

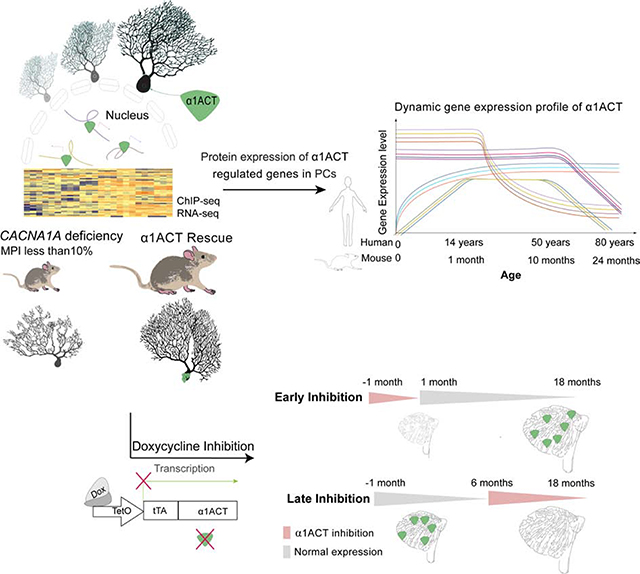
Summary
Postnatal cerebellar development is a precisely regulated process involving well-orchestrated expression of neural genes. Neurological phenotypes associated with CACNA1A gene defects have been increasingly recognized, yet the molecular principles underlying this association remain elusive. By characterizing a dose-dependent CACNA1A gene deficiency mouse model, we discovered that α1ACT, as a transcription factor and secondary protein of CACNA1A mRNA, drives dynamic gene expression networks within cerebellar Purkinje cells and is indispensable for neonatal survival. Perinatal loss of α1ACT leads to motor dysfunction through disruption of neurogenesis and synaptic regulatory networks. However, its elimination in adulthood has minimal effect on the cerebellum. These findings shed light on the critical role of α1ACT in facilitating neuronal development in both mice and humans, and support a rationale for gene therapies for calcium channel-associated cerebellar disorders. Finally, we show that bicistronic expression may be common to the VGCC gene family and help explain complex genetic syndromes.
Grpahical Abstract
Introduction
Understanding the physiological function of native polyglutamine (polyQ) proteins is critical to treatment of polyQ expansion diseases, particularly in light of gene-silencing strategies that suppress both the wildtype (WT) and disease alleles. CACNA1A mRNA transcripts encode two structurally and functionally unrelated proteins, the voltage-gated calcium channel (VGCC) α1A subunit, and a transcription factor, α1ACT expressed via an internal ribosomal entry site (IRES), both of which contain the polyQ tract (Du et al., 2014; Du et al., 2013). The polyQ-expanded α1ACT allele causes spinocerebellar ataxia type 6 (SCA6) (Bavassano et al., 2017; Du and Gomez, 2018b; Du et al., 2013; Ishida et al., 2016; Paulson et al., 2017). Although homozygous deletion of the Cacna1a gene is lethal in mice (Jun et al., 1999), it may be feasible to target the IRES and selectively suppress α1ACT expression, as shown recently in a microRNA study (Miyazaki et al., 2016; Pastor et al., 2018). It is thought that WT α1ACT is involved in Purkinje cell (PC) development. However, the role and importance of WT α1ACT in PC development and motor function over time is not fully understood. Fundamental insight into the physiological role of α1ACT is crucial to decode the pathogenesis of polyQ expansion diseases. Furthermore, these insights will provide the theoretical support for safe and tolerable gene-silencing therapy.
Studies in mice show delayed motor maturation or death in some spontaneous C terminal Cacna1a gene mutations present from postnatal p14 to 1 month (Fletcher et al., 1996; Herrup and Wilczynski, 1982). Increasing evidence from clinical studies have associated severe motor and developmental delays with cerebellar atrophy in children with large deletions/copy number change or mutations encoding either the CACNA1A IRES region or the C terminus (Coe et al., 2014; Cooper et al., 2011; Damaj et al., 2015; Fletcher et al., 1996; Riant et al., 2008; Welham et al., 2015).
In a previous study, we provided evidence that α1ACT is a transcription factor involved in regulating neuronal gene expression (Du et al., 2013). However, the role of α1ACT-mediated gene expression in cerebellar development has not been not clearly defined. Here, we demonstrate that in the cerebellum, α1ACT-regulated gene expression patterns change dynamically across the lifespan of the organism. Furthermore, α1ACT exhibits a complex transcriptional regulation profile and plays a crucial role in a perinatal cerebellar developmental window. These insights may lead to new approaches for diagnosis and genetic therapy in calcium channel gene related developmental deficiencies. Precise inhibition of α1ACT in the cerebellum at different intervals reveals that suppression of α1ACT can be well tolerated in adult mice suggesting a potential therapeutic strategy for SCA6, a polyQ expansion disease. Interestingly, we also found that bicistronic expression is not unique to the CACNA1A gene, and appears to be at play in other gene members of the VGCC family.
Dynamic gene regulation by α1ACT
Previous studies have shown that prolonged expression of α1ACT in cultured, undifferentiated neural crest-derived pheochromocytoma (pc12) cells promotes features of a neuronal phenotype (Du et al., 2013). To explore the physiological gene-regulatory properties of α1ACT, we performed chromatin immunoprecipitation-sequencing (ChIP-Seq) on pc12 cell lines stably expressing pcDNA3-α1ACT or control pcDNA3 vectors at 3 days post-plating (Figure 1A). The majority of α1ACT-occupied genomic regions were located within ±3000 bp of annotated 5′-ends of transcripts (Figure 1B). 24.6% of binding sites were located within transcribed regions and 5.7% were at the promoters of annotated transcripts (Figure 1C, Figure S1A). As we show in our transgenic mouse studies below these gene targets are also bound by α1ACT in Purkinje cells in vivo.
Figure 1. Time-dependent regulation of neuronal differentiation and development genes by α1ACT, See also Figure S1, Figure S2, and Table S1.

(A) Scheme of ChIP-Seq and RNA-Seq time points with time-dependent morphological changes.
(B) α1ACT putative binding site distribution ± 3000bp relative to the transcription start site (TSS).
(C) Pie chart of distribution of 161,535 α1ACT putative binding sites based on Rattus norvegicus (UCSC version rn5) gene annotation.
(D) Scatter plots of integrated ChIP-Seq/RNA-Seq DEGs expression values at log2 counts per million in pc12 cells expressing α1ACT compare to pcDNA3 at 4 time points.
(E) 39 α1ACT specific RNA-Seq persistent DEGs across the 4 time points.
(F) Ten top selected highly enriched GOs related to synaptic signaling and neuron development identified from 39 persistent DEGs genes at FDR ≤ 0.1 (Benjamini-Hochberg).
(G) Four enriched consensus binding motifs of α1ACT identified in the binding sites of integrated ChIP-Seq/RNA-Seq genes. The enriched de novo consensus motifs of α1ACT were identified with HOMER using ChIP-Seq binding sites with 200bp summits. The motif-associated genes were annotated with HOMER (annotatePeaks.pl).
To monitor transcriptional profiles that capture transient and prolonged dynamic changes regulated by α1ACT during cell cycle and differentiation (Bar-Joseph et al., 2012), we performed a time-series RNA-seq study of the same cells used for ChIP-Seq (Figure 1A). At 6hr, 24hr, 3day, and 10day with growth stages ranging from starting log phase to differentiating and senescent cells (Figure 1A), we found that 169, 108, 449, and 261 differentially expressed genes (DEGs) were up-regulated, and 60, 58, 268, and 267 DEGs were down-regulated, respectively, in α1ACT-expressing cells (Figure S1A-B,E). The integrated ChIP-Seq and RNA-Seq data revealed that 52–67% of RNA-Seq DEGs were identified in the ChIP-Seq as bona-fide gene targets of α1ACT (Figure 1D, Figure S1B). 39 of all the DEGs detected in all 4-time points (persistent DEGs) (Figure 1E, Figure S2A) were validated by qRT-PCR (TaqMan) in cells (R2 =0.920~0.775, P=0.0001) (Figure S1F, Suppl. Table 1). All were neural-specific (Figure 1F), associated with neurogenesis (Dusp4, Efnb2, Fgfr3, Gfra2, Ntn1, Ptger3, Penk, Odc1), synaptic transmission (Hcn4, Slc18A3, Syn2), and cell adhesion (L1cam) (gene ontology (GO) enrichment analysis, Figure 1F, Figure S2A) (Ashburner et al., 2000; Burazin and Gundlach, 1999; Dominici et al., 2017; Nakamura et al., 2010; Sergaki and Ibanez, 2017). 21 of the 39 were ChIP-Seq targets (Figure S1C,D,H). Using the integrated data for analysis with the Hypergeometric Optimization of Motif EnRichment (HOMER) wrapper program (Heinz et al., 2010), we identified four DNA binding motifs for α1ACT, which were matched with corresponding associated persistent DEGs (Figure 1G).
To define how transcriptional network modules correlated with the biological functions of the α1ACT-modulated DEGs across different time intervals, we applied weighted gene co-expression network analysis (WGCNA) (Langfelder and Horvath, 2008) at 6–24 hr, 24 hr-3days, and 3–10days. Interestingly, WGCNA data revealed a continuous trajectory of transcriptional changes. A number of common biological pathways were present in all of the modules, but with time-dependent enrichment. Genes from significantly-overlapped DEG modules from 6hr to 24hr were highly enriched in neurogenesis, synaptic function, and cell adhesion, while those from 24hr to 3day were slightly enriched in neurogenesis and cell adhesion and those from 3day to 10day modules were slightly enriched in neurogenesis, absent of synaptic function genes, but highly enriched in cell death genes (Figure S2A-D). Furthermore, α1ACT-targeted, up-regulated DEGs were significantly correlated with genes involved in neurogenesis and synaptic function at all four time points (FDR ≤ 0.2), particularly at early time points. Conversely, the α1ACT-targeted down-regulated DEGs were associated with the cell cycle at all four time points, particularly in late time points (Figure S2E).
α1ACT is essential for neonatal survival
Transgenic expression of normal α1ACT in PCs of Cacna1a−/− mice leads to slight phenotypic improvement, but they remain non viable past the neonatal period (Du et al., 2013). To gain a better understanding of the normal function of this protein throughout mouse cerebellar development, we studied a mouse mutant, Cacna1ahEx47, bearing a normal, “humanized” CACNA1A exon 47 (encoding a normal polyQ allele of Q14), originally developed as a control for an SCA6 mouse model (Figure 2A) (Watase et al., 2008). As in the initial report, we found that in homozygous Cacna1ahEx47/Ex47 mice (Figure 2B and C) the expression of the longest mRNA isoform (MPI), which generates α1ACT protein, was decreased by 80%, despite the mice being neurologically normal. The expression of full-length α1A protein was attenuated 40% (Figure 2C) and α1ACT transcription factor was undetectable by immunoblot in these mice (Watase et al., 2008). In addition, Cacna1a+/− heterozygote mice, bearing a single mouse Cacna1a allele, appear normal, while Cacna1a−/− mice are non-viable. As such, we reasoned that normal cerebellar motor control might depend on precise levels of Cacna1a gene products. This is further supported by in vivo lentiviral-based RNAi studies that target the α1A subunit, leading to similar motor abnormalities as seen in other Cacna1a mutants (Damaj et al., 2015; Labrum et al., 2009; Riant et al., 2010; Salvi et al., 2014). The humanized CACNA1A exon 47 allele is therefore an ideal model to test the relative dose-dependent contributions of the Cacna1a gene products.
Figure 2. Reintroduction of α1ACT in cerebellum during perinatal period restores survival, improves motor function, and restores normal growth of KIKO mice, See also Figure S3, Figure S7 and Movies S1–3.

(A) Mouse breeding strategy to generate KIKO and KIKO:PC-α1ACT mice.
(B)KIKO mice and KIKO:PC-α1ACT mice have reduced proportion of the endogenous MPI splice form. Quantitative real-time RT-PCR (Taqman) of Cacna1a mRNA using cerebellar mRNA from Ctr, KIKO, KIKO:PC-α1ACT, WT, Cacna1a +/−, mice to compare abundance of Ex21–22, Ex39–40, and Ex46–47 of Cacna1a mRNA (n=20, p<0.05).
(C) Expression of full-length α1A protein is decreased in KIKO and KIKO:PC-α1ACT mice compared to that in WT and Cacana1a +/− mice (n=10, p<0.005). Left. Western blot. Right. Quantitation.
(D) Left. Only KIKO and Cacna1a−/− have reduced survival compared with controls. Middle. KIKO mice have reduced survival compared with Ctr and KIKO:PC-α1ACT. Survival is normalized by α1ACT expression. Only 20% of KIKO mice could survive to age of 40 days. Littermate KIKO:PC-α1ACT had the same phenotype as Ctr mice and both mice had 100% survival rate. Kaplan–Meier survival curves comparing the survival of Cacna1a−/−, Cacna1a+/−, Ctr, KIKO, and KIKO:PC-α1ACT mice (n=100, p<0.001). Right. The surviving KIKO mice had lower body weight in the first month of life compared to Ctr and KIKO: PC-α1ACT mice. (n=20).
(E) Schedule for doxycycline (Dox) treatments to block expression of α1ACT in KIKO:PC-α1ACT mice at indicated time.
(F) The appearance of littermate Ctr, KIKO, KIKO: PC-α1ACT with or without Dox (−1/+1M) treatment at p20 days.
(G) Left. Mouse body weight in the first 28 days after birth in Dox treatment. Prenatal treatment with Dox to inhibit α1ACT expression prevented the rescue of body weights of KIKO:PC-1ACT mice from birth to P28 days. Middle. Mouse body weight from 1 month to 24 month with and without Dox (−1/+1M) treatment. Right. Prenatal inhibition of α1ACT in KIKO persists into adult life as shown by decreased total distance at indicated ages after Dox (−1/+1M) treatment in open field test.
(H) Reduced treadmill performance of KIKO mice is corrected by α1ACT expression, and prevented by prenatal Dox treatment. Treadmill running speed and total travel distance are compared among mice with/without Dox (−1/+1M) treatment at 1 month old. Prenatal inhibition of α1ACT expression prevented the rescue of motor behaviour of KIKO:PC-α1ACT mouse in treadmill (left), rotarod (middle), and openfield (right).
(I) Prenatal inhibition of α1ACT expression offset the rescued survival of KIKO:PC-α1ACT mice. Kaplan–Meier survival curves comparing the survival of KIKO, littermate Ctr, KIKO:PC-α1ACT mice with/without Dox (−1/+1M) treatment during the first 40 days of life (n=100, p<0.0001).
(J) Inhibition of α1ACT expression eliminates motor rescue in juveniles, but has no effect in adult KIKO mice. The motor behavior analysis Ctr, KIKO, KIKO:PC-α1ACT of litter mates with different starting point of Dox treatment on the openfield at indicated ages (n=10, p<0.005).
Values are represented as mean±SEM. *p<0.05, **p<0.01; ***p<0.001.
To explore whether the reduced MPI expression of a single copy of the Cacna1ahEx47 allele would support normal neurological function in place of a single copy of the mouse Cacna1a allele, we crossed the Cacna1ahEx47/Ex47 mice with Cacna1a+/− mice. We compared the phenotypes of 5 mouse genotypes (Cacna1a+/+, Cacna1a−/−, Cacna1a+/−, Cacna1ahEx47/Ex47, Cacna1ahEx47/+ and Cacna1ahEx47/−) (Figure 2A). Cacna1−/− mice exhibited dystonia and died by day 21, while Cacna1a+/−, Cacna1ahEx47/Ex47, and Cacna1ahEx47/+ mice were indistinguishable from wildtype mice. Unexpectedly, Cacna1ahEx47/− knockin-knockout compound heterozygote mice (KIKO), bearing only a single copy of the humanized mouse Cacna1ahEx47 gene, which expresses ~10% of the MPI isoform of CACNA1A (Figure 2B and C), died between postnatal days 18 and 30 (p18 to p30) (Figure 2D), unless they received extended maternal care by delayed weaning. Delay of weaning of KIKO mice by an additional 10 days allowed 96% of KIKO to survive into adulthood. However, KIKO mice surviving after p18 exhibited several neurological abnormalities reminiscent of other Cacna1a mutants, including ataxic gait and dystonic spells (Movie S1). KIKO mice had 40% decreased weight gain (Figure 2D), significantly decreased mobility on Digigait treadmill (only 56% of KIKO could run at 25 cm/sec, and for significantly reduced times), 37% of normal time on rotarod, and 33% of normal travel in open field (Figure S3B) compared to littermate control, Cacna1ahEx47/+ mice (Ctr) at 1 month. Surprisingly, these physical and motoric differences were age-dependent and disappeared as mice matured. The body weight differences between groups disappeared by 9 months (Figure 2G), while motor behaviors in KIKO and Ctr were indistinguishable by 12 months (Figure 2G right panel, Figure S3C, and Figure S7). Since the humanized mouse Cacna1ahEx47 allele expresses reduced levels of α1A, especially the MPI splice variant that generates α1ACT, these findings suggest that sufficient and precise expression of either or both α1A and α1ACT is necessary for early development of normal motor function.
To test whether the absence of α1ACT is a critical factor responsible for poor survival and delayed motor development in KIKO mice, we generated KIKO mice with TET-off transgenic expression of normal α1ACT in PCs (Du et al., 2013) (KIKO:PC-α1ACT) (Figure 2A). Transgenic expression of α1ACT in KIKO mice successfully rescued the survival rate to 98% (n>200, Figure 2D) without special care. KIKO:PC-α1ACT mice also showed significantly improved motor function compared with KIKO, although remained less coordinated than Ctr between ages p18 to p30 (Movie S2). On treadmill 90% of mice could run at 25 cm/sec, the time on Rotorod increased to 66% of Ctr, and total travel distance in open field increased to 60% of Ctr at 1 month (Figure S3B). By 12 months differences in motor behavior between KIKO mice, and either Ctr or KIKO:PC-α1ACT had fully disappeared (Figure 2G right panel, Figure S3C, and S7). These observations demonstrate that the physiological expression of α1ACT in cerebellum is important in establishing normal development of motor function in early life, but dispensable in adulthood.
To further investigate the temporal effect of α1ACT expression, we used doxycycline (Dox) to inhibit expression of α1ACT transgene (Gossen and Bujard, 1992) (Figures 2A and S3A) in KIKO:PC-α1ACT mice at different life stages. The effects of inhibition were tested over 4 periods: 1) perinatal (Dox−1/+1M); 2) early (Dox+1/+18M); 3) middle (Dox+3/+18M); 4) late (Dox+6/+18M) (Figure 2E). With perinatal inhibition of α1ACT expression, the survival rate and body weight of Dox−1/+1MKIKO:PC-α1ACT mice were the same as those of Dox−1/+1MKIKO and KIKO. Neither Dox−1/+1MKIKO nor Dox−1/+1MKIKO:PC-α1ACT survived to p30 without special care (Figures 2F, 2G left panel, and 2I) (Movie S3). The locomotor function of mice in the Dox treatment groups was assessed at 1 month by treadmill, and every 3 months by rotarod and open field observation. For the Dox−1/+1M group, with special care after birth, the motor function of Dox−1/+1M KIKO:PC-α1ACT mice was similar to KIKO and KIKO and significantly delayed compared with that of the Dox−1/+1MCtr and KIKO:PC-α1ACT at 1 month (Figure 2H). Moreover, even after Dox treatment was discontinued after 1 month of age, allowing α1ACT expression to be re-established, phenotypes of Dox−1/+1MKIKO:PC-α1ACT mice ages 1 to 12 months were very similar to age-matched Dox−1/+1MKIKO and KIKO, and inferior to litter mates KIKO:PC-α1ACT, showing slowed weight gain and impaired motor behavior on rotarod and open field (Figures 2G middle and right panel, 2H, and S7). For Dox+1/+18M and Dox+3/+18M groups, the motor abilities of Dox+1/+18MKIKO:PC-α1ACT and Dox+3/+18MKIKO:PC-α1ACT mice were significantly impaired after Dox treatment compared with those in KIKO:PC-α1ACT, but were not as severe as those observed in Dox−1/+1MKIKO:PC-α1ACT (Figures 2J and S7). Together these results demonstrate the importance of this factor in influencing motor development in a narrow window of the neonatal period. Surprisingly, when α1ACT inhibition was initiated at 6 months, there were no significant differences in motor behaviors between KIKO:PC-α1ACT mice with and without exposure to Dox, despite continued differences between KIKO and KIKO:PC-α1ACT mice without Dox treatment (Figures 2J and S7). Thus, the presence of α1ACT specifically within the perinatal period is pivotal for early cerebellar development. Inhibition of α1ACT expression within the first to third month of life, but not at later stages of life, has significant impact on motor function. These results indicate that α1ACT operates in a highly age-dependent, temporal window, pointing to the promise of potential gene-silencing therapies initiated in adulthood for treatment of SCA6.
α1ACT drives PC gene expression
To confirm that the α1ACT-specific gene targets we identified in the cultured cell line expressing α1ACT were relevant to PCs, we used our transgenic mouse KIKO:PC-α1ACT line with PC-targeted expression of a MYC-tagged α1ACT transgene under the control of the PCP2 promoter (Du et al., 2013). We performed ChIP using anti-MYC antibody to pulldown α1ACT specifically expressed in PCs together with its gene-binding targets in KIKO:PC-α1ACT mice and performed qPCR to assess the association of the DNA-binding regions with α1ACT. These ChIP-qPCR studies confirmed that Efnb2, Fgfr3, Gfra2, Ntn1, Nrcam and Rbfox1 are directly bound by α1ACT in PCs with similarly enriched DNA-binding affinity (3–10 fold) as in cultured cells (Figure 3A).
Figure 3: α1ACT regulates target gene expression in PCs.

(A) Quantification of α1ACT target gene DNAs in PCs among mice with and without PC-specific α1ACT expression by ChIP-qPCR (n=6, p<0.005).
(B) LCM and qRT-PCR to quantify target gene mRNA expression in PCs from mouse cerebellum. Upper: images of cerebellar cortex before and after PC LCM. Lower: quantitation of target genes. (n=6, p<0.005).
(C and D) Immunofluorescence staining of target gene protein expression in PCs from mouse cerebellum.
(E) Enlarged view of target gene expression in PCs.
(F) Quantification of immunofluorescence staining signals of target gene protein expression (n=6, p<0.05).
The scale bar represent 50μm (C,D). Values are represented as mean±SEM. *p<0.05.
The timing of neuronal gene expression during the critical period of cerebellar development and maturation is programmed and modifies subsequent physiological development (Ingram et al., 2016; Kuhn et al., 2012; Serra et al., 2006). Therefore, we investigated the underlying relationships between changes in KIKO motor function and the temporal patterns of α1ACT-regulated gene expression across the mouse lifespan. We examined the expression of the 39 verified persistent DEGs, plus Ncam2 and Nrcam, in cerebellar mRNA from Ctr, KIKO, and KIKO:PC-α1ACT mice with and without Dox treatment, at ages p15 and p22, and 1, 3, 6, 12 and 18 months, using qRT-PCR. Expression of 23 of these was decreased in KIKO, but restored in KIKO:PC-α1ACT mice. In WT and Ctr mouse cerebellum, Fgfr3, Gfra2, Penk, Hcn4, and Scl18a3 expression peaked around p15 to p30, but gradually decreased with age. Syn2, G0s2, Astn2, and Elmod1 expression increased around 1 month and remained at the same level until decreasing around 12 months (Figure S4A). In KIKO mice, Fgfr3, Gfra2, Penk, which are involved in neurogenesis (Blak et al., 2007) and Hcn4, Scl18a3 and Syn2, which mediate synaptic function (Zuniga et al., 2016), were all decreased more than 60% and lacked the peaks in expression seen from p15 to 1 month, in WT and Ctr mice. The temporal profile of these transcripts was restored in KIKO:PC-α1ACT mice (Figure S4A). The expression differences of these target genes between Ctr and KIKO mice resolve with age completely between 6 to 9 months. Western blotting of Ctr, KIKO, and KIKO:PC-α1ACT mouse cerebellum with and without Dox−1/+1M treatment at age p20 revealed protein expression patterns consistent with mRNA levels for Fgfr3, Gfra2, Hcn4, Scl18a3, Syn2, Nrcam, L1cam, (Figures S4B and S4C). These data further illustrate that the dynamic pattern of α1ACT-regulated gene expression involved in neurogenesis, synaptic function, and cell adhesion is critical for normal cerebellar development and is compatible with the time-dependent motor behavior changes of KIKO mice.
The temporal profile of these transcripts in the cerebellum of Dox−1/+1MKIKO:PC-α1ACT mice, with Dox inhibition prenatally until 1 month of age, was similar to that of Dox−1/+1MKIKO and KIKO mice and significantly decreased compared with that of the Dox−1/+1MCtr and untreated KIKO:PC-α1ACT mice at 1 month (Figure S4). Because the late Dox inhibition group, Dox+6/+18M KIKO:PC-α1ACT, did not exhibit neurological deterioration, we tested whether α1ACT target genes were similarly stable in these mice. Interestingly, we found that suppression of α1ACT protein expression in adult mice (Dox+6/+18M) did not lead to reduced expression of target genes (Fgfr3, Scl18a3) at the mRNA levels compared with littermate Dox+6/+18MCtr (Figure S4D). This suggests that α1ACT drives a cerebellar genetic program within a critical neonatal window, but that other expression controls for these target genes predominate in later life. These findings provide evidence for α1ACT-mediated gene regulation in promoting age-dependent morphological and functional changes predominantly during early development in mice.
The detailed studies of the temporal profile of the α1ACT-regulated genes were conducted on RNA isolated from whole cerebellar homogenates of transgenic mice. However, to confirm that the changes we associated with α1ACT were specific to PCs, we used two approaches. First, we performed laser-capture microdissection (LCM) of PCs in transgenic mice, using KIKO, KIKO:PC-α1ACT and control mice to extract PC mRNA. The qRT-PCR analysis showed that expression of Efnb2, Fgfr3, Gfra2, Ntn1, Nrcam, Rbfox1, Elmod1, L1Cam, Syn2 and Vacht1 were decreased (16~73%) in KIKO PCs, but were normalized in KIKO:PC-α1ACT PCs (Figure 3B).
Subsequently, we characterized the expression of the target genes at the protein level in situ using immunostaining. We characterized the distribution and expression level of α1ACT-targeted gene products Efnb2, Fgfr3, Gfra2, Ntn1, Nrcam, Rbfox1, Elmod1, L1Cam, Syn2 and Vacht1 in KIKO, KIKO:PC-α1ACT and control cerebellum. Fgfr3, Gfra2, Ntn1, Elmod1 and Vacht1 were colocalized with PC-specific protein Calbindin 28K (correlation coefficient R= 83~89%, Figure 3C, E and S4E). Nrcam and Syn2 were colocalized with climbing fiber protein vGlut2 (correlation coefficient R=78–85%, Figure 3D, E and S4E). Rbfox1 and Efnb2 were expressed in PC soma membrane and along the dendrites. L1cam was expressed along the dendrites (Figure 3C-E). Quantitative analysis of the the density and relative extent of dendritic distribution of those gene products revealed that they were decreased in KIKO PCs (18~54% and 10–46%) and restored in KIKO:PC-α1ACT PCs (Figure 3F), further confirming that these target genes were also regulated by α1ACT at the protein level in PCs. These data confirmed that α1ACT promotes PC morphology and cerebellar development by directly binding and regulating target genes, whose products are mainly expressed in the PC soma, or along the dendritic tree.
α1ACT transcriptome verified in human cerebellum
Of the 1272 DEGs in α1ACT-expressing pc12 cells, 1221 (96%) have a homologous gene in the mouse and human genomes, suggesting that a large number of human genes, ~50% of which are highly expressed in human cerebellum, might be regulated directly or indirectly by α1ACT (Figure 4A) (Nilsson et al., 2001). The mRNA levels of three splice variants of Cacna1a (including MPI) have similar expression curves, each peaking in mice at around 1 month (Figure 4B, left panel). To examine whether the patterns we observed with RNA profiling actually reflected changes that occurred within human cerebellum, we extracted mRNA from Purkinje cells and the surrounding molecular layer of human postmortem cerebellar tissues. The qRT-PCR revealed that CACNA1A mRNA expression was maximal from birth to 20 years, and decreased gradually until reaching a plateau at age 50 (Figure 4B right panel). The expression of α1ACT-targeted genes in human cerebellum can be grouped into four major dynamic expression patterns: Those involved in neurogenesis (FGFR3, EFNB2, PTGEP3, NTN1, NMNAT2, GFRA2, PENK) were highly expressed at 1 month and started decreasing with age after maturation. Those associated with cell adhesion (NRCAM, NCAM2, L1CAM), were highly expressed at term and decreased with age gradually. The genes RBFOX1 and ASTN2 were increased from birth to 20 years, and decreased gradually with age. The genes G0S2, ELMOD1, and GLDN showed increased expression with age (Figure 4C). The age-dependent profiles of the α1ACT-targeted genes in human were similar to those in mouse cerebellum. Using Ingenuity Pathway Analysis (IPA) as a tool for network analysis we found that in vivo α1ACT directly or indirectly regulates genes involved in three known neurogenesis pathways (Figure 4D). These findings are consistent with an increasing number of clinical reports of pediatric patients with developmental delay and cerebellar atrophy, associated with large deletions or mutations involving the C terminus of the CACNA1A gene (Coe et al., 2014; Cooper et al., 2011; Damaj et al., 2015; Fletcher et al., 1996; Riant et al., 2008; Welham et al., 2015). Therefore, impaired α1ACT expression or function may disrupt the gene expression network within the cerebellum.
Figure 4. α1ACT and regulated DEGs in mouse and human cerebellum temporal changes over lifespan, See also Figure S4 and Table S4.

(A) Tissue specific enrichment analysis for DEGs of α1ACT in the 4 time points.
(B) Expression of Cacna1a and CACNA1A gene in cerebellum through mouse (Left) and human (right) lifespan.
(C) The α1ACT-regulated DEGs in human cerebellum as a function of age (n=3). Left. DEGs of the neurogenesis pathway. Right. Synapse and cell adhesion-related DEGs.
(D) Pathway analysis showing 13 potential DEGs involved in 3 neurogenesis-related pathways indicated by yellow, green, and purple symbols. Blue-labeled genes are persistent DEGs regulated by α1ACT.
α1ACT drives PC development
During cerebellar development, there is close correspondence between the physiological and morphological maturation of PCs and changes in cell output and dendritic growth over a short developmental window (Goldowitz and Hamre, 1998; McKay and Turner, 2005). Using Golgi staining, we documented the changes in PC morphology in Ctr, KIKO, and KIKO:PC-α1ACT mice with and without Dox suppression (Dox−1/+1M) at postnatal p18, 1 and 12 months of age. At p18 to 1 month, the elaborate dendritic arborization of PCs was either absent/sparse or disordered in KIKO mice compared with that in Ctr. However, KIKO:PC-α1ACT mice had normal PC dendritic structure (Figures 5A and S5A). Using Sholl analysis to quantify dendritic development in KIKO mice at p18 and 1 month, we observed a significant reduction in intersection (5% and 21%, of normal, respectively) and dendritic length (11% and 33% of normal, respectively) of PCs compared with those in Ctr (Figure 5B). There were no significant differences in intersection and dendritic length between KIKO:PC-α1ACT and Ctr (Figures 5A and 5B). Prenatal inhibition of α1ACT in Dox−1/+1MKIKO:PC-α1ACT mice led to immature PC dendritic development at p18 and 1-month. However, by 12 months, all PC dendrites were similar regardless of Dox inhibition (Figures 5A–5C). Immunofluorescent staining of the cerebellar cortex at p18 of the three groups of mice revealed that PCs of KIKO had shortened primary dendrites with premature branching (Figure S5A and S5B). In KIKO:PC-α1ACT mice the thickness of the molecular layer (ML) and the relative height of the dendritic tree were significantly increased compared with those in KIKO mice, and were similar to control (Figure S5A and S5B).
Figure 5. Reintroduction of α1ACT in cerebellum of KIKO mice perinatally restores impaired cerebellar Purkinje cell morphology and electrophysiology, See also Figure S5.

(A) Representative Golgi staining and Sholl analysis of reconstructed Purkinje cells in Ctr, KIKO, KIKO:PC-α1ACT, and DOX−1/+1M KIKO:PC-α1ACT mice at the ages of 17 days, 1 month, and 12 month. The scale bar represents 50μm.
(B and C) Dendritic intersections per radial distance from soma (left) and total dendrite length (right) were impaired in KIKO and Dox−1/+1MKIKO:PC-α1ACT mice, but were restored in KIKO:PC-α1ACT mice at p17 (B). Morphology of Purkinje cells were similar in all mice at 1 year (C) (n=6). Prenatal inhibition of α1ACT prevents restoration of dendritic growth.
(D) CF-EPSC amplitude was increased in KIKO, but normalized in KIKO:PC-α1ACT mice.
(E) PC excitability (evoked action potentials) was increased in KIKO, but not significantly increased in KIKO:PC-α1ACT mice. Number of cells in brackets;
Values are represented as mean±SEM. *p<0.05, **p<0.01; ***p<0.001.
During normal cerebellar postnatal development, climbing fibers (CF) progressively extend along the dendrites of PCs toward the pial surface (Kano and Hashimoto, 2009). We compared the CF extension along PC dendrites at p18 from Ctr, KIKO, and KIKO:PC-α1ACT mice with and without DOX treatment. KIKO mice exhibited an immature pattern of CF synaptic contacts concentrated on PCs soma and proximal dendrites while reaching only 65% of the ML at p18 compared with Ctr. In contrast, in KIKO:PC-α1ACT the disrupted contacts of CF synapses from the PC soma were corrected and CF height reached to 89% of the ML at p18. The pattern of CF contacts in Dox−1/+1MKIKO:PC-α1ACT mice was similar to Dox−1/+1MKIKO and KIKO mice and significantly delayed compared with that of the Dox−1/+1MCtr and KIKO:PC-α1ACT mice at 1 month (Figure S5C and S5D). These data illustrate that the morphology and afferent innervation of KIKO:PC-α1ACT PCs resembled the patterns seen in Ctr. Interestingly L1cam and Ncam, regulated by α1ACT, are related to GAP-43 function (Korshunova et al., 2007), which was shown to regulate the morphology and trophic state of CFs (Grasselli et al., 2011). These results demonstrate that α1ACT plays an important role in establishing normal dendritic tree morphology and synapse development in PCs in vivo.
α1ACT matures CF-PC connections
The development of PC functional properties matches the time course of cerebellar structural and morphological maturation (Kuhn et al., 2012; Leto et al., 2016). To identify correlates in PC physiology of the observed behavioral and morphological abnormalities, we investigated the PC connectivity and intrinsic properties by patch-clamp electrophysiology, comparing KIKO mice to Ctr and KIKO:PC-α1ACT. We measured both PF and CF connectivity in KIKO mice. PF excitatory postsynaptic current (PF-EPSC) amplitude was significantly reduced relative to control both in KIKO and KIKO:PC-α1ACT at p18–22. The presynaptic release probability at PF-PC synapses (EPSC paired-pulse ratio) was comparable in the three groups (Figure S5E and S5F). This suggests no major role of PF transmission in the improved motor behavior in KIKO mice mediated by α1ACT expression.
α1ACT had a clearer effect on CF-PC transmission. We observed an 85% increase of CF-EPSC amplitudes in KIKO mice compared to Ctr (Figure 5D). This abnormal increase was abolished in KIKO:PC-α1ACT mice, whose CF-EPSCs were no different from controls. The abnormal increase of CF-EPSCs in KIKO mice is correlated with the reduced size of the PC dendritic arbor and a shift of CF innervation territory towards the soma. The normalization of CF-EPSC amplitudes and improved CF and PC morphology mediated by expression of α1ACT in PCs provides strong evidence for the role of α1ACT in this critical developmental step and implicates this developmental change in maturation of motor function.
Lastly, we assessed PC intrinsic excitability, a property altered in other models of SCA, by measuring the number of action potentials evoked by injections of a depolarizing current of increasingly larger amplitudes. We found that PC intrinsic excitability was increased in KIKO mice compared to Ctr and this effect was partially restored in KIKO:PC-α1ACT (Figure 5E). Overall these findings indicate that α1ACT plays a critical role in PC maturation, CF retraction and PC excitability, and implicate these changes in the motor abnormalities seen in KIKO mice.
Calcium channel genes are bicistronic
Based on the high level of conservation of the CACNA1A gene among vertebates, we hypothesized that α1ACT expression would be conserved among vertebrates. A comparable α1ACT polypeptide was was co-expressed with α1A in human, mouse, and zebrafish after transiently transfecting the α1A-expressing cDNA constructs from these species into HEK293 cells. In RNA folding models, stem-loop structures similar to what we reported in human α1A mRNA are present in the α1A transcripts of these species (Figure S6A and S6B). This finding indicates that the mechanism of bicistronic expression of one VGCC gene is conserved among vertebrate orthologs.
Additionally, a few reports have suggested that other gene members of the VGCC family express both calcium channels and separate C-terminal proteins. However, the mechanism of expression and separate function of these proteins remains to be characterized (Bannister et al., 2013; Gomez-Ospina et al., 2006; Lu et al., 2015). Further insights into these C-terminal proteins may contribute to understanding the complex phenotypes seen in spontaneous mutations in members of the VGCC family. To explore whether a bicistronic expression mechanism is common to VGCC genes in brain, we chose VGCC genes from distinct functional classes expressed in CNS (CACNA1C, Cav1.2, NM_199460.3 and CACNA1H, Cav3.2, NM_021098.2) (Figure S6C). Using C-terminal specific anti-sera to α1C (Cav1.2) and α1H (Cav3.2) subunits, we demonstrated that in both cases a 65–70 kDa, endogenous second gene product, is present and enriched in nuclear extracts of mouse brain tissue, but not control tissues (Figure 6A).
Figure 6. The C-terminal secondary proteins generated by CACNA1C, CACNA1H genes, See also Figure S6.

(A) Western blot detected C-terminal protein of α1C (left) or α1H (right) in fraction of forebrain and cerebellum using anti-sera to C-terminal peptide. Liver and spleen are the negative control for α1C (left) or α1H (right).
(B) Left. Cellular localization of C-terminally Flag-tagged α1C or α1H stained using antibodies against the C-terminal Flag-tag and the N-terminal in cells overexpressing CACNA1C or CACNA1H genes. The scale bar represents 20μm. Right. Quantitation of the ratio of nuclear and cytoplasmic density of N-terminal or C-terminal antibody signal of α1C, α1H of left panel (n=50 cells, *p<0.05).
(C and D) Western blot analysis of C-terminally Flag-tagged α1CCT, α1CCTstop, α1HCT and α1HCTstop showing a secondary protein product detected by Flag antibody, that is produced independently of the full-length channel.
(E and F) Schematic representation of the constructs CACNA1C (NM_199460.3) (E) and CACNA1H (NM_021098.2) (F) 5’ deletion truncations and stop mutations.
(G and H) Left. In vivo, Western blot analysis of HEK293 cells transiently transfected with truncated C-terminally Flag-tagged α1C(G), and α1H (H) to estimate the N-terminal of α1CCT and α1HCT. Right. In vitro, in vitro transcription and translation of truncated C-terminally Flag-tagged α1C and α1H, both of which generate the C-terminal proteins α1CCT (G) and α1HCT(H).
(I) Western blotting analysis of lysates from promoterless constructs of full-length CACNA1C, and CACNA1H genes.
To investigate the origin of these VGCC C-terminal proteins, we employed recombinant expression systems from cDNAs expressing the human α1C and α1H in transfected cells. Using anti-sera specific to N-terminal and C-terminal epitopes, immunofluorescent labeling of Hela cells showed predominant membrane localization for N-terminal antibody and predominant nuclear localization for C-terminal antibody for both α1C and α1H subunits (Figure 6B). Western blot analysis of lysates HEK293 cells transfected with 3xFlag-tagged human α1C or α1H cDNA showed distinct 65–70 kDa polypeptides that were enriched in nuclear fractions in addition to the full-length 250kDa calcium channel polypeptide (Figure 6C and 6D). We explored the independent origin of the smaller VGCC proteins by introducing a series of stop codons within the coding regions of the primary α1C and α1H subunits (Figures 6E and 6F). The expression of the full-length channel proteins was abolished, while the C-terminal proteins persisted until stop codons were inserted downstream of the internal start sites at 1869aa and 1953aa (Figure 6G and 6H). To estimate the N-terminus of those 65–70 kDa polypeptides, we generated a series of 5’ truncation cDNA constructs before or around 1869aa and 1953aa (Figures 6E and 6F). Methionine (M)1731aa (5469nt) and M1924aa (6045nt) were close to the N-terminus of the C-terminal α1C and α1H polypeptides respectively. Using expression constructs starting at ~1500bps upstream of M1731aa and M1924aa with or without stop codons, we found that the 65–70 kDa C-terminal proteins (α1CCT and α1HCT) were expressed both in vivo and in cell-free protein translation systems, despite presence of upstream stop codons, similar to those in CACNA1A (Figures S6G and S6H). However, expression of both the full-length and C-terminal VGCC proteins in transfected cells was abolished when the CMV-derived eukaryotic promoters were removed from the expression vectors prior to expression in HEK293 cells, excluding the possibility that these C-terminal proteins arise from cryptic promoters (Figure 6I). These findings suggest that bicistronic expression of proteins, together with the ion channel proteins, is a property common to multiple members of the VGCC gene family. Although the nature of the mechanism responsible for mediating expression of the second gene product remains to be elucidated, RNA folding models revealed that both CACNA1C and CACNA1H mRNA appeared to possess stem-loop structures in the region upstream of predicted start sites of these C terminal peptides (Figures S6F and S6G).
Discussion
The mammalian cerebellum is not fully developed at birth, and its structure and cerebellar-dependent behaviors continue to mature during early postnatal development (Goldowitz and Hamre, 1998; McKay and Turner, 2005). A critical window for rapid cerebellar maturation and development of motor function is p14 to 1-month in mice (Goldowitz and Hamre, 1998), while in humans this window exceeds two years (Butts et al., 2014). In this study, we showed that expression of α1ACT in the cerebellar PCs is required for normal motor and cerebellar development (Figure 7). Our data suggest that expresson of the MPI isoform, ~10% of normal, leads to perinatal death and early onset severe motor impairment as seen in the CACNA1A deficient KIKO mouse. However, reintroduction of α1ACT into PCs of KIKO mice in the neonatal period improves mouse survival and early motor development. α1ACT regulates a large ensemble of developmentally-controlled genes, 15 of which are directly involved in the development of different cerebellar compartments. α1ACT orchestrates a network of pathways driving neurogenesis, synapse formation and cell adhesion to mediate morphological and functional maturation of PCs and their connections (Figure 7) (Damaj et al., 2015; Oh et al., 2003; Senturk et al., 2011; Wilson et al., 2010). In cerebellum, Gfra2 and Efnb2 genes regulate neuronal migration (Senturk et al., 2011; Widenfalk et al., 1997), Fgfr3 gene regulates the onset of oligodendrocyte terminal differentiation, Rbfox1 (A2BP1) is a splicing regulator required for normal neural development, while Ntn1 proteins play a role in the developing nervous system, by promoting both axonal outgrowth and axonal guidance in pathfinding, and in the cerebellar system repels both parallel fibres and migrating granule cells (Li et al., 2004; Liu et al., 2004; Wamsley et al., 2018). The L1cam and Nrcam genes are required for normal cerebellar formation (Nakamura et al., 2010; Sakurai et al., 2001). α1ACT appears to have a pronounced effect on establishing mature CF connections, and this process, together with the normalization of the CF-PC EPSCs, is most closely associated with the resolution of the dystonic spells and ataxia in a CACNA1A loss-of-function mutant. Gene regulation plays an important role in cell/tissue-specific expression (Lee and Young, 2013). Several studies have elucidated detailed mechanisms in specific tissues wherein transcription factors functionally act by dynamic regulation (Bonev et al., 2017; Cacchiarelli et al., 2015; Wyler et al., 2015). Our study demonstrates that α1ACT acts not only on individual genes, but on gene networks within PCs, as revealed by shifts in the cluster of GO terms in a time-dependent manner. Combined with clinical genetic data, this further implies that insights into temporal gene expression patterms may provide valuable information on disease diagnosis and therapeutic strategies.
Figure 7. Summary of α1ACT-regulated DEGs temporal expression profile from pc12 cells to mouse and human cerebellum.

Top. Graphical representation of α1ACT DEGs with associated GO terms. RNA-seq only DEGs across 4 time points (outer ring, pink), RNA-seq and ChIP-seq integrated DEGs across 3 early time points (middle ring, blue) and 4 time points (inner ring, yellow) were divided into four GO term categories: Synapse, Cell Size/Cycle, Neurogenesis, Other. DEG gene names are labeled according to persistence across time points and whether they were verified in mice. Bottom. Left. Temporal expression pattern of persistent DEGs in mice and humans. Four general patterns of expression: high early expression with exponential decrease, stable middle expression with decrease, initial increase with plateau and subsequent decrease later in life, and initial increase with stable middle plateau. Right. Graphic of portion of CACNA1A cDNA showing location of α1ACT coding region and the CACNA1A IRES. The locations of several large CACNA1A deletions associated with developmental delay are depicted below.
Myriad mutations in the CACNA1A gene give rise to a diverse set of neurological syndromes from newborn to old age, and it is as yet unclear in most cases whether different phenotypes can be attributed to their differential effects on the two distinct CACNA1A gene products (Du and Gomez, 2018a), α1A and α1ACT. Some of these effects could be attributable to our finding that decreased α1ACT expression disrupts the gene expression network within the cerebellum. Our mouse rescue studies suggest that, in the case of perinatally decreased CACNA1A gene expression, the reintroduction of α1ACT might be a potential early intervention therapy. On the other hand the progressive neurodegenerative disease, SCA6, is mediated by α1ACT bearing an expanded polyQ tract. In agreement with the time-dependent gene regulation and expression profile of α1ACT, its suppression in adult life does not result in neurological or morphological deterioration. Therefore, these findings suggest that suppression of both mutant and disease alleles of α1ACT by targeting the CACNA1A IRES in adulthood may be a viable therapeutic strategy. This study provides a framework useful to establish safety and tolerability for other ASO-based gene suppression strategies in the treatment of late-onset neurodegenerative diseases.
Complex clinical phenotypes, poorly explained by simple disturbances of channel gating, are typical of genetic defects of several members of the VGCC family (Berger and Bartsch, 2014; Craddock and Sklar, 2013; Fernandes-Rosa et al., 2017; Liao and Soong, 2010; Schmunk and Gargus, 2013; Splawski et al., 2004; Yalcin, 2012). The distinct role of the second gene product of CACNA1A, α1ACT, in perinatal motor development may help explain the complex phenotypes associated with diverse CACNA1A mutations. CACNA1C gene variants can cause Timothy syndrome or Brugada syndrome, and confer the risk of autism and cognitive decline (Li et al., 2015; Zanos et al., 2015). CACNA1H gene mutations are associated with hyperaldosteronism, familial type IV epilepsy, childhood absence VI, and autism spectrum disorder (Fernandes-Rosa et al., 2017; Schmunk and Gargus, 2013). Our initial studies suggest that these two voltage-gated calcium channel genes also operate by a bicistronic expression mechanism in which two proteins, an ion channel and a nuclear protein, are expressed within a single gene. Further characterization of this genetic arrangement may also help in interpreting some complicated genotype-phenotype relationships in genetic disease (Damaj et al., 2015; Liao and Soong, 2010; Schmunk and Gargus, 2013) Given the potential importance of these insights to developing therapies it will be critical to define the mechanism of bicistronic expression (Karginov et al., 2017).
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Christopher Gomez (cgomez@neurology.bsd.uchicago.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ethics Statement
All animal experiments were approved and carried out in accordance with the regulations and guidelines for the care and use of experimental animals at the Institutional Animal Care and Use Committee at the University of Chicago.
Cacna1a mouse experiments
All mice were housed in groups of up to five littermates in special pathogen-free facility at the University of Chicago with a 12 h light/dark cycle. Mouse health was checked daily. The null mice defective in voltage-dependent calcium channel α1A subunit (C57BL/6j: α1A−/−) were obtained from Dr. Shin. Equal numbers of male and female were assigned to each experimental group. The age of the mice varied from P20 to 18 months as indicated for each experiment. The Cacna1ahEx47/Ex47 knockin mice (C57BL/6j) that carry human CACNA1A gene exon 47 with 14 CAG repeat were generated by H. Zoghbi Lab and obtained from Jackson lab. We used the PCP2 promoter and Tet-off system to generate transgenic mice (FVB: PC-α1ACT) as described previously that have selective Purkinje cell expression of C terminus of human α1A subunit carrying normal range of polyQ under control of the tetracycline response element (Du et al., 2013). Cacna1ahEx47/Ex47 mice were crossed with Cacna1a+/− mice to obtain 4 mouse lines Cacna1a+/−, Cacna1ahEx47/Ex47, Cacna1ahEx47/+, and Cacna1ahEx47/− (KIKO) (all of C57BL/6j background). KIKO mice were bred with PC-α1ACT to obtain KIKO:PC-α1ACT mice (C57BL/6j; FVB) and to investigate the contribution of α1ACT to the motor development in KIKO mice. Doxycycline hydrochloride was supplied in food at desired concentrations to suppress tTA-activated α1ACT expression at the different time points of mouse life. All mice were genotyped before experimentation to validate their inclusion in different experiment groups.
Cell Lines
HEK293 and Hela cells were maintained in EMEM medium (ATCC, Manassas, VA, USA) containing 10% fetal bovine serum (FBS) (GIBCO, Grand Island, NY, USA) in a 37°C, 5% CO2 incubator. pc12 cells (rat pheochromocytoma) were cultured in F-12K medium (ATCC, Manassas) supplemented with 10% horse serum and 2.5% FBS in a 37 °C, 5% CO2 incubator. HEK293, Hela, and pc12 cells were authenticated by ATCC prior to purchase and use. Cells were transiently transfected with pcDNA3 vectors expressing full-length cDNA of human CACNA1C (Cav1.2, NM_199460.3) and CACNA1H (Cav3.2, NM_021098.2), a series of 5’ truncation cDNA constructs close to the N-terminus of the C-terminal α1C or α1H, and empty pcDNA3 vector when they were ~60% confluent using Lipofectamine 2000 (Thermo Fisher Scientific, Madison, WI, USA). For stable cell line establishment, pc12 cells were transfected with pcDNA3 vectors expressing C terminal 3xFlag tag with Q11 (α1ACT) and empty pcDNA3 vector when they were~60% confluent using TransIT 1 (Mirus, Pittsburgh, PA, USA) according to the manufacturer’s instructions. Transfected cells were selected with 150 μg/ml and 300μg/ml G418 (GIBCO) and then stable clones were obtained. Hela and HEK293 cells are female, and pc12 cells are male.
Human Tissues
This study involves de-identified autopsy tissues analyzed in accordance with Institutional Review Board of the University of Chicago regulations. The paraffin blocks of human cerebellar tissue was obtained from the Department of Pathology University of Chicago. Four age groups were used; Term female, Term male, Term female; 20y male, 23y male, 24y female; 50y male, 50y female, 53y female; 80y female, 81y male, 83y female. The cases from each age group were examined by IHC and RT-PCR.
METHOD DETAILS
RNA Isolation and Quantitative Real-Time PCR
Total cellular RNA, mouse cerebellum RNA and human formalin-fixed paraffin-embedded (FFPE) cerebellum RNA was isolated by RNeasy mini Kit, RNeasy lipid mini Kit, and RNeasy FFPE mini Kit (QIAGEN, Valencia, CA, USA) according to the manufacturer’s instructions. cDNA was generated with SuperScriptIII reverse transcriptase (Invitrogen, Grand Island, NY, USA). The Taqman gene expression assays and probes were obtained from Invitrogen. Real-time PCR was performed as triplicates on AB7900 HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) and repeated as separate experiment in triplicate (n=3). Each 10 μl reaction contained 0.5 μL Taqman gene expression Assay, 5ul Taqman gene expression Master Mix (Invitrogen, NY, USA), and was ran under the following conditions: 50°C 2 min; 95°C 10min; 95°C 15 s, 60°C 1min for 40 cycles, followed by melting curve. As a relative quantification, fold changes were measured using the ΔΔCt method, GAPDH as an internal control.
Western blot
Immunoblotting was performed on cell extracts and tissues as described previously (Kordasiewicz et al., 2006). Nuclear and cytoplasmic proteins were extracted from either cell lines or mouse cerebellum using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Waltham, MA, USA). 50μg of total protein or fraction were subjected to SDS–PAGE (6%, 8% or 12% Tris–Glycine gel, Invitrogen, Grand Island, NY, USA) and transferred to a PVDF membrane (Millipore, Billerica, MA, USA). The transblotted membrane was blocked with TBST containing 5% nonfat milk for 60 min and then incubated with the primary antibody at 4°C overnight. The membrane was then probed with HRP-conjugated secondary antibody for 1 hr at room temperature and washed with TBST three times. Finally, the immunoblots were detected using chemiluminescent substrate (Thermo Scientific Pierce Protein Biology Products, Rockford, IL, USA) and visualized by autoradiography. Quantification of western blot protein signal was done with ImageJ (NIH, Bethesda, VA, USA) with replication of three or more experiments.
Immunolabeling
Immunohistochemistry was performed as previously reported except as modified below (Du et al., 2013). Briefly, paraffin-embedded sections of perfused brains were de-waxed and rehydrated, then steamed for 20 min in antigen retrieval solution (Reveal; Biocare Medical, Walnut Creek, CA, USA). Sections were blocked and exposed to primary antibody for 12 hr at 4°C. After washing, fluorescent secondary antibody in PBS-T (phosphate buffered saline and 0.05% Tween-20) was added for 1 hr at room temperature. Confocal fluorescence microscopy was carried out under a TCS laser scanning microscope (Leica, Buffalo Grove, IL, USA). Optical sections of 0.4 mm were scanned at the z-axis. Image J was used to quantify the percentage of specific expression of antibody in each sample.
Height and density of Purkinje dendrite trees were calculated as previously described (Miyazaki and Watanabe, 2011). Purkinje cells selected for the measurements spanned the molecular layer, were well-stained, and were not obscured by adjacent cells. The dendritic trees of the captured Purkinje cell images and the area enclosed were outlined and measured using NIH Image J software. The cell body was excluded in the height measurements. The scale was consistently measured from the point where the cell body narrows into the primary dendrite. Height of the Purkinje cells (perpendicular height) was measured from the cell body in an axis perpendicular to the edge of the molecular layer. Following measurement of height, the identical Purkinje cell was used for density calculation, in which the same fluorescence pixel was used for all the Purkinje cells identified. For both measurements, 50–100 Purkinje cells were analyzed per animal to calculate the mean. Each experiment involved more than three mice (n>3).
Golgi Staining and Sholl analysis
We used FD Rapid GolgiStaining Kit, a commercial Golgi-Cox staining system Cerebellum (n = 6 mice/genotype per age) immersed in the Golgi solution according to the manufacturer’s instructions. Cerebellar samples were cut with a vibratome (Campden Instruments, Loughborough, UK; MA752 motorized advance vibroslice) in 200 μm sagittal sections.
A Z-series of images from 20 Purkinje Cells (each mice) were collected at 20X magnification with Zeiss Axioskop microscope (Zeiss Axiocam digital color camera). At least 10 Purkinje Cells per animal were traced. Only Purkinje Cells showing a completely impregnated dendritic tree and that were relatively isolated from neighboring cells were selected for the analysis. Drawings were scaled and analyzed with NIH software ImageJ running the Sholl Analysis Plugin v1.0 (http://biology.ucsd.edu/labs/ghosh/software/index.html). The Sholl analysis was performed separately for the basal and apical dendrites with concentric circles of 10 μm. The experimenter was blind to the genotype of the mice involved in the assay.
Rotarod test
Using an accelerating computer-controlled Rotamex 5 (Columbus Instruments, Columbus, OH, USA) the rotarod test was performed by placing mice on rotating drums (7 cm in diameter) and measuring retention time on the rod. The speed of the rotarod was set to accelerate from 4 to 40 revolutions per minute rpm over 300 s, and recorded time to fall. Mice received five consecutive trials per session, one session per day (30 s between trials). Each experiment involved more than three mice (n≥3) and the tester was blind to the genotype.
Open Field
The open-field apparatus was used to test multiple processes including anxiety/hyperactivity and habituation to a novel environment. Open field chambers were 40 × 40 cm (Med Associates, St. Albans, VT, USA) with lighting at 21 lux. The Chambers were divided into a 10 × 10 cm grid. Mice were brought to the experimental room 5–20 min before testing. The total activity (as path length in cm) was recorded. Each experiment involved more than three mice (n≥3) and the tester was blind to the genotype.
Treadmill performance
For the treadmill performance, a plexiglass enclosure was placed over the treadmill to force the mice to remain on the track during the trials (enclosed treadmill space, 5 × 20 cm). Mice were provided three 20 s trials per day for 3 days at incrementing speeds (10, 15, and 20; 15, 15, and 20; 15, 20, and 20 cm/s, days 1–3, respectively). Their performance was assessed on the final day at 15 and 20 cm/s by recording the amount of time the animals remained in the forward two-thirds of the track versus falling back into the back third. Each experiment involved more than three mice (n≥6) and the tester was blind to the genotype.
Electrophysiology
Slice preparation:
Experiments were performed on KIKO, KIKO:PC-α1ACT, and Ctr mice age P16–18. The animals were anesthetized with halothane and rapidly decapitated. The cerebellar vermis was then removed and cooled to 4°C in artificial CSF (ASCF) containing the following (in mM): 124 NaCl, 5 KCl, 1.25 Na2HPO4, 2 CaCl2, 26 NaHCO3, and 10 D-glucose, bubbled with 95% O2 and 5% CO2. Parasagittal slices of the cerebellar vermis (220 μm) were prepared with a vibratome (VT-1000S; Leica Microsystems). Slices were then incubated for at least 1 hr at room temperature (RT) in oxygenated ACSF.
Somatic whole-cell patch clamp recordings: Slices were held at RT and continuously perfused with ACSF throughout the recordings. Patch-clamp recordings from Purkinje cells were performed using an EPC-10 amplifier (HEKA Electronics, Lambrecht-Pfalz, Germany). Currents were filtered at 3 kHz, digitized at 5–10 kHz, and acquired using Patchmaster software. To record PF EPSCs, patch pipettes (2–5 MΩ) were filled with a solution containing the following (in mM): 120 K-gluconate, 9 KCl, 10 KOH, 3.48 MgCl2, 10 HEPES, 4 NaCl, 4 Na2ATP, 0.4 Na3GTP, and 17.5 Sucrose (pH 7.25–7.35). While cells were held in voltage-clamp mode at −80 mV, PF-EPSCs were elicited by a stimulating electrode placed in the upper third of the molecular layer. To record CF EPSCs, patch pipettes (2–5 MΩ) were filled with a solution containing the following (in mM): 100 CsMeSO4, 50 CsCl, 1 MgCl2, 0.2 EGTA, 10 HEPES, 2 Na2ATP, 0.3 Na3GTP, and 4 mM QX-314 (pH 7.25–7.35). Cells were held in voltage-clamp mode at multiple potentials. CF-EPSCs were elicited by a stimulating electrode placed in the granule cell layer. The presence of multiple CF innervations was ascertained by gradually increasing the stimulus intensity and looking for discrete changes, or steps, in EPSC amplitude, as has been described previously (Hashimoto et al., 2011). Picrotoxin (200 μM; Sigma-Aldrich, St. Louis, MO) was added to the ACSF throughout all recordings. Each experiment involved more than three mice (n≥3).
In vivo Chromatin Immunoprecipitation
For Chromatin Immunoprecipitation (ChIP) using anti-myc antibody, cerebellum were taken from P30 mice (n=3) from each group. Brain samples were cut into small pieces and then dounced in 1% formaldehyde (wt/vol) PBS buffer. Samples were kept at 25 °C for 15 min, washed twice with co ld PBS, then processed by following the protocol as previously described (Du et al 2013). DNAs obtained from ChIP were used for downstream quantitative real time PCR (ChIP-qPCR) to confirm α1ACT target genes. Template DNAs included input (10% of starting chromatin as positive control), α1ACT-ChIP, and immunoglobulin G (IgG)-ChIP as negative control. The signals obtained from the ChIP were divided by signals obtained from an input sample. Standard errors were indicated for each gene. All reported genes had a minimum significance of FDR p < 0.05.
Laser-capture microdissection (LCM) of PCs and mRNA isolation
Frozen cerebellar sections (10 um thick) were cut on a Cryostat NX50 (Leica, Milton Keynes, UK). Sections were attached to RNase free PEN-membrane slides (Leica, Milton Keynes, UK) and were stained with fast HE staining. 10 PCs were isolated from each slice, from 3 animals, for each group, and collected in the tube using Leica LMD 6500 for subsequent RNA isolation. Total RNAs were extracted using Arcturus Picopure RNA isolation Kit (Thermo fisher, VA, USA). All samples passed Bioanalyzer RNA QC analysis (Agilent, CA, USA) with a RNA integrity Number (RIN) of at least 7.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Statistical analyses were conducted using GraphPad Prism 7 software (GraphPad Software Inc., La Jolla, CA, USA) and RStudio (Boston, MA) For single comparisons, Student’s t-test was used to establish statistical significance. For multiple sample comparisons, One-Way ANOVA was performed, followed by Student-Newman-Keuls Method post hoc tests. ANOVA on Ranks was used for morphological analysis of Golgi staining. For the DOX treatment experiments, statistical analysis was performed using a two-way ANOVA, followed by Bonferroni’s post hoc test. Data were expressed as the mean ± SEM, and written with the identification of n under appropriate figure legends, with *p<0.05, **p<0.01; ***p<0.001.
DATA AND SOFTWARE AVAILABILITY
Data
The sequencing data were deposited in the Gene Expression Omnibus (GEO, NCBI). GEO accession numbers: GSE126089. Data was submitted to Mendeley Data with the link (doi:10.17632/m7wk7d9t49.1) to access.
Software for Analysis of RNA-seq and ChIP-seq
RNA-seq and ChIP-seq analysis were done with FastQC v0.11.2, RSeQC, BWA v0.7.5a, Picard tools v1.117, Tophat v2.0.10, edgeR, samtool v0.1.19, SPP, Bioconductor package ChIPseeker, and HOMER wrapper program (findMotifsGenome.pl); R with Bioconductor package cluster Profiler, QIAGENs Ingenuity® Pathway Analysis, ReactomeFIViz in Cytoscape were used for ChIP-seq and RNA-seq integration and analysis; NIH software ImageJ running the Sholl Analysis Plugin v1.0(http://biology.ucsd.edu/labs/ghosh/software/index.html) was used for Sholl Analysis; Electrophysiology experiments were completed using the Patchmaster software.; Statistical analysis GraphPad Prism 7 software and RStudio were used.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R01NS082788, R01NS094665, R21NS094872–01(C.M.G) and University of Chicago Big Vision Grant (C.M.G). We thank Dr. Peter Pytel at the Department of Pathology for preparing human cerebellum tissue. We thank Drs. Sally Rowland, Aaron Gitler, Xiaoxi Zhuang, Roy Sillitoe, Kathy Millen, Sara London, and Brian Popko for comments on the manuscript, Dr. Pieter Faber at University of Chicago Functional Genomics Facility for ChIP and RNA-Seq Service, and Christine Labno at the University of Chicago Microscopy Core Facility for the imaging service.
Footnotes
Author Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister JP, Leo MD, Narayanan D, Jangsangthong W, Nair A, Evanson KW, Pachuau J, Gabrick KS, Boop FA, and Jaggar JH (2013). The voltage-dependent L-type Ca2+ (CaV1.2) channel C-terminus fragment is a bi-modal vasodilator. J Physiol 591, 2987–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Joseph Z, Gitter A, and Simon I (2012). Studying and modelling dynamic biological processes using time-series gene expression data. Nat Rev Genet 13, 552–564. [DOI] [PubMed] [Google Scholar]
- Bavassano C, Eigentler A, Stanika R, Obermair GJ, Boesch S, Dechant G, and Nat R (2017). Bicistronic CACNA1A Gene Expression in Neurons Derived from Spinocerebellar Ataxia Type 6 Patient-Induced Pluripotent Stem Cells. Stem Cells Dev 26, 1612–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SM, and Bartsch D (2014). The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res 357, 463–476. [DOI] [PubMed] [Google Scholar]
- Blak AA, Naserke T, Saarimaki-Vire J, Peltopuro P, Giraldo-Velasquez M, Vogt Weisenhorn DM, Prakash N, Sendtner M, Partanen J, and Wurst W (2007). Fgfr2 and Fgfr3 are not required for patterning and maintenance of the midbrain and anterior hindbrain. Dev Biol 303, 231–243. [DOI] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, et al. (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572 e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burazin TC, and Gundlach AL (1999). Localization of GDNF/neurturin receptor (c-ret, GFRalpha-1 and alpha-2) mRNAs in postnatal rat brain: differential regional and temporal expression in hippocampus, cortex and cerebellum. Brain Res Mol Brain Res 73, 151–171. [DOI] [PubMed] [Google Scholar]
- Butts T, Green MJ, and Wingate RJ (2014). Development of the cerebellum: simple steps to make a ‘little brain’. Development 141, 4031–4041. [DOI] [PubMed] [Google Scholar]
- Cacchiarelli D, Trapnell C, Ziller MJ, Soumillon M, Cesana M, Karnik R, Donaghey J, Smith ZD, Ratanasirintrawoot S, Zhang X, et al. (2015). Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell 162, 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LE, et al. (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. (2011). A copy number variation morbidity map of developmental delay. Nat Genet 43, 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, and Sklar P (2013). Genetics of bipolar disorder. Lancet 381, 1654–1662. [DOI] [PubMed] [Google Scholar]
- Damaj L, Lupien-Meilleur A, Lortie A, Riou E, Ospina LH, Gagnon L, Vanasse C, and Rossignol E (2015). CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet 23, 1505–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici C, Moreno-Bravo JA, Puiggros SR, Rappeneau Q, Rama N, Vieugue P, Bernet A, Mehlen P, and Chedotal A (2017). Floor-plate-derived netrin-1 is dispensable for commissural axon guidance. Nature 545, 350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, and Gomez CM (2018a). Spinocerebellar [corrected] Ataxia Type 6: Molecular Mechanisms and Calcium Channel Genetics. Adv Exp Med Biol 1049, 147–173. [DOI] [PubMed] [Google Scholar]
- Du X, and Gomez CM (2018b). Spinocerebellum Ataxia Type 6: Molecular Mechanisms and Calcium Channel Genetics. Adv Exp Med Biol 1049, 147–173. [DOI] [PubMed] [Google Scholar]
- Du X, Semler BL, and Gomez CM (2014). Revelations from a bicistronic calcium channel gene. Cell cycle 13, 875–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Wang J, Zhu H, Rinaldo L, Lamar KM, Palmenberg AC, Hansel C, and Gomez CM (2013). Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 154, 118–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Rosa FL, Boulkroun S, and Zennaro MC (2017). Somatic and inherited mutations in primary aldosteronism. J Mol Endocrinol 59, R47–R63. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Lutz CM, O’Sullivan TN, Shaughnessy JD Jr., Hawkes R, Frankel WN, Copeland NG, and Jenkins NA (1996). Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell 87, 607–617. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, and Hamre K (1998). The cells and molecules that make a cerebellum. Trends Neurosci 21, 375–382. [DOI] [PubMed] [Google Scholar]
- Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, and Dolmetsch R (2006). The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell 127, 591–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, and Bujard H (1992). Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proceedings of the National Academy of Sciences of the United States of America 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasselli G, Mandolesi G, Strata P, and Cesare P (2011). Impaired sprouting and axonal atrophy in cerebellar climbing fibres following in vivo silencing of the growth-associated protein GAP-43. PLoS One 6, e20791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, and Wilczynski SL (1982). Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience 7, 2185–2196. [DOI] [PubMed] [Google Scholar]
- Ingram M, Wozniak EAL, Duvick L, Yang R, Bergmann P, Carson R, O’Callaghan B, Zoghbi HY, Henzler C, and Orr HT (2016). Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways. Neuron 89, 1194–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Kawakami H, Kitajima H, Nishiyama A, Sasai Y, Inoue H, and Muguruma K (2016). Vulnerability of Purkinje Cells Generated from Spinocerebellar Ataxia Type 6 Patient-Derived iPSCs. Cell Rep 17, 1482–1490. [DOI] [PubMed] [Google Scholar]
- Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW, et al. (1999). Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proceedings of the National Academy of Sciences of the United States of America 96, 15245–15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, and Hashimoto K (2009). Synapse elimination in the central nervous system. Curr Opin Neurobiol 19, 154–161. [DOI] [PubMed] [Google Scholar]
- Karginov TA, Pastor DPH, Semler BL, and Gomez CM (2017). Mammalian Polycistronic mRNAs and Disease. Trends Genet 33, 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordasiewicz HB, Thompson RM, Clark HB, and Gomez CM (2006). C-termini of P/Q-type Ca2+ channel alpha1A subunits translocate to nuclei and promote polyglutamine-mediated toxicity. Human molecular genetics 15, 1587–1599. [DOI] [PubMed] [Google Scholar]
- Korshunova I, Novitskaya V, Kiryushko D, Pedersen N, Kolkova K, Kropotova E, Mosevitsky M, Rayko M, Morrow JS, Ginzburg I, et al. (2007). GAP-43 regulates NCAM-180-mediated neurite outgrowth. J Neurochem 100, 1599–1612. [DOI] [PubMed] [Google Scholar]
- Kuhn A, Kumar A, Beilina A, Dillman A, Cookson MR, and Singleton AB (2012). Cell population-specific expression analysis of human cerebellum. BMC Genomics 13, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrum RW, Rajakulendran S, Graves TD, Eunson LH, Bevan R, Sweeney MG, Hammans SR, Tubridy N, Britton T, Carr LJ, et al. (2009). Large scale calcium channel gene rearrangements in episodic ataxia and hemiplegic migraine: implications for diagnostic testing. J Med Genet 46, 786–791. [DOI] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, and Young RA (2013). Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto K, Arancillo M, Becker EB, Buffo A, Chiang C, Ding B, Dobyns WB, Dusart I, Haldipur P, Hatten ME, et al. (2016). Consensus Paper: Cerebellar Development. Cerebellum 15, 789–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao L, You Y, Lu T, Jia M, Yu H, Ruan Y, Yue W, Liu J, Lu L, et al. (2015). Schizophrenia Related Variants in CACNA1C also Confer Risk of Autism. PLoS One 10, e0133247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Lee J, Vikis HG, Lee SH, Liu G, Aurandt J, Shen TL, Fearon ER, Guan JL, Han M, et al. (2004). Activation of FAK and Src are receptor-proximal events required for netrin signaling. Nature neuroscience 7, 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao P, and Soong TW (2010). CaV1.2 channelopathies: from arrhythmias to autism, bipolar disorder, and immunodeficiency. Pflugers Arch 460, 353–359. [DOI] [PubMed] [Google Scholar]
- Liu G, Beggs H, Jurgensen C, Park HT, Tang H, Gorski J, Jones KR, Reichardt LF, Wu J, and Rao Y (2004). Netrin requires focal adhesion kinase and Src family kinases for axon outgrowth and attraction. Nature neuroscience 7, 1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Sirish P, Zhang Z, Woltz RL, Li N, Timofeyev V, Knowlton AA, Zhang XD, Yamoah EN, and Chiamvimonvat N (2015). Regulation of gene transcription by voltage-gated L-type calcium channel, Cav1.3. J Biol Chem 290, 4663–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BE, and Turner RW (2005). Physiological and morphological development of the rat cerebellar Purkinje cell. J Physiol 567, 829–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki Y, Du X, Muramatsu S, and Gomez CM (2016). An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. Sci Transl Med 8, 347ra394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Lee S, Haddox CL, Weaver EJ, and Lemmon VP (2010). Role of the cytoplasmic domain of the L1 cell adhesion molecule in brain development. J Comp Neurol 518, 1113–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson S, Helou K, Walentinsson A, Szpirer C, Nerman O, and Stahl F (2001). Rat-mouse and rat-human comparative maps based on gene homology and high-resolution zoo-FISH. Genomics 74, 287–298. [DOI] [PubMed] [Google Scholar]
- Oh LY, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, and Bansal R (2003). Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci 23, 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor PDH, Du X, Fazal S, Davies AN, and Gomez CM (2018). Targeting the CACNA1A IRES as a Treatment for Spinocerebellar Ataxia Type 6. Cerebellum 17, 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson HL, Shakkottai VG, Clark HB, and Orr HT (2017). Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat Rev Neurosci 18, 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riant F, Lescoat C, Vahedi K, Kaphan E, Toutain A, Soisson T, Wiener-Vacher SR, and Tournier-Lasserve E (2010). Identification of CACNA1A large deletions in four patients with episodic ataxia. Neurogenetics 11, 101–106. [DOI] [PubMed] [Google Scholar]
- Riant F, Mourtada R, Saugier-Veber P, and Tournier-Lasserve E (2008). Large CACNA1A deletion in a family with episodic ataxia type 2. Arch Neurol 65, 817–820. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Lustig M, Babiarz J, Furley AJ, Tait S, Brophy PJ, Brown SA, Brown LY, Mason CA, and Grumet M (2001). Overlapping functions of the cell adhesion molecules Nr-CAM and L1 in cerebellar granule cell development. J Cell Biol 154, 1259–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi J, Bertaso F, Mausset-Bonnefont AL, Metz A, Lemmers C, Ango F, Fagni L, Lory P, and Mezghrani A (2014). RNAi silencing of P/Q-type calcium channels in Purkinje neurons of adult mouse leads to episodic ataxia type 2. Neurobiol Dis 68, 47–56. [DOI] [PubMed] [Google Scholar]
- Schmunk G, and Gargus JJ (2013). Channelopathy pathogenesis in autism spectrum disorders. Front Genet 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senturk A, Pfennig S, Weiss A, Burk K, and Acker-Palmer A (2011). Ephrin Bs are essential components of the Reelin pathway to regulate neuronal migration. Nature 472, 356–360. [DOI] [PubMed] [Google Scholar]
- Sergaki MC, and Ibanez CF (2017). GFRalpha1 Regulates Purkinje Cell Migration by Counteracting NCAM Function. Cell Rep 18, 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra HG, Duvick L, Zu T, Carlson K, Stevens S, Jorgensen N, Lysholm A, Burright E, Zoghbi HY, Clark HB, et al. (2006). RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 127, 697–708. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. [DOI] [PubMed] [Google Scholar]
- Wamsley B, Jaglin XH, Favuzzi E, Quattrocolo G, Nigro MJ, Yusuf N, Khodadadi-Jamayran A, Rudy B, and Fishell G (2018). Rbfox1 Mediates Cell-type-Specific Splicing in Cortical Interneurons. Neuron 100, 846–859 e847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, Barrett CF, Miyazaki T, Ishiguro T, Ishikawa K, Hu Y, Unno T, Sun Y, Kasai S, Watanabe M, et al. (2008). Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proceedings of the National Academy of Sciences of the United States of America 105, 11987–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welham A, Barth B, Moss J, Penhallow J, Sheth K, Wilde L, Wynn S, and Oliver C (2015). Behavioral characteristics associated with 19p13.2 microdeletions. Am J Med Genet A 167A, 2334–2343. [DOI] [PubMed] [Google Scholar]
- Widenfalk J, Nosrat C, Tomac A, Westphal H, Hoffer B, and Olson L (1997). Neurturin and glial cell line-derived neurotrophic factor receptor-beta (GDNFR-beta), novel proteins related to GDNF and GDNFR-alpha with specific cellular patterns of expression suggesting roles in the developing and adult nervous system and in peripheral organs. J Neurosci 17, 8506–8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PM, Fryer RH, Fang Y, and Hatten ME (2010). Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J Neurosci 30, 8529–8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyler SC, Donovan LJ, Yeager M, and Deneris E (2015). Pet-1 Controls Tetrahydrobiopterin Pathway and Slc22a3 Transporter Genes in Serotonin Neurons. ACS Chem Neurosci 6, 1198–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin O (2012). Genes and molecular mechanisms involved in the epileptogenesis of idiopathic absence epilepsies. Seizure 21, 79–86. [DOI] [PubMed] [Google Scholar]
- Zanos P, Bhat S, Terrillion CE, Smith RJ, Tonelli LH, and Gould TD (2015). Sex-dependent modulation of age-related cognitive decline by the L-type calcium channel gene Cacna1c (Cav 1.2). Eur J Neurosci 42, 2499–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga R, Gonzalez D, Valenzuela C, Brown N, and Zuniga L (2016). Expression and cellular localization of HCN channels in rat cerebellar granule neurons. Biochem Biophys Res Commun 478, 1429–1435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data
The sequencing data were deposited in the Gene Expression Omnibus (GEO, NCBI). GEO accession numbers: GSE126089. Data was submitted to Mendeley Data with the link (doi:10.17632/m7wk7d9t49.1) to access.
Software for Analysis of RNA-seq and ChIP-seq
RNA-seq and ChIP-seq analysis were done with FastQC v0.11.2, RSeQC, BWA v0.7.5a, Picard tools v1.117, Tophat v2.0.10, edgeR, samtool v0.1.19, SPP, Bioconductor package ChIPseeker, and HOMER wrapper program (findMotifsGenome.pl); R with Bioconductor package cluster Profiler, QIAGENs Ingenuity® Pathway Analysis, ReactomeFIViz in Cytoscape were used for ChIP-seq and RNA-seq integration and analysis; NIH software ImageJ running the Sholl Analysis Plugin v1.0(http://biology.ucsd.edu/labs/ghosh/software/index.html) was used for Sholl Analysis; Electrophysiology experiments were completed using the Patchmaster software.; Statistical analysis GraphPad Prism 7 software and RStudio were used.