

Abstract
Activation, differentiation and expansion of alloreactive CD8+ T cells, the dominant effectors that mediate murine heart allograft rejection, requires allorecognition, costimulation, and cytokine-initiated signals. While previous work showed that alloreactive CD4+ T cell immunity entails immune cell-produced and locally activated complement, whether and how C3a receptor 1 (C3aR1) signaling impacts transplant outcomes and the mechanisms linking C3aR1 to alloreactive CD8+ T cell activation/expansion remain unclear. Herein we show that recipient C3aR1 deficiency or pharmacological C3aR1 blockade synergizes with tacrolimus to significantly prolong allograft survival vs. tacrolimus-treated controls (median survival time 21 vs. 14 days, p<0.05). Recipient C3aR1-deficiency reduced the frequencies of post-transplant, donor-reactive CD8+ T cells 2-fold. Reciprocal adoptive transfers of naive WT or C3ar1−/− CD8+ T cells into syngeneic WT or C3ar1−/− allograft recipients showed that T cell-expressed C3aR1 induces CD8+ T proliferation, mTOR activation and transcription factor T-bet expression. Host C3aR1 indirectly facilitates alloreactive CD8+ T cell proliferation/expansion by amplifying antigen presenting cell costimulatory molecule expression and innate cytokine production. In addition to expanding mechanistic insight, our findings identify C3aR1 as a testable therapeutic target for future studies aimed at improving human transplant outcomes.
INTRODUCTION
CD4+ and CD8+ T cells contribute to transplant rejection (1, 2), with CD4+ T cell-derived helper signals guiding differentiation of CD8+ T cells into effector cytotoxic lymphocytes and pro-inflammatory cytokine producers that dominantly mediate graft injury (1, 3, 4). Both CD4+ and CD8+ T cell subsets undergo activation, differentiation and expansion under circumstances that simultaneously provide at least three distinct signals: a) cognate interactions between their surface-expressed heterodimeric T cell receptor (TCR) and donor allogeneic major histocompatibility molecules (MHC), b) costimulatory signals that include ligation of T cell-expressed CD28 and CD154 by antigen presenting cell-(APC)-expressed CD80/CD86 or CD40, respectively (5–7) (among others), and c) APC-derived cytokines binding to T cell-expressed cytokine receptors. As one example of the latter, APC-derived IL-12 induces CD4+ and CD8+ effector T cell (TEFF) expansion and in part, differentiation into IFNγ producers, processes that are dependent upon the transcription factor T-bet (8–14).
The complement system, traditionally considered a component of innate immunity is now recognized as a crucial modulator of murine and human CD4+ T cell immunity (15–19). Our cumulative work since 2005 has shown that autocrine C5a/C5aR1 and C3a/C3aR1 ligations on CD4+ T cells and on dendritic cells (DCs) are vital signals that activate CD4+ effector T cells (TEFF)(18–22) and inhibit generation, function and stability of CD4+ regulatory T cells (TREG)(23–26). Absence/blockade of these signals inhibits CD4+ TEFF and enhances generation, function and stability of TREG, favoring immune tolerance (18–28); the concepts apply to CD4+ T cells responding to model antigens, autoantigens, infectious pathogens and transplant antigens. Currently understood molecular mechanisms are that ligation of C3aR1 and/or C5aR1, seven transmembrane-spanning, G-protein coupled receptors, on murine and human CD4+ T cells, transmits signals that activate phosphoinositol-4,5-bisphosphate-3-kinase-γ (PI-3Kγ) and cause phosphorylation of phosphokinase B (PKB; AKT), and thereby induce cell proliferation and survival (18, 19). The same signals also result in phosphorylation of Foxo1/3a, sequestering these molecules within the cell cytoplasm (25) and as a consequence preventing generation, stability and function of CD4+ TREG. While these mechanisms clearly apply to alloreactive CD4+ T cells, and while complement-induced CD4+ T cell help can indirectly amplify alloreactive CD8+ T cell responses (4), whether C3aR1 signaling on CD8+ T cells directly impacts their ability to respond to alloantigens in vivo is unclear. Additionally, whether targeting C3aR1, without incapacitating the entire complement system that could engender increased infectious risk, can delay T cell-dependent allograft rejection has not been determined. Herein, using murine models, we provide evidence that blocking C3aR1 signaling synergizes with calcineurin inhibition to prolong murine cardiac allograft survival, and we show that C3aR1 signaling is linked to alloreactive CD8+ T cell responses in vivo through several distinct mechanisms.
MATERIALS AND METHODS
Mice
C57BL/6 (B6, H-2b), B6 CD45.1, B6 C3−/−, BALB/c (H-2d), congenic BALB/c C3ar1−/− mice 8 weeks of age were purchased from Jackson Laboratory (Bar Harbor, ME) or bred from Jackson-derived animals at Mount Sinai. Congenic B6 C3ar1−/− mice were obtained from Dr. Craig Gerard (Children’s Hospital, Boston, MA) and backcrossed >6 additional generations to B6 at Mount Sinai. All animals were housed in the Center for Comparative Medicine and Surgery at the Icahn School of Medicine at Mount Sinai under Institutional Animal Care in accordance with guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International. Experiments were performed with age- and sex- matched mice and using animals that were littermates or were maintained in the same room and/or were co-housed within the same cages for >2 weeks to limit potential effects of microbiome differences.
Heterotopic Heart Transplantation
Murine heterotopic heart transplantation was performed by the microsurgery core in the Icahn School of Medicine at Mount Sinai as previously described (29–31). In some experiments recipients were treated with 1 mg/kg/day tacrolimus (Prograf, Astellas, Northbrook, IL) i.p. daily starting on the day of surgery until cessation of heartbeat. Rejection was defined as the day on which a palpable heartbeat was no longer detectable and was confirmed by histological examination. In selected experiments, animals were treated with a pharmacological C3aR1 antagonist (SB 290157, Calbiochem, San Diego, CA, USA) (32), administered beginning the day prior to transplantation via a subcutaneous osmotic pump (Alzet, Cupertino, CA) containing 1mM of the C3aR1 antagonist (10% DMSO).
Cell Isolations
Spleens were passed through a 40μm strainer (BD Falcon, Corning, Tewksbury, MA) to make a single-cell suspension and subsequently lysed with red blood cell (RBC) lysis buffer (Life Technologies, Carlsbad, CA). Splenic APC enrichment (CD90.2+ T cell bead depletion, kit #18951) and naïve CD8+ T cell isolations (kit # 19858, negative isolation) were performed by magnetic bead isolation as per manufacturer’s instructions (STEMCELL, Vancouver, BC, Canada). For dendritic cell (DC) analyses, spleens were minced with razor blades and treated with collagenase D (Roche, Basel, Switzerland, 1mg/ml in 1x PBS) for 30 minutes in a 37°C water bath, vortexing every 5 minutes, prior to 40μm strainer filtration and RBC lysis. For heart graft infiltrating lymphocyte (GIL) analysis, grafts were minced with scissors and treated with collagenase A (Sigma, St. Louis, MO, 1mg/ml in 1x PBS) for 30 minutes in a 37°C water bath prior to 40μm strainer filtration and RBC lysis.
T Cell Adoptive Transfers
Recipient mice were anaesthetized by intra-peritoneal ketamine/xylazine administration. In each individual transplant experiment, equal numbers of CellTrace Violet (CTV, Life Technologies, Carlsbad, CA)-labeled, naïve, CD8+ T cells were adoptively transferred into congenic recipient mice via retro-orbital injection using an insulin syringe one day prior to the heterotopic heart transplantation procedure.
Mixed Lymphocyte Reaction (MLR)
Spleen cells from transplant recipient mice were isolated in single-cell suspension and co- cultured in 96-well cell culture-treated round-bottom plates in 1:1 ratio (200,000:200,000) with T-cell-depleted spleen cells from donor-origin mice (or syngeneic control APCs) in complete RPMI (RPMI (Gibco, Thermo Fisher, Waltham, MA) + 10% fetal bovine serum + L-glutamine + sodium pyruvate + nonessential amino acids + penicillin/streptomcyin + β-mercaptoethanol) in a 37°C incubator (5% CO2) overnight (18–22 hours). The following morning GolgiPlug (Life Technologies, Carlsbad, CA) was added (final dilution 1:1000) and incubated for 4 hours. Positive control wells were additionally treated with phorbol 12-myristate 13-acetate (PMA, 10ug/ml) and ionomycin (100ug/ml, Sigma, St. Louis, MO) during GolgiPlug incubation. Cells were incubated with fluorophore-conjugated antibodies and analyzed by flow cytometry.
CD8+ T cell Degranulation (CD107) Assay
Following degranulation, CD8+ T cells transiently express cell surface CD107a/b (33). In order to detect alloantigen-induced CD8+ T cell degranulation, spleen cells from transplant recipient mice were isolated in single-cell suspension and co- cultured in 96-well cell culture-treated round-bottom plates in 1:1 ratio (200,000:200,000) with T-cell-depleted spleen cells from donor-origin mice in complete RPMI in the presence of eFluor 660-conjugated CD107a (eBio1D4B), and CD107b (eBioABL-93, (eBioscience, Affymetrix, Santa Clara, CA) followed by a 4 hour incubation (37°C, 5% CO2). Positive control wells were additionally treated with phorbol 12-myristate 13-acetate (PMA, 10ug/ml) and ionomycin (100ug/ml) during the 4 hour incubation. Cells were then incubated with fluorophore-conjugated antibodies and analyzed by flow cytometry.
mTOR Signaling Assay
Single-cell suspensions of magnetic bead enriched, splenic, naïve, CD8+ T cells were plated in 96-well round bottom plates [2×105 cells per well in 100 μl HL-1 medium (Lonza, Basel, Switzerland)] supplemented with L-glutamine and penicillin/streptomycin) and rested in a 37°C (5% CO2) incubator for 2 hours prior to stimulation. The cells were stimulated with Mouse T-Activator CD3/CD28 Dynabeads™ (Gibco, Gaithersburg, MD, 2.5 μl per 2×105 cells). Recombinant C3a was purchased from Novoprotein (Summit, NJ) and added at a final concentration of 200ng/ml where indicated at 23 hours post-stimulation.
Serum Cytokine Analysis
Blood was collected by cheek bleed from mice on days 1, 4, 6 post-transplantation. Blood was clotted at room temperature for 2 hours and spun down at 5,000 x g for 10 minutes, and serum was subsequently collected and frozen at −80°C until analysis. Sera were diluted 1:4 in recommended assay buffer and analyzed by Mesoscale V-PLEX Custom Mouse Cytokine array on Quickplex SQ 120 analyzer as per manufacturer’s instructions (Mesoscale Discovery, Rockville, MD). Serum cytokine concentrations were calculated by program software in conjunction with provided standard curve samples that were reconstituted per manufacturer’s instructions.
Antibodies and Reagents
Flow cytometry antibodies against CD3 (17A2), CD4 (GK1.5), CD40 (3/23), CD45.1 (A20), and CD45.2 (104) were purchased from BioLegend (San Diego, CA); CD8 (2.43) and CD62L (MEL-14) were purchased from Tonbo (San Diego, CA); CD11b (M1/70), CD44 (IM7), CD45 (30-F11), FoxP3 (FJK-16s), IFNγ (XMG1.2), and eFluor 780 fixable viability dye were purchased from Invitrogen (Carlsbad, CA); CD11c (N418), H-2Kd (SF1–1.1.1), IL-4 (11B11), IL-17a (eBio17B7), T-bet (eBio4B10), TNFa (MP6-XT22) were purchased from eBioscience (Affymetrix, Santa Clara, CA); CD80 (16–10A1) and CD86 (GL1) were purchased from BD Biosciences (San Jose, CA); phospho-S6 (S235/236, REA454) was purchased from Miltenyi (Bergisch Gladbach, Germany). All antibodies or dyes were used at a dilution of 1:400 except: eFluor 780 fixable viability dye 1:1000, phospho-S6 1:100. Intracellular cytokine and transcription factor detection was performed using FoxP3 staining buffer set per manufacturer’s instructions (Affymetrix, Santa Clara, CA). Phospho-protein detection was performed using 1% paraformaldehyde fixation buffer (Electron Microscopy Sciences, Hatfield, PA), BD Phosflow Perm Buffer III (BD Biosciences, San Jose, CA), and wash/staining buffer consisting of 1% w/v BSA (Sigma, St. Louis, MO) in 1x PBS (Life Technologies, Carlsbad, CA). All incubations were performed in the dark on ice for 20 minutes except p-S6, 1 hour at room temperature.
Flow Cytometry
Flow cytometric analysis was performed on FACS CANTO II (BD Biosciences, San Jose, CA) and analyzed using Cytobank (Cytobank, Santa Clara, CA) or FlowJo software (Becton Dickinson, Franklin Lakes, NJ).
Statistical Analysis
Statistical analysis was performed with GraphPad Prism software version 5 (La Jolla, CA). Differences between graft survival rates were assessed by Mantel-Cox log rank test. Group comparisons were analyzed by Student’s t test (unpaired, two-tailed). All experiments were repeated at least twice. Data are presented as mean values with SD. Statistical significance is expressed as follows: *p<0.05, **p<0.01, ***p<0.001, and ns (p>0.05) indicates not significant.
RESULTS
Absence of C3aR1 limits CD8+ T cell responses to allografts
To initially discern the effects of recipient C3aR1 on alloimmunity, we transplanted groups of untreated, congenic, age- and sex-matched WT and C3ar1−/− B6 recipients with fully MHC-disparate, BALB/c hearts, documenting graft survival and quantifying donor-reactive T cell responses. The WT and C3ar1−/− recipients each rejected BALB/c hearts with a median survival time (MST) of 8 days (n=3–4/group, p=ns, not shown) indicating that C3aR1 deficiency alone is insufficient to prolong allograft survival. Nonetheless, immune analyses performed on day 6 post-transplant (separate sets of recipients) showed smaller spleens (WT 191.1±5.1×106, n=15 vs. C3ar1−/− 148.1±9.4×106, n=8, p<0.001, not shown) and 2-fold reduced frequencies and absolute numbers of donor-reactive, IFNγ-producing (Figure 1A top, 1B) and TNFα-producing (Figure 1A middle, 1C) CD8+ T cells in C3ar1−/− recipients vs. WT recipients. We did not detect intracellular IFNγ or TNFα within splenic CD8+ T cells from transplanted mice upon in vitro re-stimulation with syngeneic APCs (not shown), verifying that the above detected responses were donor-reactive. Nor did we detected donor-reactive IL-17- or IL-4-producing CD8+ T cells in spleens of either genotype (Figure S1). We also performed degranulation assays that detect surface-expressed CD107a/b after cytotoxic granule release as a surrogate of cytotoxicity (33). These assays showed a similar reduction in the percentages and total numbers of donor-reactive CD107+ CD8+ T cells in the absence of C3aR1 (Figure 1A bottom, 1D).
Figure 1: C3aR1 is required for optimal expansion of alloreactive CD8+ T cells induced by allogeneic heterotopic heart transplantation.

A. Representative flow plots of recipient spleen cells from WT B6 and C3ar1−/− recipients of BALB/c heart grafts isolated on day 6 post-transplant, stimulated with BALB/c (donor) APCs, and stained for intracellular expression of IFNγ (top), TNFγ (middle) or extracellular CD107a/b (bottom), gated on CD8+ T cells. Numbers depict the % of cytokine+ or CD107+ cells within the outlined region. No IFNγ or TNFγ was detected following stimulation with syngeneic B6 APCs (data not shown). B-D. Quantified percentages (left panels) and total numbers (right panels) of donor-reactive IFNγ-producers (B), TNFγ-producers (C) or CD107a/b+ (D) CD8+ T cells are shown. n=8/group. E. Quantified frequencies of IFNγ-producing CD4+ and CD8+ T cells from GIL (see gating strategy in Figure S2) obtained on day 6 post-transplant from WT and C3ar1−/− recipients of BALB/c hearts. n=7/group. *p<0.05, **p<0.01, ***p<0.001; ns, not significant.
Analyses of the percentages of the day 6 post-transplant, splenic, donor-reactive IFNγ-producing CD4+ T cells (WT 1.7±0.1% vs. C3ar1−/− 1.3±0.2%, p=0.13, not shown) and TNFα-producing CD4+ T cells (WT 1.1±0.1% vs. C3ar1−/− 1.1±0.2%, p=0.79, not shown) did not differ between groups and were markedly and significantly lower than the frequencies of donor-reactive CD8+ T cells, consistent with CD8+ T cells being the dominant effectors in this system.
When we analyzed graft-infiltrating lymphocytes (GIL) on day 6 post-transplant in untreated recipients we observed that >80% of the donor-reactive IFNγ-producing T cells were CD8+ in both genotypes (Figure 1E, gating strategy shown in Figure S2), further supporting the dominance of CD8+ T cells as effector cells and as mediators of allograft injury. We did not observe differences in frequencies of intragraft IFNγ-producing CD4+ or CD8+ T cells between WT and C3ar1−/− recipients on day 6 post-transplant (Figure 1E), reflecting the similar allograft rejection kinetics between the genotypes in the absence of immunosuppression.
When we reversed the strain combination and transplanted B6 heart grafts into allogeneic WT or C3ar1−/− BALB/c recipients, the grafts also rejected with similar kinetics (MST 8 days for each, not shown) and the day 6 post-transplant analyses of splenic alloimmunity similarly revealed a ~50% reduction in donor-reactive CD8+ IFNγ-producers (WT 3.2±0.8 vs. C3ar1−/− 0.7±0.2, n=4/group p=0.03) and TNFα-producers (WT 1.1±0.1 vs. C3ar1−/− 0.3±0.1, n=4/group p<0.001, not shown).
Analysis of baseline immune phenotypes in naïve WT and C3ar1−/− deficient animals showed no differences (Figure S3) between the strains with the exception of slightly but statistically significantly lower frequencies of naïve CD4+ T cells in the C3ar1−/− mice. Additional control experiments revealed that spleen cells from naïve WT and C3ar1−/− mice contained >10-fold lower frequencies of donor-reactive IFNγ-producing CD8+ T cells compared to the spleens obtained from allograft recipients on day 6 post-transplant (indicative of transplant-induced T cell expansion) and showed no differences between the genotypes (Figure S4).
Blockade of C3aR1 signaling synergizes with tacrolimus to prolong allograft survival
To next test whether preventing C3aR1 signaling interacts with calcineurin inhibition to prolong allograft survival, we transplanted groups of WT and C3ar1−/− recipients with BALB/c hearts and treated them with daily tacrolimus. Tacrolimus administration prolonged graft survival in WT recipients from a MST of 7 to 14 days (p<0.05 vs. untreated controls), but remarkably synergized with the absence of recipient C3aR1 to prolong allograft survival to a MST of 21 days (p<0.05 vs. WT, Figure 2A).
Figure 2. Absence of recipient C3aR1 or C3aR1-blockade synergizes with tacrolimus to prolong cardiac allograft survival.

A. Kaplan–Meier survival curves of WT BALB/c allografts transplanted into WT or C3ar1−/− B6 mice treated with vehicle control or tacrolimus (1 mg/kg/day tacrolimus i.p. daily starting on the day of surgery until rejection) ± C3aR1-A administered by osmotic pump (see methods). **p<0.01, ***p<0.001; n = 5–6/condition. Statistics calculated by log-rank (Mantel–Cox) test. B-E. Spleen cells were isolated on day 10 post-transplant from tacrolimus-treated WT and C3ar1−/− B6 recipients of BALB/c hearts ± C3aR1-A. Representative flow plots from WT and C3ar1−/− recipients (gated on CD8+ T cells) (B) and absolute numbers of splenic donor-reactive CD8+ T cells that are IFNγ +(C), TNFα +(D) or CD107+ (E) are shown. F-H. Absolute numbers of splenic Foxp3+CD4+ TREG (F), and calculated ratios of donor-reactive, IFNγ-producing CD8+ T cells: TREG (G) and of donor-reactive, TNFγ -producing CD8+ T cells: TREG (H) in the day 10 post-transplant spleen cells (n=6/group). I-J. GIL were isolated on day 10 post-transplant from tacrolimus-treated WT, C3ar1−/− or WT+C3aR1-A-treated recipients of BALB/c hearts. Percentages of CD8+ CD107+ CD8+ T cells (I) and IFN γ + CD8+ T cells (J) within the CD45+ gate of the GILs are shown (n=4/group). *p<0.05, **p<0.01; ns, not significant.
To provide translational relevance (and to account for other baseline differences between WT and C3ar1−/− animals that could influence outcomes), we transplanted groups of WT B6 recipients with BALB/c hearts and treated the recipients with tacrolimus ± a pharmacological C3aR1 antagonist (C3aR1-A) administered via subcutaneous osmotic pump (Figure 2A). These survival experiments showed that while C3aR1-A alone (without tacrolimus) had no impact on allograft survival, C3aR1-A administration synergized with tacrolimus to prolong graft survival to MST of 20 days, a result that was not different from the survival of tacrolimus-treated C3ar1−/− recipients.
To test the effects of the tacrolimus plus absence of C3aR1 signaling on the donor-reactive T cell responses, we transplanted groups of WT and C3ar1−/− mice, treating them with tacrolimus, and sacrificed them on day 10 post-transplant (all grafts beating) to perform ex vivo immune analyses. These assays showed ~10-fold fewer splenic IFNγ+, TNFα+ and CD107+ CD8+ T cells vs. the untreated recipients on day 6 (regardless of C3aR1 expression, compare to Figure 1) and demonstrated that spleens obtained from the tacrolimus-treated C3ar1−/− recipients contained significantly fewer donor-reactive IFNγ+ (Figure 2B top, 2C), TNFα+ (Figure 2B bottom, 2D), and CD107a/b+ (Figure 2E) CD8+ T cells than tacrolimus-treated WT controls. While we observed no differences in TREG numbers between groups, (Figure 2F), calculated donor-reactive, IFNγ- or TNFα-producing TEFF:TREG ratios (Figure 2G-H) were lower in the C3ar1−/− recipients, indicative of enhanced TREG function (34). We similarly observed 2-fold lower numbers of IFNγ+, TNFα+ and CD107+ splenic CD8+ T cells in the day 10 post-transplant tacrolimus+C3aR1-A treated recipients vs. tacrolimus treated controls (p<0.05 for each, n=4/group, data not shown). When we analyzed CD8+ T cells within the GILs on day 10 post-transplant (Figure 2I-J) we observed fewer IFNγ -producing and fewer CD107a/b+ CD8+ T cells in the tacrolimus-treated recipients in which C3aR1 signaling was genetically or pharmacologically impeded.
C3aR1 expressed on CD8+ T cells intrinsically amplifies their responses to alloantigens
To discern additional mechanistic insight on how C3aR1 affects alloreactive CD8+ T cells in vivo, we tested the hypothesis that T cell-expressed C3aR1 signaling intrinsically modulates CD8+ T cell alloimmunity, independent of any potential indirect effects of C3aR1 signaling on other cell types, including (but not limited to) activation of antigen presenting cells (APCs), provision of CD4+ T cell help, and augmentation of TREG-mediated suppression. We purified naïve CD44−CD62L+CD8+ T cells from WT and C3ar1−/− mice, labeled them with the proliferation dye CTV, adoptively transferred them into congenic CD45.1 WT recipients, and then transplanted the adoptive hosts with BALB/c hearts (Figure 3A, schematic). On day 6 post-transplant we analyzed splenic CD8+ T cell responses gated on the congenic CD45.2+ donor population. These analyses showed significantly lower percentages and absolute numbers of donor-reactive (CTV−, indicative of proliferation in response to donor antigen), CD45.2+ CD8+ C3ar1−/− T cells (Figure 3B-D). Control experiments showed that <2% of the adoptively transferred cells proliferated in mice that underwent sham surgery (data not shown).
Figure 3. Absence of C3aR1 on CD8+ T cells limits alloreactivity in vivo.

A. Schematic outline of experiment design (see text). B. Representative flow cytometry plots of recipient spleen cells identifying the adoptively transferred CD45.2+ WT or C3ar1−/− B6 CD8+ T cells (circle) in the congenic CD45.1+ hosts 6 days post-transplant. C-D. Representative flow plots (C) and quantification of proliferating (CTV−) cells (D, top: percentages, bottom: numbers per spleen), gated on the transferred CD45.2+ CD8+ T cells. E-F. Representative histograms (E) and quantified MFI (F) of p-pS6 protein within the proliferating population of transferred cells; quantification of cumulative results were normalized to WT group average MFI (adjusted to a value of 100) in each individual experiment G. Representative flow plots demonstrating T-bet expression within the proliferating CD45.2+ CD8+ T cells. Numbers within each plot are percentages within the outlined region. H-I. Quantified percentages (H) and absolute numbers (I) of T-bet+ cells within the CTV− CD45.2+ CD8+ T cell population. *p<0.05, **p<0.01, ***p<0.001, n=6–8/group.
Activation of the mammalian target of rapamycin (mTOR) and subsequent mTOR-dependent expression of T-bet is required for optimal CD8+ T cell differentiation in response to pathogens and well as to transplant and model antigens (12–14, 35). Our previous work showed that C3a/C3aR1 ligation on CD4+ T cells initiates PI-3Kγ/AKT-dependent mTOR activation (19, 36). When we quantified the activated form of the mTOR substrate p-S6 (p-pS6) by phosphoflow in the adoptively transferred, CTV− T cells, we observed significantly less p-pS6 within the C3ar1−/− population (Figure 3E-F). The alloreactive C3ar1−/− CD8+ T cells also expressed less T-bet than the WT control CD8+ T cells (Figure 3G-I).
In previous publications we showed that activation of CD4+ T cells induces upregulation and release of alternative pathway complement components yielding C3a and C5a, which through an autocrine loop ligate and activate T cell-expressed C3aR1 and C5aR1 receptors (19, 36). To test how this mechanism applies to CD8+ T cells and in particular to link C3aR1 signaling to mTOR, we purified, naïve (CD62LhiCD44lo) WT and C3ar1−/− CD8+ T cells, stimulated them in vitro with anti-CD3/CD28 and quantified phosphorylation of the mTOR substrate pS6 by phospho-flow cytometry (Figure 4A-B). These assays showed rapid phosphorylation of p-S6 in WT and C3ar1−/− CD8 T cells but significantly less p-pS6 in the C3ar1−/− CD8+ T cells at 1 h and 24 h post-stimulation. When we added recombinant C3a to the cultures (Figure 4C) we observed increased p-pS6 expression in WT CD8+ T cells, without detectable increases in p-pS6 in the C3ar1−/− CD8+ T cells, the latter confirming that the effect is mediated via C3aR1. To test the hypothesis that the C3a/C3aR1-induced induction of p-pS6 involves immune cell-derived (and locally activated) complement we cultured WT and C3−/− spleen cells in serum free medium with anti-CD3 and quantified CD8+ T cell expression of p-pS6 1 h later (Figure 4D-F). These assays demonstrated significantly fewer p-pS6+ CD8+ T cells and lower p-pS6 expression (MFI) per cell in the C3−/− cultures. Together, the data support the conclusion that immune cell-derived and locally activated complement results in production of C3a, which drives alloreactive CD8+ T cell responses, in part by ligating T cell-expressed C3aR1.
Figure 4. C3a/C3aR1 ligations activate mTOR signaling in CD8+ T cells.
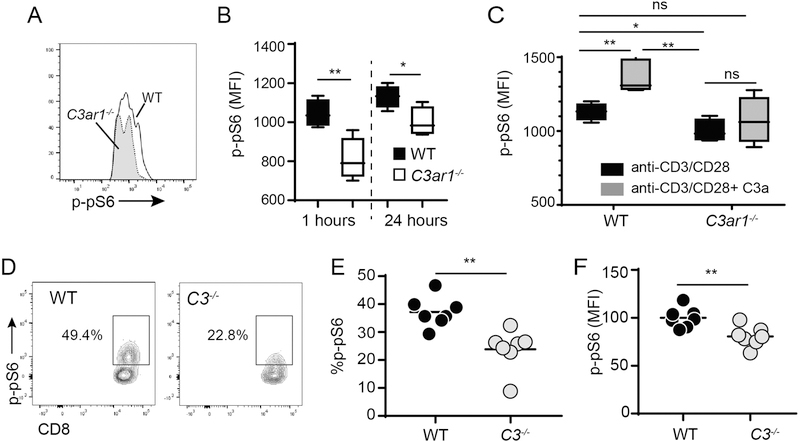
A-C. Magnetic bead enriched naïve (CD62LhiCD44lo) WT or C3ar1−/− CD8+ T cells were stimulated with anti-CD3/CD28 Dynabeads for 1 or 24 h with or without addition of recombinant C3a (200ng/ml) and p-pS6 expression was quantified by phospho-flow cytometry. Representative flow cytometry histogram of anti-CD3/CD28 stimulated cells at 1 h (A), quantification of results at 1 and 24 h (B and quantification of effects of adding C3a during the last h of a 24 h stimulation (C) are shown. D-F. Spleen cells from WT or C3−/− mice were stimulated with anti-CD3 and intracellular p-pS6 was quantified 1 h later. Representative flow cytometry plots gated on CD8+ T cells (D) and quantification of % (E) and MFI (F) of p-pS6+ CD8+ T cells in the spleens; quantification of cumulative results were normalized to WT group average MFI (adjusted to a value of 100) in each individual experiment, n=6–8/group, all experiments were repeated with similar results. *p<0.05, **p<0.01.
C3aR1 also alters APC phenotypes that indirectly amplify CD8+ T cell responses to alloantigens
To test the effects of absent host C3aR1 on responses of WT (C3aR1+) CD8+ T cells alloresponses in vivo, we isolated naïve, splenic CD8+ T cells from WT CD45.1 mice, labeled them with CTV and adoptively transferred them into CD45.2 WT or C3ar1−/− recipients (Figure 5A, schematic). We then transplanted the adoptive hosts with BALB/c hearts. Analyses performed on day 6 post-transplant showed significantly less proliferation and expansion of the WT CD8+ T cells within the C3aR1-deficient environment (Figures 5B-D), but in contrast to the findings from Figure 3, we did not observe differences in p-pS6 expression within WT alloreactive CD8+ T cells responding in the WT vs. C3ar1−/− hosts (Figure 5E-F). These findings indicate that in addition to a direct effect of CD8+ T cell-expressed C3aR1 on the responding CD8+ T cells (Figures 3–4), C3aR1 signaling on other, non-CD8+ T cells indirectly contributes to the strength of the alloreactive CD8+ T cell responses in vivo.
Figure 5. Absence of host C3aR1 limits CD8+ T cell limits alloreactivity in vivo.

A. Schematic outline of experiment design (see text). B. Representative flow cytometry plots of recipient spleen cells identifying the adoptively transferred CD45.1+ WT B6 CD8+ T cells (circle) in the congenic CD45.2+ WT or C3ar1−/− hosts 6 days post-transplant. C-D. Representative flow plots (C) and quantification of proliferating (CTV−) cells (D, top percentages, bottom numbers per spleen), gated on the transferred CD45.1+ CD8+ T cells. E-F. Representative histograms (E) and quantified MFI (F) of p-pS6 protein within the proliferating population of transferred cells; quantification of cumulative results were normalized to WT group average MFI (adjusted to a value of 100) in each individual experiment. G. Representative flow plots demonstrating T-bet expression within the proliferating CD45.1+ CD8+ T cells. Numbers within each plot are percentages within the outlined region. H-I. Quantified percentages (top) and absolute numbers (bottom) of T-bet+ cells within the CTV− CD45.1+ CD8+ T cell population. *p<0.05, **p<0.01, ***p<0.001, n=6–8/group.
Our previous work on CD4+ T cells showed that during cognate interactions with APCs, locally produced C3a can ligate C3aR1 on APCs to cause upregulation of costimulatory molecules and release of innate cytokine, including IL-12 (18, 37). These previous findings raised the possibility that C3aR1 signaling on APCs in the host could also contribute to CD8+ T cell expansion through upregulation of costimulatory molecule and/or cytokine production. To address these putative mechanisms (potentially accounting for the results observed in Figure 5) we measured costimulatory molecule expression levels on DCs (Figures 6A-D) and quantified serum cytokines by ELISA (Figures 6E-H) in the transplanted, WT and C3ar1−/− adoptive hosts. These analyses demonstrated lower CD40, CD80 and CD86 expression on CD11chiCD11b− DCs from the C3ar1−/− recipients. We observed similar lower levels of CD80, CD86 and CD40 on CD11cintCD11b−DCs from C3ar1−/− mice, observed lower CD40 expression on the CD11b+CD11c− monocytes from C3ar1−/− mice and did not observe differences in expression of these molecules on the CD11b+CD11c+ subset between the genotypes (Figure S5). We also detected lower serum levels of IL-12 in the C3ar1−/− recipients, without differences in serum levels of TNFα, IL-1β and IL-6 between groups. As IL-12 functions as a “third” signal for CD8+ T cell activation (38–40), and induces T-bet expression (41), we compared T-bet expression in the responding WT CD8+ T cells within the WT and C3ar1−/− hosts (Figure 5G-H). These analyses showed less intracellular T-bet in the WT alloreactive CD8+ T cells responding within the C3ar1−/− host environment.
Figure 6. Absence of recipient C3aR1 limits APC costimulatory molecule expression and IL-12 production.
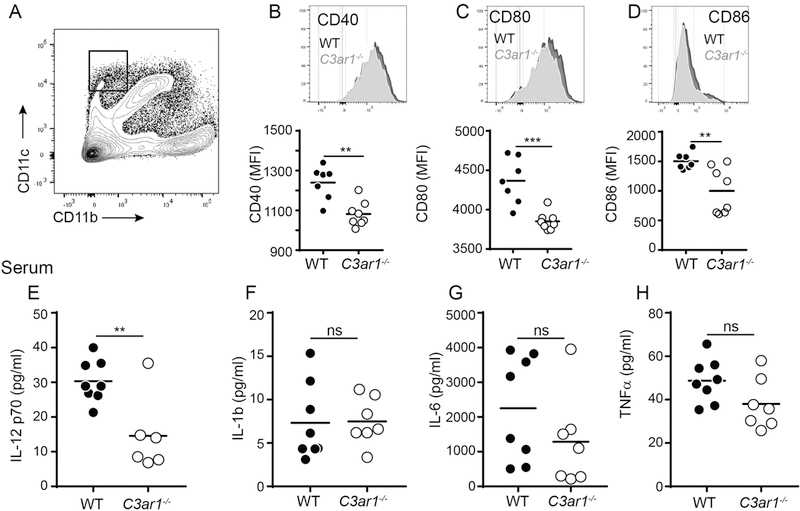
(A) Representative flow plot of post-transplant day 1 spleen cells identifying CD11chiCD11b− dendritic cells (outlined boxed area). B-D. Representative histograms (top) and quantification of MFI (bottom) showing expression of costimulatory molecules CD40 (B), CD80 (C), and CD86 (D), gated on the CD11chiCD11b− splenic dendritic cells in WT or C3ar1−/− B6 recipients of BALB/c heart grafts 1 day post-transplantation. **p<0.01, ***p<0.001, n = 8/condition. (E-H) ELISA results for IL-12 (E), IL-1b (F), IL-6 (G) and TNFα (H) performed on serum obtained on day 6 post-transplant from groups of WT and C3ar1−/− recipients of BALB/c allografts. **p<0.01; ns, not significant, n = 6–8/condition.
DISCUSSION
As alloreactive CD8+ T cells are crucial effector cells that mediate rejection of class I MHC-disparate allografts, an in-depth understanding of the molecular signals that modulate CD8+ T cell activation, expansion, and effector differentiation is paramount to devising strategies that could reduce transplant injury. Herein we demonstrate that absence or blockade of recipient C3aR1 reduces expansion of donor-reactive CD8+ T cells ~2-fold in untreated allograft recipients (Figure 1) and synergizes with tacrolimus to prolong allograft survival (Figure 2). The ex vivo analyses performed prior to rejection in the transplanted and tacrolimus-treated recipients (Figure 2) demonstrated that tacrolimus plus C3aR1 deficiency/blockade reduced the frequencies of donor-reactive IFN γ -producing and degranulating CD8+ T cells compared to tacrolimus-treated WT controls (in the spleen and in the graft), results that support the hypothesis that the prolonged allograft survival is due, at least in part, to inhibitory effects on the donor-reactive T cell repertoire. This proof-of-concept, clinically relevant finding suggests that C3aR1 inhibition could be a useful adjuvant immunosuppressive therapy potentially capable of limiting tacrolimus exposure while maintaining/improving outcomes in human transplant recipients.
Our findings additionally provide new mechanistic insight into T cell-intrinsic and extrinsic effects of C3aR1 signaling on alloreactive effector CD8+ T cells. Current concepts are that activation and expansion of naïve alloreactive CD8+ T cells requires cognate interactions with MHC, costimulation (the “second signal”) (5–7, 42) which is amplified by CD4+ help transmitted through a common APC (43), and IL-2, which together activate intracellular signaling cascades that among other effects, activate mTOR-dependent proliferation; inhibition or absence of mTOR, specifically mTORC1, limits differentiation of naïve CD8+ into TEFF (44, 45). Evidence also indicates that optimal expansion and differentiation of naïve CD8+ T cells into CD8+ TEFF cells requires a cytokine-initiated signal to guide context-specific differentiation. Type 1 interferons can provide such signals in various contexts, including viral infections (46, 47), but IL-12 is the dominant, “third signal,” cytokine in many settings (8, 38, 39, 48), including transplantation. IL-12 ligates the CD8+ T cell-expressed IL-12 receptor (IL-12R), one consequence of which is to upregulate surface CD25 and thereby enhance responsiveness to IL-2 (39, 47). IL-12/IL-12R ligations on CD8+ T cells additionally a) regulate TEFF differentiation via induction of the transcription factor T-bet (11, 49) and b) limit development of T cell exhaustion (50). T-bet initiates a genetic program that includes production of IFNγ as well as the surface marker KLRG1 that is characteristically expressed on short term CD8+ TEFF (41). The fact that tacrolimus blocks calcineurin activity downstream of the T cell receptor (51) while C3aR1 blockade predominantly inhibits mTOR signaling and T-bet expression, two distinct pathways required for T cell activation/differentiation, provides an explanation for why tacrolimus plus absent C3aR1 signaling have synergistic effects on delaying T cell-dependent graft rejection (Figure 2).
Our new data add to the understanding of this process by identifying C3aR1 signaling on CD8+ T cells and on APCs as key intermediary events that amplify mTOR- and IL-12-dependent differentiation pathways to culminate in pathogenic transplant-reactive CD8+ TEFF responses. The results add to previous literature in which we, among others, showed that C3aR1 (and C5aR1) expressed on CD4+ T cells transmit signals via PI-3Kγand AKT that are required for activation/differentiation (18, 21, 37). We demonstrate herein that CD8+ T cell-expressed C3aR1 signaling regulates mTOR activity and T-bet upregulation required for optimal alloreactive CD8+ T cell proliferation, differentiation and expansion (Figure 3). Our new data also show that C3aR1 signaling on non-T cells (principally APCs) augments donor-reactive CD8+ T cell proliferation independent of C3aR1 ligation on the responding T cells, an effect associated with upregulation of APC costimulatory molecules and increased production of IL-12 (Figures 5–6).
Our previous studies showed during cognate T cell/APC interactions, immune cell-derived complement results in locally produced C3a (and C5a) that bind to their respective receptors on both partners via an autocrine signaling loop (19), and that immune cell-derived complement is both necessary and sufficient for CD4+ TEFF differentiation (18, 19, 28, 37). While we have not formally tested the source(s) of the complement that results in C3a/C3aR1 signaling on the CD8+ T cells in vivo, our new in vitro findings (Figure 4) support the conclusion that immune cell-derived C3 is sufficient to function as an autocrine/paracrine source of C3a that ligates CD8+ T cell expressed C3aR1.
In addition to our previous documentation of a complement-dependent effect on activation/expansion of CD4+ TEFF responses, we previously showed that absence of C3aR1/C5aR1 augments TREG generation, stability and function, promoting a pro-tolerogenic environment (25, 26). Thus, while tacrolimus prolongs allograft survival and decreases donor-reactive CD8+ T cell responses in C3ar1−/− recipients (and in C3aR1-A-treated WT recipients, Figure 2), the interactive effects are likely the end result of several mechanisms through which C3aR1 regulates alloimmunity, including a) diminished CD4+ TEFF expansion (which could limit T cell help for CD8+ T cell activation), b) augmented TREG induction and function (which can suppress CD8+ T cell responses), c) limited APC activation and d) direct inhibition of CD8+ T cell differentiation/expansion.
Our mechanistic analyses indicate that C3aR1 is linked to CD8+ T cell function in part via activation of the mTOR pathway (Figures 3–4). Work by others using non-transplant model systems revealed that mTOR inhibition and specifically mTORC1 inhibition limits TEFF differentiation but paradoxically enhances development of memory T cells (44). Intriguingly, when we quantified frequencies of splenic donor-reactive memory CD8+ T cells from WT and C3ar1−/− recipients of BALB/c hearts 6-months after rejection we observed no differences (2.6±0.46% in WT vs. 2.6±0.50% in the C3ar1−/−, p=ns, n=10/group, data not shown) despite the significantly fewer TEFF in C3ar1−/− recipients at the height of the effector response (Figure 1), consistent with a memory-sparing effect mediated through limited C3aR1-initiated mTOR activation. Biochemical mechanisms that underlie links between C3aR1 and memory T cell function, including whether C3aR1 inhibition impacts expression of eomesodermin shown to be essential for memory T cell survival (52), as well as understanding whether and if so how C3ar1 impacts the function of these long-term, donor-reactive memory CD8+ T cells are issues that remain to be fully elucidated.
In summary, our studies newly highlight CD8+ T-cell-intrinsic and T-cell-extrinsic roles of C3aR1 signaling in the optimal development of anti-donor CD8+ T cell responses induced by an allogeneic heart graft. In addition to gaining molecular insight, our findings, along with the pharmaceutical industry’s interest in targeting complement activation (53), support testing C3aR1 inhibition in human transplant recipients as an adjuvant immunosuppressive strategy to spare calcineurin inhibitor exposure and to ultimately improve allograft survival and function.
Supplementary Material
ACKNOWLEDGMENTS
The work was supported by NIH grant R01 AI071185 awarded to PSH. DM was supported by NIH T32 AI007605. The authors thank the microsurgery core facility (P Boros, Y. Li, J Liu) at the Icahn School of Medicine at Mount Sinai for their contributions to the studies.
Abbreviations:
- AKT
also known as phosphokinase B, PKB
- APC
antigen presenting cell
- CFSE
Carboxyfluorescein succinimidyl ester
- C3aR1
C3a receptor 1
- C3aR1-A
C3aR1 antagonist
- C5aR1
C5a receptor 1
- CTV
CellTrace™ Violet
- DC
dendritic cell
- GILgraft
infiltrating lymphocytes
- IFNγ
interferon gamma
- mAb
monoclonal antibody
- MFI
median fluorescence intensity
- MHC
major histocompatibility complex
- MST
median survival time
- mTOR
mammalian target of rapamycin
- PBS
phosphate buffered saline
- PI-3Kγ
phosphoinositol-4,5-bisphosphate-3-kinase-gamma
- RBC
red blood cell
- TCR T
cell receptor
- TREG
regulatory T cell
- TEFF
effector T cell
- TNFγ
tumor necrosis factor alpha
Footnotes
DISCLOSURE
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
REFERENCES
- 1.Krieger NR, Yin DP, Fathman CG. CD4+ but not CD8+ cells are essential for allorejection. J Exp Med 1996;184(5):2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halamay KE, Kirkman RL, Sun L, Yamada A, Fragoso RC, Shimizu K et al. CD8 T cells are sufficient to mediate allorecognition and allograft rejection. Cell Immunol 2002;216(1–2):6–14. [DOI] [PubMed] [Google Scholar]
- 3.Taylor AL, Negus SL, Negus M, Bolton EM, Bradley JA, Pettigrew GJ. Pathways of helper CD4 T cell allorecognition in generating alloantibody and CD8 T cell alloimmunity. Transplantation 2007;83(7):931–937. [DOI] [PubMed] [Google Scholar]
- 4.Vieyra M, Leisman S, Raedler H, Kwan WH, Yang M, Strainic MG et al. Complement regulates CD4 T-cell help to CD8 T cells required for murine allograft rejection. The American journal of pathology 2011;179(2):766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forster E, Krenger W, Joergensen J, Hof R, Geha RS, Hollander GA. Contribution of CD40-CD154-mediated costimulation to an alloresponse in vivo. Transplantation 1999;67(9):1284–1287. [DOI] [PubMed] [Google Scholar]
- 6.Hancock WW, Sayegh MH, Zheng XG, Peach R, Linsley PS, Turka LA. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc Natl Acad Sci U S A 1996;93(24):13967–13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kishimoto K, Dong VM, Issazadeh S, Fedoseyeva EV, Waaga AM, Yamada A et al. The role of CD154-CD40 versus CD28-B7 costimulatory pathways in regulating allogeneic Th1 and Th2 responses in vivo. J Clin Invest 2000;106(1):63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol 2003;171(10):5165–5171. [DOI] [PubMed] [Google Scholar]
- 9.Filatenkov AA, Jacovetty EL, Fischer UB, Curtsinger JM, Mescher MF, Ingulli E. CD4 T cell-dependent conditioning of dendritic cells to produce IL-12 results in CD8-mediated graft rejection and avoidance of tolerance. J Immunol 2005;174(11):6909–6917. [DOI] [PubMed] [Google Scholar]
- 10.Kieper WC, Prlic M, Schmidt CS, Mescher MF, Jameson SC. Il-12 enhances CD8 T cell homeostatic expansion. J Immunol 2001;166(9):5515–5521. [DOI] [PubMed] [Google Scholar]
- 11.Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. Cutting Edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J Immunol 2006;177(11):7515–7519. [DOI] [PubMed] [Google Scholar]
- 12.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000;100(6):655–669. [DOI] [PubMed] [Google Scholar]
- 13.Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci U S A 2003;100(26):15818–15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juedes AE, Rodrigo E, Togher L, Glimcher LH, von Herrath MG. T-bet controls autoaggressive CD8 lymphocyte responses in type 1 diabetes. J Exp Med 2004;199(8):1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathern DR, Heeger PS. Molecules Great and Small: The Complement System. Clin J Am Soc Nephrol 2015;10(9):1636–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA et al. Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity 2015;42(6):1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science 2016;352(6292):aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 2008;112(5):1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 2008;28(3):425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N et al. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med 2005;201(10):1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J, Lin F, Strainic MG, An F, Miller RH, Altuntas CZ et al. IFN-gamma and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. J Immunol 2008;180(9):5882–5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheen JH, Strainic MG, Liu J, Zhang W, Yi Z, Medof ME et al. TLR-Induced Murine Dendritic Cell (DC) Activation Requires DC-Intrinsic Complement. J Immunol 2017;199(1):278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nat Immunol 2013;14(2):162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D AV, Demir M, Chun N, Fribourg M, Cravedi P, Llaudo I et al. T Cell Expression of C5a Receptor 2 Augments Murine Regulatory T Cell (TREG) Generation and TREG-Dependent Cardiac Allograft Survival. J Immunol 2018. [DOI] [PMC free article] [PubMed]
- 25.Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med 2013;210(2):257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Touw W, Cravedi P, Kwan WH, Paz-Artal E, Merad M, Heeger PS. Cutting Edge: Receptors for C3a and C5a Modulate Stability of Alloantigen-Reactive Induced Regulatory T Cells. J Immunol 2013. [DOI] [PMC free article] [PubMed]
- 27.Esposito A, Suedekum B, Liu J, An F, Lass J, Strainic MG et al. Decay accelerating factor is essential for successful corneal engraftment. Am J Transplant 2010;10(3):527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin M, Yin N, Murphy B, Medof ME, Segerer S, Heeger PS et al. Immune cell-derived c3 is required for autoimmune diabetes induced by multiple low doses of streptozotocin. Diabetes 2010;59(9):2247–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valujskikh A, Pantenburg B, Heeger PS. Primed allospecific T cells prevent the effects of costimulatory blockade on prolonged cardiac allograft survival in mice. Am J Transplant 2002;2(6):501–509. [DOI] [PubMed] [Google Scholar]
- 30.Raedler H, Vieyra MB, Leisman S, Lakhani P, Kwan W, Yang M et al. Anti-complement component C5 mAb synergizes with CTLA4Ig to inhibit alloreactive T cells and prolong cardiac allograft survival in mice. Am J Transplant 2011;11(7):1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chun N, Fairchild RL, Li Y, Liu J, Zhang M, Baldwin WM 3rd, et al. Complement Dependence of Murine Costimulatory Blockade-Resistant Cellular Cardiac Allograft Rejection. Am J Transplant 2017;17(11):2810–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ames RS, Lee D, Foley JJ, Jurewicz AJ, Tornetta MA, Bautsch W et al. Identification of a selective nonpeptide antagonist of the anaphylatoxin C3a receptor that demonstrates antiinflammatory activity in animal models. J Immunol 2001;166(10):6341–6348. [DOI] [PubMed] [Google Scholar]
- 33.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 2003;281(1–2):65–78. [DOI] [PubMed] [Google Scholar]
- 34.Llaudo I, Fribourg M, Medof ME, Conde P, Ochando J, Heeger PS. C5aR1 regulates migration of suppressive myeloid cells required for costimulatory blockade-induced murine allograft survival. Am J Transplant 2018. [DOI] [PMC free article] [PubMed]
- 35.Yuan X, Paez-Cortez J, Schmitt-Knosalla I, D’Addio F, Mfarrej B, Donnarumma M et al. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med 2008;205(13):3133–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lalli PN, Zhou W, Sacks S, Medof ME, Heeger PS. Locally produced and activated complement as a mediator of alloreactive T cells. Front Biosci (Schol Ed) 2009;1:117–124. [DOI] [PubMed] [Google Scholar]
- 37.Lalli PN, Strainic MG, Lin F, Medof ME, Heeger PS. Decay accelerating factor can control T cell differentiation into IFN-gamma-producing effector cells via regulating local C5a-induced IL-12 production. J Immunol 2007;179(9):5793–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt CS, Mescher MF. Peptide antigen priming of naive, but not memory, CD8 T cells requires a third signal that can be provided by IL-12. J Immunol 2002;168(11):5521–5529. [DOI] [PubMed] [Google Scholar]
- 39.Valenzuela J, Schmidt C, Mescher M. The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T cells. J Immunol 2002;169(12):6842–6849. [DOI] [PubMed] [Google Scholar]
- 40.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med 2003;197(9):1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 2007;27(2):281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turka LA, Linsley PS, Lin H, Brady W, Leiden JM, Wei RQ et al. T-cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proc Natl Acad Sci U S A 1992;89(22):11102–11105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee RS, Grusby MJ, Glimcher LH, Winn HJ, Auchincloss H Jr. Indirect recognition by helper cells can induce donor-specific cytotoxic T lymphocytes in vivo. J Exp Med 1994;179(3):865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF et al. mTOR regulates memory CD8 T-cell differentiation. Nature 2009;460(7251):108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J et al. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest 2015;125(5):2090–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol 2005;174(8):4465–4469. [DOI] [PubMed] [Google Scholar]
- 47.Xiao Z, Casey KA, Jameson SC, Curtsinger JM, Mescher MF. Programming for CD8 T cell memory development requires IL-12 or type I IFN. J Immunol 2009;182(5):2786–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD et al. Signals required for programming effector and memory development by CD8+ T cells. Immunological reviews 2006;211:81–92. [DOI] [PubMed] [Google Scholar]
- 49.Chowdhury FZ, Ramos HJ, Davis LS, Forman J, Farrar JD. IL-12 selectively programs effector pathways that are stably expressed in human CD8+ effector memory T cells in vivo. Blood 2011;118(14):3890–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerner MY, Heltemes-Harris LM, Fife BT, Mescher MF. Cutting edge: IL-12 and type I IFN differentially program CD8 T cells for programmed death 1 re-expression levels and tumor control. J Immunol 2013;191(3):1011–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today 1992;13(4):136–142. [DOI] [PubMed] [Google Scholar]
- 52.Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T et al. Cutting edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol 2010;185(9):4988–4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ricklin D, Lambris JD. New milestones ahead in complement-targeted therapy. Semin Immunol 2016;28(3):208–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.