

Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease (AD), and the most prevalent movement disorder. PD is characterized by dopaminergic neurodegeneration in the substantia nigra, but its etiology has yet to be established. Among several genetic variants contributing to PD pathogenesis, α-synuclein and leucine-rich repeat kinase (LRRK2) are widely associated with neuropathological phenotypes in familial and sporadic PD. α-Synuclein and LRRK2 found in Lewy bodies, a pathogenetic hallmark of PD, are often posttranslationally modified. As posttranslational modifications (PTMs) are key processes in regulating the stability, localization, and function of proteins, PTMs have emerged as important modulators of pathogenic mechanisms of α-synuclein and LRRK2. Aberrant PTMs altering phosphorylation, ubiquitination, nitration and truncation of these proteins promote PD pathogenesis, while other PTMs such as sumoylation may be protective. Although the causes of many aberrant PTMs are unknown, environmental risk factors may contribute to their aberrancy. Environmental toxicants such as rotenone and paraquat have been shown to interact with these proteins and promote their abnormal PTMs. Notably, manganese (Mn) exposure leads to PD-like neurological disorder referred to as manganism—and induces pathogenic PTMs of α-synuclein and LRRK2. In this review, we highlight the role of PTMs of LRRK2 and α-synuclein in PD pathogenesis and discuss the impact of environmental risk factors on their aberrancy.
Keywords: α-synuclein, LRRK2, Parkinson’s disease, manganese, posttranslational modifications
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder in the United States, characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the presence of protein aggregates known as Lewy bodies [1]. The primary components of Lewy bodies are α-synuclein and, often, leucine-rich repeat kinase 2 (LRRK2), whose genetic mutations have been historically implicated in cases of autosomal dominant PD [2, 3]. There are various genetic factors contributing to the pathogenesis of PD, but only mutations in α-synuclein and LRRK2 cause clinical and neuropathological phenotypes closely resembling the sporadic cases [3].
In addition to the genetic mutations of α-synuclein and LRRK2, an increasing body of evidence implicates that posttranslational modifications (PTMs) are also critically involved in PD pathogenesis [4, 5] (Fig. 1). While PTMs serve to facilitate the normal functions of α-synuclein and LRRK2 in the healthy brain [6, 7], abnormal modifications may be associated with PD pathogenesis. Additionally, environmental toxicants such as rotenone, paraquat, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and manganese (Mn) have been shown to interact with α-synuclein and LRRK2 and cause pathogenesis [8, 9]. Mn is particularly of interest since chronic exposure to Mn causes a neurological disorder resembling idiopathic PD, referred to as manganism [10]. Although the underlying mechanisms of aberrant PTMs are not well understood, pathogenic modifications following environmental exposure are known to promote aggregation and oligomerization of α-synuclein [11] and abnormal increases in LRRK2 kinase activity [12].
Figure 1.
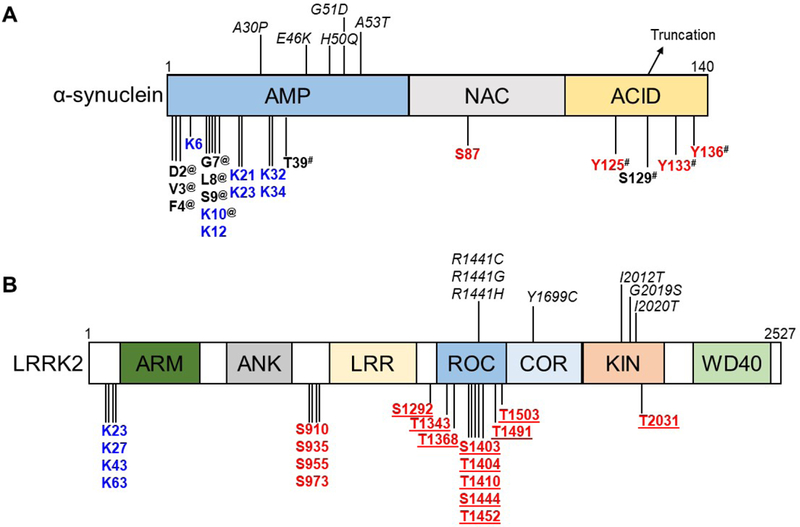
Schematic diagram of the protein domains and posttranslational modification sites of α-synuclein (A) and LRRK2 (B). Pathogenic mutations are italicized, whereas posttranslational modification sites are in bold. (A) The domains of α-synuclein are N-terminal amphiphatic (AMP), non-amyloid component (NAC) and acidic (ACID) domains. (B) The domains of LRRK2 are armadillo (ARM), ankyrin (ANK), leucine-rich repeats (LRR), Ras of complex (ROC), C-terminal of ROC (COR), kinase (KIN) and WD40. Posttranslational modification sites are depicted in red for phosphorylation and blue for ubiquitination. Other posttranslational modifications in α-synuclein are labelled (#) for nitration and (@) for acetylation. Truncation of α-synuclein occurs in its C-terminal (ACID) domain. Autophosphorylation sites in LRRK2 are underlined.
Therefore, elucidating the mechanisms of PTMs that influence pathogenesis may greatly expand our understanding of PD and leads to the identification of therapeutic targets. This review will discuss the PTMs of α-synuclein and LRRK2, the impact of the aberrant PTMs on PD pathogenesis and the influence of environmental toxins on pathogenic modifications.
α-Synuclein structure and its genetic variants
α-Synuclein is an acidic protein of 140 kD encoded by the SNCA gene, consisted of 3 domains: the N-terminal amphipathic (AMP), non-amyloid component (NAC), and acidic (ACID) domains (Fig. 1A) [13]. α-Synuclein is abundant in the human brain, comprising nearly 1% of all cytosolic neuronal proteins [14]. Despite its abundance, the physiological function of α-synuclein is not well understood. Alternately, cytoplasmic- or membrane-associated α-synuclein is localized primarily in the presynaptic nerve terminals and is thought to modulate vesicular trafficking, serving as a chaperone during SNARE complex formation [15]. Its pleiotropic functions are supported by a dynamic conformation—an unstructured, monomeric form or an α-helical oligomer with three domains [4]. As the first protein discovered to be causally linked to PD, studies have indicated a distinct role for each domain of α-synuclein in PD pathogenesis [16]. Though the mechanisms are poorly understood, all currently known mutations occur in the N-terminal AMP domain (A53T, G46L, G51A, etc.) and result in an autosomal dominant syndrome resembling sporadic PD [17]. The N-terminal domain also contains four repeats with a mitochondrial targeting signal, potentially underscoring the mitochondrial dysfunction seen in PD [17]. The second NAC domain is a hydrophobic core strongly associated with α-synuclein aggregation and fibrillization [18]. The third, acidic C-terminal domain is thought to facilitate protein-protein interactions and promote the formation of pathogenic fibrils [19].
LRRK2 structure and its genetic variants
LRRK2 is a multi-domain protein of 2,527 amino acids of approximately 280 kDa encoded by the PARK8 gene [20]. Alternately known as dardarin, LRRK2 functions both as a GTPase and kinase and is primarily localized in the cytoplasm and along the plasma membrane [21]. LRRK2 is highly expressed in the cortex and striatum, particularly in the pyramidal and medium spiny neurons [22], playing a role in vesicular trafficking, synaptic function and neurite development [23–25]. High levels of LRRK2 were also found in immune cells such as microglia and macrophages, indicating its role in inflammation, phagocytosis, and tissue repair [26, 27].
LRRK2 contains several domains: (1) armadillo (ARM), (2) ankyrin (ANK), (3) leucine-rich repeats (LRR), (4) Ras of complex (ROC), (5) C-terminal of ROC (COR), (6) kinase (KIN) and (7) WD40 domains [28] (Fig. 1B). The ROC-COR (ROCO) and KIN domain are critical for GTPase binding and kinase activity, respectively. Among 2,527 amino acid sequences of LRRK2, 20 amino acid substitutions are reported to be pathogenic, while other amino acids substitutions are associated with polymorphisms [28]. Most pathogenic mutations occur within the ROCO or KIN domain and result in a pathology resembling sporadic PD [29] (Fig. 1B). Mutations in the LRRK2 gene are the greatest genetic contributors to PD and cause autosomal-dominant PD, accounting for 13% of all familial PD cases and up to 40% of familial PD in some populations [30]. LRRK2 mutations have also been implicated in late-onset PD, constituting 1–2% of all sporadic cases [31].
The amino acid substitution of glycine for serine at position 2019 (G2019S) is the most common pathogenic mutation of LRRK2 [32]. Lewy body pathology is not present in all patients with LRRK2 mutations, but the G2019S is the most common mutation found in Lewy body-associated LRRK2 [29]. Other pathogenic mutations are found within the KIN (I2020T, I2012T) and ROCO domains (R1441C, R1441G, R1441H, Y1699C) [28]. Mutations within these domains affect the GTPase and kinase activity of LRRK2 and cause pathogenic inflammation [33] and neurodegeneration [34]. These mutations are also associated with abnormal PTMs such as phosphorylation and ubiquitination, leading to atypical LRRK2 localization and pathogenic instability [35].
The interplay of α-synuclein and LRRK2
The genetic mutations of α-synuclein and LRRK2 in autosomal dominant PD have been extensively characterized [36], but their interplay in sporadic PD is not well understood. Lewy bodies, the pathogenic hallmark of PD, are comprised primarily of phosphorylated α-synuclein—though LRRK2 is often found accumulated within their core [37]. This may indicate that beyond genetic mutations, aberrant PTMs of α-synuclein and LRRK2 may contribute to their pathogenicity.
Despite their co-localization in Lewy bodies, few studies have found direct interactions between α-synuclein and LRRK2 under physiological conditions [3]. In addition, α-synuclein and LRRK2 may interact indirectly through [34] their common pathways and functions. Both α-synuclein and LRRK2 complex with the 14–3-3 protein family after phosphorylation, a PTM contributing to pathogenesis of both α-synuclein and LRRK2 [38, 39]. α-synuclein and LRRK2 also modulate the dynamic cytoskeleton [3] and mediate vesicular transport [2], suggesting a common functionality that could be disrupted in PD pathogenesis.
Although their interactions under physiological conditions are currently unknown, α-synuclein and LRRK2 have been co-immunoprecipitated in postmortem PD brain samples [40]. It appears that LRRK2 regulates localization and clearance of α-synuclein in the brain via lysosomal degradation and microglial endocytosis [41–45]. G2019S LRRK2 mutation increases the formation of α-synuclein aggregates and exacerbates α-synuclein pathology [46], indicating that LRRK2 regulates the expression and proper clearance of α-synuclein. LRRK2 knockout attenuated α-synuclein aggregation in a transgenic mouse model as well as in vitro cell culture model [40]. Interestingly, one study reported that knockdown of LRRK2 resulted in the aggregation of α-synuclein [34]. This indicates that the interplay between LRRK2 and α-synuclein might be complex and regulated by many factors.
A recent study has shown that LRRK2 promoted α-synuclein aggregation by phosphorylating RAB35 [43], a RAB GTPase known to regulate vesicular trafficking and sorting [47]. This relationship was observed in both in vitro and in vivo settings [43]. Moreover, G2019S LRRK2 mutant kinase activity correlated with RAB35 hyperphosphorylation, resulting in α-synuclein aggregation, altered endosomal trafficking and lysosomal degradation in the brains of PD patients and in mice [43]. Alternately, studies found that α-synuclein overexpression increased LRRK2 kinase activity, with concomitant dopaminergic neurodegeneration in mice [44], while inhibition of LRRK2 attenuated α-synuclein toxicity in rats [48, 49].
Taken together, these findings suggest that LRRK2 kinase activity and α-synuclein are regulated bi-directionally in PD pathogenesis. Moreover, much of the PD-associated pathogenicity seen in α-synuclein and LRRK2 involve aberrant PTMs. For example, studies have shown that aberrant phosphorylation of both proteins has been associated with PD pathogenesis [38], indicating that understanding the role of PTMs on α-synuclein and LRRK2 is critical in furthering our knowledge of PD pathogenesis.
Posttranslational modifications of α-synuclein
Phosphorylation
An increasing body of evidence links phosphorylation of α-synuclein to Lewy body formation [50]. α-Synuclein phosphorylated at the S129 residue has been identified as the primary component of Lewy bodies [51]. Though phosphorylation of other serine (S87) [52] and tyrosine (Y125, Y133, Y136) [53] residues has been observed, phosphorylation of S129 remains the predominant PTMs associated with aggregated α-synuclein [51] (Fig. 1A).
Phosphorylation of S129 is mediated by kinases such as Polo-like kinase 2 (PLK2) [54]. Studies have shown increased PLK2 expression in the brains of aging primates, with concomitant increases in phosphorylated S129 [55]. In transgenic mice overexpressing WT α-synuclein, PLK2 was found to co-localize with phosphorylated α-synuclein [54]. Further, phosphoprotein phosphatase 2A (PP2A), a phosphatase which dephosphorylates residue S129, has been shown to attenuate the formation of pathogenic aggregates in the aging mouse brain. [56]. In the human brain and in vitro studies, α-synuclein accumulation was associated with a reduction in PP2A activity [57]. These findings suggest a significant role of S129 phosphorylation in the formation of α-synuclein aggregates.
The effects of α-synuclein phosphorylation extend beyond inclusion body formation. While the mechanisms of pathogenicity remain unclear, S129 phosphorylation has been implicated in other pathogenic hallmarks of PD [58]. Phosphorylation of S129 accelerated neuronal loss and axonal degeneration in transgenic mice expressing α-synuclein as compared to wild-type and phosphorylation-deficient mice [50, 59]. It has been reported that S129 phosphorylation of α-synuclein is linked to the formation of reactive oxygen species (ROS), superoxide formation and mitochondrial dysfunction [60]. Other studies have shown that S129 phosphorylation is associated with neuronal cell death, mitochondrial damage and potential endoplasmic reticulum-Golgi trafficking disruption [61, 62]. Together, these studies demonstrate the importance of α-synuclein phosphorylation in PD-associated pathogenicity.
Ubiquitination
Despite extensive characterization of phosphorylated α-synuclein within Lewy bodies, studies demonstrate that synucleinopathic lesions also contain monoubiquitinated α-synuclein [63], and phosphorylated α-synuclein polypeptides migrate with ubiquitinated α-synuclein in vitro [64]. Yet the mechanisms of α-synuclein ubiquitination are not well understood. Only a small fraction of aggregated α-synuclein is ubiquitinated, despite the presence of ubiquitin chains in Lewy body inclusions [64]. Instead, ubiquitination of α-synuclein in vitro induces significant aggregation [65], suggesting that ubiquitination is not directly responsible for the formation of Lewy bodies, but may instead promote the aggregative properties of α-synuclein. In the healthy brain, soluble α-synuclein is degraded via the ubiquitin-proteasome system, while insoluble aggregates are degraded by autophagy [66]. α-synuclein is thought to be targeted for degradation by the addition of a ubiquitin chain covalently linked at lysine residues [67]. The conjugation of ubiquitin chains is facilitated by E3 ubiquitin ligases, which have decreased activity in patients with sporadic PD [68]. Further, mutation of the Parkin gene, which encodes the E3 ligase Parkin [69], and ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) have been strongly associated with sporadic PD [70].
As the levels of α-synuclein mRNA do not increase in PD as compared to control patients [71], it is plausible that the inhibition of degradation plays a role in the pathological accumulation of α-synuclein. In vitro studies have demonstrated that E3 ubiquitin ligases such as E6-associated protein (E6-AP) [72] and carboxyl terminus Hsp70-interacting protein (CHIP) [73] are localized in Lewy bodies. Additionally, the E3 ligase seven in absentia homolog 1 (SIAH1) has been shown to monoubiquitinate α-synuclein at lysine residues K12, K21, K23, K32, and K34, sites which were previously found to be ubiquitinated in Lewy bodies [65]. After mono-ubiquitination by SIAH1, α-synuclein aggregation increased significantly both in vitro and in vivo. Monoubiquitinated α-synuclein showed increased aggregation and inclusion body formation in dopaminergic neurons [74].
Though little is known about the pathogenic mechanisms of α-synuclein ubiquitination, recent studies have indicated a protective role for this PTM. Ubiquitination of α-synuclein by neuronal precursor cell-expressed, developmentally down-regulated gene 4 (Nedd4) promoted the degradation of α-synuclein via the endosomal-lysosomal pathway [63]. In support of this finding, transgenic mice expressing a form of α-synuclein unable to undergo Nedd4-associated ubiquitination showed increased α-synuclein aggregation and un-ubiquitinated synucleinopathy lesions [75]. Moreover, knockdown of ubiquitin carboxyl-terminal hydrolase 8 (USP8), which co-localizes with membrane-associated α-synuclein and cleaves ubiquitin chains from K63, increased the rate of α-synuclein aggregate clearance both in vitro and in vivo [76]. However, WT deubiquitinase USP19 promoted in vitro secretion of misfolded α-synuclein and protein “seeding” in a dose-dependent manner [77]. Altogether, these findings underscore the need for elucidation of the mechanism and effects of ubiquitination on α-synuclein and PD pathology.
Nitration
Many genes associated with PD pathogenesis have been implicated in mitochondrial dysfunction. Lewy bodies isolated from patients with PD have been found to contain both nitrated and phosphorylated forms of α-synuclein [78], and the presence of nitrated α-synuclein has been used as a biomarker of neurodegeneration [79]. Reactive nitrogen species (RNS), along with their oxygen counterparts, are thought to significantly contribute to neurodegeneration and pathogenicity of PD [80]. Studies have demonstrated that the oxidative stress associated with PD leads to the nitration of tyrosine residues on α-synuclein, primarily Y39, Y125, Y133, and Y136 [81]. Further, research has shown that nitration of Y39 decreases vesicular binding of α-synuclein [82] and promotes the formation of α-synuclein oligomers via tyrosine crosslinking [79]. α-Synuclein nitration has also been shown to induce both dopaminergic neuronal loss in the substantia nigra of rats and motor dysfunctions seen in sporadic PD [83].
Interestingly, studies diverge on the role of nitration in α-synuclein pathogenicity. A recent study showed that nitration of α-synuclein induces the formation of soluble oligomers and prevents its fibrillation [84]. On the other hand, nitrated Y125 has been shown to induce the dimerization of α-synuclein, which promotes its accumulation and eventual aggregation [82]. Additionally, incubation of Y125-nitrated α-synuclein with un-nitrated α-synuclein resulted in the seeding of nitrated α-synuclein into previously un-nitrated fibrils [82]. While the consequences of α-synuclein nitration are not well understood, the diverging inhibition of fibrillation, oligomer stabilization, and neuronal loss indicate a potentially significant role of nitration in the α-synuclein modification. As the pathogenic effects of nitrated α-synuclein show similarities with the toxicity induced by the A30P point mutation [4], it remains to be determined whether nitration is the PTM partially responsible for pathogenicity or the downstream result of PD-associated oxidative stress and neuronal injury.
Truncation
Analysis of Lewy bodies has indicated the presence of truncated α-synuclein species [85]. Thus, a focus of recent research has been clarification of the role of truncation in α-synuclein pathology. Truncation at the C-terminus has been associated with aggregation of α-synuclein [85], but the pathogenicity of this modification is still controversial. While the cleavage of α-synuclein has been shown to induce PD pathology in transgenic mice [86], in vitro fragmentation did not result in the formation of α-synuclein fibrils [87]. Further, protein sequence studies have shown truncated α-synuclein bands in both control and PD patients, indicating a constitutive truncation in the normal human brain [85].
Conversely, others have found that insoluble aggregates contain truncated α-synuclein variates that are toxic to neurons [88]. Overexpression of truncated α-synuclein led to increased α-synuclein aggregation and synucleinopathic lesions in transgenic mice [89] and cross-linking of α-synuclein fibrils in vitro [90]. Further, blocking of C-terminus truncation in mThy1-α-syn mouse model attenuated PD pathology and reversed accumulation of α-synuclein [91]. C-terminally truncated α-synuclein has been shown to seed the aggregation of full-length α-synuclein in vitro [92] and truncated variants show an increased rate of fibrillization compared to full-length α-synuclein [88]. Together, these findings demonstrate the need for further investigation of the role of truncation in α-synuclein-associated pathogenesis.
Sumoylation
Though many PTMs result in a pathogenic phenotype, sumoylation of α-synuclein has been found to serve as a protective modification. In vitro, sumoylation reversed the accumulation and fibrillation of α-synuclein [93]. Inhibition of sumoylation in a His6-SUMO2 transgenic mouse model resulted in α-synuclein inclusion body formation and neurotoxicity [93]. The protective effect of sumoylation is thought to be due to the enhancement of protein solubility, a finding consistent in studies of many aggregation-prone proteins [94]. In contradiction to these findings, an in vitro study linked sumoylation to impaired α-synuclein ubiquitination and reduced proteasomal degradation [95]. Increased sumoylation was also associated with α-synuclein aggregation in postmortem human brain samples [95]. These contradictory findings highlight the need for further exploration of the role of sumoylation in PD pathogenicity.
Acetylation
Studies have also indicated a protective role for acetylation against α-synuclein pathogenesis. N-terminal acetylation was found to induce helical folding of α-synuclein, a conformational change which decreased its propensity to aggregate [96]. Molecular simulation studies have also shown that N-terminal acetylation disrupted the internal hydrogen bonds of α-synuclein and reduced its aggregative properties [97], suggesting a possible mechanism of protection. Though most studies on the protective role of α-synuclein acetylation are spectroscopy or simulation-based [97, 98], the protective role of α-synuclein acetylation may present an exciting avenue for future research.
LRRK2
Posttranslational modifications of LRRK2
Phosphorylation
Of the PTMs associated with LRRK2, phosphorylation is the most extensively studied. However, the mechanisms undergirding its pathogenicity are complex and not fully understood [99]. Under physiological conditions, LRRK2 is phosphorylated at multiple serine (S) and threonine (T) residues across several domains [100] (Fig. 1B). LRRK2 phosphorylation can be classified into constitutive and auto-phosphorylation, both of which are modulated in LRRK2-associated PD [100].
Constitutive phosphorylation of LRRK2 has been identified at multiple residues located between the ANK and LRR domains [100] (Fig. 1B). Studies have shown that constitutive phosphorylation regulates the subcellular localization, stability, and function of LRRK2 [101]. Mutations in the KIN and ROCO domains alter constitutive phosphorylation of LRRK2 and are associated with PD pathogenesis [102]. The G2019S mutation of LRRK2 increased phosphorylation in dopaminergic cell lines, resulting in abnormal neurite growth and increased neuronal vulnerability [103]. Mutation in G2019S also increased LRRK2 kinase activity [104, 105], while inhibition of kinase activity reduced its constitutive phosphorylation [103]. This indicates that G2019 mutations increase LRRK2 constitutive phosphorylation and promote LRRK2 kinase activity. Inflammation may also induce abnormalities in constitutive phosphorylation. Overexpression of LRRK2 in HEK293T cells increased constitutive phosphorylation and induced microglial inflammation in response to the inflammatory agent lipopolysaccharide (LPS) [106], implicating abnormal constitutive phosphorylation in inflammatory pathogenesis.
Conversely, ROCO domain mutations decreased constitutive phosphorylation of LRRK2, particularly in R1441C, R1441G, and Y1699C mutations [107, 108]. These mutations also led to LRRK2 aggregation [101]. Decreased constitutive phosphorylation has been associated with LRRK2 aggregation in the substantia nigra of postmortem PD brains and the presence of restricted Lewy bodies [38]. Moreover, decreased constitutive phosphorylation, particularly at the S935 residue, has been associated with abnormal localization of LRRK2 and clinical PD diagnosis [6]. Though the mechanisms are unclear, alterations in constitutive phosphorylation appear to promote LRRK2 pathogenicity. Interestingly, mutations in ROCO domains (such as G2019S) enhanced the kinase activity of LRRK2 but decreased its constitutive phosphorylation [109, 110]. These findings suggest a need for further investigation of the complex and bidirectional mechanisms of constitutive phosphorylation.
LRRK2 kinase activity is also regulated by autophosphorylation [111]. Autophosphorylation occurs along multiple residues (S1292, S1403, T1404, T1410, T1491), primarily in the ROCO domains (Fig. 1B). Although the function of autophosphorylation at each site is not yet known, autophosphorylation in this domain appears to be correlated with LRRK2 kinase activity [109, 110]. Accordingly, mutations in the ROCO domains such as R1441C or Y1699C increased autophosphorylation and LRRK2 kinase activity [28, 112, 113]. Changes in autophosphorylation and kinase activity resulted in defects in the lysosomal degradation pathway [114].
Additionally, KIN domain mutations, such as G2019S and I2020T, also increased autophosphorylation and kinase activity of LRRK2 [113]. G2019S mutation induced autophosphorylation and kinase activity, resulting in altered lysosomal pH, lysosomal defects and elevated expression of the lysosomal ATPase ATP13A2, a gene linked to a parkinsonian syndrome (Kufor–Rakeb syndrome) in both in vivo and in vitro settings [114]. Inhibition of LRRK2 kinase activity rescued these pathogenic effects, indicating that the defects associated with these mutations are dependent on both kinase activity and autophosphorylation of LRRK2.
Mutations in the KIN and ROCO domains of LRRK2 play a critical role in PD, exhibiting a wide range of effects through changes in LRRK2 phosphorylation. Thus, further investigation and analysis of LRRK2 phosphorylation may help to further our understanding of the complex pathways of LRRK2-associated PD pathogenesis.
Ubiquitination of LRRK2
The ubiquitination of LRRK2 regulates its stability and clearance [100]. LRRK2 ubiquitination occurs at multiple N-terminal ARM lysine (K) residues of LRRK2 (K27, K29, K48, K63) [100]. Ubiquitination at K48 and K63 residues signal the degradation of LRRK2 by ubiquitin-proteasome and autophagy-lysosome pathways, respectively [100, 102].
Members of the 14–3-3 protein family bind to LRRK2 and regulate its ubiquitination, a process closely associated with constitutive phosphorylation [101]. Decreased constitutive phosphorylation of LRRK2 disrupted 14–3-3-LRRK2 binding both in vitro and in a mouse model, resulting in cytoplasmic accumulation of LRRK2 without disruption of kinase activity [101]. Additionally, PD-associated LRRK2 mutations such as R1441C, R1441G, Y1699C and I2020T decreased constitutive phosphorylation of LRRK2 in vitro, resulting in its hyperubiquitination and instability [115]. These unstable mutants formed abnormal protein complexes [102] and promoted cytosolic aggregation of LRRK2 [115] as compared to the WT. Knockdown of LRRK2 impaired autophagy-lysosomal degradation in transgenic mice, resulting in increased protein ubiquitination, oxidative stress, inflammation and apoptosis [34]. Additionally, studies found that LRRK2 present in Lewy bodies is highly ubiquitinated [116]. Aggregated LRRK2 has been found to co-localize with SOCS box-containing protein 1 (WSB1), a ubiquitinase also found in Lewy bodies, while co-expression of WSB1 and LRRK2 resulted in increased LRRK2 aggregation and insolubility [116]. Taken together, these results suggest that that alteration in LRRK2 ubiquitination may significantly impact PD-associated pathogenesis, perhaps via inhibition of normal degradative pathways.
Gene-environment interaction: α-synuclein, LRRK2, and environmental toxicants
As only a small percentage of PD cases can be attributed to genetic mutations [2], researchers have begun to explore the contribution of environmental exposure to PD onset and pathogenesis. While many pathogenic PTMs occur sporadically, environmental toxins have been implicated in the pathogenic modification of α-synuclein and LRRK2. An increasing body of evidence links toxicants such as MPTP, rotenone, paraquat, and Mn to PD pathogenesis, though the mechanisms involved in pathogenesis remain to be elucidated.
α-Synuclein and environmental toxins
Perhaps the best known of all parkinsonian neurotoxins is MPTP, which induces dopaminergic neurodegeneration and inclusion body formation after its metabolism to 1-methyl-4-phenylpyridinium (MPP+) in the brain [117–119]. MPTP toxicity mimics the hallmarks of sporadic PD in humans, primates, and some rodents via inhibition of mitochondrial complex I [118] as well as post-translational modifications [118]. In a non-human primate model, exposure to MPTP resulted in S129-phosphorylation of α-synuclein [55]. These modifications occurred alongside elevated neuronal levels of PLK2, a serine/threonine kinase known to phosphorylate α-synuclein [55]. Additionally, mice treated with MPTP showed nitration of α-synuclein in the striatum and midbrain [120]. Moreover, α-synuclein null mice were resistant to the effects of MPTP [121], suggesting that MPTP preferentially targets α-synuclein to exert its neurodegenerative effects. These findings contradict previous in vitro studies, which implicated dysfunctional dopamine transporters in MPTP toxicity [122].
Rotenone is an insecticide that preferentially targets the brain and exerts PD-like neuropathology in experimental animal models [123]. Like MPTP, rotenone contributes to mitochondrial dysfunction and is associated with increased α-synuclein phosphorylation and aggregation in in vitro settings [124]. Moreover, tandem mass spectrometry and pull-down assays have indicated that rotenone exposure induces nitration of α-synuclein [125]. Paraquat, an herbicide, also induces significant fibrillation and aggregate formation in α-synuclein after exposure [126]. Interestingly, mice overexpressing α-synuclein showed α-synuclein aggregation and inclusion body formation after paraquat exposure, but they did not show signs of the neurodegeneration associated with these pathogenic hallmarks [127]. These divergent results suggest a need for continued exploration of the mechanisms undergirding environmentally-associated pathogenesis.
α-Synuclein and Mn
Chronic exposure to Mn has been linked to a parkinsonian-like syndrome known as manganism, which is characterized by motor symptoms resembling parkinsonism [128]. Mn exposure has been associated with α-synuclein aggregation, oligomerization and oxidative stress in vitro and in transgenic mouse models [129]. Studies have also found that Maneb, a fungicide containing Mn, increased α-synuclein aggregation, oxidative stress and proteasomal dysfunction in vitro [130].
α-Synuclein contains several metal binding sites in the C-terminal region [131]. While α-synuclein demonstrates a low binding affinity for Mn [132], Mn exposure significantly increased α-synuclein aggregation [133]. These findings may indicate that Mn does not directly interact with α-synuclein, but instead may induce aberrant PTMs of α-synuclein leading to aggregate. Studies have shown that Mn induces nitrosylation and cleavage of α-synuclein, resulting in oligomerization and fibrillation in a rat brain model [77]. In support of these findings, it has been demonstrated that WT α-synuclein protected against Mn-induced neurodegeneration and oxidative stress in a C. elegans model of PARK2- and PARK7-homolog knockout [134]. As WT α-synuclein attenuated Mn neurotoxicity in this model, suggesting that Mn exerts its toxic effects in part by inducing aberrant PTMs. These findings suggest that Mn may induce pathogenic PTMs of α-synuclein similar to those seen in sporadic PD, underscoring a need for further investigation.
LRRK2 and environmental toxins
The G2019S mutation of LRRK2 is heavily linked to PD pathogenesis. Intriguingly, MPTP exacerbated dopaminergic neuronal loss in G2019S-transgenic mice [8], suggesting the potential interaction of environmental toxins with PD-associated genes. LRRK2 mutation increased mitochondrial dysfunction in C. elegans after exposure to rotenone and paraquat [135]. LRRK2 G2019S transgenic flies also showed dopaminergic neurodegeneration and mitochondrial defects after exposure to rotenone [136]. Conversely, LRRK2 knockout mice did not exhibit dopaminergic neurodegeneration after MPTP exposure, despite its known inhibition of mitochondrial complex I [137]. This may suggest that environmental toxins exert pathogenic effects via different mechanisms.
Importantly, mounting evidence demonstrates that LRRK2 is involved in neural inflammation [138] although the role of PTMs in the LRRK2-inflammation process is not much studied. Knockdown of LRRK2 attenuated LPS-induced expression of inflammatory cytokines such as TNF-α and IL-1β in murine microglia [106]. Given that inflammatory processes are a risk factor for PD [139], the involvement of LRRK2 in the brain inflammation process would lead to a neuroinflammatory risk of developing PD. LRRK2 is also known to play a role in the apoptotic pathway by triggering the extrinsic cell death pathway and FADD/caspase-8 signaling [140]. The exact mechanisms of LRRK2 in mitochondrial dysfunction and inflammation and how its deletion or mutation modulates environmental toxins in PD pathogenesis are currently under intensive investigations as the gene-environment interactions aggravate and enhance PD pathology.
LRRK2 and Mn
Mn causes neurotoxicity by several mechanisms including inflammation [141]. Notably, studies demonstrate that LRRK2 plays a role in Mn toxicity. Studies demonstrated that Mn increased LRRK2 protein levels in microglia, regulating the autophagy process by increasing autophagy-related proteins such as Atg5 and Beclin-1 [12]. Moreover, inhibition of LRRK2 attenuated the Mn-induced autophagy dysfunction and inflammation, suggesting that LRRK2 activity exacerbates Mn-induced inflammation [12]. Interestingly, G2019S mutation of LRRK2 enhanced KIN domain-Mn interactions, prolonging and increasing LRRK2 kinase activity [142]. In contradiction to these findings, studies showed that Mn-induced oxidative stress and neurotoxicity are exacerbated after LRRK2 downregulation in HEK293 cells, suggesting a protective role of LRRK2 in Mn toxicity [143]. These findings indicate that Mn toxicity may involve alterations in the kinase activity of LRRK2 and its regulation of the autophagy-lysosomal pathway, warranting further exploration of the gene-environmental interaction in pathogenic PTMs.
Advances and limitations in studying PTMs
Although studying PTMs is critical to understanding the pathogenic processes of α-synuclein and LRRK2 for developing strategies and therapeutics of PD, it is extremely challenging due to limitations in the scope of current technology and the availability of resources.
Methods such as Western blotting and radiolabeling provide specific and quantitative information regarding PTMs, but require knowledge of the exact PTM of interest [144]. Further, these assays are often limited by a lack of antibody availability and/or specificity[144]. Advances in mass spectrometry (MS)-based proteomics, such as orbitrap based high-resolution MS or quadrupole/time-of-flight (Q-TOF) MS, allow for analysis without prior knowledge of the PTMs [145]. Additionally, these methods allow for the simultaneous detection of all PTM sites within a target protein. Advances in the field now enable researchers to use MS-based proteomics to understand not only the sites of PTMs but to better characterize the dynamics and consequences of these modifications [146, 147]. In PD pathogenesis, MS-based methods may also serve as a tool for examining the protein-protein interactions of LRRK2 and α-synuclein [39, 146]. Further, combining MS with additional assays such as the pull-down assay may further detect the protein-protein interactions undergirding PD pathogenesis [145].
Although MS-based methods have significantly improved the study of PTMs, several limitations remain. First, spectrometric assays require a robust cell sample for optimal analysis, limiting opportunities for sampling among PD patients of animal models with significant neurodegeneration. Moreover, PTMs are dynamic and extremely complex, limiting the analytical power of mass spectrometry to only a brief ‘snapshot’ of a rapidly changing system [145]. Proximity ligation assays and proximity ligation imaging cytometry have been developed to counter this limitation, allowing for the direct analysis of PTMs and protein-protein interactions in situ [44, 148], However, the development of additional advanced strategies will allow for the further examination of the aberrant PTMs and protein-protein interactions of α-synuclein and LRRK2 involved in PD pathogenesis.
Conclusion
Whether genetically-, sporadically- or environmentally-induced aberrant PTMs of α-synuclein and LRRK2 have been strongly linked to PD pathogenesis (Fig. 2). While PTMs support the diversity and pleiotropic functions of α-synuclein and LRRK2 in the healthy brain, pathogenic PTMs contribute to the onset and progression of PD. Pathogenic PTMs such as phosphorylation, ubiquitination, nitration and/or truncation can affect the structure, localization, and function of α-synuclein and LRRK2, while others, such as sumoylation in α-synuclein, may protect against PD pathogenesis. Altogether, the findings presented in this review suggest a need for the ongoing examination of PTMs. Likewise, the impact of gene-environment interactions in aberrant PTMs constitutes an exciting direction for exploration—extending our understanding of PD pathogenesis and identifying potential therapeutic targets.
Figure 2.
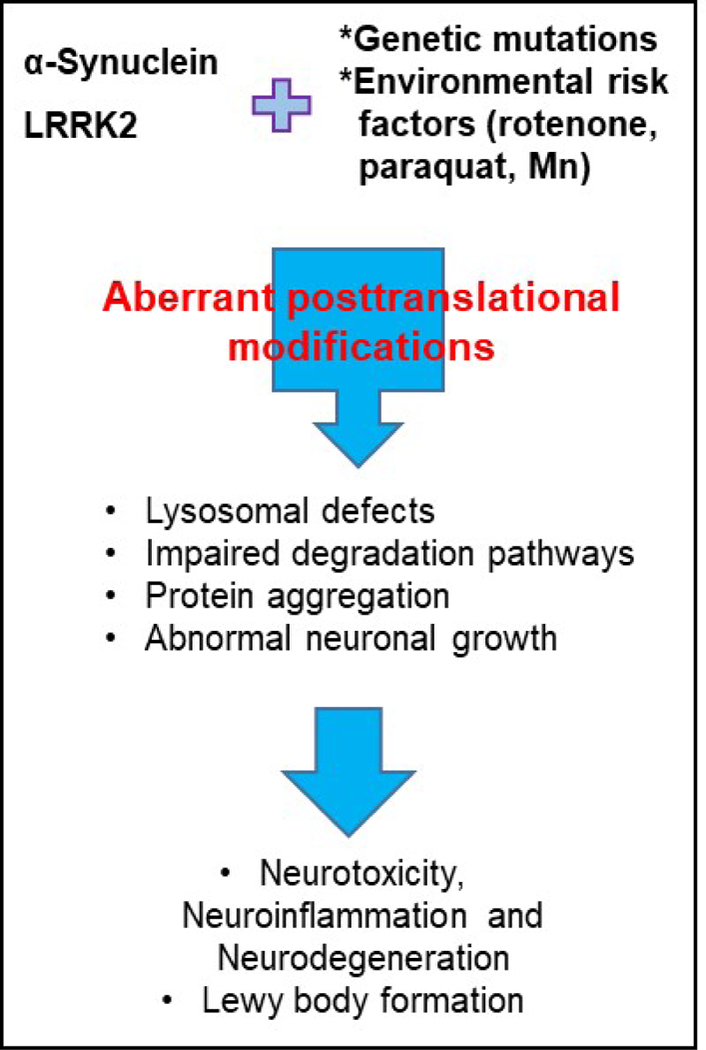
Schematic representation of the potential interplay of genetic and environmental factors influencing aberrant posttranslational modification of LRRK2 and α-synuclein in PD.
Highlights.
PTMs of α-synuclein play a critical role in its aggregation and neurotoxicity
PTMs of LRRK2 are disrupted by its genomic mutations which cause autosomal dominant PD
Environmental toxicants such as rotenone and Mn alter PTMs of both α-synuclein and LRRK2
Acknowledgment
The present study was supported in part by R01 ES024756 (EL), R01 ES10563 (MA), 1R03 ES024849 (MA), R01 ES07331 (MA) and 1R21 ES025415 (MA).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Ozansoy M and Basak AN, The central theme of Parkinson’s disease: alpha-synuclein. Mol Neurobiol, 2013. 47(2): p. 460–5. [DOI] [PubMed] [Google Scholar]
- 2.Greggio E, et al. , Leucine-rich repeat kinase 2 and alpha-synuclein: intersecting pathways in the pathogenesis of Parkinson’s disease? Mol Neurodegener, 2011. 6(1): p. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu G, Aliaga L, and Cai H, alpha-synuclein, LRRK2 and their interplay in Parkinson’s disease. Future Neurol, 2012. 7(2): p. 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrett PJ and Timothy Greenamyre J, Post-translational modification of alpha-synuclein in Parkinson’s disease. Brain Res, 2015. 1628(Pt B): p. 247–253. [DOI] [PubMed] [Google Scholar]
- 5.Sheng Z, et al. , Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med, 2012. 4(164): p. 164ra161. [DOI] [PubMed] [Google Scholar]
- 6.Dzamko N, et al. , LRRK2 levels and phosphorylation in Parkinson’s disease brain and cases with restricted Lewy bodies. Mov Disord, 2017. 32(3): p. 423–432. [DOI] [PubMed] [Google Scholar]
- 7.Hirai Y, et al. , Phosphorylated alpha-synuclein in normal mouse brain. FEBS Lett, 2004. 572(1–3): p. 227–32. [DOI] [PubMed] [Google Scholar]
- 8.Karuppagounder SS, et al. , LRRK2 G2019S transgenic mice display increased susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-mediated neurotoxicity. J Chem Neuroanat, 2016. 76(Pt B): p. 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mirzaei H, et al. , Identification of rotenone-induced modifications in alpha-synuclein using affinity pull-down and tandem mass spectrometry. Anal Chem, 2006. 78(7): p. 2422–31. [DOI] [PubMed] [Google Scholar]
- 10.Calne DB, et al. , Manganism and idiopathic parkinsonism: similarities and differences. Neurology, 1994. 44(9): p. 1583–6. [DOI] [PubMed] [Google Scholar]
- 11.Testa CM, Sherer TB, and Greenamyre JT, Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Res Mol Brain Res, 2005. 134(1): p. 109–18. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, et al. , Role of LRRK2 in manganese-induced neuroinflammation and microglial autophagy. Biochem Biophys Res Commun, 2018. 498(1): p. 171–177. [DOI] [PubMed] [Google Scholar]
- 13.Nussbaum RL and Polymeropoulos MH, Genetics of Parkinson’s disease. Hum Mol Genet, 1997. 6(10): p. 1687–91. [DOI] [PubMed] [Google Scholar]
- 14.Polymeropoulos MH, et al. , Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 1997. 276(5321): p. 2045–7. [DOI] [PubMed] [Google Scholar]
- 15.Alim MA, et al. , Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J Alzheimers Dis, 2004. 6(4): p. 435–42; discussion 443–9. [DOI] [PubMed] [Google Scholar]
- 16.Emamzadeh FN, Alpha-synuclein structure, functions, and interactions. J Res Med Sci, 2016. 21: p. 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazaro DF, et al. , The effects of the novel A53E alpha-synuclein mutation on its oligomerization and aggregation. Acta Neuropathol Commun, 2016. 4(1): p. 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu L and Pu J, Alpha-Synuclein in Parkinson’s Disease: From Pathogenetic Dysfunction to Potential Clinical Application. Parkinsons Dis, 2016. 2016: p. 1720621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Afitska K, et al. , Modification of C Terminus Provides New Insights into the Mechanism of alpha-Synuclein Aggregation. Biophys J, 2017. 113(10): p. 2182–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alegre-Abarrategui J, et al. , LRRK2 is a component of granular alpha-synuclein pathology in the brainstem of Parkinson’s disease. Neuropathol Appl Neurobiol, 2008. 34(3): p. 272–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berger Z, Smith KA, and Lavoie MJ, Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry, 2010. 49(26): p. 5511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West AB, et al. , Differential LRRK2 expression in the cortex, striatum, and substantia nigra in transgenic and nontransgenic rodents. J Comp Neurol, 2014. 522(11): p. 2465–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan PY, et al. , Parkinson’s Disease-Associated LRRK2 Hyperactive Kinase Mutant Disrupts Synaptic Vesicle Trafficking in Ventral Midbrain Neurons. J Neurosci, 2017. 37(47): p. 11366–11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meixner A, et al. , A QUICK screen for Lrrk2 interaction partners--leucine-rich repeat kinase 2 is involved in actin cytoskeleton dynamics. Mol Cell Proteomics, 2011. 10(1): p. M110 001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piccoli G, et al. , LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci, 2011. 31(6): p. 2225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marker DF, et al. , LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J Neuroinflammation, 2012. 9: p. 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moehle MS, et al. , LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci, 2012. 32(5): p. 1602–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cardona F, Tormos-Perez M, and Perez-Tur J, Structural and functional in silico analysis of LRRK2 missense substitutions. Mol Biol Rep, 2014. 41(4): p. 2529–42. [DOI] [PubMed] [Google Scholar]
- 29.Ross OA, et al. , Lrrk2 and Lewy body disease. Ann Neurol, 2006. 59(2): p. 388–93. [DOI] [PubMed] [Google Scholar]
- 30.Zimprich A, et al. , Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 2004. 44(4): p. 601–7. [DOI] [PubMed] [Google Scholar]
- 31.Kumari U and Tan EK, LRRK2 in Parkinson’s disease: genetic and clinical studies from patients. FEBS J, 2009. 276(22): p. 6455–63. [DOI] [PubMed] [Google Scholar]
- 32.Lesage S, et al. , LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med, 2006. 354(4): p. 422–3. [DOI] [PubMed] [Google Scholar]
- 33.Gillardon F, Schmid R, and Draheim H, Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience, 2012. 208: p. 41–8. [DOI] [PubMed] [Google Scholar]
- 34.Tong Y, et al. , R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci U S A, 2009. 106(34): p. 14622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.West AB, et al. , Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet, 2007. 16(2): p. 223–32. [DOI] [PubMed] [Google Scholar]
- 36.Taymans JM and Cookson MR, Mechanisms in dominant parkinsonism: The toxic triangle of LRRK2, alpha-synuclein, and tau. Bioessays, 2010. 32(3): p. 227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vitte J, et al. , Leucine-rich repeat kinase 2 is associated with the endoplasmic reticulum in dopaminergic neurons and accumulates in the core of Lewy bodies in Parkinson disease. J Neuropathol Exp Neurol, 2010. 69(9): p. 959–72. [DOI] [PubMed] [Google Scholar]
- 38.Dzamko N, et al. , Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14–3-3 binding and altered cytoplasmic localization. Biochem J, 2010. 430(3): p. 405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McFarland MA, et al. , Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol Cell Proteomics, 2008. 7(11): p. 2123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guerreiro PS, et al. , LRRK2 interactions with alpha-synuclein in Parkinson’s disease brains and in cell models. J Mol Med (Berl), 2013. 91(4): p. 513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maekawa T, et al. , Leucine-rich repeat kinase 2 (LRRK2) regulates alpha-synuclein clearance in microglia. BMC Neurosci, 2016. 17(1): p. 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volpicelli-Daley LA, et al. , G2019S-LRRK2 Expression Augments alpha-Synuclein Sequestration into Inclusions in Neurons. J Neurosci, 2016. 36(28): p. 7415–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bae EJ, et al. , LRRK2 kinase regulates alpha-synuclein propagation via RAB35 phosphorylation. Nat Commun, 2018. 9(1): p. 3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Maio R, et al. , LRRK2 activation in idiopathic Parkinson’s disease. Sci Transl Med, 2018. 10(451). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eguchi T, et al. , LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci U S A, 2018. 115(39): p. E9115–E9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schapansky J, et al. , Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble alpha-synuclein in neurons. Neurobiol Dis, 2018. 111: p. 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grant BD and Donaldson JG, Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol, 2009. 10(9): p. 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daher JP, et al. , Leucine-rich Repeat Kinase 2 (LRRK2) Pharmacological Inhibition Abates alpha-Synuclein Gene-induced Neurodegeneration. J Biol Chem, 2015. 290(32): p. 19433–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daher JP, et al. , Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc Natl Acad Sci U S A, 2014. 111(25): p. 9289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rieker C, et al. , Neuropathology in mice expressing mouse alpha-synuclein. PLoS One, 2011. 6(9): p. e24834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujiwara H, et al. , alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol, 2002. 4(2): p. 160–4. [DOI] [PubMed] [Google Scholar]
- 52.Paleologou KE, et al. , Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci, 2010. 30(9): p. 3184–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L, et al. , Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J Clin Invest, 2009. 119(11): p. 3257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mbefo MK, et al. , Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem, 2010. 285(4): p. 2807–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCormack AL, Mak SK, and Di Monte DA, Increased alpha-synuclein phosphorylation and nitration in the aging primate substantia nigra. Cell Death Dis, 2012. 3: p. e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee KW, et al. , Enhanced phosphatase activity attenuates alpha-synucleinopathy in a mouse model. J Neurosci, 2011. 31(19): p. 6963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu J, et al. , Lewy-like aggregation of alpha-synuclein reduces protein phosphatase 2A activity in vitro and in vivo. Neuroscience, 2012. 207: p. 288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou RM, et al. , Molecular interaction of alpha-synuclein with tubulin influences on the polymerization of microtubule in vitro and structure of microtubule in cells. Mol Biol Rep, 2010. 37(7): p. 3183–92. [DOI] [PubMed] [Google Scholar]
- 59.Schell H, et al. , Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience, 2009. 160(4): p. 796–804. [DOI] [PubMed] [Google Scholar]
- 60.Perfeito R, et al. , Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol Cell Neurosci, 2014. 62: p. 51–9. [DOI] [PubMed] [Google Scholar]
- 61.Karampetsou M, et al. , Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci Rep, 2017. 7(1): p. 16533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sugeno N, et al. , Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J Biol Chem, 2008. 283(34): p. 23179–88. [DOI] [PubMed] [Google Scholar]
- 63.Tofaris GK, et al. , Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc Natl Acad Sci U S A, 2011. 108(41): p. 17004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hasegawa M, et al. , Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J Biol Chem, 2002. 277(50): p. 49071–6. [DOI] [PubMed] [Google Scholar]
- 65.Engelender S, Ubiquitination of alpha-synuclein and autophagy in Parkinson’s disease. Autophagy, 2008. 4(3): p. 372–4. [DOI] [PubMed] [Google Scholar]
- 66.Engelender S, alpha-Synuclein fate: proteasome or autophagy? Autophagy, 2012. 8(3): p. 418–20. [DOI] [PubMed] [Google Scholar]
- 67.Ebrahimi-Fakhari D, Wahlster L, and McLean PJ, Protein degradation pathways in Parkinson’s disease: curse or blessing. Acta Neuropathol, 2012. 124(2): p. 153–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mandel SA, Fishman-Jacob T, and Youdim MB, Targeting SKP1, an ubiquitin E3 ligase component found decreased in sporadic Parkinson’s disease. Neurodegener Dis, 2012. 10(1–4): p. 220–3. [DOI] [PubMed] [Google Scholar]
- 69.Shimura H, et al. , Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet, 2000. 25(3): p. 302–5. [DOI] [PubMed] [Google Scholar]
- 70.Zhang J, et al. , Association between a polymorphism of ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) gene and sporadic Parkinson’s disease. Parkinsonism Relat Disord, 2000. 6(4): p. 195–197. [DOI] [PubMed] [Google Scholar]
- 71.Quinn JG, et al. , alpha-Synuclein mRNA and soluble alpha-synuclein protein levels in post-mortem brain from patients with Parkinson’s disease, dementia with Lewy bodies, and Alzheimer’s disease. Brain Res, 2012. 1459: p. 71–80. [DOI] [PubMed] [Google Scholar]
- 72.Mulherkar SA, Sharma J, and Jana NR, The ubiquitin ligase E6-AP promotes degradation of alpha-synuclein. J Neurochem, 2009. 110(6): p. 1955–64. [DOI] [PubMed] [Google Scholar]
- 73.Kalia LV, et al. , Ubiquitinylation of alpha-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLoS One, 2011. 6(2): p. e14695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rott R, et al. , Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J Biol Chem, 2008. 283(6): p. 3316–28. [DOI] [PubMed] [Google Scholar]
- 75.Periquet M, et al. , Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci, 2007. 27(12): p. 3338–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alexopoulou Z, et al. , Deubiquitinase Usp8 regulates alpha-synuclein clearance and modifies its toxicity in Lewy body disease. Proc Natl Acad Sci U S A, 2016. 113(32): p. E4688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu Y, et al. , DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell Discov, 2018. 4: p. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chavarria C and Souza JM, Oxidation and nitration of alpha-synuclein and their implications in neurodegenerative diseases. Arch Biochem Biophys, 2013. 533(1–2): p. 25–32. [DOI] [PubMed] [Google Scholar]
- 79.Souza JM, Peluffo G, and Radi R, Protein tyrosine nitration--functional alteration or just a biomarker? Free Radic Biol Med, 2008. 45(4): p. 357–66. [DOI] [PubMed] [Google Scholar]
- 80.Rocha EM, De Miranda B, and Sanders LH, Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis, 2018. 109(Pt B): p. 249–257. [DOI] [PubMed] [Google Scholar]
- 81.Duda JE, et al. , Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am J Pathol, 2000. 157(5): p. 1439–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hodara R, et al. , Functional consequences of alpha-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J Biol Chem, 2004. 279(46): p. 47746–53. [DOI] [PubMed] [Google Scholar]
- 83.Yu Z, et al. , Nitrated alpha-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS One, 2010. 5(4): p. e9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yamin G, Uversky VN, and Fink AL, Nitration inhibits fibrillation of human alpha-synuclein in vitro by formation of soluble oligomers. FEBS Lett, 2003. 542(1–3): p. 147–52. [DOI] [PubMed] [Google Scholar]
- 85.Li W, et al. , Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci U S A, 2005. 102(6): p. 2162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Masliah E, et al. , Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science, 2000. 287(5456): p. 1265–9. [DOI] [PubMed] [Google Scholar]
- 87.Mishizen-Eberz AJ, et al. , Cleavage of alpha-synuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry, 2005. 44(21): p. 7818–29. [DOI] [PubMed] [Google Scholar]
- 88.Wang W, et al. , Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein alpha-synuclein. Proc Natl Acad Sci U S A, 2016. 113(34): p. 9587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Giasson BI, et al. , Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron, 2002. 34(4): p. 521–33. [DOI] [PubMed] [Google Scholar]
- 90.Serpell LC, et al. , Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A, 2000. 97(9): p. 4897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Games D, et al. , Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci, 2014. 34(28): p. 9441–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Murray IV, et al. , Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry, 2003. 42(28): p. 8530–40. [DOI] [PubMed] [Google Scholar]
- 93.Krumova P, et al. , Sumoylation inhibits alpha-synuclein aggregation and toxicity. J Cell Biol, 2011. 194(1): p. 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Panavas T, Sanders C, and Butt TR, SUMO fusion technology for enhanced protein production in prokaryotic and eukaryotic expression systems. Methods Mol Biol, 2009. 497: p. 303–17. [DOI] [PubMed] [Google Scholar]
- 95.Rott R, et al. , SUMOylation and ubiquitination reciprocally regulate alpha-synuclein degradation and pathological aggregation. Proc Natl Acad Sci U S A, 2017. 114(50): p. 13176–13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bartels T, et al. , N-alpha-acetylation of alpha-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS One, 2014. 9(7): p. e103727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bu B, et al. , N-Terminal Acetylation Preserves alpha-Synuclein from Oligomerization by Blocking Intermolecular Hydrogen Bonds. ACS Chem Neurosci, 2017. 8(10): p. 2145–2151. [DOI] [PubMed] [Google Scholar]
- 98.Iyer A, et al. , The Impact of N-terminal Acetylation of alpha-Synuclein on Phospholipid Membrane Binding and Fibril Structure. J Biol Chem, 2016. 291(40): p. 21110–21122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.De Wit T, Baekelandt V, and Lobbestael E, LRRK2 Phosphorylation: Behind the Scenes. Neuroscientist, 2018: p. 1073858418756309. [DOI] [PubMed] [Google Scholar]
- 100.Nichols RJ, LRRK2 Phosphorylation. Adv Neurobiol, 2017. 14: p. 51–70. [DOI] [PubMed] [Google Scholar]
- 101.Nichols RJ, et al. , 14–3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem J, 2010. 430(3): p. 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhao J, et al. , LRRK2 dephosphorylation increases its ubiquitination. Biochem J, 2015. 469(1): p. 107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang L, et al. , Development of inducible leucine-rich repeat kinase 2 (LRRK2) cell lines for therapeutics development in Parkinson’s disease. Neurotherapeutics, 2013. 10(4): p. 840–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Smith WW, et al. , Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci, 2006. 9(10): p. 1231–3. [DOI] [PubMed] [Google Scholar]
- 105.Greggio E, et al. , Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis, 2006. 23(2): p. 329–41. [DOI] [PubMed] [Google Scholar]
- 106.Kim B, et al. , Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One, 2012. 7(4): p. e34693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Haugarvoll K, et al. , Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology, 2008. 70(16 Pt 2): p. 1456–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hatano T, et al. , Identification of a Japanese family with LRRK2 p.R1441G-related Parkinson’s disease. Neurobiol Aging, 2014. 35(11): p. 2656 e17–2656 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guo L, et al. , The Parkinson’s disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res, 2007. 313(16): p. 3658–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu Z, et al. , LRRK2 autophosphorylation enhances its GTPase activity. FASEB J, 2016. 30(1): p. 336–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang S, et al. , Elevated LRRK2 autophosphorylation in brain-derived and peripheral exosomes in LRRK2 mutation carriers. Acta Neuropathol Commun, 2017. 5(1): p. 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liao J, et al. , Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc Natl Acad Sci U S A, 2014. 111(11): p. 4055–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.West AB, et al. , Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A, 2005. 102(46): p. 16842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Henry AG, et al. , Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet, 2015. 24(21): p. 6013–28. [DOI] [PubMed] [Google Scholar]
- 115.Ding X and Goldberg MS, Regulation of LRRK2 stability by the E3 ubiquitin ligase CHIP. PLoS One, 2009. 4(6): p. e5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nucifora FC Jr., et al. , Ubiqutination via K27 and K29 chains signals aggregation and neuronal protection of LRRK2 by WSB1. Nat Commun, 2016. 7: p. 11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jan C, et al. , Dopaminergic innervation of the pallidum in the normal state, in MPTP-treated monkeys and in parkinsonian patients. Eur J Neurosci, 2000. 12(12): p. 4525–35. [PubMed] [Google Scholar]
- 118.Jenner P, The MPTP-treated primate as a model of motor complications in PD: primate model of motor complications. Neurology, 2003. 61(6 Suppl 3): p. S4–11. [DOI] [PubMed] [Google Scholar]
- 119.Smeyne RJ and Jackson-Lewis V, The MPTP model of Parkinson’s disease. Brain Res Mol Brain Res, 2005. 134(1): p. 57–66. [DOI] [PubMed] [Google Scholar]
- 120.Przedborski S, et al. , Oxidative post-translational modifications of alpha-synuclein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J Neurochem, 2001. 76(2): p. 637–40. [DOI] [PubMed] [Google Scholar]
- 121.Dauer W, et al. , Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A, 2002. 99(22): p. 14524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lee FJ, et al. , Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J, 2001. 15(6): p. 916–26. [DOI] [PubMed] [Google Scholar]
- 123.Richardson JR, et al. , Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol Sci, 2005. 88(1): p. 193–201. [DOI] [PubMed] [Google Scholar]
- 124.Yuan YH, et al. , The molecular mechanism of rotenone-induced alpha-synuclein aggregation: emphasizing the role of the calcium/GSK3beta pathway. Toxicol Lett, 2015. 233(2): p. 163–71. [DOI] [PubMed] [Google Scholar]
- 125.Xu B, et al. , Alpha-synuclein oligomerization in manganese-induced nerve cell injury in brain slices: a role of NO-mediated S-nitrosylation of protein disulfide isomerase. Mol Neurobiol, 2014. 50(3): p. 1098–110. [DOI] [PubMed] [Google Scholar]
- 126.Manning-Bog AB, et al. , The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem, 2002. 277(3): p. 1641–4. [DOI] [PubMed] [Google Scholar]
- 127.Manning-Bog AB, et al. , Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J Neurosci, 2003. 23(8): p. 3095–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Remelli M, et al. , Manganism and Parkinson’s disease: Mn(II) and Zn(II) interaction with a 30-amino acid fragment. Dalton Trans, 2016. 45(12): p. 5151–61. [DOI] [PubMed] [Google Scholar]
- 129.Peres TV, et al. , Untangling the Manganese-alpha-Synuclein Web. Front Neurosci, 2016. 10: p. 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou Y, et al. , Proteasomal inhibition induced by manganese ethylene-bis-dithiocarbamate: relevance to Parkinson’s disease. Neuroscience, 2004. 128(2): p. 281–91. [DOI] [PubMed] [Google Scholar]
- 131.Rasia RM, et al. , Structural characterization of copper(II) binding to alpha-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proc Natl Acad Sci U S A, 2005. 102(12): p. 4294–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Binolfi A, et al. , Interaction of alpha-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J Am Chem Soc, 2006. 128(30): p. 9893–901. [DOI] [PubMed] [Google Scholar]
- 133.Bates CA, et al. , Expression and Transport of alpha-Synuclein at the Blood-Cerebrospinal Fluid Barrier and Effects of Manganese Exposure. ADMET DMPK, 2015. 3(1): p. 15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bornhorst J, et al. , The effects of pdr1, djr1.1 and pink1 loss in manganese-induced toxicity and the role of alpha-synuclein in C. elegans. Metallomics, 2014. 6(3): p. 476–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Saha S, et al. , LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci, 2009. 29(29): p. 9210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ng CH, et al. , Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci, 2009. 29(36): p. 11257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Andres-Mateos E, et al. , Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). J Neurosci, 2009. 29(50): p. 15846–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Russo I, Bubacco L, and Greggio E, LRRK2 and neuroinflammation: partners in crime in Parkinson’s disease? J Neuroinflammation, 2014. 11: p. 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Whitton PS, Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br J Pharmacol, 2007. 150(8): p. 963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ho CC, et al. , The Parkinson disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J Neurosci, 2009. 29(4): p. 1011–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tjalkens RB, Popichak KA, and Kirkley KA, Inflammatory Activation of Microglia and Astrocytes in Manganese Neurotoxicity. Adv Neurobiol, 2017. 18: p. 159–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lovitt B, et al. , Differential effects of divalent manganese and magnesium on the kinase activity of leucine-rich repeat kinase 2 (LRRK2). Biochemistry, 2010. 49(14): p. 3092–100. [DOI] [PubMed] [Google Scholar]
- 143.Roth JA and Eichhorn M, Down-regulation of LRRK2 in control and DAT transfected HEK cells increases manganese-induced oxidative stress and cell toxicity. Neurotoxicology, 2013. 37: p. 100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mahmood T and Yang PC, Western blot: technique, theory, and trouble shooting. N Am J Med Sci, 2012. 4(9): p. 429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pagel O, et al. , Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev Proteomics, 2015. 12(3): p. 235–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Steger M, et al. , Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Manzoni C, et al. , Computational analysis of the LRRK2 interactome. PeerJ, 2015. 3: p. e778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Avin A, et al. , Quantitative analysis of protein-protein interactions and post-translational modifications in rare immune populations. Nat Commun, 2017. 8(1): p. 1524. [DOI] [PMC free article] [PubMed] [Google Scholar]