

Abstract
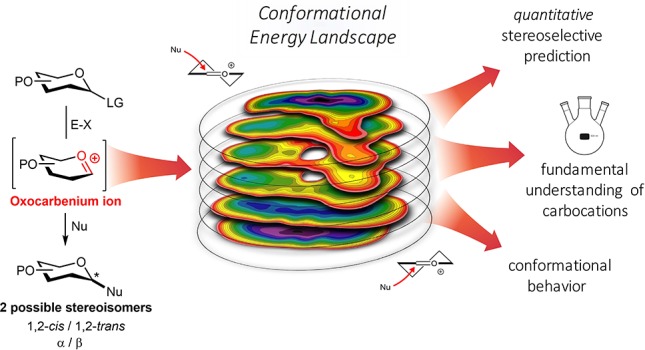
The broad application of well-defined synthetic oligosaccharides in glycobiology and glycobiotechnology is largely hampered by the lack of sufficient amounts of synthetic carbohydrate specimens. Insufficient knowledge of the glycosylation reaction mechanism thwarts the routine assembly of these materials. Glycosyl cations are key reactive intermediates in the glycosylation reaction, but their high reactivity and fleeting nature have precluded the determination of clear structure–reactivity-stereoselectivity principles for these species. We report a combined experimental and computational method that connects the stereoselectivity of oxocarbenium ions to the full ensemble of conformations these species can adopt, mapped in conformational energy landscapes (CEL), in a quantitative manner. The detailed description of stereoselective SN1-type glycosylation reactions firmly establishes glycosyl cations as true reaction intermediates and will enable the generation of new stereoselective glycosylation methodology.
Short abstract
The energy of all possible conformers of substituted 6-membered ring oxocarbenium ions has been mapped to account for the stereoselectivity in SN1-glycosylation reactions.
Carbohydrates play numerous roles in living organisms, as key players in energy housekeeping, as structural components, and as signaling molecules. To unravel the roles carbohydrates play in biological processes, well-defined, single molecules are indispensable, and organic synthesis has been one of the major suppliers for pure oligosaccharide specimens to fuel glycobiological and glycomedical research. Although significant progress has been made in the field, the generation of sufficient amounts of synthetic (complex) oligosaccharides remains a difficult and time-consuming undertaking.1−5 The main obstacle in the construction of oligosaccharides is the stereoselective construction of 1,2-cis-glycosidic linkages.6,7 While 1,2-trans linkages can be reliably installed using a neighboring group participation approach, there is no general solution for the construction of 1,2-cis linkages. Different reaction pathways can be followed during a glycosylation reaction, and these can lead to different diastereomeric products. Figure 1 depicts the current understanding of the continuum of mechanisms that is operational during a glycosylation reaction.8−10 The activation of a donor glycoside (I) leads to an array of reactive (electrophilic) intermediates (II–VIII), formed from the donor glycoside and the activator-derived counterion. In case a participating group is present at the C2 (such as an O-acyl function), these reactive intermediates are intramolecularly trapped to provide a relatively stable dioxolenium ion, that is stereoselectively substituted from the opposite side of the ring to deliver the 1,2-trans glycoside product. In the absence of a C2-participation functionality, the situation is more complex, and it has been proposed that both covalent reactive intermediates (II and VIII) and the reactive oxocarbenium ion (like) species (III–VII) can be the product-forming intermediates. The covalent intermediates on the SN2 side of the reaction mechanism continuum can be studied using low-temperature NMR techniques, and over the years, hundreds of reactive intermediates (triflates, oxosulfonium ions, among others) have been characterized.11−18 Substitution of these species with reactive nucleophiles (such as primary carbohydrate alcohols) defines the SN2 side of the reaction mechanism continuum. In contrast, the oxocarbenium ions on the SN1 side of the continuum remain ill understood, and the intermediacy of these species in glycosylation reactions is heavily debated.19−36 Because the lifetime of these intermediates in conventional reaction media is extremely short, there is currently no (spectroscopic) technique available to study these species in a direct manner and assess their behavior.37−39 It is clear that the substitution pattern on the carbohydrate ring plays an all-important role in determining the stability and reactivity of these species, but it has been impossible to establish clear structure–reactivity-stereoselectivity relationships because of the conformational freedom and short lifetime of these reactive intermediates in classical solutions. Thus, the course of SN1-type glycosylation can, at present, not be properly understood (let alone predicted), leaving a major gap in the mechanistic conception of glycosylation reactions.
Figure 1.

The reaction mechanism manifold operational during glycosylation reactions. Glycosylation reactions are best considered as taking place at a continuum between two formal extremes of the mechanisms, including the SN1 and SN2 mechanism. (I) Donor substrate; (II) reactive covalent α-intermediate; (III) contact ion pair, with the leaving group associated at the α-face; (IV) solvent-separated ion pair, with the leaving group that has departed from the α-face; (V) solvent-separated oxocarbenium ion; (VI) solvent-separated ion pair, with the leaving group that has departed from the β-face; (VII) contact ion pair, with the leaving group associated at the β-face; (VIII) reactive covalent β-intermediate; and (IX) addition product. P, protection group; E-X, promoter system; and Nu, nucleophile.
To investigate the stability and reactivity of glycosyl oxocarbenium ions as product forming intermediates in glycosylation reactions, we here report the development of a computational method that maps the stability of these species as a function of their overall shape. We show that the stereoselectivity of glycosylation reactions employing weak nucleophiles can be directly related to the conformational energy landscape (CEL) of the glycosyl oxocarbenium ions, as mapped in silico, and in doing so, define the SN1 side of the glycosylation reaction mechanism manifold. Direct spectroscopic evidence for the computed conformers is obtained by the generation of the oxocarbenium ions under superacid conditions, and it is here revealed that fully substituted glycopyranosyl oxocarbenium ions react in a highly stereoselective 1,2-cis manner.
We have mapped the energy of glycopyranosyl oxocarbenium ions as a function of their shape to understand the reactivity of these species following the strategy outlined in Figure 2. To generate the CEL maps, plotted on the Cremer–Pople sphere (a spherical representation describing all possible conformations a six-membered ring can adopt), we have generated a suite of conformations by scanning the three dihedral angles (C1–C2–C3–C4, C3–C4–C5–O5, and C5–O5–C1–C2) from −60° to 60° in 15° increments, to fill the complete conformation space (Figure 2,1). The geometry of all these conformers was optimized, and the associated energies were computed by utilizing DFT as the level of theory, B3LYP as hybrid functional,40 and 6-311G(d,p) as the basis set. Solvation of CH2Cl2 was taken into account using a polarizable continuum model, and energies are expressed in Gibbs free energy (for more information, see the Supporting Information).41 The energy landscapes were then generated by visualizing the relative energy in contour plots on “slices” of the pseudo rotational sphere.42
Figure 2.

Overview of the workflow to map the conformational and stereoselective preference of pyranosyl oxocarbenium ions. (1) The complete conformational space of a six-membered ring was scanned by computing 729 prefixed structures. A few canonical conformations (chair, half-chair, envelope, and boat) are depicted. (2) The associated energies were graphed on slices dividing the Cremer–Pople sphere. (3) Top- and bottom-face selective conformers lie in separate areas of the sphere. The family of the top face-selective (3E, 3H4, E4, and B2,5)-like structures is found in the area contoured with the red-dashed line, while the bottom face-selective family of (4E, 4H3, E3, and 2,5B)-like conformers is found on the opposite side of the sphere, grouped within the blue-dashed line. (4) On the basis of the Boltzmann distribution of the top- and bottom-face selective structures, the stereochemical outcome of nucleophilic addition reactions to pyranosyl oxocarbenium ions can be computed.
Inspection of the generated energy maps revealed that two families of structures are most relevant, the continuum of (3E, 3H4, E4, and B2,5)-like structures is grouped on the northwest side of the spheres, and these form an ensemble of structures that are preferentially attacked from the top-face. The “opposite” family of structures, located on the southeast side of the sphere, is composed of the range of (4E, 4H3, E3, and 2,5B)-like conformers, which will likely be approached by an incoming nucleophile from the bottom-face (see Figure 2,3 and the Supporting Information).20,43 The relative population of all conformational states can be calculated, on the basis of their relative energies as computed above, utilizing the Boltzmann equation (see the Supporting Information for more information). Accordingly, we determined the population of the top- and bottom-face selective families, which should be a measure for the relative stereoselectivity of addition reactions with weak nucleophiles to the glycosyl oxocarbenium ions.
To put this workflow to practice, we first investigated a set of 13 monosubstituted pyranosyl oxocarbenium ions differing by the nature of the substituent (BnO–, TBDPSO–, N3–, F–, Cl–, Br–, I–, PhS–, MeS–, and Me−) as well as the position on the ring. Their structures, the computed theoretical reaction stereoselectivity, and the experimentally determined stereoselectivity obtained in reactions with triethylsilane-d (TES-D)19,44−46 or allyl-trimethyl silane (AllylTMS) are summarized in Table 1 (entries 1–13). The CEL maps (see Figure 3a for three representative examples, all other CEL maps are provided in Figures S3–18) revealed that only a limited region of the full conformational space is accessible for the monosubstituted ions, in which local minima are found at both “poles”, centered around the 3H4- and the 4H3-like conformations. Depending on the nature of the substituents, one of these families is favored, placing the substituent either axially or equatorially. At the C4-position, electronegative substituents (BnO–, F–, TBDPSO–, N3–, Cl–, and Br−) favor an axial position to stabilize the oxocarbenium ion by through space electrostatic interactions, preferentially adopting the 4H3-like conformation.31,32,47−49 Decreasing electronegativity and increasing size of the substituent (I–, PhS–, MeS–, and Me−) translates to a preference to adopt an equatorial position (3H4-like conformations) to minimize steric interactions (Figure 3a). This trend is similar for substituents at the C3-position. An electronegative BnO-substituent at C-2 is preferentially placed in a pseudo-equatorial position, as this enables the hyperconjugative stabilization of the oxocarbenium ion by the pseudo-axial C2–H2 bond. When the population of the conformational families, as revealed in the CEL maps, are translated to a calculated stereoselectivity and compared to the stereoselectivity obtained in the experiments,31,32,50 it becomes apparent that there is an excellent agreement between theory and practice. Importantly, not only can highly stereoselective glycosylations be reliably predicted from the CEL maps, but also the condensation reactions that proceed with moderate selectivity (Table 1, entries 6, 7, and 13 for example) are accurately matched by the computed data.
Table 1. Computed and Experimentally Found Stereoselectivity for Glycosylation Reactions on Mono- and Multi-Substituted Pyranosyl Oxocarbenium Ions56.
| Entry | oxocarbenium ion | computed stereoselectivity (cis:trans)a | experimental stereoselectivity (cis:trans)a | yield (%) |
|---|---|---|---|---|
| 1 | 1 (4-OBn) | <2:98 | <2:98 | 75 |
| 2 | 2 (4-F) | <2:98 | 4:96 | 45 |
| 3 | 3 (4-OTBDPS) | 8:92 | 6:94 | 99 |
| 4 | 4 (4-N3) | 12:88 | 12:88 | 95 |
| 5 | 5 (4-Cl) | 10:90 | 14:86 | 90 |
| 6 | 6 (4-Br) | 32:68 | 29:71 | 87 |
| 7 | 7 (4-I) | 73:27 | 72:28 | 90 |
| 8 | 8 (4-SPh) | 81:19 | 78:22 | 87 |
| 9 | 9 (4-SMe) | 88:12 | 84:16 | 75 |
| 10 | 10 (4-Me) | 95:5 | 94:6 | 74 |
| 11 | 11 (3-OBn) | 90:10 | 92:8 | 95 |
| 12 | 12 (3-Me) | 4:96 | 3:97 | 41 |
| 13 | 13 (2-OBn) | 66:34 | 66:34 | 85 |
| 14 | 14 (d-lyxose) | >98:2 | >98:2 | 81 |
| 15 | 15 (d-arabinose) | >98:2 | >98:2 | 79 |
| 16 | 16 (d-xylose) | >98:2 | >98:2 | 86 |
| 17 | 17 (2-deoxy-d-xylose) | >98:2 | >98:2 | 74 |
| 18 | 18 (d-ribose) | >98:2 | >98:2 | 69 |
| 19 | 19 (l-fucose) | >98:2 | >98:2 | 74 |
| 20 | 20 (2-deoxy-l-fucose) | <2:98 | <2:98 | 89 |
| 21 | 21 (2-azido-l-fucose) | >98:2 | >98:2 | 65 |
| 22 | 22 (l-rhamnose) | >98:2 | >98:2 | 79 |
| 23 | 23 (2-deoxy-l-rhamnose) | 71:29 | 66:34 | 96 |
| 24 | 24 (d-glucose) | >98:2 | >98:2 | 70 |
| 25 | 25 (d-glucuronic acid) | >98:2 | >98:2 | 43 |
| 26 | 26 (2-deoxy-d-glucose) | 52:48 | 52:48 | 76 |
| 27 | 27 (2-azido-d-glucose) | >98:2 | >98:2 | 52 |
| 28 | 28 (d-mannose) | 97:3 | 97:3 | 93 |
| 29 | 29 (d-mannuronic-acid) | >98:2 | >98:2 | 76 |
| 30 | 30 (2-azido-d-mannuronic-acid) | >98:2 | >98:2 | 53 |
| 31 | 31 (d-galactose) | >98:2 | >98:2 | 86 |
| 32 | 32 (2-deoxy-d-galactose) | <2:98 | <2:98 | 91 |
For the monosubstituted pyranosides (entries 1–13), the cis:trans ratio is expressed as the relationship between the substituent and the coupled nucleophile. For the 2-deoxy-glycosides (entries 17, 20, 23, and 32), the cis:trans ratio is expressed as the relationship between the substituent on C-3 and the coupled nucleophile. For the other glycopyranosides (entries 14–16, 18–19, 21–22, and 27–31), the cis:trans ratio is expressed as the relationship between the substituent on C-2 and the coupled nucleophile. The names in the table relate to the carbohydrate studied. For the computational studies, per-O-methylated oxocarbenium ions are used, where the experimental glycosylation used per-O-benzylated substrates.57
Figure 3.

CEL maps of selected pyranosyl oxocarbenium ions in which the found local minima are indicated with their respective energy. (A) CEL map of monosubstituted-pyranosyl oxocarbenium ions 2, 3, and 7. (B) CEL map of multi-substituted-pyranosyl oxocarbenium ions 16, 19, 22–26, and 28–29.
Next, CEL maps of multisubstituted pyranosyl oxocarbenium ions were generated, and the theoretical stereoselectivity of these species were computed. The results of these studies are summarized in the second half of Table 1 (entries 14–32). A selection of CEL maps is depicted in Figure 3b (all CEL maps are provided in Figures S22–40). Table 1 also reports the experimental stereoselectivity and yield of the reactions of the thioglycoside donors, obtained by preactivation of the donors using the diphenyl sulfoxide (Ph2SO)/triflic anhydride (Tf2O) activator51 and TES-D as the nucleophile.52 Again, an excellent agreement is found for the calculated and experimentally obtained stereoselectivity. The stereoselectivity of all these condensation reactions can now be traced back to the families of low-energy conformers of the oxocarbenium ions, as revealed by the CEL maps. Some maps show a very localized energy minimum for a particular conformational family, such as the CEL map for the l-fucosyl oxocarbenium ion 19 (Figure 3b). In the most favorable 3H4-, 3E-, and E4-like conformations of this ion, the ring substituents at C2 and C4 take up an electronically favorable orientation, leading to the localized energy minimum around the 3H4-pole. Nucleophilic addition to these conformers stereoselectivity provides the 1,2-cis-linked products, and the generated CEL map thus provides an explanation for the high 1,2-cis-selectivity generally observed with fucosyl donors.53−55
Similarly, the mannosyl oxocarbenium ion 28 can place its C2, C3, and C4 substituents in stabilizing positions when adopting a 3H4/3E-like structure (Figure 3b), as alluded to by Woerpel and co-workers.24 These structures are selectively substituted from the top face to provide the mannosyl product, a result that is indeed born out in the glycosylation experiment (Table 1, entry 28). Glycosylations of mannuronic acid ester 29 proceed with exceptional 1,2-cis stereoselectivity, and the generated CEL map (Figure 3b) provides an adequate explanation for this reaction outcome, as a very localized energy minimum is determined for the 3H4-like conformational family. The additional stabilization from the axial C5-CO2Me in 29 with respect to the axial C5-CH2OMe group in the mannosyl oxocarbenium ion (28) becomes very clear from the comparison of the CEL maps of 28 and 29.
The CEL maps of pyranosyl oxocarbenium ions bearing substituents that have “conflicting positional interests” reveal that noncanonical conformations can become important, and that broader conformational families or families around the different poles can become equally relevant. For example, the d-xylosyl oxocarbenium ion 16 preferentially adopts a noncanonical flattened (skew)-boat-like structure (see Figure 3b). The CEL map for the 2-deoxy-l-rhamnose ion 23 reveals two conformational families of similar energy, leading to a mixture of α- and β-products in the condensation reaction (Table 1, entry 23). The CEL maps in the gluco-series illustrate how point mutations in the structure of the parent donor translate to differently shaped oxocarbenium ions and a different stereochemical outcome in the glycosylation reactions. The glucopyranosyl cation 24 is most stable when adopting a 4H3/E4-like shape, while its glucuronic acid counterpart (25), bearing a C5-carboxylic acid ester, prefers to adopt a structure in between the E4 and 2SO-conformations. Both ions are preferentially attacked from the bottom-face to selectively provide the α-product (Table 1, entries 24 and 25). For 2-deoxyglucose 26, two families of oxocarbenium ion conformers are equally stable, and the population of 4H3/4E-like and 3H4/E4-like states leads to an unselective addition reaction, leading to the formation of α- and β-products in almost equal amounts.
Overall, there is an excellent agreement between the calculated and experimentally established α/β-selectivity of the multi-substituted glycosides, providing very compelling evidence for (families of) glycopyranosyl oxocarbenium ion conformers as product-forming intermediates in the substitution reactions; thereby defining the SN1 side of the glycosylation reaction mechanism manifold.
To obtain direct experimental evidence for the conformations computed using our CEL mapping method, we studied two 2-deoxy diacetylated oxocarbenium ions derived from l-fucose 33 and l-rhamnose 34 in “non-nucleophilic” superacidic media (Figure 4a).37 The choice of the acetyl group and 2-deoxy position is guided by the fact that methoxy groups are prone to elimination, and so far, C-2 substituents able to provide the corresponding glycosyl cation are limited. As the acetyl groups at C-3 and C-4 of the oxocarbenium ions generated from donors 33 and 34 will be protonated under the superacid conditions used, polycationic oxocarbenium ions 35 and 36 were subjected to the CEL mapping method. The CEL map for 2-deoxy-fucosyl oxocarbenium ion 35 (Figure 4c) shows a strong preference for the 3H4 and closely related E4 conformations. The CEL map for the 2-deoxy-rhamnosyl oxocarbenium ion 36, on the other hand, features multiple local minima, and both the 3H4 and the 4H3 family are relatively low in energy, resulting in a conformational mixture in solution. In parallel, 2-deoxy-l-fucose and 2-deoxy-l-rhamnose acetates 33 and 34 were dissolved in HF/SbF5 to generate the polycationic structures 35 and 36, of which the NMR spectra (Figure 4b) clearly indicated the presence of an oxocarbenium ion as the main species (carboxonium signal 35: δC1 = 223.4 ppm and δH1 = 8.76 ppm; 36: δC1 = 224.2 ppm and δH1 = 8.84 ppm).37,58 Both ester groups were indeed protonated, as revealed by the presence of two proton singlets at δH = 13.27 and 13.35 ppm. Because of the sufficient lifespan of 35 and 36 in the superacid media, full conformational characterization of these species could be performed (see the SI for more information). The coupling constants of the ring protons of 35 indicate that it adopts a stable 3H4-like conformation. The NMR spectrum of 2-deoxy-l-rhamnosyl oxocarbenium 36 (Figure 4d), on the other hand, showed significant line broadening as a result of conformational flexibility of this species. Using the relevant conformations, obtained from the CEL maps for these ions, we reconstituted the NMR spectrum using the Boltzmann weighted averaged coupling constants of ions 35 and 36. Perfect agreement between the experimental NMR spectra for these peracetylated polyprotonated glycosyl cations and their simulated spectra show that conformational dynamics of these ions are well captured by the CEL mapping method.
Figure 4.

Generation and NMR spectra of 2-deoxy-pyranosyl oxocarbenium ions in HF/SbF5 at −40 °C. (A) Generation of ion 35 and 36 in HF/SbF5. (B) Experimental 1H- and 13C DEPT NMR of 2-deoxy-l-rhamnose oxocarbenium 34. (C) The generated 1H NMR spectrum of the oxocarbenium 35 compared to the simulated spectrum based on the computed CEL. (D) The generated 1H NMR spectrum of the oxocarbenium 36 compared to the simulated spectrum based on the computed CEL.
In conclusion, we have benchmarked the SN1 side of the glycosylation reaction mechanism. The stability, reactivity, and conformational mobility of glycosyl oxocarbenium ions can be fully understood by mapping the complete conformational energy landscape of these ions, and the preference of the cations can be directly related to the experimental stereochemical outcome of addition reactions to these. The maps show, in detail, how the stereoelectronic effects of various ring substituents (halogens, chalcogens, azides, and carbon-based substituents) determine the overall shape of the cations and thereby the stereochemical course of the reactions. In addition, the simulated NMR spectra of selected ions, reconstituted by using the Boltzmann weighted averaged coupling constants determined by the CEL mapping method, perfectly fit with the experimental ones observed by low-temperature NMR in superacid. Where glycosyl oxocarbenium ions were previously thought to be at the basis of nonselective coupling reactions because of their high reactivity, we here show that these species, including the ions derived from l-fucose, l-rhamnose, d-glucose, d-mannose, and d-galactose, have an intrinsic preference to generate the challenging 1,2-cis-linkages. This will enable the stereoselective synthesis of C-glycosides and open up new avenues to develop stereoselective O-glycosylation reactions.59 The mechanistic insight offered here will be instrumental in the interpretation of future glycosylation results and serve as the basis to further explore the glycosylation reaction mechanism. The uncovered stereoelectronic substituent effects will be relevant in many other transformations, involving carbocationic intermediates, and the strategy that we developed to grasp the full conformational space of these flexible intermediates can be a blueprint for the study of other flexible reactive intermediates.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00042.
Experimental and computational procedures, CEL maps of all studied ions, construction of CEL maps, additional experiments with methyl ether-protected substrates, different solvent systems and donor types, coordinates of all most relevant low energy oxocarbenium ion conformers, and NMR spectra of all new and selected compounds (PDF)
Author Contributions
T.H. and J.D.C.C. conceived the project, designed the experiments, and composed the manuscript. T.H. generated the CEL maps and analyzed the data. T.H. carried out the donor synthesis and the coupling experiments. W.A.R., S.V., and D.P.A.W. contributed to the donor synthesis. Y.B. and S.T. designed the superacid experiments. L.L and A.M. performed all superacid experiments. All authors commented on the manuscript.
Computing resources have been provided by BigGrid.nl and SURFsara. We thank the European Research Council (ERC-CoG-726072-“GLYCONTROL”, to J.D.C.C.) and EP Nuffic (Van Gogh grant to T.H.) for financial support. We also thank the CNRS, the University of Poitiers, the National Research Agency (ANR project SWEETCAT), and Campus France (PHC Van Gogh 2017) for financial support to L.L., A.M., J.D., Y.B., and S.T.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhu X.; Schmidt R. R. New Principles for Glycoside-Bond Formation. Angew. Chem., Int. Ed. 2009, 48 (11), 1900–1934. 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- Seeberger P. H. The Logic of Automated Glycan Assembly. Acc. Chem. Res. 2015, 48 (5), 1450–1463. 10.1021/ar5004362. [DOI] [PubMed] [Google Scholar]
- Wang C. C.; Lee J. C.; Luo S. Y.; Kulkarni S. S.; Huang Y. W.; Lee C. C.; Chang K. L.; Hung S. C. Regioselective One-Pot Protection of Carbohydrates. Nature 2007, 446 (7138), 896–899. 10.1038/nature05730. [DOI] [PubMed] [Google Scholar]
- Leng W.; Yao H.; He J.; Liu X. Venturing beyond Donor-Controlled Glycosylation: New Perspectives toward Anomeric Selectivity. Acc. Chem. Res. 2018, 51 (3), 628–639. 10.1021/acs.accounts.7b00449. [DOI] [PubMed] [Google Scholar]
- Nigudkar S. S.; Demchenko A. V. Stereocontrolled 1,2-Cis Glycosylation as the Driving Force of Progress in Synthetic Carbohydrate Chemistry. Chem. Sci. 2015, 6 (5), 2687–2704. 10.1039/C5SC00280J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demchenko A. V.Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Wiley, 2008. [Google Scholar]
- Bennett C. S.Selective Glycosylations with Deoxy Sugars. In Selective Glycosylations: Synthetic Methods and Catalysts; Bennett C. S., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA, 2017; pp 277–295. [Google Scholar]
- Bohé L.; Crich D. A Propos of Glycosyl Cations and the Mechanism of Chemical Glycosylation. C. R. Chim. 2011, 14 (1), 3–16. 10.1016/j.crci.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crich D. Mechanism of a Chemical Glycosylation Reaction. Acc. Chem. Res. 2010, 43 (8), 1144–1153. 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- Adero P. O.; Amarasekara H.; Wen P.; Bohé L.; Crich D. The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1–SN2 Interface. Chem. Rev. 2018, 118 (17), 8242–8284. 10.1021/acs.chemrev.8b00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vorm S.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. Stereoselectivity of Conformationally Restricted Glucosazide Donors. J. Org. Chem. 2017, 82 (9), 4793–4811. 10.1021/acs.joc.7b00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frihed T. G.; Bols M.; Pedersen C. M. Mechanisms of Glycosylation Reactions Studied by Low-Temperature Nuclear Magnetic Resonance. Chem. Rev. 2015, 115 (11), 4963–5013. 10.1021/cr500434x. [DOI] [PubMed] [Google Scholar]
- Frihed T. G.; Walvoort M. T. C.; Codée J. D. C.; van der Marel G. A.; Bols M.; Pedersen C. M. Influence of O6 in Mannosylations Using Benzylidene Protected Donors: Stereoelectronic or Conformational Effects?. J. Org. Chem. 2013, 78 (6), 2191–2205. 10.1021/jo302455d. [DOI] [PubMed] [Google Scholar]
- Kaeothip S.; Yasomanee J. P.; Demchenko A. V. Glycosidation of Thioglycosides in the Presence of Bromine: Mechanism, Reactivity, and Stereoselectivity. J. Org. Chem. 2012, 77 (1), 291–299. 10.1021/jo2019174. [DOI] [PubMed] [Google Scholar]
- Walvoort M. T. C.; Lodder G.; Mazurek J.; Overkleeft H. S.; Codée J. D. C.; van der Marel G. A. Equatorial Anomeric Triflates from Mannuronic Acid Esters. J. Am. Chem. Soc. 2009, 131 (34), 12080–12081. 10.1021/ja905008p. [DOI] [PubMed] [Google Scholar]
- Liu J.; Gin D. Y. C2-Amidoglycosylation. Scope and Mechanism of Nitrogen Transfer. J. Am. Chem. Soc. 2002, 124 (33), 9789–9797. 10.1021/ja026281n. [DOI] [PubMed] [Google Scholar]
- Garcia B. A.; Gin D. Y. Dehydrative Glycosylation with Activated Diphenyl Sulfonium Reagents. Scope, Mode of C(1)-Hemiacetal Activation, and Detection of Reactive Glycosyl Intermediates. J. Am. Chem. Soc. 2000, 122 (18), 4269–4279. 10.1021/ja993595a. [DOI] [Google Scholar]
- Crich D.; Sun S. Are Glycosyl Triflates Intermediates in the Sulfoxide Glycosylation Method? A Chemical and 1 H, 13C, and 19F NMR Spectroscopic Investigation. J. Am. Chem. Soc. 1997, 119 (46), 11217–11223. 10.1021/ja971239r. [DOI] [Google Scholar]
- van der Vorm S.; Hansen T.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. The Influence of Acceptor Nucleophilicity on the Glycosylation Reaction Mechanism. Chem. Sci. 2017, 8 (3), 1867–1875. 10.1039/C6SC04638J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moumé-Pymbock M.; Crich D. Stereoselective C-Glycoside Formation with 2-O-Benzyl-4,6-O-Benzylidene Protected 3-Deoxy Gluco- and Mannopyranoside Donors: Comparison with O-Glycoside Formation. J. Org. Chem. 2012, 77 (20), 8905–8912. 10.1021/jo3011655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codée J. D. C.; Walvoort M. T. C.; de Jong A. R.; Lodder G.; Overkleeft H. S.; van der Marel G. A. Mannuronic Acids: Reactivity and Selectivity. J. Carbohydr. Chem. 2011, 30 (7–9), 438–457. 10.1080/07328303.2011.624284. [DOI] [Google Scholar]
- Beaver M. G.; Woerpel K. A. Erosion of Stereochemical Control with Increasing Nucleophilicity: O-Glycosylation at the Diffusion Limit. J. Org. Chem. 2010, 75 (4), 1107–1118. 10.1021/jo902222a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walvoort M. T. C.; Dinkelaar J.; van den Bos L. J.; Lodder G.; Overkleeft H. S.; Codée J. D. C.; van der Marel G. A. The Impact of Oxacarbenium Ion Conformers on the Stereochemical Outcome of Glycosylations. Carbohydr. Res. 2010, 345 (10), 1252–1263. 10.1016/j.carres.2010.02.027. [DOI] [PubMed] [Google Scholar]
- Lucero C. G.; Woerpel K. A. Stereoselective C-Glycosylation Reactions of Pyranoses: The Conformational Preference and Reactions of the Mannosyl Cation. J. Org. Chem. 2006, 71 (7), 2641–2647. 10.1021/jo0522963. [DOI] [PubMed] [Google Scholar]
- Crich D.; Vinogradova O. On the Influence of the C2–O2 and C3–O3 Bonds in 4,6-O-Benzylidene-Directed β-Mannopyranosylation and α-Glucopyranosylation. J. Org. Chem. 2006, 71 (22), 8473–8480. 10.1021/jo061417b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumper J. R.; Salamant W. A.; Woerpel K. A. Continuum of Mechanisms for Nucleophilic Substitutions of Cyclic Acetals. Org. Lett. 2008, 10 (21), 4907–4910. 10.1021/ol8019956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Kawatkar S.; Rao Y.; Boons G. J. Practical Approach for the Stereoselective Introduction of β-Arabinofuranosides. J. Am. Chem. Soc. 2006, 128 (36), 11948–11957. 10.1021/ja0629817. [DOI] [PubMed] [Google Scholar]
- Ishiwata A.; Akao H.; Ito Y. Stereoselective Synthesis of a Fragment of Mycobacterial Arabinan. Org. Lett. 2006, 8 (24), 5525–5528. 10.1021/ol062198j. [DOI] [PubMed] [Google Scholar]
- Nukada T.; Bérces A.; Wang L.; Zgierski M. Z.; Whitfield D. M. The Two-Conformer Hypothesis: 2,3,4,6-Tetra-O-Methyl-Mannopyranosyl and -Glucopyranosyl Oxacarbenium Ions. Carbohydr. Res. 2005, 340 (5), 841–852. 10.1016/j.carres.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Larsen C. H.; Ridgway B. H.; Shaw J. T.; Smith D. M.; Woerpel K. A. Stereoselective C-Glycosylation Reactions of Ribose Derivatives: Electronic Effects of Five-Membered Ring Oxocarbenium Ions. J. Am. Chem. Soc. 2005, 127 (31), 10879–10884. 10.1021/ja0524043. [DOI] [PubMed] [Google Scholar]
- Ayala L.; Lucero C. G.; Romero J. A. C.; Tabacco S. A.; Woerpel K. A. Stereochemistry of Nucleophilic Substitution Reactions Depending upon Substituent: Evidence for Electrostatic Stabilization of Pseudoaxial Conformers of Oxocarbenium Ions by Heteroatom Substituents. J. Am. Chem. Soc. 2003, 125 (50), 15521–15528. 10.1021/ja037935a. [DOI] [PubMed] [Google Scholar]
- Romero J. A. C.; Tabacco S. A.; Woerpel K. A. Stereochemical Reversal of Nucleophilic Substitution Reactions Depending upon Substituent: Reactions of Heteroatom-Substituted Six-Membered-Ring Oxocarbenium Ions through Pseudoaxial Conformers. J. Am. Chem. Soc. 2000, 122 (1), 168–169. 10.1021/ja993366o. [DOI] [PubMed] [Google Scholar]
- Parent J. F.; Deslongchamps P. Bent Bonds (τ) and the Antiperiplanar Hypothesis, and the Reactivity at the Anomeric Center in Pyranosides. Org. Biomol. Chem. 2016, 14 (47), 11183–11198. 10.1039/C6OB02263D. [DOI] [PubMed] [Google Scholar]
- Ardèvol A.; Rovira C. The Molecular Mechanism of Enzymatic Glycosyl Transfer with Retention of Configuration: Evidence for a Short-Lived Oxocarbenium-Like Species. Angew. Chem., Int. Ed. 2011, 50 (46), 10897–10901. 10.1002/anie.201104623. [DOI] [PubMed] [Google Scholar]
- Iglesias-Fernández J.; Hancock S. M.; Lee S. S.; Khan M.; Kirkpatrick J.; Oldham N. J.; McAuley K.; Fordham-Skelton A.; Rovira C.; Davis B. G. A Front-Face “SNi Synthase” Engineered from a Retaining “Double-SN2” Hydrolase. Nat. Chem. Biol. 2017, 13 (8), 874–881. 10.1038/nchembio.2394. [DOI] [PubMed] [Google Scholar]
- Beenakker T. J. M.; Wander D. P. A.; Offen W. A.; Artola M.; Raich L.; Ferraz M. J.; Li K.-Y.; Houben J. H. P. M.; van Rijssel E. R.; Hansen T.; et al. Carba-Cyclophellitols Are Neutral Retaining-Glucosidase Inhibitors. J. Am. Chem. Soc. 2017, 139 (19), 6534–6537. 10.1021/jacs.7b01773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A.; Arda A.; Désiré J.; Martin-Mingot A.; Probst N.; Sinaÿ P.; Jiménez-Barbero J.; Thibaudeau S.; Blériot Y. Catching Elusive Glycosyl Cations in a Condensed Phase with HF/SbF5 Superacid. Nat. Chem. 2016, 8 (2), 186–191. 10.1038/nchem.2399. [DOI] [PubMed] [Google Scholar]
- Elferink H.; Severijnen M. E.; Martens J.; Mensink R. A.; Berden G.; Oomens J.; Rutjes F. P. J. T.; Rijs A. M.; Boltje T. J. Direct Experimental Characterization of Glycosyl Cations by Infrared Ion Spectroscopy. J. Am. Chem. Soc. 2018, 140 (19), 6034–6038. 10.1021/jacs.8b01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha E.; Marianski M.; Xu F. F.; Thomas D. A.; Meijer G.; von Helden G.; Seeberger P. H.; Pagel K. Unravelling the Structure of Glycosyl Cations via Cold-Ion Infrared Spectroscopy. Nat. Commun. 2018, 9 (1), 4174. 10.1038/s41467-018-06764-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novel corrections and functionals, including B3LYP-D3 and ωB97XD, were also used and showed comparable results (see the SI for full details).
- The effect of the solvent on the outcome of the addition reactions to the fuco- and rhamno-configured oxocarbenium ions 19 and 22, respectively, has been investigated by performing the reactions in Et2O and MeCN. The outcome of these reactions was similar to the outcome of the reactions in DCM, with the 1,2-cis-addition products being formed as the sole anomer. These results are in line with the structures of the intermediate oxocarbenium ions, as revealed by the CEL maps of these ions in the respective solvents (see the SI for full details).
- For ease of visualization, the Cremer–Pople globe is turned 180° with respect to its common representation, and both poles (the 4C1 and 1C4 structures) are omitted as these conformations are very high in energy.
- Deslongchamps P.; Dory Y. L.; Li S. 1994 R.U. Lemieux Award Lecture Hydrolysis of Acetals and Ketals. Position of Transition States along the Reaction Coordinates, and Stereoelectronic Effects. Can. J. Chem. 1994, 72 (10), 2021–2027. 10.1139/v94-258. [DOI] [Google Scholar]
- van Rijssel E. R.; van Delft P.; Lodder G.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V.; Codée J. D. C. Furanosyl Oxocarbenium Ion Stability and Stereoselectivity. Angew. Chem., Int. Ed. 2014, 53 (39), 10381–10385. 10.1002/anie.201405477. [DOI] [PubMed] [Google Scholar]
- van Rijssel E. R.; van Delft P.; van Marle D. V.; Bijvoets S. M.; Lodder G.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V.; Codée J. D. C. Stereoselectivity in the Lewis Acid Mediated Reduction of Ketofuranoses. J. Org. Chem. 2015, 80 (9), 4553–4565. 10.1021/acs.joc.5b00419. [DOI] [PubMed] [Google Scholar]
- Madern J. M.; Hansen T.; van Rijssel E. R.; Kistemaker H. A. V.; van der Vorm S.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V.; Codée J. D. C. Synthesis, Reactivity, and Stereoselectivity of 4-Thiofuranosides. J. Org. Chem. 2019, 84 (3), 1218–1227. 10.1021/acs.joc.8b02536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen H. H.; Lyngbye L.; Jensen A.; Bols M. Stereoelectronic Substituent Effects in Polyhydroxylated Piperidines and Hexahydropyridazines. Chem. - Eur. J. 2002, 8 (5), 1218–1226. . [DOI] [PubMed] [Google Scholar]
- Jensen H. H.; Bols M. Stereoelectronic Substituent Effects. Acc. Chem. Res. 2006, 39 (4), 259–265. 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]
- Bucher C.; Gilmour R. Fluorine-Directed Glycosylation. Angew. Chem., Int. Ed. 2010, 49 (46), 8724–8728. 10.1002/anie.201004467. [DOI] [PubMed] [Google Scholar]
- Beaver M. G.; Billings S. B.; Woerpel K. A. C-Glycosylation Reactions of Sulfur-Substituted Glycosyl Donors: Evidence against the Role of Neighboring-Group Participation. J. Am. Chem. Soc. 2008, 130 (6), 2082–2086. 10.1021/ja0767783. [DOI] [PubMed] [Google Scholar]
- Codée J. D. C.; Litjens R. E. J. N.; den Heeten R.; Overkleeft H. S.; van Boom J. H.; van der Marel G. A. Ph2SO/Tf2O: A Powerful Promotor System in Chemoselective Glycosylations Using Thioglycosides. Org. Lett. 2003, 5 (9), 1519–1522. 10.1021/ol034312t. [DOI] [PubMed] [Google Scholar]
- Addition reactions, in which l-fucose, l-rhamnose, and d-glucose N-phenyl trifluoroacetimidate donors were used in conjunction with a catalytic amount of TMSOTf (0.1 equiv.) and TES-D, proceeded with a similar stereochemical outcome (see the SI for full details).
- Hagen B.; Ali S.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. Mapping the Reactivity and Selectivity of 2-Azidofucosyl Donors for the Assembly of N-Acetylfucosamine-Containing Bacterial Oligosaccharides. J. Org. Chem. 2017, 82 (2), 848–868. 10.1021/acs.joc.6b02593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemineau M.; Auzanneau F. I. Challenging Deprotection Steps During the Synthesis of Tetra- and Pentasaccharide Fragments of the LeaLex Tumor-Associated Hexasaccharide Antigen. J. Org. Chem. 2012, 77 (20), 8864–8878. 10.1021/jo301644w. [DOI] [PubMed] [Google Scholar]
- Mong K. K. T.; Wong C. H. Reactivity-Based One-Pot Synthesis of a Lewis Y Carbohydrate Hapten: A Colon–Rectal Cancer Antigen Determinant. Angew. Chem. 2002, 114 (21), 4261–4264. . [DOI] [PubMed] [Google Scholar]
- The experimental data of the model glycosylations of the monosubstituted pyranosyl donors were obtained from the work of Woerpel and co-workers (experimental conditions: allyltrimethylsilane (4.0 equiv), Lewis acid (BF3·OEt2 or SnBr4; 1.0 equiv), pyranosyl-1-O-acetyl donor in DCM (0.15 M), −78 °C to room temperature.). The model glycosylations of the multisubstituted pyranosyl donors were done with preactivation conditions (experimental conditions: preactivation conditions; TES-D (2 equiv), Tf2O (1.3 equiv), Ph2SO (1.3 equiv), TTBP (2.5 equiv), pyranosyl-1-S-thiophenyl donors, DCM (0.05 M), −80 to −60 °C, 96 h.).
- To keep the calculation time manageable, large protection groups (BnO−) were substituted with electronic comparable smaller groups (MeO−) (see the SI for full details).
- Olah G. A.; Parker D. G.; Yoneda N.; Pelizza F. Oxyfunctionalization of Hydrocarbons. 1. Protolytic Cleavage-Rearrangement Reactions of Tertiary Alkyl Hydroperoxides with Magic Acid. J. Am. Chem. Soc. 1976, 98 (8), 2245–2250. 10.1021/ja00424a038. [DOI] [Google Scholar]
- van der Vorm S.; van Hengst J. M. A.; Bakker M.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. Mapping the Relationship between Glycosyl Acceptor Reactivity and Glycosylation Stereoselectivity. Angew. Chem., Int. Ed. 2018, 57 (27), 8240–8244. 10.1002/anie.201802899. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.