

Abstract
Histone deacetylases (HDAC) are therapeutic targets in multiple cancers. ACY241, an HDAC6 selective inhibitor, has shown anti-multiple myeloma (MM) activity in combination with immunomodulatory drugs and proteasome inhibitors. Here we show ACY241 significantly reduces the frequency of CD138+ MM cells, CD4+CD25+FoxP3+ regulatory T cells, and HLA-DRLow/-CD11b+CD33+ myeloid-derived suppressor cells; and decreases expression of PD1/PD-L1 on CD8+ T cells and of immune checkpoints in bone marrow cells from myeloma patients. ACY241 increased B7 (CD80, CD86) and MHC (Class I, Class II) expression on tumor and dendritic cells. We further evaluated the effect of ACY241 on antigen-specific cytotoxic T lymphocytes (CTL) generated with heteroclitic XBP1unspliced184–192 (YISPWILAV) and XBP1spliced367–375 (YLFPQLISV) peptides. ACY241 induces co-stimulatory (CD28, 41BB, CD40L, OX40) and activation (CD38) molecule expression in a dose- and time-dependent manner, and anti-tumor activities, evidenced by increased perforin/CD107a expression, IFN-γ/IL-2/TNF-α production, and antigen-specific central memory CTL. These effects of ACY241 on antigen-specific memory T cells were associated with activation of downstream AKT/mTOR/p65 pathways and upregulation of transcription regulators including Bcl-6, Eomes, HIF-1 and T-bet. These studies therefore demonstrate mechanisms whereby ACY241 augments immune response, providing the rationale for its use, alone and in combination, to restore host anti-tumor immunity and improve patient outcome.
Introduction
Successful active immunotherapy requires specific transcriptional regulation of the generation and sustained efficacy of effector T cell subsets, which eliminate tumor cells expressing the targeted tumor-associated antigens [1–3]. Accumulating evidence indicates that tumor cells evade immune recognition by creating a suppressive tumor microenvironment, due to down-regulation of MHC and costimulatory molecules as well as up-regulation of immune suppressive and checkpoint molecules. Therefore, well-designed immunotherapeutic strategies are needed to define immune mechanisms mediating clinical responses on the one hand; and importantly, to define mechanisms accounting for resistance to immunotherapy on the other, in order to overcome tumor-induced immune escape mechanisms and thereby enhance efficacy of anti-tumor immune responses in cancer patients.
Among therapeutic options, epigenetic manipulation has emerged as an attractive novel treatment modality. Genetic instability in a developing tumor leads to accumulation of genomic alterations and modifications in the epigenetic code, which are dynamic and generally reversible. Epigenetic alterations seen in tumor cells regulate the heritable patterns of gene expression, leading to gene expression profiles promoting tumorigenesis and cancer progression [4–7]. The epigenetic pathways include histone modification, DNA methylation, nucleosome remodeling, and small non-coding regulatory RNAs. Among the pathways, post-translational modifications of histones are widespread and diverse, and histone acetylation has been correlated with transcriptional activation [8]. HDACs, a family of 18 enzymes, play a critical role in the epigenetic regulation of gene expression by catalyzing the removal of functional acetyl groups from proteins and by deacetylation of histone lysine residues during chromatin remodeling. Aberrant HDAC activity and over-expression in several forms of cancer can suppress anti-cancer immunity, associated with altering the acetylation status of histone and non-histone proteins that regulate various cellular processes including survival, differentiation, and apoptosis in tumor cells [9, 10]. Conversely, HDAC inhibitors, which broadly and/or selectively target various isoforms of HDAC enzymes that catalyze the removal of functional acetyl groups from proteins, exhibit strong anti-inflammatory and anti-tumor activities and have therefore emerged as new therapeutic agents [11–14]. In particular, combination epigenetic therapies with cancer immunotherapy represents a promising novel treatment paradigm for cancer.
We have previously demonstrated mechanisms whereby an HDAC selective inhibitor ACY241 mediates anti-multiple myeloma (MM) activity, both alone and in combination with immunomodulatory drugs and proteasome inhibitors [15–19]. Here we show that ACY241 significantly reduced the frequency of CD138+ MM cells, CD4+CD25+FoxP3+ T regulatory cells, and HLA-DRLow/- CD11b+CD33+ myeloid-derived suppressor cells (MDSC), as well as decreased expression of PD1/PD-L1 on immune suppressor and CD8+ T cells. ACY241 also increased B7 (CD80, CD86) and MHC (Class I, Class II) expression on both dendritic and tumor cells, and induced co-stimulatory (CD28, 41BB, CD40L, 0X40) and activation (CD38) molecule expression on XBP1 peptides-specific CTL (XBP1-CTL). Finally, ACY241 in a dose- and time-dependent fashion enhanced anti-tumor activity of antigen-specific CTL, evidenced by triggering increased perforin/CD107a, IFN-γ/IL-2/TNF-α production, and antigen-specific central memory CD8+ T cells. These studies therefore provide the rationale for clinical evaluation of ACY241, alone and in combination with cancer vaccine, to restore host antitumor immunity and improve patient outcome.
Materials and methods
Cell lines
Multiple myeloma (U266), breast cancer (MDA-MB231) and colon cancer (SW480) cell lines were obtained from the ATCC (Manassas, VA). The T2 cell line was provided by Dr. J. Molldrem (University of Texas M. D. Anderson Cancer Center, Houston, TX). The U266, MDA-MB231, SW480, and T2 cell lines were cultured in DMEM or RPMI (Gibco-Life Technologies) media supplemented with 10% fetal calf serum (BioWhittaker, Walkersville, MD), 2mM L-glutamine (Gibco-Life Technologies), 100IU/ml penicillin, and 100μ/ml streptomycin (Gibco).
Synthetic peptides
Immunogenic HLA-A2-specific heteroclitic XBP1 US184–192 (YISPWILAV) and heteroclitic XBP1 SP367–375 (YLFPQ-LISV) peptides were synthesized by standard fmoc (9-fluorenylmethyl-oxycarbonyl) chemistry, purified to >95% using reverse-phase chromatography, and validated by mass-spectrometry for molecular weight (Biosynthesis, Lewisville, TX) (Bae et al. 2015 (1), Bae et al. 2014, Bae et al. 2012, Bae et al. 2011). Lyophilized peptides were dissolved in DMSO (Sigma, St. Louis, MO), diluted in AIM-V medium (Gibco), and stored at –140 °C until needed.
Reagents
Fluorochrome conjugated anti-human mAbs specific to CD3, CD4, CD8, CD25, CD28, CD38, CD40L, CCR7, CD45R0, CD69, CD80, CD83, CD86, CD137 (41BB), CD138, FoxP3, HLA-A2, HLA-ABC, HLA-DP/DQ/DR, IFN-γ, IL-2, TNF-α, PD1, PD-L1, 0X40, AKT (pS473), mTOR (pS2448), NF-κB p65 (pS529), Bcl-6, HIF-1 or T-bet were purchased from Becton Dickinson (San Diego, CA). Fluorochrome-conjugated anti-human Eomes mAb was purchased from eBioscience (San Diego, CA). Recombinant human IL-2, IL-4, IFN-α and TNF-α were purchased from R&D Systems, and human GM-CSF was obtained from Immunex (Seattle, WA). ACY241 was purchased from AdooQ Bioscience (Irvine, CA), and reconstituted in 1% DMSO and stored at – 30 °C until needed.
Evaluation of HDAC inhibitor effects on antigen expression on MM patients’ cells and various cancer cell lines
Multiple myeloma patients’ tumor cells, bone marrow mononuclear cells (BMMC) and peripheral blood mononuclear cells, monocyte-derived dendritic cells, and cancer cell lines including MM (U266), breast cancer (MDA-MB231), and colon cancer (SW480) cells were treated with ACY241 (0.5, 1, 2 μm) for 3h—6 days. The treated cells were then evaluated for phenotypic changes in the expression of various cell surface markers including CD138, HLA-A2, HLA-ABC, HLA-DP/DQ/DR, CD80, CD86, CD3, CD4, CD8, CD25, FoxP3, PD1, and PD-L1. After staining with each specific antibody for 30 min at room temperature, the cells were washed, fixed in 2% paraformaldehyde, and analyzed using a LSRII Fortessa™ flow cytometer and DIVA™ v8.0 software (BD).
Generation of XBP1 peptides-specific CTL (XBP1-CTL)
Normal HLA-A2+ donors’ CD3+ T cells were obtained by negative selection from the non-adherent cell fraction post-monocyte adherence using the EasySep® magnet and Robosep® (StemCell Technologies). The XBP1-CTL were generated ex vivo by repeated stimulation of CD3+ T lymphocytes with antigen-presenting cells (APC; mature dendritic cells, T2 cells) pulsed with a cocktail (50 μ/ml) of heteroclitic XBP1 US184–192 (YISPWILAV) and heteroclitic XBP1 SP367–375 (YLFPQLISV) peptides at a 1:20 APC/peptide-to-CD3+ T cell ratio. The T cells were re-stimulated weekly with XBP1 peptides pulsed-APC for up to 6 cycles prior to use in the assays.
Evaluation of HDAC inhibitor effects on critical costimulatory and activation markers, memory T cell phenotype, CD8+ T cell proliferation, and anti-tumor activities of XBP1-CTL
XBP1-CTL treated with different doses of ACY241 were evaluated for their viability and expression of costimulatory (CD28), activation (CD38) and memory (CCR7, CD45RO) T cell subsets. For measuring proliferation, untreated control or ACY241 treated XBP1-CTL were labeled with CFSE (Molecular Probes) and stimulated with irradiated (20 Gy) T2 cells loaded with HLA-A2-specific XBP1 peptides or HLA-A2+ XBP1 expressing tumor cell lines. On day 4 or 6 of stimulation, the cells were harvested, stained with mAbs, and analyzed. To evaluate functional activities including anti-tumor and peptide-specific responses, XBP1-CTL were co-cultured with tumor cells or antigen-presenting cells pulsed with the specific peptides for 1 h, in the presence or absence of CD107a mAb, followed by an additional 5 h incubation in the presence of the protein transport inhibitors Brefeldin A and Monensin (BD). Following incubation, the cells were stained with mAbs specific to surface antigens including CD3, CD8, CD45RO and CCR7, fixed/permeabilized, stained intracellularly with mAbs specific to IFN-γ, IL-2, TNF-α, and/or perforin, and analyzed using a LSRFortessa™ flow cytometer with DIVA™ v8.0 software.
Evaluation of HDAC inhibitor effects on XBP1-CTL transcriptional regulators and signal integrator expression
XBP1-CTL treated with HDAC inhibitor were evaluated for any modifications in transcriptional regulators and signal integrator expression. In brief, the CTL were stimulated with HLA-A2+ myeloma cells for 5 h, collected, washed, and stained with mAbs against surface antigens, fixed, permeabilized, and then stained with mAbs specific to AKT (pS473), mTOR (pS2448) and NF-κB p65 (pS529) or Bcl-6, HIF-1, Eomes and T-bet. The cells were analyzed using a LSRFortessa™ flow cytometer and DIVA™ v8.0 software.
Statistical analysis
Results are presented as the mean ± SE. Groups were compared using an unpaired Student’s t-test. Differences were considered significant when *p < 0.05.
Results
ACY241 treatment decreases the frequency and expression of immune checkpoints on CD138+ myeloma cells, regulatory T cells (Treg) and MDSC
The effect of ACY241 on BMMC from MM patients was first evaluated. ACY241 treatment (0.5 uM, 1.0 uM; 48 h) significantly reduced the frequency of myeloma cells (CD138+) (Fig. 1a), Treg (CD3+CD4+ CD25+FOXP3+) (Fig. 1b), and MDSC (HLA-DRLow/- CD11b+CD33+) (Fig. 1c) in BMMC (n = 3, p < 0.05) in a dose-dependent manner, without effecting the frequency of CD3+CD4+ (Supplemental Fig. 1) or CD3+CD8+ T cells (Supplemental Fig. 2). We next evaluated the effect of ACY241 on expression of immune checkpoints on various cell subsets. ACY241 treatment (1.0 uM; 48 h) significantly decreased PD-L1 expression on CD138+ tumor (Fig. 2a) and Treg (Fig. 2b) in myeloma patients’ BMMC (n = 3, p < 0.05) as compared to untreated control, without changing the PD1 expression level on Treg (data not shown). Additionally, ACY241 treatment decreased PD1 expression on CD3+ T cells, which was seen to a greater extent on CD8+ T cells subset (n = 3, p < 0.05) (Fig. 2c) rather than CD4+ T cells (Supplemental Fig. 3). These effects by ACY241 on the frequency and checkpoint expression on the tumor and immune suppressor cells suggest its role as an immune modulator.
Fig. 1.
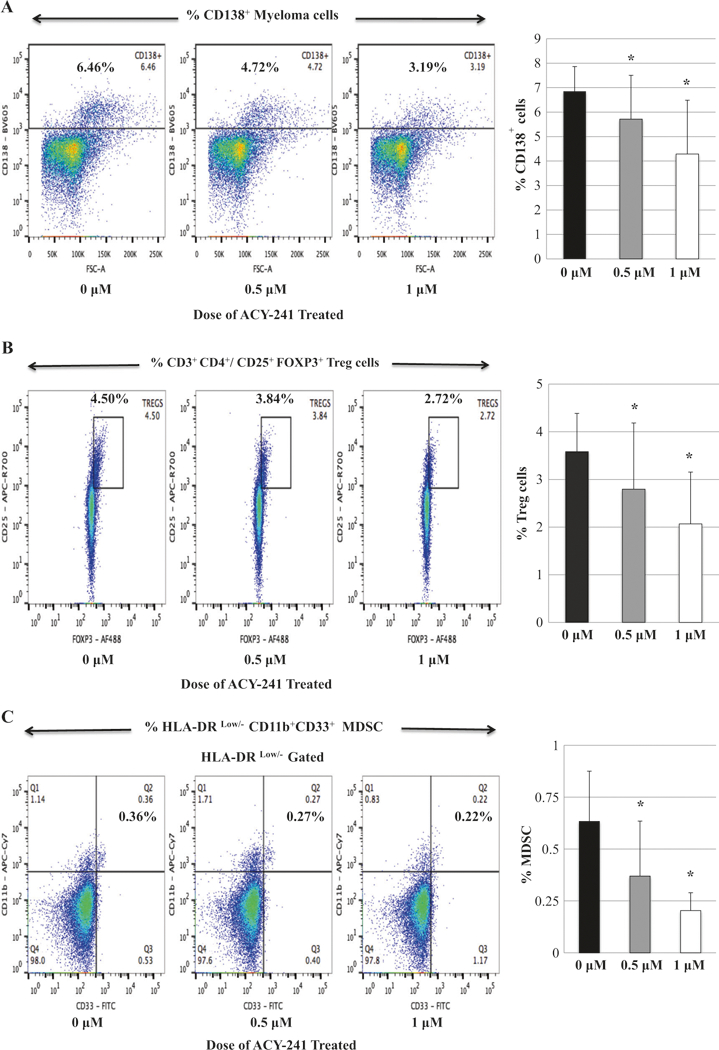
Decreased frequency of CD138+ tumor cells, regulatory T cells and myeloid-derived suppressor cells in myeloma patients’ bone marrow mononuclear cells with ACY241 treatment. Myeloma patients’ bone marrow mononuclear cells were treated with the HDAC inhibitor, ACY241 (0, 0.5, 1 μM) for 48 h, and then evaluated for changes in their phenotypic profiles. ACY241 treatment decreased the frequency of CD138+ MM cells (Fig. 1a), regulatory T cells (Fig. 1b), and myeloid-derived suppressor cells (Fig. 1c) in a dose-dependent manner (p < 0.05)
Fig. 2.
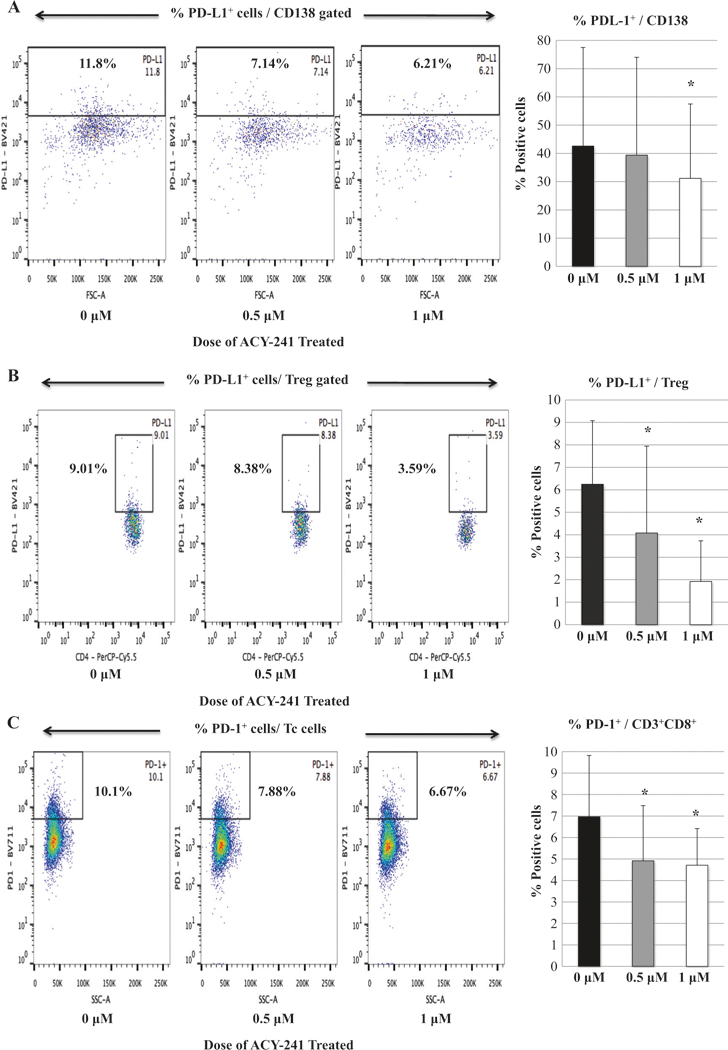
Decreased expression of immune checkpoints in myeloma patients’ bone marrow mononuclear cells with ACY241 treatment. The expression levels of immune checkpoints were evaluated following treatment of myeloma patients’ bone marrow mononuclear cells with ACY241 (0, 0.5, 1 μM; 48 h). ACY241 treatment decreased PD-L1 expression on CD138+ MM cells (Fig. 2a) and regulatory T cells (Fig. 2b) as well as PD-1 expression on CD8+ Tc (Fig. 2c) in a dose-dependent manner (p < 0.05)
ACY241 increases the expression of B7 and MHC on tumor and dendritic cells, and enhances the proliferation of CD4+ Th and CD8+ Tc cells
We next evaluated changes in key surface antigens on tumor and immune cells triggered by ACY241 treatment (0 μM, 0.5 μM, 1 μM, 2μM). The expression of B7 family proteins (CD80, CD86) (Fig. 3a), as well as MHC Class I (HLA-A2, HLA-ABC) and MHC Class II (HLA-DP/DQ/DR) (Fig. 3b), was significantly increased by ACY241 in a dose-dependent manner on MM (U266), breast cancer (MDA-MB231), and colon cancer (SW480) cells (n = 3, p < 0.05). We observed a dose-dependent upregulation of B7 and MHC molecules on dendritic cells following ACY241 treatment (n = 3, p < 0.05) (Supplemental Fig. 4). In addition, ACY241 (1 μM)-treated dendritic cells induced a greater T cell proliferation (treated 72.2% vs. untreated 50.9%) in mixed lymphocytes reactions, including both CD4+ (treated 79.9% vs. untreated 52.2%) and CD8+ T cells (treated 76.3% vs. untreated 49.2%) (Fig. 3c). Thus, these results demonstrate that ACY241 treatment upre-gulates expression of key antigens on both tumor and dendritic cells and induction of T cell proliferation, suggesting a role of ACY241 in enhancing activities on antigen-presentation and effector cells.
Fig. 3.
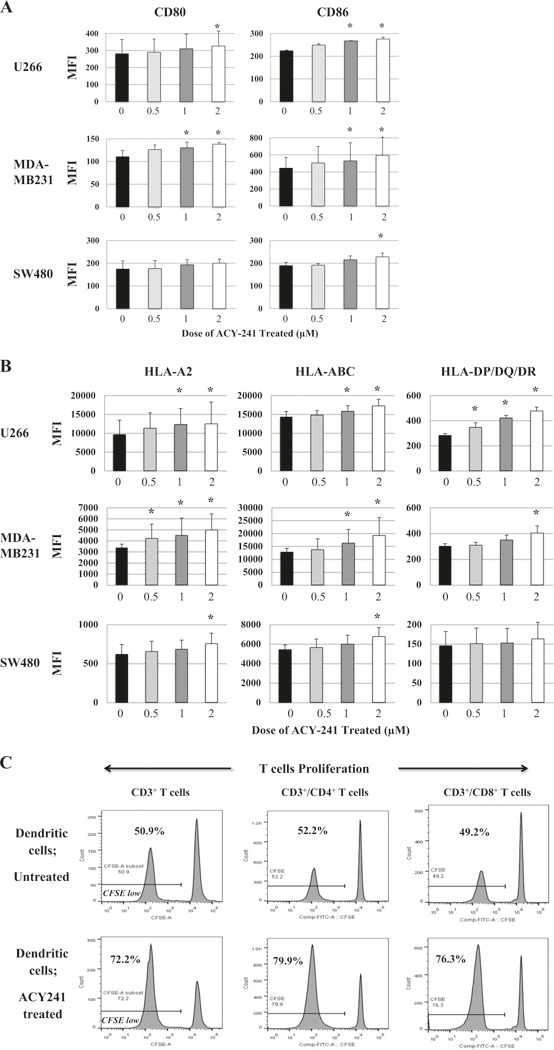
Upregulation of CD80, CD86, HLA-ABC and HLA-DP/DQ/DR on tumor and dendritic cells and induction of T cell proliferation with ACY241 treatment. ACY241 treatment increased expression of CD80/CD86 (Fig. 3a) and HLA-A2, HLA-ABC, and HLA-DP/DQ/DR (Fig. 3b) on myeloma U266, breast cancer MDA-MB231, and/or colon cancer SW480 cells in a dose-dependent manner (p < 0.05). ACY241 (1 (μM)-treated dendritic cells induced a high level of proliferation of CD3+ T cells, including both CD4+ and CD8+ cells, in mixed lymphocytes reactions (Fig. 3c)
ACY241 upregulates co-stimulatory and activation molecules on XBP1-CTL
The immune modulatory potential of ACY241 was further evaluated using XBP1-CTL. Overall, ACY241 (0.5–2 μM; 48 h) did not alter the viability or T cell subset frequency within the antigen-specific CTL (Supplemental Fig. 5); however, it did increase their expression levels of co-stimulatory (CD28) and activation (CD38) markers (Fig. 4a). Indeed, ACY241 induced a dose-dependent (0.5 μM, 1 μM, 2 μM) increase in the frequency of CD28+, CD38+, and CD28+CD38+ expressing CD8+ T cells within XBP1-CTL (n = 3, p < 0.05) (Fig. 4a). Treatment also triggered a gradual time-dependent (0 h <6 h <18 h <24 h <48 h) increase in CD28 expression, assessed by median fluorescence intensity (Fig. 4b). Moreover, increased CD28 expression was maintained up to 6 days post-treatment. Besides CD28, ACY241 increased expression of secondary costimulatory markers including CD40L (912 > 788 > 619), 41BB (1030 > 923 > 772) and 0X40 (1933 > 1523 >1381) in a dose-dependent manner (1>0.5>0μM). The increased expression level for each of these markers (histograms; Fig. 4c) directly correlated with their increased frequency (Fig. 4c). These results therefore provide additional support for the activity of ACY241 in enhancing multiple co-stimulatory pathways and CD8+ T cell activation, thereby augmenting antigen-specific CD8+ CTL activity.
Fig. 4.
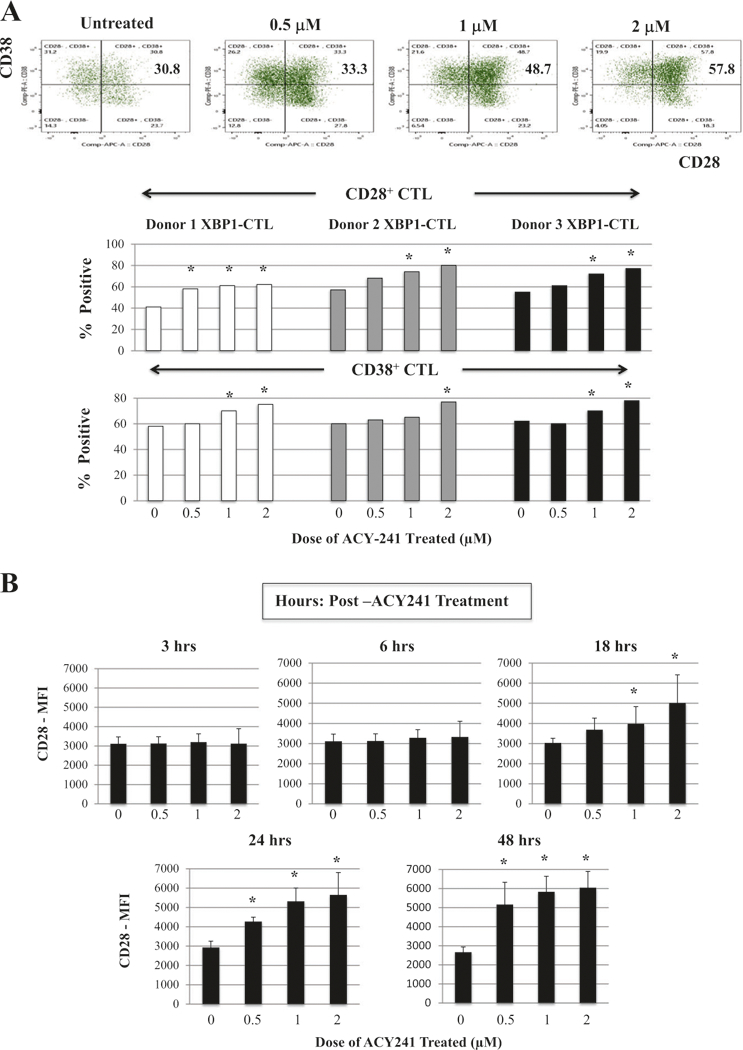
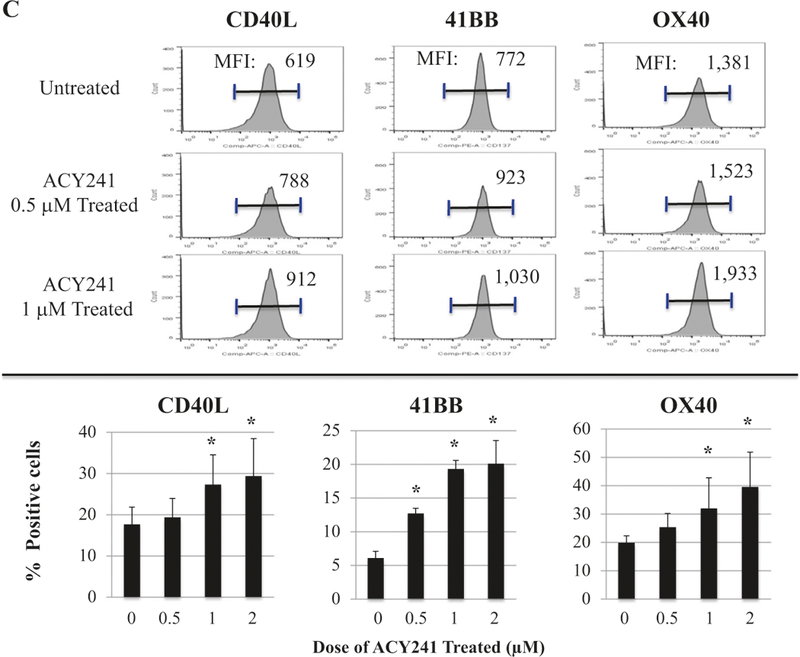
Increased expression of costimulatory and activation markers on XBP1-CTL with ACY241 treatment. XBP1 antigen-specific CTL were generated from HLA-A2+ normal donors’ CD3+ T cells by repeated weekly stimulation with a cocktail of heteroclitic unspliced XBP1184-192 (YISPWILAV) and spliced XBP1 SP196–204 (YLFPQLISV) peptides. After the fourth cycle of peptides stimulation, CTL were treated with ACY241 for 48 h and evaluated for their cell viability and cell surface profile. The frequency of CD28+ or CD38+ single positive CTL, as well as CD28+CD38+ double positive CTL, was enhanced in a dose-dependent manner (Fig. 4a). CD28 co-stimulatory molecule expression on the XBP1-CTL was also increased in a time-dependent manner (Fig. 4b) by ACY241 treatment. The expression levels (MFI) and frequency of cells expressing CD40L, 41BB and 0X40 were increased upon ACY241 treatment in a dose-dependent manner (Fig. 4c) (p < 0.05)
ACY241 increases central memory CD8+ T cells within XBP1-CTL and augments their functional activities against tumor
Next, we evaluated the effects of selective HDAC inhibition on antigen-specific memory CTL and their antitumor activities. ACY241 increased the proportion of antigen-specific central memory (CM) CD8+ T cells in a dose-dependent manner (2.0 μM: 46.3% >1.0 μM: 38.2% > 0.5 μM: 29.6% >0 μM: 21.8%). The change of memory CTL subset was associated with a corresponding decrease in the effector memory (EM) subset (2.0 μM: 53.2% >1.0 μM: 61.2% >0.5 μM: 69.6% >0 μM: 77.8%) (n = 3, p < 0.05) (Fig. 5a). In addition, we observed an increase in CD28+/CD8+ T cells and a decrease in PD1+/CD8+ T cells in both the CM and EM CTL subsets (Fig. 5b). The phenotypic changes and increased CM frequency induced by ACY241 were associated with increased antitumor activities, evidenced by upregulation of CD107a degranulation (28.0% treated vs. 18.2% untreated), IFN-γ production (24.4% treated vs. 16.5% untreated) and 41BB expression (27.1% treated vs. 17.9% untreated) upon recognition of HLA-A2+ U266 myeloma cells (n = 3, p < 0.05). These anti-tumor activities were further enhanced when both tumor and antigen-specific CTL were treated with ACY241 (Supplemental Fig. 6), and the anti-myeloma effects were mainly enhanced within the cell subsets of CM (Supplemental Fig. 7) and EM (Supplemental Figure 8). ACY241 also upregulated antitumor activity of XBP1-CTL against solid tumors, evidenced by increased CD107a expression and IFN-γ/IL-2/ TNF-α production against HLA-A2+ breast cancer (MDA-MB231) and colon cancer (SW480) cells (n = 3, p < 0.05) (Fig. 5c). These results therefore provide further evidence for the potential of selective HDAC inhibition to increase anti-tumor activities of antigen-specific CTL by inducing expression of multiple co-stimulatory molecules on CD8+ T cells and B7, as well as MHC molecules on tumor and antigen-presenting cells.
Fig. 5.
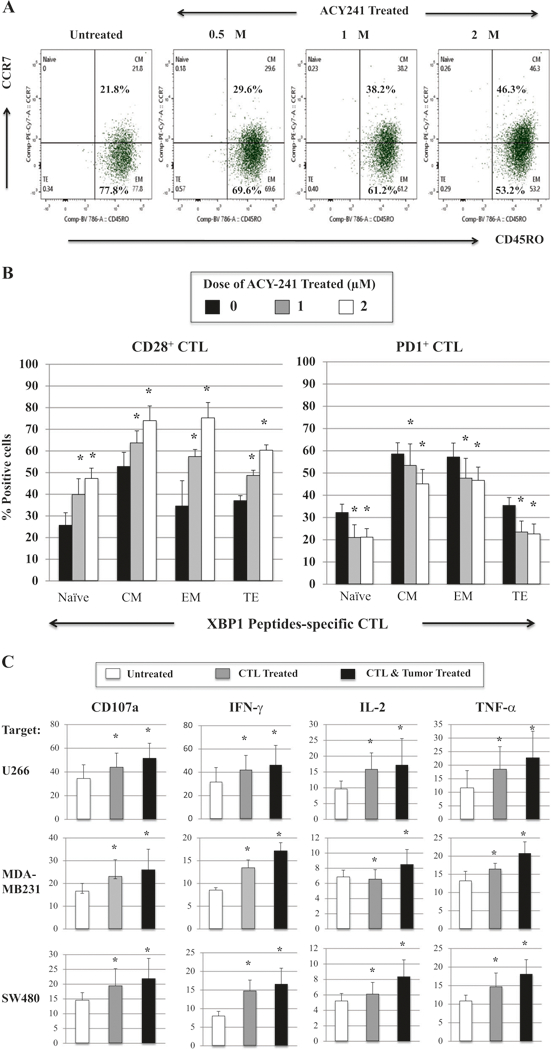
XBP1-CTL memory cell subset expansion and their increased anti-tumor activities with ACY241 treatment. ACY241 treatment (1 μM) of XBP1-CTL increased the central memory (CD45RO+CCR7+) and decreased the effector memory (CD45RO+CCR7-) cell subsets (Fig. 5a), and it increased CD28 expression and decreased PD1 expression in all of Naive:Memory cell subsets (Fig. 5b). The ACY241 treatment increased anti-tumor activities of XBP1-CTL against myeloma cells (U266), breast cancer cells (MDA-MB231) and colon cancer cells (SW480) (Average + SE; N = 3) (p < 0.05) (Fig. 5c)
Fig. 6.
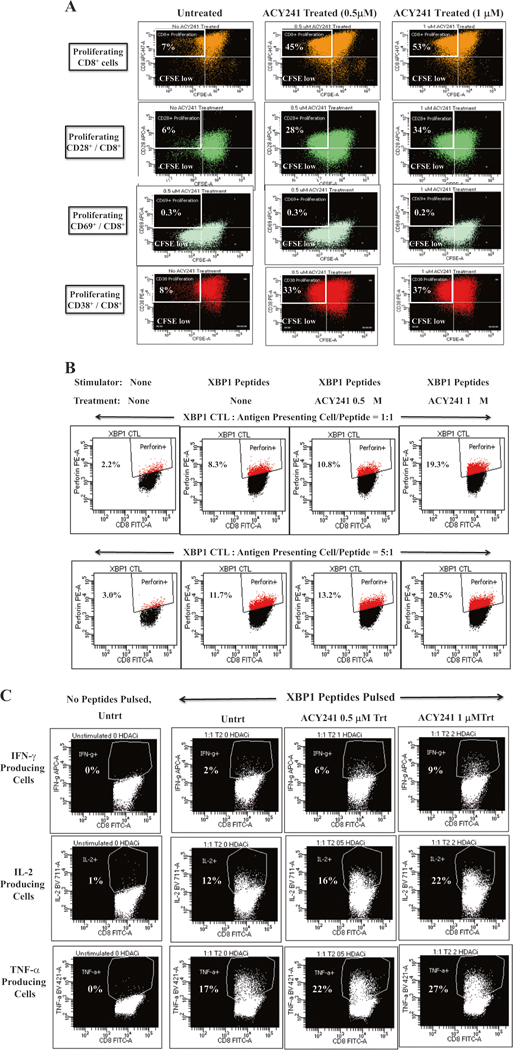
Increases peptides-specific responses of XBP1 antigen-specific CTL upon ACY241 treatment. XBP1 US184–192 (YISPWILAV) and XBP1 SP367–375 (YLFPQLISV) peptides-specific CTL activities were investigated following treatment with ACY241 (0 μM, 0.5 μM, 1 μM) and by examining CD8+ T cells proliferation, perforin upregulation, and Th-1 type cytokines production. ACY241 treatment induced dose-dependent T cell proliferation of CD8+ T cells, primarily those expressing CD28 or CD38 (Fig. 6a). ACY241 also increased perforin upregulation (Fig. 6b) and IFN-γ/IL-2/TNF-α production (Fig. 6c) in response to XBP1 specific peptides (p <0.05) in a dose-dependent manner
ACY241 increases the peptides-specific CD8+ T cell proliferation of XBP1-CtL
Next we showed that ACY241 treatment enhances antigen-specific activities, assessed by specific CD8+ T cells proliferation in response to the cognitive XBP1 US184–192 (YISPWILAV) and XBP1 SP367–375 (YLFPQLISV) peptides, in a dose-dependent manner (untreated: 7%, 0.5 μM ACY241 treated: 45%, 1.0 μM ACY241 treated: 53%) (top panel; Fig. 6a). Further studies demonstrated HDAC inhibition-induced proliferative responses in XBP1-CTL, which highly express the CD28 co-stimulatory molecule (untreated: 6%, 0.5 μM ACY241 treated: 28%, 1.0 μM ACY241 treated: 34%) or CD38 late activation marker (untreated: 8%, 0.5 μM ACY241 treated: 33%, 1.0 μM ACY241 treated: 37%). In contrast, proliferation was not induced in XBP1-CTL expressing the early activation marker (CD69) (untreated: 0.3%, 0.5 μM ACY241 treated: 0.3%, 1.0 μM ACY241 treated: 0.2%) (Fig. 6a). We also observed increased perforin production to the cognitive XBP1 peptides triggered by ACY241 treatment in a dose-dependent manner (Ratio of [E]: [S] = 1:1—untreated 8.3%, 1 μM treated 10.8%, 2 μM treated 19.3%; Ratio of [E]: [S] = 5:1—untreated 11.7%, 1 μM treated 13.2%, 2 μM treated 20.5%) (Fig. 6b). As compared to CTL alone (0~1% cytokines production), CTL stimulated with the cognitive XBP1 peptides increased Th1-type cytokines production (IFN-γ: 2%, IL-2: 12%, TNF-: 17%), which was further enhanced by ACY241 in a dose-dependent manner (Fig. 6c). Taken together, these data demonstrate that selective HDAC inhibition by ACY241 enhances the tumor-specific immune responses of XBP1-CTL via enhanced recognition of the specific peptides, thereby providing additional evidence for the ability of combination immunotherapy to increase the efficacy of peptide-based vaccines in patients.
ACY241 contributes to activation of the AKT (pS473)/mTOR (pS2448)/NF-KB p65 (pS529) pathways and increases Bcl-6+, HIF1+, Eomes+ and T-bet+ frequency within memory XBP1 peptides-specific CTL
Finally, to further delineate the effect of ACY241 on antigen-specific memory CTL, we evaluated signaling pathways important in regulating cell cycle, T cell proliferation, cytokine production, and/or survival [20–22]. We defined the effect of ACY241 on mechanisms which modulate antigen-specific memory CD8+ T cells, and specifically determined whether drug treatment induced changes in NF-κB activation, since a failure to maintain NF-κB signaling is associated with defective maintenance of memory CD8+ T cells due to impaired expression of Eomes, a critical transcriptional regulator in memory T cells [23]. We found that ACY241 enhanced AKT, mTOR and p65 activation within CD45RO+ memory CD8+ T cells, but not within CD45RO- non-memory CD8+ T cells in the antigen-specific CTL (n = 5, p < 0.05) (Fig. 7a). To further understand the mechanism of ACY241-induced HDAC inhibition on the antigen-specific CTL, we analyzed key transcriptional regulators Bcl-6, HIF-1, Eomes and T-bet, which control critical functions relating to effector and memory T cell differentiation [24–26]. As seen in the AKT, mTOR and p65 pathways, ACY241 enhanced upregulation of the key transcriptional regulators in the antigen-specific CTL (Fig. 7b), which was mainly detected within the memory CTL subset (n = 5, p < 0.05) (Fig. 7c). These results indicate that ACY241 has a role in regulation of critical pathways including AKT/mTOR/p65 and key transcriptional regulators Bcl-6/HIF-1/Eomes/T-bet, which contribute to induction and maintenance of antigen-specific memory CD8+ T cells with anti-tumor activities.
Fig. 7.
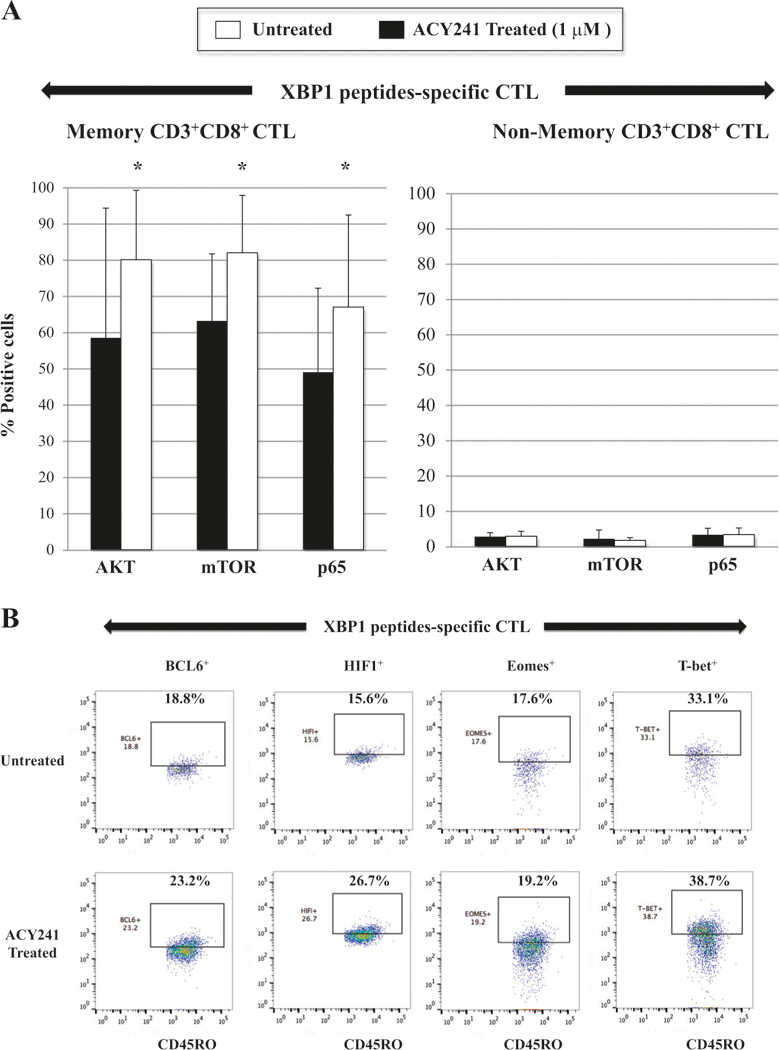
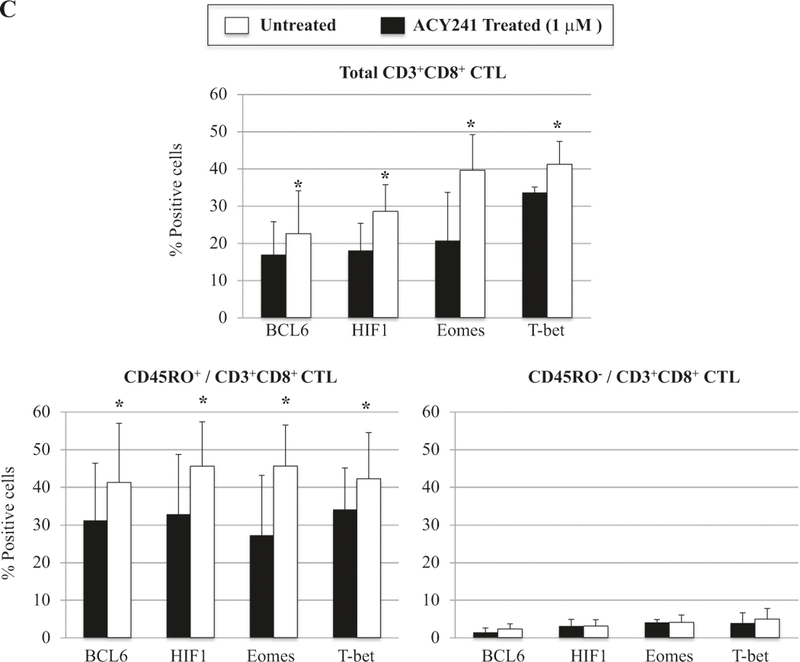
Increased AKT/mTOR/NF-κB p65 activation and Bcl-6, HIF1, Eomes and T-bet expression in memory CD8+ T cells of XBP1 antigen-specific CTL triggered by ACY241 treatment. XBP1-CTL were evaluated for the effects of ACY241 treatment on the AKT (pS473)/mTOR (pS2448)/NF-κB p65 pathways and the Bcl-6, HIF1, Eomes and T-bet expression. ACY241 treatment enhanced the activation levels of AKT, mTOR and p65, mainly within the memory cell subsets in the XBP1-CTL (Fig. 7a). In addition, ACY241 treatment enhanced the expression of key transcriptional regulators Bcl-6, HIF1, Eomes and T-bet in XBP1-CTL (Fig. 7b), which was detected mainly within memory T cell subset (p <0.05) (Fig. 7c)
Discussion
To date, four HDAC inhibitors, Vorinostat (SAHA; Pan-HDACi), Romidepsin (FK-228), Belinostat (PXD-101), and Panobinostat (LBH-589) have been granted FDA approval for treatment of various cancers. Additional HDAC inhibitors are currently in clinical trials, and multiple efforts are ongoing to develop novel HDAC inhibitors that can increase anti-tumor responses. However, to date the immune modulatory activity of HDAC inhibitors on tumor-specific immunity has not been well demonstrated or characterized. Here, we report on the ability of a selective HDAC6 inhibitor ACY241 to reduce CD138+ tumor cells, CD4+CD25+FoxP3+ Treg, and HLA-DRLow/-CD11b+CD33+ MDSC; decrease expression of PD1/PD-L1 on both immune suppressor and CD8+ T cells; increase B7 (CD80, CD86) and MHC (Class I, Class II) expression on tumor and dendritic cells; induce co-stimulatory (CD28, 41BB, CD40L, 0X40) and activation (CD38) molecules expression on CD8+ T lymphocytes; and enhance antigen-specific central memory CTL with anti-tumor activity. ACY241, alone and in combination, may therefore be useful to restore host anti-tumor immunity in patients.
We particularly examined whether ACY241 may enhance the efficacy of a cancer vaccine through direct modulation of anti-tumor activities of antigen-specific CD8+ CTL. Previously, we have demonstrated the immu-nogenicity of XBP1 US184–192 (YISPWILAV) and XBP1 SP367–375 (YLFPQLISV) peptides as a cancer vaccine capable of inducing antigen-specific CTL, and have shown that immunomodulatory drug lenalidomide can increase functional activities of antigen-specific CTL [27–30]. In this study, we examined the effect of the selective HDAC6 inhibitor ACY241 on regulating XBP1-CTL function, and demonstrated its ability to modulate the MM patients’ bone marrow microenvironment by targeting and decreasing both CD138+ MM cells and immune suppressors, as well as down-regulating immune checkpoints on BMMs. Moreover, ACY241 induced phenotypic changes on the tumor and dendritic cells, as well as increased co-stimulatory markers on XBP1-CTL, thereby enhancing their anti-tumor activity against myeloma, breast cancer, and colon cancer cells expressing the XBP1 antigen. These observations both complement and contradict previous reports by other investigators. For example, the Class I HDAC inhibitor, Depsipeptide, induced a higher level of tumor antigen gp100 expression in murine melanoma cells, which was associated with increased tumor cell killing by gp100-specific CTL [31]. On the other hand, the pan-HDAC inhibitor Valproic acid downregulated the expression of tumor antigen MUC1 and upregulated another tumor-associated antigen, NY-ESO-1, in mesothelioma cells in vitro [32]. Additionally, other investigators have demonstrated that HDAC inhibitors enhance the function of CD8+ T cells and induce hyper-acetylation of the IFN-γ locus in memory CD8+ T cells, but not in CD4+ T cells, allowing for the rapid production of IFN-γ by CTL [33]. More recently, it was demonstrated that the function of exhausted CD8+ T cells in chronic viral infection is restored upon exposure to pan-HDAC inhibitors, but the effect of Class II HDAC inhibitors on CD4+ and CD8+ T cells has to date not yet been reported [34–36]. Coupled with the present study, these reports suggest that specific isoforms of HDAC inhibitors may regulate critical markers involved in antigen recognition machinery, co-stimulatory molecule expression, and tumor-associated antigen expression.
The major goal of a cancer vaccine is to effectively induce antigen-specific memory CD8+ T cells; thus understanding the basis of memory T cell differentiation is critical for rational vaccine design. Despite the complexity of multiple interrelated signaling pathways involved in memory T cell differentiation, several pathways critically influence T cell memory development. Recent studies have identified AKT as a critical signal integrator that links distinct facets of antigen-specific CTL differentiation to FOXO, mTOR and Wnt/β-catenin signaling pathways [37, 38]. For example, mTOR is a critical regulator of CD8+ T cells: mTOR complex 1 (mTORC1) promotes CD8+ T cell effector responses whereas mTORC2 activity enhances CD8+ T cell memory [39, 40]. Thus, both AKT and mTOR have distinct roles as a rheostat in orchestrating the differentiation of CD8+ T cells and generation of memory CTL [41, 42]. In addition, specific activation of AKT and mTOR is associated with CD28 costimulatory molecule, a critical regulator of T cell proliferation and antigen-specific effector T cell development [43, 44]. In this context, we in the present study hypothesized that modulation of AKT and mTOR pathways is critical to maintaining memory CTL and anti-tumor activities induced by XBP1 specific peptides. In previous studies, we have demonstrated that lenalidomide enhanced AKT expression on antigen-specific CTL, associated with CD28 upregulation and antigen-specific memory CTL expansion [45]. In current study, we demonstrate that ACY241 triggers activation of AKT via phosphorylation at Ser473, a phosphorylation site shared by all three AKT isoforms; and induces activation of mTOR via phosphorylation at Ser2448, the only phosphorylation site of mTOR [46–50]. Specifically, we demonstrated the ability of the selective HDAC6 inhibitor ACY241 to activate the AKT and mTOR pathways in XBP1-CTL, resulting in increased expansion and function of antigen-specific memory CD8+ CTL with anti-tumor activities. These findings were repeatedly observed in XBP1-CTL generated from different donors and targeting various tumors including not only multiple myeloma, but also breast cancer and colon cancer.
Having shown activation of AKT and mTOR and immunologic sequelae triggered by ACY241, we next delineated mechanisms mediating these effects. Both AKT and mTOR activation profoundly affect the expression and activity of many immunologically relevant transcription factors, which in turn promote immune signaling and mediate effector functions. These transcription factors orchestrate cell metabolism (HIF1), lineage differentiation (T-bet, Eomes, Bcl-6), and immune activation and functions (NF-κB) [51]. T-bet and Eomes play major roles in generation of effector and memory CD8+ T cells during early stages of CD8+ T cell activation, as well as upregulation of IFN-γ and granzyme B in antigen-specific T cells [52–54]. Moreover, increased CD8+ T cell expression of T-bet and Eomes has been associated with longer patients’ survival [55–57]. Our previous reports also demonstrated that increased T-bet and Eomes expression in antigen-specific CTL were associated with greater anti-tumor activity against myeloma and solid tumors [45]. In addition, the Bcl-6 and Blimp-1 regulatory axis controls effector and memory T cell differentiation and function; importantly, increased Bcl-6 and Blimp-1 expression promotes development of tumor antigen-specific CD8+ T cells into memory cells [58–60]. We here in particular examined the effect of ACY241 on these T cell transcriptional regulators including Bcl-6, HIF-1, T-bet and Eomes, due to their critical roles in effector and memory CD8+ T cell differentiation. Our results demonstrate that ACY241 treatment affects and modulates these transcriptional regulators and AKT/mTOR signaling, within antigen-specific memory CD8+ T cell population, thereby enhancing their anti-tumor activities. This ability of ACY241 to promote antigen-specific CD8+ memory CTL with anti-tumor activity suggests that it may promote sustained clinical responses.
In summary, the selective HDAC6 inhibitor ACY241 mediates anti-MM activity both by decreasing CD138+ tumor cells and tumor-promoting immune cells and their expression of immune checkpoints, as well as by promoting the activation of antigen-specific CD8+ T cells. Importantly, it increases activation of AKT/mTOR/p65 pathways and upregulates transcription regulators Bcl-6, Eomes, HIF-1 and T-bet, thereby promoting development of antigen-specific central memory CD8+ CTL and their anti-tumor activities. Our studies therefore delineate immune modulatory effects of ACY241 and provide the rationale for its clinical evaluation to promote development and function of antigen-specific CD8+ memory T cells with anti-tumor activity, and thereby improve patient outcome.
Supplementary Material
Acknowledgements
The authors acknowledge Mr. John Daley and Ms. Suzan Lazo-Kallanian from the flow cytometry facility at Dana-Farber Cancer Institute for providing flow cytometry assistance. This work was supported in part by grants from the National Institutes of Health Grants RO1–124929 to Dr. Nikhil C. Munshi, P50–100007, PO1–78378 and PO1–155258 to Drs. Kenneth C. Anderson and Nikhil C. Munshi, and RO1–50947 to Dr. Kenneth C. Anderson. Dr. Kenneth C. Anderson is an American Cancer Society Clinical Research Professor.
Footnotes
Compliance with ethical standards
Conflict of interest N.C. Munshi is on advisory board of Celgene, Millennium and Novartis; K.C. Anderson is on advisory board of Gilead, Millennium and Bristol Myers Squibb; and P.G. Richardson is on advisory board of Celgene, Millennium and Johnson & Johnson. N. C. Munshi, K.C. Anderson and J. Bae have ownership interest and are advisory board members in OncoPep Inc. The remaining authors declare that they have no conflict of interest.
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41375–018-0062–8) contains supplementary material, which is available to authorized users.
References
- 1.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin K, Schreiner J, Zippelius A. Modulation of APC function and anti-tumor immunity by anti-cancer drugs. Front Immunol. 2015;6:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kersten K, Salvagno C, de Visser KE. Exploiting the immunomodulatory properties of chemotherapeutic drugs to improve the success of cancer immunotherapy. Front Immunol. 2015;6:516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroesen M, Gielen P, Brok IC, Armandari I, Hoogerbrugge PM, Adema GJ. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget. 2014;5:6558–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cicek MS, Koestler DC, Fridley BL, Kalli KR, Armasu SM, Larson MC, et al. Epigenome-wide ovarian cancer analysis identifies a methylation profile differentiating clear-cell histology with epigenetic silencing of the HERG K + channel. Hum Mol Genet. 2013;22:3038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336:736–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maolanon AR, Madsen AS, Olsen CA. Innovative strategies for selective inhibition of histone deacetylases. Cell Chem Biol. 2016;23:759–68. [DOI] [PubMed] [Google Scholar]
- 10.Batchu SN, Brijmohan AS, Advani A. The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin Sci (Lond). 2016;130:987–1003. [DOI] [PubMed] [Google Scholar]
- 11.Hideshima T, Mazitschek R, Qi J, Mimura N, Tseng J-C, Kung AL, Bradner JE, et al. HDAC 6 inhibitor WT 161 downregulates growth factor receptors in breast cancer. Oncotarget. 2017;8:80109–23. in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hideshima T, Qi J, Paranal RM, Tang W, Greenberg E, West N, et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc Natl Acad Sci USA. 2016;113:13162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009;10:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. 2005;11:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogl DT, Raje N, Jagannath S, Richardson P, Hari P, Orlowski R, et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin Cancer Res. 2017;23:3307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yee AJ, Bensinger WI, Supko JG, Voorhees PM, Berdeja JG, Richardson PG, et al. Ricolinostat plus lenalidomide, and dex-amethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol. 2016;17:1569–78. [DOI] [PubMed] [Google Scholar]
- 17.Mishima Y, Santo L, Eda H, Cirstea D, Nemani N, Yee AJ, et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br J Haematol. 2015;169:423–34. [DOI] [PubMed] [Google Scholar]
- 18.Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray A, Das DS, Song Y, Hideshima T, Chauhan D, Anderson KC Combination of a novel HDAC6 inhibitor ACY-241 and anti-PD-L1 antibody enhances anti-tumor immunity and cytotoxicity in multiple myeloma. Leukemia 2017. 10.1038/leu.2017.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogel A, Willoughby JE, Buchan SL, Leonard HJ, Thirdborough SM, Al-Shamkhani A. Akt signaling is critical for memory CD8 + T-cell development and tumor immune surveillance. Proc Natl Acad Sci USA. 2017;114:E1178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araki K, Morita M, Bederman AG, Konieczny BT, Kissick HT, Sonenberg N, Ahmed R. Translation is actively regulated during the differentiation of CD8 + effector T cells. Nat Immunol. 2017;18:1046–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11:114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knudson KM, Pritzl CJ, Saxena V, Altman A, Daniels MA, Teixeiro E. NFKB-Pim-1-Eomesoderminaxis is critical for maintaining CD8 T-cell memory quality. Proc Natl Acad Sci USA. 2017;114:E1659–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gnanaprakasam JNR, Sherman JW, Wang R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 2017;35:63–70. [DOI] [PubMed] [Google Scholar]
- 25.Larbi A, Zelba H, Goldeck D, Pawelec G. Induction of HIF-1alpha and the glycolytic pathway alters apoptotic and differentiation profiles of activated human T cells. J Leukoc Biol. 2010;87:265–73. [DOI] [PubMed] [Google Scholar]
- 26.Li G, Yang Q, Zhu Y, Wang HR, Chen X, Zhang X, et al. T-Bet and eomes regulate the balance between the effector/central memory T cells versus memory stem like T cells. PLoS One. 2013;8:e67401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bae J, Prabhala R, Voskertchian A, Brown A, Maguire C, Richardson P, et al. A multiepitope of XBP1, CD138 and CS1 peptides induces myeloma-specific cytotoxic T lymphocytes in T cells of smoldering myeloma patients. Leukemia. 2015;29:218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bae J, Samur M, Munshi A, Hideshima T, Keskin D, Kimmelman A, et al. Heteroclitic XBP1 peptides evoke tumor-specific memory cytotoxic T lymphocytes against breast cancer, colon cancer, and pancreatic cancer cells. OncoImmunology. 2014;3:e970914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bae J, Smith R, Daley J, Mimura N, Tai YT, Anderson KC, et al. Myeloma-specific multiple peptides able to generate cytotoxic T lymphocytes: a potential therapeutic application in multiple myeloma and other plasma cell disorders. Clin Cancer Res. 2012;18:4850–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bae J, Carrasco R, Lee AH, Prabhala R, Tai YT, Anderson KC, et al. Identification of novel myeloma-specific XBP1 peptides able to generate cytotoxic T lymphocytes: a potential therapeutic application in multiple myeloma. Leukemia. 2011;25:1610–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murakami T, Sato A, Chun NA, Hara M, Naito Y, Kobayashi Y, et al. Transcriptional modulation using HDACi depsipeptide promotes immune cell-mediated tumor destruction of murine B16 melanoma. J Invest Dermatol. 2008;128:1506–16. [DOI] [PubMed] [Google Scholar]
- 32.Roulois D, Blanquart C, Panterne C, Gueugnon F, Gregoire M, Fonteneau JF. Downregulation of MUC1 expression and its recognition by CD8(+) T cells on the surface of malignant pleural mesothelioma cells treated with HDACi. Eur J Immunol. 2012;42:783–9. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Q, Zhang L, Li L, Wang Z, Ying J, Fan Y, et al. Interferon regulatory factor 8 functions as a tumor suppressor in renal cell carcinoma and its promoter methylation is associated with patient poor prognosis. Cancer Lett. 2014;354:227–34. [DOI] [PubMed] [Google Scholar]
- 34.Woods DM, Woan K, Cheng F, Wang H, Perez-Villarroel P, Lee C, et al. The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res. 2013;23:341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan AN, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother. 2008;57:647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–24. [DOI] [PubMed] [Google Scholar]
- 37.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, et al. mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J Clin Invest. 2015;125:2090–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rao RR, Li Q, Shrikant PA. Fine-tuning CD8(+) T cell functional responses: mTOR acts as a rheostat for regulating CD8(+) T cell proliferation, survival and differentiation? Cell Cycle. 2010;9:2996–3001. [DOI] [PubMed] [Google Scholar]
- 41.Nanjappa SG, Hernández-Santos N, Galles K, Wuthrich M, Sur-esh M, Klein BS. Intrinsic MyD88-Akt1-mTOR signaling coordinates disparate Tc17 and Tc1 responses during vaccine immunity against fungal pneumonia. PLoS Pathog. 2015;11: e1005161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veliça P, Zech M, Henson S, Holler A, Manzo T, Pike R, et al. Genetic regulation of fate decisions in therapeutic T cells to enhance tumor protection and memory formation. Cancer Res. 2015;75:2641–52. [DOI] [PubMed] [Google Scholar]
- 43.Cheng J, Hamilton KS, Kane LP. Phosphorylation of Carma1, but not Bcl10, by Akt regulates TCR/CD28-mediated NF-KB induction and cytokine production. Mol Immunol. 2014;59:110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kane LP, Andres PG, Howland KC, Abbas AK, Weiss A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not TH2 cytokines. Nat Immunol. 2001;2:37–44. [DOI] [PubMed] [Google Scholar]
- 45.Bae J, Keskin D, Cowens K, Lee AH, Dranoff G, Munshi NC, et al. Lenalidomide polarizes Th1-specific anti-rumor immune response and expands XBP1 antigen-specific central memory CD3+CD8+ T cells against various solid tumors. J Leuk. 2015;3:178–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. [DOI] [PubMed] [Google Scholar]
- 47.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoi-nositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–71. [DOI] [PubMed] [Google Scholar]
- 49.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. [DOI] [PubMed] [Google Scholar]
- 50.Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature. 2014;508:541–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng H, Chi H. mTOR signaling and transcriptional regulation in T lymphocytes. Transcription. 2014;5:e28263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity. 2012;36:374–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glimcher LH, Townsend MJ, Sullivan BM, Lord GM. Recent developments in the transcriptional regulation of cytolytic effector cells. Nat Rev Immunol. 2004;4:900–11. [DOI] [PubMed] [Google Scholar]
- 54.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–3. [DOI] [PubMed] [Google Scholar]
- 55.Li G, Yang Q, Zhu Y, Wang HR, Chen X, Zhang X, et al. T-bet and Eomes regulate the balance between the effector/central memory T cells versus memory stem like T cells. PLoS One. 2013;8:e67401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pipkin M, Sacks J, Cruz-Guilloty F, Lichtenheld M, Bevan M, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen LJ, Zheng X, Shen YP, Zhu YB, Li Q, Chen J, et al. Higher numbers of T-bet(+) intratumoral lymphoid cells correlate with better survival in gastric cancer. Cancer Immunol Immunother. 2012;62:553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Z, Liu JQ, Talebian F, Wu LC, Li S, Bai XF. IL-27 enhances the survival of tumor antigen-specific CD8 + T cells and programs them into IL-10-producing, memory precursor-like effector cells. Eur J Immunol. 2013;4:468–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11:114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.D’Cruz LM, Rubinstein MP, Goldrath AW. Surviving the crash: transitioning from effector to memory CD8+ T cell. Semin Immunol. 2009;21:92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.