

Abstract
Data from a large autopsy series were analyzed to address questions pertinent to primary age- related tauopathy (PART) and Alzheimer’s disease (AD): what factors are associated with increased severity of neurofibrillary degeneration in brains that lack neuritic amyloid plaques?; is there an association between Apolipoprotein E (APOE) alleles and PART pathologic severity independent of Aβ deposits?; and, how do the stains used to detect AD-type lesions impact the experimental results? Neuropathologic data were evaluated from elderly research volunteers whose brain autopsies were performed at University of Kentucky Alzheimer’s Disease Center (UK-ADC; N=145 subjects). All of the included subjects’ brains lacked neuritic amyloid plaques according to the CERAD diagnostic criteria and the overall final MMSE score before death was 26.8±4.6. The study incorporated evaluation of tissue with both silver histochemical stains and immunohistochemical stains to compare results; the immunohistochemical stains (Aβ and phospho-Tau) were scanned and quantified using digital pathologic methods. Immunohistochemical stains provided important advantages over histochemical stains due to sensitivity and detectability via digital methods. Even when AD-type pathology was in its presumed earliest phases, lacking detected neuritic amyloid plaques, neocortical parenchymal Aβ deposits were associated with increased medial temporal lobe neurofibrillary tangles (NFTs). The observation confirms the NIA-AA recommendation that even diffuse Aβ deposits signal AD pathobiologic mechanisms are occurring. Further, the data are most compatible with the hypothesis that the APOE ε4 allele exerts its effect via driving Aβ deposition, i.e. “upstream” AD-type mechanisms, rather than being associated directly with the development of tau NFTs or PART pathology.
Keywords: neuropathology, SNAP, hippocampus, Abeta, ScanScope, MAPT, aging, A-beta
Introduction
There is increasing appreciation of the complex nature of brain pathologies seen in aged humans. When evaluating brains that would previously have been designated “pure” Alzheimer’s disease (AD) based on pathologic features, neuropathologists are currently recognizing the presence of additional, often comorbid, pathologies [1, 2]. Primary age-related tauopathy (PART) is a relatively recently recognized pathologic phenomenon that is common in old age [3]. In brains with Definite PART pathology, amyloid plaques are not detected, but tau neurofibrillary tangles (NFTs) are observed (for a more textured description, see below). The NFTs of PART are not associated with underlying frontotemporal lobar degeneration (FTLD) or chronic traumatic encephalopathy [3]. The paper that presented PART diagnostic features [3] states that using Aβ immunohistochemistry to detect the amyloid plaques is preferable, but leaves open the possibility that “laboratories using the CERAD neuritic plaque density scores may classify subjects with neuritic plaque frequency of ‘None’ as ‘Definite’ [PART]”.
The presence of age-related tauopathy in the absence of substantial Aβ amyloid plaques has long been appreciated [4–6], but we are now far more aware of the pathology’s prevalence and public health impact. For example, hippocampal NFTs are practically universal in the 20– 30% of centenarians whose brains lack detectable Aβ amyloid plaques [7, 8]. There is inter- individual variation in the severity of PART-type pathology [3], but the mechanism(s) responsible for this variation are mostly unknown. In large autopsy series, the distribution of PART pathology has been shown to be associated with antemortem cognitive impairment [3, 9], and PART also is implicated in the clinical conditions of subjective memory complaints [10], and Mild Cognitive Impairment [11]. There also is abundant evidence of PART (medial temporal lobe tauopathy lacking Aβ amyloidogenesis) in living individuals, according to biomarker studies [12, 13]. Overall, since its publication in 2014, more than 300 papers in peer- reviewed journals have cited the paper describing PART [3].
Although it has become a recognized disease entity, PART has generated some controversy among experts in the field, and divergent opinions have been expressed [14–16]. There remains much unknown about the disease itself, including how it can best be operationalized for research [17, 18], and how we should define the ‘border zones’ between AD and PART, in theory and in diagnostic practice. Consideration of these unresolved challenges may yield new insights that are fundamental to both PART and AD. Before addressing them, we acknowledge the lack of universally applied terminology to refer to “amyloid plaques” of AD. Here (as in our prior work; see Refs [1, 19, 20]), we make a distinction between “neuritic” amyloid plaques, which include a dense amyloidogenic core and/or surrounding degenerating neurites that can be detected with silver stains, and “diffuse” amyloid plaques, that contain Aβ but lack the core and/or degenerating neurites. Terms such as “amyloid plaque” or “Aβ plaque” would encompass both of these entities.
A study of cases that lack neuritic amyloid plaques may thus provide a basis to address unresolved, and interrelated, questions pertaining to tau/NFT and Aβ/plaque pathologies. Two general questions are: what factors are associated with increased severity of neurofibrillary degeneration in brains that lack abundant Aβ?; and, when evaluating hypotheses related to PART, how do the neuropathologist’s choices of stains used in detecting the brain lesions affect his/her conclusions? More specific topical questions are: 1. Is it acceptable to define PART according to negativity for “neuritic amyloid plaques” (which tend to be detected using a variety of different means including histochemical techniques), or is it necessary to rely on negativity for Aβ immunohistochemically, as commonly operationalized via Thal Aβ stages [21]?; 2. Does the burden of tau NFTs in medial temporal lobe structures, in the absence of any detected neuritic amyloid plaques, have any association with the amount of detected “diffuse” Aβ deposits?; and, 3. In the absence of detected neuritic amyloid plaques, is there any association between APOE genotype and the severity of PART pathology that is independent of detected Aβ deposits?
The goals of the present study were to address the abovementioned questions using analyses of data gathered from research volunteers followed to autopsy. Clinical and neuropathological data were drawn from the University of Kentucky Alzheimer’s Disease Center (UK-ADC) cohort. The results of these analyses indicated that even when AD-type pathology is in its presumed earliest stages, with topographically limited Aβ deposits but lacking detected neuritic plaques, the Aβ deposits were still associated with more severe medial lobe tau NFTs. The data are also compatible with the hypothesis that APOE-ε4 alleles drive Aβ deposition rather than directly being associated with the severity of NFT pathology in the medial temporal lobe.
Methods
Details of UK-ADC research volunteers’ recruitment, inclusion/exclusion criteria, and clinical and pathological assessments have been described previously [1, 22, 23]. Briefly, community-based older adult volunteers agreed to be followed annually for cognitive, physical, and neurological examination and to donate their brain at the time of death. Protocols were approved by the UK Institutional Review Board, and all participants provided written informed consent. Included subjects were ≥70 years of age at death and had no detected neuritic plaques (NPs) – i.e., the CERAD score [24] was “none” – and Braak NFT stages [7] of 0 to IV. Research subjects with relatively rare dementia syndromes (e.g. prions, trinucleotide repeat diseases, or FTLD), or any brain tumor were excluded. Additionally, since our research questions focused on the association between APOE and tau pathology, cases also had to have APOE genotyping available.
Cases meeting inclusion and exclusion criteria formed two groups based on the year of autopsy, which corresponded to distinct neuropathological assessment methods used for evaluation of Aβ pathology. The first group comprised cases autopsied between 1997–2011 (n=98), where Bielschowsky silver stains were performed to detect NPs and diffuse plaques (DPs), and Gallyas silver stains [25] were used to detect NFTs in medial temporal lobe structures, in accordance with the 1997 NIA-Reagan Institute consensus-based criteria for AD diagnosis [26]. The second group comprised cases autopsied between 2012–2017 (n=51), during which time immunohistochemical (IHC) stains were performed to detect Aβ deposits and phospho-Tau antibody used to detect NPs and NFTs (in accordance with the 2012 NIA-AA consensus-based criteria for diagnosis of AD neuropathologic changes [27]), with subsequent digital pathologic methods for lesion detection and counting (see below).
Pathological assessments and staining methods.
The following regions were sampled from the left hemisphere: middle frontal gyrus (Brodmann area [BA] 9), superior and middle temporal gyri (BAs 21 and 22), inferior parietal lobule (BAs 39 and 40), and occipital neocortex including the primary visual area (BAs 17 and 18), hippocampus at the level of the lateral geniculate nucleus, entorhinal cortex, and amygdala. The tissue was fixed in 10% formaldehyde and then processed in paraffin blocks. All specimens were cut at 8-micron thickness. The neocortical regions were used to assess Thal staging because the first location for Aβ deposition (Thal phases > 0) involve neocortex [21]. We did not limit to Thal Aβ stages <3 (“Possible PART” according to the consensus criteria [3]) because the basic goal was to explore the biological variation in this community-based sample.
1997–2011 cases: Silver stains, with visual detection and manual counting of DPs, NPs, and NFTs.
Methods of silver stains and manual counting at the UK-ADC have been previously described in detail [1, 28]. Briefly, sections were cut and stained using a modified Bielschowsky silver method and a Gallyas silver impregnation stain were performed without pretreatment [1, 25]. Amyloid plaques were counted using Bielschowsky silver stained sections of the neocortex, and the Gallyas-stained sections were used to count NFTs in the hippocampal formation (CA1 and subiculum) and entorhinal cortex. The most severely affected fields were determined by studying the whole section and marking those areas. Amyloid plaques were separated into DPs (plaques without silver-impregnated dark neurites) and NPs (cored plaques with dark, dystrophic-appearing neurites) using the Bielschowsky stain. An arithmetic mean was calculated from the count of the 5 most involved fields for DPs (number of DPs per 2.35 mm2), NPs (number of NPs per 2.35 mm2), and NFTs (number of NFTs per 0.586 mm2) for each region. Plaque counts were capped at 250 for each area representing an extremely high density of plaques.
2012–2017 cases: Immunohistochemical stains, with digital pathology for lesion detection and quantification.
In these cases, the lesions were detected using an Aperio/Leica ScanScope and quantified using the Genie pattern recognition software as previously described in detail [29]. For each case, Aβ IHC stains were scored using previously described methods, with pretreatment with 97% formic acid (Thermo) for 3 min followed by pepsin treatment (Thermo Fisher) for 10min [29]. Slides were then loaded into an Aperio ScanScope XT™, scanned at 40× magnification via the semi-automated method, and the images stored on a dedicated server. To enable a focused analysis that could be reproducible between users, a square analysis region was created (4 mm2) and placed within the gray matter at the site of highest concentration of pathology. Subsequent boxes (ten total boxes) were then placed as far from the existing boxes as possible without overlapping other analysis regions as described [29]. Once the analysis regions were selected, different quantitation algorithms were applied.
Beta-Amyloid detection and quantitation:
The monoclonal anti-Aβ (amino acid residues 1–11) NAB 228 [30] was used for Aβ IHC. Two different parameters were calculated for amyloid pathologies: an overall amyloid burden and an amyloid plaque density. For the overall amyloid burden, the Aperio Image Analysis ToolboxTM Positive Pixel Count (PPC), was used, with the following modifications to optimize for our in house staining protocol: hue value 0.1, hue width 0.5, color saturation threshold 0.24, intensity threshold weak (upper limit) 220, intensity threshold weak (lower limit) 118, intensity threshold moderate (upper limit) 118, intensity threshold moderate (lower limit) 70, intensity threshold strong (upper limit) 70, intensity threshold strong (lower limit) 0, and intensity threshold negative pixels −1. The modified PPC was then run on the ten 4mm2 boxes selected on each slide as specified above. The amyloid burden was calculated by adding up all the ‘weak’ (1+), ‘moderate’ (2+), and ‘strong’ (3+) pixels from the data and dividing by the overall analysis area (40 mm2).
For the amyloid plaque density, the Aperio Image Analysis ToolboxTM Nuclear algorithm was used with the following modifications: curvature threshold 23.5, segmentation type 2, threshold type 1, lower intensity threshold 0, upper intensity threshold 230, minimum nuclear size (um2) 100, minimum nuclear size (pixels) 1638, minimum roundness 0.1, minimum compactness 0, minimum elongation 0.1, remove light objects 0, weak (1+) threshold 207, moderate (2+) threshold 188, strong (3+) threshold 162, and black threshold 0. This modified algorithm was then run on the same 10 boxes chosen for the amyloid burden analysis. The amyloid plaque density was calculated by adding up all the ‘weak’ (1+), ‘moderate’ (2+), and ‘strong’ (3+) positive nuclei and dividing by the overall analysis area (40 mm2).
Phospho-Tau detection and quantitation:
The PHF-1 antibody recognizes an epitope on phosphorylated tau at or near amino acids ser396 and ser404 [31, 32]. Two parameters were quantified for tau pathologies in selected regions of interest: NFT density and overall tau burden. For the NFT quantitation, a single Genie algorithm was first developed to separate NFTs from other tau immunoreactive structures. Methods for developing the NFT/NP Genie algorithm was described previously [29]. The NFT density was calculated via a modified nuclear algorithm with the NFT/NP Genie classifier limited to NFTs and the following additional modifications: curvature threshold 24, segmentation type 0, threshold type 1, lower intensity threshold 75, upper intensity threshold 230, minimum nuclear size (um2) 40, minimum nuclear size (pixels) 655, minimum roundness 0.1, minimum compactness 0, minimum elongation 0.1, remove light objects 0, weak (1+) threshold 222, moderate (2+) threshold 166, strong (3+) threshold 147, and black threshold 0. This modified algorithm was run on the ten 4 mm2 boxes within the gray matter on the PHF-1 stain, selected by the method stated above. The NFT density was calculated by dividing the total number of all the ‘weak’ (1+), ‘moderate’ (2+), and ‘strong’ (3+) nuclei counted by the overall analysis area (40 mm2).
The overall tau burden was determined using a PPC algorithm, similar to the amyloid burden quantitation, but with the following modifications: hue value 0.1, hue width 0.5, color saturation threshold 4.e-002, intensity threshold weak (upper limit) 132, intensity threshold weak (lower limit) 123, intensity threshold moderate (upper limit) 123, intensity threshold moderate (lower limit) 130, intensity threshold strong (upper limit) 130, intensity threshold strong (lower limit) 0, and intensity threshold negative pixels −1. The modified PPC was then run on the same 10 4mm2 boxes selected for NFT density analyses. The tau burden was calculated by dividing the ‘strong’(3+) pixels by the overall analysis area (40 mm2), and this parameter includes detection and quantification of neuropil threads and other tauopathic lesions including NFTs.
Analyses and statistical methods
Descriptive and exploratory data analyses were conducted using χ2 tests and non- parametric tests. Non-parametric tests were used given the skewed distributions of the untransformed pathology variables. Mediation analysis was performed to test the hypothesis that APOE genotype is associated with tau pathology through Aβ deposition rather than a direct effect of APOE on tau. Three sets of analyses were completed using negative binomial regression. The negative binomial model, which uses a log link function for the outcome variable, was chosen to reflect that the measures of Aβ and tau represent count data, and the Poisson distribution requires a strict assumption that the mean and variance of the outcome are equal, which we did not find evidence to support. No offsets were used in the models given the ascertainment methods ensured all cases had the same amount of tissue sampled, and goodness of fit was assessed by the Pearson χ2/DF ratio. All analyses were adjusted for age at death and sex.
For the cases autopsied between 1997 and 2011, the dependent variable was mean NFTs in the medial temporal lobe, which included NFT counts from the hippocampal formation and entorhinal cortex. Study variables were APOE-ε4 carrier status (1=yes, 0=no) and mean DPs in the neocortex, which included the middle frontal gyrus, superior and middle temporal gyri, inferior parietal lobule, and occipital neocortex. To assess whether the association of APOE carrier status with tau pathology was mediated through amyloid deposition, we first fit the model with APOE but not DPs, and then a model with both APOE and DPs.
For the cases autopsied between 2012 and 2017, two dependent tau/tangle variables were analyzed: mean tau burden and mean NFT density in the medial temporal lobe measured with digital pathology, which included the same areas as above. The same modelling strategy was used to assess the association between APOE and medial temporal lobe tau pathology as above. For tau burden, neocortical amyloid burden was used as the independent amyloid variable. For NFT density, neocortical amyloid density was used as the independent amyloid variable. To convey how the pathology and numerical outputs correlate, we show data in Supplemental Figs. 1 and 2.
Sensitivity analyses were conducted to assess the robustness of the model results and to check for an independent association between cerebral amyloid angiopathy (CAA) and tau/tangle pathology since CAA is known to be associated with APOE genotype in this autopsy series [33]. In the first set of sensitivity analyses, we included measures of medial temporal amyloid pathology (the hippocampal formation and entorhinal cortex) in the models; these data were collected and coded in the same manner as for the neocortical regions. In the second set of sensitivity analyses we included a measure of neocortical CAA, which was assessed with Aβ IHC stains in each neocortical region, and graded on a scale from 0 (no CAA detected) to 3 (severe CAA). For these statistical models, CAA was operationalized as the mean of the ratings in parenchymal blood vessels for each of the 4 neocortical regions.
Results
Descriptive and exploratory results.
A perspective on the overall cohort and included cases, and the reasons for exclusions, are depicted in Fig. 1. Overall work flow is depicted in Fig. 2. Distributions of participant age at death, sex, education, final MMSE score, and Braak NFT stage were similar for the cases autopsied between 1997–2011 (n=95) and 2012–2017 (n=51), but the participants who were autopsied in the later period had more severe cognitive impairment based on final clinical diagnosis (χ2=14.68, 3 df, p=0.0016) (Table 1). Age range was 70–105. Note that Thal Aβ staging was performed on 43/51 (84%) of the cases and 25 (58%) were Thal Aβ stage <3, whereas 13 cases (30%) were Thal Aβ stage 3 and 5 (12%) were Thal Aβ stage 4. The observed proportion of cases who were APOE-ε4 carriers was also higher in the latter group, but the difference was marginally significant (χ2=3.74, 1 df, p=0.053). Overall, only 18/145 cases were positive for APOE-ε4 (12.4%).
Fig. 1. Flowchart of cases included, and bases for exclusions.
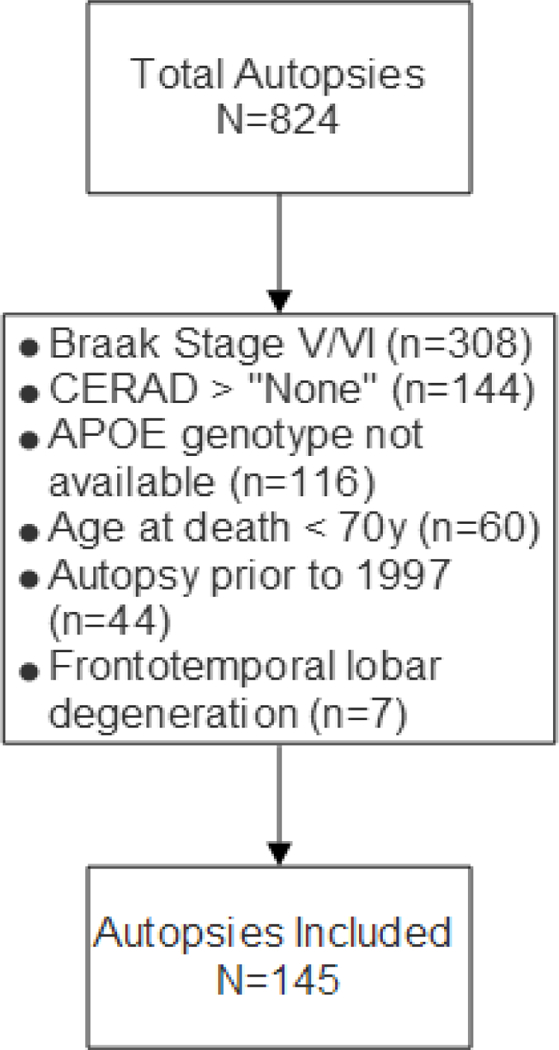
The University of Kentucky AD Center (UK-ADC) autopsy cohort is community-based and most patients were recruited while cognitively intact. Only 11% of the study participants were demented at final examination (See Table 1).
Fig. 2. Workflow.
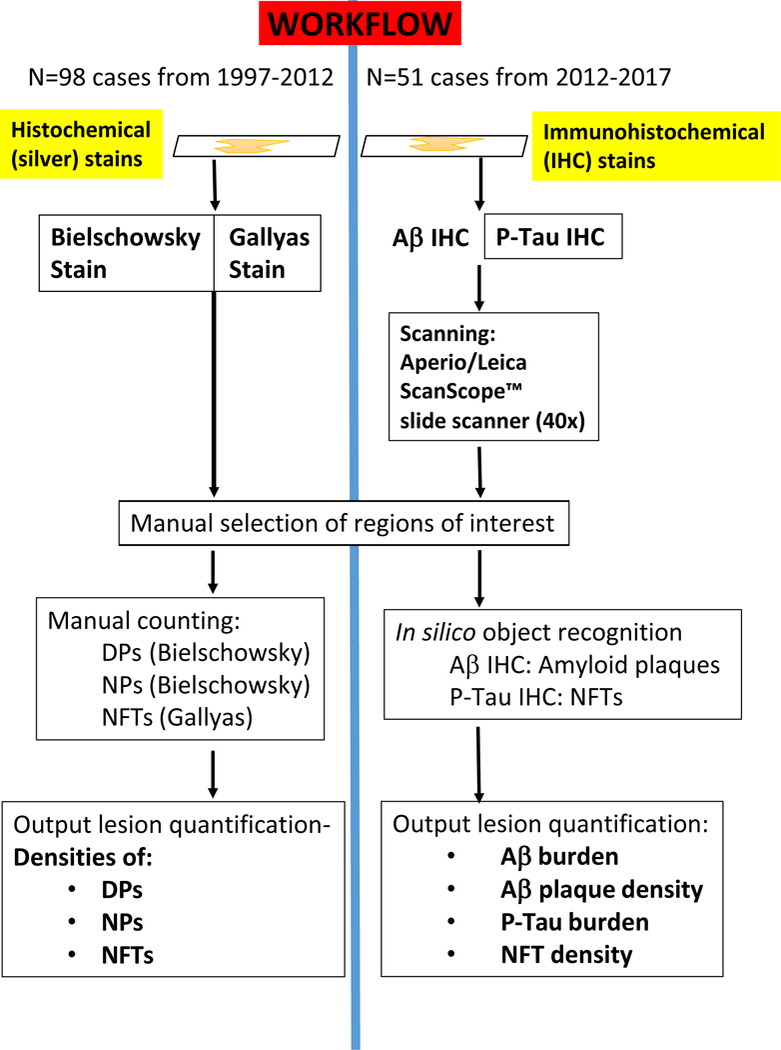
Schematic to depict the study design of the present study that included different UK-ADC cohorts and methods applied. Notably, different stains were used and the neuropathologic lesions were detected and counted separately. For the 1997–2012 cohort, histochemical stains were used with silver impregnation. Bielschowsky stains were used for diffuse amyloid plaques (DPs) and neuritic amyloid plaques (NPs), whereas the Gallyas silver impregnation technique was used for NFTs; for these cases, the output comprised manually tabulated density counts for DPs, NPs, and NFTs. For the 2012–2017 cohort, immunohistochemical (IHC) stains were used -- Aβ and P-Tau IHC – and slides scanned using a digital slide scanner, followed by in silico object recognition, with output including Aβ burden, Aβ plaque density, P-Tau burden, and P-Tau NFT density.
Table 1.
Characteristics of autopsied participants from the University of Kentucky Alzheimer’s Disease Center by year of death. All participants have CERAD neuritic plaque rating of “None”.
| All (N=145) |
Year of death, 1997–2011 (n=94) |
Year of death, 2012–2017 (n=51) |
|
|---|---|---|---|
| Age at death, y | 85.6±7.5 | 85.1±7.8 | 86.6±6.7 |
| Female | 79 (54.5) | 51 (54.3) | 28 (54.9) |
| Education, y | 16.0±2.8 | 15.9±2.6 | 16.1±3.0 |
| Last MMSE | 26.8±4.6 | 26.9±4.5 | 26.7±4.9 |
| Last Clinical Diagnosis | |||
| Normal | 102 (70.3) | 73 (77.7) | 29 (56.9) |
| Impaired, not MCI | 9 (6.2) | 8 (8.5) | 1 (2.0) |
| MCI | 18 (12.4) | 6 (6.4) | 12 (23.5) |
| Dementia | 16 (11.0) | 7 (7.5) | 9 (17.7) |
| Braak NFT stage | |||
| 0 | 17 (11.7) | 15 (16.0) | 2 (9.5) |
| I | 50 (34.5) | 32 (34.0) | 18 (35.3) |
| II | 51 (35.2) | 27 (28.7) | 24 (47.1) |
| III | 15 (10.3) | 11 (11.7) | 4 (7.8) |
| IV | 12 (8.3) | 9 (9.6) | 3 (5.9) |
| Thal Aβ stage | |||
| Absent (A0) | - | 12 | |
| Grade 1–2 (A1) | - | 14 | |
| Grade 3 (A2) | - | 13 | |
| Grade 4 (A3) | - | 5 | |
| Missing | 145 | 8 | |
| Medial temporal LBs | |||
| Present | 21 (14.5) | 11 (11.70) | 10 (19.6) |
| Hippocampal Sclerosis | |||
| Present | 14 (9.7) | 8 (8.5) | 6 (11.8) |
| APOE ε4 positive | 18 (12.4) | 8 (8.5) | 10 (19.6) |
The absence of DPs was strongly associated with the absence of the APOE-ε4 allele, such that just two of 68 cases with no detected DPs carried the ε4 allele (χ2=11.83, 1 df, p=0.0006) (Table 2). These two cases were both from the 1997–2011 autopsy series. The IHC stains were provided increased sensitivity and specificity in comparison to the silver stains, as can be appreciated in Fig. 3 where silver stains and IHC were applied to the same case. Examples of findings in quantitative pathology are depicted in Supplemental Figs. 1–3, including an evaluation of a sample of cases (n=103) with a broader spectrum of pathologies, evaluated with both manually counted DPs and Aβ burden in the same temporal cortex blocks (Supplemental Fig. 1). To illustrate the differences between DPs and NPs using silver stains, Fig. 4 shows representative results in cases with and without NPs. Representative results of digitally quantified immunohistochemically detectable pathologies are shown in Fig. 5 (Aβ) and Fig. 6 (P-Tau). Showing the distribution of pathologies in the subsample of cases any detectable plaques, Fig. 7 depicts the severity of DP/Aβ pathologies in the frontal, temporal, parietal, and occipital neocortical regions. Among cases with detected amyloid plaques (n=77, Table 2), there was no evidence that APOE was associated with more severe Braak NFT stages (χ2=4.84, 4 df, p=0.30). We note that 38/51 cases (74.5%) from the 2012–2017 series had detected Aβ plaques (i.e., Thal Aβ phase > 0), while just 35/94 cases (37.2%) from the 1997–2011 series had detected DPs (χ2=28.3, 1 df, p<0.0001) (data not shown), again attesting to the increased sensitivity of IHC staining results.
Table 2.
Braak NFT stage by diffuse plaques (present vs. absent) and APOE ε4 carrier status. All participants have CERAD neuritic plaque rating of “None”. Absence of diffuse plaques was characterized by diffuse plaque rating of “None” or Thal phase “0”.
| Any diffuse plaques | No diffuse plaques | |||
|---|---|---|---|---|
| APOE ε4 positive | APOE ε4 negative | APOE ε4 positive | APOE ε4 negative | |
| Braak NFT Stage | N=16 | N=61 | N=2 | N=66 |
| 0 | 1 (6.3) | 6 (9.8) | 0 | 10 (15.2) |
| I | 5 (31.3) | 20 (32.8) | 1 (50.0) | 24 (36.4) |
| II | 4 (25.0) | 26 (42.6) | 0 | 21 (31.8) |
| III | 4 (25.0) | 5 (8.2) | 0 | 6 (9.1) |
| IV | 2 (12.5) | 4 (6.6) | 1 (50.0) | 5 (7.6) |
Fig. 3. To illustrate characteristics of the different stains used, photomicrographs of stained sections from the hippocampus and nearby cortical structures are displayed.
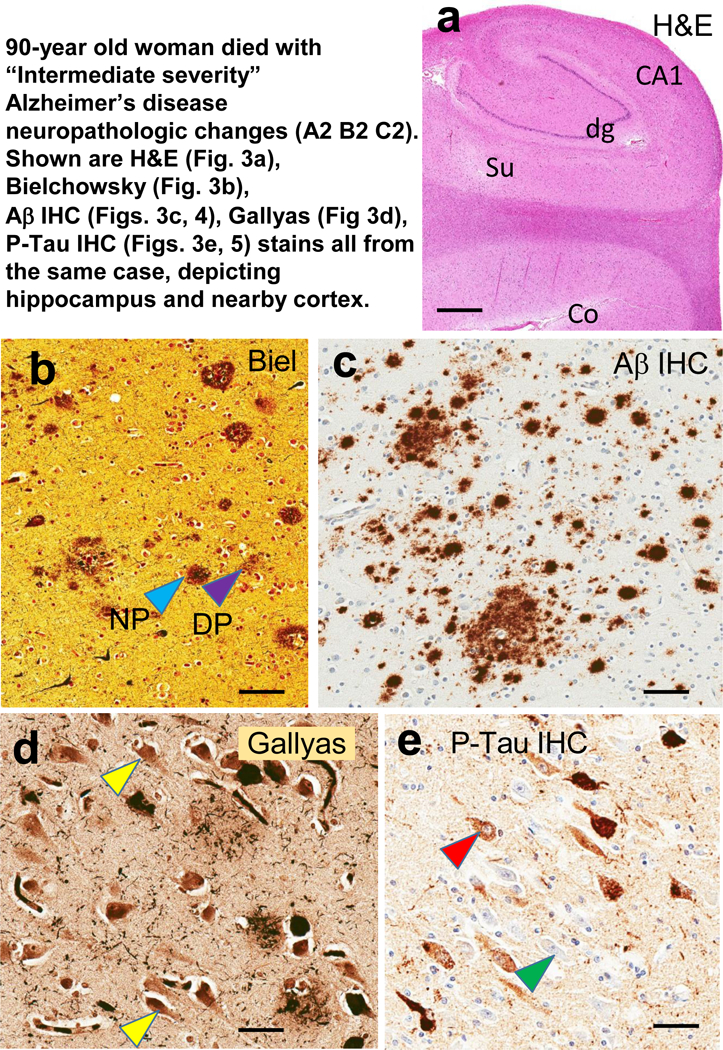
These photomicrographs all depict brain sections from a woman who died at age 90 years with “Intermediate severity” Alzheimer’s disease neuropathologic changes (Thal Aβ stage 3, Braak NFT stage III, CERAD “moderate” density NPs). This case is only for illustrative purposes and was not included in the other analyses due to the presence of NPs. Adjacent sections from the same case were used for Figs. 3 and 4 also. On the hematoxylin and eosin (H&E)-stained section are indicated the structures in the tissue block corresponding to the hippocampal formation and adjacent structures: dentate granule cells (dg), CA1, subiculum (Su), and peri- allocortical cortex (Co). From the Co region is shown immunohistochemical stains used to characterize amyloid plaques: Bielschowsky silver stain (b) and Aβ IHC (c). Note in the Bielschowsky stain that one can discriminate NPs (pale blue arrowhead) from DPs (purple arrowhead), whereas this discrimination is not possible in the Aβ IHC (c). Nonetheless, note that there is increased sensitivity and enhanced signal:noise ratio in the Aβ IHC stain compared to silver stains. For staining NFTs in CA1, the Gallyas silver stain (d) and P-Tau IHC (e) were used. Note that the signal: noise ratio is not high for the Gallyas stain (d); some of the structures may or may not represent NFTs (yellow arrowheads). This signal:noise ratio confounds digital assessment. By contrast, in the P-Tau IHC (e), even the pre-tangles (red arrowhead) are far easier to discriminate than the non-affected neurons (green arrowhead). Scale bars = 3mm (a); = 100 microns (b,c); and, = 50 microns (d,e).
Fig. 4. Depiction of stains in a case that lack neuritic amyloid plaques (Panels A-C) in comparison to a case with neuritic amyloid plaques (D-F).
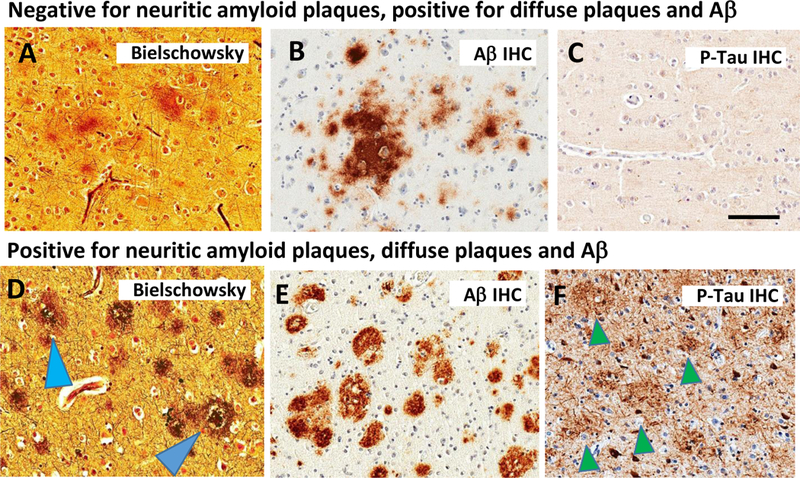
The Bielschowsky silver stain labels amorphous, fleecy material in the brain of an 80 year old woman; immunohistochemistry confirms the presence of Aβ (B; Aβ IHC), whereas P-Tau IHC is negative for dystrophic neurites. In AD, by contrast, the Bielschowsky stain highlights plaques invested with degenerating neurites (pale blue arrowhead) and/or ringing a discrete core (dark blue arrowhead). Note that Aβ IHC is immunopositive (E), and the P-Tau IHC in this case highlights the profiles of neuritic amyloid plaques (green arrowheads in panel F).
Fig. 5. Photomicrographs depict an area used to evaluate Aβ IHC using digital pathology.
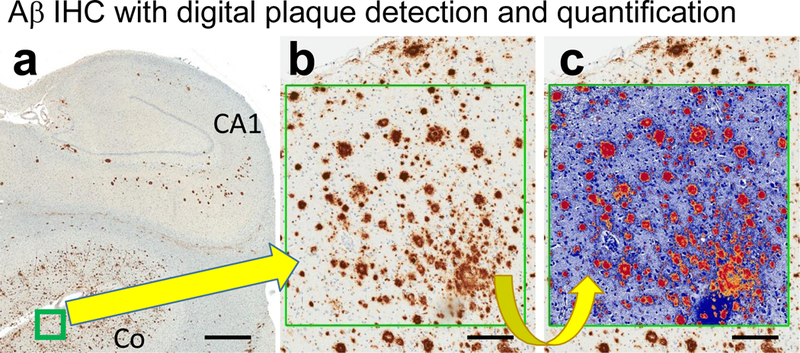
Low-power photomicrograph of Aβ IHC stained section (a) shows where the boxed area of interest was chosen. At higher magnification (b), the Aβ immunostained structures can be easily observed. The digitally false-colored box (c) shows how the Aβ plaques are digitally recognized; all of the orange, red, and yellow areas are quantified automatically. Scale bars =3mm (a); = 150 microns (b,c).
Fig. 6. Photomicrographs depict an area used to evaluate P-Tau IHC using digital pathology.
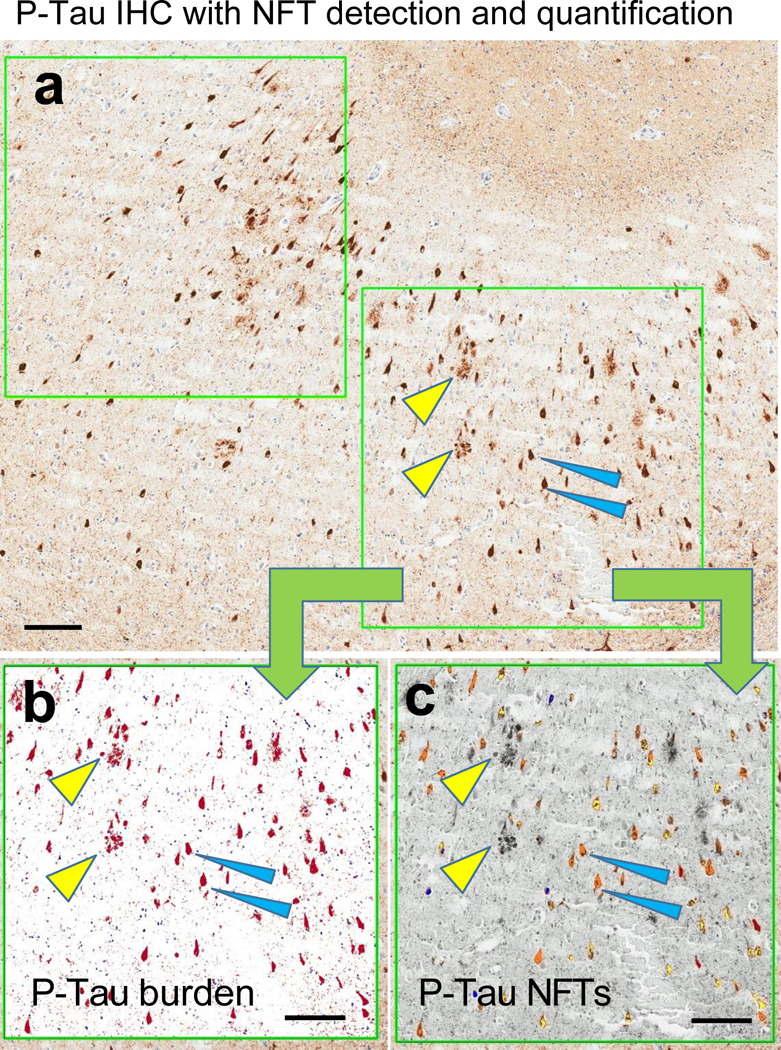
Low-power photomicrograph of P-Tau IHC stained section (a) depicts the size of the boxes (two such boxes are shown here, up to ten are used per slide) that are used to indicate regions of interest for the digital quantificaiton. At higher power (b-c), digital renderings of the lower-right box in panel (a) are presented to show the areas recognized for P-Tau burden (red in panel b); and P-Tau NFTs (orange in panel c). The yellow arrowheads in all 3 panels show the same NPs, and blue arrowheads show two specific NFTs. Note that the P-Tau burden image recognition algorithm (b) incorporated the NP structures, whereas the P-Tau NFTs highlights only the NFTs, not the NPs. Scale bars = 200 microns (a-c).
Fig. 7. Quantitative pathology in four different neocortical brain regions for cases that were CERAD-negative for neuritic amyloid plaques.
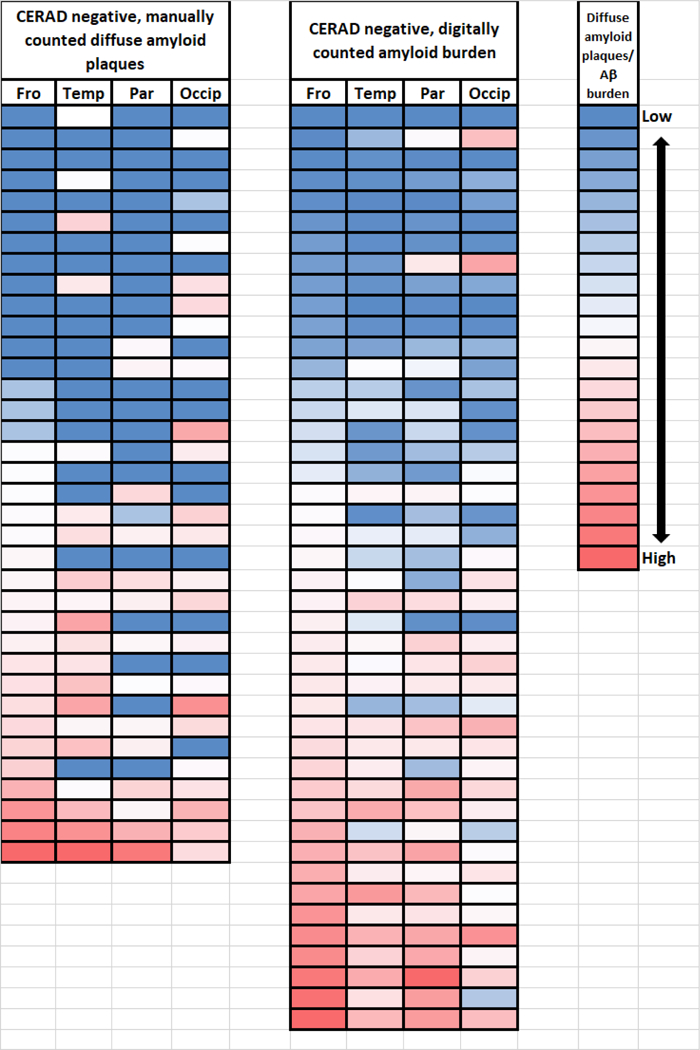
Shown are color-coded (red=high; white=middle; blue=low) representations of the cases counted manually as diffuse amyloid plaques (1997–2012; n=36) and digitally quantified Aβ amyloid burden (2012–2017; n=44), depicting the severity and distribution of those pathologies in the frontal (Fro; Brodmann areas 39/40); superior and middle temporal (Temp; Brodmann areas 2½2); parietal (Par; Brodmann area xx); and, occipital (Occip; Brodmann areas 17/18) neocortical regions. Each row represents one brain. The cases are ranked from top to bottom for both cohorts in the order from lowest to highest amount of detected pathology in the frontal cortical sections.
The association between neocortical amyloid pathology and medial temporal tau pathology was markedly different based on the autopsy series (1997–2011 vs. 2012–2017) (Table 3). For cases evaluated using silver stains, the presence of DPs was not associated with higher burden of NFTs: the observed NFT burden trended lower in cases with any DPs vs. no DPs, but this difference was not significant (Wilcoxon rank-sum test, p=0.67). On the other hand, there was an association between increasing Aβ pathology and increasing tau/tangle pathology in the 2012–2017 cases. Although the pattern of results was similar for both tau burden and NFT density (Table 3), the association was not significant (F=2.03, p=0.12). For NFT density, ANOVA on ranks showed significantly increased NFTs with quantile of amyloid density (F=3.35, p=0.027).
Table 3.
Quantitative medial temporal tau pathology by quantile of amyloid pathology (range). All participants have CERAD neuritic plaque rating of “None”.
| N | Tau pathology (median [IQR]) |
|
|---|---|---|
| Year of death, 1997–2011 | ||
| Diffuse plaque counts | NFT counts | |
| None | 60 | 2.9 (0.8–10.2) |
| > None (0.05–39.1) | 34 | 2.3 (0.6–17.4) |
| Year of death, 2012–2017 | ||
| Neocortical amyloid density | Medial temporal NFT density | |
| Q1 (0.01–0.27) | 12 | 4.1 (2.4–7.3) |
| Q2 (0.45–9.03) | 13 | 7.2 (3.6–13.2) |
| Q3 (9.56–42.3) | 13 | 8.2 (2.4–11.2) |
| Q4 (42.9–114.3) | 13 | 12.1 (8.5–24.8) |
| Neocortical amyloid burden | Medial temporal tau burden | |
| Q1 (0.03–0.73) | 12 | 8.3 (4.8–24.9) |
| Q2 (0.97–9.03) | 13 | 17.3 (12.1–37.0) |
| Q3 (10.72–36.4) | 13 | 15.4 (7.2–44.5) |
| Q4 (36.5–110.35) | 13 | 31.3 (21.3–58.7) |
Negative binomial results.
In the 1997–2011 cases, which were evaluated using silver stains, APOE carrier status was not significantly associated with mean medial temporal NFTs (β=0.85, p=0.08), but this trend was strengthened when mean neocortical DPs were added to the model (β=1.08, p=0.053). For the 2012–2017 cases, which were evaluated using IHC (Figs 5, 6), APOE carrier status was significantly associated with mean NFT density (β=0.65, p=0.032), but this association was attenuated when neocortical amyloid density was added to the model (β=0.30, p=0.35). Amyloid density was significantly associated with NFT density (β=0.011, p=0.013). Similarly, when tau burden was the outcome, without amyloid burden in the model, APOE status was significantly associated with tau burden (β=0.79, p=0.017). However, when amyloid burden was included in the model, APOE was no longer significantly associated with NFT burden (β=0.59, p=0.08) while amyloid burden was marginally significantly associated with tau burden (β=0.009, p=0.057).
Table 4 shows least squares mean estimates of tau pathology by APOE status based on the negative binomial models. As above, while APOE-ε4 was associated with higher predicted mean tau pathology in all of the analyses, the differences were only statistically significant when amyloid pathology was not in the model. For the cases from 2012–2017, the inclusion of amyloid pathology clearly attenuated the association between APOE and tau. For NFT density, the inclusion of amyloid density resulted in a 29% reduction in the estimated mean density for APOE-ε4 carriers (12.0 vs 16.8). For tau burden, the inclusion of amyloid burden resulted in a 17% reduction in the estimated mean burden for APOE-ε4 carriers (44.8 vs. 54.3). These results are consistent with the hypothesis that Aβ pathology at least partly mediates the impact of APOE ε4 on early NFT pathology. Model results remained stable for all sensitivity analyses (data not shown), which did not support the hypothesis that effect(s) of APOE ε4 on NFT pathology are mediated through CAA pathology. Point estimates changed little and the significance of model predictors was unchanged.
Table 4.
Predicted mean (95% CI)* tau pathology by APOE ε4 carrier status
| Year of death, 1997–2011 | Year of death, 2012–2017 | ||
|---|---|---|---|
| Adjusted Mean NFTs |
Adjusted Mean NFT density |
Adjusted Mean tau burden |
|
| Model 1 | |||
| APOE ε4 positive | 16.0 (6.5–39.3) | 16.8 (9.8–18.6) | 54.3 (30.2–97.6) |
| APOE ε4 negative | 6.8 (5.1–9.1) | 8.7 (6.8–11.3) | 24.6 (18.7–32.3) |
| Model 2 | |||
| APOE ε4 positive | 19.4 (7.0–53.9) | 12.0 (6.9–21.0) | 44.8 (24.7–81.2) |
| APOE ε4 negative | 6.6 (5.0–8.8) | 8.9 (7.0–11.4) | 24.7 (18.9–32.3) |
Results are based on negative binomial regression. Bolded results are significantly different between APOE positive and negative at 0.05. Model 1 includes APOE, Model 2 includes both APOE and amyloid pathology. For NFTs, the amyloid measure was diffuse plaque counts; amyloid density was used for NFT density; and amyloid burden was used for tau burden. All models were adjusted for age at death and sex.
Discussion
Collectively, these findings (Fig. 8) indicate that even when AD-type pathology is in its presumed earliest phase(s), the “diffuse” Aβ deposits are associated with increased medial lobe tau NFTs. Moreover, the observed association between APOE ε4 alleles and tau pathology appears to be at least partly mediated via Aβ deposition. However, we did find that cases with APOE ε4 alleles tended to have higher levels of tau pathology, even after controlling for amyloid pathology, although the differences were not statistically significant. It is also noteworthy that almost 90% of the cases were not ε4 carriers, as in the general population. Our analytic results were modified by the staining method, such that cases with IHC staining revealed higher levels of amyloid pathology in the absence of neuritic plaques and analyses suggested that the effect of APOE ε4 on tau pathology was diminished after controlling for amyloid. Thus, our findings underscore the importance of using IHC-based staining of brain tissue to detect the pathologic biomarkers for research purposes, in order to provide the most sensitive and specific indication of their presence.
Fig. 8. Overview of the results.
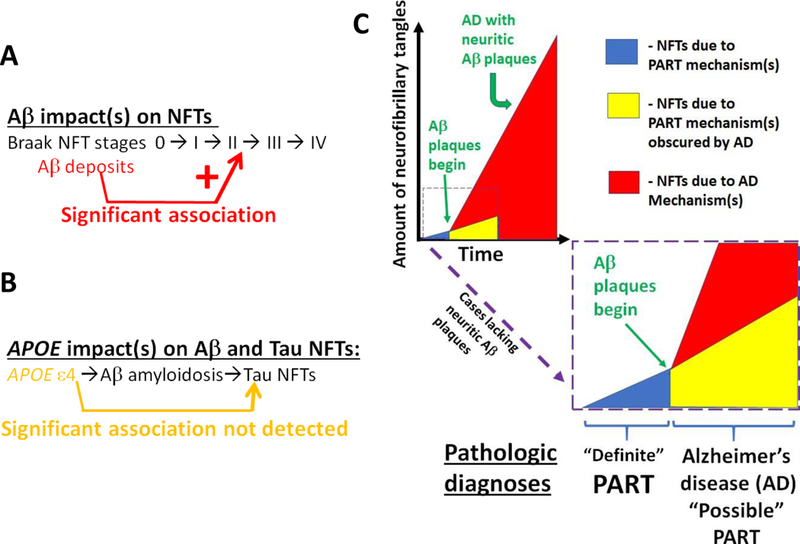
The data and analyses provide support for the hypothesis that Aβ deposits, even in the earliest “diffuse” phase, are associated with increased severity of medial temporal lobe NFT pathology as assessed by digital pathology of immunohistochemical stains (A). By contrast, when Aβ pathology was taken into account, there was not evidence for direct and robust association between APOE genotype and the severity of tau/NFT pathology (B). An over-arching point, as depicted in schematic form (C), is that in cases with early AD, both AD- related and PART-related pathogenetic mechanisms are probably occurring at the same time. Whereas it may be most specific to use Aβ immunohistochemistry for Definite PART, our analyses indicate it still is accurate to assume that some PART pathology is probably present despite the presence of Aβ plaques.
The current study has some limitations. While the UK-ADC autopsy cohort sample was drawn from a community-based group of research volunteers who mostly enrolled while cognitively intact [22], the neuropathologic findings are not representative of a population-based cohort in terms of ethnicity, comorbidities, or socioeconomic variables. We also limited our analyses to specific areas of the brain, and the observed relationships may or may not have implications about other brain regions. Further, the sample sizes may have been too small to provide adequate statistical power to test the hypothesis that APOE alleles and tauopathy independently are associated with each other. We also acknowledge technical challenges that pertain to digital pathologic methods, which we have discussed before [29, 34–36], but which still apply to efforts in this field: aspects of standardization, calibration, thresholding, segmentation, as well as how one masks or ignores sectioning “chatter” (see Fig. 3). There also may be artifacts inserted by the technical characteristics of the antibodies – for example, NAB228 may not recognize all subtypes of Aβ.
In a broader sense, autopsy data are intrinsically cross-sectional, and, some of the diagnostic and clinical practices have changed over time. For example, it wasn’t until mid-2013 that systematic Thal Aβ staging was performed on each case at the University of Kentucky AD Center. On the other hand, an enormous amount of neuropathologic data have been gathered across a wide spectrum of diseases, and the impact of the evolution of the staining techniques used to operationalize pathologic biomarkers was one of the focal points of the current study.
Despite the pertinent caveats, the results of the current study have implications about neurodegenerative disease biology and neuropathologic practices. We highlight four areas where our findings are directly relevant: the clinical-biological impact of “diffuse” Aβ deposits; the pathogenetic mechanism(s) of APOE; the “amyloid cascade hypothesis”; and, the neuropathologic diagnosis of PART.
It has long been appreciated that parenchymal plaques in AD can be differentiated into at least two subtypes: diffuse Aβ deposits (DPs) and neuritic (“senile”) amyloid plaques (NPs) [37– 40]. The latter is distinguished by the presence of misshapen axons and dendrites which often contain tau paired helical filaments and/or phosphorylated neurofilament proteins [41]. Animal models of AD have addressed some of these issues[38, 42–44]. In terms of clinical-pathologic correlation, the presence and density of NPs are more strongly correlated with cognitive impairment than is the case for DPs: the brains of many cognitively “normal” individuals harbor DPs [45–50]. However, when large autopsy series are evaluated that include sensitive antemortem cognitive tests and cases with both DP+ and DP- pathology, the presence of DPs is associated with (relatively subtle) cognitive manifestations [20, 45, 51]. The mechanism(s) of those cognitive changes, associated with DPs, is not well understood. While Aβ may exert a direct impact on cognition, the current study indicates that the Aβ deposited in neocortical DPs is attended by (perhaps causes) increased severity of medial temporal lobe tauopathy. Prior studies have been quite compatible with this hypothesis [45, 51].
Research on the neuropathologic sequelae associated with APOE polymorphisms is a related area of study. In terms of impact on public health, APOE is the strongest known genetic risk factor for late-onset AD, and a large number of mechanisms have been proposed to help explain how the APOE ε4 allele contributes to the clinical and pathological phenotype [52–57]. Without extending our focus deeply into the molecular mechanisms, we note that there is extensive evidence that APOE alleles affect the rate of Aβ deposition and/or clearance [58]. In autopsy series, there has been a consistently observed strong correlation between APOE allele status and the amount of detected Aβ plaques and CAA pathology [33, 59–61]. However, there also has been a smaller set of published studies, including animal model experiments, interpreted to indicate that the APOE ε4 allele may contribute directly to tauopathy [62–64]. There is not necessarily an “either/or” answer, and APOE may work through both as well as through completely different mechanisms [65]. However, the current study helps to illustrate an important consideration: even if APOE status is associated with tau pathology, it could be exerting its impact on Aβ, upstream of the tauopathy. This potent confounder is absolutely necessary to take into account, preferably using quantitatve metrics of the pathology. Indeed, when quantitative Aβ pathology was factored into our analyses, there was no statistical evidence that APOE genotype had any correlative impact on the amount of detected tau pathology.
The analyses of quantitative neuropathology also led to results compatible with the “amyloid cascade hypothesis” [66] which posits a directional progression in which Aβ promotes accelerated tauopathy and not the reverse. As a rule, NFTs are seen before Aβ deposition in human brains [8], yet NFTs are not pathognomonic for AD: rather, NFTs are seen in many different diseases that lack Aβ, including tauopathy-inducing mutations in the MAPT gene which do not induce Aβ pathology (by contrast, APP mutations lead to tau pathology) [67–71]. In the present study, we once again do not find evidence that tauopathy “causes” Aβ deposition or foments the progression to full-blown AD. There is abundant evidence that the brains of 15–25% of centenarians -- at the apparent limit of our species’ chronological longevity -- harbor PART pathology but lack Aβ deposits [7, 8, 72, 73]. By contrast, the data in the current study are compatible with the hypothesis that, as soon as Aβ fibrils start to appear in the parenchyma, those Aβ deposits are associated with accelerated tauopathy.
Another implication of the current study relates to the stains that are used for making the neuropathologic diagnoses. In the paper that established the diagnostic criteria for PART, the following was written: “In keeping with the current guidelines for AD, mild Aβ plaques defined using the Thal grading system, consistent with low AD neuropathologic changes, preclude the diagnosis of “Definite” PART...” [3]. However, it also was noted that “laboratories using the CERAD neuritic plaque density scores may classify subjects with neuritic plaque frequency of “None” as “Definite” [PART] and “Sparse” as “Possible” [PART]” [3]. We note that there is no consensus-based definition of a neuritic amyloid plaque, although it is arguably the lesion that comes closest to representing a pathognomonic feature of AD [74]. This is a problem that should be addressed, but the lack of consensus should not lead to stasis in the research field. We have observed that individual pathology labs prefer to visualize NPs using quite different methods: silver stains, thioflavin, or immunohistochemical stains. Presumably, even when there are substantial densities of Aβ and NPs, and/or additional comorbid pathologies, the biologic factors that affect PART severity may be still occurring (Fig. 8c), so, the concept of “Possible PART” has some merit. Yet the data and analyses presented in the current paper add support for the practice of relying on the presence or absence of Aβ deposits, rather than NPs, as a more specific criterion to distinguish between AD and PART.
Supplementary Material
Acknowledgments and funding:
We are profoundly grateful to the research volunteers and also to our colleagues at the UK-ADC. The study was supported by NIH grants P30 AG028383, R01 AG042419, R01 AG042475. Thanks to Dr. Peter Davies for generously providing PHF-1 antibody and Dr. Eddie Lee for generously providing Nab228 antibody.
Footnotes
Conflict of Interest/Disclosure Statement:
The authors have no conflict of interest to report
References Cited
- [1].Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS, Abner EL, Markesbery WR (2007) Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 66, 1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kovacs GG, Milenkovic I, Wohrer A, Hoftberger R, Gelpi E, Haberler C, Honigschnabl S, Reiner-Concin A, Heinzl H, Jungwirth S, Krampla W, Fischer P, Budka H (2013) Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 126, 365–384. [DOI] [PubMed] [Google Scholar]
- [3].Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Gearing M, Grinberg LT, Hof PR, Hyman BT, Jellinger K, Jicha GA, Kovacs GG, Knopman DS, Kofler J, Kukull WA, Mackenzie IR, Masliah E, McKee A, Montine TJ, Murray ME, Neltner JH, Santa-Maria I, Seeley WW, Serrano-Pozo A, Shelanski ML, Stein T, Takao M, Thal DR, Toledo JB, Troncoso JC, Vonsattel JP, White CL 3rd, Wisniewski T, Woltjer RL, Yamada M, Nelson PT(2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bouras C, Hof PR, Morrison JH (1993) Neurofibrillary tangle densities in the hippocampal formation in a non-demented population define subgroups of patients with differential early pathologic changes. Neurosci Lett 153, 131–135. [DOI] [PubMed] [Google Scholar]
- [5].Arriagada PV, Marzloff K, Hyman BT (1992) Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 42, 1681–1688. [DOI] [PubMed] [Google Scholar]
- [6].Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK (1992) Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging 13, 179–189. [DOI] [PubMed] [Google Scholar]
- [7].Neltner JH, Abner EL, Jicha GA, Schmitt FA, Patel E, Poon LW, Marla G, Green RC, Davey A, Johnson MA, Jazwinski SM, Kim S, Davis D, Woodard JL, Kryscio RJ, Van Eldik LJ, Nelson PT (2016) Brain pathologies in extreme old age. Neurobiol Aging 37, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70, 960–969. [DOI] [PubMed] [Google Scholar]
- [9].Josephs KA, Murray ME, Tosakulwong N, Whitwell JL, Knopman DS, Machulda MM, Weigand SD, Boeve BF, Kantarci K, Petrucelli L, Lowe VJ, Jack CR Jr., Petersen RC, Parisi JE, Dickson DW (2017) Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol 133, 705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kryscio RJ, Abner EL, Jicha GA, Nelson PT, Smith CD, Van Eldik LJ, Lou W, Fardo DW, Cooper GE, Schmitt FA (2015) Self-reported memory complaints: a comparison of demented and unimpaired outcomes. Journal of Prevention of Alzheimer’s Disease ePub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Abner EL, Kryscio RJ, Schmitt FA, Fardo DW, Moga DC, Ighodaro ET, Jicha GA, Yu L, Dodge HH, Xiong C, Woltjer RL, Schneider JA, Cairns NJ, Bennett DA, Nelson PT (2017) Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann Neurol 81, 549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jack CR Jr. (2014) PART and SNAP. Acta Neuropathol 128, 773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jack CR Jr., Wiste HJ, Weigand SD, Therneau TM, Knopman DS, Lowe V, Vemuri P, Mielke MM, Roberts RO, Machulda MM, Senjem ML, Gunter JL, Rocca WA, Petersen RC (2017) Age- specific and sex-specific prevalence of cerebral beta-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross-sectional study. Lancet Neurol 16, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Braak H, Del Tredici K (2014) Are cases with tau pathology occurring in the absence of Abeta deposits part of the AD-related pathological process? Acta Neuropathol 128, 767–772. [DOI] [PubMed] [Google Scholar]
- [15].Duyckaerts C, Braak H, Brion JP, Buee L, Del Tredici K, Goedert M, Halliday G, Neumann M, Spillantini MG, Tolnay M, Uchihara T (2015) PART is part of Alzheimer disease. Acta Neuropathol 129, 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jellinger KA, Alafuzoff I, Attems J, Beach TG, Cairns NJ, Crary JF, Dickson DW, Hof PR, Hyman BT, Jack CR Jr., Jicha GA, Knopman DS, Kovacs GG, Mackenzie IR, Masliah E, Montine TJ, Nelson PT, Schmitt F, Schneider JA, Serrano-Pozo A, Thal DR, Toledo JB, Trojanowski JQ, Troncoso JC, Vonsattel JP, Wisniewski T (2015) PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 129, 757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Crary JF (2016) Primary age-related tauopathy and the amyloid cascade hypothesis: the exception that proves the rule? J Neurol Neuromedicine 1, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nelson PT, Trojanowski JQ, Abner EL, Al-Janabi OM, Jicha GA, Schmitt FA, Smith CD, Fardo DW, Wang WX, Kryscio RJ, Neltner JH, Kukull WA, Cykowski MD, Van Eldik LJ, Ighodaro ET (2016) “New Old Pathologies”: AD, PART, and Cerebral Age-Related TDP-43 With Sclerosis (CARTS). J Neuropathol Exp Neurol 75, 482–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nelson PT, Abner EL, Scheff SW, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Patel E, Markesbery WR (2009) Alzheimer’s-type neuropathology in the precuneus is not increased relative to other areas of neocortex across a range of cognitive impairment. Neurosci Lett 450, 336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Davis DG, Poduska JW, Patel E, Mendiondo MS, Markesbery WR (2010) Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol 20, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. [DOI] [PubMed] [Google Scholar]
- [22].Schmitt FA, Nelson PT, Abner E, Scheff S, Jicha GA, Smith C, Cooper G, Mendiondo M, Danner DD, Van Eldik LJ, Caban-Holt A, Lovell MA, Kryscio RJ (2012) University of Kentucky Sanders-Brown Healthy Brain Aging Volunteers: Donor Characteristics, Procedures, and Neuropathology. Curr Alzheimer Res 9, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Smith VD, Bachstetter AD, Ighodaro E, Roberts K, Abner EL, Fardo DW, Nelson PT (2017) Overlapping but distinct TDP-43 and tau pathologic patterns in aged hippocampi. Brain Pathol 28, 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. [DOI] [PubMed] [Google Scholar]
- [25].Gallyas F (1971) Silver staining of Alzheimer’s neurofibrillary changes by means of physical development. Acta Morphol Acad Sci Hung 19, 1–8. [PubMed] [Google Scholar]
- [26].(1997) Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging 18, S1–2. [PubMed] [Google Scholar]
- [27].Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, National Institute on A, Alzheimer’s A (2012) National Institute on Aging- Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lamy C, Duyckaerts C, Delaere P, Payan C, Fermanian J, Poulain V, Hauw JJ (1989) Comparison of seven staining methods for senile plaques and neurofibrillary tangles in a prospective series of 15 elderly patients. Neuropathol Appl Neurobiol 15, 563–578. [DOI] [PubMed] [Google Scholar]
- [29].Neltner JH, Abner EL, Schmitt FA, Denison SK, Anderson S, Patel E, Nelson PT (2012) Digital pathology and image analysis for robust high-throughput quantitative assessment of Alzheimer disease neuropathologic changes. J Neuropathol Exp Neurol 71, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee EB, Skovronsky DM, Abtahian F, Doms RW, Lee VM (2003) Secretion and intracellular generation of truncated Abeta in beta-site amyloid-beta precursor protein-cleaving enzyme expressing human neurons. J Biol Chem 278, 4458–4466. [DOI] [PubMed] [Google Scholar]
- [31].Greenberg SG, Davies P, Schein JD, Binder LI (1992) Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem 267, 564–569. [PubMed] [Google Scholar]
- [32].Lang E, Szendrei GI, Lee VM, Otvos L, Jr. (1992) Immunological and conformation characterization of a phosphorylated immunodominant epitope on the paired helical filaments found in Alzheimer’s disease. Biochem Biophys Res Commun 187, 783–790. [DOI] [PubMed] [Google Scholar]
- [33].Nelson PT, Pious NM, Jicha GA, Wilcock DM, Fardo DW, Estus S, Rebeck GW (2013) APOE- epsilon2 and APOE-epsilon4 correlate with increased amyloid accumulation in cerebral vasculature. J Neuropathol Exp Neurol 72, 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Attems J, Neltner JH, Nelson PT (2014) Quantitative neuropathological assessment to investigate cerebral multi-morbidity. Alzheimers Res Ther 6, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Neltner JH, Abner EL, Baker S, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Hammack E, Kukull WA, Brenowitz WD, Van Eldik LJ, Nelson PT (2014) Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain 137, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ, Patel E, Abner EL, Kryscio RJ, Nelson PT (2015) Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun 3, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yamaguchi H, Hirai S, Morimatsu M, Shoji M, Ihara Y (1988) A variety of cerebral amyloid deposits in the brains of the Alzheimer-type dementia demonstrated by beta protein immunostaining. Acta Neuropathol 76, 541–549. [DOI] [PubMed] [Google Scholar]
- [38].Wisniewski HM, Ghetti B, Terry RD (1973) Neuritic (senile) plaques and filamentous changes in aged rhesus monkeys. J Neuropathol Exp Neurol 32, 566–584. [DOI] [PubMed] [Google Scholar]
- [39].Terry RD, Katzman R (1983) Senile dementia of the Alzheimer type. Ann Neurol 14, 497–506. [DOI] [PubMed] [Google Scholar]
- [40].Masliah E, Mallory M, Hansen L, Alford M, DeTeresa R, Terry R (1993) An antibody against phosphorylated neurofilaments identifies a subset of damaged association axons in Alzheimer’s disease. Am J Pathol 142, 871–882. [PMC free article] [PubMed] [Google Scholar]
- [41].Masliah E, Mallory M, Deerinck T, DeTeresa R, Lamont S, Miller A, Terry RD, Carragher B, Ellisman M (1993) Re-evaluation of the structural organization of neuritic plaques in Alzheimer’s disease. J Neuropathol Exp Neurol 52, 619–632. [DOI] [PubMed] [Google Scholar]
- [42].Bennett RE, DeVos SL, Dujardin S, Corjuc B, Gor R, Gonzalez J, Roe AD, Frosch MP, Pitstick R, Carlson GA, Hyman BT (2017) Enhanced Tau Aggregation in the Presence of Amyloid beta. Am J Pathol 187, 1601–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].DeVos SL, Corjuc BT, Commins C, Dujardin S, Bannon RN, Corjuc D, Moore BD, Bennett RE, Jorfi M, Gonzales JA, Dooley PM, Roe AD, Pitstick R, Irimia D, Frosch MP, Carlson GA, Hyman BT (2018) Tau reduction in the presence of amyloid-beta prevents tau pathology and neuronal death in vivo. Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, Zhang B, Gathagan RJ, Yue C, Dengler C, Stieber A, Nitla M, Coulter DA, Abel T, Brunden KR, Trojanowski JQ, Lee VM (2018) Amyloid-beta plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med 24, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Price JL, McKeel DW Jr., Buckles VD, Roe CM, Xiong C, Grundman M, Hansen LA, Petersen RC, Parisi JE, Dickson DW, Smith CD, Davis DG, Schmitt FA, Markesbery WR, Kaye J, Kurlan R, Hulette C, Kurland BF, Higdon R, Kukull W, Morris JC (2009) Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging 30, 1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Crystal HA, Dickson DW, Sliwinski MJ, Lipton RB, Grober E, Marks-Nelson H, Antis P (1993) Pathological markers associated with normal aging and dementia in the elderly. Ann Neurol 34, 566–573. [DOI] [PubMed] [Google Scholar]
- [47].Malek-Ahmadi M, Perez SE, Chen K, Mufson EJ (2016) Neuritic and Diffuse Plaque Associations with Memory in Non-Cognitively Impaired Elderly. J Alzheimers Dis 53, 1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR (2006) Neuropathologic substrate of mild cognitive impairment. Arch Neurol 63, 38–46 [DOI] [PubMed] [Google Scholar]
- [49].Morris JC, Storandt M, McKeel DW Jr., Rubin EH, Price JL, Grant EA, Berg L (1996) Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 46, 707–719. [DOI] [PubMed] [Google Scholar]
- [50].Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kovari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Besser LM, Crary JF, Mock C, Kukull WA (2017) Comparison of symptomatic and asymptomatic persons with primary age-related tauopathy. Neurology 89, 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tai LM, Thomas R, Marottoli FM, Koster KP, Kanekiyo T, Morris AW, Bu G (2016) The role of APOE in cerebrovascular dysfunction. Acta Neuropathol 131, 709–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kanekiyo T, Xu H, Bu G (2014) ApoE and Abeta in Alzheimer’s disease: accidental encounters or partners? Neuron 81, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rebeck GW (2017) The role of APOE on lipid homeostasis and inflammation in normal brains. J Lipid Res 58, 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mahley RW, Huang Y (2012) Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron 76, 871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bennett DA, Schneider JA, Wilson RS, Bienias JL, Berry-Kravis E, Arnold SE (2005) Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. J Neurol Neurosurg Psychiatry 76, 1194–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Serrano-Pozo A, Qian J, Muzikansky A, Monsell SE, Montine TJ, Frosch MP, Betensky RA, Hyman BT (2016) Thal Amyloid Stages Do Not Significantly Impact the Correlation Between Neuropathological Change and Cognition in the Alzheimer Disease Continuum. J Neuropathol Exp Neurol 75, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kim J, Basak JM, Holtzman DM (2009) The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD (1993) Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A 90, 9649–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Poduri A, Gearing M, Rebeck GW, Mirra SS, Tigges J, Hyman BT (1994) Apolipoprotein E4 and beta amyloid in senile plaques and cerebral blood vessels of aged rhesus monkeys. Am J Pathol 144, 1183–1187. [PMC free article] [PubMed] [Google Scholar]
- [61].Mungas D, Tractenberg R, Schneider JA, Crane PK, Bennett DA (2014) A 2-process model for neuropathology of Alzheimer’s disease. Neurobiol Aging 35, 301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Alzheimer’s Disease Neuroimaging I, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM (2017) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q, Dee Fish J, Wyss-Coray T, Buttini M, Mucke L, Mahley RW, Huang Y (2004) Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci 24, 2527–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Thaker U, McDonagh AM, Iwatsubo T, Lendon CL, Pickering-Brown SM, Mann DM (2003) Tau load is associated with apolipoprotein E genotype and the amount of amyloid beta protein, Abeta40, in sporadic and familial Alzheimer’s disease. Neuropathol Appl Neurobiol 29, 35–44. [DOI] [PubMed] [Google Scholar]
- [65].Wolf AB, Valla J, Bu G, Kim J, LaDu MJ, Reiman EM, Caselli RJ (2013) Apolipoprotein E as a beta-amyloid-independent factor in Alzheimer’s disease. Alzheimers Res Ther 5, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hardy J (2006) Alzheimer’s disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis 9, 151–153. [DOI] [PubMed] [Google Scholar]
- [67].Ferrer I, Lopez-Gonzalez I, Carmona M, Arregui L, Dalfo E, Torrejon-Escribano B, Diehl R, Kovacs GG (2014) Glial and neuronal tau pathology in tauopathies: characterization of disease- specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 73, 81–97. [DOI] [PubMed] [Google Scholar]
- [68].Goedert M, Spillantini MG (2011) Pathogenesis of the tauopathies. J Mol Neurosci 45, 425–431. [DOI] [PubMed] [Google Scholar]
- [69].Williams DR (2006) Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J 36, 652–660. [DOI] [PubMed] [Google Scholar]
- [70].Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38, 24–26. [DOI] [PubMed] [Google Scholar]
- [71].Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, Del-Favero J, Cruts M, van Duijn CM, Van Broeckhoven C (2006) APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 129, 2977–2983. [DOI] [PubMed] [Google Scholar]
- [72].Imhof A, Kovari E, von Gunten A, Gold G, Rivara CB, Herrmann FR, Hof PR, Bouras C, Giannakopoulos P (2007) Morphological substrates of cognitive decline in nonagenarians and centenarians: A new paradigm? J Neurol Sci 257, 72–79. [DOI] [PubMed] [Google Scholar]
- [73].Nelson PT, Head E, Schmitt FA, Davis PR, Neltner JH, Jicha GA, Abner EL, Smith CD, Van Eldik LJ, Kryscio RJ, Scheff SW (2011) Alzheimer’s disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol 121, 571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.