

Abstract
Oxidative post-translational modifications affect the structure and function of many biomolecules. Herein we examine the biophysical and functional consequences of oxidative post-translational. modifications to human calprotectin (CP, S100A8/S100A9 oligomer, MRP8/MRP14 oligomer, calgranulins A/B oligomer). This abundant metal-sequestering protein contributes to the innate immunity by starving invading microbial pathogens of transition metal nutrients in the extracellular space. It also participates in the inflammatory response. Despite many decades of study, little is known about the fate of CP at sites of infection and inflammation. We present compelling evidence for methionine oxidation of CP in vivo, supported by using 15N-labeled CP-Ser (S100A8(C42S)/S100A9(C3S)) to monitor for adventitious oxidation following human sample collection. To elucidate the biochemical and functional consequences of oxidative post-translational modifications, we examine recombinant CP-Ser with methionine sulfoxide modifications generated by exposing the protein to hydrogen peroxide. These oxidized species coordinate transition metal ions and exert antibacterial activity. Nevertheless, oxidation of M81 in the S100A9 subunit disrupts Ca(II)-induced tetramerization and, in the absence of a transition metal ion bound at the His6 site, accelerates proteolytic degradation of CP. We demonstrate that native CP, which contains one Cys residue in each full-length subunit, forms disulfide bonds within and between S100A8/S100A9 heterodimers when exposed to hydrogen peroxide. Remarkably, disulfide bond formation accelerates proteolytic degradation of CP. We propose a new extension to the working model for extracellular CP where post-translational oxidation by reactive oxygen species generated during the neutrophil oxidative burst modulates its lifetime in the extracellular space.
Graphical Abstract
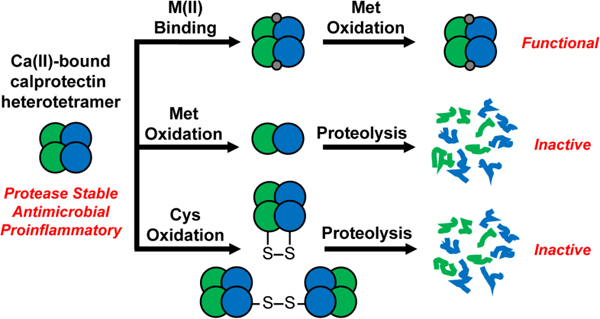
Introduction
Reactive oxygen species (ROS) are important players in the host/pathogen interaction. Neutrophils, white blood cells that are first responders during the innate immune response, generate and release ROS at infection sites in an attempt to kill invading microbial pathogens. These reactive small molecules also have the capacity to post-translationally oxidize host biomolecules. Motivated by the need to further understand the molecular complexity of infection sites, this work examines the biophysical and functional consequences of post-translational oxidation of an abundant neutrophil protein named calprotectin (CP, S100A8/S100A9 oligomer, MRP8/MRP14 oligomer, calgranulins A/B oligomer). These studies provide a new conceptual framework for considering the speciation and lifetime of CP at sites of infection and inflammation, and the role of methionine sulfoxide and disulfide bonds in directing the fate of CP during the innate immune response.
CP is a metal-sequestering protein that contributes to the innate immune response.1–3 The protein is produced by several types of epithelial cells and white blood cells, and it is particularly abundant in neutrophils where it constitutes ≈40% of cytosolic protein.4,5 In the current working model, CP is released by neutrophils into the extracellular space at concentrations that can exceed 40 µM (1 mg/mL).6 In this milieu, CP limits microbial growth by competing with invading microbes for bioavailable transition metal ions that are essential nutrients.1,3,7–10 Over the past decade, biophysical and functional studies have illuminated the molecular basis for this metal-sequestration model as described below.7–9,11,12 Recent investigations have also probed how microbial pathogens such as Staphylococcus aureus,13 Salmonella enterica serovar Typhimurium,14 Acinetobacter baumannii,15 and Neisseria spp.16,17 adapt to metal limitation caused by CP. In contrast, to the best of our knowledge, no reports have specifically addressed what happens next for CP, either metal-free or metal-bound, in the context of this model.
In addition to its antimicrobial properties, CP contributes to the inflammatory response. Data suggesting that CP is an activator of toll-like receptor 4 (TLR4) have been reported.18–20 Moreover, murine model studies indicated that CP-mediated proinflammatory signaling can lead to negative outcomes for the host, including promotion of lethal endotoxin-induced shock and generation of autoreactive T cells.18,19 Taken together, these studies highlight that CP is multi-functional, and suggest that CP can become toxic. In considering this duality, we reasoned that negative outcomes associated with CP-mediated signaling may result from a failure to clear CP, and this line of reasoning highlighted the shortcomings of the working model in describing what happens to CP after release.
Previously, we approached questions of CP function and fate by examining the molecular basis for metal sequestration by CP. In the apo form, CP exists as a 24-kDa heterodimer of S100A8 (93 amino acids, 10.8 kDa, α subunit) and S100A9 (114 amino acids, 13.2 kDa, β subunit) (Figure 1).1,22 The CP heterodimer contains six different sites for coordinating cations. Each subunit contains two Ca(II)-binding EF-hand domains, and two binding sites for transition metal ions form at the S100A8/S100A9 interface.8,23 Site 1 is a His3Asp site that binds Zn(II) with high affinity,8 and site 2 is a His6 site that binds Mn(II),24 Fe(II),10 Ni(II),25 and Zn(II)8,11 with high affinity (Figure 1). Ca(II) binding to CP has important structural and functional consequences. In the presence of excess Ca(II) ions, two CP heterodimers self-associate to form an α2β2 heterotetramer.8,23,26,27 Conditions of high Ca(II) that promote heterotetramer formation also result in enhanced transition metal affinities, protease stability, and antimicrobial activity.8,10,12,24 Because Ca(II) ion concentrations are ≈100 nM in the cytoplasm of resting cells and extracellular Ca(II) ion concentrations are ≈2 mM,28 these observations provided a working model where the heterodimer is the major cytosolic species and the Ca(II)-bound heterotetramer is the major extracellular species.8 This Ca(II) effect allows CP to sequester metals and persist in the harsh extracellular space that contains host and bacterial proteases.
Figure 1.
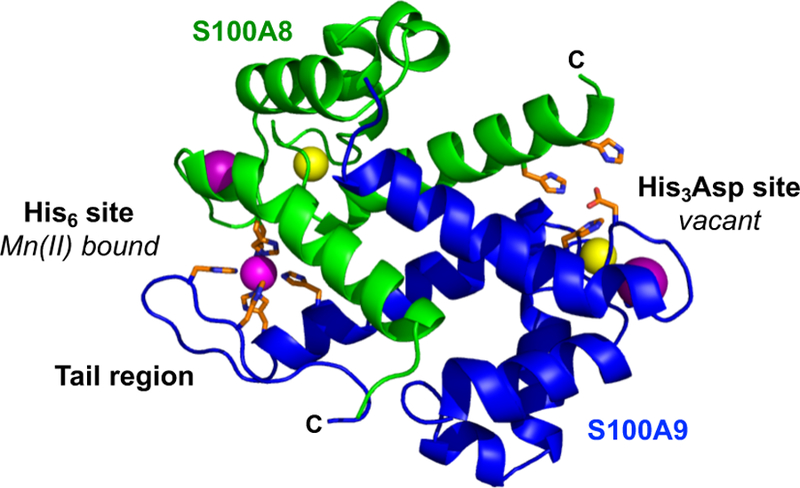
A heterodimer unit from the crystal structure (PDB: 1XK4) of Mn(II)-, Ca(II)-, and Na(I)-bound CP-Ser.21 The green and blue chains are S100A8 and S100A9, respectively. Mn(II), Ca(II), and Na(I) ions are shown in magenta, yellow, and purple, respectively. The transition-metal binding residues are shown with orange sticks.
Over the course of our studies that addressed the biological coordination chemistry of CP, several reports identified the methionine sulfoxide (MetO) post-translational modification on the CP subunits during ex vivo analyses of human or mouse specimens by mass spectrometry.29–32 A variety of oxidants, including hydrogen peroxide (H2O2), superoxide (O2•–), and hypochlorous acid (HOCl), are generated by the neutrophil oxidative burst.33 These oxidants can oxidize methionine sidechains to MetO.34–36 To us, these observations were intriguing and suggested an as-yet unappreciated complexity to the molecular speciation of CP at biological sites. The Met/MetO redox couple is often considered to serve as an antioxidant because the MetO post-translational modification can be reversed by the action of methionine sulfoxide reductases MsrA and MsrB, which are found in many cell types, including neutrophils.37–40 On the other hand, accumulation of MetO post-translational modifications is associated with protein dysfunction.41 There are few studies on the consequences arising from MetO modification of CP,30,42 and we reasoned that understanding these consequences would inform the function and fate of CP.
In this work, we identify oxidized CP species in human specimens, and we test a new hypothesis that oxidative post-translational modifications affect the function and fate of CP in the biological milieu. Our biochemical and functional results support a model where methionine oxidation and disulfide bond formation attenuate the proteolytic stability of Ca(II)-bound CP. In the absence of a divalent transition metal ion bound at the His6 site, oxidizing M81 of S100A9 disfavors Ca(II)-induced tetramerization, which leads to accelerated proteolysis by a variety of proteases. Remarkably, disulfide bonds within and between CP heterodimers also caused accelerated proteolysis, an effect that is particularly apparent when the disulfide bond includes C42 of S100A8. In total, these results support a new extension to the working model where extracellular CP can be oxidized by neutrophil-derived ROS and subsequently degraded by proteases. We further hypothesize that accelerated degradation caused by oxidative modification serves to dampen the proinflammatory signal of CP.
Experimental Methods
Complete experimental methods are provided as Supporting Information.
Results
Oxidized CP Subunits are Observed in Human Mucus and Pus.
Subunits of CP containing MetO have been observed in murine kidney abscesses,29 human cystic fibrosis bronchoalveolar fluid,30 human sputum from asthmatic and control subjects,31 and human kidney stones.32 Because methionine side chains can be easily oxidized, it is often unclear whether the oxidation occurred in vivo, following sample collection, or during analysis. Nevertheless, we were intrigued by these findings and therefore sought to ascertain whether methionine oxidation of CP occurs in vivo and whether this post-translational modification has physiological relevance. We focused on the collection and ex vivo analyses of two types of readily available human samples, nasal mucus and pimple pus, where we expected to find CP. We selected LC-MS as an analytical method to identify unmodified and modified CP subunits. Moreover, in prior biophysical studies of CP, we overexpressed and purified 15N-labeled CP-Ser (Table 1) where both subunits are globally labeled with the 15N isotope.21 We reasoned that we could use 15N-labeled CP-Ser as an internal standard by adding it to human specimens immediately after collection and subsequently monitoring for whether methionine oxidation of CP occurs before or after sample collection. The N-terminal Met residue of S100A9 is cleaved by the methionine aminopeptidase during overexpression in Escherichia coli; thus, full-length 15N-S100A9(C3S) was not observed in these experiments.
Table 1.
List of abbreviations used for proteins.
| Abbreviation | Construct a |
|---|---|
| CP | S100A8/S100A9 |
| A9(ΔM1) | S100A9 lacking Met1 |
| A9(ΔM1-M5) | S100A9 lacking Met1, Thr2, Cys3, Lys4, and Met5 |
| A9(C3S) | S100A9(C3S) |
| A8+O | S100A8 with 1 additional oxygen atom |
| A9+nO | S100A9 with n additional oxygen atoms |
| CP-Ser | S100A8(C42S)/S100A9(C3S) |
| 15N-CP-Ser | [15N]-S100A8(C42S)/[15N]-S100A9(C3S) |
| 15N-A8(C42S) | [15N]-S100A8(C42S) |
| 15N-A9(C3S) | [15N]-S100A9(C3S) |
| M63A | S100A8(C42S)/S100A9(C3S)(M63A) |
| M81A | S100A8(C42S)/S100A9(C3S)(M81A) |
| M83A | S100A8(C42S)/S100A9(C3S)(M83A) |
| CP-Ser O4 | CP-Ser with 4 oxidized Met residues b |
| CP-Ser O5 | CP-Ser with 5 oxidized Met residues b |
| MetO-disulfide-linked CP | CP with disulfide bonds and oxidized Met residues |
All recombinant S100A9 lacked the N-terminal Met residue to due excision by methionine aminopeptidase during overexpression in E. coli.
These forms are the predominant species in the CP-Ser O4 and CP-Ser O5 preparations.
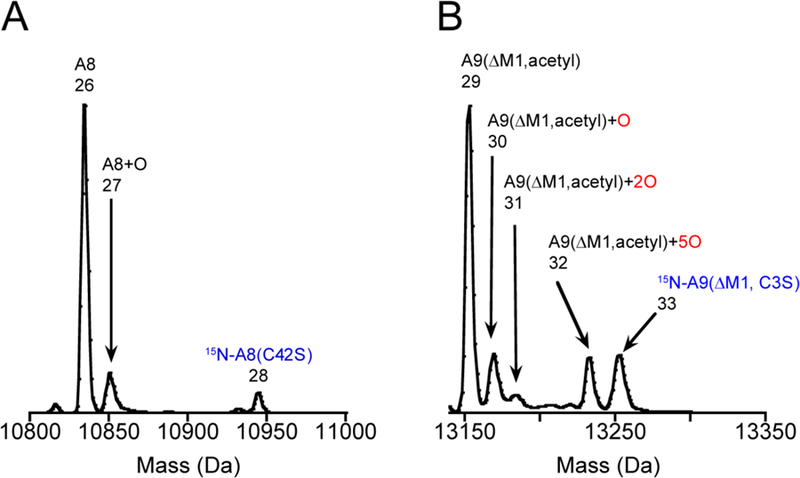
We collected and analyzed 26 nasal mucus samples from patients prior to surgery and detected both CP subunits in 15 of these samples by LC-MS (Figure 2 and S1, Table S2, Supporting Information). These analyses revealed full-length S100A8 and two truncated forms of S100A9 bearing N-terminal acetylation. Full-length S100A9 was not observed. One truncated S100A9 species lacked the initiator methionine residue and the other lacked the first five amino acids of S100A9, indicating that translation was initiated at Met5 rather than Met1 (Figure 2B,C). These truncated, N-acetylated forms of S100A9 have been previously observed in human kidney stones and bronchoalveolar fluid of cystic fibrosis patients.30,32 The longer form of S100A9 has five Met residues, the shorter form has four Met residues, and S100A8 has two Met residues. LC-MS also revealed that four of the CP-containing mucus samples exhibited deconvoluted masses consistent with oxidized CP subunits. Although the extent of CP oxidation varied depending on the sample, some common species were observed (Figure 2 and S1). In two of the four samples, a population of S100A8 that bore a single additional oxygen atom (+16 Da) was found. All three samples contained S100A9 with at least one additional oxygen atom (+16 Da). In three of the four samples, S100A9 with five additional oxygen atoms (+80 Da) was observed. One sample exhibited a series of +16 Da species ending with a molecule that appeared to have eight additional oxygen atoms (+128 Da) (Figure 2B). This S100A9 subunit contains five Met residues, and the locations of the three other oxygen atoms are unknown. We observed neither oxidized 15N-(S100A8)C42S nor oxidized 15N-S100A9(C3S) in a sample that contained oxidized CP and 15N-CP-Ser (Figure 2D,E), which provided strong evidence that the oxidation in the mucus samples occurred in vivo. We note that one of the mucus samples (Figure 2E) contained a species that had a molecular weight close to that of 15N-S100A9(C3S) with one additional oxygen atom; however, we were hesitant to assign it as such due to the error of 3 Da.
Figure 2.
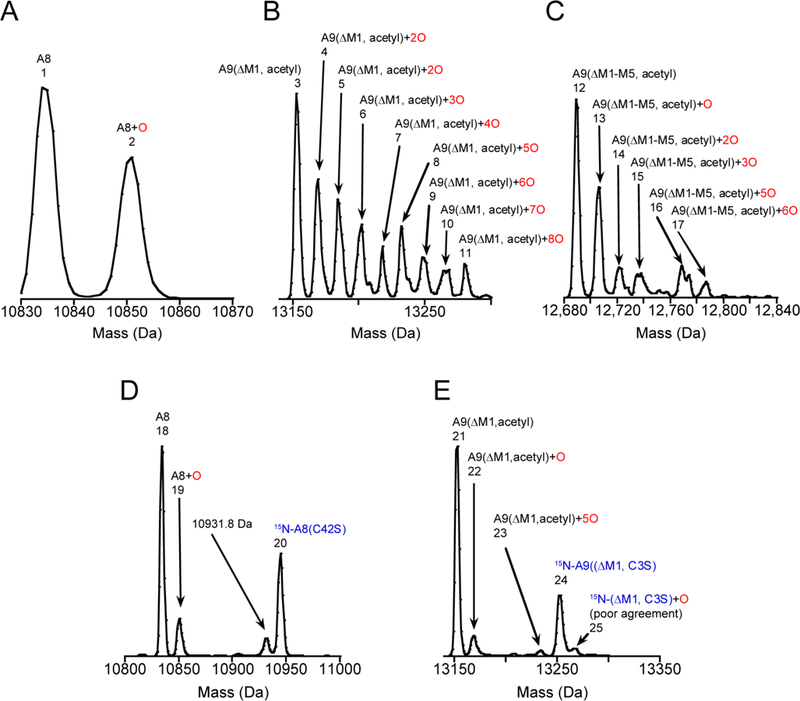
Deconvoluted mass spectra of human nasal mucus. The S100A8 panels are normalized to the unmodified S100A8 peak and S100A9 panels are normalized to the most abundant S100A9 peak. The observed and theoretical masses for each numbered peak are listed in Table S1. Data from (A) mucus sample 2 expanded around S100A8, (B) mucus sample 2 expanded around S100A9, (C) mucus sample 2 expanded around S100A9(ΔM1–5), (D) mucus sample 22 expanded around S100A8 that shows 15N-A8(C42S), (E) mucus sample 22 expanded around S100A9 that shows 15N-A9(ΔM1, C3S).
During the course of the mucus studies, we found that many samples were viscous and that CP subunits were only detected in ca. 60% of the samples. Thus, we extended the analyses to two human pus samples isolated from pimples taken from a single donor, which we found to be easier to handle than mucus. LC-MS revealed the presence of the CP subunits in the two pus samples (Figures 2 and S1). Similar to the mucus samples, we observed full-length S100A8 and truncated, N-acetylated forms of S100A9. Both pus samples also contained oxidized S100A8 and S100A9 species. The oxidized S100A8 species contained one additional oxygen atom (+16 Da) and up to five additional oxygen atoms were observed for the oxidized S100A9 species. We added 15N-CP-Ser to both of these pus samples with and observed no oxidized 15N-S100A8(C42S) species in either sample, and a small fraction of 15N-S100A9 bearing a single additional oxygen atom (+16 Da) was detected in in one of the two samples (Figures 2 and S1). Overall, these ex vivo studies of nasal mucus and pus provide further support for the existence of oxidized CP subunits in different human fluids. Importantly, the lack of oxidation of the 15N-CP-Ser internal standard indicates that the oxidative post-translational modifications of the CP subunits occurred in vivo and not during sample collection, storage, or analysis. We interpreted these data to indicate that one Met of S100A8 and up to five Met of S100A9 can be oxidized to MetO. Taken together, these observations strengthen the notion that methionine oxidation is a physiologically relevant post-translational modification of CP.
Methionine Oxidation of CP-Ser Causes Heterotetramer Dissociation.
To investigate the consequences of methionine oxidation on the biophysical properties of CP, we first treated the Ca(II)-bound CP-Ser heterotetramer with H2O2, an oxidant that oxidizes Cys and Met residues with high specificity,43 and monitored the reaction by analytical size-exclusion chromatography (SEC) and LC-MS (Figure 4A,B). We chose to begin our studies with the Cys→Ser variant to avoid complications that could arise from disulfide bond formation. SEC revealed that the CP-Ser heterotetramer peak (10.9 mL, 45.9 kDa) decreased in intensity and a new peak corresponding to the CP-Ser heterodimer (11.5 mL, 34.9 kDa) appeared (Figure 4A). In these chromatograms, tetramer dissociation was readily apparent at the 7-h time point, and the dimer became the major species at the 23-h time point. LC-MS of the corresponding samples demonstrated that, as heterotetramers dissociated into heterodimers, the number of oxygen atoms on the CP subunits increased (Figure 4B). S100A8 had gained a single additional oxygen atom (+16 Da) within 1 h of incubation, and no further oxidative modification was observed until the 23-h time point at which time S100A8 with two additional oxygen atoms (+32 Da) became the major species. In contrast, the extent of oxidation of S100A9 increased at each time point. Because the dissociation of the Ca(II)-bound heterotetramer appeared to coincide with the extent of methionine oxidation on S100A9, we reasoned that the trigger for heterotetramer dissociation was oxidation of one or more Met residues on S100A9.
Figure 4.
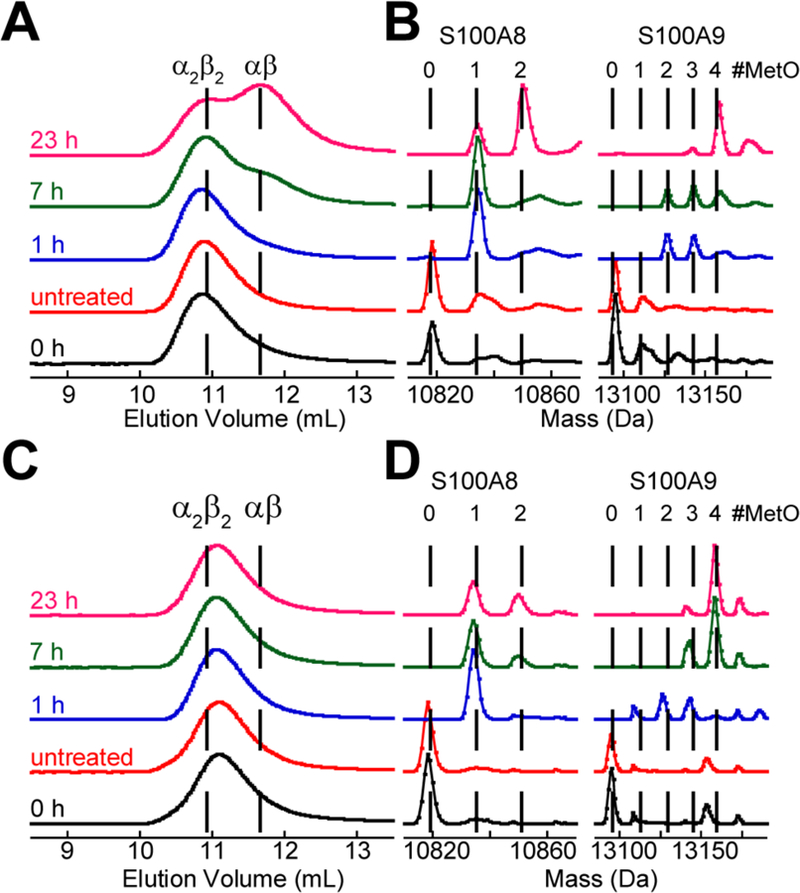
SEC chromatograms and corresponding deconvoluted mass spectra of 30 µM CP-Ser treated with 100 mM H2O2 in the presence of 1.5 mM Ca(II) or both 1.5 mM Ca(II) and 30 µM Mn(II) (75 mM HEPES, 100 mM NaCl, pH 7.5). In the mass spectra, the dashed lines represent the expected masses of the CP subunits with additional oxygen atoms. The chromatograms and mass spectra were normalized to a maximum value of 1. The listed times correspond to the duration of treatment with H2O2 prior to sample analysis. The untreated sample was incubated at 37 °C for 23 h. (A) SEC chromatograms for the Ca(II) only samples. (B) Regions of the deconvoluted mass spectra showing the S100A8 and S100A9 subunits for the Ca(II) only samples. (C) +Ca(II)+Mn(II) SEC. (C) SEC chromatograms for the Ca(II) and Mn(II) samples. (D) Regions of the deconvoluted mass spectra showing the S100A8 and S100A9 subunits for the Ca(II) and Mn(II) samples. In the SEC chromatograms, the Mn(II)- and Ca(II)-bound CP-Ser tetramer elutes slightly later than the Ca(II)-bound CP-Ser tetramer.
In prior work, we demonstrated that coordination of Mn(II) or Fe(II) at the His6 site of CP-Ser in the absence of Ca(II) causes two heterodimers to self-associate into a heterotetramer as well as tetramerization of CP-Ser variants harboring a I60K or I60E point mutation in S100A8 that could not tetramerize with Ca(II) alone.12 Thus, we questioned whether a Ca(II)- and transition-metal-bound heterotetramer dissociates into heterodimers upon methionine oxidation. We oxidized CP-Ser with H2O2 in the presence of excess Ca(II) and one equivalent of Mn(II), which only binds to the His6 site under these conditions,21 and monitored the reaction by SEC and LC-MS (Figure 4C,D). In contrast to the oxidation in the presence of only Ca(II), a peak with a maximum elution volume consistent with the heterotetramer (11.0 mL, 43.8 kDa) was only observed, indicating that the Ca(II)-and Mn(II)-bound protein remained a heterotetramer over the 23-h time course. As has been observed previously, coordination of a transition metal ion at the His6 site of CP increases the SEC elution volume and thus decreases the apparent molecular weight of the heterotetramer compared to the Ca(II)-only complex.8,10,24,25 We attribute this difference to decreased flexibility of the S100A9 C-terminal tail resulting from M(II) coordination at the His6 site. Analysis of samples by LC-MS revealed that both subunits were oxidized in the presence of Ca(II) and Mn(II) at a rate that appeared comparable to that observed for the Ca(II)-only sample. These data indicate that coordination of both Ca(II) and Mn(II) stabilizes the heterotetramer, and that Mn(II) chelation at the His6 site does not protect Met residues from oxidation by H2O2.
Oxidation of Met81 in S100A9 Serves as the Determinant for Heterotetramer Dissociation.
In order to better understand the molecular basis of H2O2-induced dissociation of the Ca(II)-bound CP-Ser heterotetramer, we investigated the contributions of individual S100A9 Met residues to this process. We overexpressed and purified three new CP-Ser variants that each contain a single Met→Ala mutation at position 63, 81, or 83 of S100A9(C3S). M63 of S100A9 is close to the C-terminal EF-hand, M81 of S100A9 appears to participate in the dimer-dimer interface, and M83 of S100A9 is close to the dimer-dimer interface (Figure S2). These variants were obtained as pure heterodimers (Table S3, Figure S4). Circular dichroism (CD) spectroscopy in the absence and presence of Ca(II) demonstrated that the α-helicity of the proteins was not perturbed by the mutations (Figure S5). SEC performed in the presence of Ca(II) indicated that all three Ca(II)-bound CP-Ser variants eluted as tetramers (Figure 5); therefore, the Met→Ala mutations in S100A9(C3S) did not perturb Ca(II)-induced tetramerization, at least as ascertained by this technique. In contrast, SEC revealed that treatment of the Met→Ala variants with H2O2 in the presence of Ca(II) caused the M63A and M83A variants to dissociate into dimers, whereas the M81A variant remained a tetramer (Figure 5). LC-MS of the Met→Ala variants after H2O2 treatment displayed oxidation comparable to CP-Ser (Figure S6). These results show that H2O2-induced dissociation of the Ca(II)-bound heterotetramer is dependent on oxidizing M81 of S100A9; however, these data do not inform whether oxidation of one or both M81 residues in the heterotetramer is required to initiate dissociation.
Figure 5.
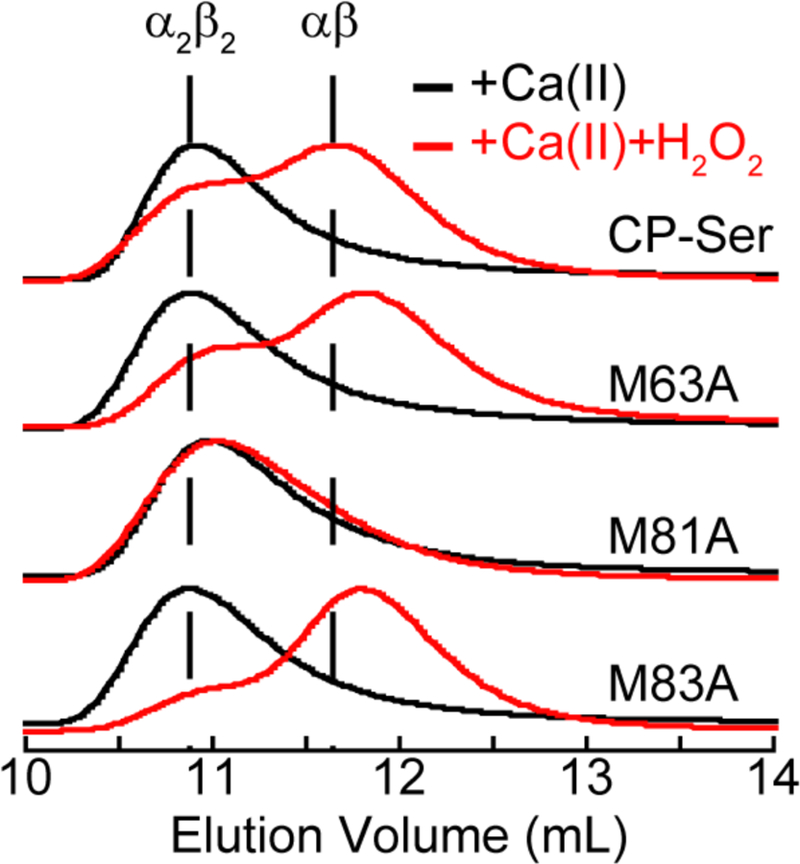
SEC chromatograms of 30 µM CP-Ser and the Met-to-Ala variants in the presence of 1.5 mM Ca(II) before (black traces) and after (red traces) treatment with 500 mM H2O2 for 7 h (75 mM HEPES, 100 mM NaCl, pH 7.5). Each chromatogram was normalized to a maximum absorbance of 1.
Preparation of Oxidized CP-Ser.
In order to further study CP-Ser bearing MetO modifications, we optimized a protocol to obtain milligram quantities of oxidized CP-Ser (Supporting Discussion, Figure S3). By incubating crude CP-Ser with H2O2, we identified conditions that provided either CP-Ser O4, oxidized CP-Ser predominantly comprised of S100A8 with one additional oxygen atom and S100A9 with three additional oxygen atoms, or CP-Ser O5, predominantly oxidized CP-Ser comprised of S100A8 with one additional oxygen atom and S100A9 with four additional oxygen atoms (Table S3 and Figure S7). Analysis of CP-Ser O4 and CP-Ser O5 by CD spectroscopy in the absence and presence of excess Ca(II) ions revealed that the α-helicity of the CP scaffold was unaffected by methionine oxidation (Figure S4).
CP-Ser O4 and CP-Ser O5 Exhibit Defective Ca(II)-Induced Tetramerization.
To further examine the consequences of methionine oxidation on the biophysical properties of CP, we investigated the oligomeric states of CP-Ser O4 and CP-Ser O5. We first examined the proteins by using analytical SEC to determine if they recapitulated the tetramerization-deficient behavior that occurred after treating CP-Ser with H2O2 (Figure 6A). In the absence of added metal ions, CP-Ser O4 and CP-Ser O5 each had an elution volume of ≈11.6 mL (33.4 kDa), consistent with the apo heterodimer (Figure 6A). In the presence of excess Ca(II) ions, the CP-Ser O4 and CP-Ser O5 peaks maintained a maximum peak height consistent with the heterodimer elution volume. However, the peaks broadened, suggesting that both dimeric and tetrameric species exist under these high-Ca(II) conditions (Figure 6A). When Mn(II) (Figure S8) or both Ca(II) and Mn(II) (Figure 6A) were present, CP-Ser O4 and CP-Ser O5 tetramerized. Thus, the SEC experiments with CP-Ser O4 and CP-Ser O5 demonstrated that the proteins displayed oligomeric properties consistent with H2O2-treated CP-Ser, and would be useful for further studying the effects of Met oxidation on CP-Ser.
Figure 6.
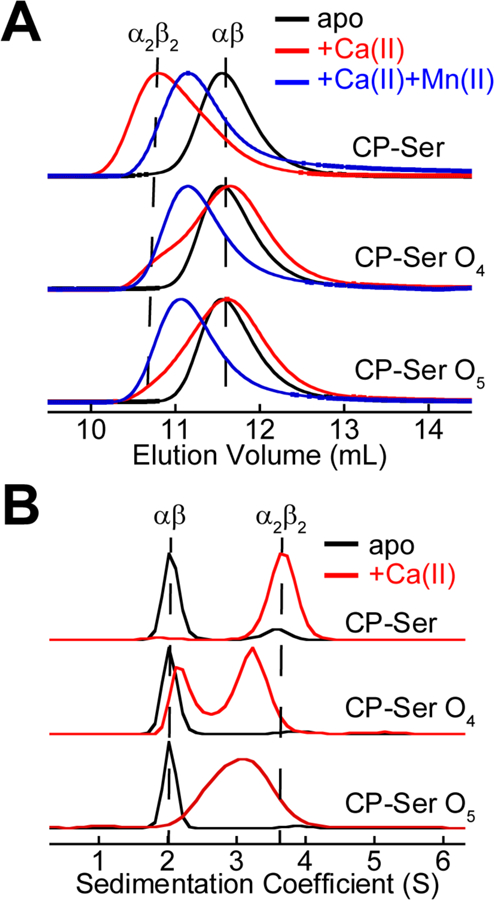
Biophysical characterization of oxidized CP-Ser. (A) SEC chromatograms of 30 µM CP-Ser, CP-Ser O4, and CP-Ser O5 in the absence and presence of 1.5 mM Ca(II) only or 1.5 mM Ca(II) and 30 µM Mn(II) (75 mM HEPES, 100 mM NaCl, pH 7.5, 20 °C). Each chromatogram was normalized to a maximum absorbance of 1. The dashed lines represent the elution volumes of the tetramer and dimer. (B) Sedimentation velocity distributions of 30 µM CP-Ser, CP-Ser O4, and CP-Ser O5 in the absence and presence of 600 µM Ca(II) analyzed by SEDFIT using the c(s) model (75 mM HEPES, 100 mM NaCl, pH 7.5, 20 °C). The peaks were normalized to a maximum value of 1. The dashed lines represent the S value of the dimer and tetramer. In the experiments with apo protein (black traces), 30 µM EDTA was added to the samples.
Anion exchange chromatography allows separation of CP-Ser heterodimers from S100A9(C3S) homodimers, and we employed this technique to ascertain whether Met oxidation caused conversion of the S100A8/S100A9 heterodimer to the S100A8 and S100A9 homodimers (Figure S9). We attempted to include the S100A8(C42S) homodimer in this analysis; however, its peak profile and retention time were highly variable between runs. The chromatograms for CP-Ser, CP-Ser O4, and CP-Ser O5 each revealed a single peak with an elution volume of 29.3 mL consistent with the S100A8(C42S)/S100A9(C3S) heterodimer, and no peak corresponding to the S100A9(C3S) homodimer (37.2 mL) was observed. Thus, we concluded that methionine oxidation does not cause CP heterooligomers to convert into S100A8 and S100A9 homodimers.
CP-Ser O4 and CP-Ser O5 Interconvert Between Heterodimers and Heterotetramers.
To further elucidate the oligomerization behavior of CP-Ser O4 and CP-Ser O5 in the presence of excess Ca(II) ions, we employed velocity analytical ultracentrifugation (AUC), a powerful technique for studying protein oligomerization that is capable of differentiating between interacting and non-interacting systems (Supporting Discussion).44,45 Along these lines, two scenarios that would give rise to the observed SEC peak shapes for CP-Ser O4 and CP-Ser O5 can be considered: (i) the oxidized proteins containing a population of tetramers and a population of dimers that do not interconvert or (ii) the oxidized proteins are in a dynamic equilibrium between heterodimer and heterotetramer.
We repeated an earlier AUC analysis of CP-Ser in the absence and presence of excess Ca(II) ions, and we used SEDFIT to obtain peak profiles and S values in good agreement with our prior data for the apo heterodimer (s20,w = 2.2 S) and the Ca(II)-bound heterotetramer (s20,w = 3.7 S) (Figures 6B and S10 and Table S4).12 We also examined two independent preparations of CP-Ser O4 and CP-Ser O5. In the absence of Ca(II) ions, the sedimentation profiles of CP-Ser O4 and CP-Ser O5 were indistinguishable from that of CP-Ser (Figures 6B and S10), which was consistent with the AXC and SEC studies. In the presence of Ca(II) ions, the sedimentation profiles of CP-Ser O4 and CP-Ser O5 displayed the hallmarks of dynamically interconverting systems (Figures 6B and S10). In particular, the peaks in the sedimentation profiles broadened, in some cases to the point where the heterodimer and heterotetramer peaks could not be resolved. Accordingly, the calculated S values for the observed peaks were in between those of the heterodimer and heterotetramer. In the profiles shown here (Figure 6B), CP-Ser O4 (+Ca(II)) displayed two broad peaks with S values between the dimer and tetramer (s20,w = 2.5 S and 3.5 S), and CP-Ser O5 exhibited a single broad peak (s20,w = 3.3 S). We also analyzed the AUC data using DCDT+,46,47 which yielded results consistent with the SEDFIT analyses (Table S5 and Figure S11). Taken together, we concluded that Met oxidation of CP-Ser does not fully prevent Ca(II)-induced tetramerization. Rather, the heterodimer-heterodimer equilibrium is strongly shifted to the heterodimer in the presence of excess Ca(II) ions.
CP-Ser Retains Antimicrobial Activity after Methionine Oxidation.
To evaluate the effect of methionine oxidation on the antimicrobial activity (AMA) of CP, we performed AMA assays against Escherichia coli ATCC 25922 and Staphylococcus aureus ATCC 25923 using CP-Ser, CP-Ser O4 and CP-Ser O5 (Figure S12). The assays were performed in Tris:TSB medium supplemented with 2 mM Ca(II) to mimic the extracellular Ca(II) concentration. Under these growth conditions, CP-Ser and the oxidized species displayed concentration-dependent growth inhibition of both bacterial species. A comparison of CP-Ser to CP-Ser O4 and CP-Ser O5 indicates that methionine oxidation caused some attenuation of antibacterial activity. For E. coli, this trend is most evident at 250 µg/mL of protein where CP-Ser completely inhibited growth and CP-Ser O4 and CP-Ser O5 inhibited growth to a lesser degree. For S. aureus, CP-Ser O4 and O5 were less active than CP-Ser at 250 and 500 µg/mL. Nevertheless, ex vivo analyses of human fluids has indicated that CP concentrations can reach >1 mg/mL at infection sites,6 and both CP-Ser O4 and O5 fully inhibit bacterial growth at these concentrations. We note that our finding that methionine oxidation does not markedly attenuate the activity of CP refutes conclusions from prior work where oxidation of (A8)M1 was reported to abolish the antimicrobial activity of CP.30 This study used HOCl as the oxidant, and other Met and non-Met residues were also reported to be oxidized in these reactions. Thus, we reason that the further oxidation of the protein scaffold was responsible for the observed loss of antibacterial activity following HOCl treatment.
Oxidized CP-Ser Binds Transition Metals with High Affinity.
To provide an initial evaluation of the metal-withholding capabilities of oxidized CP-Ser, we compared the abilities of CP-Ser, CP-Ser O4, and CP-Ser O5 to compete with the Ca(II)-insensitive fluorescent metal-ion sensor Zinpyr-1 (ZP1)48 for Mn(II), Fe(II), Ni(II), and Zn(II) (Figure S13). Under conditions of excess Ca(II) ions, CP-Ser, CP-Ser O4 and CP-Ser O5 outcompeted ZP1 for these four transition metal ions. Although these results do not rule out the possibility that methionine oxidation perturbs the binding affinities of CP for transition metal ions, these results demonstrate that, in the presence of excess Ca(II), CP-Ser O4 and CP-Ser O5 coordinate transition metals with sufficiently high affinity to outcompete ZP1. The ability of CP-Ser O4 and CP-Ser O5 to chelate transition metals with high affinity is consistent with the observed growth inhibitory activity (Figure S12).
Oxidized CP-Ser is Protease Sensitive.
In prior work, we observed that two Ca(II)-bound tetramer-deficient variants of CP-Ser that do not form heterotetramers in the presence of excess Ca(II) were more easily degraded by host proteases than Ca(II)-bound CP-Ser.12 We therefore questioned whether methionine oxidation enhances the protease susceptibility of CP-Ser and performed protease digestion assays with CP-Ser, CP-Ser O4, and CP-Ser O5 using trypsin, chymotrypsin, human neutrophil elastase (HNE), and proteinase K. These assays were performed in the presence of excess Ca(II) or in the presence of both excess Ca(II) and one equivalent of Mn(II) (Figure 7 and S14–S17). The reactions were analyzed by HPLC using a gradient that separated S100A8 (37.9 min), S100A9 (39.4 min), S100A8+O (37.3 min), S100A9+3O (37.8 min), and S100A9+4O (36.1 min). With this gradient, doubling of the oxidized S100A9 peaks occurred. Although the precise origin of the peak doubling is unknown, the phenomenon may arise from a mixture of MetO diastereomers and/or MetO regioisomers.
Figure 7.
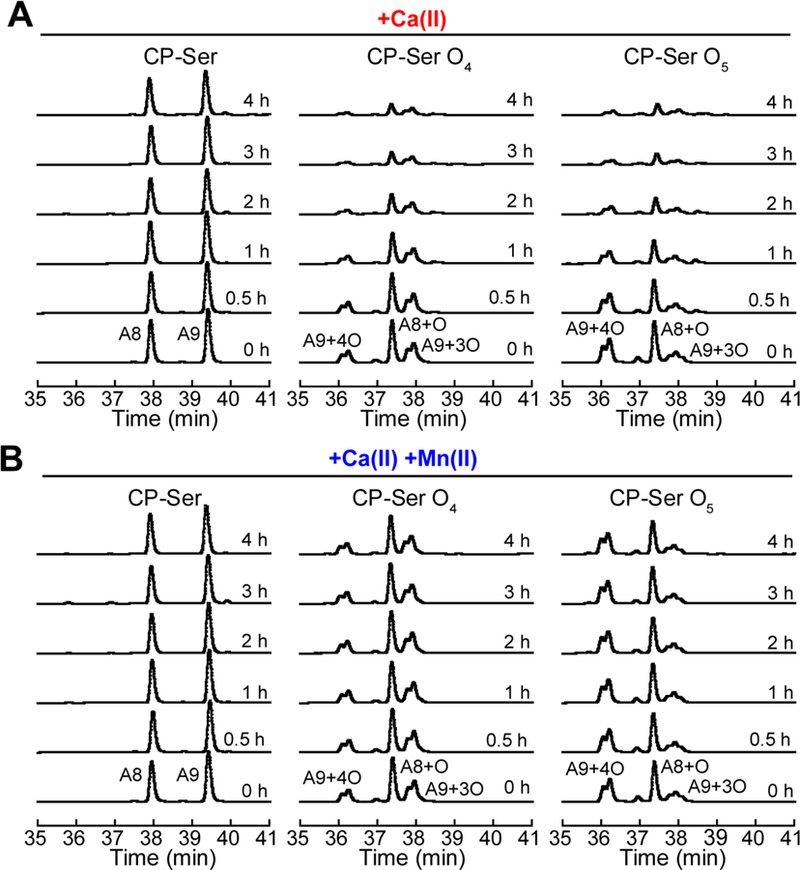
Representative HPLC traces from trypsin (0.45 µM) digestions of 30 µM CP-Ser, CP-Ser O4, and CP-Ser O5 (75 mM HEPES, 100 mM NaCl, pH 7.5, 37 °C). (A) In the presence of 1.5 mM Ca(II). (B) In the presence of 1.5 mM Ca(II) and 30 µM Mn(II).
In agreement with our prior work,12 inhibiting Ca(II)-induced tetramerization resulted in accelerated proteolysis of CP-Ser (Figure 7 and S14–S17, and Supporting Discussion). For instance, in the presence of trypsin and excess Ca(II), we did not observe cleavage of CP-Ser, whereas CP-Ser O4 and CP-Ser O5 were nearly quantitatively degraded after 3 h (Figure 7A). In the presence of chymotrypsin and Ca(II) ions, CP-Ser exhibited slow cleavage of the S100A9 tail whereas CP-Ser O4 and CP-Ser O5 were cleaved to peptides (Figure S14). Although all of the proteases displayed greater activity against CP-Ser O4 and CP-Ser O5 than CP-Ser in the presence of Ca(II), HNE degraded the oxidized proteins much more slowly than the other proteases (Figure S15). Additionally, HNE was the only protease for which we observed a marked difference in the degradation rate between S100A9+3O and S100A9+4O (Figures S15 and S16). When we performed the same experiments in the presence of 1 eq. Mn(II), we observed no degradation of CP-Ser, CP-Ser O4, or CP-Ser O5 (Figure 7B and Figures S14–S17). To ascertain whether this effect was Mn(II)-specific or a general consequence of having a divalent metal ion coordinated at the His6 site, we treated CP-Ser, CP-Ser O4, and CP-Ser O5 with trypsin in the presence of excess Ca(II) and 1 eq. Fe(II) under anaerobic conditions. The Fe(II)-bound protein was also protected from degradation (Figure S18). Based on our prior studies of Mn(II)- and Fe(II)-induced tetramerization of CP-Ser and variants,12 and the current work showing that CP-Ser O4 and CP-Ser O5 form tetramers in the presence of Ca(II) and 1 eq. Mn(II) (Figure 6), we attribute the recovery of protease stability observed for Mn(II)- and Fe(II)-bound oxidized CP species to transition-metal-induced tetramerization.
Oxidation of CP Yields Disulfide-Linked Oligomers.
CP contains two Cys residues at position 42 of S100A8 and position 3 of S100A9. Similar to methionine, Cys residues are susceptible to oxidation by H2O2.43 Thus, our studies with CP-Ser did not take the possibility of H2O2-mediated oxidation of C42 of S100A8 or C3 of S100A9 into account. With an understanding of the consequences of methionine oxidation for CP-Ser in hand, we shifted our focus to studying the more complex Cys-containing protein.
We began by examining whether treating CP with 100 µM H2O2 resulted in disulfide-bond formation in the absence or presence of excess Ca(II) ions by non-reducing SDS-PAGE and western blot (Figure 8A). Under our conditions, we observed minimal formation of disulfide bonds when CP was incubated without H2O2 (Figure 8A). In the absence of Ca(II), we observed bands consistent with disulfide-linked S100A8–S100A9 and S100A9–S100A9 species at the 1-h time point (Figure 8A). We also observed a weak band assigned to a disulfide-linked S100A8–S100A8 species appeared at 7 h and 23 h. In the presence of excess Ca(II), the disulfide reactivity changed. At the 1-h time point, the S100A9–S100A9 species was dominant and negligible S100A8–S100A9 species was detected. Over time, the S100A9–S100A9 species became less abundant, while the amount of the S100A8–S100A9 species increased over the 23-h time course. Moreover, in the presence of Ca(II), no band attributed to a disulfide-linked S100A8–S100A8 species was detected. We reason that the different disulfide reactivity likely results from Ca(II)-induced structural changes that impact the accessibility of C42 of S100A8, which is located in the linker region between the N- and C-terminal EF-hands. In contrast, C3 of S100A9 is fully solvent exposed. Moreover, the conversion of S100A9–S100A9 species to the S100A8–S100A9 species at later time points indicates that disulfide bonds involving C42 of S100A8 are more stable. We also performed reactions with a CP variant that lacks C3 of S100A9 [S100A8/S100A9(C3S)] to mimic the truncated CP species observed in the human samples that results from translation initiation at M5 of S100A9. Non-reducing SDS-PAGE afforded no evidence for disulfide bond formation following treatment of the S100A8/S100A9(C3S) variant with 100 µM H2O2 (Figure S19). Because oxidation of CP in vivo is presumed to occur in the extracellular space, we further characterized CP after treatment with 100 µM H2O2 for 23 h in the presence of Ca(II), and we name this protein disulfide-linked CP.
Figure 8.
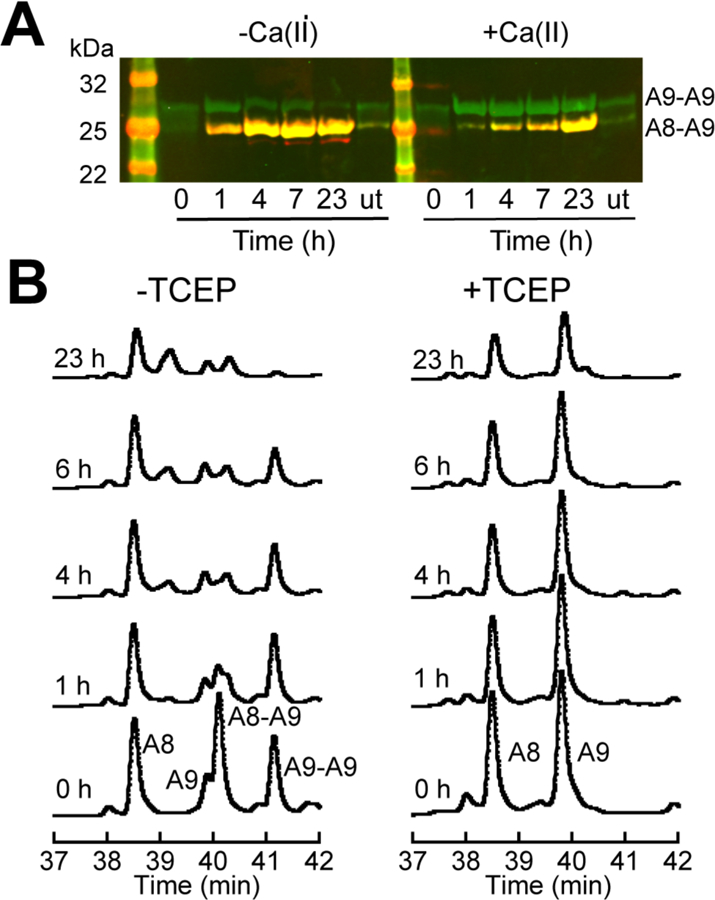
Evaluation of disulfide-linked CP. The abbreviation u.t. stands for untreated. (A) A non-reducing western blot of 30 µM CP treated with 100 µM H2O2 at various points in time in the presence and absence of 1.5 mM Ca(II) (75 mM HEPES, 100 mM NaCl, pH 7.5). S100A8 is red (mouse anti-S100A8), S100A9 is green (goat anti-S100A9), and yellow indicates both subunits. Representative HPLC chromatograms of 30 µM disulfide-linked CP treated with 0.45 µM trypsin in the presence of 1.5 mM Ca(II) (75 mM HEPES, 100 mM NaCl, pH 7.5). Samples were quenched in the absence (left panel) and presence (right panel) of TCEP.
To further decipher the speciation of CP after exposure to 100 µM H2O2 for 23 h in the presence of excess Ca(II), we treated disulfide-linked CP with TCEP to reduce the disulfide bonds prior to mass spectrometry. The reaction product displayed the expected masses for unmodified S100A8 and S100A9, and no peaks that corresponded to species containing additional oxygen atoms were found (Figure S20). This analysis indicated that 100 µM H2O2 is insufficient to oxidize the methionine residues of CP under our reaction conditions. Next, to assess the effects of Ca(II) ions and disulfide bond formation on protein oligomerization, we treated aliquots of the sample from the 23-h time point in three different ways and analyzed the samples by SEC and SDS-PAGE (Figure S21 and Supporting Discussion). We (i) removed Ca(II) ions from the protein using EDTA to examine the disulfide-containing species without bound Ca(II) ions, (ii) reduced the disulfide bonds using TCEP to examine the Ca(II)-bound reduced protein, and (iii) analyzed an untreated sample where the disulfide bonds remained intact and the protein was Ca(II)-bound. These experiments provided several new insights: (i) all species in the disulfide-linked CP mixture displayed Ca(II)-induced oligomerization, (ii) the major species in the disulfide-linked CP mixture was the CP heterotetramer that contained at least one (S100A9)C3–(S100A8)C42 “intradimer” disulfide bond, and (iii) the minor product was a small disulfide-linked CP polymer. To the best of our knowledge, the existence of an intradimer disulfide bond has not been considered to date. Examination of CP-Ser crystal structures revealed that the N-terminus of S100A9 is in close proximity to (S100A8)C42 (Figure S22), indicating that formation of an (S100A9)C3–(S100A8)C42 intradimer disulfide bond is reasonable.
Disulfide-Linked CP is Protease Sensitive.
We assayed the susceptibility of disulfide-linked CP to degradation by trypsin in the presence of Ca(II). We quenched reaction aliquots in the absence and presence of TCEP at varying time points and analyzed the mixtures by HPLC (Figure 8B). At the 0 h time point, the chromatogram of the TCEP-treated sample exhibited two major peaks corresponding to S100A8 and S100A9, and the peak intensities decreased over time, indicating trypsin-catalyzed degradation of each subunit (Figure 8B). The chromatogram of the sample that was not treated with a reductant was more complex and exhibited three major peaks along with a minor peak (Figure 8B). One of the major peaks (38.5 min) was assigned to the S100A8 monomer, and the identities of the other two major peaks were determined by mass spectrometry (Figure 8B, Table S3). The 40.0 min peak was identified as the disulfide-linked S100A8–S100A9 dimer and the 41.1 min peak was identified as the disulfide-linked S100A9–S100A9 dimer. The minor peak (39.9 min) was assigned to the S100A9 monomer. Analysis of the samples obtained at later time points revealed several features about the trypsin susceptibility of these species: (i) CP containing an intradimer disulfide bond was almost completely degraded by trypsin after only 1 h, (ii) the S100A9–S100A9 dimer was almost completely degraded after 23 h, and (iii) the CP subunits that had not formed disulfide bonds were digested to a lesser extent than the disulfide-linked protein (Figure 8B). From these results, we concluded that disulfide bond formation between CP subunits sensitizes CP to proteolysis by trypsin, and that disulfides that involve C42 of S100A8 are particularly destabilizing.
Disulfide-Linked CP and MetO-Disulfide-Linked CP are Degraded by Trypsin at Comparable Rates.
With the knowledge that methionine oxidation sensitized CP to proteolysis by weakening noncovalent oligomerization, we sought to compare the stability of disulfide-linked CP and MetO-disulfide-linked CP. We therefore developed and optimized a two-step protocol to obtain MetO-disulfide-linked CP (Figures S20 and S23 and Supporting Discussion). We found that treating disulfide-linked CP for 7 h with 100 mM H2O2 afforded the sufficient Met oxidation to resemble CP-Ser O4 and CP-Ser O5 while minimally perturbing the disulfide bonds, and we named this reaction product MetO-disulfide-linked CP. We performed trypsin degradation assays, and at each time point, quenched samples in the absence and presence of TCEP. Without TCEP reduction, the HPLC trace of MetO-disulfide-linked CP exhibited a broad peak, and we were unable to resolve individual species (Figure S24). In contrast, treatment of the protein with TCEP during the quench gave rise to a chromatogram reminiscent of CP-Ser O4 and CP-Ser O5 (Figure S24). We assigned the peaks at 36.5 min, 37.2 min, and 37.7 min to S100A9+4O, S100A9+3O, and S100A8+O by LC-MS, respectively. The fraction containing S100A9+3O also contained a detectable amount of S100A8+2O.
We anticipated that MetO-disulfide-linked CP would be degraded more quickly than disulfide-linked CP given our observations with CP-Ser O4 and CP-Ser O5. Trypsin hydrolyzed MetO-disulfide-linked CP; however, comparison of the degradation of disulfide-linked CP and MetO-disulfide-linked CP revealed that they were degraded at comparable rates (Figure S22). We also noted that rapid degradation of disulfide-linked CP and MetO-disulfide-linked CP occurred during the first hour of digestion, and then markedly slowed.
Discussion
This work demonstrates that post-translational oxidation of CP has structural and functional consequences. In particular, methionine oxidation and disulfide-bond formation alter the stability of CP to proteases. This work also highlights the complex speciation of CP in the biological milieu. Prior studies have considered speciation of CP primarily from the standpoint of the metalation.8,10,24,25,49 When one considers the potential diversity of CP arising from combinations of metalation and post-translational modifications, it becomes clear that the CP present at an infection site is likely highly heterogeneous. Our results support hypotheses for biological fates of CP that are directed by oxidation and metal speciation.
In agreement with prior studies that reported methionine-oxidized CP subunits in samples from mice and humans,29–32 we detected oxidized CP subunits in samples of human nasal mucus and human pus. Moreover, by using 15N-labeled CP-Ser as an internal standard to report on adventitious methionine oxidation, we obtained compelling support for the formation and relevance of methionine-oxidized CP in vivo. CP is an abundant neutrophil protein that is released into the extracellular space, and methionine oxidation of CP likely results from the neutrophil oxidative burst. This process occurs after neutrophil activation, and leads to production of ROS including O2•-, H2O2, and HOCl.33 Indeed, neutrophil activation increases methionine oxidation of neutrophil proteins and surrounding tissues,35,50 and α1-proteinase inhibitor is inactivated by methionine oxidation caused by neutrophil-derived ROS in vivo.34 Under certain circumstances, microbes may also cause oxidation of CP as an immune-evasion strategy. For instance, Helicobacter pylori can induce ROS generation in epithelial cells and macrophages,51,52 which could modify CP and result in its premature degradation.
In this study, we employed H2O2 to oxidize CP and CP-Ser because it is more selective for Met and Cys oxidation than other oxidants.43 We recognize that the time and H2O2 concentrations required to oxidize CP in our experiments raise questions of kinetic competence in vivo. Indeed, an estimate of H2O2 in a neutrophil phagosome is between 1 to 4 µM,53 far below the concentrations we employed to obtained oxidized CP(-Ser) species. It is important to note that the primary oxidant during the neutrophil oxidative burst is HOCl produced by MPO. Although H2O2 is thermodynamically a strong oxidant, it is kinetically slow with a rate constant of 6×10−3 M-1s−1 for oxidizing Met to MetO.36 In contrast, the rate constant for HOCl reacting with Met to form MetO is 3.8×107 M−1s-1.36 Due to the extremely fast reactivity of HOCl with Met, we reason that this oxidant may be responsible for producing the majority of the oxidized methionine residues observed in CP. With the results reported here as a foundation for future work, investigations of the consequences of HOCl and chloramines for the structure and function of CP-Ser and CP are warranted.
By using H2O2, we found that oxidation of M81 of S100A9 perturbed the dimer-tetramer equilibrium in the presence of Ca(II). Crystal structures of the CP heterotetramer show that the side chain of M81 of S100A9 points directly into the dimer-dimer interface (Figure S2). Thus, we expect that conversion of Met to MetO decreases the driving force for tetramerization due to the hydrophilic nature of MetO, which is considerably more hydrophilic than Met. Indeed, MetO was calculated to be approximately as hydrophilic as lysine.54 Protease susceptibility assays performed in the presence of excess Ca(II) ions revealed that both CP-Ser O4 and CP-Ser O5 were more rapidly degraded by trypsin, chymotrypsin, and proteinase K, which we attribute to incomplete tetramerization caused by oxidation of M81 of S100A9. Protease resistance was rescued by addition of one equivalent of Mn(II) to CP-Ser O4 and CP-Ser O5, which coordinated to the His6 site and caused the proteins to tetramerize. Thus, the increased protease resistance conferred by Mn(II) or Fe(II) binding results from the metal ion converting CP-Ser O4 and CP-Ser O5 to the tetrameric state.12 The protease susceptibility assays also revealed that, in the presence of excess Ca(II) ions, CP-Ser O4 and CP-Ser O5 were poor substrates for HNE. It is possible that the low activity of HNE against CP-Ser O4 and CP-Ser O5, as well as CP-Ser, reflects a selected trait of CP. HNE and CP are both released by neutrophils, and we can imagine that that resisting degradation by HNE allows CP to perform its function without being immediately degraded.
Native CP presents a more complex scenario because each full-length subunit contains a single cysteine residue that is susceptible to oxidation. By investigating the consequences of oxidative post-translational modifications to the native protein, we demonstrated that disulfide-bond formation between CP molecules also increased the rate of protease degradation. This effect was especially pronounced for the species containing the S100A8–S100A9 disulfide linkage, but the S100A9–S100A9 disulfide-linked protein was also degraded faster than CP-Ser. We propose that disulfide bonds involving S100A8 are destabilizing because C42 of S100A8 is located in the “hinge” region between the N- and C- terminal EF-hands. Considering the oxidative nature of the extracellular space and the ROS generated during the neutrophil response, it is reasonable to imagine that CP can form disulfide bonds with other CP molecules as well as other cysteine-containing biomolecules. Along these lines, human S100A7 was found to form high-molecular weight disulfide-linked oligomers in wounds.55 Amino acid sequence alignment of S100A8 polypeptides from 36 available genomes shows that C42 of S100A8 is highly conserved (Figure S25). This residue is surface exposed in human CP, which is unusual for Cys residues,56 and is likely surface exposed in orthologues from other mammals. Thus, it is possible that disulfide bond formation with S100A8 enhances the protease susceptibility of CP in many organisms.
Although disulfide bonding increased protease susceptibility more than Met oxidation, we expect that Met oxidation can influence the lifetime of CP at biological sites. Ex vivo analyses of human kidney stones32 and our work indicate that there is a significant population of S100A9 that lacks the first 5 amino acids and thus the Cys residue at position 3. This truncated S100A9 species cannot form disulfides that afford enhanced protease susceptibility, and we reason that methionine oxidation may accelerate clearance of CP containing this truncated S100A9 subunit.
Taken together, the investigations of CP and CP-Ser presented in this work afford an extension to the working model of CP (Figure 9). Previously, CP was thought to reside in the extracellular space as the Ca(II)-bound tetramer, with some fraction also coordinated to one or more transition metals. We propose a more dynamic picture where methionine oxidation and disulfide bonds modulate the quaternary structure and thus lifetime of CP in this milieu by enhancing the susceptibility of Ca(II)-bound CP to proteolytic degradation. CP is multi-functional, and after its release, the host must attempt to balance its nutrient withholding role against its contributions to proinflammatory signaling. We hypothesize that Met oxidation helps the host achieve this balance. Because the concentration of CP at an infection site is expected to exceed the concentration of bioavailable transition metal ions, we expect that the majority of CP exists in the Ca(II)-bound state and is thus susceptible to proteolytic degradation after post-translational oxidation. We reason that recovery of protease stability after binding a metal at the His6 site is advantageous for the host because it prevents premature release of transition metal ions. This scenario would allow the host to preferentially degrade excess or “unnecessary” CP while preserving CP that is actively withholding transition metal ions. Removing CP is likely important for preventing it from participating in inflammatory shock and the development of autoreactive CD8+ T cells.18,19
Figure 9.
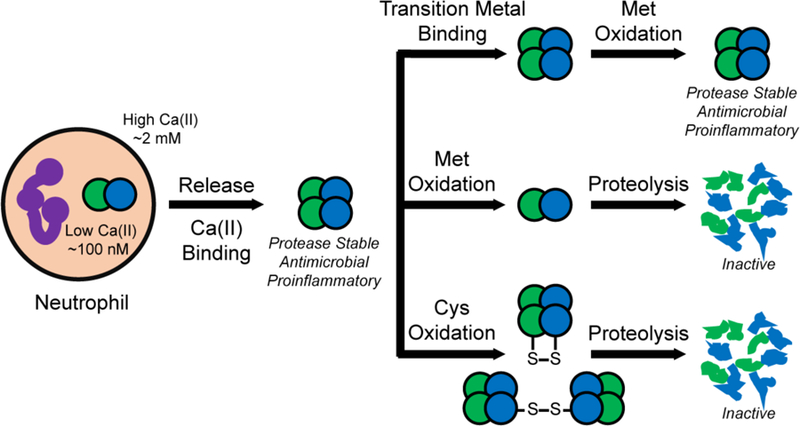
Working model for the function and fate of extracellular CP considering the consequences of post-translational oxidation. Although not depicted, we expect that CP species containing disulfide bonds and MetO modifications without and with bound transition metal ions also exist.
Beyond the context of CP in the host/microbe interaction and inflammation, this work agrees with previous work on the MetO post-translational modification. Various structural consequences of methionine oxidation have been described. For instance, the presence of MetO can disrupt interactions between protein domains,57,58 hinder formation of noncovalent heterocomplexes,34,59 and destabilize protein folds.60 In addition, the connection between MetO and accelerated proteolysis has precedence. Met oxidation of calmodulin targeted it to the proteasome in an ubiquitin-independent fashion;61 in contrast, our work demonstrated that Met oxidation of CP accelerated non-targeted proteolysis. Lastly, the Met/MetO redox couple in the context of ROS have been considered to serve an antioxidant role.38,39 The hypothesis is that surface Met residues are oxidized to MetO, which can be reduced to methionine by Msr proteins, thereby protecting critical residues from ROS. Although we agree that Met can play an important sacrificial antioxidant function, our data and those of others also support a regulatory role for Met oxidation.57,62 Considering the observation that ROS enable neutrophil proteases to be active in the extracellular space by oxidizing Met to MetO,34 the necessity for ROS to form neutrophil extracellular traps,63 and our finding that MetO modification sensitizes CP to proteolysis argues in favor of ROS and MetO also serving as signals during the immune response.
In closing, the results presented in this work support a model where methionine oxidation and disulfide bond formation promote proteolytic degradation of CP, providing new molecular-level insights into the fate of CP after release into the extracellular space. We propose that the host uses oxidative modification of CP to accelerate its proteolysis. We envision that, by sensitizing CP to proteolysis, the host can dispose of CP in a timely manner to prevent negative outcomes from CP-mediated proinflammatory signaling. Moreover, because transition-metal binding rescues tetramerization and protease resistance, it appears that the host strategically avoids degrading transition-metal-bound CP to prevent release of nutrients to microbes.
Supplementary Material
Figure 3.
Deconvoluted mass spectra of human pus with 15N-CP-Ser added immediately after collection. The observed and theoretical masses for each numbered peak are contained in Table S1. Data from pus sample 2 (A) expanded around S100A8 and (B) expanded around S100A9.
Acknowledgements
We thank the NIH (R01GM118695), the MIT Research Support Committee (Wade Award), and the MIT Department of Chemistry for financial support. The MIT Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems is supported by NSF grant 0070319. We thank Prof. B. L. Pentelute for generously providing access to mass spectrometry instrumentation, Dr. A. Vinogradov for his assistance with mass spectrometry, and Ms. A. Nocera for technical assistance with the human nasal mucus samples.
Footnotes
Supporting Information
Complete experimental methods, Tables S1–S9, Figures S1–S25, Supporting Discussion on AUC and oxidized protein preparation protocols. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Steinbakk M; Naess-Andresen C-F; Lingaas E; Dale I; Brandtzaeg P; Fagerhol MK Lancet 1990, 336, 763–765. [DOI] [PubMed] [Google Scholar]
- (2).Hood MI; Skaar EP Nat. Rev. Microbiol 2012, 10, 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Corbin BD; Seeley EH; Raab A; Feldmann J; Miller MR; Torres VJ; Anderson KL; Dattilo BM; Dunman PM; Gerads R; Caprioli RM; Nacken W; Chazin WJ; Skaar EP Science 2008, 319, 962–965. [DOI] [PubMed] [Google Scholar]
- (4).Johne B; Fagerhol MK; Lyberg T; Prydz H; Brandtzaeg P; Naess-Andresen CF; Dale I J. Clin. Pathol.: Mol. Pathol 1997, 50, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Edgeworth J; Gorman M; Bennett R; Freemont P; Hogg N J. Biol. Chem 1991, 266, 7706–7713. [PubMed] [Google Scholar]
- (6).Fagerhol MK; Dale I; Andersson T Scand. J. Haemotol 1980, 24, 393–398. [Google Scholar]
- (7).Damo SM; Kehl-Fie TE; Sugitani N; Holt ME; Rathi S; Murphy WJ; Zhang Y; Betz C; Hench L; Fritz G; Skaar EP; Chazin WJ Proc. Natl. Acad. Sci. U.S.A 2013, 110, 3841–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Brophy MB; Hayden JA; Nolan EM J. Am. Chem. Soc 2012, 134, 18089–18100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Brophy MB; Nakashige TG; Gaillard A; Nolan EM J. Am. Chem. Soc 2013, 135, 17804–17817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Nakashige TG; Zhang B; Krebs C; Nolan EM Nat. Chem. Biol 2015, 11, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nakashige TG; Stephan JR; Cunden LS; Brophy MB; Wommack AJ; Keegan BC; Shearer JM; Nolan EM J. Am. Chem. Soc 2016, 138, 12243–12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Stephan JR; Nolan EM Chem. Sci 2016, 7, 1962–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Garcia YM; Barwinska-Sendra A; Tarrant E; Skaar EP; Waldron KJ; Kehl-Fie TE PLOS Pathog 2017, 13, e1006125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Liu JZ; Jellbauer S; Poe AJ; Ton V; Pesciaroli M; Kehl-Fie TE; Restrepo Nicole A.; Hosking MP; Edwards RA; Battistoni A; Pasquali P; Lane TE; Chazin WJ; Vogl T; Roth J; Skaar EP; Raffatellu M Cell Host & Microbe 2012, 11, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nairn BL; Lonergan ZR; Wang J; Braymer JJ; Zhang Y; Calcutt MW; Lisher JP; Gilston BA; Chazin WJ; de Crécy-Lagard V; Giedroc DP; Skaar EP Cell Host & Microbe 2016, 19, 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Stork M; Grijpstra J; Bos MP; Mañas Torres C; Devos N; Poolman JT; Chazin WJ; Tommassen J PLOS Pathog 2013, 9, e1003733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jean S; Juneau RA; Criss AK; Cornelissen CN Infect. Immun 2016, 84, 2982–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vogl T; Tenbrock K; Ludwig S; Leukert N; Ehrhardt C; van Zoelen MAD; Nacken W; Foell D; van der Poll T; Sorg C; Roth J Nat. Med 2007, 13, 1042–1049. [DOI] [PubMed] [Google Scholar]
- (19).Loser K; Vogl T; Voskort M; Lueken A; Kupas V; Nacken W; Klenner L; Kuhn A; Foell D; Sorokin L; Luger TA; Roth J; Beissert S Nat. Med 2010, 16, 713–717. [DOI] [PubMed] [Google Scholar]
- (20).Austermann J; Friesenhagen J; Fassl SK; Ortkras T; Burgmann J; Barczyk-Kahlert K; Faist E; Zedler S; Pirr S; Rohde C; Müller-Tidow C; von Köckritz-Blickwede M; von Kaisenberg CS; Flohé SB; Ulas T; Schultze JL; Roth J; Vogl T; Viemann D Cell Reports 2014, 9, 2112–2123. [DOI] [PubMed] [Google Scholar]
- (21).Gagnon DM; Brophy MB; Bowman SEJ; Stich TA; Drennan CL; Britt RD; Nolan EM J. Am. Chem. Soc 2015, 137, 3004–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hunter MJ; Chazin WJ J. Biol. Chem 1998, 273, 12427–12435. [DOI] [PubMed] [Google Scholar]
- (23).Korndörfer IP; Brueckner F; Skerra AJ Mol. Biol 2007, 370, 887–898. [DOI] [PubMed] [Google Scholar]
- (24).Hayden JA; Brophy MB; Cunden LS; Nolan EM J. Am. Chem. Soc 2013, 135, 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Nakashige TG; Zygiel EM; Drennan CL; Nolan EM J. Am. Chem. Soc 2017, 139, 8828–8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Vogl T; Roth J; Sorg C; Hillenkamp F; Strupat K J. Am. Soc. Mass Spectrom 1999, 10, 1124–1130. [DOI] [PubMed] [Google Scholar]
- (27).Vogl T; Leukert N; Barczyk K; Strupat K; Roth J Biochim. Biophys. Acta 2006, 1763, 1298–1306. [DOI] [PubMed] [Google Scholar]
- (28).Brini M; Ottolini D; Calì T; Carafoli E Met. Ions Life Sci 2013, 13, 81–137. [DOI] [PubMed] [Google Scholar]
- (29).Spraggins JM; Rizzo DG; Moore JL; Rose KL; Hammer ND; Skaar EP; Caprioli RM J. Am. Soc. Mass Spectrom 2015, 26, 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Magon NJ; Turner R; Gearry RB; Hampton MB; Sly PD; Kettle AJ Free Radic. Biol. Med 2015, 86, 133–144. [DOI] [PubMed] [Google Scholar]
- (31).Gomes LH; Raftery MJ; Yan WX; Goyette JD; THomas PS; Geczy CL Free Radic. Biol. Med 2013, 58, 170–186. [DOI] [PubMed] [Google Scholar]
- (32).Martelli C; Marzano V; Iavarone F; Huang L; Vincenzoni F; Desiderio C; Messana I; Beltrami P; Zattoni F; Ferraro PM; Buchholz N; Locci G; Faa G; Castagnola M; Gambaro G J. Urol 2016, 196, 911–918. [DOI] [PubMed] [Google Scholar]
- (33).Nathan C Nat. Rev. Immunol 2006, 6, 173–182. [DOI] [PubMed] [Google Scholar]
- (34).Weiss SJ N. Engl. J. Med 1989, 320, 365. [DOI] [PubMed] [Google Scholar]
- (35).Maier K; Leuschel L; Costabel U Am. Rev. Respir. Dis 1991, 143, 271–274. [DOI] [PubMed] [Google Scholar]
- (36).Davies MJ Biochim. Biophys. Acta 2005, 1703, 93–109. [DOI] [PubMed] [Google Scholar]
- (37).Levine RL; Mosoni L; Berlett BS; Stadtman ER Proc. Natl. Acad. Sci. U.S.A 1996, 93, 15036–15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Moskovitz J; Berlett BS; Poston JM; Stadtman ER Proc. Natl. Acad. Sci. U.S.A 1997, 94, 9585–9589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Levine RL; Berlett BS; Moskovitz J; Mosoni L; Stadtman ER Mech. Ageing Dev 1999, 107, 323–332. [DOI] [PubMed] [Google Scholar]
- (40).Achilli C; Ciana A; Rossi A; Balduini C; Minetti G J. Leukoc. Biol 2007, 83, 181–189. [DOI] [PubMed] [Google Scholar]
- (41).Stadtman ER; Van Remmen H; Richardson A; Wehr NB; Levine RL Biochim. Biophys. Acta 2005, 1703, 135–140. [DOI] [PubMed] [Google Scholar]
- (42).Lim SY; Raftery MJ; Goyette J; Hsu K; Geczy CL J. Leukoc. Biol 2009, 86, 577–587. [DOI] [PubMed] [Google Scholar]
- (43).Winterbourn CC Methods Enzynol 2013, 528, 3–25. [DOI] [PubMed] [Google Scholar]
- (44).Brown PH; Balbo A; Schuck P In Current Protocols in Immunology; John Wiley & Sons, Inc: 2001, 1815.1–18.15.39. [Google Scholar]
- (45).Lebowitz J; Lewis MS; Schuck P Protein Sci 2002, 11, 2067–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Stafford WF Anal. Biochem 1992, 203, 295–301. [DOI] [PubMed] [Google Scholar]
- (47).Philo JS Anal. Biochem 2006, 354, 238–246. [DOI] [PubMed] [Google Scholar]
- (48).Walkup GK; Burdette SC; Lippard SJ; Tsien RY J. Am. Chem. Soc 2000, 122, 5644–5645. [DOI] [PubMed] [Google Scholar]
- (49).Hadley RC; Gagnon DM; Brophy MB; Gu Y; Nakashige TG; Britt RD; Nolan EM J. Am. Chem. Soc 2018, 140, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Fliss H; Weissbach H; Brot N Proc. Natl. Acad. Sci. U.S.A 1983, 80, 7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Obst B; Wagner S; Sewing KF; Beil W Carcinogenesis 2000, 21, 1111–1115. [PubMed] [Google Scholar]
- (52).Chaturvedi R; Cheng Y; Asim M; Bussière FI; Xu H; Gobert AP; Hacker A; Casero RA; Wilson KT J. Biol. Chem 2004, 279, 40161–40173. [DOI] [PubMed] [Google Scholar]
- (53).Imlay JA EcoSal Plus 2009, 3, ecosalplus 5.4.4. [DOI] [PubMed] [Google Scholar]
- (54).Black SD; Mould DR Anal. Biochem 1991, 193, 72–82. [DOI] [PubMed] [Google Scholar]
- (55).Lee KC; Eckert RL J. Investig. Dermatol 2007, 127, 945–957. [DOI] [PubMed] [Google Scholar]
- (56).Janin J; Miller S; Chothia C J. Mol. Biol 1988, 204, 155–164. [DOI] [PubMed] [Google Scholar]
- (57).Ciorba MA; Heinemann SH; Weissbach H; Brot N; Hoshi T Proc. Natl. Acad. Sci. U.S.A 1997, 94, 9932–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Snijder J; Rose RJ; Raijmakers R; Heck AJR J. Struct. Biol 2011, 174, 187–195. [DOI] [PubMed] [Google Scholar]
- (59).Bertolotti-Ciarlet A; Wang W; Lownes R; Pristatsky P; Fang Y; McKelvey T; Li Y; Li Y; Drummond J; Prueksaritanont T; Vlasak J Mol. Immunol 2009, 46, 1878–1882. [DOI] [PubMed] [Google Scholar]
- (60).Gao J; Yin DH; Yao Y; Sun H; Qin Z; Schöneich C; Williams TD; Squier TC Biophys. J 1998, 74, 1115–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Balog EM; Lockamy EL; Thomas DD; Ferrington DA Biochemistry 2009, 48, 3005–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Erickson JR; Joiner M.-l. A.; Guan X; Kutschke W; Yang J; Oddis CV; Bartlett RK; Lowe JS; O’Donnell SE; Aykin-Burns N; Zimmerman MC; Zimmerman K; Ham A-JL; Weiss RM; Spitz DR; Shea MA; Colbran RJ; Mohler PJ; Anderson ME Cell 2008, 133, 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Brinkmann V; Zychlinsky A Nat. Rev. Microbiol 2007, 5, 577–582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.