

Abstract
Progressive skeletal muscle wasting in cancer cachexia involves a process of dysregulated protein synthesis and breakdown. This catabolism may be the result of mal-nutrition, and an upregulation of both pro-inflammatory cytokines and the ubiquitin proteasome pathway (UPP), which can subsequently increase myostatin and activin A release. The skeletal muscle wasting associated with cancer cachexia is clinically significant, it can contribute to treatment toxicity or the premature discontinuation of treatments resulting in increases in morbidity and mortality. Thus, there is a need for further investigation into the pathophysiology of muscle wasting in cancer cachexia to develop effective prophylactic and therapeutic interventions. Several studies have identified a central role for chronic-systemic inflammation in initiating and perpetuating muscle wasting in patients with cancer. Interestingly, while exercise has shown efficacy in improving muscle quality, only recently have investigators begun to assess the impact that exercise has on chronic-systemic inflammation. To put this new information into context with established paradigms, here we review several biological pathways (e.g. dysfunctional inflammatory response, hypothalamus pituitary adrenal axis, and increased myostatin/activin A activity) that may be responsible for the muscle wasting in patients with cancer. Additionally, we discuss the potential impact that exercise has on these pathways in the treatment of cancer-related muscle wasting. Exercise is an attractive intervention for muscle wasting in this population, partially because it disrupts chronic-systemic inflammation mediated catabolism. Most importantly, exercise is a potent stimulator of muscle synthesis, and therefore this therapy may reverse muscle damage caused by cancer cachexia.
Keywords: Cancer Cachexia, Muscle Wasting, Chronic-Systemic Inflammation, Exercise
Introduction
Cancer cachexia is a multifactorial condition that results in loss of skeletal muscle with or without loss of fat mass. Many interventions, including maintenance of energy balance are not effective in reversing the symptoms of cancer cachexia [1–6]. Skeletal muscle wasting associated with cancer cachexia is clinically significant because it contributes to a reduction or discontinuation of cancer treatment and is associated with increased morbidity and mortality [7, 8, 2, 9]. Because muscle function may be directly proportional to the amount of muscle mass, decreases in muscle mass lead to weakness and impaired function, which adversely affect quality of life (QOL) [2, 9, 4, 10]. Cancer cachexia is a deleterious syndrome that affects 31–87% of all cancer patients, most of whom have advanced disease [10–12], and is responsible for more than 20% of all cancer deaths [13, 14]. Currently, the underlying mechanisms of cancer cachexia are not clearly understood, and there are no FDA-approved treatments.
Recent research has revealed several emerging mechanisms that may contribute to cancer-related muscle wasting, including malnutrition [15], an upregulation of cytokines that lead to the down-regulation of genes that promote protein synthesis [16–19], and an up-regulation of the ubiquitin proteasome pathway (UPP) [20]. Based on these mechanisms, treatments for cancer cachexia have focused on the maintenance and recovery of skeletal muscle mass through the use of nutritional support [21, 22], proteasome inhibition [23], and manipulation of cytokine signaling [24]. Such interventions have shown limited efficacy in clinical trials, and in some cases, have exacerbated side effects associated with cancer and its treatments [23, 21]. Other possible mechanism of cancer cachexia involves the immune system and systemic cytokines, which is chronically activated by cancer and its treatments [25, 26], the hypothalamic-pituitary-adrenal (HPA) axis, and mitochondria in muscle cells [25], all of which regulate hormone release and cell respiration [25]. The result of this pathophysiology appears to upregulate myostatin and activin A release, and skeletal muscle metabolism (Figure 1).
Figure 1. The role of tumor cell derived cytokines and cancer treatment-induced systemic inflammation in cancer-related muscle wasting.
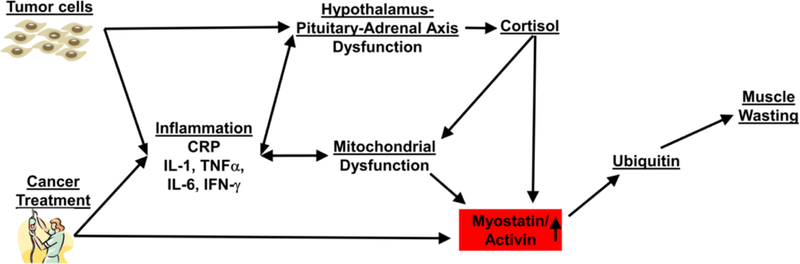
Cancer and its treatments cause chronic systemic inflammation that leads to Hypothalamus-Pituitary-Adrenal axis dysfunction that results in increased cortisol production. Increased cortisol production coupled with chronic inflammation causes mitochondrial dysfunction in muscle cells. This mitochondrial dysfunction and chronic inflammation leads to an increase in circulating myostatin/activing, which upregulates the ubiquitin proteasome pathway in muscle cells, and leads to uncontrolled muscle wasting
Recent research suggests that exercise may prevent or reverse the muscle wasting experienced by patients with cancer cachexia. Certainly, exercise is a potent modulator of skeletal muscle mass and function. Exercise preserves and increases muscle mass and function [27], while regulating both catabolic [28, 29] and anabolic [30] pathways in healthy individuals and those with disease [31]. Research investigating the association between exercise and cancer cachexia, however, is limited. Despite a strong rationale for the use of exercise to mitigate cancer cachexia, there is insufficient evidence to evaluate the safety and effectiveness of exercise in these patients [32].
Further investigation into the pathophysiology that leads to muscle wasting in cancer cachexia is needed to develop effective preventive and therapeutic measures. While the etiology of cancer cachexia is multifactorial, the purpose of this paper is to review the effect of chronic-systemic inflammation on mechanisms that lead to muscle wasting in patients with cancer cachexia and examine the potential therapeutic impact that exercise could have on these mechanisms.
Regulatory Pathways of Skeletal Muscle
The maintenance of skeletal muscle is governed by dynamic processes that regulate protein synthesis and proteolysis [33]. Numerous extracellular signals, which can be hypertrophic or atrophic, activate discrete intracellular signaling pathways. In a homeostatic environment both muscle protein synthesis and breakdown occur simultaneously, influenced by nutrient availability, mechanical loading and hormone regulation [34]. In cancer cachexia, there is a chronic net muscle protein breakdown [35].
Skeletal muscle mass and muscle fiber size differ based on physiological and pathological conditions. Changes in muscle mass result from net changes in muscle protein as well as organelles and cytoplasm. Increases in muscle mass are observed during development and as a direct result of mechanical overload or anabolic hormonal stimulation [36]. Insulin, insulin-like growth factor-1 (IGF-1), testosterone, and β2-adrenergic agonists provide anabolic hormonal stimulation to skeletal muscle [37]. The binding of insulin and IGF-1 to specific membrane receptors activates the Akt/mTOR-mediated signal propagation system that promotes muscle protein synthesis and inhibits proteolysis [37, 36, 38, 39].
On the other hand, the UPP is one of the major proteolytic processes [36]. In fact, hyperactivity of this pathway underlies the pathogenesis of muscle wasting that is characteristic of many muscular diseases, including cancer cachexia [33] (Figure 2). These proteolytic systems are activated by a number of different stimuli such as inflammatory cytokines, glucocorticoids, oxidative stress, and growth factors (myostatin and activin A) [38]. Evidence suggests that myostatin and activin A play a central role in the stunted muscle growth and muscle loss in various disease states [16, 40].
Figure 2. Uncontrolled activation of myostatin/activing causes muscle wasting in patients with cancer.
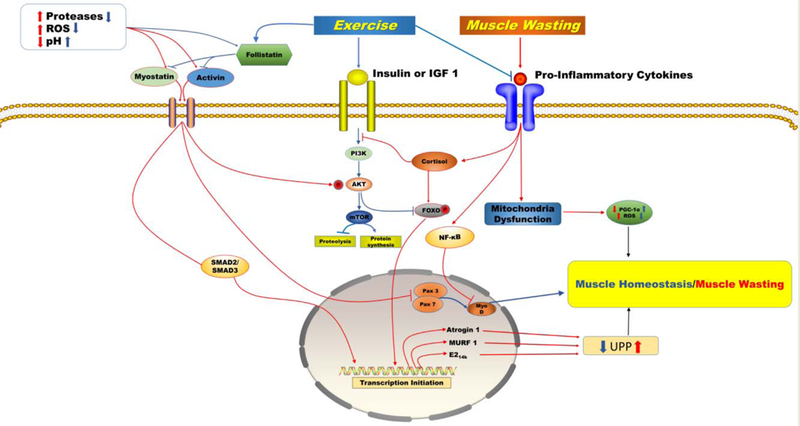
An increase in extracellular proteases and free radicals accompanied by a severe decrease in pH and follistatin activates myostatin/activin factors. In myofibers, binding of myostatin/activin to their receptors activates downstream processes that inhibit the IGF-1/PI3K/Akt hypertrophy pathway through phosphorylation of Akt, resulting in the translocation of smad 2/3 and FoxO1 into the nucleus and the upregulation of transcription of proteolytic genes, and inhibits myoblast growth and myogenic differentiation through suppression of Pax3/7 and myoD. This cycle of events initiates muscle wasting by upregulating transcription of Atrogin 1, MURF 1, and E2 ligases, which are all active in the ubiquitin proteasome pathway. A chronic inflammatory response leads to altered Hypothalamus Pituitary Adrenal axis signalling causing a dysregulated cortisol response that induces muscle wasting through a resistance to insulin and IGF-1, activation and translocation of NF-κB into the nucleus, and mitochondria dysfunction. Exercise improves muscle wasting by mediating key pathways. Exercise regulates myostatin/activin by reducing proteases and free radicals while increasing pH and follistatin. Exercise activates the PI3K/Akt/mTOR pathway. This pathway inhibits FOXO through phosphorylation. Exercise reduces local and systemic inflammation, regulates cortisol production, and enhances mitochondria biogenesis and myocellular regeneration while reducing production of reactive oxygen species which results in muscle homeostasis. Blue arrows describe the pathways to muscle homeostasis under conditions of chronic exercise. Red arrows describe the pathways to muscle wasting in patients with advanced staged cancer
In muscle, homeostatic pathways that regulate growth and breakdown function to prevent unnecessary protein cycling. For example, in response to activation by insulin growth factor-1 (IGF-1) or insulin, Akt phosphorylates FoxO, which prevents its translocation into the nucleus in myocytes, thereby inhibiting the proteolytic effects on FoxO [37]. In contrast, nuclear FoxO transcription factors upregulate the expression of ubiquitin ligases that are active in proteolysis [41]. Additionally, during anabolic conditions, phosphorylation of mTOR suppresses the activity of proteolytic pathways (i.e. autophagy/UPP), while activating the translation and building of proteins [42]. Inversely, the activation of FoxO proteins by Smad or nuclear factor kappa B (NF-κB) promotes the transcription of proteolytic proteins while inhibiting protein translation and Akt/mTOR activity [37, 36] (see Figure 2). The maintenance of skeletal muscle requires efficient interplay of the anabolic and catabolic systems that are responsible for protein breakdown and synthesis. Dysfunction in these systems may play an important role in the muscle wasting experienced by patients with cancer cachexia. Furthermore, the evolution of cancer cachexia may differentially affect patients with cancer based on the type of cancer diagnosis. Research to elucidate the mechanisms responsible for muscle wasting in patients with cancer cachexia should focus on the role that sustained low-grade systemic inflammation plays in this cascade.
Muscle wasting among different types of Cancer
Muscle Wasting is a prominent feature of cancer cachexia. However, a diagnosis of cancer cachexia involves both the loss of adipose tissue and lean body mass that results in significant weight loss [43]. Although weight loss is common in patients with cancer, occurring at a rate of 50%, the incidence of cachexia is not distributed equally among all cancer diagnosis [44]. Patients with pancreatic or gastric cancers suffer the highest degree of weight loss (83–87%), followed by colon, prostate, and lung cancer (48–61%). Patients with breast cancer, various types of leukemia and sarcomas lose the least amount of weight (31–40%) [45]. Even without the loss of adipose tissue, skeletal muscle depletion is a meaningful prognostic factor of cancer [46]. Cancer-related skeletal muscle wasting has been observed in both human and animal models of cancer cachexia; however, the mechanism(s) of wasting seem to differ based on tumor types [47]. Clinical studies show that the prevalence of muscle wasting ranged from 14%−25% in patients with breast cancer [9, 48–50], 39% in colorectal cancer [51], 47% in non-small cell lung cancer (NSLC) [52], and 56–63% in patients with pancreatic cancer [53, 54]. Because clinical research on cancer-related muscle wasting is sparse, the underlying mechanisms that cause this condition remain unknown [47]. However, available research in animal models and humans substantiate an interaction among the tumor diagnosis [55–57] and treatments for the tumor [58–60] and chronic low-grade inflammation, as possible culprits in the onset and pathogenesis of cancer-related muscle wasting.
Cancers of the gastrointestinal (GI) tract, including pancreatic cancer, may cause or exacerbate cancer-related muscle wasting by causing a decrease in appetite, nausea, vomiting, and dysphagia that leads to negative energy balance [61]. However, the muscle wasting that is experienced by this population cannot be fully explained by a negative energy balance. Research suggest that the magnitude of nutrient deficits do not correlate with the degree of muscle wasting [62]. Furthermore, studies in animal models of cachexia show skeletal muscle wasting in the presence of sustained energy balance [63]. Patients with newly diagnosed lung cancer experienced increased protein turnover while consuming a normal diet [64]. A key factor in patients that experience cancer-related muscle wasting without a decrease in energy consumption may relate to an increase in resting energy expenditure (REE) (caused by tumor-related glucose cycling) [65] and acute phase protein response (APPR) [66]. Together, the pathological increase in REE and APPR result in the synthesis of C-reactive protein (CRP) [67] which is correlated with muscle wasting in patients with GI cancers. The factors contributing to an elevated CRP may include systemic inflammation that result from the tumor burden or increased bacterial translocation in the gut [14]. While the increase in APPR is not fully understood, it may result from decreased amino acid availability caused by insufficient dietary protein intake caused by anorexia.
In patients diagnosed with non-metastatic pancreatic adenocarcinomas (PDAC), the only possible cure is surgical resection [68]. However, most patients experience cancer recurrence due to the aggressive and metastatic nature of the disease [69, 70]. Adjuvant chemo and/or radiation therapy improves survival in patients who undergo resection and is therefore necessary to maximize outcomes [71–73]. The combination of surgery, chemo, and radiation therapy are the primary treatments for metastatic pancreatic cancer because together, they reduce the size of or slow tumor growth for a time and may increase survival. However, with increases in survival come the burden of treatment toxicities such as skeletal muscle wasting [74]. In patients with PDAC, the presence of chronic systemic inflammation and the loss of muscle mass may be the result of the cancer, chemotherapy and/or the surgical procedure [75, 3, 76]. Chronic systemic inflammation is associated with debilitating muscle wasting that reduces quality of life and treatment tolerance in patients with pancreatic cancer [76, 3] and is correlated with variations in lean tissue and body fat [77] in patients with cancer.
Treatments for cancer that alter endocrine function also play a significant role in cancer-related muscle wasting. In clinical studies, treatment with Androgen Deprivation Therapy (ADT) in male patients with non-metastatic prostate cancer resulted in significant decreases in lean body mass [60, 59]. This reduction in lean body mass was associated with decreased serum testosterone and estradiol, which are positively correlated with lean muscle mass [78]. The reduction in anabolic activity coupled with catabolism initiated by systemic low-grade inflammation may explain the muscle wasting experienced by this population.
The occurrence of muscle wasting is rare in patients diagnosed with breast cancer and its development most often results from metastatic nature of the tumor. Cancer cells rely on production of pro-inflammatory mediators for growth, defense against cell death, and promotion of metastasis. Therefore, as the tumor grows it may initiate a sharp upregulation of cytokines that leads to skeletal muscle wasting. A case study performed by Consul et al concluded that an increased metastatic breast cancer tumor burden was strongly correlated with muscle wasting [79]. This finding is in line with a study that showed significant muscle wasting in breast cancer patients during the 12-month period before death [80]. This research suggests that muscle wasting may be a product of tumor progression. As the tumor grows, it may activate an uncontrolled increase in pro-inflammatory cytokines that initiates cancer-related muscle wasting during end-stage disease. In agreement with this, clinically, cancer cachexia is a continuum that develops from pre-cachexia, cachexia, and finally refractory cachexia [2]. Research studies have implicated pro-inflammatory cytokines in the etiology of cancer cachexia [81, 82] . In patients with NSLC, it was found that a high percentage of patients suffered muscle wasting consistent with end-stage disease [52]. The etiology of muscle wasting may differ based on the type of cancer; however, chronic systemic inflammation may be the factor that links them together.
Inflammation and the Hypothalamic-Pituitary-Adrenal Axis
Multiple factors contribute to the development of cancer cachexia and the uncontrolled muscle wasting patients with this condition experience. The anatomical distance between tumor cells and sites of muscle wasting suggests that intermediary signals link the two. It is possible that inflammatory cytokines transmit systemic signals that initiate increased protein degradation while reducing protein synthesis. Chronic-systemic inflammation may potentiate muscle wasting through the alteration of myofibrillar intracellular pathways regulated by both hormones and cytokines that slow protein synthesis and accelerate catabolism [83]. Research suggests that the enhanced catabolism experienced by cancer patients with cachexia is mediated primarily by increases in pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) [84, 85], interleukin 1 (IL-1), interleukin 6 (IL-6) [86], and interferon gamma (IFN-γ) [87, 88] and/or decreases in anti-inflammatory cytokines such as interleukin 10 (IL-10) [83].
TNF-α is produced primarily by macrophages and is involved in physiological processes such as systemic inflammation, apoptosis, and the initiation of the acute phase response [89]. In addition to enhancing its own secretion, TNF-α also stimulates the production of other inflammatory cytokines and chemokines [89]. TNF-α triggers the activation of NF-κB and mitogen-activated protein kinase (MAPK) pathways, both of which are essential for the expression of pro-inflammatory cytokines [89]. For example, TNF-α administration to HepG2 cells induces rapid proteolysis of IκB (α and β), the protein that inhibits NF-κB, which results in its translocation into the nucleus [90]. Although pre-clinical data suggests that chronic exposure to low doses of TNF-α may contribute to pro-inflammatory signaling detrimental to muscle [91], blocking this pathway has failed to show clinical significance in the treatment of cancer cachexia [92] and incurable malignancies [93] in humans with advanced cancer. Indeed, the role of TNF-α in cancer is controversial. Some early studies in animal models of cancer seemed to show promise in TNF-α as a therapy. One study concluded that exogenous administration of bolus amounts of rTNF-α resulted in hemorrhagic necrosis of SA1 tumor cells in immunocompetent mice [94]. However, this treatment with non-physiological amounts of TNF-α did not result in complete tumor regression, was highly toxic, and in some cases, fatal. Other research in animal models suggests that the binding of TNF-α to its receptors plays a role in protecting cells from apoptosis and induces a range of inflammatory mediators [95, 96]. Conversely, in studies in which animals are genetically modified to overexpress TNF-α the results in some cases were chronic systemic inflammation and autoimmune disease [97, 98]. Furthermore, in biopsies from human cancer, TNF-α production by tumors has been associated with a poor prognosis, loss of treatment response, and cachexia [99, 100]. Differences in these findings may be related to the test species and/or differences in origin of TNF-α production. Regardless, these data seem to suggest that targeting TNF-α alone is not sufficient to prevent cancer cachexia. However, a therapy directed against multiple potentially harmful pro-inflammatory cytokines may provide greater efficacy. Serum levels of IL-6 and IL-1 are also elevated in animal models of cancer cachexia [101]. IL-1 and IFN-γ have been shown to induce weight loss and anorexia [102] in rat cancer models. In particular, in rats, IL-1β seems to regulate food intake and feeding behavior by causing early satiety and suppressing hunger [103]. In this animal model, IL-1 also elicits the production of IL-6, a cytokine that increases lipolysis and contributes to weight loss. Although IL-6 is thought to be important in the development of cachexia, its effects are indirect [103]. These data suggest that a clinical intervention that targets inflammation in the treatment of cancer cachexia may reduce or prevent muscle wasting in this population.
The inflammatory immune response is mediated in part by neuronal pathways and hormone secretions, and the hypothalamic-pituitary-adrenal (HPA) axis is at the center of this response. In patients with cancer , a chronic inflammatory response may be the result of dysfunctional HPA signaling, resulting in altered glucocorticoid (GC) release or disturbed receptor function [104]. Cortisol, secreted by the adrenal glands is a potent pro-inflammatory GC hormone and its production is regulated by the HPA axis [104]. The HPA axis is activated by internal and external stimuli that are recognized by higher-order cognitive areas as a threat to homeostasis. The centrally localized regions that identify these threats send excitatory signals to key areas of the hypothalamus causing the synthesis and release of corticotrophin-releasing factor (CRF), which acts on the pituitary gland to initiate a signaling cascade inducing the release of GCs. In healthy humans, GCs initiate a negative feedback system that inhibits their own release [105]. The HPA-axis disturbance observed in several different disease states [106–109] may alter this negative feedback response leading to augmented GC release and enhanced protein degradation. Basal levels of GC are needed to maintain homeostatic organ and tissue function; however, even small deviations in plasma concentrations of these steroids produce vast changes in a variety of physiological and biochemical factors. Previous research in rats suggests that not only do GCs regulate immune activity, but that the reverse is also true [109]. This research, along with human studies [110, 111], present evidence that an immune-neuroendocrine feedback mechanism exists in which immune cells secrete molecules that stimulate the release of adrenal GCs to limit their own activity. In fact, exogenous administration of IL-1 [112], IL-6 [113], TNF-α [114], and IFN-γ [115] result in an increase in plasma concentrations of cortisol in cancer patients [116] and healthy humans [117]. IL-6 is a particularly potent activator of the HPA axis. It is well established that IL-1 and IL-6 are two cytokines responsible for stimulating the secretion of adrenocorticotropic hormone (ACTH) [118], which is produced by the pituitary gland and is the primary physiologic regulator of adrenal GC secretion [119]. Particularly, the hypothalamus mediates communication between the HPA axis and IL-1 [120]. In addition, IL-1 affects several other hormones that are under neuroendocrine control [110].
Chronic inflammation is a burdensome side effect experienced by cancer patients because of malignancy and cancer treatments such as chemotherapy and radiotherapy [25, 121, 26, 122]. There may be a correlation between chronic inflammation and hyperactivity of the HPA axis in patients with cancer. The result is an overproduction and secretion of ACTH and GC. Increased levels of GC may induce muscle atrophy through upregulation of proteasomes and FoxO transcription factors [123]. In addition to the regulation of proteasomes and FoxO, GCs also contribute to muscle atrophy by inducing resistance to the anabolic regulators insulin, insulin-like growth factor-1 (IGF-1), and leucine, resulting in inhibited protein synthesis [123]. In patients with cachexia, dysregulation at the level of the hypothalamus combined with resistance to growth factors may contribute to poor appetite and IGF-1R function. This dysregulation can lead to reduced nutrient availability that exacerbates muscle wasting driven by processes described above. Given the adverse effects on muscle that a dysfunctional HPA can pose, patients with cachexia may benefit from treatments that target this axis.
Mitochondrial Dysfunction
Reduced energy intake and increased energy expenditure contribute to the debilitating weight loss that patients with cancer cachexia suffer [124]. In other words, not only do patients with cancer cachexia eat less due to changes in appetite and a multitude of other factors, they also utilize more energy during rest and activity than do their non-cachectic counterparts, thus accelerating their weight loss. Indeed, these patients are in a hypermetabolic state characterized by altered regulation of molecular mechanisms, specifically, in mitochondria. Mitochondria in tumors are few in number, have an altered shape, a diluted mitochondrial matrix [125], and a decreased ability to synthesize adenosine triphosphate (ATP). Thus, tumor cells depend heavily on glucose as a primary energy source [126]. Moreover, systemic effects of cancer can cause mitochondrial dysfunction in distant tissues, leading to systemic energy inefficiency, muscle atrophy, and ultimately whole-body weight loss. Mitochondrial dysfunction in liver [127] and skeletal muscle cells [128] has been discovered in animal models [127, 129] and in human [128] cancer cachexia patients. Mitochondrial dysfunction is associated with reduced oxidative phosphorylation, uncoupling of electron transport from adenosine triphosphate (ATP) production, and loss of skeletal muscle structural integrity [126, 130]. In a rat model of cancer cachexia, significant decreases in mitochondrial phosphatidic acid, phosphatidylglycerol, and cardiolipin are counteracted by increases in phosphatidylcholine levels. The net result of these changes is increased uncoupling and a reduced ability of muscles to produce ATP [131].
The exact cause(s) of mitochondrial dysfunction in muscle wasting have yet to be fully described, but it has been suggested that alterations in mitochondrial morphology are the result of oxidative damage and cytokine activity. In response to structural alterations, mitochondria experience a reduced capacity to produce antioxidants that defend against reactive oxygen species (ROS) (Figure 2). When this occurs, a destructive cycle begins that increases the net production of ROS thereby further damaging mitochondria [132]. Increased ROS leads to accumulation of mitochondrial machinery responsible for maintaining the cellular redox state and not surprisingly, increases in production of inflammatory cytokines. In a murine model of muscle wasting, TNF-α is associated with increased oxidative stress in skeletal muscle in the presence of cancer [133], and ROS-induced IL-6 alters mitochondrial morphology, decreases mitochondrial content, and reduces cellular respiration [134]. In a mouse model, IL-6 was necessary to induce muscle wasting and its overexpression led to cachexia in pre-cachectic animals [135]. Thus, damage to mitochondria caused by cancer and its treatments leads to increased ROS production. The result is increased cell damage and a chronic inflammatory response that may result in muscle wasting.
Myostatin and Activin A
Myostatin is a protein that inhibits muscle growth and its overexpression may contribute to muscle wasting in patients with cancer cachexia. Myostatin, also known as growth differentiating factor-8 (GDF-8), is a member of the transforming growth factor-beta (TGF-β) family. Expressed primarily in skeletal muscle, myostatin acts as a negative regulator of myocellular development [136, 137]. Muscle cells secrete myostatin through paracrine and autocrine mechanisms. Extracellular myostatin binds to receptors on the surface of muscle cells, activates downstream targets, and causes protein degradation [136]. Systemically, myostatin circulates through the blood and is stored in skeletal muscle in a latent complex bound to a pro-peptide [137]. In addition to the pro-peptide, follistatin and the follistatin-like proteins also bind to and inhibit myostatin [138]. Follistatin, also a member of the TGF-β super family, binds to myostatin and activin A, and inhibits their signaling capacity [138]. While the mechanism of activation of the myostatin/activin A complexes is unknown, it has been suggested that mature myostatin is cleaved from the latent complex by proteases, low pH, and ROS [139, 140] (Figure 2).
Once activated, myostatin binds to its receptor ActRIIB with high affinity and regulates the expression of downstream target genes [141]. Myostatin initiates muscle wasting by activating UPP. Indeed, proteolysis is an integral component of muscle wasting during disease, both in animals [142] and humans [143, 144]. In fact, the ubiquitin-associated genes atrogin-1, MuRF-1, and E214k are upregulated during myostatin treatment [142] (Figure 2). In addition to increasing proteolysis, myostatin signaling also inhibits the IGF-1/PI3K/Akt hypertrophy pathway through phosphorylation of Akt thereby increasing translocation of active FoxO1 into the nucleus and increasing expression of atrophy-related genes [142] (Figure 2). In vitro, myostatin has been shown to inhibit myoblast growth and myogenic differentiation through the suppression of myoD and Pax3 [142]. In adult muscle tissue myostatin alters satellite cell activation by preventing Pax7 from entering the cell cycle, thereby reducing its potential for self-renewal [145].
Activins exert a broad range of effects on the differentiation, proliferation, and functions of numerous cells and may play a pivotal role in the muscle wasting that is experienced by patients with cancer cachexia. Activins are members of the Activin-Inhibin subfamily of the TGF-β superfamily [146]. Activins play a role in stimulating the release of follicle-stimulating hormone (FSH) from the pituitary gland and in regulating follistatin [147, 148]. Activin A is a major form of activin and, like myostatin, functions as a paracrine/autocrine factor in skeletal muscle. It also assumes a latent complex and is regulated by follistatin and follistatin-like proteins [149]. Activin A regulates biological activities such as cell proliferation and differentiation [150], the immune response, and angiogenesis [151].
Myostatin and Activin A exert their biological effects by binding to the same surface receptor complex. Binding to this receptor activates downstream processes that result in the translocation of Smad2/3 and FoxO1 into the nucleus and the upregulation of transcription of proteolytic genes [42, 152] (Figure 2). Myostatin signaling can also engage in cross talk with the exercise induced transcription coactivator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1-α), believed to be a marker of mitochondria biogenesis, in muscle. Its inhibition has been associated with increased production of PGC1-α and mitochondria biogenesis in muscle, which enhanced exercise performance [153]. Growing evidence suggests that signaling by myostatin [154–157] and Activin A [158–161] is upregulated in catabolic diseases and contributes to muscle wasting.
Treatment of Cancer Cachexia
Treatments capable of preventing, halting and/or reversing muscle atrophy in cancer cachexia may improve physical function, quality of life, and tolerance to cancer treatments. Pharmaceutical and nutritional [162–166] therapies have been developed to treat muscle atrophy. These designed therapies target the very pathways reviewed in the prior sections: proteasome inhibition [167, 23, 168], cytokine signaling [169–171], and myostatin inhibition [172, 173, 152, 174]. While targeting these systems held some promise biologically, initially efficacy in clinical trials has been limited. Due to the complex nature of cancer cachexia and the many factors and signaling pathways that contribute to muscle wasting, it is likely that treatments targeting only one aspect of the syndrome will not result in clinically meaningful benefits.
Exercise possesses a sound rationale for treatment of cancer cachexia as it promotes muscle growth via multiple pathways and is typically safe and cost-effective. Moreover, recent research suggests that exercise may alter pathogenesis and progression of the uncontrolled muscle atrophy experienced by cancer patients with cachexia [175–183] (Figure 2). For these reasons, exercise, which is typically safe and cost-effective, should be further explored as a therapeutic strategy for cancer cachexia. The following sections review the impact of exercise on inflammation, the HPA axis, mitochondrial dysfunction, myostatin/activin A, and cancer cachexia.
Exercise and Inflammation
Both resistance and aerobic exercise training have the ability to reduce inflammation [184]. Ironically, acute exercise upregulates C-reactive protein (CRP) in diseased patients, initially; however, after four months of exercise training, resting CRP levels were significantly lower and there was a significant attenuation of exercise-induced CRP [185]. In addition, one study showed that American adults who engaged in frequent exercise tended to have lower CRP than adults who were less active [186]. Single bouts of exercise augment the production of cytokines involved in the acute-phase anti-inflammatory response [187–190]. For example, contracting muscle fibers induce the production of the myokine IL-6, which promotes anti-inflammatory actions in response to exercise. At the onset of exercise, IL-6 is upregulated and increases exponentially up to 100-fold depending on exercise intensity, mode, duration, and amount of muscle mass recruited; it is then downregulated, at the cessation of the exercise bout [190, 191]. Specifically, IL-6 inhibits the production of the pro-inflammatory cytokine TNF-α while increasing plasma concentrations of anti-inflammatory cytokines such as (IL-1 receptor agonist IL-1ra) and IL-10 [192, 193]. Moreover, IL-6 may be involved in the protection against chronic diseases associated with systemic low grade inflammation [193]. The positive effects of exercise-induced increases in circulating IL-6 are quite surprising given its known relationship with muscle wasting in diseased states. More research should focus on delineating the relationship between exercise and disease related upregulation of IL-6. Exercise also directly increases the anti-inflammatory cytokine IL-10 and cytokine inhibitors IL-1ra, sTNF-r1, and sTNF-r2 (TNF receptors) that antagonize inflammatory cytokines by interfering with their ability to signal [189, 194]. It is plausible that exercise can prevent and reverse the muscle atrophy associated with cancer cachexia by altering the production and/or behavior of inflammatory cytokines and affecting systemic inflammation.
Exercise and the HPA axis
Chronic aerobic exercise affects key parameters that regulate HPA axis function. Voluntary wheel running in rodents increased the size and mass of the right adrenal medulla, which facilitated adaptive changes in ACTH levels [195] and normalized GC levels [196, 197]. Exercise can also attenuate the HPA axis response to other physiological [198] and psychological [199] stressors in a dose-dependent manner. These findings suggest that exercise might be capable of mediating the HPA axis response to the physiological and psychological stress caused by cancer and its treatments. Research on the hypothalamus has also explored intermittent exercise-induced adaptations of the HPA axis. In rats, voluntary and intermittent wheel running resulted in a significant reduction in expression of C-fos [199], a proto-oncogene expressed in the paraventricular (PVN) nucleus of the hypothalamus that has been implicated as a predictor of cancer progression and survival [200]. This finding suggests that voluntary and intermittent running may minimize excitatory input from the PVN to the pituitary gland, possibly resulting in a reduction in ACTH release. This reduction in ACTH would reduce cortisol production, immune response, and inflammation. Regulation of the HPA axis and thus cortisol production, immune response and inflammation may help mediate the muscle wasting experienced by patients with cancer cachexia. Future research should investigate the relationship between exercise related changes in HPA axis function and inflammatory response.
Exercise and Mitochondrial Dysfunction
Muscles contain the highest mitochondrial density of any tissue in the body in order to provide high amounts of ATP for movement and exercise [201]. Among the various types of muscle fibers, Type I fibers contain the largest number of mitochondria; this feature provides for them a greater capacity to resist fatigue. Early studies on the effects of aerobic exercise on skeletal muscle mitochondria determined that chronic aerobic exercise increases mitochondrial biogenesis. In addition, aerobic exercise enhances mitochondrial function by enhancing the coupling of oxidative phosphorylation with ATP production [202–204]. In a mouse study, changes in mitochondrial function were driven by increases in mitochondrial respiratory enzymes such as succinate dehydrogenase (SDH) and increases in electron transport chain activity in association with decreased oxidative damage [204]. Human studies of diseased populations concluded that exercise improved mitochondrial biogenesis and function. These improvements were observed in patients with diabetes mellitus [205, 206] and in elderly individuals [207]. Increased expression of peroxisome proliferator-activated receptor gamma coactivator (PGC-1α), cardiolipin, and creatine kinase (CK) [208] with associated decreases in uncoupling proteins (UPS) have also been implicated in exercise-induced improvements in mitochondrial status [206]. The transcriptional coactivator PGC-1α, a potent regulator of metabolism in numerous tissues, is widely believed to be required for exercise-induced mitochondrial biogenesis. However, enhanced mitochondrial function and biogenesis were unperturbed in a cohort of mice engineered to lack PGC-1α that were exposed to two weeks of in-cage voluntary wheel running [203]. Therefore, exercise may be as effective in improving mitochondrial function in disease states as it is in healthy states. Decreases in UPS may lead to a downregulation of ROS production, which improves mitochondrial function. Indeed, studies completed in mice show that regular aerobic exercise exerts beneficial neuroprotection through regulation of cytokine and ROS production. This effect of exercise on improvements in mitochondrial function and biogenesis may prove effective in treating the muscle wasting and dysfunction experienced by patients with cancer cachexia. Research should clarify the relationship between exercise-induced modulation of cytokine and ROS production and signaling.
Exercise, Myostatin, and Activin A
Myostatin is a potent inhibitor of skeletal muscle growth and is upregulated in several diseases characterized by muscle wasting and cachexia. Exercise is effective in inhibiting myostatin and activin A [209] in healthy individuals [210, 211], individuals with kidney failure [212], diabetes [213], chronic heart failure [214], and the elderly [215, 183]; however, the mechanisms by which exercise exerts its effects on myostatin and activin A are unclear.
One potential mechanism is through the upregulation of follistatin. Follistatin, a member of the TGF-β super family, which binds to myostatin and activin A and inhibits their signaling capacity [138]. Neutralization of myostatin by follistatin has profound effects on skeletal muscle growth [216]. Thus, in mice with follistatin overexpression, marked increases in muscle mass are observed [137]. Exercise has been shown to increase systemic plasma follistatin in humans and mice [217, 218]; however, Hansen et al [217] found that the increased production of follistatin was not derived from working muscle. In fact, these investigators determined that the increase in plasma follistatin originated from the liver of both human subjects and mice. Other groups have investigated the effects of exercise on the expression of follistatin in skeletal muscle [210, 219, 220] and the results were conflicting. In two of these experiments, intramuscular follistatin was not increased in response to an acute bout of exercise [219, 220]. Conversely, when expression of follistatin in skeletal muscle was assessed in postmenopausal women who were using hormone replacement therapy and engaged in resistance exercise training , an up-regulation was observed in both the intervention and control groups [210]. Neither of these studies assessed systemic levels of follistatin, which may have led to the discrepancy in findings. The exercise-induced increase in follistatin may positively impact the muscle wasting experienced by patients with cancer cachexia by increasing the amount of bound/inactive myostatin and activin A, thereby reducing their signaling capacity.
A primary signaling pathway by which myostatin exerts its deleterious effects on muscle is through the activation of Smad proteins which lead [221] to the downregulation of myogenic regulating factors (MRFs) [222]. Hyperplasia and hypertrophy mechanisms in muscle growth are controlled by the sequential expression of MRFs [223]. Both acute and chronic exercise is effective in reducing myostatin while subsequently up-regulating myogenin and myogenic differentiation factor D (MyoD) [224, 225, 223, 226, 31]. Furthermore, 16 weeks of resistance training, three days/week, resulted in increased skeletal muscle mass, myogenin, and MyoD in healthy men and women [227]. Therefore, exercise may be effective in preventing or halting muscle wasting in patients with cancer cachexia via regulation of the myostatin/activin A pathway.
Exercise in Human Cancer Cachexia
In recent years, the study of exercise as a behavioral intervention in oncology and supportive care research is growing [228]. There is a tremendous need for further exploration into exercise as a therapeutic strategy for cancer cachexia [229]. In addition to its many well-described psychological and physical benefits [230], exercise interventions are easily paired with anti-neoplastic therapies and could potentially improve physical performance, a task that has proven difficult with pharmacologic anti-cachexia agents [231, 232]. Safety concerns certainly arise when prescribing exercise to patients with cachexia. For this reason, it is vital that exercise duration, mode, and intensity are prescribed based on an individual’s medical history, exercise tolerance, and current performance status. Additionally, the type of exercise could range from resistance training to low-intensity activities like waking, yoga, or tai chi [228]. Most importantly, the literature supports that exercise is feasible and safe even in the sickest of population cohorts. For example, in a study of 66 terminally ill cancer patients where the definition of cachexia is not a criterion for inclusion, subjects were randomized to a resistance or a cardiovascular exercise regimen [233]. The resistance training exercise group performed 1 set of 8 to 15 repetitions for all major muscle groups, using free weights or circuit weight training equipment. Unless a lifting restriction was imposed, resistance was set to a level where the participant felt he or she needed a short (1–2 min) rest at the end of the set. Amount of resistance, repetitions, and sets were increased as tolerated. The cardiovascular exercise group performed exercise on aerobic exercise equipment (i.e. stationary bike, treadmill, arm ergometer). Respiratory rate, pulse rate, and perceived exertion were monitored to ensure safe exercise intensities. Participants were asked to exercise at a rate of 10 to 12 (or fairly light) on the Borg Rating of Perceived Exertion Scale for 30–60 minutes. Participants in both groups attended exercise sessions twice weekly for 10 weeks. In this large randomized control trail (RCT) study participant’s mean age was 62 years. The group was made up of an even mix of males and females who were all undergoing treatment at the time of the exercise intervention (chemotherapy, radiotherapy, hormone therapy, other therapies). A weakness of this study is that weight is not defined. Research has shown that obesity is linked to chronic low-grade inflammation characterized by elevated levels of circulating pro-inflammatory mediators such as TNF-α and IL-6 [234]. In this condition, muscle wasting may be more severe and exercise more difficult or less effective. Given these factors, almost 80% of the patients completed the study and there were no withdrawals because of exercise-related adverse events. There was also significant improvement in the Short Physical Performance Battery (SPPB) score (Table 1) suggesting exercise may improve physical performance while being safe and attainable in cancer patients. In this study, no significant differences were found in any outcome measures based on mode of exercise.
Table 1.
Exercises outcomes on cachexia in different cancers
| Article (year of publication | Patients in Exercise Group N (M/F) | Age (years; mean or median) | Tumor, Stage | Human/Animal | Exercise Type | Outcome |
|---|---|---|---|---|---|---|
| Litterini et al. 2013 [233] | 66 (30/36) | 62, mean | Breast, Colorectal, Lung, Prostate, Gynecologic, Lymphoma/Hodgkin disease – All stage IV | Human | Aerobic/Resistance | Significant increase in SPPB score in both groups. |
| Oldervoll et al. 2011 [235] | 121 (40/81) | 62.6 mean | GI, Breast, Lung, Urological, Gynecological, Hematological, Other - All Stage IV | Human | Circuit Training | Significant improvement in physical performance (SWT and HGS test) |
| Solheim et al 2017 [238] | 25 (15/10) | 63 median | Lung – Stages III and IV Pancreatic – Stages III and IV | Human | Aerobic/Resistance | No adverse effects of exercise in this feasibility study |
In a larger study of 231 cancer patients with a prognosis of less than 2 years, subjects were randomized to exercise versus usual care [235]. The mean age of participants was 62 years. The exercise group was comprised of 67% female, 33% male. There were 57% female, 43% male in the usual care group. There was no significant difference in baseline bodyweight between the groups (exercise = 70.4kg, usual care = 72.2kg). In the exercise group 70% of participants reported a previous low level of physical activity ( 1hour/wk), 30% reported high levels (1–2 or 3 hours/wk). In the usual care group 62% reported low levels of physical activity, 32% reported high levels. Interestingly, these numbers mirror the US national averages for physical activity in adults. All participants were currently undergoing treatment for their disease at the time of the intervention. In this study, the exercise group had two exercise sessions per week for 8 weeks. Each session lasted 50–60 minutes and included a warm-up (10–15 minutes), circuit training with six exercise stations (30 minutes) and stretching/relaxation (10–15 minutes). The warm-up exercises were made up of aerobic exercises using large muscle groups. During circuit training each participant completed resistance exercise at six different stations for two minutes, with a one-minute interval in which patients moved to the next station, continuing for 30 minutes in total. The primary focus of each exercise was lower and upper limb strength, standing balance, and aerobic endurance. The stretching and relaxation sessions included five minutes of static stretching for each major muscle group. The usual care group received no exercise intervention.
Only 35% of the enrolled subjects dropped out of the exercise group during the course of the study, which includes 10% who dropped out prior to starting the study. The majority of withdrawals were due to progression of disease. It is important to note that, the participants in these large RCT’s all were diagnosed with advanced cancer, but were in good physical condition as indicated by receiving clearance from their primary care physician to exercise and/or a Karnofsky performance status score ≥60. Large percentages of each cohort had received previous treatment for their disease and were undergoing treatment (chemotherapy, radiotherapy, hormonal therapy, and/or targeted therapy) at the time of the exercise intervention. In this study, individuals in the exercise group showed a significant improvement in physical performance when compared to the usual care group (Table 1). No significant difference was found in ability to complete or benefit from exercise based on previous or current treatment status or activity level in either study. Also, a number of the participants were diagnosed with co-morbidities such as heart disease, lung, disease, diabetes, and other chronic diseases. No significant differences were found in ability to exercise or benefit from exercise based on these conditions, in either study.
Despite being an intuitive therapeutic strategy, a systematic review published in 2013 reported finding no controlled studies examining exercise in pre-cachectic or cachectic patients [236]. Many of the exercise studies evaluated in this review were completed in early stage cancer patients without muscle wasting or weight loss as inclusion criteria. Furthermore, a Cochrane review reported in 2015 determined that there have been no randomized studies examining exercise interventions in cachexia [237]. It was noted that there have been many trials investigating exercise in cancer that likely included subjects with cachexia, but lack of a consistent clinical definition of cancer cachexia, until recently, has made it difficult to assess safety and efficacy of exercise specifically in this population. Most recently, a randomized phase II feasibility trial was published in 2017 evaluating a multi-modality intervention that included exercise specifically in those with (<20%) weight loss [238]. The participants were 60% male, 40% female in the exercise group and 50% male and 50% in the non-exercise, control group. Median ages in the exercise and control groups were 63 and 59 years, respectively. All were undergoing treatment for their disease. However, individuals were excluded if they had received anti-cancer treatment therapy in the preceding four weeks before the start of the research study. BMI for both groups were in the normal range (24 kg/m2). In this study the home-based resistance exercise was a combination of resistance exercises for all major muscle groups, performed using resistance bands of different tension, 3 times per week for 20 minutes. Subjects were required to start with one set of 15 reps and increase week to week based on their capabilities. The aerobic exercise regimen consisted of self-paced walking for 30 minutes 2 times per week at the patience leisure. The trial showed a early stages, this trial is significant as it provides evidence 60% compliance rate with the prescribed 10-week home-based resistance and aerobic exercise regimen. Although studying exercise in humans with cachexia remains in the that exercise trials in this population are feasible and will hopefully, lead to large, well-designed phase III trials in the years to come.
Conclusion
Cancer cachexia is a multifactorial condition characterized by weight loss resulting primarily from loss of muscle mass. The precise mechanisms by which cancer cachexia occurs are unknown, but may involve inflammation, the HPA axis, mitochondria, and myostatin and activin A pathways. Chronic activation of the immune response by cancer and its treatments may be responsible for dysfunction of the HPA axis and mitochondria that results in an upregulation of active myostatin and activin A, both of which can augment protein degradation and reduce protein synthesis. Exercise is a promising intervention and a logical next step in the treatment of muscle wasting that plagues advanced cancer patients. Exercise mitigates protein degradation while enhancing protein synthesis and mitochondrial function, while reducing systemic levels of myostatin and activin A in healthy and diseased populations. However, research on these biological mechanisms of cancer cachexia is sparse; additional research should investigate the impact of exercise on these pathways specifically in patients with cancer cachexia and determine whether or not exercise mediated regulation of the inflammatory response system can alter its downstream targets.
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle - Clinical Reports (von Haehling S, Ebner N, Morley JE, Coats AJS, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle - Clinical Reports. J Cachexia Sarcopenia Muscle Clinical Reports 2016; 1;e28:1–2.) This work was supported by a grant from the National Institutes of Health PHS awards (P30 AR069655).
Footnotes
Conflict of Interest
Calvin Cole, Ian Kleckner, Aminah Jatoi, Edward Schwarz and Richard Dunne declare that they have no conflict of interest.
References
- 1.Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, Jatoi A, Kalantar-Zadeh K, Lochs H, Mantovani G, Marks D, Mitch WE, Muscaritoli M, Najand A, Ponikowski P, Rossi Fanelli F, Schambelan M, Schols A, Schuster M, Thomas D, Wolfe R,Anker SD. Cachexia: a new definition. Clin Nutr 2008;27:793–9. [DOI] [PubMed] [Google Scholar]
- 2.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, Davis M, Muscaritoli M, Ottery F, Radbruch L, Ravasco P, Walsh D, Wilcock A, Kaasa S,Baracos VE. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–95. [DOI] [PubMed] [Google Scholar]
- 3.Fearon K, Arends J,Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nature Reviews Clinical Oncology 2013;10:90. [DOI] [PubMed] [Google Scholar]
- 4.Argiles JM, Busquets S, Stemmler B,Lopez-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer 2014;14:754–62.. [DOI] [PubMed] [Google Scholar]
- 5.Al-Majid S,Waters H. The biological mechanisms of cancer-related skeletal muscle wasting: the role of progressive resistance exercise. Biol Res Nurs 2008;10:7–20. [DOI] [PubMed] [Google Scholar]
- 6.Fearon Kenneth CH, Glass David J,Guttridge Denis C. Cancer Cachexia: Mediators, Signaling, and Metabolic Pathways. Cell Metabolism 2012;16:153–66. [DOI] [PubMed] [Google Scholar]
- 7.Andreyev HJ, Norman AR, Oates J,Cunningham D. Why do patients with weight loss have a worse outcome when undergoing chemotherapy for gastrointestinal malignancies? Eur J Cancer 1998;34:503–9. [DOI] [PubMed] [Google Scholar]
- 8.Attaix D, Ventadour S, Codran A, Bechet D, Taillandier D,Combaret L. The ubiquitin-proteasome system and skeletal muscle wasting. Essays Biochem 2005;41:173–86. [DOI] [PubMed] [Google Scholar]
- 9.Prado CM, Baracos VE, McCargar LJ, Reiman T, Mourtzakis M, Tonkin K, Mackey JR, Koski S, Pituskin E,Sawyer MB. Sarcopenia as a determinant of chemotherapy toxicity and time to tumor progression in metastatic breast cancer patientsreceivingcapecitabine treatment. Clin Cancer Res 2009;15:2920–6. [DOI] [PubMed] [Google Scholar]
- 10.Muscaritoli M, Bossola M, Aversa Z, Bellantone R,Rossi Fanelli F. Prevention and treatment of cancer cachexia: new insights into an old problem. Eur J Cancer 2006;42:31–41. [DOI] [PubMed] [Google Scholar]
- 11.Fielitz J Cancer cachexia-when proteasomal inhibition is not enough. J Cachexia Sarcopenia Muscle 2016;7:239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Utech AE, Tadros EM, Hayes TG,Garcia JM. Predicting survival in cancer patients: the role of cachexia and hormonal, nutritional and inflammatory markers. Journal of Cachexia, Sarcopenia and Muscle 2012;3:245–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inagaki J, Rodriguez V,Bodey GP. Proceedings: Causes of death in cancer patients. Cancer 1974;33:568–73. [DOI] [PubMed] [Google Scholar]
- 14.Skipworth RJ, Stewart GD, Dejong CH, Preston T,Fearon KC. Pathophysiology of cancer cachexia: much more than host-tumour interaction? Clin Nutr 2007;26:667–76. [DOI] [PubMed] [Google Scholar]
- 15.Bosaeus I Nutritional support in multimodal therapy for cancer cachexia. Supportive Care in Cancer 2008;16:447. [DOI] [PubMed] [Google Scholar]
- 16.Han HQ, Zhou X, Mitch WE,Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol 2013;45:2333–47. [DOI] [PubMed] [Google Scholar]
- 17.Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S,Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol 2009;296:C1258–70. [DOI] [PubMed] [Google Scholar]
- 18.Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M, Ringel MD, Skipworth RJ, Fearon KC, Hollingsworth MA, Muscarella P, Burghes AH, Rafael-Fortney JA,Guttridge DC. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005;8:421–32. [DOI] [PubMed] [Google Scholar]
- 19.Loumaye A, de Barsy M, Nachit M, Lause P, Frateur L, van Maanen A, Trefois P, Gruson D,Thissen JP. Role of Activin A and myostatin in human cancer cachexia. J Clin Endocrinol Metab 2015;100:2030–8. [DOI] [PubMed] [Google Scholar]
- 20.Williams A, Sun X, Fischer JE,Hasselgren P-O. The expression of genes in the ubiquitin-proteasome proteolytic pathway is increased in skeletal muscle from patients with cancer. Surgery 1999;126:744–50. [PubMed] [Google Scholar]
- 21.Kotler DP. Cachexia. Annals of Internal Medicine 2000;133:622–34. [DOI] [PubMed] [Google Scholar]
- 22.Bosaeus I Nutritional support in multimodal therapy for cancer cachexia. Support Care Cancer 2008;16:447–51. [DOI] [PubMed] [Google Scholar]
- 23.Penna F, Bonetto A, Aversa Z, Minero VG, Rossi Fanelli F, Costelli P,Muscaritoli M. Effect of the specific proteasome inhibitor bortezomib on cancer-related muscle wasting Journal of Cachexia, Sarcopenia and Muscle 2016;7:345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaughan VC, Martin P,Lewandowski PA. Cancer cachexia: impact, mechanisms and emerging treatments. J Cachexia Sarcopenia Muscle 2013;4:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saligan LN, Olson K, Filler K, Larkin D, Cramp F, Yennurajalingam S, Escalante CP, del Giglio A, Kober KM, Kamath J, Palesh O,Mustian K. The biology of cancer-related fatigue: a review of the literature. Support Care Cancer 2015;23:2461–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Illman J, Corringham R, Robinson D Jr., Davis HM, Rossi JF, Cella D,Trikha M Are inflammatory cytokines the common link between cancer-associated cachexia and depression? J Support Oncol 2005;3:37–50. [PubMed] [Google Scholar]
- 27.Adamsen L, Quist M, Andersen C, Moller T, Herrstedt J, Kronborg D, Baadsgaard MT, Vistisen K, Midtgaard J, Christiansen B, Stage M, Kronborg MT,Rorth M. Effect of a multimodal high intensity exercise intervention in cancer patients undergoing chemotherapy: randomised controlled trial. Bmj 2009;339:b3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donges CE, Burd NA, Duffield R, Smith GC, West DW, Short MJ, Mackenzie R, Plank LD, Shepherd PR, Phillips SM,Edge JA. Concurrent resistance and aerobic exercise stimulates both myofibrillar and mitochondrial protein synthesis in sedentary middle-aged men. J Appl Physiol (1985). 2012;112:1992–2001. [DOI] [PubMed] [Google Scholar]
- 29.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM,Moraes CT. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A 2009;106:20405–10. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Camera DM, Edge J, Short MJ, Hawley JA,Coffey VG. Early time course of Akt phosphorylation after endurance and resistance exercise. Med Sci Sports Exerc 2010;42:1843–52. [DOI] [PubMed] [Google Scholar]
- 31.Hulmi JJ, Ahtiainen JP, Kaasalainen T, Pollanen E, Hakkinen K, Alen M, Selanne H, Kovanen V,Mero AA. Postexercise myostatin and activin IIb mRNA levels: effects of strength training. Med Sci Sports Exerc 2007;39:289–97. [DOI] [PubMed] [Google Scholar]
- 32.Grande AJ, Silva V,Maddocks M. Exercise for cancer cachexia in adults: Executive summary of a Cochrane Collaboration systematic review. Journal of Cachexia, Sarcopenia and Muscle 2015;6(3):208–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonaldo P,Sandri M. Cellular and molecular mechanisms of muscle atrophy. Disease Models & Mechanisms 2013;6:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Atherton PJ,Smith K. Muscle protein synthesis in response to nutrition and exercise. The Journal of Physiology 2012;590:1049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bowen TS, Schuler G,Adams V. Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J Cachexia Sarcopenia Muscle 2015;6:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schiaffino S, Dyar KA, Ciciliot S, Blaauw B,Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS Journal 2013;280:4294–314. [DOI] [PubMed] [Google Scholar]
- 37.Schiaffino S,Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skeletal Muscle 2011;1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCarthy JJ,Esser KA. Anabolic and catabolic pathways regulating skeletal muscle mass. Current opinion in clinical nutrition and metabolic care 2010;13:230–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egerman MA,Glass DJ. Signaling pathways controlling skeletal muscle mass. Critical Reviews in Biochemistry and Molecular Biology 2014;49:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Argiles JM, Orpi M, Busquets S,Lopez-Soriano FJ. Myostatin: more than just a regulator of muscle mass. Drug Discov Today 2012;17:702–9. [DOI] [PubMed] [Google Scholar]
- 41.Milan G, Romanello V, Pescatore F, Armani A, Paik J-H, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, Blaauw B, DePinho RA,Sandri M. Regulation of autophagy and the ubiquitin– proteasome system by the FoxO transcriptional network during muscle atrophy 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R,Sandri M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol 2009;296:C1248–57. [DOI] [PubMed] [Google Scholar]
- 43.Fearon KC, Voss AC,Hustead DS. Definition of cancer cachexia: effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am J Clin Nutr 2006;83:1345–50. [DOI] [PubMed] [Google Scholar]
- 44.Tisdale MJ. Molecular pathways leading to cancer cachexia. Physiology (Bethesda) 2005;20:340–8. [DOI] [PubMed] [Google Scholar]
- 45.Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass HO Jr., Engstrom PF, Ezdinli EZ, Horton J, Johnson GJ, Moertel CG, Oken MM, Perlia C, Rosenbaum C, Silverstein MN, Skeel RT, Sponzo RW,Tormey DC. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med 1980;69:491–7. [DOI] [PubMed] [Google Scholar]
- 46.Martin L, Birdsell L, Macdonald N, Reiman T, Clandinin MT, McCargar LJ, Murphy R, Ghosh S, Sawyer MB,Baracos VE. Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J Clin Oncol 2013;31:1539–47. [DOI] [PubMed] [Google Scholar]
- 47.Johns N, Stephens NA,Fearon KC. Muscle wasting in cancer. Int J Biochem Cell Biol 2013;45:2215–29. [DOI] [PubMed] [Google Scholar]
- 48.Villasenor A, Ballard-Barbash R, Baumgartner K, Baumgartner R, Bernstein L, McTiernan A,Neuhouser ML. Prevalence and prognostic effect of sarcopenia in breast cancer survivors: the HEAL Study. J Cancer Surviv 2012;6:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Del Fabbro E, Parsons H, Warneke CL, Pulivarthi K, Litton JK, Dev R, Palla SL, Brewster A,Bruera E. The relationship between body composition and response to neoadjuvant chemotherapy in women with operable breast cancer. Oncologist 2012;17:1240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.George SM, McTiernan A, Villasenor A, Alfano CM, Irwin ML, Neuhouser ML, Baumgartner RN, Baumgartner KB, Bernstein L, Smith AW,Ballard-Barbash R. Disentangling the body weight-bone mineral density association among breast cancer survivors: an examination of the independent roles of lean mass and fat mass. BMC Cancer 2013;13:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lieffers JR, Bathe OF, Fassbender K, Winget M,Baracos VE. Sarcopenia is associated with postoperative infection and delayed recovery from colorectal cancer resection surgery. Br J Cancer 2012;107:931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baracos VE, Reiman T, Mourtzakis M, Gioulbasanis I,Antoun S. Body composition in patients with non-small cell lung cancer: a contemporary view of cancer cachexia with the use of computed tomography image analysis. Am J Clin Nutr 2010;91:1133s–7s. [DOI] [PubMed] [Google Scholar]
- 53.Dalal S, Hui D, Bidaut L, Lem K, Del Fabbro E, Crane C, Reyes-Gibby CC, Bedi D,Bruera E. Relationships among body mass index, longitudinal body composition alterations, and survival in patients with locally advanced pancreatic cancer receiving chemoradiation: a pilot study. J Pain Symptom Manage 2012;44:181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan BH, Birdsell LA, Martin L, Baracos VE,Fearon KC. Sarcopenia in an overweight or obese patient is an adverse prognostic factor in pancreatic cancer. Clin Cancer Res 2009;15:6973–9. [DOI] [PubMed] [Google Scholar]
- 55.Norton JA, Moley JF, Green MV, Carson RE,Morrison SD. Parabiotic transfer of cancer anorexia/cachexia in male rats. Cancer Res 1985;45:5547–52. [PubMed] [Google Scholar]
- 56.Wigmore SJ, Todorov PT, Barber MD, Ross JA, Tisdale MJ,Fearon KCH. Characteristics of patients with pancreatic cancer expressing a novel cancer cachectic factor. British Journal of Surgery 2000;87:53–8. [DOI] [PubMed] [Google Scholar]
- 57.Cariuk P, Lorite MJ, Todorov PT, Field WN, Wigmore SJ,Tisdale MJ. Induction of cachexia in mice by a product isolated from the urine of cachectic cancer patients. British Journal of Cancer 1997;76:606–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gilliam LA,St Clair DK. Chemotherapy-induced weakness and fatigue in skeletal muscle: the role of oxidative stress. Antioxid Redox Signal 2011;15:2543–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang D, Joseph DJ, Ebert MA, Galvao DA, Taaffe DR, Denham JW, Newton RU,Spry NA. Effect of androgen deprivation therapy on muscle attenuation in men with prostate cancer. J Med Imaging Radiat Oncol 2014;58:223–8. [DOI] [PubMed] [Google Scholar]
- 60.Smith MR, Finkelstein JS, McGovern FJ, Zietman AL, Fallon MA, Schoenfeld DA,Kantoff PW. Changes in body composition during androgen deprivation therapy for prostate cancer. J Clin Endocrinol Metab 2002;87:599–603. [DOI] [PubMed] [Google Scholar]
- 61.DD T, FI D, KL S, CL O, BG P,MJ L. Energy expenditure in malnourished gastrointestinal cancer patients. Cancer 1984;53:1265–73. [DOI] [PubMed] [Google Scholar]
- 62.MacDonald N, Easson AM, Mazurak VC, Dunn GP,Baracos VE. Understanding and managing cancer cachexia. J Am Coll Surg 2003;197:143–61. [DOI] [PubMed] [Google Scholar]
- 63.al-Majid S,McCarthy DO. Resistance exercise training attenuates wasting of the extensor digitorum longus muscle in micebearing the colon-26 adenocarcinoma. Biol Res Nurs 2001;2:155–66. [DOI] [PubMed] [Google Scholar]
- 64.Melville S, McNurlan MA, Calder AG,Garlick PJ. Increased Protein Turnover Despite Normal Energy Metabolism and Responses to Feeding in Patients with Lung Cancer. Cancer Research 1990;50:1125–31. [PubMed] [Google Scholar]
- 65.Moses AWG, Slater C, Preston T, Barber MD,Fearon KCH. Reduced total energy expenditure and physical activity in cachectic patients with pancreatic cancer can be modulated by an energy and protein dense oral supplement enriched with n-3 fatty acids. British Journal Of Cancer 2004;90:996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wigmore SJ, Plester CE, Ross JA,Fearon KC. Contribution of anorexia and hypermetabolism to weight loss in anicteric patients with pancreatic cancer. Br J Surg 1997;84:196–7. [PubMed] [Google Scholar]
- 67.Stuart FJ, H. FKC, A. RJ, Robert E, J. WS, James JO,C. CD. Acute-phase protein response and survival duration of patients with pancreatic cancer. Cancer 1995;75:2077–82. [DOI] [PubMed] [Google Scholar]
- 68.Bergquist JR, Ivanics T, Shubert CR, Habermann EB, Smoot RL, Kendrick ML, Nagorney DM, Farnell MB,Truty MJ. Type of Resection (Whipple vs. Distal) Does Not Affect the National Failure to Provide Post-resection Adjuvant Chemotherapy in Localized Pancreatic Cancer. Annals of Surgical Oncology 2017:1–8. [DOI] [PubMed] [Google Scholar]
- 69.Chang DK, Johns AL, Merrett ND, Gill AJ, Colvin EK, Scarlett CJ, Nguyen NQ, Leong RW, Cosman PH, Kelly MI, Sutherland RL, Henshall SM, Kench JG,Biankin AV. Margin clearance and outcome in resected pancreatic cancer. J Clin Oncol 2009;27:2855–62. [DOI] [PubMed] [Google Scholar]
- 70.Schnelldorfer T, Ware AL, Sarr MG, Smyrk TC, Zhang L, Qin R, Gullerud RE, Donohue JH, Nagorney DM,Farnell MB. Long-term survival after pancreatoduodenectomy for pancreatic adenocarcinoma: is cure possible? Ann Surg 2008;247:456–62. [DOI] [PubMed] [Google Scholar]
- 71.Yeo CJ, Abrams RA, Grochow LB, Sohn TA, Ord SE, Hruban RH, Zahurak ML, Dooley WC, Coleman J, Sauter PK, Pitt HA, Lillemoe KD,Cameron JL. Pancreaticoduodenectomy for pancreatic adenocarcinoma: postoperative adjuvant chemoradiation improves survival. A prospective, single-institution experience. Ann Surg 1997;225:621–33; discussion 33–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ueno H, Kosuge T, Matsuyama Y, Yamamoto J, Nakao A, Egawa S, Doi R, Monden M, Hatori T, Tanaka M, Shimada M,Kanemitsu K. A randomised phase III trial comparing gemcitabine with surgery-only in patients with resected pancreatic cancer: Japanese Study Group of Adjuvant Therapy for Pancreatic Cancer. Br J Cancer 2009;101:908–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oettle H, Neuhaus P, Hochhaus A, Hartmann JT, Gellert K, Ridwelski K, Niedergethmann M, Zulke C, Fahlke J, Arning MB, Sinn M, Hinke A,Riess H. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. Jama 2013;310:1473–81. [DOI] [PubMed] [Google Scholar]
- 74.Thota R, Pauff JM,Berlin JD. Treatment of metastatic pancreatic adenocarcinoma: a review. Oncology (Williston Park) 2014;28:70–4. [PubMed] [Google Scholar]
- 75.Knight BC, Kausar A, Manu M, Ammori BA, Sherlock DJ,O’Reilly DA. Evaluation of surgical outcome scores according to ISGPS definitions in patients undergoing pancreatic resection. Dig Surg 2010;27:367–74. [DOI] [PubMed] [Google Scholar]
- 76.Fearon KCH,Baracos VE. Cachexia in pancreatic cancer: new treatment options and measures of success. HPB 2010;12:323–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fouladiun M, Körner U, Bosaeus I, Daneryd P, Hyltander A,Lundholm KG. Body composition and time course changes in regional distribution of fat and lean tissue in unselected cancer patients on palliative care—Correlations with food intake, metabolism, exercise capacity, and hormones. Cancer 2005;103:2189–98. [DOI] [PubMed] [Google Scholar]
- 78.van den Beld AW, de Jong FH, Grobbee DE, Pols HA,Lamberts SW. Measures of bioavailable serum testosterone and estradiol and their relationships with muscle strength, bone density, and body composition in elderly men. J Clin Endocrinol Metab 2000;85:3276–82. [DOI] [PubMed] [Google Scholar]
- 79.Consul N, Guo X, Coker C, Lopez-Pintado S, Hibshoosh H, Zhao B, Kalinsky K,Acharyya S. Monitoring Metastasis and Cachexia in a Patient with Breast Cancer: A Case Study. Clinical Medicine Insights Oncology 2016;10:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kubo Y, Naito T, Mori K, Osawa G,Aruga E. Skeletal muscle loss and prognosis of breast cancer patients. Supportive Care in Cancer 2017;25:2221–7. [DOI] [PubMed] [Google Scholar]
- 81.Wigmore SJ, Fearon KC, Maingay JP, Garden OJ,Ross JA. Effect of interleukin-2 on peripheral blood mononuclear cell cytokine production and the hepatic acute phase protein response. Clin Immunol 2002;104:174–82. [DOI] [PubMed] [Google Scholar]
- 82.Wigmore SJ, Fearon KC, Sangster K, Maingay JP, Garden OJ,Ross JA. Cytokine regulation of constitutive production of interleukin-8 and −6 by human pancreatic cancer cell lines and serum cytokine concentrations in patients with pancreatic cancer. Int J Oncol 2002;21:881–6. [DOI] [PubMed] [Google Scholar]
- 83.Costamagna D, Costelli P, Sampaolesi M,Penna F. Role of Inflammation in Muscle Homeostasis and Myogenesis. Mediators of Inflammation 2015;2015:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Costelli P, Carbó N, Tessitore L, Bagby GJ, Lopez-Soriano FJ, Argilés JM,Baccino FM. Tumor necrosis factor-alpha mediates changes in tissue protein turnover in a rat cancer cachexia model. Journal of Clinical Investigation 1993;92:2783–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gelin J, Moldawer LL, Lönnroth C, Sherry B, Chizzonite R,Lundholm K. Role of Endogenous Tumor Necrosis Factor α and Interleukin 1 for Experimental Tumor Growth and the Development of Cancer Cachexia. Cancer Research 1991;51:415–21. [PubMed] [Google Scholar]
- 86.Baltgalvis KA, Berger FG, Pena MMO, Davis JM, Muga SJ,Carson JA. Interleukin-6 and cachexia in ApcMin/+ mice. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 2008;294:R393–R401. [DOI] [PubMed] [Google Scholar]
- 87.Seruga B, Zhang H, Bernstein LJ,Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nature Reviews Cancer 2008;8:887–99. [DOI] [PubMed] [Google Scholar]
- 88.Mantovani G, Macciò A, Mura L, Massa E, Mudu MC, Mulas C, Lusso MR, Madeddu C,Dessì A. Serum levels of leptin and proinflammatory cytokines in patients with advanced-stage cancer at different sites. Journal of Molecular Medicine 2000;78:554–61. [DOI] [PubMed] [Google Scholar]
- 89.Chu W-M. Tumor necrosis factor. Cancer letters 2013;328:222–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Han Y, Weinman S, Boldogh I, Walker RK,Brasier AR. Tumor necrosis factor-alpha-inducible IkappaBalpha proteolysis mediated by cytosolic m-calpain. A mechanism parallel to the ubiquitin-proteasome pathway for nuclear factor-kappab activation. J Biol Chem 1999;274:787–94. [DOI] [PubMed] [Google Scholar]
- 91.Tisdale MJ. Biology of cachexia. Journal of the National Cancer Institute 1997;89:1763–73. [DOI] [PubMed] [Google Scholar]
- 92.Jatoi A, Ritter HL, Dueck A, Nguyen PL, Nikcevich DA, Luyun RF, Mattar BI,Loprinzi CL. A placebo-controlled, double-blind trial of infliximab for cancer-associated weight loss in elderly and/or poor performance non-small cell lung cancer patients (N01C9). Lung Cancer 2010;68:234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jatoi A, Dakhil SR, Nguyen PL, Sloan JA, Kugler JW, Rowland KM Jr., Soori GS, Wender DB, Fitch TR, Novotny PJ,Loprinzi CL. A placebo-controlled double blind trial of etanercept for the cancer anorexia/weight loss syndrome: results from N00C1 from the North Central Cancer Treatment Group. Cancer 2007;110:1396–403. [DOI] [PubMed] [Google Scholar]
- 94.Havell EA, Fiers W,North RJ. The antitumor function of tumor necrosis factor (TNF), I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent, and limited by severe toxicity. The Journal of Experimental Medicine 1988;167:1067–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Varfolomeev EE,Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie? Cell 2004;116:491–7. [DOI] [PubMed] [Google Scholar]
- 96.Hehlgans T,Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology 2005;115:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hildebrandt GC, Olkiewicz KM, Corrion LA, Chang Y, Clouthier SG, Liu C,Cooke KR. Donor-derived TNF-alpha regulates pulmonary chemokine expression and the development of idiopathic pneumonia syndrome after allogeneic bone marrow transplantation. Blood 2004;104:586–93. [DOI] [PubMed] [Google Scholar]
- 98.Feldmann M Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol 2002;2:364–71. [DOI] [PubMed] [Google Scholar]
- 99.Szlosarek PW,Balkwill FR. Tumour necrosis factor alpha: a potential target for the therapy of solid tumours. Lancet Oncol 2003;4:565–73. [DOI] [PubMed] [Google Scholar]
- 100.Tisdale MJ. Cancer cachexia. Langenbecks Arch Surg 2004;389:299–305. [DOI] [PubMed] [Google Scholar]
- 101.Catalano MG, Fortunati N, Arena K, Costelli P, Aragno M, Danni O,Boccuzzi G. Selective up-regulation of tumor necrosis factor receptor I in tumor-bearing rats with cancer-related cachexia. Int J Oncol 2003;23:429–36. [PubMed] [Google Scholar]
- 102.Patra SK,Arora S. Integrative role of neuropeptides and cytokines in cancer anorexia-cachexia syndrome. Clin Chim Acta 2012;413:1025–34. [DOI] [PubMed] [Google Scholar]
- 103.Tijerina AJ. The biochemical basis of metabolism in cancer cachexia. Dimens Crit Care Nurs 2004;23:237–43. [DOI] [PubMed] [Google Scholar]
- 104.Seruga B, Zhang H, Bernstein LJ,Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer 2008;8:887–99. [DOI] [PubMed] [Google Scholar]
- 105.Stranahan AM, Lee K,Mattson MP. Central Mechanisms of HPA axis Regulation by Voluntary Exercise. Neuromolecular medicine 2008;10:118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.O’brien J, Ames D, Schweitzer I, Mastwyk M,Colman P. Enhanced adrenal sensitivity to adrenocorticotrophic hormone (ACTH) is evidence of HPA axis hyperactivity in Alzheimer’s disease. Psychological medicine 1996;26:7–14. [DOI] [PubMed] [Google Scholar]
- 107.Crofford LJ. The hypothalamic-pituitary-adrenal axis in the pathogenesis of rheumatic diseases. Endocrinol Metab Clin North Am 2002;31:1–13. [DOI] [PubMed] [Google Scholar]
- 108.Gold SM, Mohr DC, Huitinga I, Flachenecker P, Sternberg EM,Heesen C. The role of stress-response systems for the pathogenesis and progression of MS. Trends Immunol 2005;26:644–52. [DOI] [PubMed] [Google Scholar]
- 109.TURNBULL AV, RIVIER CL. Regulation of the Hypothalamic-Pituitary-Adrenal Axis by Cytokines: Actions and Mechanisms of Action. Physiological Reviews 1999;79:1–71. [DOI] [PubMed] [Google Scholar]
- 110.Turnbull AV,Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev 1999;79:1–71. [DOI] [PubMed] [Google Scholar]
- 111.Albertus B,Lambertus GT. Review: Endotoxin and the hypothalamo-pituitary-adrenal (HPA) axis. Journal of Endotoxin Research 2003;9:3–24. [DOI] [PubMed] [Google Scholar]
- 112.Curti BD, Urba WJ, Longo DL, Janik JE, Sharfman WH, Miller LL, Cizza G, Shimizu M, Oppenheim JJ,Alvord WG. Endocrine Effects of IL-1 [alpha] and [beta] Administered in a Phase I Trial to Patients with Advanced Cancer. Journal of Immunotherapy 1996;19:142–8. [DOI] [PubMed] [Google Scholar]
- 113.Mastorakos G, Chrousos GP,Weber JS. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. The Journal of Clinical Endocrinology&Metabolism 1993;77:1690–4. [DOI] [PubMed] [Google Scholar]
- 114. .Nolten W, Goldstein D, Lindstrom M, McKenna M, Carlson I, Trump D, Schiller J, Borden EC,Ehrlich E. Effects of cytokines on the pituitary–adrenal axis in cancer patients. Journal of interferon research 1993;13:349–57. [DOI] [PubMed] [Google Scholar]
- 115.Gisslinger H, Svoboda T, Clodi M, Gilly B, Ludwig H, Havelec L,Luger A. Interferon-α stimulates the hypothalamic-pituitary-adrenal axis in vivo and in vitro. Neuroendocrinology 1993;57:489–95. [DOI] [PubMed] [Google Scholar]
- 116.Mastorakos G, Weber JS, Magiakou M, Gunn H,Chrousos GP. Hypothalamic-pituitary-adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin-6 in humans: potential implications for the syndrome of inappropriate vasopressin secretion. The Journal of Clinical Endocrinology & Metabolism 1994;79:934–9. [DOI] [PubMed] [Google Scholar]
- 117.Tsigos C, Papanicolaou DA, Defensor R, Mitsiadis CS, Kyrou I,Chrousos G. Dose effects of recombinant human lnterleukin-6 on pituitary hormone secretion and energy expenditure. Neuroendocrinology 1997;66:54–62. [DOI] [PubMed] [Google Scholar]
- 118.Woloski B, Smith E, Meyer W, Fuller G,Blalock J. Corticotropin-releasing activity of monokines. Science 1985;230:1035–7. [DOI] [PubMed] [Google Scholar]
- 119.Smith EM,Blalock JE. Human lymphocyte production of corticotropin and endorphin-like substances: association with leukocyte interferon. Proc Natl Acad Sci U S A 1981;78:7530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Berkenbosch F, van Oers J, del Rey A, Tilders F,Besedovsky H. Corticotropin-releasing factor-producing neurons in the rat activated by interleukin-1. Science 1987;238:524–6. [DOI] [PubMed] [Google Scholar]
- 121.Zargar-Shoshtari K,Hill AG. Postoperative fatigue: a review. World J Surg 2009;33:738–45. [DOI] [PubMed] [Google Scholar]
- 122.Mueller TC, Bachmann J, Prokopchuk O, Friess H,Martignoni ME. Molecular pathways leading to loss of skeletal muscle mass in cancer cachexia--can findings from animal models be translated to humans? BMC Cancer 2016;16:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Menconi M, Fareed M, O’Neal P, Poylin V, Wei W,Hasselgren P-O. Role of glucocorticoids in the molecular regulation of muscle wasting. Critical care medicine 2007;35:S602–S8. [DOI] [PubMed] [Google Scholar]
- 124.Argilés JM, Fontes-Oliveira CC, Toledo M, López-Soriano FJ,Busquets S. Cachexia: a problem of energetic inefficiency. Journal of Cachexia, Sarcopenia and Muscle 2014;5:279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gaude E,Frezza C. Defects in mitochondrial metabolism and cancer. Cancer & Metabolism 2014;2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Argiles JM, Lopez-Soriano FJ,Busquets S. Muscle wasting in cancer: the role of mitochondria. Curr Opin Clin Nutr Metab Care 2015;18:221–5. [DOI] [PubMed] [Google Scholar]
- 127.Dumas JF, Goupille C, Julienne CM, Pinault M, Chevalier S, Bougnoux P, Servais S,Couet C. Efficiency of oxidative phosphorylation in liver mitochondria is decreased in a rat model of peritoneal carcinosis. J Hepatol 2011;54:320–7. [DOI] [PubMed] [Google Scholar]
- 128.Shum AM, Mahendradatta T, Taylor RJ, Painter AB, Moore MM, Tsoli M, Tan TC, Clarke SJ, Robertson GR,Polly P. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging (Albany NY) 2012;4:133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McLean JB, Moylan JS,Andrade FH. Mitochondria dysfunction in lung cancer-induced muscle wasting in C2C12 myotubes. Front Physiol 2014;5:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fontes-Oliveira CC, Busquets S, Toledo M, Penna F, Aylwin MP, Sirisi S, Silva AP, Orpí M, García A, Sette A, Genovese MI, Olivan M, López-Soriano FJ,Argilés JM. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: Altered energetic efficiency? Biochimica et Biophysica Acta (BBA) - General Subjects 2013;1830:2770–8. [DOI] [PubMed] [Google Scholar]
- 131.Antunes D, Padrão AI, Maciel E, Santinha D, Oliveira P, Vitorino R, Moreira-Gonçalves D, Colaço B, Pires MJ, Nunes C, Santos LL, Amado F, Duarte JA, Domingues MR,Ferreira R. Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2014;1841:896–905. [DOI] [PubMed] [Google Scholar]
- 132.Lin MT,Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787–95. [DOI] [PubMed] [Google Scholar]
- 133.Buck M,Chojkier M. Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. The EMBO Journal 1996;15:1753–65. [PMC free article] [PubMed] [Google Scholar]
- 134.White JP, Puppa MJ, Sato S, Gao S, Price RL, Baynes JW, Kostek MC, Matesic LE,Carson JA. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle 2012;2:14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Baltgalvis KA, Berger FG, Pena MM, Davis JM, Muga SJ,Carson JA. Interleukin-6 and cachexia in ApcMin/+ mice. Am J Physiol Regul Integr Comp Physiol 2008;294:R393–401. [DOI] [PubMed] [Google Scholar]
- 136.Lee SJ,McPherron AC. Myostatin and the control of skeletal muscle mass. Curr Opin Genet Dev 1999;9:604–7. [DOI] [PubMed] [Google Scholar]
- 137.Lee SJ,McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A 2001;98:9306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sidis Y, Mukherjee A, Keutmann H, Delbaere A, Sadatsuki M,Schneyer A. Biological Activity of Follistatin Isoforms and Follistatin-Like-3 Is Dependent on Differential Cell Surface Binding and Specificity for Activin, Myostatin, and Bone Morphogenetic Proteins. Endocrinology 2006;147:3586–97. [DOI] [PubMed] [Google Scholar]
- 139.Annes JP, Munger JS,Rifkin DB. Making sense of latent TGFβ activation. Journal of Cell Science 2003;116:217–24. [DOI] [PubMed] [Google Scholar]
- 140.Wolfman NM, McPherron AC, Pappano WN, Davies MV, Song K, Tomkinson KN, Wright JF, Zhao L, Sebald SM, Greenspan DS,Lee SJ. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci U S A 2003;100:15842–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Elkasrawy MN,Hamrick MW. Myostatin (GDF-8) as a key factor linking muscle mass and bone structure. J Musculoskelet Neuronal Interact 2010;10:56–63. [PMC free article] [PubMed] [Google Scholar]
- 142.McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, Smith H, Sharma M,Kambadur R. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-κB-independent, FoxO1-dependent mechanism. Journal of Cellular Physiology 2006;209:501–14. [DOI] [PubMed] [Google Scholar]
- 143.Bossola M, Muscaritoli M, Costelli P, Bellantone R, Pacelli F, Busquets S, Argiles J, Lopez-Soriano FJ, Civello IM, Baccino FM, Rossi Fanelli F,Doglietto GB. Increased muscle ubiquitin mRNA levels in gastric cancer patients. Am J Physiol Regul Integr Comp Physiol 2001;280:R1518–23. [DOI] [PubMed] [Google Scholar]
- 144.DeJong CH, Busquets S, Moses AG, Schrauwen P, Ross JA, Argiles JM,Fearon KC. Systemic inflammation correlates with increased expression of skeletal muscle ubiquitin but not uncoupling proteins in cancer cachexia. Oncol Rep 2005;14:257–63. [PubMed] [Google Scholar]
- 145.McFarlane C, Hennebry A, Thomas M, Plummer E, Ling N, Sharma M,Kambadur R. Myostatin signals through Pax7 to regulate satellite cell self-renewal. Experimental Cell Research 2008;314:317–29. [DOI] [PubMed] [Google Scholar]
- 146.Bilezikjian LM, Blount AL, Donaldson CJ,Vale WW. Pituitary actions of ligands of the TGF-beta family: activins and inhibins. Reproduction 2006;132:207–15. [DOI] [PubMed] [Google Scholar]
- 147.Bilezikjian LM, Blount AL, Leal AMO, Donaldson CJ, Fischer WH,Vale WW. Autocrine/paracrine regulation of pituitary function by activin, inhibin and follistatin. Molecular and Cellular Endocrinology 2004;225:29–36. [DOI] [PubMed] [Google Scholar]
- 148.Bilezikjian LM, Corrigan AZ, Vaughan JM,Vale WM. Activin-A regulates follistatin secretion from cultured rat anterior pituitary cells. Endocrinology 1993;133:2554–60. [DOI] [PubMed] [Google Scholar]
- 149.Tsuchida K, Nakatani M, Hitachi K, Uezumi A, Sunada Y, Ageta H,Inokuchi K. Activin signaling as an emerging target for therapeutic interventions. Cell Communication and Signaling 2009;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Clotman F, Lemaigre FP. Control of Hepatic Differentiation by Activin/TGFβ Signaling. Cell Cycle 2006;5:168–71. [DOI] [PubMed] [Google Scholar]
- 151.Poulaki V, Mitsiades N, Kruse FE, Radetzky S, Iliaki E, Kirchhof B,Joussen AM. Activin A in the Regulation of Corneal Neovascularization and Vascular Endothelial Growth Factor Expression. The American Journal of Pathology 2004;164:1293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL,Han HQ. Reversal of Cancer Cachexia and Muscle Wasting by ActRIIB Antagonism Leads to Prolonged Survival. Cell 2010;142:531–43. [DOI] [PubMed] [Google Scholar]
- 153.LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM,Brown TA. Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. J Gerontol A Biol Sci Med Sci 2009;64:940–8. [DOI] [PubMed] [Google Scholar]
- 154.Aversa Z, Bonetto A, Penna F, Costelli P, Di Rienzo G, Lacitignola A, Baccino FM, Ziparo V, Mercantini P, Rossi Fanelli F,Muscaritoli M. Changes in Myostatin Signaling in Non-Weight-Losing Cancer Patients. Annals of Surgical Oncology 2012;19:1350–6. [DOI] [PubMed] [Google Scholar]
- 155.Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M, Nair G, Arver S,Bhasin S. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci U S A 1998;95:14938–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sun DF, Chen Y,Rabkin R. Work-induced changes in skeletal muscle IGF-1 and myostatin gene expression in uremia. Kidney Int 2006;70:453–9. [DOI] [PubMed] [Google Scholar]
- 157.Ju C-R,Chen R-C. Serum myostatin levels and skeletal muscle wasting in chronic obstructive pulmonary disease. Respiratory Medicine 2012;106:102–8. [DOI] [PubMed] [Google Scholar]
- 158.Risbridger GP, Mellor SL, McPherson SJ,Schmitt JF. The contribution of inhibins and activins to malignant prostate disease. Molecular and Cellular Endocrinology 2001;180:149–53. [DOI] [PubMed] [Google Scholar]
- 159.Terpos E, Kastritis E, Christoulas D, Gkotzamanidou M, Eleutherakis-Papaiakovou E, Kanellias N, Papatheodorou A, Dimopoulos MA. Circulating activin-A is elevated in patients with advanced multiple myeloma and correlates with extensive bone involvement and inferior survival; no alterations post-lenalidomide and dexamethasone therapy. Annals of Oncology 2012;23:2681–6. [DOI] [PubMed] [Google Scholar]
- 160.Wildi S, Kleeff J, Maruyama H, Maurer CA, Buchler MW,Korc M. Overexpression of activin A in stage IV colorectal cancer. Gut 2001;49:409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kleeff J, Ishiwata T, Friess H, Buchler MW,Korc M. Concomitant overexpression of activin/inhibin beta subunits and their receptors in human pancreatic cancer. Int J Cancer 1998;77:860–8. [DOI] [PubMed] [Google Scholar]
- 162.Evans WK, Makuch R, Clamon GH, Feld R, Weiner RS, Moran E, Blum R, Shepherd FA, Jeejeebhoy KN,DeWys WD. Limited impact of total parenteral nutrition on nutritional status during treatment for small cell lung cancer. Cancer Res 1985;45:3347–53. [PubMed] [Google Scholar]
- 163.Loprinzi CL, Schaid DJ, Dose AM, Burnham NL,Jensen MD. Body-composition changes in patients who gain weight while receiving megestrol acetate. Journal of Clinical Oncology 1993;11:152–4. [DOI] [PubMed] [Google Scholar]
- 164.Simons JP, Schols AM, Hoefnagels JM, Westerterp KR, ten Velde GP,Wouters EF. Effects of medroxyprogesterone acetate on food intake, body composition, and resting energy expenditure in patients with advanced, nonhormone-sensitive cancer: a randomized, placebo-controlled trial. Cancer 1998;82:553–60. [PubMed] [Google Scholar]
- 165.Morley JE, Thomas DR,Wilson MM. Cachexia: pathophysiology and clinical relevance. Am J Clin Nutr 2006;83:735–43. [DOI] [PubMed] [Google Scholar]
- 166.Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y,Fearon KC. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials. Lancet Oncol 2016;17:519–31. [DOI] [PubMed] [Google Scholar]
- 167.Tawa NE Jr., Odessey R,Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest 1997;100:197–203.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang L, Tang H, Kou Y, Li R, Zheng Y, Wang Q, Zhou X,Jin L. MG132-mediated inhibition of the ubiquitin-proteasome pathway ameliorates cancer cachexia. J Cancer Res Clin Oncol 2013;139:1105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tisdale MJ. Biology of cachexia. J Natl Cancer Inst 1997;89:1763–73. [DOI] [PubMed] [Google Scholar]
- 170.Lissoni P, Paolorossi F, Tancini G, Barni S, Ardizzoia A, Brivio F, Zubelewicz B,Chatikhine V. Is there a role for melatonin in the treatment of neoplastic cachexia? Eur J Cancer 1996;32a:1340–3. [DOI] [PubMed] [Google Scholar]
- 171.Goldberg RM, Loprinzi CL, Mailliard JA, O’Fallon JR, Krook JE, Ghosh C, Hestorff RD, Chong SF, Reuter NF,Shanahan TG. Pentoxifylline for treatment of cancer anorexia and cachexia? A randomized, double-blind, placebo-controlled trial. J Clin Oncol 1995;13:2856–9. [DOI] [PubMed] [Google Scholar]
- 172.Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG,Zimmers TA. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 2010;391:1548–54. [DOI] [PubMed] [Google Scholar]
- 173.Bonetto A, Penna F, Minero VG, Reffo P, Bonelli G, Baccino FM,Costelli P. Deacetylase inhibitors modulate the myostatin/follistatin axis without improving cachexia in tumor-bearing mice. Curr Cancer Drug Targets 2009;9:608–16. [DOI] [PubMed] [Google Scholar]
- 174.Busquets S, Toledo M, Orpi M, Massa D, Porta M, Capdevila E, Padilla N, Frailis V, Lopez-Soriano FJ, Han HQ,Argiles JM. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J Cachexia Sarcopenia Muscle 2012;3:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Keller C, Keller P, Giralt M, Hidalgo J,Pedersen BK. Exercise normalises overexpression of TNF-alpha in knockout mice. Biochem Biophys Res Commun 2004;321:179–82. [DOI] [PubMed] [Google Scholar]
- 176.Starkie R, Ostrowski SR, Jauffred S, Febbraio M,Pedersen BK. Exercise and IL-6 infusion inhibit endotoxin-induced TNF-alpha production in humans. Faseb j 2003;17:884–6. [DOI] [PubMed] [Google Scholar]
- 177.Wittert GA, Livesey JH, Espiner EA,Donald RA. Adaptation of the hypothalamopituitary adrenal axis to chronic exercise stress in humans. Medicine and science in sports and exercise 1996;28:1015–9. [DOI] [PubMed] [Google Scholar]
- 178.Fediuc S, Campbell JE,Riddell MC. Effect of voluntary wheel running on circadian corticosterone release and on HPA axis responsiveness to restraint stress in Sprague-Dawley rats. Journal of Applied Physiology 2006;100:1867–75. [DOI] [PubMed] [Google Scholar]
- 179.White JP, Puppa MJ, Sato S, Gao S, Price RL, Baynes JW, Kostek MC, Matesic LE,Carson JA. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the Apc Min/+ mouse. Skeletal muscle 2012;2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Psilander N The effect of different exercise regimens on mitochondrial biogenesis and performance. Inst för fysiologi och farmakologi/Dept of Physiology and Pharmacology 2014. [Google Scholar]
- 181.Tonkonogi M,Sahlin K. Physical exercise and mitochondrial function in human skeletal muscle. Exerc Sport Sci Rev 2002;30:129–37. [DOI] [PubMed] [Google Scholar]
- 182.Bueno PG, Bassi D, Contrera DG, Carnielli HM, Silva RN, Nonaka KO, Selistre-de-Araujo HS,Leal AM. Post-exercise changes in myostatin and actRIIB expression in obese insulin-resistant rats. Mol Cell Endocrinol 2011;339:159–64. [DOI] [PubMed] [Google Scholar]
- 183.Hittel DS, Axelson M, Sarna N, Shearer J, Huffman KM,Kraus WE. Myostatin decreases with aerobic exercise and associates with insulin resistance. Med Sci Sports Exerc 2010;42:2023–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS,Nimmo MA. The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol 2011;11:607–15. [DOI] [PubMed] [Google Scholar]
- 185.Lara Fernandes J, Serrano CV Jr., Toledo F, Hunziker MF, Zamperini A, Teo FH, Oliveira RT, Blotta MH, Rondon MU,Negrao CE. Acute and chronic effects of exercise on inflammatory markers and B-type natriuretic peptide in patients with coronary artery disease. Clin Res Cardiol 2011;100:77–84. [DOI] [PubMed] [Google Scholar]
- 186.Ford ES. Does exercise reduce inflammation? Physical activity and C-reactive protein among U.S. adults. Epidemiology 2002;13:561–8. [DOI] [PubMed] [Google Scholar]
- 187.Pedersen BK,Hoffman-Goetz L. Exercise and the immune system: regulation, integration, and adaptation. Physiol Rev 2000;80:1055–81. [DOI] [PubMed] [Google Scholar]
- 188.Nieman DC. Exercise effects on systemic immunity. Immunol Cell Biol 2000;78:496–501. [DOI] [PubMed] [Google Scholar]
- 189.Ostrowski K, Rohde T, Asp S, Schjerling P,Pedersen BK. Pro- and anti-inflammatory cytokine balance in strenuous exercise in humans. J Physiol 1999;515:287–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Steensberg A, Keller C, Starkie RL, Osada T, Febbraio MA,Pedersen BK. IL-6 and TNF-alpha expression in, and release from, contracting human skeletal muscle. Am J Physiol Endocrinol Metab 2002;283:E1272–8. [DOI] [PubMed] [Google Scholar]
- 191.Febbraio MA,Pedersen BK. Muscle-derived interleukin-6: mechanisms for activation and possible biological roles. Faseb j 2002;16:1335–47. [DOI] [PubMed] [Google Scholar]
- 192.Pedersen BK. The anti-inflammatory effect of exercise: its role in diabetes and cardiovascular disease control. Essays Biochem 2006;42:105–17. [DOI] [PubMed] [Google Scholar]
- 193.Petersen AM,Pedersen BK. The anti-inflammatory effect of exercise. J Appl Physiol (1985) 2005;98:1154–62. [DOI] [PubMed] [Google Scholar]
- 194.Helmark IC, Mikkelsen UR, Borglum J, Rothe A, Petersen MC, Andersen O, Langberg H,Kjaer M. Exercise increases interleukin-10 levels both intraarticularly and peri-synovially in patients with knee osteoarthritis: a randomized controlled trial. Arthritis Res Ther 2010;12:R126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Droste SK, Gesing A, Ulbricht S, Muller MB, Linthorst AC,Reul JM. Effects of long-term voluntary exercise on the mouse hypothalamic-pituitary-adrenocortical axis. Endocrinology 2003;144:3012–23. [DOI] [PubMed] [Google Scholar]
- 196.Campbell JE, Kiraly MA, Atkinson DJ, D’Souza A M, Vranic M,Riddell MC. Regular exercise prevents the development of hyperglucocorticoidemia via adaptations in the brain and adrenal glands in male Zucker diabetic fatty rats. Am J Physiol Regul Integr Comp Physiol 2010;299:R168–76.. [DOI] [PubMed] [Google Scholar]
- 197.Fediuc S, Campbell JE,Riddell MC. Effect of voluntary wheel running on circadian corticosterone release and on HPA axis responsiveness to restraint stress in Sprague-Dawley rats. Journal of applied physiology 2006;100:1867–75. [DOI] [PubMed] [Google Scholar]
- 198.Dishman RK, Bunnell B, Youngstedt SD, Yoo H, Mougey E,Meyerhoff J. Activity wheel running blunts increased plasma adrenocorticotrophin (ACTH) after footshock and cage-switch stress. Physiology & behavior 1998;63:911–7. [DOI] [PubMed] [Google Scholar]
- 199.Campeau S, Nyhuis TJ, Sasse SK, Kryskow EM, Herlihy L, Masini CV, Babb JA, Greenwood BN, Fleshner M,Day HE. Hypothalamic pituitary adrenal axis responses to low-intensity stressors are reduced after voluntary wheel running in rats. J Neuroendocrinol 2010;22:872–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mahner S, Baasch C, Schwarz J, Hein S, Wölber L, Jänicke F,Milde-Langosch K. C-Fos expression is a molecular predictor of progression and survival in epithelial ovarian carcinoma. British Journal of Cancer 2008;99:1269–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Arsalan Damirchi PB, Gholamali Meysam and Ranjbar Kamal. Mitochondrial Biogenesis in Skeletal Muscle: Exercise and Aging, Skeletal Muscle Julianna Cseri, IntechOpen; 2012:24. [Google Scholar]
- 202.Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem 1967;242:2278–82. [PubMed] [Google Scholar]
- 203.Rowe GC, El-Khoury R, Patten IS, Rustin P,Arany Z. PGC-1α is Dispensable for Exercise-Induced Mitochondrial Biogenesis in Skeletal Muscle PLOS ONE 2012;7:e41817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Silva LA, Pinho CA, Scarabelot KS, Fraga DB, Volpato AMJ, Boeck CR, De Souza CT, Streck EL,Pinho RA. Physical exercise increases mitochondrial function and reduces oxidative damage in skeletal muscle. European Journal of Applied Physiology 2009;105:861–7. [DOI] [PubMed] [Google Scholar]
- 205.Phielix E, Meex R, Moonen-Kornips E, Hesselink MKC,Schrauwen P. Exercise training increases mitochondrial content and ex vivo mitochondrial function similarly in patients with type 2 diabetes and in control individuals. Diabetologia 2010;53:1714–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lumini JA, Magalhães J, Oliveira PJ,Ascensão A. Beneficial Effects of Exercise on Muscle Mitochondrial Function in Diabetes Mellitus. Sports Medicine 2008;38:735–50. [DOI] [PubMed] [Google Scholar]
- 207.Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE,Goodpaster BH. Effects of Exercise on Mitochondrial Content and Function in Aging Human Skeletal Muscle. The Journals of Gerontology: Series A 2006;61:534–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE,Goodpaster BH. Effects of Exercise on Mitochondrial Content and Function in Aging Human Skeletal Muscle. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 2006;61:534–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hansen J, Rinnov A, Brandt C,Plomgaard P. Activin A suppression during acute exercise. Signaling Originating from Membrane Receptors. Endocrine Society 2013. p. MON-398-MON-. [Google Scholar]
- 210.Dieli-Conwright CM, Spektor TM, Rice JC, Sattler FR,Schroeder ET. Hormone Therapy and Maximal Eccentric Exercise Alters Myostatin-Related Gene Expression in Postmenopausal Women. The Journal of Strength & Conditioning Research 2012;26:1374–82. [DOI] [PubMed] [Google Scholar]
- 211.Matsakas A, Friedel A, Hertrampf T,Diel P. Short-term endurance training results in a muscle-specific decrease of myostatin mRNA content in the rat. Acta Physiol Scand 2005;183:299–307. [DOI] [PubMed] [Google Scholar]
- 212.Kopple JD, Cohen AH, Wang H, Qing D, Tang Z, Fournier M, Lewis M, Casaburi R,Storer T. Effect of exercise on mRNA levels for growth factors in skeletal muscle of hemodialysis patients. J Ren Nutr 2006;16:312–24. [DOI] [PubMed] [Google Scholar]
- 213.Bassi D, Bueno PdG, Nonaka KO, Selistre-Araujo HS, Leal AMdO. Exercise alters myostatin protein expression in sedentary and exercised streptozotocin-diabetic rats. Archives of Endocrinology and Metabolism 2015;59:148–53. [DOI] [PubMed] [Google Scholar]
- 214.Lenk K, Schur R, Linke A, Erbs S, Matsumoto Y, Adams V,Schuler G. Impact of exercise training on myostatin expression in the myocardium and skeletal muscle in a chronic heart failure model. European Journal of Heart Failure 2009;11:342–8. [DOI] [PubMed] [Google Scholar]
- 215.Konopka AR, Douglass MD, Kaminsky LA, Jemiolo B, Trappe TA, Trappe S,Harber MP. Molecular adaptations to aerobic exercise training in skeletal muscle of older women. J Gerontol A Biol Sci Med Sci 2010;65:1201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Haidet AM, Rizo L, Handy C, Umapathi P, Eagle A, Shilling C, Boue D, Martin PT, Sahenk Z, Mendell JR,Kaspar BK. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci U S A 2008;105:4318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hansen J, Brandt C, Nielsen AR, Hojman P, Whitham M, Febbraio MA, Pedersen BK,Plomgaard P. Exercise Induces a Marked Increase in Plasma Follistatin: Evidence That Follistatin Is a Contraction-Induced Hepatokine. Endocrinology 2011;152:164–71. [DOI] [PubMed] [Google Scholar]
- 218.Dieli-Conwright CM, Spektor TM, Rice JC, Sattler FR,Schroeder ET. Influence of hormone replacement therapy on eccentric exercise induced myogenic gene expression in postmenopausal women. J Appl Physiol (1985) 2009;107:1381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Jensky NE, Sims JK, Rice JC, Dreyer HC,Schroeder ET. The influence of eccentric exercise on mRNA expression of skeletal muscle regulators. Eur J Appl Physiol 2007;101:473–80. [DOI] [PubMed] [Google Scholar]
- 220.Jensky NE, Sims JK, Dieli-Conwright CM, Sattler FR, Rice JC,Schroeder ET. Exercise does not influence myostatin and follistatin messenger RNA expression in young women. J Strength Cond Res 2010;24:522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Watts R, McAinch AJ, Dixon JB, O’Brien PE,Cameron-Smith D. Increased Smad signaling and reduced MRF expression in skeletal muscle from obese subjects. Obesity (Silver Spring) 2013;21:525–8. [DOI] [PubMed] [Google Scholar]
- 222.Zhu X, Topouzis S, Liang L-f, Stotish RL. Myostatin signaling through Smad2, Smad3 and Smad4 is regulated by the inhibitory Smad7 by a negative feedback mechanism. Cytokine 2004;26:262–72. [DOI] [PubMed] [Google Scholar]
- 223.Yin H, Li D, Wang Y, Zhao X, Liu Y, Yang Z,Zhu Q. Myogenic regulatory factor (MRF) expression is affected by exercise in postnatal chicken skeletal muscles. Gene 2015;561:292–9. [DOI] [PubMed] [Google Scholar]
- 224.Costa A, Dalloul H, Hegyesi H, Apor P, Csende Z, Racz L, Vaczi M,Tihanyi J. Impact of repeated bouts of eccentric exercise on myogenic gene expression. European Journal of Applied Physiology 2007;101:427–36. [DOI] [PubMed] [Google Scholar]
- 225.Raue U, Slivka D, Jemiolo B, Hollon C,Trappe S. Myogenic gene expression at rest and after a bout of resistance exercise in young (18–30 yr) and old (80– 89 yr) women. Journal of Applied Physiology 2006;101:53–9. [DOI] [PubMed] [Google Scholar]
- 226.Psilander N, Damsgaard R,Pilegaard H. Resistance exercise alters MRF and IGF-I mRNA content in human skeletal muscle. Journal of Applied Physiology 2003;95:1038–44. [DOI] [PubMed] [Google Scholar]
- 227.Kosek DJ, Kim J-s, Petrella JK, Cross JM,Bamman MM. Efficacy of 3 days/wk resistance training on myofiber hypertrophy and myogenic mechanisms in young vs. older adults. Journal of Applied Physiology 2006;101:531–44. [DOI] [PubMed] [Google Scholar]
- 228.Kleckner IR, Dunne RF, Asare M, Cole C, Fleming F, Fung C, Lin P, Mustian KM Exercise for Toxicity Management in Cancer: A Narrative Review. Oncology & Hematology Review 2018;14. [PMC free article] [PubMed] [Google Scholar]
- 229.Dunne RF, Mustian KM, Garcia JM, Dale W, Hayward R, Roussel B, Buschmann MM, Caan BJ, Cole CL, Fleming FJ, Chakkalakal JV, Linehan DC, Hezel AF,Mohile SG. Research priorities in cancer cachexia: The University of Rochester Cancer Center NCI Community Oncology Research Program Research Base Symposium on Cancer Cachexia and Sarcopenia. Curr Opin Support Palliat Care 2017;11:278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Mustian KM, Sprod LK, Palesh OG, Peppone LJ, Janelsins MC, Mohile SG,Carroll J. Exercise for the management of side effects and quality of life among cancer survivors. Curr Sports Med Rep 2009;8:325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y,Fearon KC. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials. Lancet Oncol 2016. [DOI] [PubMed] [Google Scholar]
- 232.Ebner N,von Haehling S. Unlocking the wasting enigma: Highlights from the 8th Cachexia Conference. J Cachexia Sarcopenia Muscle 2016;7:90–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Litterini AJ, Fieler VK, Cavanaugh JT,Lee JQ. Differential effects of cardiovascular and resistance exercise on functional mobility in individuals with advanced cancer: a randomized trial. Arch Phys Med Rehabil 2013;94:2329–35. [DOI] [PubMed] [Google Scholar]
- 234.Ouchi N, Parker JL, Lugus JJ,Walsh K. Adipokines in inflammation and metabolic disease. Nature reviews Immunology 2011;11:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Oldervoll LM, Loge JH, Lydersen S, Paltiel H, Asp MB, Nygaard UV, Oredalen E, Frantzen TL, Lesteberg I, Amundsen L, Hjermstad MJ, Haugen DF, Paulsen O,Kaasa S. Physical exercise for cancer patients with advanced disease: a randomized controlled trial. Oncologist 2011;16:1649–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Stene GB, Helbostad JL, Balstad TR, Riphagen II, Kaasa S,Oldervoll LM. Effect of physical exercise on muscle mass and strength in cancer patients during treatment--a systematic review. Crit Rev Oncol Hematol 2013;88:573–93. [DOI] [PubMed] [Google Scholar]
- 237.Grande AJ, Silva V,Maddocks M. Exercise for cancer cachexia in adults: Executive summary of a Cochrane Collaboration systematic review. J Cachexia Sarcopenia Muscle 2015;6:208–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Solheim TS, Laird BJA, Balstad TR, Stene GB, Bye A, Johns N, Pettersen CH, Fallon M, Fayers P, Fearon K,Kaasa S. A randomized phase II feasibility trial of a multimodal intervention for the management of cachexia in lung and pancreatic cancer. J Cachexia Sarcopenia Muscle 2017;8:778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]