

Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disease in which heterotopic bone forms in muscle and soft tissue, leading to joint dysfunction and significant disability. FOP is progressive and many patients are wheelchair‐bound by the 3rd decade of life. FOP is caused by an activating mutation in the ACVR1 gene, which encodes the activin A Type 1 receptor. Aberrant signalling through this receptor leads to abnormal activation of the pSMAD 1/5/8 pathway and triggers the formation of bone outside of the skeleton. There is no curative therapy for FOP; however, exciting advances in novel therapies have developed recently. Here, we review the clinical and translational pharmacology of three drugs that are currently in clinical trials (palovarotene, REGN 2477 and rapamycin) as well as other emerging treatment strategies for FOP.
Keywords: children, drug development, immunology, immunosuppression, paediatrics
Introduction
Fibrodysplasia ossificans progressiva (FOP) is a rare genetic condition characterized by massive heterotopic ossification of the soft connective tissues, leading to progressive joint immobilization and disability 1. FOP affects approximately one in 1.3–2 million people worldwide and is typically diagnosed within the 1st decade of life 1, 2. It is a progressive disease and is characterized by episodic flare‐ups in which patients develop painful, localized soft tissue inflammation, either spontaneously or in response to various triggers including trauma, injury, immunization or illness. These flare‐ups lead to the progressive formation of heterotopic endochondral bone in the muscle, joints, tendons, and ligaments resulting in severe joint restrictions and loss of mobility over time. The majority of FOP patients are wheelchair‐bound by the 3rd decade of life 3.
FOP is most commonly caused by a highly recurrent autosomal dominant gain‐of‐function missense mutation in the ACVR1/ALK2 gene (c.617G > A; p.R206H), which encodes the activin receptor type Ia 4. This mutation affects the GS activation domain of ACVR1, leading to aberrant signalling through the kinase receptor and over‐activation of the downstream SMAD 1/5/8 signalling pathway 4. Over‐activation of these intracellular signalling pathways occurs when canonical bone morphogenetic proteins (BMPs) bind to the mutated ACVR1 receptor 4. In addition, the R206H mutation appears to change the signalling specificity of the ACVR1 receptor to activin A, leading to phosphorylation of SMAD 1/5/8 instead of SMAD 2/3 in response to activin A binding 5, 6. These signalling changes are thought to trigger the formation of excessive heterotopic endochondral bone 7. Multiple cells types have been implicated in the formation of heterotopic ossification in FOP, including Mx+ cells in the muscle interstitium and scleraxis (Scl+) cells in tendons and ligaments, but it is not yet clear whether these cells are also responsible for initiating the process of heterotopic ossification 8.
Currently, there are no curative treatments for FOP. Standard‐of‐care therapy focuses on supportive measures including the judicious use of glucocorticoids and nonsteroidal anti‐inflammatory drugs within 24–48 h of a flare‐up to decrease the excessive inflammation present in early FOP lesions 1. However, these therapies are not particularly effective in preventing heterotopic ossification, and they do not mitigate the progressive nature of this disease. Furthermore, there are notable side effects with long‐term corticosteroid use, making this a less favourable drug for chronic treatment. Mast‐cell inhibitors and leucotriene inhibitors are also often used on a chronic basis to empirically address the inflammatory aspect of early FOP lesions 9, 10. Bisphosphonates are occasionally used for refractory flares that do not respond to glucocorticoids; however, concrete clinical data for these treatments are sparse 10. Avoidance of trauma and prevention of injury remain the mainstays of therapy. Surgical resection, which can be used in some nongenetic causes of heterotopic ossification (e.g. trauma, burns, spinal cord injuries and post‐surgical), is contraindicated in patients with FOP as it can trigger a cascade of unrelenting, excessive bone formation at the both the surgical site and at distant locations 1, 11, 12, 13.
The significant disabilities caused by FOP and the lack of suitable pharmacological treatment options make this a critical area for drug development. ACVR1 is widely expressed in humans, and both BMP and activin A signalling play critical roles in multiple tissues types; therefore, there are inherent challenges in designing drugs that target these pathways. In response to this need, several promising new drugs have emerged over the past decade with the goal of decreasing the formation and progression of heterotopic bone formation in FOP patients. This has been greatly facilitated as new knowledge about FOP disease pathogenesis has emerged. In this review, we discuss the clinical and translational pharmacology of three drug compounds (palovarotene, REGN 2477 and rapamycin) that are currently in clinical trials to evaluate their efficacy in decreasing heterotopic endochondral bone formation in patients with FOP. We will also briefly discuss several other compounds that target important pathways involved in heterotopic ossification pathogenesis in FOP and may be moving towards clinical trials in the near future.
Palovarotene
Retinoid signalling plays a critical biological role in many tissues and organ systems 14 and is essential for chondrogenesis and the proper formation of the skeleton 15, 16, 17, 18, 19, 20. Retinoic acid signalling is mediated through nuclear hormone receptors known as retinoic acid receptors (RAR) which act as key regulators of gene transcription. There are three RARs (α, β and γ) that form heterodimers with retinoic X receptors and subsequently bind to retinoic acid response elements in the enhancer regions of target genes 15, 17. In the presence of their endogenous ligand, all‐trans retinoic acid (atRA), the RAR–retinoic X receptor heterodimer acts as a transcriptional activator. However, in its un‐bound form, this receptor complex acts as a transcriptional repressor 15. The relationship between retinoid signalling and chondrogenesis is antagonistic: several elegant studies have shown that skeletal precursor cells treated with atRA, and with subsequent activation of retinoid signalling, have repressed chondrogenesis and cartilage formation 21, 22. By allowing chondrogenesis to proceed only in the absence of retinoid signalling, this mechanism helps keep mesenchymal condensations in their prechondrogenic state and blocks differentiation into chondroblasts 23.
These advances in our understanding of the biological role of retinoid signalling in chondrogenesis, and the knowledge that both genetic and nongenetic causes of heterotopic ossification can proceed through a cartilage intermediate via endochondral ossification, have made this pathway an attractive pharmacological target for FOP and other forms of heterotopic ossification 18. While direct treatment with atRA and active retinoids does inhibit chondrogenesis, the effects of nonselective retinoids are too broad to be a feasible treatment for heterotopic ossification due to their direct action in many other tissues and organ systems. However, in the 1990s, several synthetic retinoid agonists specific for RARα, ‐β or ‐γ were developed for other indications. Since RARα is expressed in mesenchymal stem cell progenitors at the earliest stage of chondrogenic commitment, Pacifici et al. 21 tested the RARα agonist NRX195183 in a mouse model of heterotopic ossification in which mice received subcutaneous implantations of rhBMP‐2/Matrigel scaffolds. The mice treated with NRX195183 showed significantly less heterotopic ossification compared to the nontreated control mice. Furthermore, they had lower expression of chondrogenic genes, including Sox9, collagen XI and X, and Runx2 15, 21, 22. However, high doses of the agonist were required to achieve this result. The group then noted that while RARα was expressed only during early mesenchymal stem cell commitment, RARγ was expressed in both mesenchymal precursor cells and mature chondrocytes. Therefore, they hypothesized that the use of an RARγ agonist might be more effective at inhibiting heterotopic ossification as it would act on both early and late steps of chondrogenesis 22.
To evaluate whether a RARγ agonist would be effective in blocking heterotopic ossification in FOP patients, Pacifici et al. tested the highly‐specific RARγ agonist, R667 (subsequently renamed palovarotene), in a transgenic mouse model of FOP expressing a Cre‐inducible, constitutively active ALK2Q207D mutant protein 24. Mice induced with Cre recombinase showed overactive BMP signalling through the SMAD 1/5/8 pathway and subsequent heterotopic ossification formation 24 with local injection of adeno‐Cre and cardiotoxin into the anterior tibial muscles. The mice showed significant skeletal muscle injury, inflammation and, ultimately, heterotopic ossification at the site of injury within 2–4 weeks. In contrast, the mice who were treated with R667 had almost no heterotopic bone formation. Palovarotene was further tested in a conditional‐on knock‐in mouse model containing the classical ALK2R206H mutation and again demonstrated significant reduction in heterotopic bone formation as well as a decrease in mast cells. 25
At the time of these studies, palovarotene had already been evaluated in a Phase 2 trial for α‐1‐antitrypsin‐induced emphysema, and its safety profile had been well‐characterized. The drug is well‐tolerated in humans, with mucocutaneous side effects being the major concern 26. These safety data, in combination with the supportive preliminary results in mouse models of FOP, prompted interest in this drug as a potential treatment for FOP. Phase 2 clinical trials were initiated in 2014 by Clementia Pharmaceuticals to evaluate the safety and efficacy of palovarotene for treatment of FOP (Clinicaltrials.gov registration NCT02190747). Forty patients were enrolled in the trial, and the primary outcome was to assess whether the volume of heterotopic ossification formation decreased over time in treated versus untreated subjects. Top line results from this trial were publicly released in October 2016 and suggested that palovarotene decreased the percentage of FOP patients who develop heterotopic ossification, the time to flare‐up resolution, and patient‐reported pain 27, 28. These results have led to a Phase 3 clinical trial in adults and children (Clinicaltrials.gov registration NCT03312634), which is currently in progress.
Based on positive animal data in another rare bone disease (multiple osteochondroma, also known as hereditary multiple exostoses), palovarotene is also being evaluated in a Phase 2 trial as a treatment to block the formation of osteochondromas throughout the skeleton, which lead to skeletal deformities, pain and disability (Clinicaltrials.gov registration NCT03442985) 29.
Palovarotene, like other retinoic acid derivatives, is a known teratogen and causes limb malformations in the developing fetus. The use of retinoic acid receptor agonists in children could potentially affect the growth plates, hearing and vision 26. ACVR1‐R206H mice treated with palovarotene notably had preservation of their growth plates and long bone function, providing supportive data that the potential skeletal effects of palovarotene might be tolerable in FOP 25. Other potential risks include pancreatitis and hypertriglyceridaemia, as well as pseudotumor cerebri when co‐administered with tetracycline derivatives 30, 31. In addition, like other retinoids, palovarotene can have significant mucocutaneous side effects including dry skin, sensitivity to sunlight, and paronychia 26. All of these systems are being monitored closely during these trials.
Anti‐activin A antibody (REGN2477)
A significant breakthrough in our understanding of FOP and ACVR1R206H signalling occurred in 2015 when Hatsell et al. demonstrated that activin A, a noncanonical ligand for ACVR1, can bind to the mutant ACVR1R206H receptor and activate signalling through the SMAD 1/5/8 pathway 5. In contrast, when activin A binds to ACVR1WT receptors it does not activate SMAD 1/5/8 signalling but rather functions as an antagonist by preventing other BMP ligands from binding to the receptor. These studies were complemented by work from Hino et al. in which induced pluripotent stem cell‐derived mesenchymal stem cells from patients with FOP were co‐injected with activin A into nude mice and triggered heterotopic endochondral ossification 6. Activin A is a ligand in the BMP/TGFβ signalling pathway and typically binds to the type 1 receptors ACVR1B and ACVR1C and signals through the SMAD 2/3 pathway. The R206H mutation causes the ACVR1 receptor to misinterpret activin A and generate a signal as if a BMP ligand were present. Thus, the discovery that activin A could activate SMAD1/5/8 through ACVR1R206H but not ACVR1WT changed our understanding of the basic biology underlying ACVR1R206H signalling.
This neo‐function of the ACVR1‐R206H receptor was further tested in vivo in a physiologically relevant, genetically humanized conditional‐on knock‐in mouse model of FOP (Acvr1 [R206H]FlEx ) 5. When treated with tamoxifen, these mice undergo genetic recombination to express the ACVR1R206H protein. These mice developed progressive heterotopic ossification at anatomic locations similar to those seen in FOP, including the jaw, skull, paraspinal region, sternum, ribs and limbs. The heterotopic ossification occurred spontaneously in these mice but was also noted to be more prominent in areas where the mice were handled more frequently. When injected with activin A, the mutant mice developed more heterotopic ossification throughout the skeleton. Additionally, mutant mice treated with a blocking antibody directed against activin A did not develop heterotopic bone either spontaneously or in response to localized injury to muscles or tendons. The authors concluded that activin A is an obligatory secreted factor that is required for at least the initiation of heterotopic ossification in FOP, and that blocking activin A could prevent the formation of heterotopic bone.
As a result of these preclinical studies, an anti‐activin A antibody (REGN2477) is now in a Phase 2 trial to examine the safety, tolerability and efficacy on abnormal bone formation in adults with FOP, and is actively enrolling patients (Clinicaltrials.gov registry NCT03188666). Activin A is widely expressed in multiple organ systems, most notably in the gallbladder, ovary, endometrium, lung and placenta 32, and plays important roles in the development and maturation of the ovarian follicle and spermatogenesis and steroidogenesis in the testes 33, 34, 35. Furthermore, multiple studies have established that activin A is a critical regulator of inflammation and immunity both systemically and in specific tissues such as the lung 36, 37. There is also evidence that activin A plays a role in neuronal differentiation 38. The potential effects of activin A inhibition in these tissues, and the effects on inflammation and immune response, must be carefully monitored.
Rapamycin
Rapamycin is a commonly‐used immunosuppressant that exerts its biological effect by complexing with FKBP12 and inhibiting mechanistic target of rapamycin complex 1 (mTORC1). mTORC1 is a serine–threonine protein kinase that is responsible for activating multiple intracellular signalling pathways related to cell proliferation, including HIF1α. Interestingly, mTORC1 inhibition has been shown to decrease heterotopic ossification in FOP and other forms of heterotopic ossification, acting downstream of activin A signalling 39.
Hino et al. developed a high‐throughput screening system using FOP induced pluripotent stem cell‐derived mesenchymal stem cells and activin‐A‐induced chondrogenesis to screen a library of ~7000 small molecules 40. Using this method, they identified mTOR signalling as a critical pathway in chondrogenesis and heterotopic ossification formation in FOP. They then tested rapamycin in two in vivo systems, ACVR1R206H mice and an FOP‐iPSC‐based heterotopic ossification model in which ectopic bones derived from FOP patient‐derived cells are formed in mice. In both models, heterotopic ossification decreased after treatment with rapamycin. Agarwal et al. [41] also tested the efficacy of rapamycin in traumatic and genetic mouse models of heterotopic ossification. Rapamycin abrogated heterotopic ossification in both of these models 41.
Rapamycin use has been reported in two patients with FOP 42. One patient was a 3‐year‐old girl with the classic FOP‐ACVR1R206H mutation who was treated with rapamycin (1 mg m–2) for 3 months for persistent, unrelenting flare‐ups of her neck and back. No clinical changes in disease course were noted while on the drug, and she continued to develop new flares throughout treatment. Rapamycin trough levels had remained at the lower limit of normal for the duration of her treatment. The second patient was a 9‐year‐old boy with classic FOP (ACVR1R206H mutation) who also had undergone liver transplantation 4 years prior as a result of liver failure secondary to cytomegalovirus infection. He was placed on rapamycin for an episode of minor rejection, and continued on this for 18 years. However, he continued to experience flares and the formation of heterotopic bone despite daily use of rapamycin. These two cases did not show clear responses to pharmacological doses of rapamycin; however, it is difficult to assess the response to therapy in the absence of a well‐controlled clinical trial since the therapeutic threshold may not have been achieved in these case reports. A clinical trial evaluating the efficacy of rapamycin recently opened at Kyoto University Hospital and aims to evaluate the efficacy of rapamycin in 20 FOP patients in Japan (UMIN000028429) 43, 44.
Two key advantages of rapamycin are the well‐established therapeutic thresholds for immunosuppression in transplants, and the extensive experience with rapamycin in children. Whether the same therapeutic thresholds are needed to suppress bone formation in FOP is unknown. Potential concerns that have been reported with rapamycin use in patients with kidney transplants include proteinuria, dyslipidaemia, peripheral oedema, oral ulcerations, cytopenias and acne 45. In patients treated with rapamycin for vascular anomalies, notable reported side effects included blood/bone marrow and gastrointestinal toxicities 46. Rapamycin currently carries a black‐box warning for increased risk of infection, lymphoma and other malignancies due to immunosuppression, and is contraindicated in liver transplant patients 32. Additionally, male but not female mice that were treated with chronic rapamycin for 54 weeks developed diabetes mellitus that was partially reversible with cessation of the drug 47. The frequency of incident or worsening diabetes mellitus has not been reported. These potential safety issues will need careful monitoring.
Emerging therapeutic strategies for the treatment of FOP
There are several emerging treatment strategies for FOP that have been trialled as off‐label treatments or are currently in preclinical development.
Imatinib is a tyrosine kinase inhibitor that was approved in the 1990s for treatment of chronic myeloid leukaemia 48. The drug is well‐tolerated in both adults and children and has a minimal side effect profile but does require careful monitoring since it is an immunosuppressant 49. In addition to its primary role as an inhibitor of the BCR‐ABL fusion protein in chronic myeloid leukaemia, imatinib also interacts with multiple pathways that are important in FOP, including PDGFRα, c‐KIT, HIF1α and other MAP kinase signalling pathways 50. Additionally, imatinib has an immunomodulatory effect in lymphocytes, macrophages and mast cells, and downregulates critical pathways in these cells that are known to play a key role in FOP heterotopic ossification pathogenesis 50, 51. Imatinib is also a potent HIF1α inhibitor. Wang et al. [52] demonstrated that early inflammatory FOP lesions in both humans and mice are profoundly hypoxic. They used patient‐derived stem cells from exfoliated deciduous teeth (SHED cells) to show that HIF1α activity prolongs the duration and intensity of BMP signalling through downregulation of RABEP1/Rabaptin5, causing the mutant ACVR1 receptor to be retained within the endosomal compartment 52. Furthermore, they showed that inhibition of HIF1α through treatment with imatinib normalized BMP signalling in FOP SHED cells and decreased the formation of heterotopic ossification in their constitutively active mouse model of FOP (Acvr1 Q207D/+ ) 52.
These preclinical studies, in addition to the other off‐label experience with imatinib in paediatric inflammatory conditions, prompted off‐label use of this medication in seven paediatric FOP patients with severe FOP disease marked by relentless, continuous flares in the axial skeleton that were refractory to standard‐of‐care treatments 50. Six of the seven patients successfully took daily imatinib (range of 4–36 months duration), and all six patients reported a decrease in the intensity and frequency of their flare‐ups. Additionally, none of the children developed loss of movement at a new site while taking imatinib. Five out of seven children developed gastrointestinal irritation while on the drug, which improved when the medication was taken with food. No other side effects were reported, and patients were monitored closely with complete blood counts and liver function tests, neither of which demonstrated any abnormalities while taking imatinib. Since this was not a randomized controlled trial, and relied on retrospective anecdotal reports, we can not make any conclusive statements about the efficacy of imatinib, as these patients may have improved without the use of this drug. However, these results do provide some support for developing a well‐designed clinical trial to ascertain whether imatinib has efficacy in preventing heterotopic ossification in FOP 50.
Another key strategy has been to target the increased receptor kinase activity caused by the ACVR1R206H mutation. Saracatinib is a small molecule kinase inhibitor that has the potential to directly target ACVR1 kinase activity. Promising preliminary in vitro and in vivo results were presented at the International Fibrodysplasia Ossificans Progressiva Association Drug Development Forum in 2017 53. Additionally, several pharmaceutical companies are independently developing directed ACVR1 kinase inhibitors, with the goal of slowing or preventing heterotopic ossification. Candidate drugs include BCX9250, BCX9499 and BLU‐782 54, which have been selected based on the combination of kinase potency, selectivity and safety profile. Many other promising compounds continue to be discovered.
One additional strategy has been to use small molecule BMP Type 1 receptor inhibitors to block signalling through the ACVR1 receptor. These compounds are based on the chemical structure of dorsomorphin, which was discovered by employing a phenotypic screen in zebrafish embryos to identify which compounds could cause dorsalization 55. Several compounds have been developed 55, 56, but LDN‐212854 has shown the most specificity for ACVR1 57.
Finally, novel therapeutic methods such as small interfering RNAs (siRNA) have been tested in other diseases. This approach shows promise in FOP SHED cells transfected with allele‐specific siRNA; however, it remains to be seen whether sufficiently specific targeting of ACVR1 and a pharmacological response can be achieved in vivo 58, 59.
Conclusions
Over the past several years, there have been rapid and exciting advances in FOP drug development (Table 1, Figure 1), several of which are currently in ongoing clinical trials. Additionally, there are promising new therapies on the horizon, which target multiple levels and mechanisms in the FOP pathogenic pathway. These diverse strategies will allow for possible combinatorial pharmacological management which has proven effective for complex medical conditions, including transplant rejection, human immunodeficiency virus infection, and cancer. While most of these therapies have focused on targeting bone‐related pathways, other aspects of FOP will become important as more knowledge is gained.
Table 1.
Ongoing clinical trials for pharmacological treatments in fibrodysplasia ossificans progressiva (FOP) as of July 2018
| Drug name | Title | Phase | Sponsor |
|---|---|---|---|
| Palovarotene (RARγ agonist) | MOVE: an efficacy and safety study of palovarotene for the treatment of FOP | Phase 3 clinical trial (NCT03312634) | Clementia Pharmaceuticals |
| REGN2477 (anti‐activin A antibody) | A study to examine the safety, tolerability and effects on abnormal bone formation of REGN2477 in Patients with FOP (LUMINA‐1) | Phase 2 clinical trial (NCT03188666) | Regeneron |
| Rapamycin (mTOR1 kinase inhibitor) | Multicenter randomized double‐blind comparison test followed by open‐label continuous administration test of NPC‐12 T for FOP | Phase 2,3 Clinical Trial (Japan) | Kyoto University |
Figure 1.
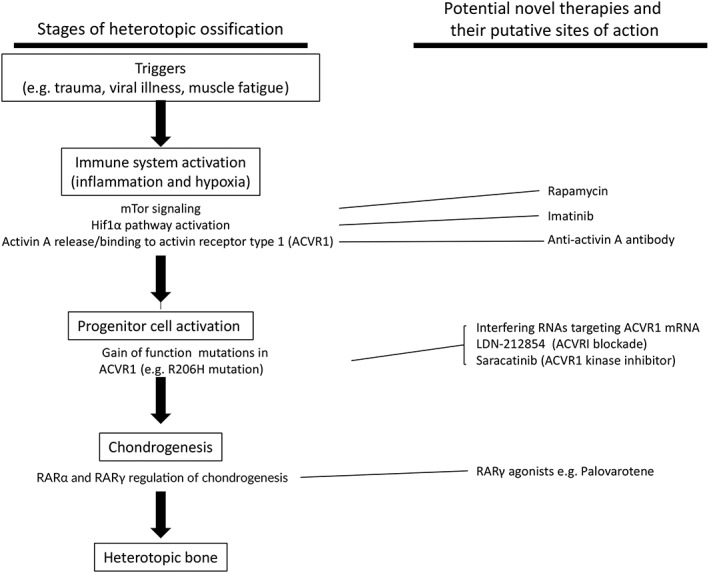
Pharmacological targets in fibrodysplasia ossificans progressive. RAR, retinoic acid receptor
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 60, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 61, 62, 63.
Competing Interests
Research funding: Clementia Pharmaceuticals (K.L.W., U.M. and E.C.H.); International FOP Association Medical Registry Advisory Board (E.C.H., unpaid); Fibrous Dysplasia Foundation Medical Advisory Board (E.C.H., unpaid); International Clinical Council on FOP (E.C.H., unpaid); Regeneron Pharmaceuticals (E.C.H.).
This work was supported by the National Institute of Arthritis, Musculoskeletal, and Skin Disorders grant (R01 AR066735 to E.C.H.) and Eunice Kennedy Shriver National Institute of Child Health and Human Development Grant (F32HD091025 to K.L.W.); the University of California at San Francisco Department of Medicine (to E.C.H.); and the Radiant Hope Foundation (to E.C.H.).
Wentworth K. L., Masharani U., and Hsiao E. C. (2019) Therapeutic advances for blocking heterotopic ossification in fibrodysplasia ossificans progressiva, Br J Clin Pharmacol, 85, 1180–1187. doi: 10.1111/bcp.13823.
Contributor Information
Kelly L. Wentworth, Email: kelly.wentworth@ucsf.edu.
Edward C. Hsiao, Email: edward.hsiao@ucsf.edu.
References
- 1. Pignolo RJ, Bedford‐Gay C, Liljesthrom M, Durbin‐Johnson BP, Shore EM, Rocke DM, et al The natural history of flare‐ups in fibrodysplasia ossificans progressiva (FOP): a comprehensive global assessment. J Bone Miner Res 2016; 31: 650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baujat G, Choquet R, Bouee S, Jeanbat V, Courouve L, Ruel A, et al Prevalence of fibrodysplasia ossificans progressiva (FOP) in France: an estimate based on a record linkage of two national databases. Orphanet J Rare Dis 2017; 12: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS. Age‐ and joint‐specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Relat Res 1994; 243–248. [PubMed] [Google Scholar]
- 4. Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006; 38: 525–527. [DOI] [PubMed] [Google Scholar]
- 5. Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, et al ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med. 2015; 7: 303ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, et al Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci U S A 2015; 112: 15438–15443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alessi Wolken DM, Idone V, Hatsell SJ, Yu PB, Economides AN. The obligatory role of activin A in the formation of heterotopic bone in fibrodysplasia ossificans progressiva. Bone 2018; 109: 210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dey D, Bagarova J, Hatsell SJ, Armstrong KA, Huang L, Ermann J, et al Two tissue‐resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci Transl Med. 2016; 8: 366ra163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brennan TA, Lindborg CM, Bergbauer CR, Wang H, Kaplan FS, Pignolo RJ. Mast cell inhibition as a therapeutic approach in fibrodysplasia ossificans progressiva (FOP). Bone 2018; 109: 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaplan FS, Shore EM, Pignolo RJE, Hsiao EC, FOP TICCo . The medical management of Fibrodysplasia ossificans progressiva: current treatment considerations. Clinc Proce Intl Clin Consort FOP 2011; 4: 1–100. [Google Scholar]
- 11. Wu XB, Yang MH, Zhu SW, Cao QY, Wu HH, Wang MY, et al Surgical resection of severe heterotopic ossification after open reduction and internal fixation of acetabular fractures: a case series of 18 patients. Injury 2014; 45: 1604. [DOI] [PubMed] [Google Scholar]
- 12. Kornhaber R, Foster N, Edgar D, Visentin D, Ofir E, Haik J, et al The development and impact of heterotopic ossification in burns: a review of four decades of research. Scars Burn Heal 2017; 3: 2059513117695659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eekhoff EMW, Netelenbos JC, de Graaf P, Hoebink M, Bravenboer N, Micha D, et al Flare‐up after maxillofacial surgery in a patient with fibrodysplasia ossificans progressiva: an [(18)F]‐NaF PET/CT study and a systematic review. JBMR Plus 2018; 2: 55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mark M, Ghyselinck NB, Chambon P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol 2006; 46: 451–480. [DOI] [PubMed] [Google Scholar]
- 15. Pacifici M. Retinoid roles and action in skeletal development and growth provide the rationale for an ongoing heterotopic ossification prevention trial. Bone 2018; 109: 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Underhill TM, Cash DE, Linney E. Constitutively active retinoid receptors exhibit interfamily and intrafamily promoter specificity. Mol Endocrinol 1994; 8: 274–285. [DOI] [PubMed] [Google Scholar]
- 17. Weston AD, Rosen V, Chandraratna RA, Underhill TM. Regulation of skeletal progenitor differentiation by the BMP and retinoid signaling pathways. J Cell Biol 2000; 148: 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Underhill TM, Sampaio AV, Weston AD. Retinoid signalling and skeletal development. Novartis Found Symp 2001; 232: 171–185 discussion 85–8. [DOI] [PubMed] [Google Scholar]
- 19. Weston AD, Hoffman LM, Underhill TM. Revisiting the role of retinoid signaling in skeletal development. Birth Defects Res C Embryo Today 2003; 69: 156–173. [DOI] [PubMed] [Google Scholar]
- 20. Weston AD, Blumberg B, Underhill TM. Active repression by unliganded retinoid receptors in development: less is sometimes more. J Cell Biol 2003; 161: 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shimono K, Morrison TN, Tung WE, Chandraratna RA, Williams JA, Iwamoto M, et al Inhibition of ectopic bone formation by a selective retinoic acid receptor alpha‐agonist: a new therapy for heterotopic ossification? J Orthop Res 2010; 28: 271–277. [DOI] [PubMed] [Google Scholar]
- 22. Shimono K, Tung WE, Macolino C, Chi AH, Didizian JH, Mundy C, et al Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor‐gamma agonists. Nat Med 2011; 17: 454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cappato S, Giacopelli F, Ravazzolo R, Bocciardi R. The horizon of a therapy for rare genetic diseases: a "druggable" future for fibrodysplasia ossificans progressiva. Int J Mol Sci. 2018; 19: pii: E989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fukuda T, Scott G, Komatsu Y, Araya R, Kawano M, Ray MK, et al Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis 2006; 44: 159–167. [DOI] [PubMed] [Google Scholar]
- 25. Chakkalakal SA, Uchibe K, Convente MR, Zhang D, Economides AN, Kaplan FS, et al Palovarotene inhibits heterotopic ossification and maintains limb mobility and growth in mice with the human ACVR1(R206H) fibrodysplasia ossificans progressiva (FOP) mutation. J Bone Miner Res 2016; 31: 1666–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stolk J, Stockley RA, Stoel BC, Cooper BG, Piitulainen E, Seersholm N, et al Randomised controlled trial for emphysema with a selective agonist of the gamma‐type retinoic acid receptor. Eur Respir J 2012; 40: 306–312. [DOI] [PubMed] [Google Scholar]
- 27. Clementia announces top‐line results from phase 2 trial of palovarotene for treatment of patients with fibrodysplasia ossificans progressiva. 2016. Available at http://investor.clementiapharma.com/news-releases/news-release-details/clementia-announces-top-line-results-phase-2-trial-palovarotene (last accessed 23 December 2018).
- 28. Kaplan FHE, Baujat G, Keen R, Grogan D, Pignolo R. Efficacy and safety of palovarotene in fibrodysplasia ossificans progressiva (FOP): a randomized, placebo‐controlled, double‐blind study. J Bone Miner Res 32 (Suppl.1). [Plenary Presentation FR0034 Poster presentation SA 0334]. In press 2017. Available at http://www.asbmr.org/education/AbstractDetail?aid=35b09323-8ada-4562-8254-96027dda2751 (last accessed 29 July 2018). [Google Scholar]
- 29. Inubushi T, Nozawa S, Matsumoto K, Irie F, Yamaguchi Y. Aberrant perichondrial BMP signaling mediates multiple osteochondromagenesis in mice. JCI Insight 2017; 2: pii: 90049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caruana DM, Wylie G. 'Washout' period for oral tetracycline antibiotics prior to systemic isotretinoin. Br J Dermatol 2016; 174: 929–930. [DOI] [PubMed] [Google Scholar]
- 31. Opel D, Kramer ON, Chevalier M, Bigby M, Albrecht J. Not every patient needs a triglyceride check, but all can get pancreatitis: a systematic review and clinical characterization of isotretinoin‐associated pancreatitis. Br J Dermatol 2017; 177: 960–966. [DOI] [PubMed] [Google Scholar]
- 32.Available at https://www.proteinatlas.org/ENSG00000122641-INHBA/tissue (last accessed 23 December 2018).
- 33. Knight PG, Satchell L, Glister C. Intra‐ovarian roles of activins and inhibins. Mol Cell Endocrinol 2012; 359: 53–65. [DOI] [PubMed] [Google Scholar]
- 34. Jones KL, Mansell A, Patella S, Scott BJ, Hedger MP, de Kretser DM, et al Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci U S A 2007; 104: 16239–16244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hedger MP, Winnall WR. Regulation of activin and inhibin in the adult testis and the evidence for functional roles in spermatogenesis and immunoregulation. Mol Cell Endocrinol 2012; 359: 30–42. [DOI] [PubMed] [Google Scholar]
- 36. Petrakou E, Fotopoulos S, Anagnostakou M, Anatolitou F, Samitas K, Semitekolou M, et al Activin‐A exerts a crucial anti‐inflammatory role in neonatal infections. Pediatr Res 2013; 74: 675–681. [DOI] [PubMed] [Google Scholar]
- 37. Mayer K, Buchbinder A, Morty RE. Activin A: a mediator governing inflammation, immunity, and repair. Am J Respir Crit Care Med 2012; 185: 350–352. [DOI] [PubMed] [Google Scholar]
- 38. Rodriguez‐Martinez G, Molina‐Hernandez A, Velasco I. Activin A promotes neuronal differentiation of cerebrocortical neural progenitor cells. PLoS One. 2012; 7: e43797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peterson JR, Agarwal S, Brownley RC, Loder SJ, Ranganathan K, Cederna PS, et al Direct Mouse Trauma/Burn Model of Heterotopic Ossification. J Vis Exp 2015; e52880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hino K, Horigome K, Nishio M, Komura S, Nagata S, Zhao C, et al Activin‐A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J Clin Invest 2017; 127: 3339–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Agarwal S, Loder S, Brownley C, Cholok D, Mangiavini L, Li J, et al Inhibition of Hif1alpha prevents both trauma‐induced and genetic heterotopic ossification. Proc Natl Acad Sci U S A 2016; 113: E338–E347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaplan FS, Zeitlin L, Dunn SP, Benor S, Hagin D, Al Mukaddam M, et al Acute and chronic rapamycin use in patients with fibrodysplasia ossificans progressiva: a report of two cases. Bone 2018; 109: 281–284. [DOI] [PubMed] [Google Scholar]
- 43. Radke J. Can Sirolimus Help Patients with Fibrodysplasia Ossificans Progressiva? 2017. Available at https://www.raredr.com/news/sirolimus‐fop (last accessed 2 January 2019).
- 44. First‐ever trial of drug using iPS cells to begin at Kyoto University The Japan Times 2017 Available at https://www.japantimes.co.jp/news/2017/08/01/national/science‐health/first‐ever‐trial‐drug‐developed‐ips‐cells‐begin‐kyoto‐university/ ‐ .W1Tjvi01SgB (last accessed 2 January 2019).
- 45. Verhave J, Boucher A, Dandavino R, Collette S, Senecal L, Hebert MJ, et al The incidence, management, and evolution of rapamycin‐related side effects in kidney transplant recipients. Clin Transplant 2014; 28: 616–622. [DOI] [PubMed] [Google Scholar]
- 46. Adams DM, Trenor CC 3rd, Hammill AM, Vinks AA, Patel MN, Chaudry G, et al Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics 2016; 137: e20153257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schindler CE, Partap U, Patchen BK, Swoap SJ. Chronic rapamycin treatment causes diabetes in male mice. Am J Physiol Regul Integr Comp Physiol 2014; 307: R434–R443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov 2002; 1: 493–502. [DOI] [PubMed] [Google Scholar]
- 49. Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al Long‐term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med 2017; 376: 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaplan FS, Andolina JR, Adamson PC, Teachey DT, Finklestein JZ, Ebb DH, et al Early clinical observations on the use of imatinib mesylate in FOP: a report of seven cases. Bone 2018; 109: 276–280. [DOI] [PubMed] [Google Scholar]
- 51. Convente MR, Chakkalakal SA, Yang E, Caron RJ, Zhang D, Kambayashi T, et al Depletion of mast cells and macrophages impairs heterotopic ossification in an ACVR1(R206H) mouse model of fibrodysplasia ossificans progressiva. J Bone Miner Res 2018; 33: 269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang H, Lindborg C, Lounev V, Kim JH, McCarrick‐Walmsley R, et al Cellular hypoxia promotes heterotopic ossification by amplifying BMP signaling. J Bone Miner Res 2016; 31: 1652–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bullock AN. Saracatinib: an ACVR1 inhibitor for FOP. 2017. Available at http://www.fopfriends.com/sites/default/files/blog_uploads/dr_alex_bullock.pdf (last accessed 23 December 2018)
- 54. Staab T. BioCryst Advancing Potential Treatment for Rare and Severely Debilitating Fibrodysplasia Ossificans Progressiva 2018. Available at https://globenewswire.com/news‐release/2018/01/05/1284093/0/en/BioCryst‐Advancing‐Potential‐Treatment‐for‐Rare‐and‐Severely‐Debilitating‐Fibrodysplasia‐Ossificans‐Progressiva.html (last accessed 2 January 2019).
- 55. Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, et al Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol 2008; 4: 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sanvitale CE, Kerr G, Chaikuad A, Ramel MC, Mohedas AH, Reichert S, et al A new class of small molecule inhibitor of BMP signaling. PLoS One 2013; 8: e62721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Williams E, Bullock AN. Structural basis for the potent and selective binding of LDN‐212854 to the BMP receptor kinase ALK2. Bone 2018; 109: 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shrivats AR, Hollinger JO. The delivery and evaluation of RNAi therapeutics for heterotopic ossification pathologies. Methods Mol Biol 2014; 1202: 149–160. [DOI] [PubMed] [Google Scholar]
- 59. Kaplan J, Kaplan FS, Shore EM. Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele‐specific targeting. Gene Ther 2012; 19: 786–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acid Res 2018; D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 2017; 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 2017; 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]