performed in low‐ and middle‐income countries located in geographically diverse areas but there was a lack of studies conducted in Asia. We found no studies that examined the effect of including fluoroquinolones in a standard six month TB treatment regimen on treatment failure. We do not know whether adding fluoroquinolones or substituting fluoroquinolones for ethambutol in a standard six month TB treatment regimen reduces treatment failure, relapse, death, or adverse events. Substituting fluoroquinolones for isoniazid in a standard six month TB treatment regimen may have little or no difference upon death and adverse events. Currently, there are nine randomised controlled trials ongoing.
HIV‐positive participants were relatively well‐represented in the included trials but none of the included trials stratified outcomes by HIV status. Also, the primary outcomes of all the included trials were reached before initiation of antiretroviral treatment. Evidence is generally lacking on the safety and efficacy of fluoroquinolone additions or substitutions in children (< 18 years) and in pregnant and lactating women.
Summary of findings
Summary of findings for the main comparison. Fluoroquinolone plus standard regimen compared to standard regimen alone for presumed drug‐sensitive TB.
| Fluoroquinolone plus standard regimen compared to standard regimen alone for drug‐sensitive TB | ||||||
| Patient or population: Patients with presumed drug‐sensitive TB Settings: New York and Hawaii Intervention: Fluoroquinolone plus standard regimen Comparison: Standard regimen alone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Standard regimen alone | Fluoroquinolone plus standard regimen | |||||
| Treatment failure | ‐ | ‐ | ‐ | (0 studies) | ‐ | Not reported |
| Relapse | ‐ | ‐ | ‐ | (0 studies) | ‐ | Not reported |
| Death from any cause | 75 per 1000 | 20 per 1000 (2 to 185) | RR 0.27 (0.03 to 2.47) | 174 (1 study) | ⊕⊝⊝⊝ very low1,2,3 | |
| TB‐related death | 25 per 1000 | 20 per 1000 (1 to 310) | RR 0.80 (0.05 to 12.40) | 174 (1 study) | ⊕⊝⊝⊝ very low1,2,3 | |
| Sputum culture conversion at 8 weeks | 973 per 1000 | 954 per 1000 (885 to 1041) | RR 0.98 (0.91 to 1.07) | 174 (1 study) | ⊕⊕⊝⊝ very low2,4,5 | |
| Serious adverse events | 149 per 1000 | 127 per 1000 (60 to 265) | RR 0.85 (0.4 to 1.78) | 174 (1 study) | ⊕⊝⊝⊝ very low1,2,6 | |
| One or more adverse event | 172 per 1000 | 172 per 1000 (89 to 330) | RR 1 (0.52 to 1.92) | 174 (1 study) | ⊕⊝⊝⊝ very low1,2,7 | |
| *The basis for the assumed risk (eg the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded by one for risk of bias: 73/174 (42%) of trial participants were excluded. 2 Downgraded by one for indirectness: This single trial was conducted in adults with and without HIV infection in New York and Hawaii over 15 years ago. The result may not be generalized to other situations. Levofloxacin was added to the standard regimen: 500 mg daily for two weeks (induction phase); then 750 mg levofloxacin thrice weekly for six weeks; then standard regimen only (continuation phase). 3 Downgraded by one for imprecision: This trial is underpowered to detect a statistically significant result. Only four deaths were reported: one in the intervention group and three in the controls. Of these only two were deemed to be due to TB; one in each group. 4 Downgraded by one for risk of bias: 73/174 (42%) of trial participants were excluded at eight weeks analysis. 5 Downgraded by one for imprecision: This trial remains underpowered to detect difference. The result is not statistically significant. 6 Downgraded by one for imprecision: This trial is underpowered to detect rare but important adverse effects. The adverse effects are described as: nausea, vomiting, peripheral neuropathy, dermatologic reactions with fever, haematological adverse events, renal or metabolic toxicity, and hepatic toxicity.
Summary of findings 2. Fluoroquinolone substitution for ethambutol in a standard six month regimen compared to standard regimen for presumed drug‐sensitive TB.
| Fluoroquinolone substitution for ethambutol in a standard six month regimen compared to standard regimen for drug‐sensitive TB | ||||||
|
Patient or population: Patients with presumed drug‐sensitive TB
Settings: Brazil, North America, Africa, and South Africa
Intervention: Fluoroquinolone substitution for ethambutol in a standard six month regimen (fluoroquinolones + HRZ) Comparison: Standard regimen (HRZE) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Standard regimen | Fluoroquinolone substitution for ethambutol | |||||
| Treatment failure | ‐ | ‐ | ‐ | (0 studies) | ‐ | Not reported |
| Relapse | 66 per 1000 | 47 per 1000 (11 to 202) | RR 0.71 (0.17 to 3.06) | 170 (1 study) | ⊕⊝⊝⊝ very low1,2,3 | |
| Death from any cause | 32 per 1000 | 17 per 1000 (7 to 42) | RR 0.52 (0.21 to 1.32) | 723 (3 studies) | ⊕⊕⊝⊝ very low1,4,5,6 | |
| TB‐related death | 16 per 1000 | 5 per 1000 (0 to 129) | RR 0.33 (0.01 to 8.07) | 170 (1 study) | ⊕⊝⊝⊝ very low1,2,6 | |
| Sputum culture conversion at 8 weeks | 704 per 1000 | 753 per 1000 (683 to 838) | RR 1.07 (0.97 to 1.19) | 723 (3 studies) | ⊕⊕⊝⊝ very low1,4,5,7 | |
| Serious adverse events | 65 per 1000 | 60 per 1000 (34 to 105) | RR 0.93 (0.53 to 1.62) | 723 (3 studies) | ⊕⊕⊝⊝ very low1,4,5,8 | |
| One or more adverse events | ‐ | ‐ | ‐ | (0 studies) | ‐ | Not reported |
| *The basis for the assumed risk (eg the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded by one for risk of bias. All three trials were considered at high risk of bias due to high levels of exclusions from the final analysis. 2 Downgraded by one for indirectness: Only a single trial comparing moxifloxacin with ethambutol assessed this outcome. It was conducted in adults in Brazil between 2004 and 2007 and is not easily generalized to other fluoroquinolones or populations. 3 Downgraded by one for imprecision: The result is not statistically significant and the 95% CI is wide. This study was underpowered to detect an effect. 4 No serious inconsistency: None of three trials found a statistically significant difference. 5 No serious indirectness. Moxifloxacin, gatifloxacin, and ofloxacin have been compared to ethambutol and moxifloxacin in three trials, and gatifloxacin and ofloxacin in one trial each. These were conducted in adults from Brazil (between 2004 and 2007), North America, and Africa (dates not given), and South Africa (between 2004 and 2005). 6 Downgraded by two for imprecision: Only 14 deaths were reported in the three trials. Only Conde 2009 reported on TB‐related death and only one occurred. Much larger trials would be necessary to show an effect. 7 Downgraded by one for imprecision. CIs of two of three studies are wide and studies remain underpowered. 8 Downgraded by one for imprecision. All three trials were underpowered to detect difference and CIs are wide.
Summary of findings 3. Fluoroquinolone substitution for isoniazid in a standard six month regimen compared to standard regimen for presumed drug‐sensitive TB.
| Fluoroquinolone substitution for isoniazid in a standard six month regimen compared to standard regimen for presumed drug‐sensitive TB | ||||||
|
Patient or population: Patients with presumed drug‐sensitive TB
Settings: North America, Brazil, South Africa, Spain, Uganda
Intervention: Fluoroquinolone substitution for isoniazid in a standard six month regimen Comparison: Standard regimen (HRZE) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Standard regimen | Fluoroquinolone substitution for isoniazid | |||||
| Treatment failure | ‐ | ‐ | ‐ | (0 studies) | ‐ | not reported |
| Relapse | ‐ | ‐ | ‐ | (0 studies) | ‐ | not reported |
| Death from any cause | 24 per 1000 | 18 per 1000 (4 to 79) | RR 0.75 (0.17 to 3.30) | 433 (1 study) | ⊕⊕⊝⊝ low1,2 | |
| TB‐related death | 6 per 1000 | 12 per 1000 (1 to 131) | RR 2 (0.18 to 21.84) | 433 (1 study) | ⊕⊕⊝⊝ low1,2 | |
| Sputum culture conversion at 8 weeks | 549 per 1000 | 604 per 1000 (500 to 730) | RR 1.10 (0.91 to 1.33) | 433 (1 study) | ⊕⊕⊝⊝ low1,3 | |
| Serious adverse events | 37 per 1000 | 41 per 1000 (16 to 104) | RR 1.1 (0.43 to 2.8) | 433 (1 study) | ⊕⊕⊝⊝ low1,4 | |
| One or more adverse event | ‐ | ‐ | ‐ | (0 studies) | ‐ | not reported |
| *The basis for the assumed risk (eg the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded by one for risk of bias. 105/433 (24.3%) participants were excluded from the final analysis. 2 Downgraded by one for imprecision: The result is not statistically significant and the 95% CI is wide. This study was underpowered to detect an effect. Only seven deaths occurred and three were deemed related to TB. 3 Downgraded by one for imprecision: this single trial remains underpowered to detect an effect. The result is not statistically significant and the 95% CI is wide. 4 Downgraded by one for imprecision: The result is not statistically significant and the 95% CI is wide. This single study was underpowered to detect an effect. Only 14 serious adverse events occurred and they were equally distributed between comparison groups.
Background
Description of the condition
Tuberculosis (TB) is caused by Mycobacterium tuberculosis and more than two billion people, one third of the world’s total population, are believed to be latently infected (WHO 2012). Latently infected, immunocompetent people have an estimated lifetime risk of developing TB disease of 10% (WHO 2012). Although the incidence of TB has declined since 2004, the global burden of the disease remains high with an estimated 8.8 million people falling ill with TB each year (WHO 2011a). In 2010, 60% of new cases globally occurred in Asia while sub‐Saharan Africa had the highest incidence with over 270 cases per 100,000 population per year (WHO 2012). Pulmonary TB is the commonest clinical presentation and sputum smear‐positive cases are the most important source of ongoing infection in the community (Grzybowski 1975).
TB remains the most common opportunistic infection and a leading cause of death among people living with HIV and AIDS (Corbett 2003). Those co‐infected with HIV and TB are between 21 to 34 times more likely to develop active TB disease than HIV‐negative people infected with M. tuberculosis (WHO 2012) and nearly a quarter of deaths among people with HIV are due to TB. In 2010, 82% of new active TB cases among HIV‐positive people were in Africa (WHO 2012) where it has been the single most important factor determining the increased incidence of TB since 1990 (WHO 2010a).
Active TB disease may be fatal if left untreated or if treated inappropriately. In 2010, 1.4 million people died from TB (WHO 2012). It caused more adult deaths each year than any other single infectious disease in the twentieth century (Kochi 1991) and remains second only to HIV/AIDS as the greatest infectious killer worldwide (WHO 2012). Multi‐drug resistant TB (MDR‐TB: defined as resistance to both rifampicin and isoniazid) threatens the success of TB programmes with up to 510,000 people worldwide in need of specialised treatment with second‐line drugs and much lower rates of treatment success.
Description of the intervention
Effective pharmacological treatment for TB has been available since the 1940s. The first‐line antituberculous drugs are the most active agents with proven clinical efficacy that form the core of initial standardized treatment regimens. The recommended first‐line antituberculous regimen consists of isoniazid (H, 5 mg/kg, 300 mg daily or 10 mg/kg, 900 mg three times weekly), rifampicin (R, 10 mg/kg or 450 mg to 600 mg daily), pyrazinamide (Z, 25 mg/kg daily or 35 mg/kg three times weekly), and ethambutol (E, 15 mg/kg daily or 30 mg/kg three times weekly) (ie HRZE) (Blumberg 2003; WHO 2003; WHO 2006; WHO 2007a; WHO 2010b). Streptomycin, although used less commonly, is also a first‐line drug on the World Health Organization's (WHO's) list of essential anti‐TB drugs (WHO 2006; WHO 2007a; WHO 2011b). The efficacy of regimens containing rifampicin and isoniazid is well established for treatment and prevention (WHO 2007b; Ziganshina 2011), even in HIV‐positive people (WHO 2003; Woldehanna 2004). Rates of cure (defined for drug‐sensitive TB as negative sputum culture at two months and at the end of treatment) with six to nine month rifampicin‐containing regimens can approach 100%, provided the bacteria are drug‐sensitive, there are no additional co‐morbidities (especially HIV infection and diabetes), and that patients adhere to treatment (STS/BMRC 1981; Anonymous 1983). However, pyrazinamide is also essential to the current first‐line regimen and a duration of therapy of six months is not adequate for cure without it. Ethambutol is believed to be a weak drug used primarily to prevent the emergence of resistance.
WHO considers fluoroquinolones (other than ciprofloxacin) together with injectable medicines as forming the backbone of treatment for MDR‐TB and consistently recommends fluoroquinolones for drug‐sensitive TB in cases of intolerance of standard first‐line drugs, particularly hepatotoxicity (Gillespie 1998; Blumberg 2003; WHO 2003; WHO 2006; WHO 2010b). Ofloxacin is on the WHO Model List of Essential Medicines as a reserve second‐line drug for the treatment of MDR‐TB to be used in specialized centres adhering to WHO standards for TB control. Levofloxacin is included as an alternative based on availability and programme considerations (WHO 2011b). These recommendations for MDR‐TB are supported by expert opinion (Falzon 2011) and by systematic reviews of observational data (Johnston 2009) showing an association of fluoroquinolone use with treatment success (OR 2.20, 95% CI 1.19 to 4.09) in MDR‐TB. Hence equipoise has been disturbed despite the lack of randomized evidence in this area and it is unlikely that placebo‐controlled trials in MDR‐TB will be performed.
There is currently no consensus on the potential efficacy of fluoroquinolones as additions to or substitutions for established first‐line drugs in the standard regimen. Studies of substitution of ciprofloxacin for pyrazinamide and ethambutol in the first‐line regimen were associated with higher rates of treatment failure and relapse at a duration of six months in previous versions of this review (Kennedy 1993; Kennedy 1996; Ziganshina 2008). Hence the research questions for this update focus on substitution of newer fluoroquinolones for either isoniazid or ethambutol, which are thought to be more dispensable components of the first‐line regimen. Some small or uncontrolled studies have suggested that substituting ofloxacin for ethambutol in an established first‐line antituberculous regimen might make it possible to shorten TB chemotherapy from six months to five or even four months (Kohno 1992; TRC 2002). More recently, additional conflicting data suggest unclear efficacy when fluoroquinolones are included in first‐line antituberculous regimens (El‐Sadr 1998; Burman 2006; Rustomjee 2008a; Conde 2009; Dorman 2009). The rationale for fluoroquinolone substitution/addition in any first‐line antituberculous regimen is that the modified regimen might improve efficacy at a duration of six months or produce similar efficacy at a reduced duration of, for example, four months. SInce the long‐term outcomes of current first‐line therapy are excellent, with 95% or greater rates of cure, most attention has been given to developing shorter regimens.
How the intervention might work
Fluoroquinolones are fluorine‐containing nalidixic acid derivatives characterized by broad‐spectrum antimicrobial activity. The mechanism of action is inhibition of the DNA gyrase enzyme which is responsible for supercoiling of nucleic acid, an essential process for all bacteria. This mechanism is distinct from that of other antituberculous drugs, raising the possibility of synergistic activity. While initially fluoroquinolones were most useful for infections caused by gram‐negative bacteria, extensive modification of the basic pharmacophore has steadily increased the in vitro activity of newer fluoroquinolones against M. tuberculosis.
The favourable combination of pharmacodynamic and pharmacokinetic characteristics of fluoroquinolones (Ginsburg 2003) could give the following benefits when added to antituberculous regimens:
Add to the bactericidal and sterilizing effect of combination therapy.
Increase penetration into chronic TB lesions.
Improve adherence to treatment due to potentially better tolerability than first‐line drugs and by shortening treatment.
On the other hand, fluoroquinolones also have the potential to do harm. They may:
Increase liver and central nervous system (CNS) toxicity of antituberculous drugs (Yew 2001) and cause clinically significant drug interactions with antituberculous (Yew 2001), anti‐HIV (Burman 1999), and other drugs, resulting in reduced efficacy and potential toxicity (WHO 2006).
Cause additional adverse drug reactions, such as musculoskeletal damage, gastrointestinal problems (pseudo‐membranous colitis), cardiac arrhythmias, infections from fungi or bacteria, psychosis, and convulsions (Martindale 1996).
Induce resistance in M. tuberculosis (Alangaden 1997; Jacobs 1999; Wang 2006), which may rapidly become cross‐resistant to all members of the fluoroquinolone class (Ginsburg 2003).
The problem of resistance to fluoroquinolones is further complicated by the broad indications of this class of antimicrobials in treatment of various lower respiratory tract and other infections. This may at least be partially responsible for the rising resistance rates among M. tuberculosis strains to fluoroquinolones (Ginsburg 2003). Retrospective studies have shown that empiric antituberculous treatment with fluoroquinolones or fluoroquinolone use for misdiagnosed pneumonia delayed diagnosis of TB in an endemic area and impaired outcomes (Yoon 2005; Wang 2006).
Why it is important to do this review
In the light of these uncertainties, we have conducted a systematic review of trials of fluoroquinolones in people with presumed drug‐sensitive TB. These drugs are likely to be used as substitutes for existing drugs or as an addition to current treatment regimens in regimens based on rifampicin and pyrazinamide of six months duration or less. A shorter first‐line TB regimen would improve individual outcomes for TB sufferers and greatly reduce the operational burden on TB programmes.
Objectives
To assess fluoroquinolones as substitute or additional components in antituberculous drug regimens for drug‐sensitive TB.
We formulated the research questions as follows.
In presumed drug‐sensitive disease:
1. Do fluoroquinolones improve outcomes when added to the standard first‐line antituberculous regimen (HRZE)?
2. Do fluoroquinolones improve outcomes when substituted for ethambutol in the standard first‐line regimen (HRZE)?
3. Do fluoroquinolones improve outcomes when substituted for isoniazid in the standard first‐line regimen (HRZE)?
4. Are four month regimens with fluoroquinolone as good as six months of the standard regimen (HRZE)?
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCTs). Quasi‐RCTs were excluded.
Types of participants
People newly diagnosed with bacteriologically culture positive pulmonary TB, with presumed or proven drug‐sensitive disease, in areas with low prevalence of MDR‐TB (2% primary resistance) or where susceptibility testing was available.
Types of interventions
Intervention
Standard first‐line TB treatment regimens where a fluoroquinolone drug was used either:
as an addition to the standard first‐line TB treatment regimen of six months duration.
as substitution for ethambutol or isoniazid of six months duration.
as part of a shorter regimen of four months or less.
For the purpose of this review, we defined the standard first‐line TB treatment regimen as a regimen containing at least rifampicin and pyrazinamide and treatment given for six months (typically 2HRZE/4HR). We also planned to compare regimens containing fluoroquinolone drugs but given for less than six months to this standard regimen.
Control
Standard first‐line TB treatment regimens as defined above and not containing fluoroquinolones.
Types of outcome measures
Treatment failure, defined as continued or recurrent positive sputum cultures after four months of treatment, in participants in whom medication ingestion was assured.
Relapse, defined as becoming sputum smear or culture positive up to two years after being culture negative having completed therapy.
Combined endpoint of treatment failure and relapse, as defined above.
Death from any cause.
TB‐related death.
Sputum culture or smear conversion at eight weeks.
Time to sputum culture or smear conversion, defined as a continuous outcome providing an estimate of time in weeks or months needed to achieve the first negative sputum culture or smear.
Serious adverse events, defined as fatal, life‐threatening, requiring hospitalization, or change of treatment regimen.
Adverse effects specifically associated with fluoroquinolones (eg tendon rupture, QT‐interval prolongation).
Total number of people with adverse events.
Search methods for identification of studies
We attempted to identify all relevant trials regardless of language or publication status (published, unpublished, in press, and in progress).
Electronic searches
We searched the following databases using the search terms and strategy described in Appendix 1: Cochrane Infectious Diseases Group's Specialized Register (March 2013); Cochrane Central Register of Controlled Trials (CENTRAL), published in The Cochrane Library (2013, Issue 1); MEDLINE (1966 to March 2013); EMBASE (1974 to March 2013); LILACS (1982 to March 2013); Science Citation Index (1940 to March 2013); and the following Databases of Russian Publications (1988 to March 2013): Rossiyskaya medicina (http://www.scsml.rssi.ru) and Otkritiy medicinskiy club (http://www.medart.tomsk.ru). We also searched the metaRegister of Controlled Trials (March 2013) using the following search terms: tuberculosis AND (fluoroquinolones OR moxifloxacin OR ofloxacin OR gatifloxacin OR levofloxacin OR ciprofloxacin).
Searching other resources
Conference proceedings
We searched the following conference proceedings for relevant abstracts: 4th World Congress on TB, Washington, DC, USA, 3 to 5 June 2002 (published in Tubercle); International Union Against Tuberculosis Lung Disease (IUATLD) Annual Conference Proceedings (published in the International Journal of Tuberculosis and Lung Disease 1997 to 2012); American Thoracic Society Meetings Proceedings 2001 to 2012; and the British Society for Antimicrobial Therapy 2000 to 2012.
Researchers, organizations, and pharmaceutical companies
For the original reviews (Ziganshina 2005; Ziganshina 2008), we searched the current controlled trials web site and contacted individual researchers working in the field, organizations (Centers for Disease Control and Prevention (CDC), the Clinical Trials Unit of the International Union against Tuberculosis and Lung Disease (IUATLD), and the UK Medical Research Council Clinical Trials Unit), and pharmaceutical companies (Bayer, Merck Sharp & Dohme, Hoechst Marion Roussel, and Aventis Pharma) for unpublished and ongoing trials.
Reference lists
We also checked the reference lists of all included studies.
Data collection and analysis
Selection of studies
Lilia E. Ziganshina (LEZ) and Geraint Davies (GDAV) checked the citations and their abstracts to establish their relevance. We independently applied the inclusion criteria using an eligibility form and resolved any disagreements by discussion. We obtained the full text article if we agreed it was relevant and in cases of uncertainty. Finally, where we were still unsure if the study should be included because further information was necessary, we allocated the study to the list of those awaiting assessment and we then attempted to contact the study authors for clarification. We excluded studies that did not meet the inclusion criteria and gave the reason for exclusion in the 'Characteristics of excluded studies' section.
Data extraction and management
We independently (LEZ and GDAV) extracted data on trial characteristics, including methods, participants, interventions, and outcomes as well as data on dose and drug ratios of the combinations using a standardized data extraction form. We resolved any differences in the extracted data by referring to the original articles and through discussion. Where data were insufficient or missing we attempted to contact the trial authors for additional information.
For binary efficacy outcomes, we extracted the number of participants with the event and the number analyzed to allow for complete case analysis in each treatment group. Where possible, we extracted data to allow an intention‐to‐treat analysis (including all the participants in the groups to which they were originally randomly allocated). We used these data for safety outcomes and for the worst‐best case analyses used as sensitivity analyses. We calculated the percentage loss to follow‐up and exclusions from final analyses and we presented it in the 'Characteristics of included studies' section when the numbers randomized and the numbers analyzed were inconsistent. We extracted the number of serious adverse events and have presented these data in a forest plot. Where the trial data permitted we extracted the total number of participants with adverse events and with fluoroquinolone specific adverse events.
Assessment of risk of bias in included studies
LEZ and GDAV independently assessed the risk of bias for each trial using 'The Cochrane Collaboration's tool for assessing the risk of bias' (Higgins 2011). We followed the guidance to assess whether adequate steps had been taken to reduce the risk of bias across six domains: sequence generation; allocation concealment; blinding (of participants, personnel, and outcome assessors); incomplete outcome data; selective outcome reporting; and other sources of bias. We have categorized these judgments as 'low risk of bias', 'high risk of bias', or 'unclear'. Where we judged risk of bias as unclear, we attempted to contact the trial authors for clarification.
Measures of treatment effect
We presented dichotomous data and we combined them using risk ratios. We showed risk ratios accompanied by 95% confidence intervals (CIs).
Unit of analysis issues
If the same trial was included in the analysis more than once, we split the numbers of participants in the control group proportionately.
Dealing with missing data
Where data from the trial reports were insufficient, unclear, or missing, we attempted to contact the trial authors for additional information. We aimed to do an intention‐to‐treat (ITT) analysis but as there were missing data we did a complete case analysis (ie including all patients with a measured outcome). Where the number of people with a measured outcome was not reported, we extracted the number of patients in the per‐protocol analysis. The complete case analysis does not make an assumption about the outcome of missing patients. The potential effects of missing data were explored through a series of sensitivity analyses (Appendix 2). As a sensitivity analysis, we did a best‐worst case analysis; the best case analysis assumed missing patients had a positive outcome; the worst case analysis assumed they had a negative outcome.
Efficacy outcomes: The analysis of efficacy outcomes drew on the WHO's guidelines for treatment of TB (WHO 2010b). Due to the length of time needed for bacteriological confirmation of active TB disease, a high number of randomized participants are excluded from the final efficacy outcome as losses to follow‐up or exclusions for not meeting inclusion criteria, or voluntary or involuntary withdrawals. For this reason we conducted a sensitivity analysis which aimed to restore the integrity of the randomization process (as is usual in trial analysis) and test the robustness of the results to this methodology. For a summary of the methodology and sensitivity analysis see Appendix 2. Complete case analysis: We extracted the total numbers of failures, relapses, deaths, and sputum culture conversions and we used them as numerators. The denominator excludes participants for whom an outcome was not available (e.g. those who were lost to follow‐up, withdrew consent, took other antituberculous drugs, failed to complete treatment, or other reasons) and those participants who were found not to fulfil the inclusion criteria after randomization by trial report authors.
Assessment of heterogeneity
We assessed for heterogeneity amongst trials by inspecting the forest plots, applying the Chi2 test with a 10% level of statistical significance, and also using the I2 statistic with a value of 50% used to denote moderate levels of heterogeneity.
Assessment of reporting biases
We did not present funnel plots due to the small number of trials.
Data synthesis
We used Review Manager 5 to analyze the data and we grouped the trials according to comparisons. We used a fixed‐effect model to combine the data unless significant heterogeneity was present, in which case and where it was still appropriate to pool data, we used a random‐effects model.
Subgroup analysis and investigation of heterogeneity
We planned to investigate potential sources of heterogeneity through the following subgroup analyses if the number of trials permitted: HIV status, participant age, fluoroquinolone dose, length of treatment, and allocation concealment.
Sensitivity analysis
We explored the effect of missing data by doing a best‐worst case analysis (Appendix 2). We would have explored the impact of risk of bias on the results if there were more studies that met the inclusion criteria.
Results
Description of studies
Results of the search
We identified five RCTs that met the inclusion criteria out of 71 potentially relevant trials (see 'Characteristics of included studies'), and nine trials that are still in progress (see 'Characteristics of ongoing studies'). We illustrated these results in the study flow diagram (Figure 1).
1.
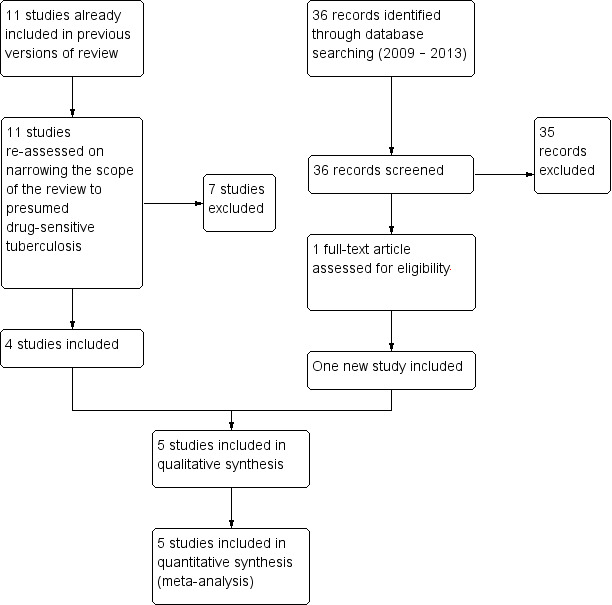
Study flow diagram
Included studies
The five RCTs that met the inclusion criteria included 1330 participants, with a range of 170 to over 400 participants per trial. The participants were aged 18 years or older. The majority of participants were male with a range of between 41% to 88% across trials. All five trials involved participants presumed to be drug‐sensitive according to treatment history, other trial exclusion criteria, and local surveillance data.
Four trials included both HIV‐positive and HIV‐negative participants as one group (El‐Sadr 1998; Burman 2006; Rustomjee 2008a; Dorman 2009), although none stratified the analysis by HIV status. One trial involved participants presumed to be HIV‐negative according to local endemicity, reference data, and exclusion criteria (Conde 2009).
Study locations were diverse and three trials included multiple centres. Trials were conducted in North America and Africa (one trial), the USA (one trial), Brazil (one trial), and North America, Brazil, South Africa, Spain and Uganda (one trial), and in South Africa (one trial). The mean duration of follow‐up ranged from eight weeks to 24 months.
Interventions
Comparison 1. Fluoroquinolones added to standard regimens (Fluoroquinolones + HRZE versus HRZE alone)
One trial (El‐Sadr 1998) compared a fluoroquinolone (levofloxacin for eight weeks) added to the standard treatment (ie isoniazid, rifampicin, pyrazinamide, and ethambutol for six or nine months) versus the standard regimen. El‐Sadr 1998 used 500 mg of levofloxacin daily orally for the first two weeks, then 750 mg orally thrice weekly for the following six weeks.
Comparison 2. Fluoroquinolone substitution for ethambutol in a standard six month regimen (Fluoroquinolones + HRZ versus HRZE)
Three trials (Burman 2006; Rustomjee 2008a; Conde 2009) substituted a fluoroquinolone for ethambutol for the first two months of treatment. The drugs tested were moxifloxacin 400mg daily (Burman 2006; Rustomjee 2008a; Conde 2009), gatifloxacin 400 mg daily (Rustomjee 2008a) or ofloxacin 800 mg daily (Rustomjee 2008a). Rustomjee 2008a was a four arm trial in which three different fluoroquinolones substituted for ethambutol were compared to the control. The control regimen included standard doses of isoniazid, rifampicin, and pyrazinamide‐(fluoroquinolones + HRZ), with standard ethambutol in the control arm (HRZE).
Comparison 3. Fluoroquinolone substitution for isoniazid in a standard six month regimen (Fluoroquinolones + RZE versus HRZE)
One trial (Dorman 2009) compared moxifloxacin 400 mg daily substituting for isoniazid. The base treatment was standard doses of rifampicin, pyrazinamide, and ethambutol (fluoroquinolones + RZE) with standard isoniazid doses in the control arm (HRZE).
Comparison 4. Fluoroquinolones as part of a four month regimen compared with a six month standard regimen
None of the included trials addressed the third research question of this review on the potential of fluoroquinolones to reduce treatment duration from six months to four months. We identified four on‐going trials that address this question and may be included in updates of this review (ISRCTN44153044 RIFAQUIN; NCT00216385; NCT00728507; NCT00864383 REMoxTB).
The treatment doses of standard antituberculous drugs (isoniazid, rifampicin, pyrazinamide, and ethambutol) were within the recommended body weight adjusted limits but varied among the trials. All of the included trials ensured the adherence of participants by administering the drugs under direct observation with special nursing facilities in outpatient settings or in hospital settings (see Characteristics of included studies).
Outcomes
The reported outcomes included treatment failure (one trial), relapse (one trial), death ‐ all cause and TB‐related (five trials), culture conversion at eight weeks (five trials), serious adverse events (five trials), and total number of people with adverse events (three trials). Trials did not report in a uniform way on time to culture conversion. Conde 2009 and Rustomjee 2008a estimated time to culture conversion by the Kaplan‐Meier method and only Conde 2009 compared the difference in time to culture conversion using the log‐rank test. Rustomjee 2008a used Cox proportional hazards models to estimate hazard ratios of culture conversion for the individual regimens. Burman 2006 used proportion of sputum culture negative at weeks 2, 4, 6, and 8. Dorman 2009 presented the probability that stable conversion has been observed at weeks 2, 4, 6, and 8. El‐Sadr 1998 reported cumulative percentage culture negative, by visit ‐ at weeks 2, 4, 6, and 8. The authors did not present the data as primary analysis in simple units of time. None of the trials reported on fluoroquinolone‐specific adverse effects.
Excluded studies
We have detailed the reasons for excluding the remaining 66 studies in the 'Characteristics of excluded studies' section.
Risk of bias in included studies
For details of risk of bias in the included trials please see Risk of bias tables of individual trials in Characteristics of included studies. For a summary of the 'Risk of bias' assessments please see Figure 2.
2.
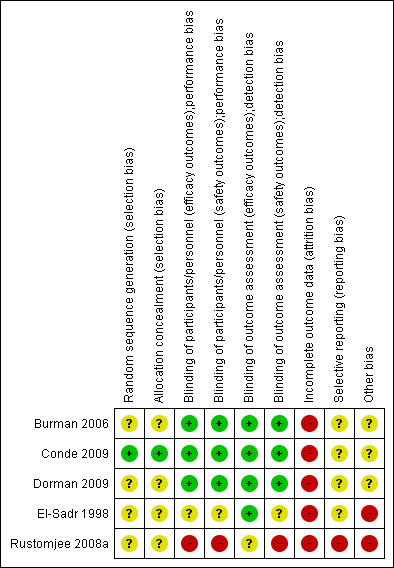
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
We judged generation of the randomized allocation sequence and of allocation concealment to be at low risk of bias for one trial (Conde 2009), and four trials were unclear regarding randomization methods (El‐Sadr 1998; Burman 2006; Dorman 2009; Rustomjee 2008a).
Blinding
Of the five included trials, one trial report stated that the trial blinded the providers, participants, and assessors (Conde 2009) and was judged to be at low risk of both performance and detection bias. One trial blinded the assessors for efficacy outcomes (El‐Sadr 1998) and was judged to be at low risk of detection bias. Blinding in the remaining three trials was not described. However, both Burman 2006 and Dorman 2009 used double‐dummy placebo controls and were therefore judged implicitly as at low risk of performance and detection bias for both efficacy and safety outcomes. Rustomjee 2008a was described as open‐label and was therefore judged generally at high risk of performance and detection bias, though laboratory‐based efficacy outcomes are likely to have been an exception and were classified as unclear.
Incomplete outcome data
We judged all five trials to be at high risk of bias due to either moderate dropout (> 15%), differential dropout between groups that had the potential to alter the result, or participants missing from the primary analysis who could not be accounted for. None of the trials included all of the randomized participants in the final analysis.
Selective reporting
We considered one trial (Rustomjee 2008a) to be at high risk of bias due to selective reporting of data on participants missing from the analyses, data on adverse events, and not specifying to which study group missing participants and participants with adverse events belonged. We judged the other four trials (El‐Sadr 1998; Burman 2006; Conde 2009; Dorman 2009) as unclear regarding reporting bias.
Other potential sources of bias
Pharmaceutical companies provided study drugs in four of the included trials. Further involvement of the pharmaceutical company in trial design, execution of trials, and analysis was only described in one study (Conde 2009). Three studies (El‐Sadr 1998; Burman 2006; Dorman 2009) did not describe potential input of pharmaceutical companies.
Two trials declared a financial relationship with a pharmaceutical entity that had an interest in the subject of the manuscript (Burman 2006, two out of 12 authors) and (Dorman 2009, three out of 19 authors). Two trial reports had no conflict of interest statements (El‐Sadr 1998; Rustomjee 2008a). The authors of one trial report (Conde 2009) declared no conflict of interest.
Effects of interventions
See: Table 1; Table 2; Table 3
Comparison 1. Fluoroquinolones plus standard regimen versus standard regimen alone (Fluoroquinolones + HRZE versus HRZE alone, one trial, 174 participants)
El‐Sadr 1998 compared levofloxacin added to first‐line antituberculous drugs (fluoroquinolones + HRZE) with the standard regimen (HRZE) (one trial, 174 randomized participants).
Death from any cause
Four deaths occurred in this single trial, and all occurred during the first eight weeks of treatment: one in the levofloxacin group (fluoroquinolones + HRZE) and three in the control (HRZE) group (Analysis 1.1).
1.1. Analysis.
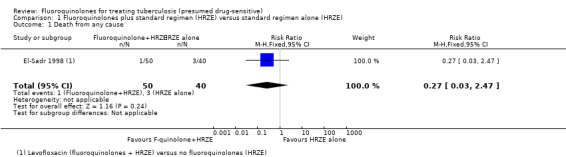
Comparison 1 Fluoroquinolones plus standard regimen (HRZE) versus standard regimen alone (HRZE), Outcome 1 Death from any cause.
TB‐related death
One participant in the levofloxacin group died of pulmonary TB after 17 days of treatment and had a pan‐susceptible isolate; one participant in the control group had MDR‐TB and died five days after admission (Analysis 1.2).
1.2. Analysis.
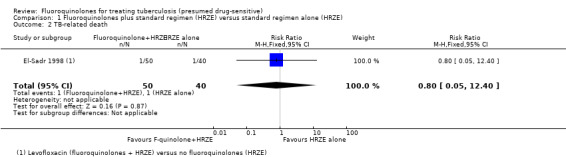
Comparison 1 Fluoroquinolones plus standard regimen (HRZE) versus standard regimen alone (HRZE), Outcome 2 TB‐related death.
Sputum culture conversion at eight weeks
For sputum conversion, no difference was detected. Sensitivity analysis for worst case and best case scenario did not alter the finding of no difference detected (one trial, 174 participants, Analysis 1.3).
1.3. Analysis.
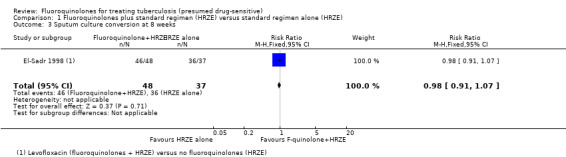
Comparison 1 Fluoroquinolones plus standard regimen (HRZE) versus standard regimen alone (HRZE), Outcome 3 Sputum culture conversion at 8 weeks.
Time to sputum culture or smear conversion
There was no reported differences in the time to culture conversion between levofloxacin added to the standard first‐line regimen and the standard regimen alone using cumulative percentage culture negative by visit.
Serious adverse events
There were 24 people with reported serious adverse events; 11 in the levofloxacin group (fluoroquinolones + HRZE) and 13 in the control (HRZE) group (Analysis 1.4).
1.4. Analysis.
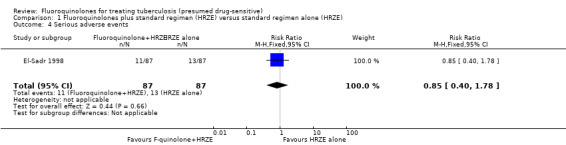
Comparison 1 Fluoroquinolones plus standard regimen (HRZE) versus standard regimen alone (HRZE), Outcome 4 Serious adverse events.
Total number of people with adverse events
El‐Sadr 1998 reported the total number of people with adverse events. There were 15 people in each group (levofloxacin and control) with one or more adverse events (Analysis 1.5). The adverse events included hepatic and hematologic toxicity, dermatologic reactions with fever, renal or metabolic toxicity, peripheral neuropathy, nausea, vomiting, and others not specified in the trial report (see Appendix 3).
1.5. Analysis.
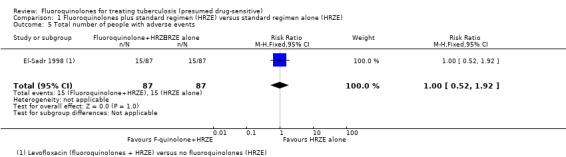
Comparison 1 Fluoroquinolones plus standard regimen (HRZE) versus standard regimen alone (HRZE), Outcome 5 Total number of people with adverse events.
Comparison 2. Fluoroquinolone substitution for ethambutol in a standard six month regimen (Fluoroquinolones + HRZ versus HRZE, three trials, 723 participants)
Three trials (Burman 2006; Rustomjee 2008a; Conde 2009) compared moxifloxacin substituting for ethambutol in isoniazid, rifampicin and pyrazinamide‐containing antituberculous regimens (fluoroquinolones + HRZ) with the standard ethambutol‐containing regimen (HRZE). Rustomjee 2008a also compared substitutions with gatifloxacin or ofloxacin in the same setting. In Conde 2009 all participants were HIV‐negative. In Burman 2006 and Rustomjee 2008a, HIV‐positive and HIV‐negative participants were equally represented in fluoroquinolones (fluoroquinolones + HRZ) and control groups (HRZE). Conde 2009 and Rustomjee 2008a reported fully‐sensitive M. tuberculosis isolates. One participant was isoniazid‐resistant in Rustomjee 2008a. Burman 2006 reported more than 90% participants to have sensitive M. tuberculosis isolates but included participants with isoniazid‐resistant organisms in the primary analysis.
Relapse
One trial (Conde 2009) reported on relapse. Three participants relapsed in the moxifloxacin group and four people relapsed in the ethambutol group (one trial, 170 participants, Analysis 2.1). Conde 2009 confirmed relapse by positive culture and compatible clinical symptoms within a year after completion of treatment. No measure of completeness of follow‐up was presented in the study report and no molecular analysis of relapse strains was performed. Much larger trials would be needed to detect a statistically significant difference.
2.1. Analysis.
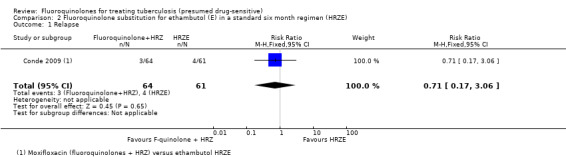
Comparison 2 Fluoroquinolone substitution for ethambutol (E) in a standard six month regimen (HRZE), Outcome 1 Relapse.
Death from any cause
All three included trials reported on death from any cause. In pooled meta‐analysis we found no overall difference in the number of deaths from any cause nor in subgroups by substituting fluoroquinolone. We did not detect any significant heterogeneity between trials (three trials, 723 participants, Analysis 2.2).
2.2. Analysis.
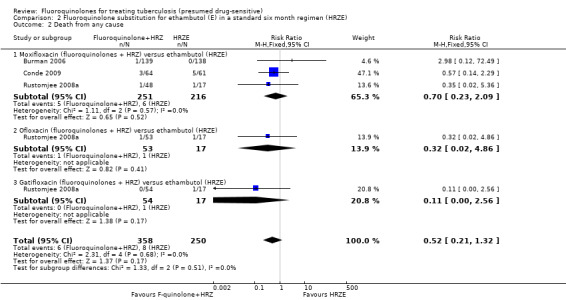
Comparison 2 Fluoroquinolone substitution for ethambutol (E) in a standard six month regimen (HRZE), Outcome 2 Death from any cause.
In Burman 2006 one participant died in the moxifloxacin group during the intensive phase. The trial authors attributed the death to pulmonary embolism unrelated to antituberculous treatment. Nobody died in the control group (HRZE). In Conde 2009 one participant in each group died during the intensive phase. The trial authors attributed the deaths of three participants in the moxifloxacin group (fluoroquinolones + HRZ) to urinary sepsis, gunshot wound, and oesophageal neoplasm (Conde 2009). Among the five participants in the ethambutol group (HRZE) who died, two had gunshot wounds, one had a subdural haemorrhage, for one participant the cause of death was unknown and for one participant death was attributed to TB (Conde 2009). Rustomjee 2008a did not present data on cause of death by study groups, or time of death: one death was due to haemoptysis, one was due to epileptic seizures, and two deaths were attributed to progression of AIDS.
TB‐related death
Only one trial (Conde 2009) reported on TB‐related death: this was one death in the control (HRZE) group which occurred during the intensive phase (on the 31st day of enrolment) (Analysis 2.3).
2.3. Analysis.
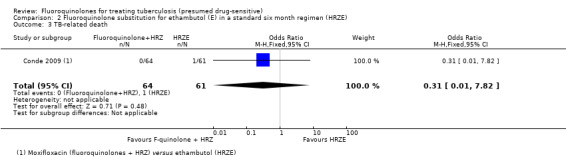
Comparison 2 Fluoroquinolone substitution for ethambutol (E) in a standard six month regimen (HRZE), Outcome 3 TB‐related death.
Sputum culture conversion at eight weeks
All three included trials reported on this outcome. We found no difference in the number of sputum culture converted participants: neither when we subgrouped by substituting fluoroquinolone nor when we pooled data together. We detected no significant heterogeneity with I2 = 32% (Analysis 2.4).
2.4. Analysis.
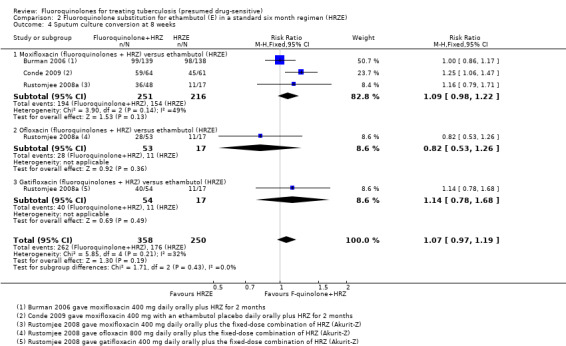
Comparison 2 Fluoroquinolone substitution for ethambutol (E) in a standard six month regimen (HRZE), Outcome 4 Sputum culture conversion at 8 weeks.
Time to sputum culture or smear conversion
Since the trials did not report on time to culture conversion uniformly we could not combine these findings in a meaningful way. Conde 2009 reported that moxifloxacin substitution for ethambutol resulted in more rapid sputum culture conversion, with a median time to consistently negative cultures of 35.5 days in the moxifloxacin group versus 48.5 days in the ethambutol group (log‐rank P = 0.005). Burman 2006 found that the proportion of sputum culture negative was higher at week 4 in the moxifloxacin group 37% (62 of 167) versus 26% (43 of 165) (P = 0.05) in the ethambutol group, but without statistically significant differences at earlier and later weeks, including week eight. Rustomjee 2008a, using Cox proportional hazards modelling to estimate time to culture conversion, found that moxifloxacin but not gatifloxacin accelerated culture conversion compared to the control (moxifloxacin HR 1.73, P = 0.009; gatifloxacin HR = 1.26, P = 0.3).
Serious adverse events
We found no difference between the regimens (fluoroquinolones + HRZ versus HRZE) in the number of people with serious adverse events. We detected no heterogeneity (Analysis 2.5).
2.5. Analysis.
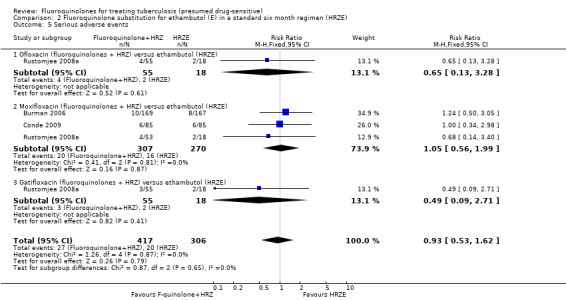
Comparison 2 Fluoroquinolone substitution for ethambutol (E) in a standard six month regimen (HRZE), Outcome 5 Serious adverse events.
Total number of people with adverse events
None of the three trials in this comparison reported on this outcome. However, Burman 2006 reported that nausea was more common among participants in the moxifloxacin group (fluoroquinolones + HRZ) than in the ethambutol group (HRZE): RR 2.4; 95% CI 1.4, to 4.2, one trial).
Adverse events across the trials included nausea and/or vomiting, diarrhoea, vision change, dizziness, paraesthesias and ataxia and peripheral neuropathy, rash and pruritis, fevers, arthralgia, and hepatotoxicity (see Appendix 3).
Comparison 3. Fluoroquinolone substitution for isoniazid in a standard six month regimen (Fluoroquinolones + RZE versus HRZE, one trial, 433 participants)
Dorman 2009 compared moxifloxacin substitution for isoniazid in the standard first‐line regimen (fluoroquinolones + RZE) with the standard isoniazid containing regimen (HRZE). More than 90% of randomized participants were HIV‐negative and had sensitive M. tuberculosis isolates.
Death from any cause
Only seven deaths occurred in this single trial: 3/219 in the moxifloxacin (fluoroquinolones + RZE) group and 4/214 in the control (HRZE) group (Analysis 3.1). Much larger trials would be needed to detect a statistically significant difference. However, the deaths occurred at different times in the two groups.
3.1. Analysis.
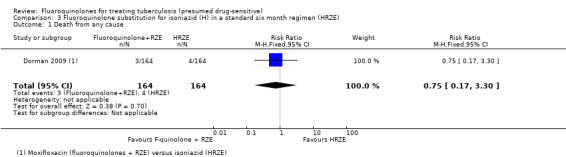
Comparison 3 Fluoroquinolone substitution for isoniazid (H) in a standard six month regimen (HRZE), Outcome 1 Death from any cause.
All three participants who died in the moxifloxacin group (fluoroquinolones + RZE) died during the intensive phase of antituberculous treatment (Dorman 2009). The authors attributed the cause of two of these deaths to advanced pulmonary TB, and the cause of death of the third participant to acute pulmonary embolus: a 48‐year old African female without known diabetes mellitus developed diabetic ketoacidosis after 14 days of study treatment and died five days later in the hospital.
All four participants who died in the isoniazid group (HRZE) died during the continuation phase of antituberculous treatment (Dorman 2009). The authors judged these deaths unrelated to study drugs: two of these died from complications of HIV infection: one from sequale of severe pulmonary TB, and one from colon cancer.
TB‐related death
Only three deaths were judged by the authors to be related to TB: two in the moxifloxacin group and one in the control group (Analysis 3.2). However, these deaths occurred at different times in the different groups.
3.2. Analysis.
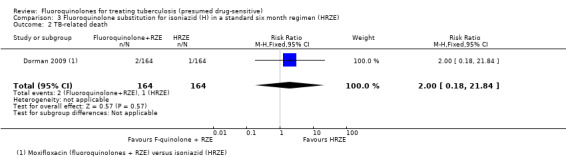
Comparison 3 Fluoroquinolone substitution for isoniazid (H) in a standard six month regimen (HRZE), Outcome 2 TB‐related death.
The two TB‐related deaths in the moxifloxacin group (fluoroquinolones + RZE) occurred during the intensive phase of treatment, and the TB‐related death in the isoniazid group (HRZE) occurred in the continuation phase of antituberculous treatment (see Analysis 3.2).
Sputum culture conversion at eight weeks
Moxifloxacin substituted for isoniazid did not have any effect on sputum culture conversion (one trial, 433 randomized participants; Analysis 3.3).
3.3. Analysis.
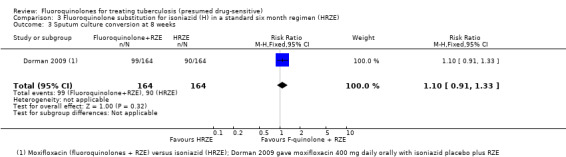
Comparison 3 Fluoroquinolone substitution for isoniazid (H) in a standard six month regimen (HRZE), Outcome 3 Sputum culture conversion at 8 weeks.
Time to culture or smear conversion
There was no difference in probability of observing stable culture conversion between the moxifloxacin and isoniazid arms using the Gehan‐Wilcoxon test (P = 0.16)
Serious adverse events
Substituting with moxifloxacin for isoniazid did not result in fewer people with serious adverse events: there were nine in the moxifloxacin group and eight in the control group (one trial, 433 randomized participants, Analysis 3.4).
3.4. Analysis.
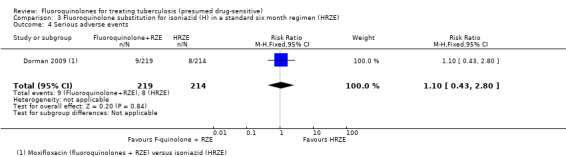
Comparison 3 Fluoroquinolone substitution for isoniazid (H) in a standard six month regimen (HRZE), Outcome 4 Serious adverse events.
Total number of people with adverse events
Dorman 2009 did not report on this outcome, but did report that nausea was more common among participants in the moxifloxacin group (fluoroquinolones + RZE) than in the isoniazid control (HRZE) (RR 1.68, 95% CI 1.05 to 2.66 (see Appendix 3).
We conducted the sensitivity analysis (as described in Appendix 2) to test the robustness of our methodology. These analyses did not substantially change the direction, magnitude, or CIs of the estimate of effect.
Discussion
WHO treatment guidelines recommend fluoroquinolones for treating MDR‐TB based on observational cohort data and expert opinion (WHO 2006; Johnston 2009; WHO 2010b; Falzon 2011) and it is now unlikely that randomized placebo‐controlled trials to support their use will be forthcoming. However, equipoise remains concerning the potential of fluoroquinolones to improve or reduce the duration of first‐line therapy. Hence, this systematic review focuses on trials conducted in the context of drug‐sensitive TB since this area is currently of greatest research interest. This review assesses the benefits and harms of fluoroquinolones when added to or substituted for isoniazid or ethambutol in the first line regimen, focusing on clinically relevant and widely accepted outcomes, and specifically excludes quantitative bacteriological methods.
Summary of main results
We identified five RCTs, involving 1330 participants, that met the inclusion criteria.
Fluoroquinolones added to standard regimens
A single trial (El‐Sadr 1998; 174 participants) evaluated the addition of levofloxacin to the standard first‐line TB treatment regimen. The trial did not report on treatment failure or relapse for this comparison, and did not demonstrate an effect on death, sputum conversion, or adverse events (all outcomes‐ very low quality evidence).
Fluoroquinolones substituted for ethambutol in standard regimens
Three trials (Burman 2006; Rustomjee 2008a; Conde 2009 ; 723 participants) evaluated substitution of moxifloxacin, gatifloxacin, and ofloxacin into the standard first‐line TB regimen. One trial (Conde 2009) reported no effect on relapse (very low quality evidence). These trials did not report on treatment failure and did not show an effect on death (very low quality evidence; Figure 3), sputum conversion (very low quality evidence; Figure 4), or serious adverse events (very low quality evidence).
3.
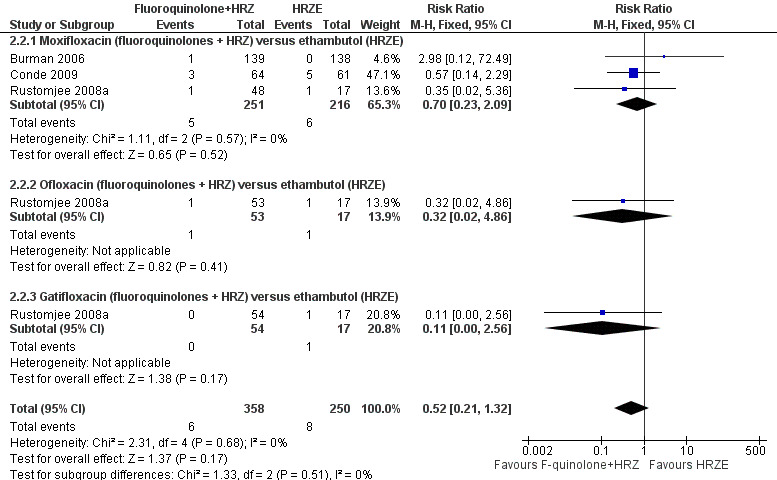
Forest plot of comparison: 2 Fluoroquinolone (F‐quinolone) substitution for ethambutol (E) in a standard six month regimen (HRZE), outcome: 2.2 Death from any cause (complete case analysis).
4.
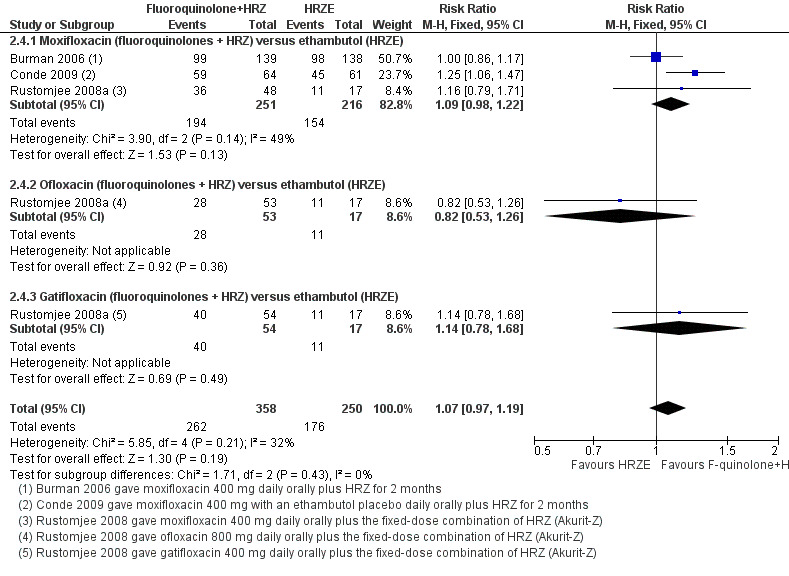
Forest plot of comparison: 2 Fluoroquinolone (F‐quinolone) substitution for ethambutol (E) in a standard six month regimen (HRZE), outcome: 2.4 Sputum culture conversion at eight weeks (complete case analysis).
Fluoroquinolones substituted for isoniazid in standard regimens
A single trial (Dorman 2009; 433 participants) evaluated substitution of moxifloxacin into the standard first‐line TB regimen. This trial did not report on treatment failure or relapse, and did not demonstrate an effect on death, sputum conversion, or serious adverse events (all outcomes‐ low quality evidence).
Overall completeness and applicability of evidence
In this review, we restricted inclusion of trials to those that recruited new cases of TBwith presumed drug‐sensitive TB. We also restricted the scope of the review to trials in which the intervention contained at least rifampicin and pyrazinamide, the drugs on which the current duration of first‐line treatment depends. The trial sites were geographically diverse but there was a lack of studies conducted in Asia. The trials were conducted in low‐income and middle‐income countries, which means the results of this review are likely to be applicable to situations where the burden of TB is high and new revised treatment strategies are most urgently needed. HIV‐positive participants were relatively well‐represented in the included trials (29.8% of included participants overall). However, one trial explicitly excluded them and the primary outcomes of all the included trials were reached before initiation of ART. None of the included trials stratified outcomes by HIV status. Given the poor prognosis of HIV‐positive patients with TB (Daley 1992; Telzak 1999; El‐Sadr 2001), further evidence relating to use of fluoroquinolones in conjunction with ART in this group would be welcome. Evidence is generally lacking on the safety and efficacy of fluoroquinolone additions or substitutions in children (< 18 years) and in pregnant and lactating women who were excluded from all of the included trials.
Within the included studies, reporting of the selected outcomes was inconsistent and at times incomplete. All of the trials that we included were Phase II trials. Most trials did not report follow‐up even to the end of the treatment regimen and only one trial reported on relapse after treatment was discontinued. While this was not unexpected, it made reporting of the combined endpoint of treatment failure/relapse impossible in the current review. Furthermore, variable reporting of data on time to culture or smear conversion and safety made meaningful data synthesis of these outcomes difficult. Harmonized reporting standards for these and other outcomes in TB trials would be welcome, given that nine ongoing trials have been identified and will soon expand the evidence base addressing the questions of this review.
All the included studies were small Phase II studies the power of this analysis for long term efficacy and safety outcomes is bound to be limited. However, this will be addressed by the ongoing Phase III trials we identified.
None of the trials reported on fluoroquinolone‐specific adverse effects, such as tendinopathy or rupture, dysglycaemia, or dysrhythmias due to QTc prolongation. However, they did report the number of adverse events, including those considered serious enough to discontinue or change treatment, and no difference in the number of patients with adverse effects in fluoroquinolone regimens was detected. Substitution of moxifloxacin for ethambutol (Burman 2006) or for isoniazid (Dorman 2009) in first‐line regimens was reported by the trial authors to result in higher incidences of nausea. For descriptive safety data, please see the adverse events table (Appendix 3).
Quality of the evidence
We assessed the quality of the evidence using the GRADE process (Guyatt 2008) and we presented the results in the 'Summary of findings tables'. For these tables we asked the following questions:
1) Should fluoroquinolone be added to standard first‐line regimen to improve outcomes in presumed drug‐sensitive TB?
We do not know from this single trial (El‐Sadr 1998) if fluoroquinolone addition to standard first‐line regimen improves treatment outcomes in people with presumed drug‐sensitive TB (Table 1).
2) Are fluoroquinolones a suitable substitution (alternative to) for ethambutol in a standard six month first‐line regimen in people with presumed drug‐sensitive TB?
From three trials (Burman 2006; Rustomjee 2008a; Conde 2009) we do not know if fluoroquinolones present a suitable substitution for ethambutol in a standard six month first‐line regimen. There is very low quality evidence that fluoroquinolones (moxifloxacin, gatifloxacin, and ofloxacin) perform no worse than ethambutol (Table 2).
3) Are fluoroquinolones a suitable substitution (alternative to) for isoniazid in a standard six month first‐line regimen in people with presumed drug‐sensitive TB?
The single trial (Dorman 2009) provided low quality evidence that moxifloxacin may perform no worse than isoniazid in treating people with presumed drug‐sensitive TB (Table 3).
Potential biases in the review process
We performed the data extraction unblinded. All of the included trials are published and we were unable to obtain further unpublished data from pharmaceutical companies. GDAV is a co‐author on the trial report of Rustomjee 2008a.
Agreements and disagreements with other studies or reviews
We asked whether fluoroquinolones have a role in the closely related goals of improving the efficacy or shortening duration of first‐line therapy in newly diagnosed TB patients with presumed drug‐sensitive organisms. The original surpassed version of this review provided clear evidence that substitution of ciprofloxacin for pyrazinamide and ethambutol did not improve efficacy or tolerability compared to the current first‐line regimen. Culture conversion during treatment appeared slower and observed relapse rates were higher (Kennedy 1993; Kennedy 1996; Ziganshina 2005; Ziganshina 2008). Another study replacing rifampicin with ciprofloxacin reported similar smear conversion but a higher incidence of combined treatment failure and relapse using this regimen, though these endpoints were not based on culture (Saigal 2001). These unfavourable results argue against a useful role for ciprofloxacin in treatment of new cases of TB and we have hence removed these trials from the review.
In this updated review, we found little evidence to support use of ofloxacin as a substitute for ethambutol in the first‐line regimen. Estimates from one trial (Rustomjee 2008a) suggest that culture conversion is not improved compared to ethambutol, and observed numbers of adverse events were not lower. Another trial, which was excluded from this review, though reported definitive outcomes, found estimates of efficacy and safety outcomes to be similar (Kohno 1992). Thus ofloxacin may be an acceptable alternative to ethambutol for individual patients in cases of individual poor tolerability of the drug but would not appear to have enough advantages to routinely replace it in first‐line regimens. Neither does addition of levofloxacin to the standard first‐line regimen improve efficacy or tolerability.
We did not find evidence to support use of the newer fluoroquinolones gatifloxacin or moxifloxacin as a component of the first‐line regimen. Four Phase II trials have to date evaluated substitution of these drugs for ethambutol or isoniazid and as yet any evidence of efficacy rests on surrogate endpoints such as different measures of culture conversion which are not universally accepted and reported inconsistently by investigators. In our meta‐analysis, we observed no statistically significant differences in culture conversion at eight weeks, the most widely supported surrogate endpoint,. Though overall numbers of adverse events were similar, two trials based on similar safety reporting protocols reported more frequent nausea in participants randomized to moxifloxacin.
Authors' conclusions
Implications for practice.
Four fluoroquinolones − ofloxacin, levofloxacin, moxifloxacin, and gatifloxacin − have been tested in RCTs for treating presumed drug‐sensitive TB in a standard six month regimen. None of the tested fluoroquinolones when added to or substituted into the first‐line regimen, either separately or in pooled meta‐analysis, improved any of the review outcomes: relapse, treatment failure, death, TB‐related death, sputum culture conversion at eight weeks, or serious adverse events. Moxifloxacin consistently contributed to more nausea in trial participants. Data are currently lacking regarding use of fluoroquinolones with early ART. Currently there is no high‐quality evidence to change existing WHO recommendations relating to fluoroquinolones.
Implications for research.
We identified several on‐going trials that have been designed to further evaluate the potential of fluoroquinolones to shorten the duration of first‐line treatment, the results of which are likely to become publicly available in the next few years. These trials are currently predicated on the inconsistently reported and variable results of time‐to‐event data which could not be meaningfully synthesized in this review and on data from quantitative bacteriology which is not well‐supported as a surrogate endpoint and was therefore not included in the scope of the review. In the absence of preliminary proof of efficacy on commonly accepted early endpoints, these trials will provide important data on the definitive endpoint of treatment failure and relapse and emerging safety concerns such as dysglycaemia for gatifloxacin, QT prolongation for moxifloxacin, or tendon rupture for high‐dose levofloxacin.
In this review, we noted that the reporting standards in these recently conducted TB trials was variable and lacking in quality. In addition, there were significant differences in terms of inclusion criteria relating to HIV‐seropositivity, initial drug resistance, and in the definition of outcomes, and the power of studies. A move towards more standardized approaches to measuring and reporting efficacy outcomes, adverse events, and more high powered studies would greatly improve comparability between TB trials and facilitate subsequent meta‐analysis.
Though HIV seropositive participants were represented in the current trials, new trials looking at the efficacy and safety of fluoroquinolones that stratify results by HIV status and use of ART would provide valuable information for future deployment of putative new fluoroquinolone‐containing regimens in high burden countries. The most vulnerable populations (pregnant women and children) were excluded from all trials, and represent a critical gap in current knowledge.
What's new
| Date | Event | Description |
|---|---|---|
| 4 March 2013 | New search has been performed | We narrowed the scope of the review to presumed drug‐sensitive tuberculosis, restructured research questions, comparisons and outcomes. We included one more comparison ‐ combined end‐point of relapse/treatment failure and one additional trial (Rustomjee 2008a). Geraint Davies joined the author team. We refined the conclusions. |
| 4 March 2013 | New citation required but conclusions have not changed | We carried out a new search, included one new trial, restructured the review and refined the conclusions. |
History
Protocol first published: Issue 2, 2004 Review first published: Issue 3, 2005
| Date | Event | Description |
|---|---|---|
| 3 May 2010 | New search has been performed | 2010, Issue 7: We updated the search and included two new trials. Albina F.Titarenko joined the author team. We refined the c onclusions. |
| 18 August 2008 | Amended | We c onverted to a new review format with minor editing. |
| 13 November 2007 | New citation required but conclusions have not changed | 2008, Issue 1: We updated the search and we included one new trial. Alexander Vizel stepped down as a co‐author. |
Acknowledgements
We acknowledge the following support provided for the original review (Ziganshina 2005): The protocol was developed during the Fellowship Programme, organized by the Cochrane Infectious Diseases Group in June‐July 2003. We finalized the original review (Ziganshina 2005) during the Fellowship Programme organized by the CIDG in July 2004. The EQUI‐TB Knowledge Programme (which receives funding from the UK Department for International Development) also provided support.
We acknowledge Stephen B. Squire for contributing as an author to previous versions of the review.
This document is an output from a project funded by the UK Department for International Development (DFID) for the benefit of developing countries. The views expressed are not necessarily those of DFID. We finalized the current version of the review during the Fellowship Programme organized by the Cochrane Infectious Diseases Group in July‐August 2012. We thank Paul Garner, David Sinclair, Sarah Donegan, Anne‐Marie Stephani, and Hannah Ryan for assistance with this update. We thank Vittoria Lutje, Trials Search Co‐ordinator for the CIDG for continuous assistance with the searches for all versions of the review, Philomena Hinds, Christianne Esparza and other staff of the CIDG for assistance during the process of developing the protocol, the first version and updating the review.
Appendices
Appendix 1. Search methods: detailed search strategies
| Search set | CIDG SRa | CENTRAL | MEDLINEb | EMBASEb | LILACSb | SCIb | Russian database |
| 1 | tuberculosis | TUBERCULOSIS | TUBERCULOSIS | TUBERCULOSIS | tuberculosis | tuberculosis | tuberculosis |
| 2 | fluoroquinolones | tuberculosis | tuberculosis | tuberculosis | fluoroquinolones | fluoroquinolones | quinolones |
| 3 | — | fluoroquinolone | 1 or 2 | 1 or 2 | ciprofloxacin | ciprofloxacin | fluoroquinolones |
| 4 | — | amifloxacin | QUINOLINES | QUINOLINE DERIVED ANTIINFECTIVE AGENTS | enoxacin | enoxacin | ciprofloxacin |
| 5 | — | balofloxacin | QUINOLONES | fluoroquinolones | fleroxacin | fleroxacin | clinafloxacin |
| 6 | — | cetefloxacin | ANTI‐INFECTIVE AGENTS, QUINOLONE | amifloxacin | norfloxacin | norfloxacin | enoxacin |
| 7 | — | ciprofloxacin | FLUOROQUINOLONES | balofloxacin | pefloxacin | pefloxacin | fleroxacin |
| 8 | — | clinafloxacin | amifloxacin | CETEFLOXACIN | 2‐7/or | 2‐7/or | gatifloxacin |
| 9 | — | enoxacin | balofloxacin | cetefloxacin | 1 and 8 | 1 and 8 | gemifloxacin |
| 10 | — | fleroxacin | cetefloxacin | CIPROFLOXACIN | — | — | grepafloxacin |
| 11 | — | gatifloxacin | CIPROFLOXACIN | ciprofloxacin | — | — | levofloxacin |
| 12 | — | gemifloxacin | ciprofloxacin | CLINAFLOXACIN | — | — | lomefloxacin |
| 13 | — | grepafloxacin | clinafloxacin | clinafloxacin | — | — | moxifloxacin |
| 14 | — | irloxacin | ENOXACIN | ENOXACIN | — | — | norfleroxacin |
| 15 | — | levofloxacin | enoxacin | enoxacin | — | — | norfloxacin |
| 16 | — | lomefloxacin | FLEROXACIN | FLEROXACIN | — | — | ofloxacin |
| 17 | — | moxifloxacin | fleroxacin | fleroxacin | — | — | pefloxacin |
| 18 | — | nordifloxacin | gatifloxacin | GATIFLOXACIN | — | — | premafloxacin |
| 19 | — | norfleroxacin | gemifloxacin | gatifloxacin | — | — | rufloxacin |
| 20 | — | norfloxacin | grepafloxacin | GEMIFLOXACIN | — | — | sparfloxacin |
| 21 | — | ofloxacin | irloxacin | gemifloxacin | — | — | temafloxacin |
| 22 | — | oxociprofloxacin | levofloxacin | GREPAFLOXACIN | — | — | trovafloxacin |
| 23 | — | pefloxacin | lomefloxacin | grepafloxacin | — | — | — |
| 24 | — | premafloxacin | moxifloxacin | IRLOXACIN | — | — | — |
| 25 | — | prulifloxacin | nordifloxacin | irloxacin | — | — | — |
| 26 | — | rufloxacin | norfleroxacin | LEVOFLOXACIN | — | — | — |
| 27 | — | sitafloxacin | NORFLOXACIN | levofloxacin | — | — | — |
| 28 | — | sparfloxacin | norfloxacin | LOMEFLOXACIN | — | — | — |
| 29 | — | temafloxacin | ofloxacin | lomefloxacin | — | — | — |
| 30 | — | tosufloxacin | oxociprofloxacin | MOXIFLOXACIN | — | — | — |
| 31 | — | trovafloxacin | PEFLOXACIN | moxifloxacin | — | — | — |
| 32 | — | 2‐31/OR | pefloxacin | NORDIFLOXACIN | — | — | — |
| 33 | — | 1 and 32 | premafloxacin | nordifloxacin | — | — | — |
| 34 | — | — | prulifloxacin | NORFLEROXACIN | — | — | — |
| 35 | — | — | rufloxacin | norfleroxacin | — | — | — |
| 36 | — | — | sitafloxacin | NORFLOXACIN | — | — | — |
| 37 | — | — | sparfloxacin | norfloxacin | — | — | — |
| 38 | — | — | temafloxacin | OFLOXACIN | — | — | — |
| 39 | — | — | tosufloxacin | ofloxacin | — | — | — |
| 40 | — | — | trovafloxacin | OXOCIPROFLOXACIN | — | — | — |
| 41 | — | — | 4‐40/or | oxociprofloxacin | — | — | — |
| 42 | — | — | 3 and 41 | PEFLOXACIN | — | — | — |
| 43 | — | — | limit 42 to human | pefloxacin | — | — | — |
| 44 | — | — | — | PREMAFLOXACIN | — | — | — |
| 45 | — | — | — | premafloxacin | — | — | — |
| 46 | — | — | — | PRULIFLOXACIN | — | — | — |
| 47 | — | — | — | prulifloxacin | — | — | — |
| 48 | — | — | — | RUFLOXACIN | — | — | — |
| 49 | — | — | — | rufloxacin | — | — | — |
| 50 | — | — | — | SITAFLOXACIN | — | — | — |
| 51 | — | — | — | sitafloxacin | — | — | — |
| 52 | — | — | — | SPARFLOXACIN | — | — | — |
| 53 | — | — | — | sparfloxacin | — | — | — |
| 54 | — | — | — | TEMAFLOXACIN | — | — | — |
| 55 | — | — | — | temafloxacin | — | — | — |
| 56 | — | — | — | tosufloxacin | — | — | — |
| 57 | — | — | — | 4‐56/or | — | — | — |
| 58 | — | — | — | 3 and 57 | — | — | — |
| 59 | — | — | — | limit 58 to human | — | — | — |
aCochrane Infectious Diseases Group Specialized Register. b Search terms used in combination with the search strategy for retrieving trials developed by The Cochrane Collaboration (Lefebvre 2011); upper case: MeSH or EMTREE heading; lower case: free text term.
Appendix 2. Efficacy outcome measures and sensitivity analyses
| Analysis | Participants | Numerator | Denominator |
| Primary analysis | Exclusions after enrolment varying by outcome | Excludeda | Excluded |
| Sensitivity analysis 1 – Worst case: Negative outcomes b |
All exclusions after enrolment | Included as failures | Included |
| Sensitivity analysis 2 – Best case: Negative outcomes |
All exclusions after enrolment | Excluded | Included |
| Sensitivity analysis 1 – Worst case: Positive outcomes (culture conversion at eight weeks) |
All exclusions after enrolment | Excluded | Included |
| Sensitivity analysis 2 – Best case: Positive outcomesc (culture conversion at eight weeks) |
All exclusions after enrolment | Included as successes | Included |
Footnotes
a'Excluded' means removed from the calculation.
bTo re‐classify all exclusions after enrolment (losses to follow‐up, withdrawn consent, other antibiotic use, or failure to complete treatment) as treatment failures. For negative outcomes this represents a true worse‐case scenario.
c To re‐classify all exclusions after enrolment (losses to follow‐up, withdrawn consent, other antibiotic use, or failure to complete treatment) as treatment successes. For positive outcomes this represents a true best‐case scenario.
Appendix 3. Adverse events table
| 1. Fluoroquinolones plus standard regimen (fluoroquinolones + HRZE) versus standard regimen alone (HRZE) | ||||
| Study ID | Fluoroquinolone | Adverse event monitoring | Blinding | Summary of adverse event findings |
|
El‐Sadr 1998 (174 participants) |
Levofloxacin | Assessed AE that occurred during receipt of study medication and for eight weeks after its discontinuation, graded on a five‐point scale | Open label Assessors blinded |
Serious adverse events: 12.6 % (11/87) in fluoroquinolones+ HRZE group versus 14.9% (13/87) in HRZE group, nature of serious adverse events not reported, described as “at least one adverse event of grade IV or higher”; Any drug permanently discontinued: 8.1% in fluoroquinolones + HRZE group versus 11.5% in HRZE group (P = 0.61) GI: nausea/vomiting observed only in fluoroquinolones + HRZE group (1.2%) CNS/PNS: peripheral neuropathy observed only in fluoroquinolones + HRZE group (1.2%) CVS/RS: none reported Dermatological: dermatologic reactions with fever (2.3% in each group) Haematological: 3.5% in fluoroquinolones + HRZE versus 2.3% in HRZE (P = 1.00) Biochemical: renal or metabolic toxicity (3.5% in fluoroquinolones + HRZE versus 1.2 in HRZE alone (P = 0.62), hepatic toxicity (1.2% in fluoroquinolones + HRZE versus 5.8% in HRZE alone) Other: not specified, any AE probably related to study drug: 4.6% in fluoroquinolones + HRZE versus 5.8% in HRZE alone (P = 1.00) |
| 2. Fluoroquinolone substitution for ethambutol in a standard six month regimen (Fluoroquinolones + HRZ versus HRZE) | ||||
| Study ID | Fluoroquinolone | Adverse event monitoring | Blinding | Summary of adverse event findings |
| Burman 2006 (336 participants) | Moxifloxacin | Adverse event monitoring not described | Single blind, further – unclear | Serious adverse events: 10/169 in fluoroquinolones + HRZ versus 8/167 in HRZE (P = 1.00), including one death and eight hospitalizations in moxifloxacin (fluoroquinolones + HRZ) group versus no deaths and six hospitalizations in HRZE GI: nausea/vomiting (grade 3 or 4) observed only in fluoroquinolones + HRZ group (4/169, P = 0.12, RR 8.9, 95% CI 0.5 to 164) Nausea (any grade, selected symptom) 36/169 in fluoroquinolones + HRZ versus 15/167 in HRZE (P = 0.002; RR 2.4, 95% CI 1.4 to 4.2) Diarrhoea (grade 3 or 4) observed only in fluoroquinolones + HRZ group (3/169, P = 0.25, RR 6.9, 95% CI 0.4 to 133) Diarrhoea (any grade, selected symptoms) 12/169 in fluoroquinolones + HRZ versus 6/167 in HRZE (P = 0.23; RR 2, 95% CI 0.8 to 5.1) Vomiting (any grade, selected symptoms) 20/169 in fluoroquinolones + HRZE versus 15/167 in HRZE (P = 0.48; RR 1.3, 95% CI 0.7 to 2.5) CNS/PNS: Vision change (grade 3 or 4) 10/169 in fluoroquinolones + HRZ versus 9/167 in HRZE (P = 1.00, RR 1.1, 95% CI 0.5 to 2.6); dizziness (any grade, selected symptoms) 24/169 in fluoroquinolones + HRZ versus 15/167 in HRZE (P = 0.17; RR 1.6, 95% CI 0.9 to 2.9) CVS/RS: none reported Dermatological: none reported Haematological: none reported Biochemical: hepatotoxicity (grade 3 or 4) 6/169 in fluoroquinolones + HRZ group versus 7/167 in HRZE group (P = 0.79; RR 0.9, 95% CI 0.3 to 2.5) Other: Fevers (any grade) 29/169 in fluoroquinolones + HRZ group versus 20/167 in HRZE group (P = 0.22; RR 1.4, 95% CI 0.9 to 2.4); joint pain 57/169 in fluoroquinolones + HRZ group versus 44/167 in HRZE group (P = 0.15; RR 1.3, 95% CI 0.9 to 1.8) Overall: Any grade 3 or 4 toxicity: 31/169 in fluoroquinolones + HRZ group versus 19/167 in HRZE group (P = 0.09; RR 1.6, 95% CI 0.95 to 2.7) |
| Conde 2009 (170 participants) | Moxifloxacin | Assessed at weekly clinic visits. Liver enzymes, serum creatinine levels and complete blood counts performed monthly. ECG obtained at weeks 2, 4, 6, and 8 of treatment. |
Double‐blind | Serious adverse events: 6/85 in fluoroquinolones + HRZ group versus 6/85 in HRZE group including three versus five deaths and one versus one hospitalizations Specifics: Fluoroquinolones + HRZE: 1 – gun‐shot wound (death) 1 – community‐acquired pneumonia and pulmonary abscess 1 – urinary sepsis (death) 1 – spontaneous aborting 1 – oesophageal neoplasm (death) 1 – proteinuria HRZE: 1 – TB (death) 2 – gun‐shot wounds (two deaths) 1 – cutaneous reaction 1 – polyneuropathy (death) 1 – subdural haemorrhage (death) Treatment discontinued (five participants): Fluoroquinolones + HRZ: 1 – Nausea /vomiting (grade 2) 1 – Parasthesias and ataxia (grade 2) HRZE: 2 – Rash and pruritis (grade 2) 1 – Peripheral neuropathy (grade 3) |
| Rustomjee 2008a (217 participants) | Moxifloxacin Gatifloxacin Ofloxacin |
Adverse event monitoring not described | No blinding | Serious adverse events: Moxi 4/53 (1 death) Gati 3/55 (no death) O 4/55 (1 death) E 7/54 (2 deaths) GI: vomiting: Moxi 1/53 Gati 3/55 O 4/55 E 2/54 CNS/PNS: none reported CVS/RS: none reported Derm: none reported Haematological: anaemia 7% Biochemical: raised amylase activity in 41% of participants, attributed to HIV infection; hypokalaemia 6% Raised transaminase: Moxi 9/53 Gati 4/55 O 3/55 E 6/54 Other: arthralgia: Moxi 3/53 Gati 6/55 O 7/55 E 3/54 Overall comment: Authors did not present data on the most frequent adverse events by study group or on cause of death by study group, or time of death: one death was due to haemoptysis, the other to epileptic seizures and two deaths were attributed to progression of AIDS. Presentation of adverse events in the text and in the table was confusing. |
| 3. Fluoroquinolone substitution for isoniazid in a standard 6 month regimen (Fluoroquinolones + RZE versus HRZE) | ||||
| Study ID | Fluoroquinolone | Adverse event monitoring | Blinding | Summary of adverse event findings |
| Dorman 2009 (433 participants) | Moxifloxacin | Assessed at baseline and weeks 2, 4, 6, and 8 of treatment: symptoms, blood tests for AST, bilirubin, creatinine, and complete blood count. | Unclear, double‐blind | Serious adverse events: 9 in fluoroquinolones +RZE group versus 8 in HRZE group, including 3 versus 4 deaths (intensive phase versus continuation phase); Serious adverse events attributed to study treatments – 3 versus 2 GI: Nausea 42 in fluoroquinolones + RZE group versus 24 in HRZE group (P = 0.03; RR 1.68; 95% CI 1.05 to 2.66); Vomiting 22 versus 20 (P = 0.86) Diarrhoea 17 versus 12 (P = 0.40) CNS/PNS: dizziness 30 versus 19 (P = 0.13) CVS/RS: none reported Derm: rash 23 versus 23 (P = 0.88) Haematological: none reported Biochemical: hepatitis (AST 3 times ULN) 7/219 versus 7/214 (P = 0.93) Other: joint pain or discomfort 68 versus 61 (P = 0.65) Overall: study drugs permanently discontinued 31/219 in fluoroquinolones + RZE group versus 22/214 in HRZE group (P = 0.25; RR 1.35; 95% CI 0.81 to 2.25), grade 3 or greater toxicity 32/219 in fluoroquinolones + RZE group versus 39/214 in HRZE group (P = 0.27) |
Data and analyses
Comparison 1. Fluoroquinolones plus standard regimen (HRZE) versus standard regimen alone (HRZE).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death from any cause | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.03, 2.47] |
| 2 TB‐related death | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.8 [0.05, 12.40] |
| 3 Sputum culture conversion at 8 weeks | 1 | 85 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.91, 1.07] |
| 4 Serious adverse events | 1 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.40, 1.78] |
| 5 Total number of people with adverse events | 1 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.52, 1.92] |
Comparison 2. Fluoroquinolone substitution for ethambutol (E) in a standard six month regimen (HRZE).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Relapse | 1 | 125 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.17, 3.06] |
| 2 Death from any cause | 3 | 608 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.52 [0.21, 1.32] |
| 2.1 Moxifloxacin (fluoroquinolones + HRZ) versus ethambutol (HRZE) | 3 | 467 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.23, 2.09] |
| 2.2 Ofloxacin (fluoroquinolones + HRZ) versus ethambutol (HRZE) | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.02, 4.86] |
| 2.3 Gatifloxacin (fluoroquinolones + HRZ) versus ethambutol (HRZE) | 1 | 71 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.00, 2.56] |
| 3 TB‐related death | 1 | 125 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.01, 7.82] |
| 4 Sputum culture conversion at 8 weeks | 3 | 608 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.97, 1.19] |
| 4.1 Moxifloxacin (fluoroquinolones + HRZ) versus ethambutol (HRZE) | 3 | 467 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.98, 1.22] |
| 4.2 Ofloxacin (fluoroquinolones + HRZ) versus ethambutol (HRZE) | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.53, 1.26] |
| 4.3 Gatifloxacin (fluoroquinolones + HRZ) versus ethambutol (HRZE) | 1 | 71 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.78, 1.68] |
| 5 Serious adverse events | 3 | 723 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.53, 1.62] |
| 5.1 Ofloxacin (fluoroquinolones + HRZ) versus ethambutol (HRZE) | 1 | 73 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.13, 3.28] |
| 5.2 Moxifloxacin (fluoroquinolones + HRZ) versus ethambutol (HRZE) | 3 | 577 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.56, 1.99] |
| 5.3 Gatifloxacin (fluoroquinolones + HRZ) versus ethambutol (HRZE) | 1 | 73 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.09, 2.71] |
Comparison 3. Fluoroquinolone substitution for isoniazid (H) in a standard six month regimen (HRZE).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death from any cause | 1 | 328 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.17, 3.30] |
| 2 TB‐related death | 1 | 328 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.18, 21.84] |
| 3 Sputum culture conversion at 8 weeks | 1 | 328 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.1 [0.91, 1.33] |
| 4 Serious adverse events | 1 | 433 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.43, 2.80] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Burman 2006.
| Methods | Trial design: multicentre RCT Follow‐up: 8 weeks Adverse event monitoring: not described Inclusion of all randomized participants in the final analysis: 59/336 (17.6%) excluded from final analysis |
|
| Participants | Number: 336 randomized; 277 evaluated Inclusion criteria: aged 18 years or older with suspected pulmonary TB and acid‐fast bacilli in an expectorated sputum sample Exclusion criteria: history of > 7 days of a fluoroquinolone antibiotic or TB treatment within the previous 6 months; pregnancy or breastfeeding; initial sputum cultures negative for M. tuberculosis or resistance to rifampicin, fluoroquinolones, or pyrazinamide (patients whose isolates were resistant to isoniazid were included) |
|
| Interventions | Fluoroquinolone (moxifloxacin) substituted into regimen (replacing ethambutol) for 2 months (8 weeks), initial 2 weeks of daily therapy under "supervision" 1. Moxifloxacin (400 mg daily) orally plus basic regimen (5 days a week or thrice a week for both dosing regimens) for 2 months 2. Ethambutol (0.8 g ‐ 40 to 55 kg; 1.2 g ‐ 56 to 75 kg; 1.6 g ‐ 76 to 90 kg) daily orally 5 days a week or (1.2 g ‐ 40 to 55 kg; 2 g ‐ 56 to 75 kg; 2.4g ‐ 76 to 90 kg) thrice weekly for 2 months plus basic regimen Basic regimen: Isoniazid (300 mg), rifampicin (450 mg if ≤ 45 kg; 600 mg if > 45 kg), and pyrazinamide (1 g ‐ 40 to 55 kg; 1.5 g ‐ 56 to 75 kg; 2 g ‐ 76 to 90 kg) given orally 5 days a week for 2 months; or isoniazid (15 mg/kg, max dose 900 mg), rifampicin (450 mg if ≤ 45 kg; 600 mg if > 45 kg), and pyrazinamide (1.5 g ‐ 40 to 55 kg; 2.5 g ‐ 56 to 75 kg; 3 g ‐ 76 to 90 kg) given thrice weekly orally for 2 months |
|
| Outcomes | 1. Death from any cause: 1/169 versus 0/167 2. Sputum culture conversion at 8 weeks: 99/169 versus 98/167 3. Serious adverse events: 10/169 versus 8/167 |
|
| Notes | Location: North America and Africa Setting: not described HIV status: HIV‐positive participants (30/169 ‐ fluoroquinolones + HRZ group, 30/167 ‐ control HRZE group) Resistance: isoniazid resistance (15/169 ‐ fluoroquinolones + HRZ group, 10/167 ‐ control HRZE group); 11 participants with resistance to rifampicin, fluoroquinolone or pyrazinamide ‐ excluded from analysis Dates: no mention in the trial report Funding: the US CDC. Bayer Pharmaceuticals donated moxifloxacin and moxifloxacin placebo tablets. Two of 12 authors had a financial relationship with the commercial entity that had an interest in the subject of the manuscript. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised in a factorial design"; "Randomisation was stratified by continent of enrolment and presence of pulmonary cavitation". |
| Allocation concealment (selection bias) | Unclear risk | Method of concealment not described. |
| Blinding of participants/personnel (efficacy outcomes);performance bias All outcomes | Low risk | Not described in study report. However the trial was double‐dummy placebo controlled. Review authors judged that the efficacy outcomes were not likely to be influenced by lack of blinding. |
| Blinding of participants/personnel (safety outcomes);performance bias All outcomes | Low risk | Not described in study report. However the trial was double‐dummy placebo controlled. Review authors judged that the safety outcomes were not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (efficacy outcomes);detection bias All outcomes | Low risk | Not described in the study report. However the trial was double‐dummy placebo controlled and outcome assessment for bacteriological outcomes was independent and almost certainly blinded. |
| Blinding of outcome assessment (safety outcomes);detection bias | Low risk | Not described in the study report. However the trial was double‐dummy placebo controlled so safety outcomes were not likely to have been influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 59/336 (17.6%) excluded from final analysis. Quote: "we excluded (1) patients who took non study therapy or required more than 70 days to complete the intensive phase, (2) patients who died during the intensive phase of therapy, and (3) patients whose sputum cultures were overgrown with bacteria or yeast. Patients who received at least one dose of study drug were included in the safety analysis". |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement |
| Other bias | Unclear risk | Two of 12 authors had a financial conflict of interest. Bayer Pharmaceuticals donated moxifloxacin and moxifloxacin placebo tablets. |
Conde 2009.
| Methods | Trial design: single‐centre randomized double‐blind double‐dummy controlled trial Follow‐up: 18 months Adverse event monitoring: at weekly clinic visits. Liver enzymes, serum creatinine levels and complete blood counts performed monthly. ECG obtained at weeks 2, 4, 6, and 8 of treatment. Inclusion of all randomized participants in the final analysis: 45/170 (26.5%) excluded from final analysis |
|
| Participants | Number: 170 randomized; 125 evaluated Inclusion criteria: aged 18 years or older with sputum smear‐positive pulmonary TB and abnormal chest radiograph, and at least one acid‐fast bacilli in an expectorated sputum sample with no previous history of treatment. Exclusion criteria: haemoglobin < 70 g/L; AST or ALT > 3 times the upper limit of normal value; serum creatinine > twice the upper limit of normal; an ECG with a QTc interval more than 450 ms; pregnancy; breastfeeding; silico‐TB; a history of severe adverse reactions to fluoroquinolones or any other study agent; seropositivity for HIV with CD4‐cell count < 200 cells per μL. Randomized patients were excluded if culture‐negative or culture resistant to isoniazid, rifampicin, or ethambutol. |
|
| Interventions | Fluoroquinolone (moxifloxacin) substituted into regimen (replacing ethambutol) for 2 months (8 weeks) under direct supervision 1. Moxifloxacin (400 mg daily) with an ethambutol placebo orally plus basic regimen (5 days a week) for 2 months (8 weeks) 2. Ethambutol (15 to 20 mg/kg) plus moxifloxacin placebo daily orally 5 days a week for 2 months (8 weeks) plus basic regimen Basic regimen: isoniazid (300 mg), rifampicin (450 mg if < 50 kg; 600 mg if > 50 kg), and pyrazinamide (20 to 25mg/kg) given orally 5 days a week for 2 months |
|
| Outcomes | 1. Relapse (within a year after treatment completion) 1/85 versus 2/85 2. Death (from any cause) 3/85 versus 5/85 3. TB‐related death 0/85 versus 1/85 4. Sputum culture negative at 8 weeks: 59/85 versus 45/85. 5. Time to culture conversion: no numerical data 6. Serious adverse events: 6/85 versus 6/85 |
|
| Notes | Location: Brazil, Rio de Janeiro Setting: Hospital Clementino Fraga Filho HIV status: HIV negative participants, HIV‐seropositivity was used as an exclusion criterion Resistance: 11 participants with isoniazid, rifampicin, or ethambutol resistance excluded from authors' analysis Dates: October 2004 to March 2007 Funding: Office of Orphan Product Development, USA FDA, Fogarty International Center of the NIH, career development grant supported Dr. Chaisson's participation. Bayer Healthcare donated moxifloxacin and matching placebo. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "permuted block randomisation with blocks of four". |
| Allocation concealment (selection bias) | Low risk | Adequate: "allocation slips sealed in opaque envelopes opened after enrolment". |
| Blinding of participants/personnel (efficacy outcomes);performance bias All outcomes | Low risk | Double dummy placebo control was used. Patients, study clinicians, and study staff were unaware of the treatment assignments of patients with the exception of the pharmacist who dispensed medication packets. |
| Blinding of participants/personnel (safety outcomes);performance bias All outcomes | Low risk | Double dummy placebo control was used. Patients, study clinicians, and study staff were unaware of the treatment assignments of patients with the exception of the pharmacist who dispensed medication packets. |
| Blinding of outcome assessment (efficacy outcomes);detection bias All outcomes | Low risk | Double dummy placebo control was used. Though not specifically stated in the study report laboratory staff were likely blinded to treatment assignment. |
| Blinding of outcome assessment (safety outcomes);detection bias | Low risk | Double dummy placebo control was used. Patients, study clinicians and study staff were unaware of the treatment assignments of patients with the exception of the pharmacist who dispensed medication packets. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 45/170 (26.5%) excluded from final analysis. Quote: "Randomised patients were excluded if their baseline culture did not grow M. tuberculosis or grew a strain of M. tuberculosis that was resistant to isoniazid, rifampicin, or ethambutol". "The primary endpoint of the trial, culture conversion, was assessed by modified ITT analysis; patients whose baseline cultures were negative, contaminated, or contained drug‐resistant M. tuberculosis were excluded and all missing 8 week results were deemed treatment failures". |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement. |
| Other bias | Unclear risk | The authors declared no conflict of interest. Bayer Healthcare donated moxifloxacin and matching placebo, but had no input into the study design, execution, or analysis. Authors described the role of the funding source in the trial from design to publication of report. |
Dorman 2009.
| Methods | Trial design: multicentre randomized placebo‐controlled double‐blind phase 2 clinical trial Follow‐up: 2 months Adverse event monitoring: assessed at baseline and weeks 2, 4, 6, and 8 of treatment: symptoms, blood tests for AST, bilirubin, creatinine, and complete blood count. Inclusion of all randomized participants in the final analysis: 105/433 (24.3%) excluded from final analysis |
|
| Participants | Number: 433 randomized; 328 evaluated Inclusion criteria: adults (age not specified) with suspected pulmonary TB and acid‐fast bacilli in a sputum specimen Exclusion criteria: history of > 7 days of antituberculous treatment within the previous 6 months or of fluoroquinolone treatment within the previous 3 months; pregnancy, or breastfeeding; initial sputum cultures negative for M. tuberculosis or resistance to isoniazid, fluoroquinolones, rifampicin, or pyrazinamide. |
|
| Interventions | Fluoroquinolone (moxifloxacin) substituted into regimen (replacing isoniazid) for 2 months (8 weeks) under direct observation 1. Moxifloxacin (400 mg daily) with an isoniazid placebo orally plus basic regimen (5 days a week or 7 days per week during the first two weeks) for 2 months (8 weeks) 2. Isoniazid (300 mg) plus moxifloxacin placebo daily orally 5 days a week (or 7 days per week during the first two weeks) for 2 months (8 weeks) plus basic regimen Basic regimen: rifampicin, pyrazinamide, ethambutol, and pyridoxine in accordance with published guidelines (Blumberg 2003) |
|
| Outcomes | 1. Death from any cause: 3/219 versus 4/214 (intensive versus continuation phase) 2. TB‐related death: 2/219 versus 1/214 3. Sputum culture negative at 8 weeks: 99/219 versus 90/214 2. Time to sputum culture conversion (no numeric data provided) 3. Serious adverse events: 9/219 versus 8/214 |
|
| Notes | Location: North America, Brazil, South Africa, Spain, and Uganda Setting: 26 Tuberculosis Trials Consortium (TBTC) sites HIV status: HIV‐positive participants (17/219 in study group fluoroquinolones + RZE, 18/214 in control group HRZE) Resistance: 13/219 in fluoroquinolones + RZE group, 14/214 in HRZE group); full susceptibility not confirmed: 13/219 in fluoroquinolones + RZE group, 6/214 in HRZE group Dates: no mention in the trial report Funding: CDC and the Global Alliance for Tuberculosis Drug Development. Bayer Pharmaceuticals provided moxifloxacin and moxifloxacin placebo tablets. Three out of 19 authors had a financial relationship with a commercial entity that had an interest in the subject of the manuscript. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study report specified "randomly assigned" but did not mention the method of randomization. "Randomisation was stratified by the presence of cavitation on baseline chest radiograph and continent of enrolment (Africa or not Africa); randomisation was not restricted within strata". |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding of participants/personnel (efficacy outcomes);performance bias All outcomes | Low risk | Double dummy placebo control was used. Review authors judged that efficacy outcomes were unlikely to have been influenced by a lack of blinding. |
| Blinding of participants/personnel (safety outcomes);performance bias All outcomes | Low risk | Double dummy placebo control was used. Review authors judged that safety outcomes were unlikely to have been influenced by a lack of blinding. |
| Blinding of outcome assessment (efficacy outcomes);detection bias All outcomes | Low risk | Double dummy placebo control was used. Laboratory staff assessing bacteriological outcomes were likely blinded to the treatment assignment. |
| Blinding of outcome assessment (safety outcomes);detection bias | Low risk | Double dummy placebo control was used. Review authors judged that safety outcomes were unlikely to have been influenced by a lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 105/433 (24.3%) excluded from final analysis. Quote: "Two efficacy analysis groups were prespecified. A modified ITT group excluded participants whose enrolment specimen failed to grow M. tuberculosis or had proven resistance to isoniazid, rifampin, pyrazinamide, ciprofloxacin, or ofloxacin; and enrollees whose treatment was incorrectly allocated. A protocol‐correct (PC) group excluded participants whose enrolment specimen failed to grow M. tuberculosis or was not proven susceptible to isoniazid, rifampin, pyrazinamide, ciprofloxacin, and ofloxacin; whose treatment was incorrectly allocated; who had contaminated week‐8 cultures; who died during intensive phase; who required more than 700 days to complete the study intensive‐phase treatment; or who took non study therapy for more than 14 days during the intensive phase. For safety analyses, all participants who received at least one dose of study treatment were included". |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement. |
| Other bias | Unclear risk | Three out of 19 authors had a financial conflict of interest. Bayer Pharmaceuticals provided moxifloxacin and moxifloxacin placebo tablets. |
El‐Sadr 1998.
| Methods | Trial design: multicentre open label RCT Follow‐up: 12 months Adverse event monitoring: regularly with two week intervals during receipt of study medication and for 8 weeks after its discontinuation, graded on a five‐point scale Inclusion of all randomized participants in the final analysis: 73/174 (42%) excluded at 8 weeks analysis; 39% lost to follow‐up in continuation phase |
|
| Participants | Number: 174 randomized; 101 evaluated Inclusion criteria: suspected human immunodeficiency virus (HIV) and pulmonary TB; age > 18 years in resistant areas or > 13 years in other areas; aspartate aminotransferase (AST) ≤ 10 times upper limit; serum bilirubin < 2.5 times upper limit; serum creatinine ≤ 3 times upper limit or creatinine clearance rate ≥ 50 mL/min Exclusion criteria: history of MDR‐TB or close contact with an MDR‐TB patient; > 3 weeks continuous antituberculous treatment immediately prior to enrolment; > 12 weeks antituberculous therapy in the past 2 years; pregnancy; exclusively extrapulmonary TB. |
|
| Interventions | Fluoroquinolone (levofloxacin) added to regimen: 1. Levofloxacin plus standard regimen 500 mg levofloxacin daily for 2 weeks (induction phase); then 750 mg levofloxacin three times weekly for 6 weeks; then standard regimen only (continuation phase) 2. Standard regimen Induction phase (2 weeks daily): isoniazid (300 mg), vitamin B6 (50 mg), rifampicin (450 mg to 600 mg; < 50 kg to > 50 kg), pyrazinamide (1.5 g to 2.0 g; < 50 kg to > 50 kg), ethambutol (20 mg/kg; rounded to the nearest 400 mg) 6 weeks (thrice weekly): isoniazid (600 to 900 mg; < 50 kg to > 50 kg), vitamin B6 (50 mg), rifampicin (600 mg), pyrazinamide (2.0 g to 2.5 g; < 50 kg to > 50 kg), ethambutol (30 mg/kg; rounded to the nearest 400 mg) Continuation phase lasting 6 or 9 months (18 or 31 weeks of total therapy) (twice weekly): isoniazid (600 mg to 900 mg; < 50 kg to > 50 kg), vitamin B6 (50 mg), rifampicin (600 mg) Study report states that "the protocol strongly recommended directly observed therapy: all units had access to such programs" |
|
| Outcomes | 1. Death from any cause: 1/87 versus 3/87 ‐ best case analysis ; 1/50 versus 3/40 ‐ complete case analysis
2. TB‐related death: 1/87 versus 1/87
5. Sputum culture negative at 8 weeks: 46/87 versus 36/87 6. Serious adverse events: 11/87 versus 13/87 |
|
| Notes | Location: New York city area and Hawaii, USA Setting: 21 clinics across the USA HIV status: HIV positive and suspected HIV positive participants Resistance: > 80% M. tuberculosis strains susceptible to both isoniazid and rifampin. Dates: 1993‐95 to August 1997 Funding: National Institute for Allergy and Infectious Disease. Drugs were supplied by manufacturers. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Stratified permuted block randomisation was used in both phases of this study, with the research unit being the stratification factor". However, the specific method of randomization was not mentioned. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not described. |
| Blinding of participants/personnel (efficacy outcomes);performance bias All outcomes | Unclear risk | Study report gave no information on blinding but study was open‐labelled. |
| Blinding of participants/personnel (safety outcomes);performance bias All outcomes | Unclear risk | Study report gave no information on blinding but study was open‐labelled. |
| Blinding of outcome assessment (efficacy outcomes);detection bias All outcomes | Low risk | Study report gave no information on blinding but laboratory staff were likely blinded to treatment allocation and "all TB endpoints were reviewed by an independent clinical events committee blinded to treatment group". |
| Blinding of outcome assessment (safety outcomes);detection bias | Unclear risk | Study report gave no information on blinding but study was open‐labelled. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 73/174 (42%) excluded at 8 weeks analysis; 39% lost to follow‐up in continuation phase. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement. |
| Other bias | High risk | No conflict of interest statement. Drugs were supplied by manufacturers. |
Rustomjee 2008a.
| Methods | Trial design: four‐arm, open label RCT Follow‐up: 2 months Adverse event monitoring: not described Inclusion of all randomized participants in the final analysis: 18 participants (8.3%) excluded |
|
| Participants | Number: 217 randomized / 199 analyzed Inclusion criteria: newly diagnosed patients with pulmonary TB, with two consecutive sputum smears positive for acid‐fast bacilli, aged between 18 to 65 years, weighing between 38 to 80 kg, and willing to co‐operate in the intensive study were eligible. The medical findings for haematology, chemistry, liver enzymes, and cardiovascular function were not to exceed grade 2 of the Division of Microbiology and Infectious Disease. Exclusion criteria: resistance to RMP of the pre‐treatment strain of M. tuberculosis, extra‐pulmonary TB, pregnancy, WHO stage 4 of HIV infection, prolongation of the cardiological QT interval 480 msec, bradycardia, any condition causing delay in drug absorption, metabolism or elimination, or other serious illness. |
|
| Interventions | Fluroquinolone (ofloxacin, or moxifloxacin, or gatifloxacin) substituted into regimen (replacing ethambutol in standard first‐line regimen ‐ HRZE). Control: Akurit‐4 (Lupin, Mumbai, India) containing 275 mg E, 75 mg H, 150 mg R, and 400 mg Z ‐ HRZE regimen. Patients weighing between 38 to 50 kg received three tablets and those weighing between 50 to 80 kg received four tablets. Fluroquinolone: Ofloxacin 800 mg daily orally for 2 months Moxifloxacin 400 mg daily orally for 2 months Gatifloxacin 400 mg daily orally for 2 months plus AKurit‐Z (Lupin) containing the same first‐line drugs without ethambutol (E) ‐ HRZ. |
|
| Outcomes | 1. Death from any cause 2. TB‐related death 3. Sputum culture conversion at 8 weeks 4. Serious adverse events 5. Rate of late phase elimination of organisms as measured by serial sputum viable colony counting 6. Hazard ratio of culture conversion in Cox regression |
|
| Notes | Location: KwaZulu Natal, South Africa Setting: four clinics in KwaZulu Natal HIV status: 53% ‐ 63% HIV positive participants Resistance: all patients confirmed not resistant to rifampicin; 1 participant was isoniazid resistant Dates: June 2004 to June 2005 Funding: European Commission, Framework 5, WHO/UNDP/UNISEF Special programme on Tropical Disease Research (TDR) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. Quote: "Patients were randomly allocated in successive blocks of 20 equally to one of four regimens for the first 8 weeks of treatment". |
| Allocation concealment (selection bias) | Unclear risk | Not described. Quote: "Patients were randomly allocated in successive blocks of 20 equally to one of four regimens for the first 8 weeks of treatment". |
| Blinding of participants/personnel (efficacy outcomes);performance bias All outcomes | High risk | Not described. Study was open‐labelled. Review authors judged that study procedures could have been affected by a lack of blinding. |
| Blinding of participants/personnel (safety outcomes);performance bias All outcomes | High risk | Not described. Study was open‐labelled. Review authors judged that assessment of safety outcomes could have been affected by a lack of blinding. |
| Blinding of outcome assessment (efficacy outcomes);detection bias All outcomes | Unclear risk | Not described. Study was open‐labelled but laboratory staff were likely blinded to treatment assignment. Review authors judged that bacteriological efficacy outcomes were unlikely to have been at high risk of bias. |
| Blinding of outcome assessment (safety outcomes);detection bias | High risk | Not described. Study was open‐labelled. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 18 participants (8.3%) excluded; however the proportion of missing outcomes compared with observed event risk enough to induce clinically relevant bias in intervention effect estimate; quote: "Of these, eight were excluded from the SSCC analysis, two due to initial resistance to RMP but no other drug, two due to severe acquired immunodeficiency syndrome (AIDS) and super‐infections during treatment caused by pan‐resistant strains, one due to death on day 2, one had no sputum cultures after day 0 (included in the culture results), one withdrew voluntarily from the study and one had only two widely different bacillary counts." "For the analysis of sputum culture results at 8 weeks, a further five patients were excluded, one due to early death, one due to heavy ethanol consumption, two had treatment changed due to drug reactions and one voluntary withdrawal". |
| Selective reporting (reporting bias) | High risk | Authors did not present data on the most frequent adverse events by study group or on cause of death by study group, or time of death. Presentation of adverse events in the text and in the table was confusing. This obscured the data. Quote: "The most frequent adverse events were raised amylase (in 41% of patients due to HIV infection), raised transaminase (10%), arthralgia (9%), anaemia (7%), hypokalaemia (6%) and vomiting (5%). Serious adverse events occurred in 18 patients. Deaths were due to progression of AIDS in two patients, and to haemoptysis and to epileptic seizure in two other patients. Of the remaining 14 patients, two had elevated liver enzymes, three had arthralgia and the remaining nine developed one of the following events: renal failure, pancytopaenia, thrombocytopaenia, deep vein thrombosis, gastroenteritis, gastritis, Pneumocystis carinii infection, spontaneous pneumothorax or worsening pulmonary TB with AIDS. There were no serious glycaemic events in any arm. Minor grades of hypoglycaemia or hyperglycaemia were no more evident during therapy than at baseline, nor were they more common in the Gati arm. There was no evidence of prolongation of the QTc interval in electrocardiograms". |
| Other bias | High risk | No conflict of interest statement. |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abdullah 1997 | Reported as an abstract only, MDR‐TB, not in review. |
| Abdullah 1998 | Reported as an abstract only, MDR‐TB, not in review. |
| Agarwal 2007 | Reported as an abstract only. No randomization. |
| Andries 2005 | Experimental animal study, plus a small section in healthy human volunteers (tolerability); not a trial report. |
| Anonymous 1997 | No randomization or control group. |
| Barmina 2009 | Not relevant research question: roncoleukin versus basic regimen. |
| Bartacek 2009 | Not relevant research question: fixed‐dose combination versus single tablets treatment, fluoroquinolones not used. |
| Carroll 2012 | A case report, XDR‐NB, another drug ‐ linezolide, not in review. |
| Chambers 1998 | The outcome, early bactericidal activity, not in review. |
| Chang 2008 | Not relevant research question: hepatotoxicity of pyrazinamide. No randomization, cohort and case‐control analysis. |
| Chang 2009 | Not relevant research question: participants with community acquired pneumonia or exacerbation of bronchiectasis. |
| Chen 2003 | No randomization and the intervention was a combination of levofloxacin plus capreomycin. |
| Chigutsa 2012 | Not a RCT, MDR‐TB, not in review. |
| Chukanov 2006 | Mixed intervention of ciprofloxacin, ofloxacin, or levofloxacin plus kanamycin or amikacin added to the basic regimen in study group versus streptomycin added to the basic regimen in control group. |
| Diacon 2009 | Not relevant comparison/research question: study drug TMC207 (investigational diarylquinoline compound), ofloxacin used in the study and control groups. |
| Diacon 2012a | Not relevant study drug TMC207 ‐ bedaquiline, MDR‐TB, not in review. |
| Diacon 2012b | The outcome, bactericidal activity, not in review. |
| Estebanez 1992 | Exclusively urogenital TB. |
| Fouad 2011 | Review paper, not RCT. |
| Gosling 2003 | The outcome, early bactericidal activity, not in review. |
| Grishin 1998 | No randomization; cohort study. |
| Heemskerk 2011 | Protocol for TB meningitis. International Standard Randomized Controlled Trial Number ISRCTN61649292. |
| Ho 2007 | Reported as an abstract only, not a trial report. |
| Huang 2000 | Participants with MDR‐TB, not in review. |
| Jenkins 2008 | Not relevant comparison/research question: ciprofloxacin compared with clarithromycin in patients with pulmonary disease caused by M. avium‐intracellulare (MAC). |
| Ji 2001 | Participants: MDR‐TB, not in review. |
| Johnson 2006 | The outcome, early bactericidal activity, not in review. |
| Kang 2009 | Mixed intervention of 3RFT AM Ofx Pto PAS‐INH/5RFT Ofx Pto PAS‐INH in study group versus 3 H3R3Z3E3S3/5 H3R3E3 in control group, that is comparison groups differed not only by fluoroquinolone use. |
| Kawahara 1992 | No randomization. |
| Kennedy 1993 | Intervention: ciprofloxacin substituted for pyrazinamide and ethambutol, not in review. |
| Kennedy 1996 | Intervention: ciprofloxacin substituted for pyrazinamide and ethambutol, not in review. |
| Kohno 1992 | Intervention: ofloxacin substituted for ethambutol in drug‐sensitive disease, basic regimen without pyrazinamide, not in review. |
| Kumar 2004 | Study in healthy volunteers, not a trial report, in which the outcome was uric acid concentration in urine samples excreted over 0 to 8 hours. |
| Lee 2011 | A retrospective study, not a RCT. |
| Li 2008 | Mixed intervention of 3RFT AM Ofx Pto PAS‐INH/5RFT Ofx Pto PAS‐INH regimen, including rifapentine (RFT), amikacin (Am), ofloxacin (Ofx), protionamide (Pto), para‐aminosalicylic acid‐isoniazid (PAS‐INH) for three months and then RFT, Ofx, Pto, and PAS‐INH for five months in study group versus 3 H3R3Z3E3S3/5 H3R3E3, including isoniazid (H), rifampin (R), pyrazinamide (Z), ethambutol (E), and streptomycin (S) for three months and then H, R, and E for five months in control group, that is comparison groups differed not only by fluoroquinolone use. |
| Lu 2000 | Participants: presumed MDR‐TB, re‐treatment; basic regimen without rifampicin, not in review. |
| Marra 2005 | Retrospective safety study; not a trial report. |
| Merle 2012 | Discussion of OFLOTUB project, not a trial report. |
| Moadebi 2007 | Review paper, not a trial report. |
| Mohanty 1993 | Intervention: ciprofloxacin substituted for rifampicin, not in review. |
| Moulding 2008 | Correspondence paper, not a trial report. |
| Nakamura 2007 | Not relevant research question: comparison of five day short‐course therapies for secondary infection in patients with chronic respiratory disease using gatifloxacin and levofloxacin. |
| Nosova 2008 | Not relevant research question: study of fluoroquinolone resistance in TB patients; not a RCT. |
| O'Brien 1994 | Communication to the Editor of Chest; not a trial report. |
| Peloquin 2008 | Not relevant research question: population pharmacokinetics of three fluoroquinolones ‐ levofloxacin, gatifloxacin and moxifloxacin. |
| Pletz 2004 | The outcome, early bactericidal activity, not in review. |
| Ruslami 2013 | TB meningitis, not in review. |
| Rustomjee 2008b | The outcome, early bactericidal activity, not in review. |
| Saigal 2001 | Intervention: ofloxacin + pyrazinamide substituted for rifampicin, not in review. |
| Sirgel 1997 | The outcome, early bactericidal activity, not in review. |
| Sirgel 2000 | The outcome, early bactericidal activity, not in review. |
| Sokolova 1998 | No randomization; cohort study. |
| Sun 2000 | Participants: all proven MDR‐TB, not in review. |
| Suo 1996 | No randomization; not a controlled study. |
| Thwaites 2011 | Participants: TB meningitis, not in review. |
| TRC 2002 | No control arm, that is, a group treated without the studied fluoroquinolone (ofloxacin), a different fluoroquinolone, or different dose. |
| Valerio 2003 | No randomization and outcomes not reported. |
| Venter 2006 | The outcome, indices of adrenocortical function, not in review; none of the included outcomes reported, too small (20 participants). |
| Wang 2006 | Retrospective study; not a trial report. |
| Wolbers 2011 | TB meningitis, not in review. Current Controlled Trials ISRCTN61649292. |
| Yoon 2005 | Retrospective case‐control study; not a trial report. |
| Yoon 2012 | Not a RCT report, fluoroquinolone substitution for rifampicin, not in review. |
| Zhang 1997 | The efficacy of bronchofibrescope and catheter intervention with ofloxacin and amikacin studied in comparison with traditional chemotherapy. |
| Zhang 2006 | The efficacy of rifabutin versus rifapentine containing antituberculous regimens studied, both regimens included levofloxacin; study question not in review. |
| Zhao 2003 | No randomization. |
| Zheng 2004 | Mixed intervention of levofloxacin plus pasiniazide plus M. vaccae. |
| Zhu 2006 | The efficacy of rifabutin versus rifapentine containing antituberculous regimens studied, both regimens included levofloxacin; study question not in review. |
| Zhu 2012 | A review paper, not a RCT report. |
| Zvada 2012 | Not a RCT report. |
Characteristics of ongoing studies [ordered by study ID]
CTRI/2012/10/003060.
| Trial name or title | Thrice weekly 4‐ months moxifloxacin or gatifloxacin regimens for pulmonary TB |
| Methods | Randomized, parallel group, active controlled trial Method of generating randomization sequence: stratified block randomization. Method of allocation concealment: sequentially numbered, sealed, opaque envelopes. Blinding and masking: open label |
| Participants | Inclusion criteria: patients aged 18 years and above, residing in or around Chennai or Madurai will be eligible for enrolment to the study. They should not have had anti‐TB treatment in the past or should have had less than one month of treatment (but less than one week in the preceding one month before enrolment in the study).
They should have sputum culture‐positive pulmonary TB (at least two cultures should be positive). Patients will be enrolled to the study when two sputum smears are positive and will be retained for analysis only if two cultures are positive.
They should consent to attend the treatment centre for supervised treatment for 6 months and for home visits by the staff of the centre. They should give written informed consent. Exclusion criteria: body weight less than 30 kg; hepatic or renal disease as evidenced by clinical or biochemical abnormalities; diabetes mellitus; a history of seizures; psychiatric illness; an abnormal electrocardiogram or those on anti‐arrhythmic medication; those in a moribund state; those sero‐positive for HIV antibodies; pregnant or lactating women. |
| Interventions | Intervention 1: 2GHRZ thrice weekly/ 2GHR thrice weekly: gatifloxacin, isoniazid, rifampicin, and pyrazinamide thrice weekly for 2 months followed by gatifloxacin, isoniazid, and rifampicin thrice weekly for 2 months (total duration 4 months). Intervention 2: 2MHRZ thrice weekly/ 2MHR thrice weekly: moxifloxacin, isoniazid, rifampicin, and pyrazinamide thrice weekly for 2 months followed by moxifloxacin, isoniazid, and rifampicin thrice weekly for 2 months (total duration 4 months). Intervention 3: Regimen 1: 2MHRZ thrice weekly/2 MHR thrice‐weekly. Regimen 2: 2 GHRZ thrice weekly/ 2 GHR thrice weekly. Regimen 1: 2 MHRZ thrice weekly/ 2 MHR thrice weekly: moxifloxacin, isoniazid, rifampicin, and pyrazinamide thrice‐weekly for 2 months followed by moxifloxacin, isoniazid, and rifampicin thrice‐weekly for 2 months (duration 4 months). Regimen 2: 2 GHRZ thrice weekly/ 2 GHR thrice weekly: gatifloxacin, isoniazid, rifampicin, and pyrazinamide thrice‐weekly for 2 months followed by gatifloxacin, isoniazid, and rifampicin thrice‐weekly for 2 months (duration 4 months). Intervention 4: Regimen 2: 2 GHRZ thrice weekly/ 2 GHR thrice weekly: Regimen 2: gatifloxacin, isoniazid, rifampicin, and pyrazinamide thrice weekly for 2 months followed by gatifloxacin, isoniazid, and rifampicin thrice weekly for 2 months (total duration 4 months). Control Intervention 1: 2EHRZ thrice weekly/ 4HR thrice weekly: ethambutol, isoniazid, rifampicin, and pyrazinamide thrice weekly for 2 months followed by isoniazid and rifampicin thrice weekly for 4 months (total duration 6 months). Control Intervention 2: 2EHRZ thrice weekly/ 4HR thrice weekly: ethambutol, isoniazid, rifampicin, and pyrazinamide thrice weekly for 2 months followed by isoniazid and rifampicin thrice weekly for 4 months (total duration 6 months). Control Intervention 3: Regimen 3: 2EHRZ thrice weekly/ 4HR thrice weekly: ethambutol, isoniazid, rifampicin, and pyrazinamide thrice weekly for 2 months followed by isoniazid and rifampicin thrice weekly. |
| Outcomes | TB recurrence rate timepoint: up to 24 months post‐treatment; secondary outcomes: a) sputum culture conversion at 2 months; b) status at end of treatment; c) treatment related adverse reaction timepoint: up to 24 months post‐treatment. |
| Starting date | 14‐05‐2004 |
| Contact information | National Institute for Research in TB, Indian Council of Medical Research, No:1, Mayor Sathyamoorthy Road, Chetpet, Chennai‐31 600 031Chennai, TAMIL NADUIndia; shaheedjawahar@gmail.com; 04428369500 |
| Notes | Location: India Source of funding: ICMR, National Institute for Research in TB, Indian Council of Medical Research, No:1, Mayor Sathyamoorthy Road, Chetpet, Chennai‐31 |
ISRCTN44153044 RIFAQUIN.
| Trial name or title | An international multi centre controlled clinical trial to evaluate high dose RIFApentine and a QUINolone in the treatment of pulmonary TB ‐ RIFAQUIN |
| Methods | — |
| Participants | Inclusion criteria: newly diagnosed pulmonary TB; 2 sputum specimens positive for tubercle bacilli on direct smear microscopy; either no previous antituberculous chemotherapy, or < 2 weeks of previous chemotherapy; aged 18 years and over; firm home address that is readily accessible for visiting and be intending to remain there during the entire treatment and follow‐up period; willing to agree to participate in the study and to give a sample of blood for HIV testing. Exclusion criteria: any condition (except HIV infection) that may prove fatal during the study period; tuberculous meningitis; pre‐existing nontuberculous disease likely to prejudice the response to, or assessment of, treatment (e.g. insulin‐dependent diabetes, liver or kidney disease, blood disorders, peripheral neuritis); female and known to be pregnant or breastfeeding; suffering from a condition likely to lead to uncooperative behaviour such as psychiatric illness or alcoholism; contraindications to any medications in the study regimens; requires antiretroviral treatment (ART) at diagnosis; history of prolonged QTc syndrome or current or planned therapy with quinidine, procainamide, amiodarone, sotalol, disopyramide, ziprasidone, or terfenadine during the intensive phase of antituberculous therapy; haemoglobin < 7g/L; aspartate aminotransferase (AST) or alanine aminotransferase (ALT) > 5 times the upper range; creatinine clearance < 30 mL/min; history of seizures; HIV positive with a CD4 count < 200/mm3; weight < 35 kg. |
| Interventions | 1. 2 months of daily ethambutol (E), moxifloxacin (M), rifampicin (R), and pyrazinamide (Z) followed by 2 months of twice weekly moxifloxacin and rifapentine (2EMRZ/2P2M2). 2. 2 months of daily ethambutol, moxifloxacin, rifampicin, and pyrazinamide followed by 4 months of once weekly moxifloxacin and rifapentine (2EMRZ/4P1M1). 3. 2 months of daily ethambutol (E), isoniazid (H), rifampicin (R), and pyrazinamide (Z) followed by 4 months of daily isoniazid and rifampicin (2EHRZ/4HR). |
| Outcomes | 1. Combined rate of failure at the end of treatment and relapse, measured at 18 months. 2. Presence of rifamycin mono‐resistance (RMR) in relapse cultures of HIV‐infected patients, measured at 5, 6, 7, 8, 9, 10, 11, 12, 15, 18 months on the 4‐month arm and 7, 8, 9, 10, 11, 12, 15, 18 months on the 6‐month arm, plus at any unscheduled visit. 3. Occurrence of serious adverse events at any time during chemotherapy, recorded as they present themselves throughout the course of the trial. 4. Sputum culture results at 2 months after the initiation of chemotherapy, measured at all visits. 5. Rate of completion of chemotherapy according to the protocol, measured at all visits. 6. Number of observed doses of chemotherapy ingested, measured at all visits. 7. Any adverse events, recorded as they present themselves throughout the course of the trial. |
| Starting date | 31 July 2007 |
| Contact information | Dr Amina Jindani (ajindani@sgul.ac.uk), Centre for Infection Department of Cellular and Molecular Medicine St. George’s University of London, UK |
| Notes | Location: South Africa, Mozambique, Zimbabwe, Zambia Registration number: ISRCTN44153044 Source of funding: European and Developing Countries Clinical Trials Partnership (EDCTP) (The Netherlands) |
NCT00216385.
| Trial name or title | A controlled trial of a 4‐month quinolone‐containing regimen for the treatment of pulmonary TB OFLOTUB III |
| Methods | — |
| Participants | Inclusion criteria: male or female; aged 18 to 65 years; currently suffering from recently diagnosed microscopically proven pulmonary TB and providing informed consent for inclusion in the study. Exclusion criteria: history of TB treatment within the last 3 years; history of diabetes mellitus or non‐insulin dependent diabetes mellitus requiring treatment; concomitant infection requiring additional anti‐infective treatment (especially antiretroviral therapy); HIV‐ infected patients with WHO stage 3 infection ‐ except those presenting with only the "loss of weight > 10% body weight" criterion ‐ and all HIV infected patients at WHO stage 4. |
| Interventions | 1. 4‐month gatifloxacin‐containing antituberculous regimen 2. Standard antituberculous regimen |
| Outcomes | 1. Percentage of relapses by 24 months following treatment cure. 2. Percentage of adverse events. 3. Time to relapse. 4. Percentage of smear and culture conversion at 8 weeks. 5. Percentage of patient cured at the end 6. of treatment. 7. Time to a composite "unsatisfactory" endpoint. 8. Distribution of type and grading of adverse events. |
| Starting date | January 2005 |
| Contact information | Christian Lienhardt (Study Director), Institut de Recherche pour le Developpement, France |
| Notes | Location: Benin, Guinea, Kenya, Senegal, South Africa Registration number: NCT00216385 Sponsors and collaborators: Institut de Recherche pour le Developpement; WHO; European Commission |
NCT00396084.
| Trial name or title | Randomized, open label, multiple dose Phase I study of the early bactericidal activity of linezolid, gatifloxacin, levofloxacin, and moxifloxacin in HIV‐non‐infected adults with Initial episodes of sputum smear‐positive pulmonary TB (DMID 01‐553) |
| Methods | — |
| Participants | Inclusion criteria: adults, male or female, aged 18 to 65 years; women with child‐bearing potential (not surgically sterilized or postmenopausal for < 1 year) must be using or agree to use an adequate method of birth control (condom: intravaginal spermicide (foams, jellies, sponge) and diaphragm: cervical cap or intrauterine device) during study drug treatment; newly diagnosed sputum smear‐positive pulmonary TB as confirmed by sputum AFB smear and chest x‐ray findings consistent with pulmonary TB; willing and able to provide informed consent; reasonably normal haemoglobin (≥ 8 gm/dL), renal function (serum creatinine < 2 mg/dL), hepatic function (serum AST < 1.5 times the upper limit of normal for the testing laboratory and total bilirubin < 1.3 mg/dL), and random blood glucose < 150 mg/dL. Exclusion criteria: HIV infection; weight < 75% of ideal body weight; presence of significant haemoptysis; patients who cough up frank blood (more than blood streaked sputum); pregnant or breastfeeding women and those who are not practicing birth control; significant respiratory impairment (respiratory rate > 35/min); clinical suspicion of disseminated TB or TB meningitis; presence of serious underlying medical illness (e.g. such as liver failure, renal failure, diabetes mellitus, chronic alcoholism, decompensated heart failure, haematologic malignancy) or patients receiving myelosuppressive chemotherapy; patients receiving any of monoamine oxidase inhibitors (phenelzine, tranylcypromine), adrenergic/serotonergic agonists such as pseudoephedrine and phenylpropanolamine (frequently found in cold and cough remedies), tricyclic antidepressants (amitriptyline, nortriptyline, protriptyline, doxepin, amoxapine, etc), antipsychotics (e.g. chlorpromazine and buspirone), serotonin re‐uptake inhibitors (fluoxetine, paroxetine, sertraline, etc), bupropion, agents known to prolong the QTc interval [erythromycin, clarithromycin, astemizole, type Ia (quinidine, procainamide, disopyramide) and III (amiodarone, sotalol) anti‐arrhythmics, carbamazepine, insulin, sulphonylureas, and meperidine; presence of QTc prolongation (> 450 msec) on baseline EKG; allergy or contraindication to use of study drugs; treatment with antituberculous medications or other antibiotics with known activity against M. tuberculosis during the preceding 6 months; inability to provide informed consent; total white blood cell count < 3000/mm3; platelet count < 150,000/mm3; patients with suspected drug‐resistant TB (e.g. contact to source patient with drug‐resistant TB, patients who have relapsed after previous treatment for TB); patients likely, in the opinion of the local investigator, to be unable to comply with the requirements of the study protocol. |
| Interventions | Participants will be randomized to receive gatifloxacin, levofloxacin, moxifloxacin, or isoniazid (control), and after these arms are enrolled, they will be randomized to receive either linezolid (600 mg once daily) or linezolid (600 mg twice daily) or isoniazid (control). After the initial treatment, all participants will receive 6 months of standard antituberculous treatment outside of the hospital. |
| Outcomes | 1. Early bactericidal activity. 2. Extended early bactericidal activity. 3. Safety evaluations including clinical examination, complete blood counts, and serum total bilirubin, aspartate aminotransferase, and creatinine, and urinalysis will be followed to monitor for drug toxicity. |
| Starting date | February 2004 |
| Contact information | John Johnson (jlj@po.cwru.edu) |
| Notes | Location: University of Espírito Santo, Vitória, Brazil Registration number: NCT00396084 Sponsors: National Institute of Allergy and Infectious Diseases (NIAID) |
NCT00728507.
| Trial name or title | A Phase II randomized, open‐label trial of a rifapentine plus moxifloxacin‐based regimen for intensive phase treatment of smear‐positive pulmonary TB |
| Methods | Treatment randomized, open label, parallel assignment, safety/efficacy study |
| Participants | Inclusion criteria: presumptive diagnosis of sputum smear‐positive pulmonary TB; age: > 18 years; seven or fewer days of multidrug therapy for TB disease in the preceding 6 months; seven or fewer days of fluoroquinolone therapy in the preceding 3 months; documentation of HIV infection status; for HIV seropositive individuals, a CD4 T lymphocyte count of greater than or equal to 200 cells/mm3; documentation of study baseline laboratory parameters done at, or 14 days prior to screening; AST less than or equal to 2.5 times upper limit of normal; total bilirubin level less than 2.5 times upper limit of normal; creatinine level less than 2 times upper limit of normal; haemoglobin level of at least 8.0 g/dL; platelet count of at least 75,000 mm3; potassium level of at least 3.5; negative pregnancy test (women of childbearing potential); Karnofsky score of at least 60 (requires occasional assistance but is able to care for most of his/her needs); male or nonpregnant, non‐nursing female; provision of informed consent. Exclusion criteria: CD4 count < 200 cells/cu mm; presence of active AIDS‐related opportunistic infection (other than TB) or active AIDS‐related malignancy; known intolerance to any of the study drugs; concomitant disorders or conditions for which any of the study drugs is contraindicated. These include severe hepatic damage, acute liver disease of any cause, and acute uncontrolled gouty arthritis; inability to take oral medication; central nervous system TB; pulmonary silicosis; current or planned therapy, during study phase (intensive phase of TB treatment) with any one or more of the following drugs: quinidine, procainamide, amiodarone, sotalol, disopyramide, terfenadine, cisapride, erythromycin, clarithromycin, phenothiazines, haloperidol, olanzapine, ziprasidone, tricyclic antidepressants, chronic corticosteroids administered either orally or intravenously, chronic fluconazole, chronic itraconazole, chronic ketoconazole, oral or intravenous tacrolimus, oral or intravenous cyclosporine, HIV protease inhibitor, HIV non‐nucleoside reverse transcriptase inhibitor; concurrent severe and/or uncontrolled medical or psychiatric condition that, in the opinion of the investigator, could cause unacceptable safety risks or compromise compliance with the protocol; unable or unwilling to receive directly observed therapy and/or adhere with follow‐up (e.g. due to residence remote from the study site); refusal of consent. |
| Interventions | Drug: rifapentine, moxifloxacin, pyrazinamide, isoniazid Drug: isoniazid, rifampin, pyrazinamide, ethambutol |
| Outcomes | Primary: to compare, by treatment group, the proportions of patients with a negative sputum culture at the end of intensive phase therapy. Week 8: no.
To compare the safety and tolerability of the 2 intensive phase regimens. Weekly or more frequent: yes. Secondary: to compare the time to respiratory culture conversion of the 2 intensive phase regimens, using data from weekly cultures. Weekly: no. To compare, by treatment group, the proportions of subjects who experience treatment failure. Month 6: no. To compare, by HIV serostatus, a) the safety of the 2 intensive phase regimens, b) the proportions of patients with negative sputum cultures at the end of intensive phase therapy, and c) the time to culture conversion using data from weekly cultures. Weekly or more frequently: yes. To compare, in subjects with versus without cavitation on baseline chest x‐ray, the proportions of patients with negative sputum cultures at the end of intensive phase therapy. Week 8: no. To store serum for future assessment of hypersensitivity to study drugs, should it occur; to store plasma for future assessment of drug concentrations. Future: yes |
| Starting date | September 2009 |
| Contact information | Susan Dorman, MD Tel: 410‐955‐1755 dsusan1@jhmi.edu |
| Notes | Not yet recruiting. Location: Brazil, Hospital Universitario Clementio Fraga Filho Rio de Janeiro; Hospital Escola Sao Francisco de Assis, Rio de Janeiro |
NCT00864383 REMoxTB.
| Trial name or title | Controlled comparison of two moxifloxacin containing treatment shortening regimens in pulmonary TB CTRI/2011/05/001745 REMoxTB version 1.3 |
| Methods | A randomized placebo controlled double blind trial comparing two treatment shortening regimens with the standard regimen (two months ethambutol, isoniazid, rifampicin, and pyrazinamide followed by four months isoniazid and rifampicin) namely 1) two months moxifloxacin, isoniazid, rifampicin, and pyrazinamide followed by two months moxifloxacin, isoniazid, and rifampicin and 2) two months ethambutol, moxifloxacin, rifampicin, and pyrazinamide followed by two months moxifloxacin and rifampicin for the treatment of adults with pulmonary TB ‐ REMoxTB. Randomized, parallel group, placebo controlled trial. Method of generating randomization sequence: random number table. Method of allocation concealment: pre‐numbered or coded identical containers. Blinding and masking: participant, investigator, outcome assessor, and data‐entry operator blinded. |
| Participants | Target sample size: 1800 Inclusion criteria: signed written consent or witnessed oral consent in the case of illiteracy, before undertaking any trial related activity; two sputum specimens positive for tubercle bacilli on direct smear microscopy at the local laboratory and confirmed at the study laboratory on a sample taken at screening; aged 18 years or over; no previous anti‐TB chemotherapy; a firm home address that is readily accessible for visiting and willingness to inform the study team of any change of address during the treatment and follow‐up period; agreement to participate in the study and to give a sample of blood for HIV testing (see appendices 1 & 2); pre‐menopausal women must be using a barrier form of contraception or be surgically sterilized or have an IUCD in place; laboratory parameters performed up to 14 days before enrolment (Serum aspartate transaminase (AST) activity less than 3 times the upper limit of normal. Serum total bilirubin level less than 2.5 times upper limit of normal. Creatinine clearance (CrCl) level greater than 30 mLs/min. Haemoglobin level of at least 7.0 g/dL. Platelet count of at least 50x109cells/L. Serum potassium greater than 3.5 mmol/L); negative pregnancy test (women of childbearing potential). Exclusion criteria: unable to take oral medication; previously enrolled in this study; received any investigational drug in the past 3 months; received an antibiotic active against M. tuberculosis in the last 14 days (fluoroquinolones, macrolides, standard anti‐TB drugs); any condition that may prove fatal during the first two months of the study period; TB meningitis or other forms of severe TB with high risk of a poor outcome; pre‐existing non‐TB disease likely to prejudice the response to, or assessment of, treatment e.g. insulin‐dependent diabetes, liver or kidney disease, blood disorders, peripheral neuritis, chronic diarrhoeal disease; pregnant or breast feeding; suffering from a condition likely to lead to uncooperative behaviour e.g. psychiatric illness or alcoholism; contraindications to any medications in the study regimens; known to have congenital or sporadic syndromes of QTc prolongation or receiving concomitant medication reported to increase the QTc interval (e.g. amiodarone, sotalol, disopyramide, quinidine, procainamide, terfenadine); known allergy to any fluoroquinolone antibiotic or history of tendinopathy associated with quinolones; patients already receiving antiretroviral therapy; patients whose initial isolate is shown to be MDR‐TB; weight less than 35 kg; HIV infection with CD4 count less than 250 cells/µL; end stage liver failure (class Child‐Pugh C). |
| Interventions | Intervention 1: moxifloxacin in combination with ethambutol, pyrazinamide, and rifampicin: moxifloxacin, ethambutol, rifampicin, pyrazinamide for 2 months and moxifloxacin, rifampicin, and isoniazid placebo for 2 months and then isoniazid and rifampicin placebo for 2 months. Dose depend upon weight of the patient. Intervention 2: moxifloxacin in combination with isoniazid, pyrazinamide, and rifampicin: moxifloxacin, isoniazid, rifampicin, pyrazinamide for 2 months and moxifloxacin, isoniazid, and rifampicin for 2 months. Isoniazid and rifampicin placebo for 2 months. Dose depend upon weight of the patient. Control intervention 1: standard anti‐TB treatment: rifampicin, isoniazid, ethambutol, pyrazinamide for 2 months. Rifampicin and isoniazid and placebo for 4 months. Dose depend upon the weight of the patient. |
| Outcomes | Primary: 1. Efficacy: combined failure of bacteriological cure and relapse within one year of completion of therapy as defined by culture using solid media. 2. Safety: both comparisons: proportion of patients with grade 3 or 4 adverse events.Timepoint: 1.5 years Secondary: Efficacy: 1. Combined failure of bacteriological cure and relapse within one year of completion of therapy as defined by culture using liquid media. The following endpoints will be measured separately using both solid and liquid media. 2. Sensitivity analyses assuming all losses to follow‐up and non‐tuberculous deaths have an unfavourable outcome. 3. Sensitivity analyses assuming all losses to follow‐up and non‐tuberculous deaths have a favourable outcome. 4. Proportion of patients who are culture negative at eight weeks. 5. Time to first culture negative sputum sample. 6. Speed of decline of sputum viable count. Timepoint: 1.5 years. |
| Starting date | January 2008 |
| Contact information | Stephen H Gillespie
Tel: +44 (0) 20 7794 0500 ext.: 33539
s.gillespie@medsch.ucl.ac.uk Kapil Arora 579, Devli East Sainik Farms 110062New Delhi, DELHIIndia 011‐24502551 Kapil. Arora@apothecaries.net |
| Notes | Recruiting locations: Kenya, Centre for Respiratory Disease Research at KEMRI Nairobi; South Africa, Unit for Clinical & Biomedical TB Research, MRC Durban, South Africa, Tiervlei Trial Center and University of Stellenbosch, Cape Town, South Africa, Centre for TB Research and Innovation, UCT Lung Institute, Cape Town; Tanzania, Kilimanjaro Christian Medical Centre, Moshi; Tanzania, NIMR Mbeya Medical Research Programme, Mbeya; Zambia, University Teaching Hospital, Lusaka; China, India, Mexico, South Africa, Thailand, Global Alliance for TB Drug Development, New York, USA Primary sponsor: Apothecaries Private Limited http://www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=2686 |
NCT01498419.
| Trial name or title | Evaluation of 8 weeks of treatment with the combination of moxifloxacin, PA‐824, and pyrazinamide in patients with drug sensitive and multi drug‐resistant pulmonary TB NC‐002‐(M‐Pa‐Z) |
| Methods | A Phase II open‐label partially randomized trial to evaluate the efficacy, safety and tolerability of the combination of moxifloxacin plus PA‐824 plus pyrazinamide after 8 weeks of treatment in adult patients with newly diagnosed drug‐sensitive or multi drug‐resistant, smear‐positive pulmonary TB. |
| Participants | Target sample size: 230; Age: minimum: 18 years; Age maximum: 65 years;
Gender: both Inclusion criteria: provide written, informed consent prior to all trial‐related procedures including HIV testing (if an HIV test was performed within 1 month prior to trial start, it should not be repeated as long as documentation can be provided (ELISA, or Western blot, or both); body weight (in light clothing and with no shoes) between 40 kg and 90 kg, inclusive; sputum smear‐positive pulmonary TB (at trial appointed laboratory). For drug sensitive TB treatment arms, subjects should be newly diagnosed and previously untreated. Exception: participants can be included in the trial if they were diagnosed and treated for TB greater than 5 years prior to screening and can provide documentation of cure for that episode. Additionally, participants who have previously received H prophylactically can be included as long as that treatment is/was discontinued at least 7 days prior to randomization into this trial. Drug‐sensitive status to be confirmed with fluoroquinolone, rifampicin, and isoniazid susceptibility testing at screening using Hain Plus rapid sputum test. For the MDR‐TB treatment arm only: subjects with smear‐positive MDR infection, defined as confirmed resistance to at least both R and H confirmed at screening for entry into this trial. Resistance to R and H will be determined using the rapid screen test (Hain Plus). If the first spot sputum shows an indeterminate result, the test must be repeated on freshly collected spot sputum or overnight sputum and that result may be used. Subjects with newly diagnosed MDR‐TB are defined as a) subjects with MDR‐TB who have never been treated for TB before, or b) subjects with MDR‐TB who have previously been treated with only one course of first‐line TB drugs (H, R, E, Z and/or S) and that treatment is/was discontinued at least 7 days prior to randomization into this trial. Additionally, MDR‐TB participants who have previously received H prophylactically can be included as long as that treatment is/was discontinued at least 7 days prior to randomization into this trial; a chest X‐ray picture which in the opinion of the Investigator is compatible with TB; sputum positive (at site laboratory) on direct microscopy for acid‐fast bacilli (at least 1+ on the IUATLD/WHO scale); ability to produce an adequate volume of sputum as estimated from a spot assessment (estimated 10 ml or more overnight production); females may participate if they are: 1) of non‐childbearing potential (have had a bilateral oophorectomy, tubal ligation or hysterectomy or both, or have been postmenopausal for at least 12 consecutive months), 2) if they are using effective birth control methods and are willing to continue practicing birth control methods throughout treatment, or 3) be non‐heterosexually active, practice sexual abstinence or have a vasectomized partner (confirmed sterile). Therefore to be eligible for this study women of childbearing potential should either: 1) use a double barrier method to prevent pregnancy (i.e. use a condom with either diaphragm or cervical cap) or 2) use hormonal‐based contraceptives in combination with a barrier contraceptive, or 3) use an intrauterine device in combination with a barrier contraceptive. They must also be willing to continue these contraceptive measures until one week after the last dose of study medication or one week after discontinuation from study medication in case of premature discontinuation. (Note: hormone‐based contraception alone may not be reliable when taking IMP; therefore, hormone‐based contraceptives alone cannot be used by female participants to prevent pregnancy); male participants who are having heterosexual intercourse with females of child‐bearing potential are required to use one of the following birth control methods during their participation in the trial and for 12 weeks after their last dose of study medication to prevent pregnancy: a double barrier method which can include a male condom, diaphragm, cervical cap, or female condom; or a barrier method combined with hormone‐based contraceptives or an intra‐uterine device for the female partner. The use of the above mentioned birth control method does not apply if the male participant has been vasectomized or has had a bilateral orchidectomy minimally three months prior to screening, or is not heterosexually active, or practices sexual abstinence, or if the female sexual partner has had a bilateral oophorectomy, tubal ligation or hysterectomy or both, or has been postmenopausal for at least 12 consecutive months. Exclusion criteria: evidence of clinically significant (as judged by the investigator), metabolic, gastrointestinal, cardiovascular, musculoskeletal, ophthalmological, pulmonary, neurological, psychiatric or endocrine diseases, malignancy, or other abnormalities (other than the indication being studied) including myasthenia gravis and malaria; end stage liver failure (class Child Pugh C); poor general condition where any delay in treatment cannot be tolerated per discretion of the Investigator; clinically significant evidence of extrathoracic TB (e.g. miliary TB, abdominal TB, urogenital TB, osteoarthritic TB, TB meningitis), as judged by the Investigator; history of allergy or hypersensitivity to any of the study IMP or related substances, including a known allergy to any fluoroquinolone antibiotic, history of tendinopathy associated with quinolones or suspected hypersensitivity to any rifamycin antibiotics; resistance to fluoroquinolones (Hain plus rapid test), or pyrazinamide, or both; participants may be included in the study prior to receipt of the susceptibility test results for fluoroquinolones or pyrazinamide, however once these are received after a participant has entered into the study and if the results show the participant is resistant to fluoroquinolones, or pyrazinamide, or both, such a participant should be removed from the trial. DS participants will not be replaced, but MDR‐TB participants taking part in the EBA sub‐study could be replaced after consultation and written approval with the sponsor; known (positive urine drug screen) or suspected, current or history of within the past 2 years, alcohol or drug abuse, that is, in the opinion of the Investigator sufficient to compromise the safety or cooperation of the participant; For HIV infected participants: having a CD4+ count < 200 cells/µL, or having received intravenous antifungal medication within the last 90 days, or with an AIDS‐defining opportunistic infection or malignancies (except pulmonary TB), or having participated in other trials. |
| Interventions | Drug: M (400 mg) Pa (100 mg) Z (1500 mg) Drug: M (400 mg) Pa (200 mg) Z (1500 mg) Drug: rifafour |
| Outcomes | Primary: the rate of change in colony forming units (CFUs) using non‐linear mixed effects modelling of the Serial Sputum Colony Counts (SSCC) over 8 weeks of treatment (time frame: over 8 weeks of treatment). Secondary: proportion of participants with adverse events and proportion of participants who discontinue due to an adverse event in each experimental arm (time frame: over 8 weeks). Proportion of patients with sputum culture conversion at 8 weeks (time frame: 8 weeks). The rate of change in time to sputum culture positivity (TTP) through 8 weeks in the MGIT system in sputum over 8 weeks in participants which may be described with linear, bi‐linear, or non‐linear regression of TTP on time (time frame: over 8 weeks). Time to sputum conversion using data from weekly cultures through 8 weeks (separately, on solid and liquid media) (time frame: over 8 weeks). |
| Starting date | February 2012 |
| Contact information | Almari Conradie: Tel 27 (12) 844‐0951; almari.conradie@tballiance.org Rodney Dawson, MD University of Cape Town http://clinicaltrials.gov/show/NCT01498419 |
| Notes | Not yet recruiting; location: Brazil, South Africa, Tanzania Primary sponsor: Global Alliance for TB Drug Development |
NCT01589497.
| Trial name or title | Early bactericidal activity (EBA) study of TB regimens with and without INH and moxifloxacin |
| Methods | Allocation: randomized; endpoint classification: efficacy study; intervention model: parallel assignment; masking: open label; primary purpose: treatment |
| Participants | Age minimum: 18 years; age maximum: N/A; gender: both Inclusion criteria: absence of HIV‐1 infection within 30 days prior to study entry or HIV‐1 infection; confirmed sputum positive for acid fast bacilli (AFB) by smear‐microscopy =1+ within 1 day prior to study entry; body weight: 40 kg to 90 kg, inclusive; age = 18 years at study entry; certain laboratory values, as defined in section 4.1.5 in the protocol, obtained within 30 days prior to entry. For HIV‐positive candidates only: CD4+ cell count of > 100 cells/mm3, obtained within 7 days prior to study entry at a DAIDS approved laboratory. For females of reproductive potential, negative serum or urine pregnancy test within 7 days prior to entry. Female participants who are participating in sexual activity that could lead to pregnancy must agree to use one reliable non‐hormonal form of contraceptive (condoms, with a spermicidal agent; a diaphragm, or cervical cap with spermicide; or an IUD) while receiving study medications. If a chest x‐ray has not been performed within 14 days prior to entry, or the results of such an x‐ray are not available, then a chest x‐ray must be performed as part of screening. Ability and willingness of subject or legal guardian/representative to provide informed consent. Willingness to be hospitalized for approximately 16 days. Exclusion criteria: receipt of INH prophylaxis or TB therapy for more than 7 cumulative days in the last 6 months, or of any fluoroquinolone in the 1 month prior to entry; currently or within 30 days on antiretroviral treatment (ART) or expected to initiate ART within 2 weeks after study entry; breastfeeding; known intolerance to any of the study drugs; resistance to rifampicin determined by GeneXpert within 7 days prior to study entry; known history of resistance to isoniazid or rifampin or known close exposure (i.e., household exposure) to someone with MDR TB or known study candidate default on previous TB treatment (ie, the study candidate was diagnosed with TB, started TB treatment but did not complete that treatment); known allergy to any fluoroquinolone antibiotic; history of prolonged QT syndrome or a QTc of > 450 ms; current or planned therapy with quinidine, procainamide, amiodarone, sotalol, or ziprasidone during the first 2 months of TB treatment; current or prior diagnosis of pulmonary silicosis; advanced disease as defined by Karnofsky score = 70 at entry; any of the following current comorbidities, complications, or underlying medical conditions: ‐ poorly controlled diabetes (definition: patients with random plasma glucose > 180 mg/dL within 2 days prior to study entry) ‐ uncontrolled hypertension (definition: requiring acute medical treatment or immediate hospitalization) ‐ miliary TB ‐ neurological TB (including TB of the spine, TB meningitis) ‐ peripheral neuropathy = Grade 2 according to the December 2004 (Clarification, August 2009) Division of AIDS (DAIDS) Toxicity table, within 90 days prior to study entry ‐ Active drug or alcohol use or dependence that, in the opinion of the site investigator, would interfere with adherence to study requirements. ‐ Estimated overnight sputum production of < 10 mL. ‐ Requirement for concomitant medications that may potentially interact with study drugs. |
| Interventions | Drug: ethambutol Drug: isoniazid Drug: moxifloxacin Drug: pyrazinamide Drug: rifampicin |
| Outcomes | Daily decrease in log10 CFU/ml sputum between day 2 and 14 since study treatment initiation (time frame: 12 days) |
| Starting date | January 2013 |
| Contact information | William Bishai, MD, PhD, KwaZulu‐Natal Research Institute for TB and HIV (K‐RITH); Susan Swindells, MBBS, University of Colorado Hospital CRS |
| Notes | Location: South Africa Primary sponsor: AIDS Clinical Trials Group SEcondary sponsor: National Institute of Allergy and Infectious Diseases (NIAID) |
NCT01785186.
| Trial name or title | Evaluation of SQ109, high‐dose rifampicin, and moxifloxacin in adults with smear‐positive pulmonary TB in a MAMS design (PanACEA‐MAMS‐TB‐01) |
| Methods | Allocation: randomized; endpoint classification: safety/efficacy study; intervention model: single group Assignment; masking: open label; primary purpose: treatment |
| Participants | Age minimum: 18 years; age maximum: N/A; gender: both Inclusion criteria 1. The patient has given free, signed written or witnessed oral informed consent for study participation prior to all trial‐related procedures, including HIV testing if HIV serostatus is not known or the last documented negative is more than four weeks ago. 2. The patient has a diagnosis of pulmonary TB from a health clinic established by sputum smear and/or GeneXpert MTB/RIF® and/or chest X‐ray. 3. An adequate sputum bacterial load is confirmed by a Ziehl‐Neelsen stained smear in the study laboratory, done from concentrated sputum found at least 1+ on the IUATLD/WHO scale. 4. The patient has a valid rapid test result (GeneXpert MTB/RIF®) from the sputum positive for M. tuberculosis complex, and indicating susceptibility to Rifampicin. This test must be done in the study laboratory. 5. The patient is aged at least 18 years at the day of informed consent. 6. The patient has a body weight in light clothing and without shoes of at least 35 kg, but not more than 90 kg. 7. Female patients of childbearing potential must have a negative serum pregnancy test, and consent to practise an effective method of birth control until week 26. Effective birth control for female patients has to include two methods, including methods that the patient's sexual partner(s) use. At least one must be a barrier method. Female patients are considered not to be of childbearing potential if they are post‐menopausal with no menses for the last 12 months, or surgically sterile (this condition is fulfilled by bilateral oophorectomy, hysterectomy, and by tubal ligation which is done at least 12 months prior to enrolment). 8. Male patients must consent to use an effective contraceptive method, if their sexual partner(s) is/are of childbearing potential, and if they are not surgically sterile (see 6). Contraception by male participants must be practised until at least week 24 to cover the period of spermatogenesis. Contraceptive methods used by male participants may include hormonal methods used by the partner(s). 9. The patient has a firm home address that is readily accessible for visiting and willingness to inform the study team of any change of address during trial participation, or will be compliant to study schedule, in the discretion of the investigator. Exclusion criteria 1. Circumstances that raise doubt about free, uncoerced consent to study participation (e.g. in a prisoner or mentally handicapped person) 2. Poor general condition where delay in treatment cannot be tolerated or death within three months is likely. 3. The patient is pregnant or breast‐feeding. 4. The patient has an HIV infection and is receiving ART, or is likely to require ART during the twelve weeks of experimental study treatment as per local guidelines, or both. 5. The patient has a known intolerance to any of the study drugs, or concomitant disorders or conditions for which SQ109, rifampicin, moxifloxacin, or standard TB treatment are contraindicated. 6. The patient has an history or evidence of clinically relevant metabolic, gastrointestinal, neurological, psychiatric, or endocrine diseases, malignancy, or any other condition that will influence treatment response, study adherence or survival in the judgement of the investigator, especially: clinically significant evidence of severe TB (e.g. miliary TB, TB meningitis. Limited lymph node involvement will not lead to exclusion); serious lung conditions other than TB or severe respiratory impairment in the discretion of the investigator; neuropathy, epilepsy, or significant psychiatric disorder; uncontrolled and/or insulin‐dependent diabetes; cardiovascular disease such as myocardial infarction, heart failure, coronary heart disease, uncontrolled hypertension (systolic blood pressure =160 mmHg, or diastolic blood pressure of =100 mmHg on two occasions, or both), arrhythmia, or tachyarrhythmia; long QT syndrome (see criterion 9), or family history of long QT syndrome or sudden death of unknown or cardiac‐related cause; Plasmodium spp. parasitaemia as indicated by thick blood smear or a positive rapid test present at screening; alcohol or other drug abuse that is sufficient to significantly compromise the safety or cooperation of the patient, includes substances prohibited by the protocol, or has led to significant organ damage at the discretion of the investigator. 7. History of previous TB within the last five years. 8. Laboratory: at screening one or more of the following abnormalities were observed for the patient in screening laboratory: serum amino aspartate transferase (AST), or serum alanine aminotransferase (ALT) activity >3x the upper limit of normal, or both; Serum total bilirubin level > 2.5 times the upper limit of normal; creatinine clearance (CrCl) level lower than 30 mLs/min; complete blood count with haemoglobin level < 7.0 g/dL; platelet count < 50,000/mm3; Serum potassium below the lower level of normal. 9. ECG findings in the screening ECG: QTcB, or QTcF, or both, of > 0.450 s; atrioventricular (AV) block with PR interval > 0.20 s; prolongation of the QRS complex over 120 milliseconds; other changes in the ECG that are clinically relevant as per discretion of the investigator. 10. The patient has had treatment with any other investigational drug within 1 month prior to enrolment, or enrolment into other clinical (intervention) trials is planned during week 1 to 26. 11. Previous anti‐TB treatment: the patient has had previous treatment with drugs active against M. tuberculosis within the last 3 months, including but not limited to INH, EMB, RIF, PZA, amikacin, cycloserine, rifabutin, streptomycin, kanamycin, para‐aminosalicylic acid, rifapentine, thioacetazone, capreomycin, fluoroquinolones, thioamides. 12. QT prolonging medications: administration within 30 days prior to study start, anticipated administration during the study period, or during the 12 weeks of experimental treatment, of any QT‐prolonging agents such as, but not limited to, azithromycin, bepridil chloroquine, chlorpromazine, cisapride, cisapride, clarithromycin, disopyramide dofetilide, domperidone, droperidol, erythromycin, halofantrine, haloperidol, ibutilide, levomethadyl, lumefantrine, mefloquine, mesoridazine, methadone, moxifloxacin, pentamidine, pimozide, procainamide, quinidine, quinine, roxithromycin, sotalol, sparfloxacin, terfenadine, thioridazine. Exceptions may be made for participants who have received 3 days or less of one of these drugs or substances, if there has been a wash‐out period. |
| Interventions | Dietary supplement: pyridoxine Drug: ethambutol Drug: isoniazid Drug: moxifloxacin Drug: pyrazinamide Drug: rifampicin Drug: SQ109 |
| Outcomes | Primary outcome: sputum culture conversion (2 negative cultures) using liquid media (time frame: 0 to 12 weeks) Secondary outcomes: changes in baseline laboratory safety parameters during treatment and follow‐up (time frame: 0 to 12 weeks) Frequency of adverse events (time frame: 0 to 12 weeks) Mycobacteriology identification and characterization by PCR and MIC (time frame: 0 to 12 weeks) Occurence of treatment failure (relapse or emergence of drug‐resistance) (time frame: 0 to 12 weeks) Pharmacodynamics including AUC0‐24/MIC (h*ng/mL) and Cmax/MIC (ng/mL) (time frame: 0 to 12 weeks) Pharmacokinetics including AUC, Cl, t1/2, Vd, and protein binding (time frame: 0 to 12 weeks) Proportion of negative sputum cultures (time frame: 0 to 12 weeks) Rate of change in quantitative PCR during therapy (time frame: 0 to 12 weeks) Rate of change in time to positivity (time frame: 0 to 12 weeks) Time to first negative culture on liquid and solid media (time frame: 0 to 12 weeks) |
| Starting date | February 2013 |
| Contact information | Michael Hoelscher; Klinikum of the University of Munich; 0049 89 2180; hoelscher@lrz.uni‐muenchen.de |
| Notes | Location: South Africa, Tanzania Secondary sponsor: European and Developing Countries Clinical Trials Partnership (EDCTP), German Federal Ministry of Education and Research, Medical Research Council, Radboud University, Sequella, Inc. |
Differences between protocol and review
2005, Issue 3 (first review version): We did not search SIGLE because we searched for conference proceedings using alternative sources. We added "sputum smear positive" to the definition of the relapse outcome, and added "total number of adverse events" to the list of outcome measures.
2008, Issue 1 (second review version): We did not incorporate changes to the structure of the previously published version of the review.
2013, Issue 1 (third review version): We focused on drug‐sensitive TB and excluded trials and comparisons for MDR‐TB. We restructured intervention comparisons to better address immediate clinical questions. We renumbered outcomes and abandoned subdivision into primary and secondary ones. We reworded "total number of adverse events" for "total number of people with adverse events". We removed methods for continuous data. Albina Titarenko and GDAV joined the team as authors and Stephen B. Squire stepped down as co‐author.
Contributions of authors
LEZ was the author of the original review and was responsible for this update. All authors were involved in the conception of this review update. Data extraction and assessment of risk of bias was performed by LEZ and GDAV. Albina F.Titarenko contributed with the updated search and analysis of the Russian language literature. LEZ conducted the data input and analysis with input from GDAV. LEZ drafted the text with input from other authors.
Sources of support
Internal sources
-
Kazan Federal University, Russian Federation.
Department of Basic and Clinical Pharmacology
Liverpool School of Tropical Medicine, UK.
University of Liverpool, UK.
External sources
Department for International Development (DFID), UK.
Wellcome Trust, UK.
Declarations of interest
GDAV is a co‐author on the trial report of Rustomjee 2008a.
Unchanged
References
References to studies included in this review
Burman 2006 {published data only}
- Burman WJ, Goldberg S, Johnson JL, Muzanye G, Engle M, Mosher AW, et al. Moxifloxacin versus ethambutol in the first two months of treatment for pulmonary tuberculosis. American Journal of Respiratory and Critical Care Medicine 2006;174(3):331‐8. [DOI] [PubMed] [Google Scholar]
Conde 2009 {published data only}
- Conde MB, Efron A, Loredo C, Souza GR, Graca NP, Cezar MC, et al. Moxifloxacin versus ethambutol in the initial treatment of tuberculosis: a double‐blind, randomised, controlled phase II trial. Lancet 2009;373(9670):1183‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dorman 2009 {published data only}
- Dorman SE, Johnson JL, Goldberg S, Muzanye G, Padayatchi N, Bozeman L, et al. Substitution of moxifloxacin for isoniazid during intensive phase treatment of pulmonary tuberculosis. American Journal of Respiratory and Critical Care Medicine 2009;180(3):273‐80. [DOI] [PubMed] [Google Scholar]
El‐Sadr 1998 {published data only}
- El‐Sadr WM, Perlman DC, Matts JP, Nelson ET, Cohn DL, Salomon N, et al. Evaluation of an intensive intermittent‐induction regimen and duration of short‐course treatment for human immunodeficiency virus‐related pulmonary tuberculosis. Terry Beirn Community Programs for Clinical Research on AIDS (CPCRA) and the AIDS Clinical Trials Group (ACTG). Clinical Infectious Diseases 1998;26(5):1148‐58. [DOI] [PubMed] [Google Scholar]
Rustomjee 2008a {published data only}
- Rustomjee R, Lienhardt C, Kanyok T, Davies GR, Levin J, Mthiyane T, et al. A Phase II study of the sterilising activities of ofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. International Journal of Tuberculosis and Lung Disease 2008;12(2):128‐38. [PubMed] [Google Scholar]
References to studies excluded from this review
Abdullah 1997 {published data only}
- Abdullah A, Abu‐Hussein AH, Al‐Akshar M, Abdel R, Ads CH, Taieb S. Ofloxacin in the treatment of primary drug resistant pulmonary tuberculosis (PDR‐TB). European Respiratory Journal 1997;10(Suppl):25214. [Google Scholar]
Abdullah 1998 {published data only}
- Abdullah A, Abu‐Hussein AH, Al‐Akshar M, Ads CH, El‐Taieb S. Ofloxacin dosage in the treatment of multidrug resistant pulmonary tuberculosis (MDR‐TB). European Respiratory Journal 1998;12(Suppl):28367. [Google Scholar]
Agarwal 2007 {published data only}
- Agarwal SK. Treatment of resistant tuberculosis in Indian patients of Eastern Uttar Pradesh having isolates resistant to isoniazid, rifampicin and quinolones. Respirology 2007;12(4):A114. [Google Scholar]
Andries 2005 {published data only}
- Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs LM, Winkler H, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005;307(5707):223‐7. [DOI] [PubMed] [Google Scholar]
Anonymous 1997 {published data only}
- Anonymous. A controlled study of rifabutin and an uncontrolled study of ofloxacin in the retreatment of patients with pulmonary tuberculosis resistant to isoniazid, streptomycin and rifampicin. Hong Kong Chest Service/British Medical Research Council. Tubercle and Lung Disease 1997;73(1):59‐67. [PubMed] [Google Scholar]
Barmina 2009 {published data only}
- Barmina NA, Burukhina LV, Shurygin AA, Archakova LI. Roncoleukin in enhancing the efficiency of complex therapy for infiltrative pulmonary tuberculosis in adolescents. Problemy Tuberkuleza i Boleznei Legkikh 2009;5:27‐31. [PubMed] [Google Scholar]
Bartacek 2009 {published data only}
- Bartacek A, Schutt D, Panosch B, Borek M, Rimstar 4‐FDC Study Group. Comparison of a four‐drug fixed‐dose combination regimen with a single tablet regimen in smear‐positive pulmonary tuberculosis. International Journal of Tuberculosis and Lung Disease 2009;13(6):760‐6. [PubMed] [Google Scholar]
Carroll 2012 {published and unpublished data}
- Carroll MW, Choi H, Min S, Hwang S, Park H, Song T, et al. Rhabdomyolysis in a patient treated with linezolid for extensively drug‐resistant (XDR) tuberculosis. Clinical Infectious Diseases 2012;54(11):1624‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chambers 1998 {published data only}
- Chambers HF, Kocagoz T, Sipit T, Turner J, Hopewell PC. Activity of amoxicillin/clavulanate in patients with tuberculosis. Clinical Infectious Diseases 1998;26(4):874‐7. [DOI] [PubMed] [Google Scholar]
Chang 2008 {published data only}
- Chang KC, Leung CC, Yew WW, Lau TY, Tam CM. Hepatotoxicity of pyrazinamide: Cohort and case‐control analyses. American Journal of Respiratory and Critical Care Medicine 2008;177(12):1391‐6. [DOI] [PubMed] [Google Scholar]
Chang 2009 {published data only}
- Chang KC, Leung CC, Yew WW, Lau TY, Leung WM, Tam CM, et al . Newer fluoroquinolones for treating respiratory infection: do they mask tuberculosis?. European Respiratory Journal 2009;35(3):606‐13. [DOI] [PubMed] [Google Scholar]
Chen 2003 {published data only}
- Chen QL, Chen L, Yin JJ. A study on the clinical efficacy of a combination regimen with levofloxacin and capreomycin in the treatment of multi‐drug resistant pulmonary tuberculosis. Zhonghua Jie He He Hu Xi Za Zhi [Chinese Journal of Tuberculosis and Respiratory Diseases] 2003;26(8):454‐7. [PubMed] [Google Scholar]
Chigutsa 2012 {published and unpublished data}
- Chigutsa E, Meredith S, Wiesner L, Padayatchi N, Harding J, Moodley P, et al. Population pharmacokinetics and pharmacodynamics of ofloxacin in South African patients With multi‐drug resistant tuberculosis. Antimicrobial Agents and Chemotherapy 2012;56(7):3857‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chukanov 2006 {published data only}
- Chukanov VI, Komissarova OG, Maishin VI, Abdullaev RI, Kononets AS. Efficiency of a new standard chemotherapy regimen in the treatment of patients with recurrent pulmonary tuberculosis. Problemy Tuberculeza i Boleznej Legkikh 2006, issue 8:9‐13. [PubMed]
Diacon 2009 {published data only}
- Diacon AH, Pym A, Grobusch M, Patientia R, Rustomjee R, Page‐Shipp L, et al. The diarylquinoline TMC207 for multidrug‐resistant tuberculosis. New England Journal of Medicine 2009;360(23):2397‐405. [DOI] [PubMed] [Google Scholar]
Diacon 2012a {published and unpublished data}
- Diacon AH, Donald PR, Pym A, Grobusch M, Patientia RF, Mahanyele R, et al. Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug‐resistant tuberculosis: long‐term outcome, tolerability and effect on emergence of drug resistance. Antimicrobial Agents and Chemotherapy 2012;56(6):3271‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Diacon 2012b {published data only}
- Diacon AH, Dawson R, Groote‐Bidlingmaier F, Symons G, Venter A, Donald PR, et al. 14‐day bactericidal activity of PA‐824, bedaquiline, pyrazinamide, and moxifloxacin combinations: a randomised trial. Lancet 2012;380(9846):986‐93. [DOI] [PubMed] [Google Scholar]
Estebanez 1992 {published data only}
- Estebanez Zarranz MJ, Martinez‐Sagarra JM, Alberte A, Amon Sesmero J, Rodriguez Toves A. Treatment of urogenital tuberculosis with ofloxacin. Preliminary study. Actas Urologicas Espanolas 1992;16(1):64‐8. [PubMed] [Google Scholar]
Fouad 2011 {published and unpublished data}
- Fouad M, Gallagher JC. Moxifloxacin as an alternative or additive therapy for treatment of pulmonary tuberculosis. Annals of Pharmacotherapy 2011;45(11):1439‐44. [DOI] [PubMed] [Google Scholar]
Gosling 2003 {published data only}
- Gosling RD, Uiso LO, Sam NE, Bongard E, Kanduma EG, Nyindo M, et al. The bactericidal activity of moxifloxacin in patients with pulmonary tuberculosis. American Journal of Respiratory and Critical Care Medicine 2003;168(11):1342‐5. [DOI] [PubMed] [Google Scholar]
Grishin 1998 {published data only}
- Grishin VK, Polunina TE. Lomefloxacin in phthisiatric practice. Antibiotiki i Khimioterapiia 1998;43(10):17‐8. [PubMed] [Google Scholar]
Heemskerk 2011 {published and unpublished data}
- Heemskerk D, Day J, Chau TT, Dung NH, Yen NT, Bang ND, et al. Intensified treatment with high dose rifampicin and levofloxacin compared to standard treatment for adult patients with tuberculous meningitis (TBM‐IT): protocol for a randomized controlled trial. Trials 2011;12(25):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ho 2007 {published data only}
- Ho CC, Chen YC, Liao WY, Tsai TH, YU CJ, Yang PC, et al. Safety of fluoroquinolone in the treatment of tuberculosis patients with hepatotoxicity induced by first‐line antituberculosis regimens [Abstract]. Respirology 2007;12((Suppl 4)):A240. [Google Scholar]
Huang 2000 {published data only}
- Huang CS, Wu CC. Observation of the clinical efficacy of sparfloxacin in the treatment of multiple drug resistance pneumonial tuberculosis. Chinese Journal of Antibiotics 2000;25(4):302‐3. [Google Scholar]
Jenkins 2008 {published data only}
- Jenkins PA, Campbell IA, Banks J, Gelder CM, Prescott RJ, Smith AP. Clarithromycin vs ciprofloxacin as adjuncts to rifampicin and ethambutol in treating opportunist mycobacterial lung diseases and an assessment of Mycobacterium vaccae immunotherapy. Thorax 2008;63(7):627‐34. [DOI] [PubMed] [Google Scholar]
Ji 2001 {published data only}
- Ji YM, Dong LH, Wang Q, Yu WQ. Short‐term observating of curative effects in treatment of multiple‐drug resistance pulmonary tuberculosis with sparfloxacin and ofloxacin. Journal of Postgraduates of Medicines 2001;24(7):32‐3. [Google Scholar]
Johnson 2006 {published data only}
- Johnson JL, Hadad DJ, Boom WH, Daley CL, Peloquin CA, Eisenach KD, et al. Early and extended early bactericidal activity of levofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. International Journal of Tuberculosis and Lung Disease 2006;10(6):605‐12. [PubMed] [Google Scholar]
Kang 2009 {published data only}
- Kang WL, Xie YG, Tan WG, Chu NH, Li L, You YH, et al. Study on the efficacy and safety of short‐term treatment including fluoroquinolones anti‐tuberculosis drugs for rifampicin resistant pulmonary tuberculosis. Zhonghua Liu Xing Bing Xue Za Zhi 2009;30(2):179‐83. [PubMed] [Google Scholar]
Kawahara 1992 {published data only}
- Kawahara S, Eirei J. Evaluation of new antitubercular agents‐ new quinolone agents. Kekkaku 1992;67(10):679‐82. [PubMed] [Google Scholar]
Kennedy 1993 {published data only}
- Kennedy N, Fox R, Uiso L, Ngowi FI, Gillespie SH. Safety profile of ciprofloxacin during long‐term therapy for pulmonary tuberculosis. Journal of Antimicrobial Chemotherapy 1993;32(6):897‐902. [DOI] [PubMed] [Google Scholar]
Kennedy 1996 {published data only}
- Kennedy N, Berger L, Curram J, Fox R, Gutmann J, Kisyombe GM, et al. Randomized controlled trial of a drug regimen that includes ciprofloxacin for the treatment of pulmonary tuberculosis. Clinical Infectious Diseases 1996;22(5):827‐33. [DOI] [PubMed] [Google Scholar]
- Kennedy N, Fox R, Kisyombe GM, Saruni AO, Uiso LO, Ramsay AR, et al. Early bactericidal and sterilizing activities of ciprofloxacin in pulmonary tuberculosis. American Review of Respiratory Disease 1993;148(6 Pt 1):1547‐51. [DOI] [PubMed] [Google Scholar]
Kohno 1992 {published data only}
- Kohno S, Koga H, Kaku M, Maesaki S, Hara K. Prospective comparative study of ofloxacin or ethambutol for the treatment of pulmonary tuberculosis. Chest 1992;102(6):1815‐8. [DOI] [PubMed] [Google Scholar]
Kumar 2004 {published data only}
- Kumar AK, Gurumurthy P. Disposition of uric acid upon administration of ofloxacin alone and in combination with other anti‐tuberculosis drugs. Indian Journal of Experimental Biology 2004;42(3):323‐5. [PubMed] [Google Scholar]
Lee 2011 {published and unpublished data}
- Lee J, Lee CH, Kim DK, Yoon HI, Kim JY, Lee SM, et al. Retrospective comparison of levofloxacin and moxifloxacin on multidrug‐resistant tuberculosis treatment outcomes. Korean Journal of Internal Medicine 2011;26(2):153‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2008 {published data only}
- Li L, Zheng SH, Chu NH, Xie YG, Yang YZ, Li Q, et al. Effects of two treatment regimens for drug‐resistant tuberculosis in tuberculosis control project areas: a comparative study. Zhonghua Yi Xue Za Zhi 2008;88(48):3387‐91. [PubMed] [Google Scholar]
Lu 2000 {published data only}
- Lu Y, Zhu L, Duan L. Antituberculosis effect of levofloxacin. Zhonghua Jie He He Hu Xi Za Zhi 2000;23(1):50‐4. [PubMed] [Google Scholar]
Marra 2005 {published data only}
- Marra F, Marra CA, Moadebi S, Shi P, Elwood RK, Stark G, et al. Levofloxacin treatment of active tuberculosis and the risk of adverse events. Chest 2005;128(3):1406‐13. [DOI] [PubMed] [Google Scholar]
Merle 2012 {published data only (unpublished sought but not used)}
- Merle CS, Sismanidis C, Sow OB, Gninafon M, Horton J, Lapujade O, et al. A pivotal registration phase III, multicenter, randomized tuberculosis controlled trial: design issues and lessons learnt from the Gatifloxacin for TB (OFLOTUB) project. Trials 2012; Vol. 13, issue 61:1‐10. [DOI: 10.1186/1745-6215-13-61] [DOI] [PMC free article] [PubMed]
Moadebi 2007 {published data only}
- Moadebi S, Harder CK, Fitzgerald MJ, Elwood KR, Marra F. Fluoroquinolones for the treatment of pulmonary tuberculosis. Drugs 2007;67(14):2077‐99. [DOI] [PubMed] [Google Scholar]
Mohanty 1993 {published data only}
- Mohanty KC, Dhamgaye TM. Controlled trial of ciprofloxacin in short‐term chemotherapy for pulmonary tuberculosis. Chest 1993;104(4):1194‐8. [DOI] [PubMed] [Google Scholar]
Moulding 2008 {published data only}
- Moulding T. A randomised controlled trial of high‐dose isoniazid adjuvant therapy for multidrug‐resistant tuberculosis. International Journal of Tuberculosis and Lung Disease 2008;12(9):1102. [PubMed] [Google Scholar]
Nakamura 2007 {published data only}
- Nakamura M, Hasegawa N, Miyao N, Nakajima T, Terashima T, Sakamaki F, et al. Comparison of five day short‐course therapies for secondary infection in patients with chronic respiratory disease using gatifloxacin and levofloxacin. Japanese Journal of Chemotherapy 2007;55(6):451‐62. [Google Scholar]
Nosova 2008 {published data only}
- Nosova EIu, Galkina KIu, Antonova OV, Garmash IuIu, Skotnikova OI, Moroz AM. Use of molecular‐biological microchip TB‐BIOCHIP‐2 for detecting of Mycobacterium tuberculosis with multidrug resistance to fluoroquinolones in patients with new detected and chronic tuberculosis. Vestnik Rossiiskoi Akademii Meditsinskikh Nauk 2008;3:16‐9. [PubMed] [Google Scholar]
O'Brien 1994 {published data only}
- O'Brien RJ, Nunn PP. Ciprofloxacin is not a component of first‐line TB. Chest 1994;106(4):1312. [DOI] [PubMed] [Google Scholar]
Peloquin 2008 {published data only}
- Peloquin CA, Hadad DJ, Molino LP, Palaci M, Boom WH, Dietze R, et al. Population pharmacokinetics of levofloxacin, gatifloxacin, and moxifloxacin in adults with pulmonary tuberculosis. Antimicrobial Agents and Chemotherapy 2008;52(3):852‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pletz 2004 {published data only}
- Pletz MW, Roux A, Roth A, Neumann KH, Mauch H, Lode H. Early bactericidal activity of moxifloxacin in treatment of pulmonary tuberculosis: a prospective, randomized study. Antimicrobial Agents and Chemotherapy 2004;48(3):780‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ruslami 2013 {published data only}
- Ruslami R, Ganiem AR, Dian S, Apriani L, Achmad TH, Ven AJ, et al. Intensified regimen containing rifampicin and moxifloxacin for tuberculous meningitis: an open‐label, randomised controlled phase 2 trial. Lancet Infectious Diseases 2013;13(1):27‐35. [DOI] [PubMed] [Google Scholar]
Rustomjee 2008b {published data only}
- Rustomjee R, Diacon AH, Allen J, Venter A, Reddy C, Patientia RF, et al. Early bactericidal activity and pharmacokinetics of the diarylquinoline TMC207 in treatment of pulmonary tuberculosis. Antimicrobial Agents and Chemotherapy 2008;52(8):2831‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Saigal 2001 {published data only}
- Saigal S, Agarwal SR, Nandeesh HP, Sarin SK. Safety of an ofloxacin‐based antitubercular regimen for the treatment of tuberculosis in patients with underlying chronic liver disease: a preliminary report. Journal of Gastroenterology and Hepatology 2001;16(9):1028‐32. [DOI] [PubMed] [Google Scholar]
Sirgel 1997 {published data only}
- Sirgel FA, Botha FJ, Parkin DP, Wal BW, Schall R, Donald PR, et al. The early bactericidal activity of ciprofloxacin in patients with pulmonary tuberculosis. American Journal of Respiratory and Critical Care Medicine 1997;156(3 Pt 1):901‐5. [DOI] [PubMed] [Google Scholar]
Sirgel 2000 {published data only}
- Sirgel FA, Donald PR, Odhiambo J, Githui W, Umapathy KC, Paramasivan CN, et al. A multicentre study of the early bactericidal activity of anti‐tuberculosis drugs. Journal of Antimicrobial Chemotherapy 2000;45(6):859‐70. [DOI] [PubMed] [Google Scholar]
Sokolova 1998 {published data only}
- Sokolova GB, Kunichan AD, Koriakin VA, Lazareva IaV. Lomefloxacin in complex treatment of acute progressive form of pulmonary tuberculosis. Antibiotiki i Khimioterapiia 1998;43(10):10‐2. [PubMed] [Google Scholar]
Sun 2000 {published data only}
- Sun W, Wenyi C, Cunzhi L, Yanhong Y, Yuzhen X, Zhaosheng S. A randomized controlled study of sparfloxacin and ofloxacin in the treatment of multiple drug resistant pulmonary tuberculosis. Chinese Journal of Antibiotics 2000;25(1):52‐4. [Google Scholar]
Suo 1996 {published data only}
- Suo J, Yu MC, Lee CN, Chiang CY, Lin TP. Treatment of multidrug‐resistant tuberculosis in Taiwan. Chemotherapy 1996;42(Suppl 3):20‐3. [DOI] [PubMed] [Google Scholar]
Thwaites 2011 {published and unpublished data}
- Thwaites GE, Bhavnani SM, Chau TT, Hammel JP, Torok ME, Wart SA, et al. Randomized pharmacokinetic and pharmacodynamic comparison of fluoroquinolones for tuberculous meningitis. Antimicrobial Agents and Chemotherapy 2011;55(7):3244‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
TRC 2002 {published data only}
- Tuberculosis Research Centre (Indian Council of Medical Research), Chennei. Shortening short course chemotherapy: a randomized clinical trial for treatment of smear positive pulmonary tuberculosis with regimens using ofloxacin in the intensive phase. Indian Journal of Tuberculosis 2002;49(1):27‐38. [Google Scholar]
Valerio 2003 {published data only}
- Valerio G, Bracciale P, Manisco V, Quitadamo M, Legari G, Bellanova S. Long‐term tolerance and effectiveness of moxifloxacin therapy for tuberculosis: preliminary results. Journal of Chemotherapy 2003;15(1):66‐70. [DOI] [PubMed] [Google Scholar]
Venter 2006 {published data only}
- Venter WDF, Panz VR, Feldman Ch, Joffe BI. Adrenocortical function in hospitalised patients with active pulmonary tuberculosis receiving a rifampicin‐based regimen ‐ a pilot study. South African Medical Journal 2006;96(1):62‐6. [PubMed] [Google Scholar]
Wang 2006 {published data only}
- Wang JY, Hsueh PR, Jan IS, Lee LN, Liaw YS, Yang PC, et al. Empirical treatment with a fluoroquinolone delays the treatment for tuberculosis and is associated with a poor prognosis in endemic areas. Thorax 2006;61(10):903‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wolbers 2011 {published and unpublished data}
- Wolbers M, Heemskerk D, Chau TT, Yen NT, Caws M, Farrar J, et al. Sample size requirements for separating out the effects of combination treatments: randomised controlled trials of combination therapy vs. standard treatment compared to factorial designs for patients with tuberculous meningitis. Trials 2011;12(26):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yoon 2005 {published data only}
- Yoon YS, Lee HJ, Yoon HI, Yoo CG, Kim YW, Han SK, et al. Impact of fluoroquinolones on the diagnosis of pulmonary tuberculosis initially treated as bacterial pneumonia. International Journal of Tuberculosis and Lung Disease 2005;9(11):1215‐9. [PubMed] [Google Scholar]
Yoon 2012 {published and unpublished data}
- Yoon HE, Jeon YJ, Chung HW, Shin SJ, Hwang HS, Lee SJ, et al. Safety and efficacy of a quinolone‐based regimen for treatment of tuberculosis in renal transplant recipients. Transplantation Proceedings 2012;44(3):730‐3. [DOI] [PubMed] [Google Scholar]
Zhang 1997 {published data only}
- Zhang Y, Qian H, Chen M. Bronchofiberscope and catheter intervention in treatment of multi‐drug resistant pulmonary tuberculosis. Zhonghua Jie He He Hu Xi Za Zhi 1997;20(6):354‐7. [PubMed] [Google Scholar]
Zhang 2006 {published data only}
- Zhang X, Li M, Hu CM, Yu J, Lin FS, Mao KJ, et al. Clinic assessment of rifabutin in the treatment of multidrug‐resistant pulmonary tuberculosis. Chinese Journal of Antibiotics 2006;31(4):223‐4, 242. [Google Scholar]
Zhao 2003 {published data only}
- Zhao RZ, Wang QM, Zhang PC, Zhang HM. Therapeutic effects of levofloxacin‐containing regimen in patients with retreated pulmonary tuberculosis. Chinese Journal of Antibiotics 2003;28(8):497‐9. [Google Scholar]
Zheng 2004 {published data only}
- Zheng XM, Li SM, Xing BC. Short‐term effect of treatment protocol utilizing levofloxacin, pasiniazide and M. vaccae on multi‐drug resistant pulmonary tuberculosis. Di Yi Jun Yi Da Xue Xue Bao [Academic Journal of the First Medical College of PLA] 2004;24(5):574‐5. [PubMed] [Google Scholar]
Zhu 2006 {published data only}
- Zhu LZ, Fu Y, Chu NH, Ye ZZ, Xiao HP, Wang W, et al. A controlled clinical trial of long course chemotherapy regimens containing rifabutin in the treatment of multi‐drug resistant pulmonary tuberculosis. Zhonghua Jie He He Hu Xi Za Zhi 2006;29(8):520‐3. [PubMed] [Google Scholar]
Zhu 2012 {published and unpublished data}
- Zhu H, Lei X, Zhang F, Zhang ZJ, Lu J, Li H, et al. Effectiveness and safety of levofloxacin for multidrug resistant pulmonary tuberculosis: A systematic review. Chinese Journal of Evidence‐Based Medicine 2012;12(2):201‐8. [Google Scholar]
Zvada 2012 {published and unpublished data}
- Zvada SP, Denti P, Geldenhuys H, Meredith S, As D, Hatherill M, et al. Moxifloxacin population pharmacokinetics in patients with pulmonary tuberculosis and the effect of intermittent high‐dose rifapentine. Antimicrobial Agents and Chemotherapy 2012;56(8):4471‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
CTRI/2012/10/003060 {published data only}
- ICMR. Thrice weekly 4‐months moxifloxacin or gatifloxacin regimens for pulmonary TB [A study of the efficacy and tolerability of moxifloxacin and gatifloxacin containing regiments in the treatment of patients with sputum‐positive pulmonary tuberculosis]. http://www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=5124 (accessed in March 2013). [DOI] [PMC free article] [PubMed]
ISRCTN44153044 RIFAQUIN {published data only}
- ISRCTN44153044. An international multicentre controlled clinical trial to evaluate high dose RIFApentine and a QUINolone in the treatment of pulmonary tuberculosis. www.controlled‐trials.com/ISRCTN44153044/ISRCTN44153044 (accessed 27 July 2007).
NCT00216385 {published data only}
- NCT00216385. A controlled trial of a 4‐month quinolone‐containing regimen for the treatment of pulmonary tuberculosis. clinicaltrials‐nccs.nlm.nih.gov/ct2/show/NCT00216385 (accessed 23 July 2007).
NCT00396084 {published data only}
- NCT00396084. Randomized, open label, multiple dose Phase I study of the early bactericidal activity of linezolid, gatifloxacin, levofloxacin, and moxifloxacin in HIV‐non‐infected adults with Initial episodes of sputum smear‐positive pulmonary tuberculosis (DMID 01‐553). clinicaltrials.gov/ct/show/NCT00396084?order=11 (accessed 27 July 2007).
NCT00728507 {published data only}
- NCT00728507. A phase II randomized, open‐label trial of a rifapentine plus moxifloxacin‐based regimen for intensive phase treatment of smear‐positive pulmonary tuberculosis. http://clinicaltrials.gov/ct2/show/NCT00728507 (accessed on 9 November 2009).
NCT00864383 REMoxTB {published data only}
- NCT00864383. Controlled comparison of two moxifloxacin containing treatment shortening regimens in pulmonary tuberculosis. http://clinicaltrials.gov/ct2/show/NCT00864383 (accessed on 9 November 2009).
NCT01498419 {published data only}
- NCT01498419. Evaluation of 8 weeks of treatment with the combination of moxifloxacin, PA‐824 and pyrazinamide in patients With drug sensitive and multi drug‐resistant pulmonary tuberculosis (TB). http://clinicaltrials.gov/ct2/show/NCT01498419 (accessed on 25 May 2012).
NCT01589497 {published data only}
- AIDS Clinical Trials Group. Early bactericidal activity (EBA) study of tuberculosis regimens with and without INH and moxifloxacin [Essentiality of isoniazid after the first two doses: a randomized phase IIa clinical trial comparing the early bactericidal activity (EBA) of a standard anti‐tuberculosis regimen (RHZE) with a regimen omitting isoniazid (RZE) or a regimen substituting moxifloxacin for isoniazid (RMZE) during days 3 to 14 of tuberculosis therapy]. http://clinicaltrials.gov/show/NCT01589497 2013.
NCT01785186 {published data only}
- Michael Hoelscher. Evaluation of SQ109, high‐dose rifampicin, and moxifloxacin in adults with smear‐positive pulmonary TB in a MAMS Design (PanACEA‐MAMS‐TB‐01) [A multiple arm, multiple stage, phase 2, OL, randomized, controlled trial to evaluate 4 treatment regimens of SQ109, increased doses of rifampicin, and Moxifloxacin in Adults with newly diagnosed, smear‐positive pulmonary tuberculosis]. http://clinicaltrials.gov/ct2/show/NCT01785186 accessed March 2013.
Additional references
Alangaden 1997
- Alangaden GJ, Lerner SA. The clinical use of fluoroquinolones for the treatment of mycobacterial diseases. Clinical Infectious Diseases 1997;25(5):1213‐21. [DOI] [PubMed] [Google Scholar]
Anonymous 1983
- Anonymous. Controlled clinical trial of 4 short‐course regimens of chemotherapy (three 6‐month and one 8‐month) for pulmonary tuberculosis. Tubercle 1983;64(3):153‐66. [DOI] [PubMed] [Google Scholar]
Blumberg 2003
- Blumberg HM, Burman WJ, Chaisson RE, Daley CL, Etkind SC, Friedman LN, et al. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. American Journal of Respiratory Critical Care Medicine 2003;167(4):603‐62. [DOI] [PubMed] [Google Scholar]
Burman 1999
- Burman WJ, Gallicano K, Peloquin C. Therapeutic implications of drug interactions in the treatment of human immunodeficiency virus‐related tuberculosis. Clinical Infectious Diseases 1999;28(3):419‐30. [DOI] [PubMed] [Google Scholar]
Corbett 2003
- Corbett EL, Watt CJ, Walker N, Maher D, Williams BG, Raviglione MC, et al. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Archives of Internal Medicine 2003;163(9):1009‐21. [DOI] [PubMed] [Google Scholar]
Daley 1992
- Daley CL, Small PM, Schecter GF, Schoolnik GK, McAdam RA, Jacobs WR Jr, et al. An outbreak of tuberculosis with accelerated progression among persons infected with the human immunodeficiency virus. An analysis using restriction‐fragment‐length polymorphisms. New England Journal of Medicine 1992;326(4):231‐5. [DOI] [PubMed] [Google Scholar]
El‐Sadr 2001
- El‐Sadr WM, Perlman DC, Denning E, Marts JP, Cohn DL. A review of efficacy studies of 6‐month short‐course therapy for tuberculosis among patients infected with human immunodeficiency virus: differences in study outcomes. Clinical Infectious Diseases 2001;32(4):623‐32. [DOI] [PubMed] [Google Scholar]
Falzon 2011
- Falzon D, Jaramillo E, Schünemann HJ, Arentz M, Bauer M, Bayona J, et al. WHO guidelines for the programmatic management of drug‐resistant tuberculosis: 2011 update. European Respiratory Journal 2011;38(3):516‐28. [DOI] [PubMed] [Google Scholar]
Gillespie 1998
- Gillespie SH, Kennedy N. Fluoroquinolones: a new treatment for tuberculosis?. International Journal of Tuberculosis and Lung Disease 1998;2(4):265‐71. [PubMed] [Google Scholar]
Ginsburg 2003
- Ginsburg AS, Grosset JH, Bishai WR. Fluoroquinolones, tuberculosis, and resistance. Lancet Infectious Diseases 2003;3(7):432‐42. [DOI] [PubMed] [Google Scholar]
Grzybowski 1975
- Grzybowski S, Barnett GD, Styblo K. Contacts of cases of active pulmonary tuberculosis. Bulletin of the International Union Against Tuberculosis 1975; Vol. 50, issue 1:90‐106. [PubMed]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist G, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors) Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011] The Cochrane Collaboration, 2011. Available from www.cochrane.org/resources/handbook/hbook.htm.
Jacobs 1999
- Jacobs MR. Activity of quinolones against mycobacteria. Drugs 1999;58(Suppl 2):19‐22. [DOI] [PubMed] [Google Scholar]
Johnston 2009
- Johnston JC, Shahidi NC, Sadatsafavi M, Fitzgerald JM. Treatment outcomes of multidrug‐resistant tuberculosis: a systematic review and meta‐analysis. PLoS One 2009;4(9):e6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kochi 1991
- Kochi A. The global tuberculosis situation and the new control strategy of the World Health Organization. Tubercle 1991;72(1):1‐6. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Search for studies. In: Higgins JPT, Green S (editors) Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011] The Cochrane Collaboration, 2011. Available from www.cochrane.org/resources/handbook/hbook.htm.
Martindale 1996
- Reynolds JEF, Parfitt K, Parsons AV, Sweetmann SC, editors. Martindale: the extra pharmacopoeia. 1st Edition. London: Royal Pharmaceutical Society, 1996:1881. [Google Scholar]
Review Manager 5 [Computer program]
- The Nordic Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Centre, The Cochrane Collaboration, 2008.
STS/BMRC 1981
- Anonymous. Clinical trial of six‐month and four‐month regimens of chemotherapy in the treatment of pulmonary tuberculosis: the results up to 30 months. Tubercule 1981;62(2):95‐102. [DOI] [PubMed] [Google Scholar]
Telzak 1999
- Telzak EE, Chirgwin KD, Nelson ET, Matts JP, Sepkowitz KA, Benson CA, et al. Predictors for multidrug‐resistant tuberculosis among HIV‐infected patients and response to specific drug regimens. Terry Beirn Community Programs for Clinical Research on AIDS (CPCRA) and the AIDS Clinical Trials Group (ACTG), National Institutes for Health. International Journal of Tuberculosis and Lung Disease 1999;3(4):337‐43. [PubMed] [Google Scholar]
WHO 2003
- WHO Global Tuberculosis Programme. Treatment of tuberculosis: guidelines for national programmes. [WHO/CDS/TB/2003.313]. 3rd Edition. Geneva: World Health Organization, 2003:1‐13. [Google Scholar]
WHO 2006
- World Health Organization. Stop TB Dept. Guidelines for the programmatic management of drug‐resistant tuberculosis. [WHO/HTM/TB/2006.361]. Geneva: World Health Organization, 2006:38‐53. [Google Scholar]
WHO 2007a
- World Health Organization. WHO model list of essential medicines: 15th edition. WHO Drug Information 2007;21(2):95‐111. [Google Scholar]
WHO 2007b
- World Health Organization. Stop TB Dept. Global tuberculosis control : surveillance, planning, financing : WHO report 2007 [WHO/HTM/TB/2007.376]. http://www.who.int/tb/publications/global_report/2007/download_centre/en/index.html. Geneva: World Health Organization, (accessed August 2007).
WHO 2010a
- World Health Organization. Fact File: 10 Facts about tuberculosis. www.who.int/features/factfiles/tb_facts/en/index.html (accessed 8 March 2010).
WHO 2010b
- World Health Organisation. Treatment of tuberculosis: guidelines: WHO/HTM/TB/2009.420. 4th Edition. Geneva, Switzerland: World Health Organization, 2009. [Google Scholar]
WHO 2011a
- World Health Organisation. Global tuberculosis control: WHO report 2011. http://www.who.int/tb/publications/global_report/2011/gtbr11_main.pdf 2011:1‐73.
WHO 2011b
- World Health Organization. WHO Model list of essential medicines: 17th edition. http://whqlibdoc.who.int/hq/2011/a95053_eng.pdf 2011.
WHO 2012
- World Health Organization. Fact sheet No 104: Tuberculosis. http://www.who.int/mediacentre/factsheets/fs104/en/ accessed 25 July 2012.
Woldehanna 2004
- Woldehanna S, Volmink J. Treatment of latent tuberculosis infection in HIV infected persons. Cochrane Database of Systematic Reviews 2004, Issue 1. [DOI: 10.1002/14651858.CD000171.pub2] [DOI] [PubMed] [Google Scholar]
Yew 2001
- Yew WW. Clinically significant interactions with drugs used in the treatment of tuberculosis. Drug Safety 2001;25(2):111‐33. [DOI] [PubMed] [Google Scholar]
Ziganshina 2011
- Ziganshina L, Eisenhut M. Tuberculosis (HIV‐negative people). Clinical Evidence 2011;03(904):1‐42. [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Ziganshina 2005
- Ziganshina LE, Vizel AA, Squire SB. Fluoroquinolones for treating tuberculosis. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD004795.pub2] [DOI] [PubMed] [Google Scholar]
Ziganshina 2008
- Ziganshina LE, Squire SB. Fluoroquinolones for treating tuberculosis. Cochrane Database of Systematic Reviews 2008, Issue 1. [DOI: 10.1002/14651858.CD004795.pub3] [DOI] [PubMed] [Google Scholar]