

Abstract
Aim: To determine the mechanisms of HEATR1 on cell survival in non-small cell lung cancer (NSCLC).
Methods: HEATR1 mRNA expression levels in 57 pairs of NSCLC tumor and adjacent normal lung tissues were analyzed using the TCGA database. The effect of HEATR1 inhibition on cell proliferation, apoptosis, and colony formation was measured in A549 and NCI-H460 cells lines. In addition, the effect of HEATR1 inhibition on tumor growth was measured using in vivo xenograft nude mouse models. Additionally, downstream signaling pathways affected by HEATR1 inhibition were analyzed using microarrays and bioinformatics analysis, and were validated using quantitative real-time polymerase chain reaction and Western blot analysis.
Results: HEATR1 levels were significantly higher in NSCLC tumor tissues compared to normal adjacent lung tissues (P<0.001). In vitro, cell proliferation was significantly reduced in both A549 and NCI-H1299 cells transduced with shHEATR1 compared to shCtrl (P<0.001). Colony formation was also significantly reduced after HEATR1 interference (P<0.01). Additionally, the percentage of apoptosis was significantly increased in cells transduced with shHEATR1 (P<0.001). In vivo, HEATR1 inhibition significantly reduced xenograft tumor growth in nude mice. HEATR1 inhibition drastically affected the p53-signaling pathway, significantly up-regulating PUMA and BAX both at the mRNA and protein levels (P<0.001), while BCL2 levels were significantly down-regulated (P<0.01). The cell proliferation and apoptosis were recovered in cell transduced with shHEATR1 and shp53 compared to shHEATR1 (P<0.05).
Conclusion: HEATR1 inhibition activated p53 by reducing ribosome biogenesis, which subsequently led to NSCLC cell apoptosis and reduced cell survival through the p53-PUMA-BAX/BCL2 axis. Our results provide a mechanism by which therapeutic modulation of HEATR1 could be a treatment strategy for NSCLC. In addition, HEATR1 could be used as a potential biomarker for the prognosis or therapeutic evaluation of NSCLC.
Keywords: HEATR1, non-small cell lung cancer, p53, PUMA, cell survival
Introduction
Lung cancer is one of the most frequently diagnosed cancers and is the leading cause of cancer deaths worldwide.1 Lung cancer is classified into two histological subtypes: small-cell lung cancer and non-small-cell lung cancer (NSCLC).2 NSCLC is the major histological type and comprises about 85% of all lung cancer. It mainly includes adenocarcinomas (AD) and squamous cell carcinomas .3 Despite the latest advancements in NSCLC diagnosis and treatment strategies, the 5-year survival rate of NSCLC patients is approximately 10–15%.4,5 It is critical to identify novel biomarkers and key signaling pathways to develop effective therapeutic, diagnostic, and prognostic strategies for NSCLC.
HEAT repeat-containing protein 1 (HEATR1) is composed of conserved HEAT repeats, that were initially found in a diverse family of proteins that includes elongation factor-3, huntingtin, and the PR65/A subunit of protein phosphatase 2A. HEAT repeats are thought to be involved in protein–protein interactions.6 The human HEATR1 gene is located on chromosome lq43 and encodes a large (236 kDa) protein consisting of 2,144 amino acids. The protein contains one HEAT repeat on its c-terminal end.7 Several reports have implicated HEATR1 in regulating cell response by activating cytotoxic T lymphocyte response in gliomas.8 In addition, HEATR1 is thought to be involved in the mTOR signaling pathway to dephosphorylate AKT in pancreatic ductal adenocarcinomas.9 However, the cellular function of HEATR1 remains to be deciphered.
In this study, the effects of HEATR1 inhibition on cell proliferation, apoptosis, and tumor growth in nude mice were evaluated. Using microarray and bioinformatics analysis, the mechanism by which HEATR1 influence NSCLC cell survival was all investigated. The results may help evaluate HEATR1 as a therapeutic target, or a potential biomarker for the prognosis or therapeutic evaluation of NSCLC.
Materials and methods
Cell culture
The NSCLC cell lines, A549 cells, and NCI-H460 cells were obtained from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. A549 cells were cultured in F-12K media (Invitrogen, Carlsbad, CA, USA) and NCI-H460 cells in 1,640 medium (Gibco, Gaithersburg, MD, USA), supplemented with 10% fetal bovine serum (Gibco, Gaithersburg, MD, USA), 100 U/mL penicillin and 0.1 mg/mL streptomycin (Gibco, Gaithersburg, MD, USA) at 37°C in 100% air.
RNA interference
NSCLC cells were seeded in six-well plates at a density of 2*105/well for RNA interference studies. NSCLC cells were transduced with either control lentivirus (shCtrl group, 5ʹ-TTCTCCGAACGTGTCACGT-3ʹ) or shHEATR1 lentivirus (shHEATR1-1 group, 5ʹ-AGAGTTGATGGAAGATGAA-3ʹ; shHEATR1-2 group, 5ʹ-TGAACAAGTCCG AATAGAA-3ʹ; shHEATR1-3 group, 5ʹ-GATGTTGTTTTGTCGGCTA-3ʹ), all of which also expressed GFP. 72 hrs post-infection, the expression of GFP was analyzed using a fluorescent microscope. The percentage of fluorescence indicated the number of cells that were infected. If greater than 70% of the cells were infected, experimental studies were performed.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from NSCLC cells using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). RNA concentration was determined using the NanoDrop 2,000 spectrophotometer (Thermo Fisher Scientific, Rockford, IL, USA). A total of 1 μL RNA was reverse-transcribed using the First-Strand cDNA Synthesis kits (Invitrogen, Carlsbad, CA, USA) based on the manufacturer's instructions. qRT-PCR was then performed on the ABI 7500 System (Applied Biosystems, Foster City, CA, USA) and was used to analyze the expression levels of the target mRNAs. Relative mRNA expression levels were calculated using GAPDH as the internal control. Each sample was run in triplicate. The primer pairs used in this study are listed as follows. HEATR1 forward: 5ʹ- TTCACTTGTCGCCTTACTTCC-3ʹ; reverse: 5ʹ-CCAGAACCATCTGTGCTTTGA −3ʹ; PUMA forward: 5ʹ- TGAAGAGCAAATGAGCCAAACG-3ʹ; reverse: 5ʹ- CAGAGCACAGGATTCACAGTCT-3ʹ; BAX forward: 5ʹ-CCCGAGAGGTCTTTT TCCGAG-3ʹ; reverse: 5ʹ- CCAGCCCATGATGGTTCTGAT-3ʹ; BCL2 forward:5ʹ- ATGTGTGTGGAGAGCGTCAA-3ʹ; reverse: 5ʹ-TTCAGAGACAGCCAGGAGAAA-3ʹ; GAPDH forward: 5ʹ-TGACTTCAACAGCGACACCCA-3ʹ; reverse: 5ʹ- CACCCTGTTGCTGTAGCCAAA-3ʹ.
Western blot analysis
Cell lysates were collected, and protein concentrations were determined using the bicinchoninic acidprotein assay kit (Boster, Wuhan, China). Equal amounts of proteins were separated using 10% SDS-polyacrylamide gels and then transferred to polyvinylidene difluoride membranes. The membranes were then incubated with primary antibodies against HEATR1 (Abcam, UK), PUMA (Cell Signaling Technology, Danvers, MA, USA), BAX (Cell Signaling Technology, Danvers, MA, USA), BCL-2 (Cell Signaling Technology, Danvers, MA, USA) and GAPDH (Santa Cruz, Dallas, TX, USA) overnight at 4°C. Afterwards, the appropriate secondary antibodies (Santa Cruz, Dallas, TX, USA) were incubated for 1 hr at room temperature. Signals were detected using the gel imaging system (ProteinSimple, San Jose, CA, USA). The ratio of target proteins to GAPDH was analyzed using the FluorChem FC3 software. All experiments were in performed in triplicate.
Cell proliferation assays
NSCLC cells transduced with control or shHEATR1 lentivirus were seeded in 96-well plates at a density of approximately 2–5*103/well in quintuplicate. Cell proliferation was measured after one, two, three, four, and five days after seeding. Fluorescent intensity was used to measure cell proliferation using Celigo Imaging Cytometer (Nexcelom Bioscience, Lawrence, MA, USA). In addition, MTT assays were performed to measure end-point proliferation rates. Briefly, 20 μL of MTT solution was added to each well and after 4 hrs of incubation at 37°C, the solution was removed and DMSO was added to dissolve the formazan crystals formed. The absorbance at 490 nm was measured using the Glomax-Multi + Detection System (Promega, Madison, WI, USA). All experiments were performed using three biological replicates.
Colony formation assay
Cells were seeded in six-well plates at a density of 400–1,000 cells per well. Culture media were changed every 72 hrs. Colonies were fixed using 4% paraformaldehyde and stained using GIEMSA solution (Dingguo, Shanghai, China) after 14 days. Cell colonies were visualized using fluorescence microscopy and then quantitated. Three independent biological experiments were performed.
Cell apoptosis assays
Apoptosis was measured using the eBioscience Annexin V Apoptosis Detection Kit APC (Thermo Fisher Scientific, Rockford, IL, USA) according to the manufacturer's instruction. Briefly, cells were harvested with trypsin, washed twice with ice-cold PBS and resuspended in 1*binding buffer. Then, 10 μL of annexin V-APC was added into 200 μL of cell suspensions. After incubation for 15 mins, the samples were analyzed using the Beckman Counter flow cytometer.
Xenograft mouse models for tumor growth
A total of 20 female BALB/C nude mice (4–6 weeks, 18-22 g) were purchased from Shanghai Lingchang Biotechnology (SCXK2013-0018). Mice were randomly divided into two groups, consisting of the NC group (n=10) and KD group (n=10). Mice were then subcutaneously inoculated with A549 cells (5*106) stably expressing either the control or shHEATR1 lentivirus. When the diameter of the tumor was larger than 5 mm, tumor volume was determined twice per week. Tumor volumes were calculated using the formulae, V=(width2 * length)/2. After euthanasia, tumors were harvested and weighed. The Ethics Committee of Shanghai Seventh People’s Hospital approved all animal studies and conformed to the ethics guidelines of the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8,023, revised 1,978).
Microarray and bioinformatics analysis
Total RNA was extracted from A549 cells (transfected with either the control or shHEATR1 lentivirus) using the Trizol reagent (n=3 for each group). After QC, RNA samples were analyzed using the Affymetrix GeneChip primeview human gene expression array. Differentially expressed genes were selected using a cut-off P-value of less than 0.05 based on statistical analysis and a twofold change cut-off value. Differentially expressed genes from microarray analyses were uploaded to Ingenuity Pathway Analysis (IPA, Ingenuity Systems) for pathway analysis and molecular pathway identification.
Statistical analysis
Statistical analysis was performed using SPSS 16.0 (Chicago, IL, USA). Data were expressed as mean ± standard deviation and analyzed using Mann–Whitney test due to non-normal distribution or heterogeneity of variance. P<0.05 was considered statistically significant. All results were confirmed using at least three independent experiments.
Results
Increased HEATR1 expression levels in NSCLC tumor tissues
To determine whether HEATR1 was dysregulated in NSCLC tumor samples, we analyzed the mRNA expression levels of HEATR1 in 57 pairs of NSCLC tumor vs anormal adjacent lung tissues obtained from the TCGA database. As shown in Figure 1A, HEATR1 levels were significantly up-regulated in tumor tissues (P<0.001), suggesting that increased HEATR1 expression levels may be associated with the occurrence and development of NSCLC. Furthermore, we analyzed the relationship between HEATR1 expression and survival time. NSCLC patients with higher HEATR1 expression had longer survival time, but it did not show a significant difference (Figure S1).
Figure 1.

The expression level of HEATR1 in NSCLC and the knockdown efficiency of HEATR1 in A549 and NCI-H460 cells. (A) The expression levels of HEATR1 in 57 pairs of NSCLC tumor and normal adjacent lung tissues were obtained from the TCGA database. HEATR1 levels were significantly up-regulated in tumor tissues (P<0.001). N=57. (B and C) The knockdown efficiency of HEATR1 was determined using qRT-PCR (B) and Western blots (C). In the mRNA level, the knockdown efficiency of shHEATR1-1 was 85.1% in A549 cells and 85.6% in NCI-H460 cells, which was the highest among three shRNAs. In protein level, HEATR1 was also significantly knocked down in both A549 and NCI-H460 cells. N=3. Values were expressed as mean ± standard deviation.
HEATR1 is required for cell proliferation and apoptosis inhibition
To determine whether HEATR1 affected NSCLC tumorigenesis, we transduced A549 and NCI-H460 cells with shCtrl or shHEATR1 lentivirus and determined cell proliferation and apoptosis. HEATR1 knockdown efficiency was determined using qRT-PCR and Western blots. Compared to cells transduced with control lentivirus, cells transduced with shHEATR1 lentivirus had reduced HEATR1 expression levels both at the mRNA and protein levels (Figure 1B and C). In the mRNA level, the knockdown efficiency of shHEATR1-1 was 85.1% in A549 cells and 85.6% in NCI-H460 cells, which was the highest among three shRNAs. Thus, shHEATR1-1 was chosen for further study and termed as shHEATR1.
Celigo and MTT assays demonstrated that cell proliferation was reduced in shHEATR1 transduced cells after five days post-transduction (Figure 2A and B). On 2nd, 3rd, 4th, and 5th day, the cell proliferation was significantly reduced in shHEATR1 transduced cells compared to cells transduced with shCtrl lentivirus (P<0.05). Similarly, colony formation was significantly reduced after HEATR1 inhibition in both A549 and NCI-H460 cell lines (P<0.01, Figure 2C). Additionally, the percentage of apoptotic cells was significantly increased in cells transduced with shHEATR1 compared to cells transduced with shCtrl lentivirus (P<0.001, Figure 2D). These results indicated that HEATR1 inhibition could reduce cell proliferation and promote apoptosis in vitro.
Figure 2.

The cell proliferation and cell apoptosis influenced by HEATR1 inhibition in A549 and HCI-H460 cells. (A) Celigo detection. Cell proliferation was reduced in shHEATR1 transduced cells after five days post-transduction. On 2nd, 3rd, 4th, and 5th day, the cell count was significantly smaller in shHEATR1 transduced cells compared to cells transduced with shCtrl lentivirus in both A549 and NCI-H460 cell lines (P<0.05). (B) MTT assay. HEATR1 inhibition significantly reduced cell proliferation in both A549 and NCI-H460 cells (P<0.05). (C) Colony formation assay. HEATR1 inhibition significantly decreased the colony number in A549 cells (P<0.01) and NCI-H460 cells (P<0.001). (D) Apoptosis assay. Y-axis represents whether the cells are infected with lentivirus which are carried with green fluorescence, and x-axis represents whether the cells are apoptosis which are dyed with red fluorescence. First quartile represents apoptosis cells infected with lentivirus. HEATR1 inhibition significantly increased the percentage of apoptosis cells in both A549 and NCI-H460 cells (P<0.001). N=3. Values were expressed as mean ± standard deviation. *P<0.05, **P<0.01, ***P<0.001, compared to shCtrl group.
HEATR1 inhibition suppressed in vivo tumor growth
To further validate the effect of HEATR1 inhibition, we performed in vivo assays using tumor xenograft mouse models with A549 cell lines (Figure 3) and NCI-460 cell lines (Figure S2). Tumor volume significantly increased in mice transplanted with cells transduced with control lentivirus, while tumors rarely formed in mice transplanted with cells transduced with shHEATR1 lentivirus (Figure 3A). Upon euthanasia, the tumors were harvested, weighed, and compared (Figure 3B and C). Noticeable tumors were formed in mice from the NC group while no tumors were observed in mice from the HEATR1 KD group. These results suggested that HEATR1 inhibition could dramatically inhibit in vivo tumor growth.
Figure 3.

The effect of HEATR1 inhibition in vivo. (A) Tumor volume significantly increased in mice transplanted with cells transduced with control lentivirus, while tumors rarely formed in mice transplanted with cells transduced with shHEATR1 lentivirus. (B) The pictures of animals and tumors were taken and compared in NC group and KD group. (C) Tumor weight was significantly increased in the NC group, while tumors rarely formed in the KD group N=10. Values were expressed as mean ± standard deviation.
HEATR1 inhibition affects the p53/puma signaling pathway
To determine the mechanism of HEATR1 inhibition on NSCLC cell survival, we performed gene expression profiling of A549 cells transduced with either control or shHEATR1 lentivirus. Six hundred and fifty-six genes were up-regulated and 524 genes down-regulated in cells transduced with shHEATR1 lentivirus compared to cells transduced with control lentivirus (Figure 4A, red denotes up-regulated genes, and green denotes down-regulated genes). Canonical pathways enriched were analyzed using IPA. The p53 signaling pathway was significantly enriched, specifically the p53/PUMA signaling pathway (Figure 4B). Subsequently, we validated our microarray data by qRT-PCR and Western blot analysis. As shown in Figure 4C, the expression levels determined by qRT-PCR and Western blots for PUMA, BAX, and BCL2 were consistent with our gene expression profiling data. PUMA and BAX expression levels were significantly up-regulated in cells transduced with shHEATR1 lentivirus both at the mRNA and protein levels (P<0.001), and BCL2 was significantly down-regulated (P<0.01).
Figure 4.

The p53/PUMA pathway influenced by HEATR1 inhibition. (A) The heatmap of differently expressed genes between shCtrl group and shHEATR1 group. Red denotes up-regulated genes, and green denotes down-regulated genes. Upper tree structure is listed according to the sample characteristics, and left tree structure is listed according to the gene characteristics. There is a higher similarity between the adjacent samples or genes. (B) The p53 signaling pathway significantly altered by HEATR1 inhibition. Red denotes up-regulated genes, and grey denotes down-regulated genes. (C) The mRNA and protein expressions of PUMA, BAX, and BCL2 determined using qRT-PCR and Western blot. GAPDH was as the internal standard. PUMA and BAX were significantly up-regulated by HEATR1 inhibition in both mRNA and protein levels (P<0.001), and BCL2 was significantly down-regulated (P<0.01). N=3. Values were expressed as mean ± standard deviation. ** P<0.01, *** P<0.001, compared to shCtrl group.
Furthermore, in order to clarify whether the suppression of HEATR1 induced cell growth suppression or cell death via p53, we also analyzed cells HEATR1 and p53 knocked down simultaneously. MTT assays demonstrated that cell proliferation was recovered in the shHEATR1+ shp53 group after five days post-transduction in both A549 and NCI-H460 cell lines (Figure 5A). On 3rd, 4th, and 5th day, the cell proliferation was significantly increased in cells transduced with both shHEATR1 and shp53 compared to cells transduced with shHEATR1 lentivirus (P<0.05). Additionally, the percentage of apoptotic cells was significantly reduced in A549 and NCI-H460 cell lines transduced with shHEATR1 and shp53 compared to cells transduced with shHEATR1 lentivirus (P<0.001, Figure 5B). This suggests that HEATR1 may influence NSCLC cells survival by regulating the p53 pathway.
Figure 5.

The cell proliferation and cell apoptosis influenced by HEATR1 and p53 inhibition in A549 and HCI-H460 cells. (A) MTT assay. Cell proliferation was significantly increased in cells transduced with both shHEATR1 and shp53 compared to cells transduced with shHEATR1 lentivirus (P<0.05). (B) Apoptosis assay. The percentage of apoptosis was significantly reduced in cells transduced with both shHEATR1 and shp53 compared to cells transduced with shHEATR1 lentivirus (P<0.001). N=3. Values were expressed as mean ± standard deviation. * P<0.05, ** P<0.01, *** P<0.001, compared to shHEATR1 group.
Discussion
In this study, we investigated the role of human HEATR1 as it was a poorly characterized protein but was thought to play a role in tumorigenesis. We are the first to report that HEATR1 is overexpressed in NSCLC and plays a role in NSCLC tumor survival and progression. HEATR1 gene is a multiply spliced 7-kb gene that encodes bap28, a protein involved in nucleolar processing of pre-18S ribosomal RNA and ribosome biosynthesis.8 We analyzed clinical data from the TCGA database and found that HEATR1 levels were significantly up-regulated in NSCLC tumor tissues compared to normal adjacent lung tissues (Figure 1A). In vitro, after HEATR1 inhibition, cell growth was significantly reduced as determined by Celigo and MTT assays. In addition, colony formation was significantly reduced while apoptosis was significantly increased (Figure 2). In vivo, HEATR1 inhibition significantly suppressed tumor growth in xenograft nude mouse models (Figure 3). These results indicate that HEATR1 inhibition may influence the occurrence and development of NSCLC.
A few previous studies demonstrated that down-regulation of HEATR1 led to cell arrest in a p53-dependent manner.7 This was consistent with our results demonstrating that the p53 signaling pathway was significantly altered after HEATR1 inhibition through microarray analysis (Figure 4B). The p53 signaling pathway has being closely associated with NSCLC.10,11 It has been reported that activation of p53 upon HEATR1 interference was mediated by ribosomal protein uL18 (RPL5), which is a component of the ribosomal precursor complex that consists of the nascent ribosomal protein uL5 (RPL11) and 5S rRNA.12,13 Upon ribosome biogenesis impairment, the ribosomal complex gets redirected from being assembled into ribosomes to binding to MDM2. This leads to the stabilization and activation of p53.12,14 This is consistent with our findings that MDM2 levels were significantly altered after HEATR1 inhibition (Figure 4B). Additionally, HEATR1 inhibition resulted in ribosome biogenesis stress.7 This specific signaling pathway was recently named the Impaired Ribosome Biogenesis Checkpoint pathway, to distinguish it from other p53-activating stress signaling pathways.15,16
Due to the intimate association between ribosome biogenesis, cell growth, and cell cycle regulation, enhanced ribosome biogenesis could inhibit differentiation and promote tumorigenesis.17–19 Conversely, aberrant low levels of ribosome biogenesis represent the hallmark of ribosomopathies. This is a disease characterized by hyperactivation of p53 and reduced cell growth and protein synthesis.20,21 Several studies have clearly demonstrated that inherited and acquired abnormalities of ribosome biogenesis were closely associated with the increased risk of cancer.12,22 Increased HEATR1 level in NSCLC tumor tissues found in this study are consistent with the concept of global deregulation of ribosome biogenesis in cancer.
In addition, we found that PUMA, BAX, and BCL2 levels were significantly altered after HEATR1 inhibition (Figure 4). PUMA, a well-known BH3-only protein, is positively regulated by p53 and plays a role in promoting apoptosis in tumor cells.23,24 PUMA is expressed at very low levels under normal physiological conditions; however, its expression levels dramatically increase in response to stress.25,26 PUMA can induce BAX-mediated mitochondrial apoptotic pathways by interacting with p53 and members of the BCL2 family.27–29 This was consistent with our results that BAX levels significantly increased and BCL2 level decreased after HEATR1 inhibition (Figure 4B).
In summary, we demonstrated that HEATR1 inhibition could activate p53 via impaired ribosome biogenesis. This subsequently leads to NSCLC cell apoptosis and reduces cell survival through the activation of the p53-PUMA-BAX/BCL2 axis (Figure 6). Our study provides insights into the crucial role of HEATR1 in regulating ribosome biogenesis and p53-induced cell apoptosis in NSCLC. Further studies are needed to determine the therapeutic value of inhibiting HEATR1 for NSCLC therapy, and whether it has value as a potential biomarker for diagnosis or prognosis.
Figure 6.
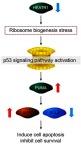
Proposed model of HEATR1-induced NSCLC cell apoptosis. HEATR1 inhibition could activate p53 signaling pathway via ribosome biogenesis stress, which subsequently leads to cell apoptosis and reduces cell survival through the activation of p53-PUMA-BAX/BCL2 axis.
Acknowledgments
The study was supported by grants from the National Natural Science Foundation of China (No. 81703791 and No. 81873178), Shanghai Municipal Commission of Health and Family Planning (No. 20174Y0044), Key Specialty Construction Project of Pudong Health and Family Planning Commission of Shanghai (No. PWZxk2017-06), Science and Technology Development Fund of Shanghai Pudong New Area (No. PKJ2017-Y14) and Talents Training Program of Seventh People's Hospital of Shanghai University of Traditional Chinese Medicine (No. XX2017-04 and No. XX2017-06).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
Supplementary materials


References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262 [DOI] [PubMed] [Google Scholar]
- 2.Jin D, Guo J, Wang D, et al. The antineoplastic drug metformin downregulates YAP by interfering with IRF-1 binding to the YAP promoter in NSCLC. EBioMedicine. 2018; 37:188–204. doi: 10.1016/j.ebiom.2018.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Wang C, Yin R, Dai J, et al. Whole-genome sequencing reveals genomic signatures associated with the inflammatory microenvironments in Chinese NSCLC patients. Nat Commun. 2018;9(1):2054. doi: 10.1038/s41467-018-04492-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Q, Ran P, Zhang X, et al. Downregulation of N-Acetylglucosaminyltransferase GCNT3 by miR-302b-3p Decreases Non-Small Cell Lung Cancer (NSCLC) cell proliferation, migration and invasion. Cell Physiol Biochem. 2018;50(3):987–1004. doi: 10.1159/000494482 [DOI] [PubMed] [Google Scholar]
- 5.Hurria A, Kris MG. Management of lung cancer in older adults. CA Cancer J Clin. 2003;53(6):325–341. [DOI] [PubMed] [Google Scholar]
- 6.Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell. 1999;96(1):99–110. [DOI] [PubMed] [Google Scholar]
- 7.Turi Z, Senkyrikova M, Mistrik M, Bartek J, Moudry P. Perturbation of RNA polymerase I transcription machinery by ablation of HEATR1 triggers the RPL5/RPL11-MDM2-p53 ribosome biogenesis stress checkpoint pathway in human cells. Cell Cycle. 2018;17(1):92–101. doi: 10.1080/15384101.2017.1403685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu ZB, Qiu C, Zhang AL, et al. Glioma-associated antigen HEATR1 induces functional cytotoxic T lymphocytes in patients with glioma. J Immunol Res. 2014;2014:131494. doi: 10.1155/2014/394127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu T, Fang Y, Zhang H, et al. HEATR1 negatively regulates akt to help sensitize pancreatic cancer cells to chemotherapy. Cancer Res. 2016;76(3):572–581. doi: 10.1158/0008-5472.CAN-15-0671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen J, Song G, An M, et al. The use of hollow mesoporous silica nanospheres to encapsulate bortezomib and improve efficacy for non-small cell lung cancer therapy. Biomaterials. 2014;35(1):316–326. doi: 10.1016/j.biomaterials.2013.09.098 [DOI] [PubMed] [Google Scholar]
- 11.Li C, Hu J, Li W, Song G, Shen J. Combined bortezomib-based chemotherapy and p53 gene therapy using hollow mesoporous silica nanospheres for p53 mutant non-small cell lung cancer treatment. Biomate Sci. 2016;5(1):77–88. doi: 10.1039/C6BM00449K [DOI] [PubMed] [Google Scholar]
- 12.Bursac S, Brdovcak MC, Donati G, Volarevic S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim Biophys Acta. 2014;1842(6):817–830. doi: 10.1016/j.bbadis.2013.08.014 [DOI] [PubMed] [Google Scholar]
- 13.Fumagalli S, Di Cara A, Neb-Gulati A, et al. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol. 2009;11(4):501–508. doi: 10.1038/ncb1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bursac S, Jurada D, Volarevic S. New insights into HEATR1 functions. Cell Cycle. 2018;17(2):143–144. doi: 10.1080/15384101.2017.1411325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gentilella A, Moron-Duran FD, Fuentes P, et al. Autogenous control of 5ʹTOP mRNA stability by 40S ribosomes. Mol Cell. 2017;67(1):55–70 e54. doi: 10.1016/j.molcel.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell. 2010;40(2):216–227. doi: 10.1016/j.molcel.2010.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Derenzini M, Montanaro L, Trere D. Ribosome biogenesis and cancer. Acta Histochem. 2017;119(3):190–197. doi: 10.1016/j.acthis.2017.01.009 [DOI] [PubMed] [Google Scholar]
- 18.Orsolic I, Jurada D, Pullen N, Oren M, Eliopoulos AG, Volarevic S. The relationship between the nucleolus and cancer: current evidence and emerging paradigms. Semin Cancer Biol. 2016;37–38:36–50. doi: 10.1016/j.semcancer.2015.12.004 [DOI] [PubMed] [Google Scholar]
- 19.Stepinski D. Nucleolus-derived mediators in oncogenic stress response and activation of p53-dependent pathways. Histochem Cell Biol. 2016;146(2):119–139. doi: 10.1007/s00418-016-1443-6 [DOI] [PubMed] [Google Scholar]
- 20.Nakhoul H, Ke J, Zhou X, Liao W, Zeng SX, Lu H. Ribosomopathies: mechanisms of disease. Clin Med Insights Blood Disord. 2014;7:7–16. doi: 10.4137/CMBD.S16952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Danilova N, Gazda HT. Ribosomopathies: how a common root can cause a tree of pathologies. Dis Model Mech. 2015;8(9):1013–1026. doi: 10.1242/dmm.020529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pelletier J, Thomas G, Volarevic S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer. 2018;18(1):51–63. doi: 10.1038/nrc.2017.104 [DOI] [PubMed] [Google Scholar]
- 23.Park SY, Jeong MS, Jang SB. In vitro binding properties of tumor suppressor p53 with PUMA and NOXA. Biochem Biophys Res Commun. 2012;420(2):350–356. doi: 10.1016/j.bbrc.2012.03.001 [DOI] [PubMed] [Google Scholar]
- 24.Li XQ, Yu Q, Chen FS, Tan WF, Zhang ZL, Ma H. Inhibiting aberrant p53-PUMA feedback loop activation attenuates ischaemia reperfusion-induced neuroapoptosis and neuroinflammation in rats by downregulating caspase 3 and the NF-kappaB cytokine pathway. J Neuroinflammation. 2018;15(1):250. doi: 10.1186/s12974-018-1220-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu T, Yuan Y, Xiao DJ. The clinical relationship between the slug-mediated Puma/p53 signaling pathway and radiotherapy resistance in nasopharyngeal carcinoma. Eur Rev Med Pharmacol Sci. 2017;21(5):953–958. [PubMed] [Google Scholar]
- 26.Peng SL, Yao DB, Zhao Y, et al. Prognostic value of PUMA expression in patients with HBV-related hepatocellular carcinoma. Eur Rev Med Pharmacol Sci. 2015;19(1):38–44. [PubMed] [Google Scholar]
- 27.Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science (New York, NY). 2005;309(5741):1732–1735. doi: 10.1126/science.1114297 [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Huang K, O’Neill KL, Pang X, Luo X. Bax/Bak activation in the absence of Bid, Bim, Puma, and p53. Cell Death Dis. 2016;7:e2266. doi: 10.1038/cddis.2016.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He K, Zheng X, Zhang L, Yu J. Hsp90 inhibitors promote p53-dependent apoptosis through PUMA and Bax. Mol Cancer Ther. 2013;12(11):2559–2568. doi: 10.1158/1535-7163.MCT-13-0284 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials