

Abstract
RNA-guided programmable nucleases from CRISPR systems generate precise breaks in DNA or RNA at specified positions. In cells, this activity can lead to changes in DNA sequence or RNA transcript abundance. Base editing is a newer genome editing approach that uses components from CRISPR systems together with other enzymes to directly install point mutations into cellular DNA or RNA without making double-stranded DNA breaks (DSBs). DNA base editors comprise a catalytically disabled nuclease fused to a nucleobase deaminase enzyme and, in some cases, a DNA glycosylase inhibitor. RNA base editors achieve analogous changes using components that target RNA. Base editors directly convert one base or base pair into another, enabling the efficient installation of point mutations in non-dividing cells without generating excess undesired editing byproducts. In this Review, we summarize base editing strategies to generate specific and precise point mutations in genomic DNA and RNA, highlight recent developments that expand the scope, specificity, precision, and in vivo delivery of base editors, and discuss limitations and future directions of base editing for research and therapeutic applications.
Introduction
The ability to precisely and efficiently edit DNA sequences within the genome of living cells has been a major goal of the life sciences since the first demonstration of restriction cloning1. Recently, RNA-programmable CRISPR-associated (Cas) nucleases have contributed to the pursuit of this goal2–4 through their ability to generate a double-stranded DNA break (DSB) at a precise target location in the genome of a wide variety of cells and organisms5–8 (reviewed extensively9–12). Catalytically inactivated Cas nucleases are also useful as programmable DNA-binding proteins that localize tethered proteins to target DNA loci2,13–16.
Generation of a DSB does not directly lead to DNA editing; rather, editing following nuclease treatment occurs as a result of cellular responses to DSBs. Processes including non-homologous end-joining (NHEJ) and microhomology-mediated end-joining (MMEJ) can lead to gene disruption through the introduction of insertions, deletions, translocations, or other DNA rearrangements at the site of a DSB17–19. Alternatively, a precise DNA edit can be made by supplying a donor DNA template encoding the desired DNA change flanked by sequence homologous to the region upstream and downstream of the DSB. Cellular homology-directed repair (HDR) then results in the incorporation of sequence from the exogenous DNA template at the DSB site20,21. Although HDR is a flexible tool with the ability to make precise insertions, deletions, or any point mutation of interest, HDR is largely restricted to the G2 and S phases of the cell cycle, limiting efficient HDR to actively dividing cells, and even in cultured cell lines HDR efficiency can be modest22–24. Moreover, NHEJ and HDR are competing processes, and under most conditions NHEJ is more efficient than HDR. Thus, a majority of edited products will usually contain small insertions or deletions24,25.
In mammalian cells, DSB-induced NHEJ is an effective way to disrupt a gene of interest. However, more reliable techniques that generate precise DNA or RNA modifications are necessary to make comparisons between alleles, study the effects of specific mutations within genes, or to treat genetic disease through gene correction. Although sampling bias due to the extensive use of short-read sequencing to analyze genomic diversity is possible26,27, the largest class of known human pathogenic mutations, by far, is the point mutation (also called single nucleotide polymorphism (SNP)) (Figure 1a)28,29. Installing or reversing pathogenic SNPs efficiently and cleanly is thus of great interest for the study and treatment of genetic disorders, and requires a method to specifically change the sequence of an individual base pair within a vast genome.
Figure 1. Distribution of human pathogenic genetic variants, including point mutations.
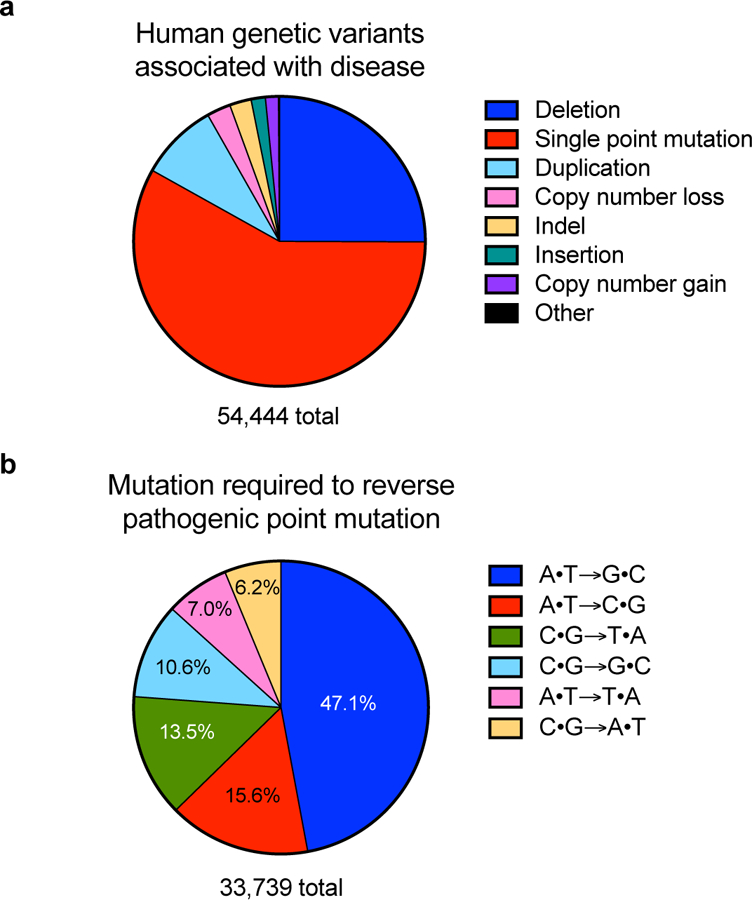
a) Classification of human pathogenic genetic variants in the ClinVar database (accessed May 29, 2018)28,29. As noted in the text, sampling bias due to the extensive use of short-read sequencing to analyze genomic diversity is possible. b) Distribution of base pair changes needed to reverse the pathogenic point mutations represented in the red wedge in (a)28,29. The percentage represented by each base pair change is noted on the pie chart; transition mutations are labeled in white and transversion mutations are labeled in black.
DSBs created by nucleases such as Cas9 result in indels, translocations, and rearrangements27,36–38 that are undesired byproducts when attempting to install a point mutation. Base editing is a genome editing method that directly generates precise point mutations in genomic DNA or in cellular RNA without directly generating DSBs, requiring a DNA donor template or relying on cellular HDR30–32. Since base editors do not normally create DSBs, they minimize the formation of DSB-associated byproducts32,39. Instead, DNA base editors (BEs) comprise fusions between a catalytically impaired Cas nuclease and a base-modification enzyme that operates on single-stranded DNA (ssDNA) but not double-stranded DNA (dsDNA). Upon binding to its target locus in DNA, base pairing between the guide RNA and target DNA strand leads to displacement of a small segment of single-stranded DNA in an “R-loop”33. DNA bases within this single-stranded DNA bubble are modified by the deaminase enzyme. To improve efficiency in eukaryotic cells, the catalytically disabled nuclease also generates a nick in the non-edited DNA strand, inducing cells to repair the non-edited strand using the edited strand as a template30–32.
Two classes of DNA base editor have been described: cytosine base editors (CBEs) convert a C•G base pair into a T•A base pair30,31,34, and adenine base editors (ABEs) convert an A•T base pair to a G•C base pair. Collectively, CBEs and ABEs can mediate all four possible transition mutations (C to T, A to G, T to C, and G to A) (Figure 1b)32,35. In RNA, targeted adenosine conversion to inosine has also been developed using both antisense40–49 and Cas13-guided35 RNA-targeting methods. In this Review, we describe the development of DNA and RNA base editors, their capabilities and limitations, and their current and future applications.
Cytosine base editors (CBEs): development and mechanism
The first DNA base editors convert a C•G base pair to a T•A base pair by deaminating the exocyclic amine of the target cytosine to generate uracil (Figure 2a). To localize deamination activity to a small target window within the mammalian genome, Liu and coworkers used an APOBEC1 cytidine deaminase, which accepts ssDNA as a substrate but is incapable of acting on dsDNA50. Fusion of APOBEC1 to dead Cas9 from Streptococcus pyrogenes (dCas9, a mutant of Cas9 containing D10A and H840A) resulted in base editor 1 (BE1) (Table 1)30. When bound to its cognate DNA, dCas9 performs local denaturation of the DNA duplex to generate an R-loop in which the DNA strand not paired with the guide RNA exists as a disordered single-stranded bubble2,33. This feature enables BE1 to perform efficient and localized cytosine deamination in a test tube, with deamination activity restricted to a ~5-bp window of ssDNA (positions ~4–8, counting the protospacer adjacent motif (PAM) as positions 21–23) generated by dCas9. Fusion to dCas9 presents the target site to APOBEC1 in high effective molarity, enabling BE1 to deaminate cytosines located in a variety of different sequence motifs, albeit with differing efficacies30 (Figure 2b).
Figure 2. Cytosine base editing.

(a) Cytosine deamination generates uracil, which base pairs as thymidine. R = 2′-deoxyribose in DNA, or ribose in RNA. (b) Cytosine base editing strategy by BE1, BE2, BE3, or BE4. R-loop formation exposes a region of single-stranded DNA to the cytidine deaminase domain. Target cytosines in this region are deaminated to uracil30,39. (c) Cellular response to cytosine base editing. Uracil DNA glycosylase-mediated excision of the uracil generated in genomic DNA is inhibited by BE2, BE3, and BE4. BE3 and BE4 are designed to nick the non-edited strand (containing the G of the original C•G target base pair), stimulating cellular DNA repair of that strand to replace the G with an A, completing the conversion of the original C•G base pair to a U•A or, following DNA replication or repair to a T•A base pair30,39.
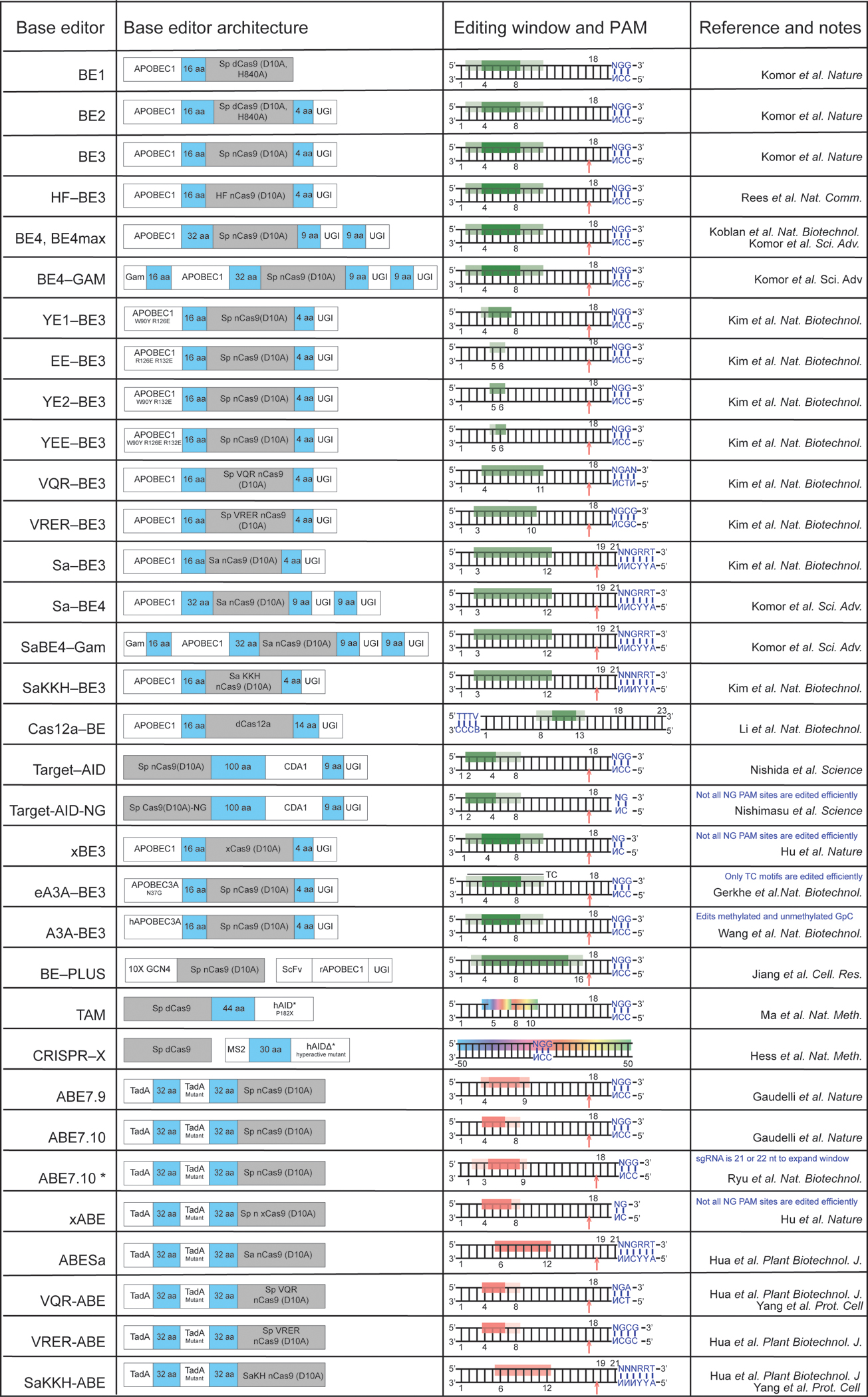
Table 1: DNA base editors and their approximate editing windows.
Cytosine base editor activity windows are shown in green (dark green indicates higher editing efficiency; light green denotes lower editing efficiency). The activity windows of editors that convert C to random mixtures of A, G or T are shown as a rainbow. Adenine base editors have windows shown in red. The location of an induced nick in the target DNA backbone is indicated by an arrow. Base positions are numbered relative to the PAM-distal end of the guide RNA; for example, the NGG PAM sequence of SpCas9 is numbered 21–23.
 |
A major challenge for the use of base editors in mammalian cells is circumventing DNA repair processes that oppose target base pair conversion. Although BE1 mediates efficient, RNA-programmed deamination of target cytosines in vitro, it is not effective in human cells (deamination efficiency fell from 25–40% in vitro to 0.8–7.7% in cells)30. This decrease is largely due to effective cellular repair of the U•G intermediate in DNA51. Base excision repair (BER) of U•G in DNA is initiated by uracil N-glycosylase (UNG), which recognizes the U•G mismatch and cleaves the glyosidic bond between uracil and the deoxyribose backbone of DNA. BER will usually result in the reversion of the U•G intermediate created by BE1 back to a C•G base pair (Figure 2c)51,52. To inhibit UNG, Liu and co-workers fused uracil DNA glycosylase inhibitor (UGI), a small protein from bacteriophage PBS, to the C-terminus of BE1, generating BE2. UGI is a DNA mimic that potently inhibits both human and bacterial UNG53. BE2 mediates efficient base editing in bacterial cells54 and moderately efficient editing in mammalian cells, enabling conversion of a C•G base pair to a T•A base pair through a U•G intermediate (Figure 2c)30.
The base editing efficiency of BE2 is limited by its ability to edit only one strand of DNA. To direct cellular replacement of the G present in the non-targeted strand of DNA with A, Liu and co-workers designed third-generation base editors (BE3) that specifically nick the non-edited DNA strand (Figure 2c). Nicking the non-edited DNA strand biases cellular repair of the U•G mismatch to favor a U•A outcome, greatly elevating base editing efficiencies in mammalian cells. Restoration of His 840 in dCas9 generates a base editor that uses Cas9 “nickase” (D10A) instead of dCas9, resulting in nicking of the non-edited DNA strand (Figure 2c). The APOBEC1– Cas9 nickase–UGI fusion (BE3) yielded efficient editing in mammalian cells, averaging 37% across six loci in the initial report30. Notably, although indels are a detectable byproduct upon treatment with BE3, their frequency is typically small relative to the base edit (indel formation averaged 1.1% across the six reported loci), and much less frequent than indels induced by DSBs30.
Nishida and co-workers described a similar system for cytosine base editing in yeast and mammalian cells, termed “Target-AID”31. In lieu of APOBEC1, they used the cytidine deaminase CDA1 in a Cas9 nickase–CDA1–UGI base editor construct named Target-AID. Target-AID displays a slightly shifted activity window relative to BE3 (Table 1). Nishida and co-workers also noted that base editing at certain bases is less precise than expected, demonstrating that C-to-G or C-to-A edits are, in some cases, significant byproducts of base editing31, as was also observed with BE330. Improvements to CBEs that minimize byproduct formation and increase editing efficiency are discussed below.
Adenine base editors (ABEs): development and mechanism
The distribution of pathogenic point mutations in living systems is not uniform across the six possible ways to exchange one base pair for another (Figure 1b). This uneven distribution is consistent with the relatively high rate of spontaneous cytosine deamination (estimated to be 100–500 deamination events per cell per day), that, if uncorrected, can mutate a G•C base pair to an A•T base pair55,56. A molecular machine capable of reversing such mutations by converting an A•T base pair into a G•C base pair is therefore of particular interest because it would enable correction of the most common type of pathogenic SNPs in the ClinVar database, representing~47% of disease-associated point mutations (Figure 1b). Like cytosine, adenine contains an exocyclic amine that can be deaminated to alter its base pairing preferences. Deamination of adenosine yields inosine (Figure 3a). Although inosine in the third position of a tRNA anticodon is well-known to pair with A, U, or C in mRNA during translation, in the context of a polymerase active site, inosine exhibits the base-pairing preference of guanosine57.
Figure 3. Adenine base editing in DNA and RNA.

(a) Adenosine deamination generates inosine, which has the same base pairing preferences as a guanosine in the active site of a polymerase. R = 2′-deoxyribose in DNA, or ribose in RNA. (b) ABE-mediated DNA base editing to convert an A•T base pair to a G•C base pair. Current ABEs contain one wild-type TadA structural monomer and one evolved TadA catalytic monomer. R-loop formation exposes a small region of ssDNA, within which A is deaminated to I by the heterodimeric wild-type TadA–evolved TadA heterodimer.32 (c-e) Antisense-RNA mediated RNA editing. ADAR variants are localized to the RNA transcript of interest through an antisense RNA with variable lengths of homology to the target transcript. The target A is specified with an A•C mismatch in the mRNA:antisense-RNA duplex. The antisense RNA is in orange, and the target mRNA is in black. (c) Antisense-directed RNA editing by covalent linkage of an ADAR deaminase domain (ADARDD)–SNAP tag fusion to a benzylguanine (BzG)-modified antisense RNA47,49,65. (d) ADAR-BoxB mediated RNA editing40,41. The ADAR2DD is fused to λN protein; λN binds to one of the two BoxB hairpins integrated into the antisense RNA, localizing ADAR-mediated deamination activity to the target adenine. (e) Full-length ADAR2-mediated RNA editing43. The antisense RNA comprises a 5’ R/G-binding motif hairpin, the native binding sequence for full length ADAR2, followed by a 19-nt antisense region complementary to the target RNA with the target adenine centrally located and specified by an A•C mismatch. Overexpression of full-length ADAR2 results in localization of the deaminase to the target base within the target transcript through the native ADAR2 double-stranded RNA binding domains. (f) RNA editing with REPAIR. A dCas13b:guide RNA complex is guided to a the target RNA by a 50-nt spacer. The target A is specified by an A•C mismatch centrally located within the 50-nt spacer.35.
The major hurdle to the development of an ABE was the lack of any known adenosine deaminase enzymes capable of acting on ssDNA. Attempts to force RNA adenosine deaminases to act on DNA by installing them in place of APOBEC1 in BE3 resulted in no detectable adenine base editing32. To overcome this problem, Liu and co-workers evolved a deoxyadenosine deaminase enzyme that accepts ssDNA starting from an Escherichia coli tRNA adenosine deaminase enzyme, TadA32. E. coli cells were equipped with TadA mutants and defective antibiotic resistance genes. To grow in the presence of antibiotic, a mutant TadA–dCas9 fusion (TadA*–dCas9) must convert a deoxyadenosine to a deoxyinosine in the defective antibiotic resistance gene. Bacteria encoding TadA–dCas9 fusions capable of repairing the mutated resistance gene were isolated and then tested in a mammalian cell context.
Although TadA*–dCas9 fusions during this evolution and engineering process were capable of efficient A-to-I conversion in E. coli, simple TadA*–Cas9 nickase fusions resulted in only modest editing rates in mammalian cells. In its native (E. coli) context, TadA acts as a homodimer, with one monomer catalyzing deamination and the other monomer contributing to tRNA substrate binding58. In the E. coli selection, endogenous wild-type TadA could form dimers with the mutated TadA*–dCas9 construct in trans; however the absence of TadA in mammalian cells precludes TadA•TadA* heterodimerization. This challenge was addressed by engineering heterodimeric proteins that incorporate a wild-type non-catalytic TadA monomer, an evolved TadA* monomer and a Cas9 nickase (TadA–TadA*–Cas9 nickase) in a single polypeptide chain (Figure 3b). The single-chain heterodimeric construct greatly improved adenine base editing efficiency in mammalian cells when compared to the corresponding homodimeric TadA*–TadA*–Cas9 nickase editor, suggesting that the mutations required to support deoxyadenosine deamination are incompatible with the structural role played by the N-terminal TadA monomer32.
As with the CBEs, ABEs catalyze deamination within a small window of exposed ssDNA generated by Cas9:guide RNA binding to the target locus. ABE7.10, containing 14 amino acid substitutions in the catalytic TadA* domain, is the most efficient and sequence context-independent ABE reported to date, and performs A•T to G•C conversion within an editing window of protospacer positions ~4–7, counting the PAM as positions 21–23. Different ABE evolutionary relatives, such as ABE7.9 or ABE6.3, can offer higher editing efficiencies at positions closer to the PAM (such as positions 8 or 9; see Table 1). Together, ABEs represent powerful new tools that enable precise conversion of a target A•T base pair to G•C in the genomic DNA of living cells32.
Base editing of RNA
Editing individual bases in RNA can also provide powerful capabilities for the life sciences and, potentially, for medicine. Due to its single-stranded nature, 12 possible base editors that operate on RNA, rather than six possible base editors that operate on dsDNA, are needed to cover all possible changes. To date, the only reported programmable oligonucleotide-directed transformation that changes Watson–Crick base pairing in RNA is deamination of A to I.
Antisense-oligonucleotide-directed A-to-I RNA editing
All RNA base editors characterized in mammalian cells thus far use adenosine deaminases from the ADAR family that natively catalyze hydrolytic adenosine deamination, converting an adenosine to an inosine59,60. Unlike most other RNA-editing enzymes, ADARs are not natively RNA-guided61. Instead, they contain a distinct RNA-binding domain that recognizes and localizes the enzyme to certain regions of double-stranded RNA62,63.
Pioneering efforts by Stafforst, Rosenthal, Nakagawa, and their respective coworkers to generate a targetable adenine RNA editor tethered the catalytic domain of an ADAR enzyme to a guiding antisense RNA oligonucleotide40–48. These RNA editors rely on Watson–Crick base pairing between an antisense RNA and the target transcript to localize an ADAR deaminase domain (ADARDD) to the target RNA. At least three strategies have been developed to establish a physical linkage between the deaminase and the antisense RNA. First, fusing a SNAP tag to the ADAR and generating a benzylguanine-modified antisense RNA (BG-RNA)64 enabled editing in vitro48,65. Delivery of the modified antisense RNA combined with overexpression of a SNAP–ADAR fusion in cells resulted in the covalent linkage between the SNAP-tagged ADAR and the antisense RNA44–49 (Figure 3c). Second, appending the RNA-binding λ-phage N protein to the ADAR deaminase domain and fusing the antisense RNA with a 17-nt “Box B” hairpin that is bound by BoxB also enabled the association of the antisense RNA and the ADAR, enabling both the guiding RNA and deaminase construct to be genetically encoded40,41 (Figure 3d). Third, Stafforst, Fukuda, and their respective coworkers showed that fusing the antisense RNA to the natural substrate for ADAR2 can localize ADAR2 to the antisense RNA for editing in cells42,43 (Figure 3e).
Two key innovations improved the efficiency and specificity of these RNA-guided deamination systems. Stafforst and Schneider exploited the natural sequence preference of human ADAR1 and ADAR2, which preferentially deaminate an adenine that is mispaired with a cytosine in a double-stranded RNA substrate66,67. They designed an 17-nt antisense RNA sequence which placed a C opposite the target A to generate an A•C mismatch upon binding to the target RNA48. This use of the A•C mismatch to direct ADAR activity improved editing in vitro at the on-target adenine and in many of the motifs they tested, with no detectable editing at nearby adenines in the same RNA48,65. Rosenthal and coworkers combined the A•C mismatch strategy40 with use of a hyperactive human ADAR2 mutant (E488Q) to further increase editing efficiency and demonstrated RNA editing in HEK293T cells40, which was improved in efficiency by using two BoxB recruitment domains41 (Figure 3d). Despite these improvements, the use of antisense–deaminase conjugates remained challenging due to high rates of off-target deamination and strong context-dependent editing of adenine bases located in sequence motifs preferred by ADARs40–42,44–48.
In the most recently reported antisense-guided RNA editing system, Stafforst and co-workers dramatically reduce off-target deamination that usually accompanies efficient RNA editing. They integrated an inducible SNAP–ADAR fusion construct into HEK293 cells and delivered chemically modified antisense 22-nt benzylguanine (BG)-linked RNAs by lipofection (Figure 3c)49. The SNAP tag spontaneously becomes covalently bound to the RNA. Editing efficiency was impressively high at six assayed endogenous target transcripts (15–90%) and could be multiplexed without efficiency loss. Significant improvements to the specificity of editing were also made through modifying all the nucleotides in the antisense RNA with a 2’-methoxy group other than the cytosine which specifies the target adenine through the previously described A•C mismatch and its two neighboring bases. This innovation minimized proximal off-target editing other than at adenine-rich triplet targets49. Distal, transcriptome-wide off-target editing was significant when hyperactive ADAR variants were used, but reduced to negligible levels with wild-type ADARs, although on-target editing rates were also lower with wild-type ADARs49.
The most notable limitation of this method is its sequence context dependence; GAN (where N is any nucleotide) target sites are not efficiently edited with any assayed variant due to the native preference of ADAR1 and ADAR2. Future work may harness ADAR mutants, such as E488Q, which show a reduced sequence preference68 into this system to overcome the targeting sequence limitation. For tolerated sequence motifs this approach represents a substantial improvement to efficiency and specificity of RNA editing when genomic integration of the RNA editor construct and delivery of a chemically modified antisense RNA can be performed49.
Cas13-directed A-to-I RNA base editing
Zhang and co-workers developed a different approach to RNA-guided RNA base editing that uses a catalytically dead RNA-guided Cas13b enzyme (dPspCas13b) to localize an ADAR to the target RNA35. dPspCas13b is fused to the deamination domain (DD) of an ADAR (ADARDD) to generate an RNA-guided editor (Table 1; Figure 3f). This approach was termed RNA Editing for Programmable A-to-I Replacement (REPAIR)35. REPAIR incorporates two aspects of ADAR-mediated RNA editing described above: use of the hyperactive ADAR2DD(E488Q) mutant, and specifying the target adenine with an A•C mismatch (Figure 3c–e)35. Notably, REPAIR may offer broad sequence context compatibility; when tested at all 16 possible NAN motifs in a luciferase reporter transcript, REPAIRv1 could edit all 16 codons, apparently overcoming the native ADAR preference through binding to the target site with high effective molarity35.
Zhang and co-workers demonstrated that REPAIRv1 offers higher editing efficiency (89%) than two antisense-mediated strategies: BoxB-ADAR2 (50%)40 and full-length ADAR2 (35%)42 when targeted to a Cluc reporter transcript. However, in two endogenous transcripts tested with REPAIRv1, editing efficiency was reduced to 15–40%35. Transcriptome-wide RNA sequencing (RNA-seq) revealed that REPAIRv1 displays off-target editing that is comparable to that of the BoxB-ADAR strategy and significantly greater than that resulting from overexpression of full-length ADAR235. Proximal off-target RNA base editing was also observed with REPAIRv1: adenine bases 50 bp up- or down-stream of the target adenine were edited at a frequency of approximately 10–20%35. Off-target RNA editing was attributed to overexpression of the hyperactive ADAR deaminase.
To improve the specificity of REPAIRv1, Zhang and coworkers introduced mutations into ADAR2DD(E488Q) designed to reduce the binding affinity between ADARDD and non-target cellular RNA. Using ADAR2DD(E488Q/T375G) in the REPAIRv1 architecture resulted in REPAIRv2. In transcriptome-wide sequencing assays using a guide programmed to edit Cluc, REPAIRv2 yielded only 20 detectable off-target editing events, a 900-fold improvement relative to REPAIRv1. Although still detected, REPAIRv2 also dramatically reduced proximal off-target editing in the 100-nt region upstream or downstream of the target adenine. As expected due to its higher specificity, on-target editing efficiencies of REPAIRv2 were reduced relative to REPAIRv1 (from 89% to approximately 45% in the Cluc reporter), and it is possible that the sequence targeting scope of REPAIRv2 is reduced compared to REPAIRv1. Nevertheless, its high specificity makes REPAIRv2 a promising tool for A-to-I RNA base editing in the mammalian transcriptome35.
Cellular decoding of inosine in mRNA
In DNA base editing of deoxyadenosine, the resulting deoxyinosine is decoded by a DNA or RNA polymerase either during DNA replication or during transcription. Inosine in RNA is functionally decoded by different machinery, such as the ribosome (when in protein-coding regions) or the spliceosome (when in splice sites). Whereas there is strong evidence that deoxyinosine in DNA is read as a G in the active site of a polymerase in human cells57, an inosine in the wobble position of a tRNA pairs with A, C or U in mRNA, enabling a single tRNA to decode multiple cognate codons69. Indeed, in miRNAs the reduced binding strength between the I:C base pair compared to the G:C base pair is thought to be biologically significant for directing mRNA decay70.
Inosine’s ability to form base pairs with multiple bases raises concern that an inosine in an mRNA might be decoded as a mixture of bases in the context of a ribosome or spliceosome active site. Known examples of natural A-to-I editing in the coding regions of mRNA suggest that editing to an inosine at codon position 1 or 2 results predominantly in the inosine being read as a guanine, both in cells71 and in vitro72. For applications involving RNA editing to modulate splicing, observations are also consistent with the spliceosome reading an inosine as a guanine, as A-to-I editing can directly generate or destroy splice sites as if the I were a G73,74.
Base editor limitations and improvements
Base editing product purity
Initial reports of CBEs identified that at some genomic loci, unanticipated C-to-non-T edits are observed, reducing base editing product purity30,31,75–77. Liu and coworkers investigated the determinants of base editing product purity by performing cytosine base editing in cells lacking various genes including UNG, encoding uracil N-glycosylase. In UNG–/– cells, product purity improved from an average of 68% to >98% across 12 target cytosines, indicating that UNG is required for byproduct formation39. This insight was used to improve base editing outcomes. Fusing a second UGI domain onto the C-terminus of BE3 improved the editing purity in UNG-containing cell lines, likely due to increased inhibition of UNG. In addition, installation of a more flexible set of linkers improved efficiency of editing to generate a fourth-generation editor, BE4 (Figure 2c; Table 1)39. Overexpression of UGI in trans with a BE3 also improves product purity and reduces indel formation in mammalian cells78, but this may be accompanied by a global increase in C to T mutation rates79,80.
In some cases, the ability of a CBE with no fused UGI to mutate a target C to a mixture of T, A, and G provides a useful system for targeted random mutagenesis. Bassik, Chang, and their respective coworkers developed two such systems that exploit C-to-non-T editing abilities of base editors for targeted mutagenesis in mammalian cells. These approaches, targeted AID-mediated mutagenesis77 and CRISPR-X75, have been reviewed extensively here81.
Adenine base editing by ABE typically exhibits very high product purity; indeed, there are no reports of significant A-to-non-G edits to date32,82–85, perhaps because of the much weaker ability of cells to remove inosine from DNA than uracil. Consistent with this potential explanation, the use of ABE in cells deficient in alkyl adenine DNA glycosylase (AAG), an enzyme known to recognize and remove inosine in DNA86, did not improve editing efficiency32.
Generation of indels
DNA base editing can yield a low but detectable rate of indel formation. Liu and co-workers noted that as well as improved product purity profiles, UNG-knockout cells displayed reduced indel formation39. This observation is consistent with a model in which UNG-mediated creation of an abasic site following C-to-U deamination can lead to nicking of the targeted strand DNA-(apurinic or apyrimidinic site) lyase (AP lyase) (Figure 2c)87. If the opposite strand has been nicked by the Cas9 nickase component of the base editor, the resulting proximity of the two nicks results in a DSB, which is likely to be resolved by indel-prone end-joining processes (Figure 2c). Liu and co-workers showed that indel formation can be substantially reduced by fusing the bacteriophage Mu-derived Gam (Mu-GAM) protein to BE4 to generate BE4-Gam, which further reduces indels in treated HEK293T cells relative to BE439. BE4-Gam treatment also resulted in higher product purity and reduced indel frequency compared to BE3 in rabbit embryos88.
ABE typically leads to very low (in some cases undetectable) indel frequencies, typically well below 1%, for treated cells in culture32,82,85,89,90, mice82 and in plants91. The lower frequency of ABE-mediated indels is consistent with the requirement of a glycosylase or other enzyme involved in DNA repair to remove inosine and induce a nick in the edited strand to form an indel32. Since the removal of inosine is thought to be substantially less efficient than removal of uracil from DNA86, fewer nicks in the target strand, fewer resulting DSBs, and fewer indels would be expected to follow adenine base editing compared to cytosine base editing.
Off-target editing with DNA base editors
As with all genome editing technologies, both cytosine and adenine DNA base editors have the potential to operate on DNA at off-target genomic loci30–32,92. Off-target base editing can be classified into “proximal off-target editing”, editing that takes place near (for example, within 200 bp of) the target locus but outside the activity window, and “distal off-target editing”, editing that takes place away from the target locus. While the off-target effects of DNA base editors continue to be investigated, early evidence suggests that distal off-target base editing generally occurs only at a subset of loci that experience off-target editing from Cas9 nuclease93. In contrast to RNA editors (see above), current data30,32 suggest that DNA base editors typically do not induce measureable proximal off-target edits, although an in-depth study of proximal off-target base editing has not yet been reported.
Since the Cas9 component mediates the DNA-targeting ability of base editors, off-target base edits have been interrogated through deep sequencing of genomic loci known to be edited by Cas9 nuclease30–32,85,89,92. As expected, off-target loci that contain a C positioned in the activity window of the editor are sometimes edited at a low but detectable frequency by CBEs. Since not all the Cas nuclease off-targets contain an editable cytosine, off-target profiles of CBEs are generally more favorable than that of the corresponding nucleases programmed with the same guide RNAs30–32,85,89,92. To improve the DNA specificity of cytosine base editing, high-fidelity versions of BE3 have been generated by incorporating mutations known to improve the editing fidelity of Cas9 nuclease into the Cas9 portion of BE3. Liu and co-workers used the mutations discovered by Joung and coworkers94 to improve the DNA specificity of Cas9 nuclease, resulting in high-fidelity BE3 (HF-BE3)94. HF-BE3 shows a substantial reduction in off-target editing, even when paired with highly promiscuous guide RNAs92 (Table 1). Soo-Kim and co-workers have generated an alternative high-fidelity base editor, called Sniper-BE3, using the same strategy with a different set of mutations95.
Kim and co-workers developed an unbiased in vitro screen for identifying off-target edits by CBEs using purified genomic DNA and BE3ΔUGI (BE3b39) ribonucleoproteins (RNPs), finding that the off-target loci deaminated by rAPOBEC1–Cas9 nickase are indeed predominantly, but not entirely, a subset of the loci edited by Cas9 nuclease93. Although off-target adenine base editing has not been broadly interrogated, examination of off-target ABE activity at known off-targets of Cas9 nuclease when programmed with the same guide RNAs suggests that ABEs exhibit substantially lower off-target activity than Cas9 nucleases, and even less than was observed from BE330,32. Further studies to investigate off-target ABE activity in cells and in vivo are needed to fully characterize and explain the apparent higher DNA specificity of the ABEs compared to the CBEs.
In addition to the off-target editing that could be directed by the DNA-binding protein component of base editors, deamination of non-target ssDNA (such as within a transient bubble of ssDNA during transcription), or in RNA, may occur from DNA base editors. Misregulation or overexpression of endogenous deaminases has been linked to elevated mutation rates96–98, and expression of the UGI component of CBEs could also lead to an elevated rate of C-to-T transitions in the genome through impeding repair of spontaneously generated uracils79,80. However, studies of CBE off-target editing to date do not report widespread C-to-T mutations upon CBE expression or treatment, and transient delivery methods such as RNP delivery are likely to further reduce the mutagenicity of UGI in the context of CBEs30,31,76,85,89,92,93,
Whole-genome sequencing (WGS), when performed on the genomic DNA from sufficient numbers of independent cells, has the potential to detect all types of off-target base editing in cells or whole higher organisms. The WGS experiments reported to date on base-edited animals, however, have not been performed with sufficient power or controls to identify such events across an entire mammalian genome. Kim, Huang, and their respective co-workers performed WGS on mutant mice generated through treatment with ABE7.10 and a guide RNA targeted to the Tyr locus82 or targeting the Hoxd13 locus in a one-cell stage embryo99. Computational analysis indicated that none of the SNPs identified in the treated mice were likely to have arisen through off-target base editing. Together, these studies further suggest high DNA specificity of ABE7.10.
We should note that these studies do not exclude the possibility of deamination from base editors that is not directed by the DNA- or RNA-binding component of the editors, but instead by random encounters between the deaminase domain of base editors and transient single-stranded DNA. More data are required to characterize this possibility, including WGS of treated and untreated littermate controls and of mice treated with base editor mutants with catalytically inactivated deaminases. The continued development of context-dependent base editors100 or future base editor variants that lack the ability to bind ssDNA without assistance from the guide RNA represent potential solutions to further minimize the possibility of random non-directed off-target base editing.
Editing window and bystander edits
In the case of BE3, which incorporates Streptococcus pyrogenes (SpCas9) as the DNA-targeting moiety, the “activity window” in which efficient editing is observed, is approximately five nucleotides wide (positions 4–8, counting the PAM as positions 21–23)30,39,92. Bases located outside the activity window but within the ssDNA R-loop region may still be edited at a lower efficiency, particularly if they are located in a favorable editing motif (see below). For many genome-editing applications, only a single nucleotide is targeted for conversion, so an ideal base editor would have a narrow activity window that focuses activity only on the target base. However, such a narrow window necessitates that the base editor be targetable to a broad range of PAM sequences. As the repertoire of natural Cas nucleases with different PAM requirements and function in human cells (including SaCas9101 LbCpf1 and AsCpf1102, CjCas9103, StCas9104,and NmCas9105, engineered CRISPR proteins106,107, and laboratory-evolved CRISPR proteins85) continues to expand, the desirability of more precise base editor variants with narrower activity windows will increase.
For some target sites, multiple editable Cs or As exist within or nearby the activity window, which can result in conversion of bases in addition to the target base. We use the term “bystander editing” to describe editing in the protospacer at a nucleotide other than the target nucleotide (Figure 4b). Bystander editing may be inconsequential, especially when base editing to disrupt promoters, splice sites, or other regulatory sequences, or when knocking out gene function by introducing premature stop codons. When editing protein-coding genes, within a canonical 5-base editing window most, but not all, base editing cases will only result in the desired single amino acid change, in part because the genetic code dictates that almost all third-position transitions in a codon are silent (see Box 1 for a detailed analysis).
Figure 4. Overcoming targeting challenges associated with base editing.

An example of a cytosine base editing site has been shown; these principles also apply to other classes of base editors (a) An ideal base editing target locus. The target base is located within the base editor activity window relative to the PAM site, there is only one target base in the activity window, and the target base is found in a motif (AC in this example) that is efficiently deaminated by most cytosine base editors30,39. (b) Example of a target site with a bystander base. If the bystander edit (deamination of the cytosine shown in yellow) is undesired, a narrowed-window108 or context-specific base editor100 may be used to preferentially edit the target base over the bystander base (c) The target base is located outside of the activity window. Base editing on this target may be possible with base editors that recognize different PAMs (see Table 1). (d) The target base is located within a sequence context that may not an efficient substrate for a particular deaminase30,39. Editing of the target may be improved by using an editor with a different deaminase31,39, or an editor more tolerant of methylated DNA115.
Box 1: Calculating the probability of a non-silent bystander edit when base editing a protein-coding gene.
Bystander editing is editing of one or more bases in addition to the target base in the activity window of a base editor (Figure 4b)100. The width of the activity window is somewhat target-and sequence-dependent, and researchers have mutated deaminases to generate editors with narrower windows108 or which only edit Cs within a particular sequence context100 thus reducing the likelihood of bystander editing. Nevertheless, bystander edits are sometimes unavoidable and are of particular concern if they lead to a coding (i.e. non-silent) mutation in a target gene.
Commonly used cytosine base editors including BE3 and BE4 have an activity window ~5 nt wide, whereas ABE7.10 has an editing window ~4 nt wide. Current base editors generate transition mutations (C to T, T to C, A to G, and G to A). In a genome with a random distribution of codons, (30/32) = 94% of transitions at codon position 3 are silent, (1/32) = 3% of transitions at codon position 2 are silent, and (2/32 = 6%) of the transitions at codon position 1, are silent. Since a transition at the target base is necessarily non-silent when mutating a protein-coding gene, the overwhelming majority (1–0.06/1.97 = 97%) of target bases are located at codon position 1 or codon position 2. With a target base at codon 1 or 2, there are, on average, 2.33 remaining non-silent mutable bases (located at codon position 1 or 2) in the CBE window or 1.75 for the ABE7.10 window (Figure 5a).
For a CBE, the likelihood that all of the non-silent bases in the activity window are non-C is: 0.75^2.33 = 0.51 or 51% (for ABE7.10 the equivalent calculation for A is 0.75^1.75 = 0.604, or 60.4%). In the remaining cases, there are 1 (39%), 2 (9%) or 3 (0.5%) editable target bases within the window (Figure 5b). Assuming that the probability of a target base lying at codon position 1 equals the probability that it is found at codon position 2, we can calculate the probability that there are zero non-silent bystander edits in the base editor window. For windows with 1 editable bystander base the probability that this bystander base edit is silent is 3/64, for cases with two editable bystander bases in the window the probability that they are both silent is (3/64)^2 and for three it is (3/64)^3.
Thus, for the 49% of cases in which there are one or more bystander Cs at codon positions 1 or 2 in the editing window for BE3, we can calculate the chance that these are all silent as ((3/64)*(0.39) + (3/64)^2*(0.09) + (3/64)^3*(0.005))*100% = 1.85%. indicating that the overall probability of a non-silent bystander mutation with BE3 is 49%−1.85% = 47% For ABE, the equivalent probability is reduced to 38%, and for base editors with narrowed activity windows ~2-nt wide the probability of a non-silent bystander mutation is reduced further to 12%. Note that several transitions at codon positions 1 and 2 lead to non-silent but conservative substitutions (such as Ile←→Val) that in many cases may not impact protein function.
To minimize bystander editing, researchers have developed base editor variants with altered activity windows. Liu and coworkers engineered CBEs with mutations in the rAPOBEC1 domain that attenuate deamination activity, resulting in editors with reduced processivity and narrower activity windows (YE1-BE3, YE2-BE3, and YEE-BE3; see Table 1)108. These narrow-window CBEs enable selective editing of a target C over a neighboring C that is located within the standard editing window of BE3. For ABE7.10, which is generally the most efficient and widely used A-base editor, the activity window is approximately located from positions 4–7 in the protospacer (counting the PAM as positions 21–23). For certain targets, ABE7.9 or ABE6.3 may be more useful due to a slightly broader activity window enabling editing from positions 4–932. Recent work by Kim and co-workers described how pairing a 5’ extended guide RNA with ABE7.10 can increase editing to positions 2–3, although editing at these positions remains modest82 (Table 1). The use of base editor variants that exhibit strong sequence context preference serves as a promising additional strategy to minimizing bystander base editing. These variants are discussed below (see “Base editing sequence context”).
Conversely, Huang and coworkers expanded the width of the editing window by eingeering “BE-PLUS”, a CBE variant in which a SunTag109 was fused to the N-terminus of Cas9(D10A) nickase. Separately expressing a scFv–APOBEC–UGI fusion allows up to ~10 UGI domains to associate with each SunTag110. This construct enabled editing from protospacer positions 4–16, with reduced indel and C-to-non-T editing compared to BE3, likely due to the recruitment of many UGI domains110 (Table 1). While base editors with enlarged editing windows are more prone to bystander editing, they also facilitate access of the target base pair, and may be especially useful when targeting non-protein-coding sites.
Targeting limitations
Successful DNA target binding by CRISPR-family nucleases requires a PAM, a conserved sequence up or downstream of the variable guide RNA protospacer sequence (Figure 4a)2,16. For base editing, the PAM must be appropriately positioned relative to the target base to ensure efficient editing. Even though SpCas9 offers the least restrictive PAM among those CRISPR enzymes reported to function with high activity in mammalian cells, due to this requirement only ~26% of known pathogenic SNPs that are of the four types of base conversions (C to T, G to A, A to G, or T to C) that can be performed can be targeted by SpCas9-derived base editors (Figure 4c)85. This limitation creates the need to develop base editors with additional PAM compatibilities.
To increase the number of targetable bases, researchers have developed base editors incorporating different CRISPR-associated nuclease enzymes (Table 1). Liu and co-workers described a set of alternative CBEs with Staphylococcus aureus Cas9 (SaCas9) and engineered variants of SpCas9 and SaCas9 capable of efficient editing with non-NGG PAMs108 including SaBE3, Sa(KKH)-BE3, VQR-BE3, VRER-BE3, and EQR-BE3. Chen and co-workers described a CBE derived from Cas12a (also known as Cpf1; PAM = TTTV, where V is A, C or G), which allows access to T-rich regions of genomic DNA34. Since there is no known mutation capable of transforming Cas12 into a nickase that cleaves only the target strand of DNA, Chen and co-workers characterized a dead LbCas12a-base editor, which nevertheless displays editing efficiencies averaging 22% across 10 target sites in HEK293T cells34 (Table 1).
Recently, Liu and co-workers used phage-assisted continuous evolution (PACE) to evolve SpCas9 to recognize a broader range of PAMs. A resulting evolved variant, xCas9(3.7), harbors mutations allowing it to access some target sequences with some NG, GAA or GAT PAMs. Replacing Cas9 in the BE3 construct with xCas9(3.7) made xBE3, a CBE capable of editing some loci with NGN, GAA and GAT PAMs (Table 1)85. While xCas9 variants are capable of mediating DNA cleavage or base editing at several non-NGG PAMs, xCas9-mediated editing efficiency varies among different target sites, and like many engineered or evolved Cas9 variants, likely requires a high degree of perfection between the guide RNA and the target sequence, including a G at the 5’ end of the guide RNA and at the corresponding first position of the protospacer100,111. Surprisingly, in addition to its expanded PAM acceptance, xCas9 also displays higher editing fidelity than SpCas985.
Nureki and co-workers used a rational design approach to develop another SpCas9 variant with broadened PAM compatability, termed “NG-Cas9”112. In mammalian cells, the relative activities of xCas9 and NG-Cas9 appears to be guide RNA-dependent; Nureki and co-workers reported that NG-Cas9 is more active than xCas9 at 15/15 NGC, 16/18 NGT and 15/19 NGA PAM sites, but NG-Cas9 exhibits a loss of efficiency at the canonical NGG PAM sites that is not observed with xCas9112. NG-Cas9 also does not exhibit the enhanced fidelity observed with xCas9, but tolerates inclusion of fidelity-enhancing mutations112. As a CBE, NG-Cas9 accepted a subset of NG PAM loci as substrates for efficient base editing (Table 1)112.
Alternative-PAM ABEs have been developed by adapting SaCas983, Sa(KKH)Cas9113,114, Sp(VQR)Cas9,113,114 and Sp(VRER)Cas9113 into the ABE7.10 architecture, resulting in efficient generation of mutant rice plants (Table 1). Additional ABE variants with altered PAM requirements would substantially augment the scope of targetable bases for adenine base editing.
Base editing sequence context
In addition to PAM- and activity window-imposed sequence restrictions, the particular deaminase enzyme variant used in a base editor may impose sequence context preferences that affect editing efficiency at a particular locus. For example, rAPOBEC1 exhibits poor processing of cytosines within some (but not all) GC motifs30,39 (Figure 4d). By contrast, other cytidine deaminases such as activation induced deaminase (AID) or cytidine deaminase 1 (CDA1) do not display this particular sequence preference but exhibit lower editing efficiencies than rAPOBEC1 in most tested sequence contexts when tested in a BE3 architecture39. Yang and co-workers identified that rAPOBEC1-mediated base editing rates are reduced by DNA methylation at CpG dinucleotides115, and that human APOBEC 3A (hA3A) can edit cytosines found in CpG dinucleotides and in GC motifs more efficiently than rAPOBEC1115.
Joung and co-workers harnessed the sequence preferences of different cytosine deaminase enzymes to engineer a mutant hA3A-based CBE that preferentially deaminates cytosines preceded by a T100 (Table 1) as a strategy to reduce bystander editing. Structure-guided design and screening of hA3A deaminase mutants resulted in an enhanced variant (eA3A) with a single mutation (N57G) that deaminates the target motif (TC) but significantly reduces activity at Cs in other sequence contexts, resulting in a context-dependent base editor that maintains a 5-nucleotide activity window100. Importantly, Joung and co-workers performed a detailed analysis of the individual alleles that were generated upon successful base editing by eA3A-BE3, BE3, and other engineered variants (YEE-BE3, YE1-BE3 and YE2-BE3) to demonstrate that eA3A can make the desired allele at a high efficiency and purity100. A high-throughput sequencing data analysis package facilitated this detailed analysis of base editing outcomes116.
Context-specific base editors such as those developed by Joung and coworkers represent an important advance that offers more precise base editing, with the trade-off of lower target site applicability since the target nucleotide must naturally exist in the preferred sequence context. Thus far, the data from mammalian-cell editing with ABE7.10 indicates that it is relatively free from motif-related sequence preferences in human cells32, but Kim and co-workers have demonstrated that there is a preference for editing at TA motifs relative to GA, CA, or AA in Arabidopsis thaliana91. The development of additional context-specific ABE and CBE variants will be enabling for applications in which editing only a single base is paramount.
Improving intracellular expression and nuclear localization of base editors
For plasmid delivery of Cas9 nuclease, optimization of codon use for mammalian cell expression improves soluble protein levels and enhances editing efficiencies111. Optimization of the nuclear localization sequence (NLS) also improves Cas9-medited editing in vivo117. Liu and co-workers identified that poor expression is also a bottleneck to the efficiency of base editors, and optimized codon usage and nuclear location sequences to generate improved cytosine and adenine base editors, resulting in BE4max and ABEmax from BE4 and ABE7.10 respectively90. The use of ancestral sequence reconstruction starting from the protein sequences of the hundreds of known APOBEC homologs, a process that has been demonstrated to improve protein expression118, resulted in AncBE4max. All three optimized base editors offered substantially improved editing efficiency, especially under suboptimal conditions such as when delivery into cells is limiting90.
In an elegant independent study, Dow and co-workers optimized CBE codon usage by removing premature poly(A) sites and rare mammalian codons, and improved CBE nuclear localization by adding a second NLS to the N-terminus of BE3, to generate an optimized FNLS-BE3 that results in much higher editing efficiencies than BE3. When packaged into lentivirus, FNLS-BE3 mediated efficient editing in murine intestinal organoids119. Hydrodynamic injection of the plasmid encoding FNLS, together with a guide RNA that programs the base editor to make a S45F mutation in Ctnnb1 lead to significantly more efficient base editing and corresponding physiological changes (tumor nodule formation) in the livers of mice than BE3 treatment119. Dow and co-workers also generated lentiviral constructs with the corresponding optimized editor versions of BE4-Gam, which enable improved editing rates with reduced indel formation119. Ensuring optimal expression of the base editor construct in the target cell type is critical for applications that require high editing efficiency, and the above developments thus represent important advances.
Delivery of base editors
DNA delivery strategies: plasmid transfection and viral delivery
Since most proteins cannot spontaneously traverse cell membranes, a delivery method is required to facilitate cell entry. A common strategy is to deliver DNA encoding the target protein through chemical transfection120, electroporation121, or viral infection122, and then rely on target cell transcription and translation to produce the desired protein.
For cell lines in culture (including HEK293T, HeLa, U2OS and murine NIH/3T3 cells), lipid-mediated transfection of plasmids encoding base editors has resulted in high editing efficiencies without selection for transfected cells30–32,34,35,39,82,85,92. For cell types resistant to plasmid lipofection, electroporation followed by fluorescence-activated cell sorting (FACS) to isolate transfected cells has yielded favorable editing efficiencies for lymphoblastoid cell lines (LCLs)32 and mouse astrocytes30. Although plasmid-based delivery is a convenient delivery strategy, DNA delivery raises the risk of exogenous DNA recombination into the genome and protracted overexpression of genome-editing agents increases off-target editing rates76,92,123–125.
The use of viruses to deliver DNA encoding base editors is a promising delivery modality for some in vivo research or therapeutic applications. Use of non-integrating vectors such as adeno-associated virus (AAV), herpes simplex virus (HSV), or adenoviral vectors reduces the potential for random integration of exogenous DNA into the host genome. Infection with adenovirus and HSV-1, however, may provoke inflammatory responses122. By contrast, AAV is thought to be both non-inflammatory and non-pathogenic126. When coupled with its broad tropism, well-studied serotypes, and ability to infect dividing cells, AAV is a particularly promising strategy for viral delivery of genome editing agents.
AAV-mediated delivery of many CRISPR genome-editing agents, including base editors, is challenging due to the 4.9 kbp packaging limit of AAV127. A CBE or ABE plus a guide RNA totals approximately 6 kbp. Kim and co-workers overcame this through use of two trans-RNA splicing AAVs (tsAAVs)128 encoding each half of ABE7.1082. Dual tsAAV-mediated delivery of ABE7.10 into skeletal muscle in a mouse model of DMD corrected a premature stop codon82. After dual infection, homologous recombination between the identical inverted terminal repeat (ITR) sequences generates the full-length ABE7.10 transcript82, enabling ABE7.10 protein production.
RNP delivery
DNA-free base editing enables precise and specific changes to genomic DNA without exposing a cell to exogenous DNA89,92. Sustained overexpression of genome-editing agents erodes DNA specificity. After successful editing, the target site is no longer a binding site for the editing agent, and residual editor can only act to mediate off-target editing. Thus, controlling the exposure to editing agents, including base editors, can greatly improve their DNA specificity25,92,123,125.
Kim and co-workers established that purified Cas9 complexed with a guide RNA, forming an RNP complex, can be efficiently delivered into mammalian cells in culture by electroporation, and that RNP delivery of Cas9 leads to improved DNA specificity relative to plasmid-based delivery125. Liu and co-workers demonstrated that cationic lipid-mediated delivery of Cas9 RNP complexes can facilitate in vivo delivery of Cas9 near the site of administration, as well as efficient delivery into cells in culture, and resulted in greatly improved DNA specificity relative to plasmid-based lipofection123,124,129.
BE3 protein has also been purified76,92, and Liu and coworkers have packaged BE3:guide RNA RNP into cationic liposomes for lipid-mediated delivery to cultured cells, zebrafish embryos, and the inner ear of postnatal mice89,92. Analogous to the delivery of Cas9, cationic lipid-mediated delivery of BE3 dramatically improves DNA specificity in human cells compared to plasmid delivery89,92. BE3 RNPs have also been delivered through electroporation into mice76, and through direct injection into Xenopus laevis embryos130. RNP delivery is also effective for alternative base editors; the engineered high-precision editor eA3A(N57Q) has been delivered as an RNP into human erythroid precursor cells via nucleofection of the RNP complex to correct a mutant HBB allele, resulting in a 4-fold increase in HBB expression100. The advantages of RNP delivery include improving editing specificity and removing the reliance on intracellular transcription and translation to generate the editing agent.
mRNA delivery of base editors
Delivery of mRNA is a commonly used delivery strategy to deliver genome-editing agents into embryos. Kim, Huang, Lin, Liu, Zhang, Li and their respective coworkers have demonstrated that in vitro transcription followed by purification of an mRNA encoding BE3, when combined with a guide RNA, can be co-delivered into single cell mouse76,131, human132,133, rabbit88, rat134 or zebrafish zygote135,136 by electroporation or direct injection to generate point mutations with high efficiency and DNA specificity. These studies establish mRNA delivery of base editors into embryos as a robust and efficient strategy for the generation of animals with tailor-made point mutations.
Applications of base editing
Base editing to install or correct pathogenic point mutations
Since point mutations are the largest class of known pathogenic genetic variants (Figure 1a) and CBEs and ABEs collectively have the potential to install or reverse up to ~60% of pathogenic point mutations (Figure 1b)28,29, a major application of base editing is the study or treatment of disease-associated point mutations.
Examples of base-editor-induced gene correction in cultured cells are already numerous. Liu and coworkers showed that plasmid nucleofection of BE3 can convert the Alzheimer’s disease associated allele APOE4 to APOE3r in mouse astrocytes, and to correct the cancer-associated p53 mutation Tyr163Cys in breast cancer cells30. Subsequently, codon-optimized CBEs were delivered as plasmids in patient-derived fibroblasts to correct the Leu119Pro mutation in MPDU190 that causes the congenital disorder of glycosylation type 1f137. Liu and coworkers also showed that plasmid delivery of ABE7.10 can correct the hereditary-haemochromatosis-causing mutation C282Y in an immortalized patient-derived LCLs, and to install a mutation known to increase fetal hemoglobin (HBG) expression in adults32. Joung, Huang, and their respective coworkers reported correction of a mutant HBB allele that causes beta-thalassemia in an engineered HEK293T cell line100 and in patient-derived primary fibroblasts133.
Direct injection of base editor-encoding mRNA along with a guide RNA has also proven effective for editing pathogenic alleles in human embryos. Direct injection of mRNA encoding BE3132,133,138, YE1-BE3138 or YEE-BE3133 together with a guide RNA can generate homozygous mutants at a rate of up to 77% of embryos that survive to the blastomere stage132,133.
Viral delivery of base editors is an effective method for correcting pathogenic mutations in mouse disease models in vivo. Kim and co-workers used AAV to deliver ABE7.10 with a guide RNA programmed to correct a premature stop codon in the DMD gene in a mouse model of muscular dystrophy. Although the correction rate was only 3.3% of sequenced cells, dystrophin expression was restored in 17% of muscle fibers82, highlighting that low levels of editing can often lead to therapeutically-relevant phenotypic change. Separately, Musunuru and co-workers generated an adenoviral vector encoding BE3 and a guide RNA programmed to make the W159Stop mutation in murine Pcsk9. They measured a median rate of 25% editing in liver cells, and show a modest reduction in plasma PCSK9 protein levels and plasma cholesterol 4 weeks post-injection139.
In vivo base editing has also been used to ascertain whether a genotype is causal for a particular phenotype. Dow and co-workers performed hydrodynamic transfection of an optimized BE3 plasmid construct, termed FNLS-BE3, with a guide RNA programmed to make the S45F cancer-associated mutation in Ctnnb1. They demonstrated efficient (nearly 100%) base editing in liver cells, and showed that treated mice treated with FNLS-BE3 plus the on-target guide RNA grew a significant number of visible tumor nodules compared to controls119. Lin and co-workers delivered of BE3 as an mRNA into one-cell-stage zebrafish embryos to generate a P302S mutation in tyr that mimics a common mutation observed in human ocular albinism. This approach enabled investigation into the effects of such a mutation on ocular pigmentation135. These studies hint at the promise of BEs as potential therapeutics, and demonstrate their efficacy for researchers interested in ascertaining the phenotypic effects of precise genetic changes in cell culture and in vivo.
Base editing in post-mitotic cells
Liu and co-workers demonstrated that base editing can occur in the non-mitotic sensory supporting and hair cells140 in vivo in the mouse inner ear89. BE3 combined with a guide RNA targeting β-catenin was used to control flux through the Wnt signaling pathway89. Blocking phosphorylation at S33 through an S33F mutation extends the cellular half-life of β-catenin, increasing Wnt signaling. For this target, maintaining low indel rates is critical as indels would likely disrupt the gene and reduce β-catenin levels, opposing the desired change89. Lipid-mediated delivery of BE3 complexed with the S33F guide RNA as an RNP into the inner ear of mice led to editing in post-mitotic somatic cells at efficiencies up to 8%. Dissection and staining of treated hair cells identified that BE3 treatment, unlike treatment with Cas9 nuclease and an HDR template, induced cellular reprogramming of other cells into cells resembling cochlear hair cells. These results establish the ability base editing to occur in post-mitotic cells that are resistant to DSB-stimulated HDR23,141.
Cytosine base editing to introduce premature stop codons
CBEs (but not ABEs) can install premature stop codons to disrupt genes in a homogenous manner by precisely converting one of four codons (CAA, CAG, or CGA in the non-coding strand; or TGG in the coding strand) into stop codons. Kim and co-workers demonstrated this possibility by using BE3 to introducing a premature stop codon in Dmd in mouse embryos76. The CRISPR-Stop142 and iSTOP143 methods use this principle to enable high throughput BE3-mediated gene inactivation without generation of DSBs and accompanying indels. Ciccia and co-workers generated a database describing a set of guide RNAs, that, when complexed with BE3, are capable of generating premature STOP codons in >98.6% of open reading frames (ORFs) in the human genome (reference genome assembly GRCh38). They published a freely accessible online database enabling researchers to find appropriate guide RNAs for iSTOP to use in eight species143. Adli and co-workers identified that this strategy results in a significant reduction in apoptosis when compared to Cas9 nuclease treatment142, possibly due to lower DSB-induced toxicity144–146. While typically efficient and widely utilized, NHEJ-mediated knock out of genes following DSBs leads to a mixed population of cells, DNA translocations and rearrangements27,147, and the induction of cell death144–146, all of which in principle are avoided through the use of base editors to install precise stop codons. Flow cytometry of CRISPR-Stop treated cells indicated that stop-codon introduction is similar in efficiency to Cas9-mediated gene knockout142.
Perez-Pinera and co-workers confirmed that base-editor induced C-to-T edits at the conserved splicing acceptor site can induce exon skipping148. Their method (termed CRISPR-SKIP) was similar in efficiency to Cas9 DSB-mediated exon skipping, but unlike nuclease treatment did not generate DSBs148.
Base editing in embryos to generate animal models
A common goal of genome editing at the single-cell embryo stage is to generate model organisms. To minimize mosaicism and maximize the chance that editing occurs in the germ line, it is critical that editing occurs quickly and efficiently. Since nuclease-mediated editing strategies often fail to generate homozygous, non-mosaic progeny in the F0 generation149,150 the high efficiency of base editing offers an attractive alternative. CBEs are particularly useful for generating loss-of-function animal models by inserting a premature stop codon into a gene of interest without generating DSBs or indels76,135.
Kim and co-workers demonstrated that microinjection of mRNA encoding BE3 together with a guide RNA, or electroporation of the BE3:guide RNA RNP complex mediates efficient generation of premature stop codons in one-cell stage mouse embryos at two target sites: Q871Stop in Dmd or Q68Stop in Tyr76. Impressively, mRNA treatment yielded the target mutation in 11/15 or 10/10 blastocysts at the Dmd or Tyr loci, respectively. RNP delivery of BE3 was also effective; 2/7 of the embryos treated with a BE3 RNP pre-complexed with a guide RNA targeted to the Tyr locus were transplanted into surrogate mothers to yield homozygous, non-mosaic progeny with the expected albino phenotype76. Independently, Songyang and coworkers used either BE3 or a high-fidelity version of BE2 (BE2-HF2) mRNA to perform base editing in mouse zygotes, resulting in up to 50% of sequenced embryos harboring a C-to-T point mutation at the target locus131. Li and coworkers made rabbit models of human disease using mRNA injection of BE3, BE4-Gam or ABE7.10 into blastocysts88. They performed ABE-mediated editing generated the point mutation T297A in Exon9 of Dmd which is associated with cardio-specific XCLM in humans88 and BE3 was used to make the mutation c.1821C>T in Lmna, generating a rabbit model of Hutchinson-Gilford progeria syndrome88.
Huang and coworkers performed multiplexed base editing through co-injection of ABE7.10 and SaBE3 mRNA along with guide RNA sequences targeting Tyr (an S. aureus guide RNA was used to generate the Q58Stop mutation) and Hoxd13 (a Streptococcus pyogenes guide generated the Q312R mutation) in one-cell mouse embryos. Impressively, A-to-G and C-to-T edits were simultaneously observed in blastocysts99. The same strategy has been used to deliver ABE mRNA and guide RNAs into rat embryos114,134. Zhang and coworkers showed that co-injection of two different guide RNAs efficiently generated two transmissible A to G point mutations in the F0 generation simultaneously134 whilst Yin and co-workers used ABE to generate a rat model of Pompe disease114. These data demonstrate that base-editing is an enabling tool for generating mutant mice, rats, and rabbits for animal studies; previous nuclease-based editing methods usually failed to generate non-mosaic mice with 100% mutation frequency in the F0 generation151.
Base editors as cellular event recorders
In addition to its applications in biomedical research to install and correct point mutations, base editing has also been used as a synthetic biology tool to record cellular signaling and exposure to stimuli. Unlike the stochastic indels that result from Cas9 DNA cleavage, base editors generate predictable single point mutations. By coupling the stimulus of interest to the activity of the BE, the resulting stimulus-dependent single point mutations can be used to record exposure to signals into the genome. Liu and co-workers developed a ligand-responsive editing system by appending a blocking sequence to a guide RNA through a ligand-dependent hammerhead ribozyme152. This system facilitated ligand-dependent base editing in mammalian cells152.
Subsequently, Liu and Tang demonstrated that controlling expression of a base editor or its accompanying guide RNA using stimulus-dependent promoters enables recording of a wide variety of stimuli—including exposure to light, nutrients, antibiotics, or virus—durably as point mutations into a cell’s genome. This recording system was termed “CRISPR-mediated analog multi-event recording apparatus” 2 (CAMERA 2)54. Control of BE expression through small-molecule-responsive promoters enabled dose- and time-dependent base editing of four small molecules (aTc, IPTG, arabinose, and rhamnose) simultaneously in bacterial cells. Through careful design of two ratcheted protospacers, in which base editing from one guide RNA edits the binding site for the second guide RNA, the order of exposure could also be recorded54. The same principles were used in mammalian cells: signals including exposure to doxycycline, tetracycline, or IPTG were recorded as base edits in the CCR5 safe-harbor locus. CAMERA 2 could also record changes in Wnt signaling in mammalian cells.
Independently, Lu and co-workers used cytosine base editors to develop a related platform for cellular reading and writing, named DOMINO (DNA-based Ordered Memory and Iteration Network Operator)153. As in CAMERA 2, expression of the base editor and guide RNAs are controlled with different small-molecule-responsive promoters in E. coli. DOMINO can directly couple stimulus-dependent base editing to a phenotypic readout. For example, successful DNA editing by two input guide RNAs could enable a third guide RNA to bind to a target DNA operator site upstream of a genomically-integrated GFP gene. Binding of the guide RNA:base editor complex to this operator resulted in GFP fluorescence reduction153.
Lu and coworkers also used DOMINO as a self-reinforcing “molecular clock” in human HEK293 cells that records stimulus exposure time. They fused a CBE with the VP64 transcriptional activator to perform sequential editing of a repetitive operator region located just upstream of GFP. The circuit was designed such that over the course of 15 days the repetitive operator region was sequentially edited to generate more guide RNA binding sites, increasing localization of the editor to the operator region and thus increasing GFP expression. Both the number of GFP-positive cells and the C-to-T editing levels in the operator region reflected the number of days of exposure between the cell population and active editor construct153. Both DOMINO and CAMERA rely on the exquisite precision of base editing, as indel-generating methods would not be expected to predictably write new protospacer sequences. We anticipate that future cellular recording applications will use both CBEs and ABEs to develop more complex recording systems, since ABEs can erase signals written by CBEs, and vice-versa.
Base editing in plants
Base editing in plants could enable researchers and agriculturalists to rapidly generate novel plant mutants with an efficiency beyond that of conventional breeding154. Generation of precise, gain-of-function point mutations can improve many agronomic traits; for example, a point mutation in the plant ALS gene confers resistance to herbicides such as sulfonylureas and imidazoliones155. Generating precise point mutations in plant cells remains challenging using DSB-induced HDR156,157.
Multiple plant species of agronomic interest have been edited with CBEs and ABEs. Gao and co-workers demonstrated that BE3 generates efficient point mutations in maize, rice and wheat158. In a separate study, Kondo and co-workers showed that the Target-AID editor is capable of efficient editing in rice and tomato159. More recently, two independent reports from the Zhou and Zhu labs demonstrated that ABE editing is highly efficient in rice83,84,113. Gao and co-workers optimized the architecture of ABE7.10 for adenine base editing in rice and wheat, and used the resulting editor in protoplasts and in regenerated plants160. Kim and co-workers recently described two phenotypic changes generated through transient Agrobacterium tumefaciens-transfection of ABE7.10 into A. thaliana and Brassica napus91. Using a plant-optimized expression system, they performed editing in A. thaliana to generate a single codon change (Y85H) in the FT protein, generating a late-flowering phenotype, or to disrupt a splice acceptor site in the PDS3 gene, generating a dwarf phenotype. After transformation, >85% of T1 plants showed >50% editing, and T2 seedlings isolated from T1 plants also displayed the same phenotypes, indicating that the editing was germline-transmissible91.
These demonstrations establish that base editing is a promising approach for rapidly engineering of polyploid plant genomes. We anticipate that RNP delivery of BEs into crop species is particularly important from a regulatory and consumer perspective, because transgene integration from plasmid delivery results in plants with genetically modified organism (GMO) status. RNP-delivery of base editors would enable DNA-free precision editing that avoids the creation of GMO crops155.
Conclusions and future perspectives
The ability to efficiently and cleanly install changes to genetic information in living systems at the highest-resolution level—that of the individual base pair—resembled science fiction even only recently. The major developments summarized in this review have rapidly established base editing of individual nucleotides as a robust technology with the potential to broadly impact the life sciences and medicine.
The two classes of DNA base editors described thus far have repeatedly proven effective for making precise point mutations in the genome of a wide variety of living cells and organisms. That said, cytosine and adenine base editors make only two of the six possible changes of one base pair to another. Much additional work is needed to develop base editors that can install transversion mutations, and possibly other DNA or RNA changes, at programmable target loci. Success will likely benefit a deep understanding and creative manipulation of cellular mechanisms controlling base modification and DNA repair in mammalian cells.
Although early examples of in vivo base editing are highly encouraging, challenges associated with delivery of large proteins into specific tissues remain an important focus of ongoing efforts, including the use of base editing to treat human genetic diseases. Thus the development of novel base editor delivery systems, including those that target specific tissues, is likely to be another major focus in the coming years. Detailed analyses of the off-target editing activities of base editors in vivo under a variety of conditions relevant to ongoing research and therapeutic applications are also needed, together with assessments of the potential biological consequences of making off-target point mutations in vivo. For example, since base editors in general do not create DSBs that can lead to indels, translocations, or large DNA rearrangements, can the clinically relevant consequences of off-target base editing be adequately assessed by monitoring the DNA sequences of a defined set of oncogenesis-associated genes and their regulatory regions? Experimentally testing such possibilities in animals would represent important steps towards advancing base editing into the clinic.
The continued development of additional editing technologies that maximize base editing efficiency and targeting scope, while minimizing off-target base editing, will continue to propel the field towards increasingly ambitious and sophisticated applications. For the vast majority of base editing applications described here, the target sequence is known in advance. Thus, the development of many distinct classes of future base editors that each convert a target DNA base pair or RNA base exclusively in a particular sequence context, or in a protospacer containing a particular PAM, is likely to play an important role in maximizing the precision and specificity of base editing.
“Figure 5” (Figure to accompany Box 1).

Illustration of the probability calculations for non-silent bystander editing in a protein-coding gene, as described in Box 1. (a) Use of base editing to generate a non-silent mutation in the protein-coding region of a gene. There are three possible orientations of the base editor activity window relative to the reading frame of the protein. Base editors currently generate transition mutations, and the vast majority (97%) of transitions at codon position 3 are silent. Thus, the target base in a coding gene is predominantly located at coding position 1 or 2 (N1 or N2), because the consequence of generating a transition at N3 is almost always to generate a silent mutation. In a 5-nt activity window, typical of a CBE, an average of 2.33 nucleotides at positions N1 or N2 are present in the window, in addition to the target nucleotide. For a 4-nt window typical of an ABE there are on average 1.75 nucleotides at codon positions 1 or 2 in addition to the target nucleotide (see Box 1). These additional editable nucleotides at N1 or N2, depending on their identity and sequence context, may contribute to bystander editing (see Box 1). (b) Flow chart to calculate the probability of finding a bystander mutation within a 5-nt window, as described in Box 1. The probability of the indicated event (p) assumes a genome with randomly distributed bases. For a description of how each probability was calculated, see Box 1. The same calculation is described for a 4-nt window typical of ABEs in Box 1.
Acknowledgements
D.R.L. gratefully acknowledges support from DARPA HR0011-17-2-0049; the Ono Pharma Foundation; NIH RM1 HG009490, R01 EB022376, and R35 GM118062; and HHMI. H.A.R. is supported by the Kilpatrick Educational fund from the Chemistry and Chemical Biology Department, Harvard University. We thank Keith Joung, Feng Zhang, Aditya Raguram, Wei-Hsi Yeh, Tony Huang, Kevin Zhao, and Weixin Tang for their helpful comments.
Glossary
- Base editor activity window
The region of DNA or RNA, typically defined by the number of nucleotides from the PAM, in which a particular base editor acts to induce efficient point mutations. The activity window for most base editors is ~4–5 nucleotides wide.
- Bystander editing
Editing of a non-target base that resides in the activity window of a particular base editor and guide RNA. Bystander editing occurs in addition to editing of the target base.
- Cas nickase
A catalytically disabled mutant of a Cas enzyme that is able to create a single-stranded DNA break but not a double stranded DNA break.
- Cas12a (also known as Cpf1)
A Class 2, Type V RNA-guided endonuclease from the CRISPR system. Variants from several species have been characterized. Catalyzes site-specific cleavage of double stranded DNA at sites with an TTTV (where V is A, C or G) PAM.
- Cas13b
A Class 2, Type VI RNA-guided RNase from the CRISPR system. Variants from several species have been characterized. Catalyzes site-specific cleavage of single-stranded RNA.
- Distal off-target editing
Unwanted editing of bases residing in locations of the genome or transcriptome unrelated to (for example, >100-nt away from) the target site of the base editor.
- Genome editing product purity
The term used to describe the spectrum of mutations induced by a particular genome editing technology. In the context of base editing, low product purity occurs when a target base is mutated to bases other than the desired point mutation or when indels are generated in addition to the desired edit; for example C-to-G or C-to-A edits, rather than the desired C-to-T edit, from a cytosine base editor.
- Guide RNA
Short RNA sequence comprising a scaffold for binding to the necessary Cas enzyme and a variable spacer region which defines the target site for the enzyme. In natural CRISPR systems the guide RNA is often made of two molecules of RNA with complementarity. Engineered “single guide” RNAs connecting the two natural guide RNA components are often accepted by Cas enzymes.
- Mosaicism
A state in which two or more cell populations with distinct genotypes present in the same organism, derived from a single fertilized egg.
- Non-target strand
The DNA strand that is not complementary to the guide RNA sequence for a Cas-enzyme. For DNA base editors, deamination occurs on the non-target DNA strand.
- Protospacer adjacent motif (PAM)
A small region of nucleotides in the target DNA sequence adjacent to the sequence specified by a guide RNA. The PAM is not specified in the guide RNA, but Cas enzymes do not bind or cleave a sequence unless they are next to the appropriate PAM.
- Protospacer Flanking Sequence (PFS)
For RNA-targeting CRISPR systems, the PFS is the region in the target RNA found 5’ to the guiding RNA which binds to the nuclease enzyme but is not specified in the guide RNA.
- Protospacer sequence
The 15-to 25-nt region in a guide RNA that specifies the target RNA or DNA locus.
- Proximal off-target editing
Unwanted editing of bases outside of the activity window, but found nearby (for example, 100-nt up-or down-stream of) the target site.
- SaCas9
An RNA-guided endonuclease variant isolated from the CRISPR system of Streptococcus aureus. Catalyzes site-specific cleavage of double stranded DNA at sites with an NNGRRT PAM.
- SpCas9
An RNA-guided endonuclease variant isolated from the CRISPR system of Streptococcus pyogenes. Catalyzes site-specific cleavage of double stranded DNA at sites with an NGG PAM.
- Target strand
The strand of DNA (or RNA) complementary to the guide RNA sequence for a Cas-enzyme. This strand hybridizes with the guide RNA.
- Wobble position
The third nucleotide in a codon.
References
- 1.Cohen SNC, A.C.Y; Boyer HW; Helling RB Construction of Biologically Functional Bacterial Plasmids In Vitro. Proc Natl Acad Sci U S A 70, 3240–3244 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821, 10.1126/science.1225829 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jansen R, Embden JD, Gaastra W & Schouls LM Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43, 1565–1575 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Garneau JE et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71, 10.1038/nature09523 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Cho SW, Kim S, Kim JM & Kim JS Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31, 230–232, 10.1038/nbt.2507 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823, 10.1126/science.1231143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jinek M et al. RNA-programmed genome editing in human cells. Elife 2, e00471, 10.7554/eLife.00471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826, 10.1126/science.1232033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu PD, Lander ES & Zhang F Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278, 10.1016/j.cell.2014.05.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komor AC, Badran AH & Liu DR CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168, 20–36, 10.1016/j.cell.2016.10.044 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doudna JA & Charpentier E Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096, 10.1126/science.1258096 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Sternberg SH & Doudna JA Expanding the Biologist’s Toolkit with CRISPR-Cas9. Mol Cell 58, 568–574, 10.1016/j.molcel.2015.02.032 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Mali P et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31, 833–838, 10.1038/nbt.2675 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bikard D et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41, 7429–7437, 10.1093/nar/gkt520 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng AW et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 23, 1163–1171, 10.1038/cr.2013.122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasiunas G, Barrangou R, Horvath P & Siksnys V Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109, E2579–2586, 10.1073/pnas.1208507109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeggo PA DNA breakage and repair. Adv Genet 38, 185–218 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Rouet P, Smih F & Jasin M Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol 14, 8096–8106 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lukacsovich T, Yang D & Waldman AS Repair of a specific double-strand break generated within a mammalian chromosome by yeast endonuclease I-SceI. Nucleic Acids Res 22, 5649–5657 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rudin N, Sugarman E & Haber JE Genetic and physical analysis of double-strand break repair and recombination in Saccharomyces cerevisiae. Genetics 122, 519–534 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouet P, Smih F & Jasin M Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci U S A 91, 6064–6068 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapman JR, Taylor MR & Boulton SJ Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47, 497–510, 10.1016/j.molcel.2012.07.029 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Cox DB, Platt RJ & Zhang F Therapeutic genome editing: prospects and challenges. Nat Med 21, 121–131, 10.1038/nm.3793 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paquet D et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129, 10.1038/nature17664 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Lin S, Staahl BT, Alla RK & Doudna JA Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3, e04766, 10.7554/eLife.04766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodwin S, McPherson JD & McCombie WR Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17, 333–351, 10.1038/nrg.2016.49 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kosicki M, Tomberg K & Bradley A Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol, 10.1038/nbt.4192 (2018). [DOI] [PMC free article] [PubMed]
- 28.Landrum MJ et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D862–868, 10.1093/nar/gkv1222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landrum MJ et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42, D980–985, 10.1093/nar/gkt1113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424, 10.1038/nature17946 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishida K et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, 10.1126/science.aaf8729 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Gaudelli NM et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471, 10.1038/nature24644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishimasu H et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949, 10.1016/j.cell.2014.02.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X et al. Base editing with a Cpf1-cytidine deaminase fusion. Nat Biotechnol 36, 324–327, 10.1038/nbt.4102 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Cox DBT et al. RNA editing with CRISPR-Cas13. Science 358, 1019–1027, 10.1126/science.aaq0180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin HY et al. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat Commun 8, 15464, 10.1038/ncomms15464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33, 187–197, 10.1038/nbt.3117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L et al. Large genomic fragment deletions and insertions in mouse using CRISPR/Cas9. PLoS One 10, e0120396, 10.1371/journal.pone.0120396 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komor AC et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci Adv 3, eaao4774, 10.1126/sciadv.aao4774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montiel-Gonzalez MF, Vallecillo-Viejo I, Yudowski GA & Rosenthal JJ Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc Natl Acad Sci U S A 110, 18285–18290, 10.1073/pnas.1306243110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montiel-Gonzalez MF, Vallecillo-Viejo IC & Rosenthal JJ An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res 44, e157, 10.1093/nar/gkw738 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fukuda M et al. Construction of a guide-RNA for site-directed RNA mutagenesis utilising intracellular A-to-I RNA editing. Sci Rep 7, 41478, 10.1038/srep41478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wettengel J, Reautschnig P, Geisler S, Kahle PJ & Stafforst T Harnessing human ADAR2 for RNA repair - Recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res 45, 2797–2808, 10.1093/nar/gkw911 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vogel P, Hanswillemenke A & Stafforst T Switching Protein Localization by Site-Directed RNA Editing under Control of Light. ACS Synth Biol 6, 1642–1649, 10.1021/acssynbio.7b00113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanswillemenke A, Kuzdere T, Vogel P, Jekely G & Stafforst T Site-Directed RNA Editing in Vivo Can Be Triggered by the Light-Driven Assembly of an Artificial Riboprotein. J Am Chem Soc 137, 15875–15881, 10.1021/jacs.5b10216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vogel P & Stafforst T Site-directed RNA editing with antagomir deaminases--a tool to study protein and RNA function. ChemMedChem 9, 2021–2025, 10.1002/cmdc.201402139 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Vogel P, Schneider MF, Wettengel J & Stafforst T Improving site-directed RNA editing in vitro and in cell culture by chemical modification of the guideRNA. Angew Chem Int Ed Engl 53, 6267–6271, 10.1002/anie.201402634 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Stafforst T & Schneider MF An RNA-deaminase conjugate selectively repairs point mutations. Angew Chem Int Ed Engl 51, 11166–11169, 10.1002/anie.201206489 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Vogel P et al. Efficient and precise editing of endogenous transcripts with SNAP-tagged ADARs. Nat Methods 15, 535–538, 10.1038/s41592-018-0017-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris RS, Petersen-Mahrt SK & Neuberger MS RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell 10, 1247–1253 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Kunz C, Saito Y & Schar P DNA Repair in mammalian cells: Mismatched repair: variations on a theme. Cell Mol Life Sci 66, 1021–1038, 10.1007/s00018-009-8739-9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pearl LH Structure and function in the uracil-DNA glycosylase superfamily. Mutat Res 460, 165–181 (2000). [DOI] [PubMed] [Google Scholar]
- 53.Mol CD et al. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell 82, 701–708 (1995). [DOI] [PubMed] [Google Scholar]
- 54.Tang W & Liu DR Rewritable multi-event analog recording in bacterial and mammalian cells. Science 360, 10.1126/science.aap8992 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krokan HE, Drablos F & Slupphaug G Uracil in DNA--occurrence, consequences and repair. Oncogene 21, 8935–8948, 10.1038/sj.onc.1205996 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Lindahl T Instability and decay of the primary structure of DNA. Nature 362, 709–715, 10.1038/362709a0 (1993). [DOI] [PubMed] [Google Scholar]
- 57.Yasui M et al. Miscoding properties of 2’-deoxyinosine, a nitric oxide-derived DNA Adduct, during translesion synthesis catalyzed by human DNA polymerases. J Mol Biol 377, 1015–1023, 10.1016/j.jmb.2008.01.033 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Losey HC, Ruthenburg AJ & Verdine GL Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat Struct Mol Biol 13, 153–159, 10.1038/nsmb1047 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Bass BL & Weintraub H An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55, 1089–1098 (1988). [DOI] [PubMed] [Google Scholar]
- 60.Matthews MM et al. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol 23, 426–433, 10.1038/nsmb.3203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu YT et al. Internal modification of U2 small nuclear (sn)RNA occurs in nucleoli of Xenopus oocytes. J Cell Biol 152, 1279–1288 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bass BL RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71, 817–846, 10.1146/annurev.biochem.71.110601.135501 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bazak L et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24, 365–376, 10.1101/gr.164749.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keppler A et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol 21, 86–89, 10.1038/nbt765 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Schneider MF, Wettengel J, Hoffmann PC & Stafforst T Optimal guideRNAs for re-directing deaminase activity of hADAR1 and hADAR2 in trans. Nucleic Acids Res 42, e87, 10.1093/nar/gku272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herbert A & Rich A The role of binding domains for dsRNA and Z-DNA in the in vivo editing of minimal substrates by ADAR1. Proc Natl Acad Sci U S A 98, 12132–12137, 10.1073/pnas.211419898 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lehmann KA & Bass BL Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39, 12875–12884 (2000). [DOI] [PubMed] [Google Scholar]
- 68.Kuttan A & Bass BL Mechanistic insights into editing-site specificity of ADARs. Proc Natl Acad Sci U S A 109, E3295–3304, 10.1073/pnas.1212548109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crick FH Codon--anticodon pairing: the wobble hypothesis. J Mol Biol 19, 548–555 (1966). [DOI] [PubMed] [Google Scholar]
- 70.Kume H, Hino K, Galipon J & Ui-Tei K A-to-I editing in the miRNA seed region regulates target mRNA selection and silencing efficiency. Nucleic Acids Res 42, 10050–10060, 10.1093/nar/gku662 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seeburg PH & Hartner J Regulation of ion channel/neurotransmitter receptor function by RNA editing. Curr Opin Neurobiol 13, 279–283 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Yang JH, Sklar P, Axel R & Maniatis T Editing of glutamate receptor subunit B pre-mRNA in vitro by site-specific deamination of adenosine. Nature 374, 77–81, 10.1038/374077a0 (1995). [DOI] [PubMed] [Google Scholar]
- 73.Nishikura K Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79, 321–349, 10.1146/annurev-biochem-060208-105251 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Athanasiadis A, Rich A & Maas S Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2, e391, 10.1371/journal.pbio.0020391 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hess GT et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat Methods 13, 1036–1042, 10.1038/nmeth.4038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim K et al. Highly efficient RNA-guided base editing in mouse embryos. Nat Biotechnol 35, 435–437, 10.1038/nbt.3816 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Ma Y et al. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat Methods 13, 1029–1035, 10.1038/nmeth.4027 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Wang L et al. Enhanced base editing by co-expression of free uracil DNA glycosylase inhibitor. Cell Res 27, 1289–1292, 10.1038/cr.2017.111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Di Noia J & Neuberger MS Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 419, 43–48, 10.1038/nature00981 (2002). [DOI] [PubMed] [Google Scholar]
- 80.Radany EH et al. Increased spontaneous mutation frequency in human cells expressing the phage PBS2-encoded inhibitor of uracil-DNA glycosylase. Mutat Res 461, 41–58 (2000). [DOI] [PubMed] [Google Scholar]
- 81.Hess GT, Tycko J, Yao D & Bassik MC Methods and Applications of CRISPR-Mediated Base Editing in Eukaryotic Genomes. Mol Cell 68, 26–43, 10.1016/j.molcel.2017.09.029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ryu SM et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat Biotechnol, 10.1038/nbt.4148 (2018). [DOI] [PubMed]
- 83.Hua K, Tao X, Yuan F, Wang D & Zhu JK Precise A.T to G.C Base Editing in the Rice Genome. Mol Plant 11, 627–630, 10.1016/j.molp.2018.02.007 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Yan F et al. Highly Efficient A.T to G.C Base Editing by Cas9n-Guided tRNA Adenosine Deaminase in Rice. Mol Plant 11, 631–634, 10.1016/j.molp.2018.02.008 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63, 10.1038/nature26155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lau AY, Wyatt MD, Glassner BJ, Samson LD & Ellenberger T Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci U S A 97, 13573–13578, 10.1073/pnas.97.25.13573 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kouzminova EA & Kuzminov A Patterns of chromosomal fragmentation due to uracil-DNA incorporation reveal a novel mechanism of replication-dependent double-stranded breaks. Mol Microbiol 68, 202–215, 10.1111/j.1365-2958.2008.06149.x (2008). [DOI] [PubMed] [Google Scholar]
- 88.Liu Z et al. Highly efficient RNA-guided base editing in rabbit. Nat Commun 9, 2717, 10.1038/s41467-018-05232-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yeh WH, Chiang H, Rees HA, Edge ASB & Liu DR In vivo base editing of post-mitotic sensory cells. Nat Commun 9, 2184, 10.1038/s41467-018-04580-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Koblan LW et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol, 10.1038/nbt.4172 (2018). [DOI] [PMC free article] [PubMed]
- 91.Kang BC et al. Precision genome engineering through adenine base editing in plants. Nat Plants, 10.1038/s41477-018-0178-x (2018). [DOI] [PubMed]
- 92.Rees HA et al. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat Commun 8, 15790, 10.1038/ncomms15790 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim D et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol 35, 475–480, 10.1038/nbt.3852 (2017). [DOI] [PubMed] [Google Scholar]
- 94.Kleinstiver BP et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495, 10.1038/nature16526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee JK et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat Commun 9, 3048, 10.1038/s41467-018-05477-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yamanaka S et al. Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. Proc Natl Acad Sci U S A 92, 8483–8487 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Okazaki IM et al. Constitutive expression of AID leads to tumorigenesis. J Exp Med 197, 1173–1181, 10.1084/jem.20030275 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burns MB et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370, 10.1038/nature11881 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu Z et al. Efficient generation of mouse models of human diseases via ABE- and BE-mediated base editing. Nat Commun 9, 2338, 10.1038/s41467-018-04768-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gehrke JM et al. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat Biotechnol, 10.1038/nbt.4199 (2018). [DOI] [PMC free article] [PubMed]
- 101.Ran FA et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191, 10.1038/nature14299 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zetsche B et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771, 10.1016/j.cell.2015.09.038 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim E et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun 8, 14500, 10.1038/ncomms14500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Muller M et al. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol Ther 24, 636–644, 10.1038/mt.2015.218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee CM, Cradick TJ & Bao G The Neisseria meningitidis CRISPR-Cas9 System Enables Specific Genome Editing in Mammalian Cells. Mol Ther 24, 645–654, 10.1038/mt.2016.8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kleinstiver BP et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 33, 1293–1298, 10.1038/nbt.3404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kleinstiver BP et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485, 10.1038/nature14592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim YB et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol 35, 371–376, 10.1038/nbt.3803 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS & Vale RD A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646, 10.1016/j.cell.2014.09.039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jiang W et al. BE-PLUS: a new base editing tool with broadened editing window and enhanced fidelity. Cell Res, 10.1038/s41422-018-0052-4 (2018). [DOI] [PMC free article] [PubMed]
- 111.Kim S, Bae T, Hwang J & Kim JS Rescue of high-specificity Cas9 variants using sgRNAs with matched 5’ nucleotides. Genome Biol 18, 218, 10.1186/s13059-017-1355-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nishimasu H et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science (2018). [DOI] [PMC free article] [PubMed]
- 113.Hua K, Tao X & Zhu JK Expanding the base editing scope in rice by using Cas9 variants. Plant Biotechnol J, 10.1111/pbi.12993 (2018). [DOI] [PMC free article] [PubMed]
- 114.Yang L et al. Increasing targeting scope of adenosine base editors in mouse and rat embryos through fusion of TadA deaminase with Cas9 variants. Protein Cell 9, 814–819, 10.1007/s13238-018-0568-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang X et al. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat Biotechnol, 10.1038/nbt.4198 (2018). [DOI] [PubMed]
- 116.Pinello L et al. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol 34, 695–697, 10.1038/nbt.3583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Suzuki K et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149, 10.1038/nature20565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wheeler LC, Lim SA, Marqusee S & Harms MJ The thermostability and specificity of ancient proteins. Curr Opin Struct Biol 38, 37–43, 10.1016/j.sbi.2016.05.015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zafra MP et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat Biotechnol, 10.1038/nbt.4194 (2018). [DOI] [PMC free article] [PubMed]
- 120.Midoux P, Pichon C, Yaouanc JJ & Jaffres PA Chemical vectors for gene delivery: a current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br J Pharmacol 157, 166–178, 10.1111/j.1476-5381.2009.00288.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bodles-Brakhop AM, Heller R & Draghia-Akli R Electroporation for the delivery of DNA-based vaccines and immunotherapeutics: current clinical developments. Mol Ther 17, 585–592, 10.1038/mt.2009.5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Thomas CE, Ehrhardt A & Kay MA Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet 4, 346–358, 10.1038/nrg1066 (2003). [DOI] [PubMed] [Google Scholar]
- 123.Zuris JA et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol 33, 73–80, 10.1038/nbt.3081 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang M et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc Natl Acad Sci U S A 113, 2868–2873, 10.1073/pnas.1520244113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim S, Kim D, Cho SW, Kim J & Kim JS Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24, 1012–1019, 10.1101/gr.171322.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Miao CH et al. Nonrandom transduction of recombinant adeno-associated virus vectors in mouse hepatocytes in vivo: cell cycling does not influence hepatocyte transduction. J Virol 74, 3793–3803 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dong JY, Fan PD & Frizzell RA Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum Gene Ther 7, 2101–2112, 10.1089/hum.1996.7.17-2101 (1996). [DOI] [PubMed] [Google Scholar]
- 128.Lai Y et al. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol 23, 1435–1439, 10.1038/nbt1153 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gao X et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221, 10.1038/nature25164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Park DS et al. Targeted Base Editing via RNA-Guided Cytidine Deaminases in Xenopus laevis Embryos. Mol Cells 40, 823–827, 10.14348/molcells.2017.0262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liang P et al. Effective gene editing by high-fidelity base editor 2 in mouse zygotes. Protein Cell 8, 601–611, 10.1007/s13238-017-0418-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li G et al. Highly efficient and precise base editing in discarded human tripronuclear embryos. Protein Cell 8, 776–779, 10.1007/s13238-017-0458-7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liang P et al. Correction of beta-thalassemia mutant by base editor in human embryos. Protein Cell 8, 811–822, 10.1007/s13238-017-0475-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ma Y et al. Highly efficient and precise base editing by engineered dCas9-guide tRNA adenosine deaminase in rats. Cell Discov 4, 39, 10.1038/s41421-018-0047-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang Y et al. Programmable base editing of zebrafish genome using a modified CRISPR-Cas9 system. Nat Commun 8, 118, 10.1038/s41467-017-00175-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tanaka S et al. In vivo targeted single-nucleotide editing in zebrafish. Sci Rep 8, 11423, 10.1038/s41598-018-29794-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schenk B et al. MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type If. J Clin Invest 108, 1687–1695, 10.1172/JCI13419 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zeng Y et al. Correction of the Marfan Syndrome Pathogenic FBN1 Mutation by Base Editing in Human Cells and Heterozygous Embryos. Molecular Therapy, 10.1016/j.ymthe.2018.08.007 (2018). [DOI] [PMC free article] [PubMed]
- 139.Chadwick AC, Wang X & Musunuru K In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler Thromb Vasc Biol 37, 1741–1747, 10.1161/ATVBAHA.117.309881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Roccio M, Hahnewald S, Perny M & Senn P Cell cycle reactivation of cochlear progenitor cells in neonatal FUCCI mice by a GSK3 small molecule inhibitor. Sci Rep 5, 17886, 10.1038/srep17886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308, 10.1038/nprot.2013.143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kuscu C et al. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat Methods 14, 710–712, 10.1038/nmeth.4327 (2017). [DOI] [PubMed] [Google Scholar]
- 143.Billon P et al. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol Cell 67, 1068–1079 e1064, 10.1016/j.molcel.2017.08.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Haapaniemi E, Botla S, Persson J, Schmierer B & Taipale J CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med, 10.1038/s41591-018-0049-z (2018). [DOI] [PubMed]
- 145.Ihry RJ et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med, 10.1038/s41591-018-0050-6 (2018). [DOI] [PubMed]
- 146.Aguirre AJ et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov 6, 914–929, 10.1158/2159-8290.CD-16-0154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Roukos V & Misteli T The biogenesis of chromosome translocations. Nat Cell Biol 16, 293–300, 10.1038/ncb2941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gapinske M et al. CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol 19, 107, 10.1186/s13059-018-1482-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hur JK et al. Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat Biotechnol 34, 807–808, 10.1038/nbt.3596 (2016). [DOI] [PubMed] [Google Scholar]
- 150.Sung YH et al. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol 31, 23–24, 10.1038/nbt.2477 (2013). [DOI] [PubMed] [Google Scholar]
- 151.Sung YH et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res 24, 125–131, 10.1101/gr.163394.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tang W, Hu JH & Liu DR Aptazyme-embedded guide RNAs enable ligand-responsive genome editing and transcriptional activation. Nat Commun 8, 15939, 10.1038/ncomms15939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Farzadfard F et al. Single-Nucleotide-Resolution Computing and Memory in Living Cells. bioRxiv, 10.1101/263657 (2018). [DOI] [PMC free article] [PubMed]
- 154.Yin K, Gao C & Qiu JL Progress and prospects in plant genome editing. Nat Plants 3, 17107, 10.1038/nplants.2017.107 (2017). [DOI] [PubMed] [Google Scholar]
- 155.Voytas DF & Gao C Precision genome engineering and agriculture: opportunities and regulatory challenges. PLoS Biol 12, e1001877, 10.1371/journal.pbio.1001877 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Svitashev S et al. Targeted Mutagenesis, Precise Gene Editing, and Site-Specific Gene Insertion in Maize Using Cas9 and Guide RNA. Plant Physiol 169, 931–945, 10.1104/pp.15.00793 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Endo M, Mikami M & Toki S Biallelic Gene Targeting in Rice. Plant Physiol 170, 667–677, 10.1104/pp.15.01663 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zong Y et al. Precise base editing in rice, wheat and maize with a Cas9-cytidine deaminase fusion. Nat Biotechnol 35, 438–440, 10.1038/nbt.3811 (2017). [DOI] [PubMed] [Google Scholar]
- 159.Shimatani Z et al. Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat Biotechnol 35, 441–443, 10.1038/nbt.3833 (2017). [DOI] [PubMed] [Google Scholar]
- 160.Li C et al. Expanded base editing in rice and wheat using a Cas9-adenosine deaminase fusion. Genome Biol 19, 59, 10.1186/s13059-018-1443-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]