

Abstract
Nickel-catalyzed decarbonylative stannylation of acyl fluorides under ligand-free conditions was disclosed. A variety of aromatic acyl fluorides are capable of reacting with silylstannanes in the presence of cesium fluoride. A one-pot decarbonylative stannylation/Migita-Kosugi-Stille reaction of benzoyl fluoride, giving rise to the direct formation of the corresponding cross-coupled products, further demonstrated the synthetic utility of the present method. This newly developed methodology with a good functional-group compatibility via C–F bond cleavage and C–Sn bond formation under nickel catalysis opens a new area for the functionalization of acyl fluorides in terms of carbon-heteroatom bond formation.
Keywords: nickel, acyl fluorides, stannylation, decarbonylation, carbon-tin bond formation
1. Introduction
Acyl fluorides as one of carboxylic acid derivatives have attracted much attention in organic synthesis, due to their great stability, easy availability, and unique intrinsic nature [1,2,3]. Conventionally, transition metal-catalyzed transformations of acyl fluorides with organometallic reagents (Zn, Si, and B) have focused on the synthesis of biaryl ketones in a carbonyl-group retentive manner [4,5,6]. In a sharp contrast, recently, decarbonylative transformations of carboxylic acid derivatives [7,8], especially acyl fluorides have been studied intensively [9]. Sakai and Ogiwara have disclosed that the auxiliary ligand of the palladium catalyst can control the reaction type of reduction of acyl fluorides [10]. However, transition-metal catalyzed decarbonylative transformations of acyl fluorides are mainly C–C bond formation, such as palladium-catalyzed trifluoromethylation [11], nickel-catalyzed Suzuki-Miyaura reaction [12], iridium-catalyzed arylation via C–H bond activation [13], and our recent work on nickel-catalyzed ethylation with BEt3 [14] and DPPM-assisted methylenation with AlMe3 [15]. Therefore, the development of carbon-heteroatom bond-forming reactions of acyl fluorides are of great importance. Very recently, we successfully demonstrated the first nickel-catalyzed decarbonylative borylation of acyl fluorides with diboron, forming the C–B bond [16], and the related decarbonylative borylation catalyzed by palladium have been reported [17,18].
Arylstannanes as one of common organometallic reagents are extensively applied in Migita-Kosugi-Stille reaction [19,20,21], which has been utilized as a powerful method for C–C bond formation, especially, in natural product synthesis [22,23,24]. Conventional synthetic methods of arylstannanes are the reactions of organometallic reagents such as arylzinc compounds with triorganotin halides [25,26,27]. Alternatively, catalytic cross-coupling reactions of aryl (pseudo)halides with tributyltin hydride [28], tributylstannyl methoxide [29], and hexaalkyldistannane [30,31] have been documented (Scheme 1a). Recent studies on arylstannanes synthesis utilizing air- and moisture-insensitive silylstannyl reagent, Bu3Sn-SiMe3, could prove that the C–O bond is also a powerful alternative to aryl halides (Scheme 1b) [32]. In addition, Rueping and co-workers developed nickel-catalyzed stannylation of aromatic esters in a decarbonylative manner (Scheme 1c) [33]. Although these methods have made a great contribution to the synthesis of arylstannanes, novel and practical methods to afford arylstannanes from more simple starting materials remain highly desirable. Herein, we report the first decarbonylative stannylation of acyl fluorides with Bu3Sn-SiMe3 catalyzed by air-stable and inexpensive nickel(II) chloride under ligand and additive-free conditions (Scheme 1d).
Scheme 1.

Various synthetic routes for arylstannanes.
2. Results and Discussion
We commenced our research by choosing benzoyl fluoride (1a) and 1.5 equiv of Bu3Sn-SiMe3 (2) as the model substrates, and the results are summarized in Table 1. Various transition metal sources were investigated to facilitate the decarbonylative stannylation reaction (entries 1–6). Among them, nickel(II) chloride displayed a superior result, affording the target product 3a in 90% yield (entry 3). When cesium carbonate was employed in place of cesium fluoride, the yield of 3a was dramatically dropped to 48% (entry 7) and no stannylation reaction occurred when potassium fluoride was used, along with silylstannane 2 recovered (entry 8). Additionally, amounts of 2 could be reduced to 1.2 equiv, which afforded 94% GC yield of 3a (entries 3, 9, and 10). The yields of 3a were slightly decreased as the reaction time was shortened (entries 9 vs. 11–13). When benzoyl chloride was employed instead of 1a, 3a was obtained in 56% yield, suggesting the unique feature of the present reaction of acyl fluorides.
Table 1.
Optimization of the reaction conditions.
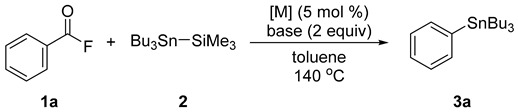
| Entry | [M] | Base | 2 (equiv) | Time (h) | Yield of 3a (%) 1 |
|---|---|---|---|---|---|
| 1 | FeCl2 | CsF | 1.5 | 24 | 29 |
| 2 | CoCl2 | CsF | 1.5 | 24 | 21 |
| 3 | NiCl2 | CsF | 1.5 | 24 | 90 |
| 4 | NiBr2 | CsF | 1.5 | 24 | 68 |
| 5 | Ni(cod)2 | CsF | 1.5 | 24 | 16 |
| 6 | PdCl2 | CsF | 1.5 | 24 | 4 |
| 7 | NiCl2 | Cs2CO3 | 1.5 | 24 | 48 |
| 8 | NiCl2 | KF | 1.5 | 24 | 0 |
| 9 | NiCl2 | CsF | 1.2 | 24 | 94 (90) |
| 10 | NiCl2 | CsF | 1.0 | 24 | 58 |
| 11 | NiCl2 | CsF | 1.2 | 18 | 91 |
| 12 | NiCl2 | CsF | 1.2 | 12 | 87 |
| 13 | NiCl2 | CsF | 1.2 | 6 | 76 |
1 Determined by GC analysis of the crude mixture using n-dodecane as an internal standard. An isolated yield is given in parentheses.
A generality of the decarbonylative stannylation was examined with the optimized reaction conditions (Table 2). Acyl fluorides bearing electron-donating groups such as alkyl (1b–1d), phenyl (1e–1g), and alkoxy (1h, 1i) groups gave the corresponding products 3b–3i in 56%–85% yields regardless of the substitution positions. Other oxygen-containing functional groups such as benzyloxy (1j) and acetal (1k) were also well tolerated during the reaction. Acyl fluorides bearing electron-withdrawing groups such as trifluoromethyl (1l) and fluoro (1m, 1n) groups were also well compatible. In particular, an aryl chloride skeleton (1o) is known to a reactive electrophile in some nickel-catalyzed cross-coupling reactions [29]. Although 4-bromo- and 4-iodobenzoyl fluorides were employed as the substrates, no trace of the desired products was detected, presumably due to the bromo and iodo groups are highly reactive under the present reaction conditions. Acyl fluorides with fused aromatic systems (1p–1r) afforded arylstannanes in moderate to good yields. Heterocycles including benzothiophene and quinoline yielded 3s and 3t in 62% and 84% yields, respectively. Unfortunately, however, the reactions employing surrogate aliphatic acyl fluorides gave no formation of the desired products.
Table 2.
Decarbonylative stannylation of acyl fluorides a,b.
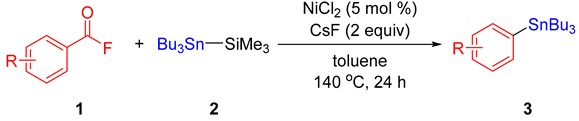
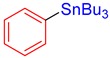 3a, 90% |
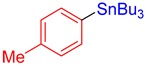 3b, 81% |
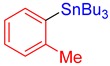 3c, 63% |
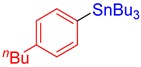 3d, 67% |
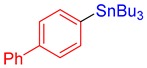 3e, 82% |
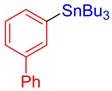 3f, 56% |
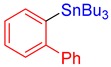 3g, 85% |
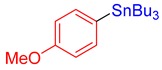 3h, 64% |
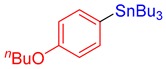 3i, 61% |
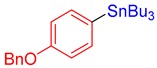 3j, 50% |
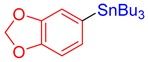 3k, 87% |
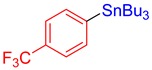 3l, 61% |
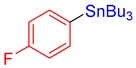 3m, 86% |
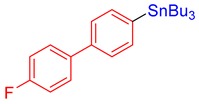 3n, 72% |
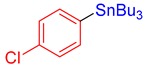 3o, 90% |
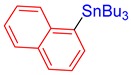 3p, 51% |
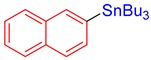 3q, 79% |
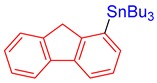 3r, 82% |
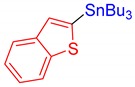 3s, 62% |
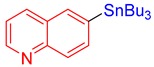 3t, 84% |
aReaction conditions: 1 (0.2 mmol), 2 (0.24 mmol), NiCl2 (0.01 mmol), CsF (0.4 mmol), toluene (1 mL), 140 °C, 24 h. b Isolated yields.
To demonstrate the synthetic utility of the present method, one-pot reaction of a successive decarbonylative stannylation/Migita-Kosugi-Stille reaction of 1a was investigated (Scheme 2) [31]. To our delight, with the aid of the additional palladium catalyst into the reaction mixture, 71% yield of compound 4 was obtained.
Scheme 2.

One-pot reaction of decarbonylative stannylation/Migita-Kosugi-Stille reaction of 1a.
To gain more detailed insights into the reaction mechanism, some control experiments were carried out (Table 3). Although arylstannane 3a was obtained in 94% yield, along with the formation of hexabutylditin (5; 13%) under the optimized reaction conditions (entry 1), indicating that nickel(II) chloride was reduced to Ni(0) species. This hypothesis was further proved by the reaction of 1a with 2 in the absence of cesium fluoride (entry 2). Without nickel(II) chloride, no target product 3a was formed, and 5 was obtained quantitatively (entry 3). In some stannylation reactions, hexabutyldistannane (5) could also be used as a stannylating reagent [30,31]. Thus, the reaction of 1a (0.2 mmol) with 5 (0.24 mmol) was evaluated under identical reaction conditions. However, neither the desired product 3a nor a viable acyl stannane was delivered, along with decomposition of 1a and the remained 5 unreacted, which suggests that the once formed 5 never be involved into the catalytic cycle because of its lower reactivity (entry 4).
Table 3.
Control experiments for Ni-catalyzed decarbonylative stannylation.
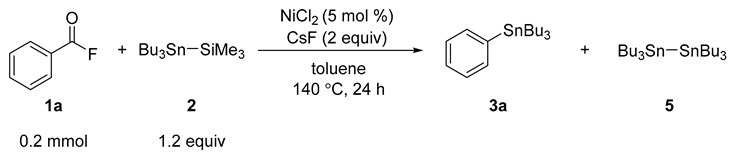
| Entry | Deviation from Standard Conditions | GC Yield (%) 1 | ||
|---|---|---|---|---|
| 2 | 3a | 5 | ||
| 1 | none | 0 | 94 | 13 |
| 2 | without CsF | 120 | 0 | 0 |
| 3 | without NiCl2 | 0 | 0 | 63 |
| 4 2 | 5 instead of 2 | - | 0 | >99 |
1 Determined by GC analysis of the crude mixture, using n-dodecane as an internal standard. 2 4 (0.24 mmol) was added.
Our proposed mechanism of the present decarbonylative stannylation is outlined in Scheme 3. Combining the related references [12,34] with our previous work [16], it is assumed that oxidative addition of acyl fluorides 1 to Ni(0) species A, derived from reduction of Ni(II) chloride with 2, yields acyl nickel(II) species B. Subsequently, decarbonylation of B delivers arylnickel(II) species C [12,16]. Transmetalation between the complex C and activated silylstannane 2 by cesium fluoride affords complex D [32]. Following reductive elimination gives the target product arylstannanes 3, regenerating Ni(0) species A.
Scheme 3.

Proposed mechanism.
3. Experimental Sections
3.1. General
Unless otherwise noted, all the reactions were carried out under an argon atmosphere using standard Schlenk techniques. Glassware was dried in an oven (150 °C) and heated under reduced pressure prior to use. Solvents were employed as eluents for all other routine operation, as well as dehydrated solvent were purchased from commercial suppliers and employed without any further purification. For thin layer chromatography (TLC) analyses throughout this work, Merck precoated TLC plates (silica gel 60 GF254, 0.25 mm) were used. Silica gel column chromatography was carried out using silica gel 60 N (spherical, neutral, 40–100 μm) from Kanto Chemicals Co., Inc. (Tokyo, Japan). NMR spectra (1H, 13C{1H} and 19F{1H}) were recorded on Varian INOVA-600 (600 MHz), Mercury-400 (400 MHz), or 300-NMR ASW (300 MHz) spectrometers (Agilent Technologies International Japan, Ltd., Tokyo, Japan). Chemical shifts (δ) are in parts per million relative to CDCl3 at 7.26 ppm for 1H and at 77.16 ppm for 13C{1H}. The 19F{1H} NMR spectra were measured by using CCl3F (δ = 0.00 ppm) as an external standard. The NMR yields were determined using dibromomethane as an internal standard. The GC yields were determined by GC analysis of the crude mixture, using n-dodecane as an internal standard. GC analyses were performed on a Shimadzu GC-14A equipped with a flame ionization detector using Shimadzu Capillary Column (CBP1-M25-025) and Shimadzu C-R6A-Chromatopac integrator (Kyoto, Japan). Infrared spectra were recorded on a SHIMADZU IRPrestige-21 spectrophotometer. Elemental analyses were carried out with a Perkin-Elmer 2400 CHN elemental analyzer (Perkin-Elmer, Waltham, MA, USA) at Okayama University. 1H NMR, 13C{1H} NMR, 19F{1H} NMR spectra of the compounds 1t, 2, 3a–t and 4 can be found at the Supplementary Materials.
3.2. Experimental Method
3.2.1. Representative Procedure for the Synthesis of Acyl Fluorides from Acyl Chlorides
To a 50 mL of Schlenk tube charged with a magnetic stir bar, were successively added acyl chloride (4.0 mmol), 18-crown-6 (52.9 mg, 0.2 mmol, 5 mol %), KF (2.32 g, 40 mmol, 10 equiv), and THF (20 mL). After the reaction was stirred at 40 °C for 24 h, insoluble inorganic solid (KF or KCl) was filtered, and the volatiles were concentrated using a rotary evaporator. The crude product was purified by bulb-to-bulb distillation to afford the corresponding acyl fluorides 1 [35].
3.2.2. Representative Procedure for the Synthesis of Acyl Fluorides from Carboxylic Acids
To a 20 mL of Schlenk tube charged with a magnetic stir bar, were successively added carboxylic acid (3.0 mmol) and CH2Cl2 (15 mL). After the mixture was stirred at 0 °C for 30 min, Deoxo-Fluor® reagent (608 μL, 3.3 mmol, 1.1 equiv) was slowly added to the reaction mixture. After the reaction mixture was stirred at 0 °C for 30 min, the solution was slowly poured into saturated NaHCO3, extracted with CH2Cl2 (3 × 15 mL), and dried over MgSO4. The crude product was purified by flash chromatography on silica gel to afford the corresponding acyl fluorides 1 [36].
3.2.3. Synthesis of Trimethyl(tributylstannyl)silane (2)
To a solution of naphthalene (51.3 mg, 0.4 mmol) in THF (16 mL), was added lithium clippings (84 mg, 12 mmol) under an argon atmosphere. During the resulting mixture was stirred at room temperature for 1 h, the color turned to dark green. Then, hexabutylditin (2.02 mL, 4 mmol) was added dropwise and the mixture was stirred at room temperature for 3 h. The resulting solution (16 mL) was transferred via cannula to a Schlenk tube under Ar and then stored at room temperature. A THF solution prepared as described above was added via a cannula into the stirred solution of chlorotrimethylsilane (951 mg, 8.8 mmol) in THF at 0 °C. The reaction was stirred at room temperature overnight followed by extraction with hexane. The organic phase was washed with brine and dried over Na2SO4. Removal of the solvent and purification by bulb-to-bulb distillation under reduced pressure provided Bu3Sn-SiMe3 as a colorless oil [37].
3.2.4. Representative Procedure for Ni-catalyzed Decarbonylative Stannylation of Acyl Fluorides
A 20 mL dried Schlenk tube containing a stirring bar and CsF (60.8 mg, 0.4 mmol, 2 equiv) was dried with a heat gun under reduced pressure and filled with Ar after cooling to room temperature. To this vessel, were added NiCl2 (1.3 mg, 0.01 mmol, 5 mol %), toluene (1 mL), acyl fluorides (1) (0.2 mmol, 1 equiv) and trimethyl(tributylstannyl)silane (2) (87.2 mg, 0.24 mmol, 1.2 equiv). The mixture was heated at 140 °C with stirring for 24 h. The solution was then cooled to room temperature, and the solvent was removed under vacuum. The decarbonylative stannylation products 3 were purified by flash column chromatography on silica gel.
3.2.5. One-Pot Decarbonylative Stannylation/Migita-Kosugi-Stille Cross-Coupling Reaction of 1a
A 20 mL dried Schlenk tube containing a stirring bar and CsF (60.8 mg, 0.4 mmol, 2 equiv) was dried with a heat gun under reduced pressure and filled with Ar after cooling to room temperature. To this vessel, were added with NiCl2 (1.3 mg, 0.01 mmol, 5 mol %), toluene (1 mL), benzoyl fluoride (1a) (24.8 mg, 0.2 mmol), and trimethyl(tributylstannyl)silane (2) (87.2 mg, 0.24 mmol, 1.2 equiv). The mixture was heated at 140 °C with stirring for 24 h. The solution was then cooled to room temperature. 6-Bromobenzo[b]thiophene (42.6 mg, 0.2 mmol, 1 equiv), palladium acetate (0.4 mg, 0.002 mmol, 1 mol %), tricyclohexylphosphine (1.1 mg, 0.004 mmol, 2 mol %), and anhydrous cesium fluoride (45.6 mg, 0.3 mmol, 1.5 equiv) were added to the reaction mixture. The mixture was heated at 110 °C with stirring. After 24 h, the reaction mixture was cooled, the volatiles were evaporated under reduced pressure. The product was purified by flash chromatography on silica gel by elution with hexane, compound 4 was obtained in 71% yield (30 mg, 0.14 mmol) as white solid [31].
3.3. Characterization Data of Starting Materials and Products
Quinoline-6-carbonyl fluoride (1t). Yield: 55% (288.8 mg); white solid; melting point: 104–105 °C; 1H NMR (400 MHz, CDCl3) δ 7.53–7.56 (m, 1H), 8.20–8.25 (m, 2H), 8.30 (dd, J = 8.4, 1.9 Hz, 1H), 8.62 (d, J = 1.9 Hz, 1H), 9.08 (dd, J = 4.3, 1.9 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 122.5, 123.1, 127.4, 129.4 (d, JC-F = 4.0 Hz), 130.8 (d, JC-F = 1.4 Hz), 133.9 (d, JC-F = 3.0 Hz), 137.6, 150.7, 153.8, 156.9 (d, JC-F = 344.2 Hz); 19F{1H} NMR (282 MHz, CDCl3) δ 18.9; FT-IR (neat, cm−1): 735 (s), 781 (s), 854 (s), 1011 (s), 1045 (s), 1171 (s), 1231 (s), 1622 (s), 1805 (s); Anal. Calcd for C14H9FO3: C, 68.57; H, 3.45; N, 8.00%. Found: C, 68.50; H, 3.23; N, 7.95%.
Trimethyl(tributylstannyl)silane (2) [37]. Yield: 92% (2.67 g); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.23 (s, JH-Sn = 26.4 Hz, 9H), 0.84–0.90 (m, 15H), 1.29 (sext, J = 7.3 Hz, 6H), 1.44–1.48 (m, 6H).
Tributyl(phenyl)stannane (3a) [33]. Yield: 90% (66.1 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.91–0.95 (m, 9H), 1.03–1.16 (m, JH-Sn = 54.6 Hz, 6H), 1.33–1.41 (m, 6H), 1.53–1.63 (m, 6H), 7.32–7.36 (m, 3H), 7.46–7.54 (m, JH-Sn = 36.4 Hz, 2H).
Tributyl(p-tolyl)stannane (3b) [33]. Yield: 81% (61.8 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.90 (t, J = 7.2 Hz, 9H), 1.00–1.10 (m, JH-Sn = 52.4 Hz, 6H), 1.31–1.38 (m, 6H), 1.48–1.59 (m, 6H), 2.35 (s, 3H), 7.17 (d, J = 7.2 Hz, 2H), 7.37 (d, J = 7.8 Hz, JH-Sn = 35.8 Hz, 2H).
Tributyl(o-tolyl)stannane (3c) [31]. Yield: 63% (48.0 mg); colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 7.2 Hz, 9H), 1.01–1.14 (m, JH-Sn = 50.4 Hz, 6H), 1.34 (sext, J = 7.2 Hz, 6H), 1.47–1.58 (m, 6H), 2.40 (s, 3H), 7.12–7.17 (m, 1H), 7.17–7.25 (m, 2H), 7.40 (d, J = 6.8 Hz, JH-Sn = 42.8 Hz, 1H).
Tributyl(4-butylphenyl)stannane (3d). Yield: 67% (56.7 mg); colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 7.6 Hz, 9H), 0.94 (t, J = 7.6 Hz, 3H), 0.99–1.12 (m, JH-Sn = 50.8 Hz, 6H), 1.29–1.40 (m, 8H), 1.50–1.64 (m, 8H), 2.60 (t, J = 7.8 Hz, 2H), 7.14–7.19 (m, 2H), 7.37 (d, J = 6.4 Hz, JH-Sn = 39.2 Hz, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 9.7 (JC-Sn = 338.7 Hz), 13.8, 14.1, 22.6, 27.6 (JC-Sn = 57.2 Hz), 29.3 (JC-Sn = 19.8 Hz), 33.8, 35.8, 128.3 (JC-Sn = 41.2 Hz), 136.5 (JC-Sn = 31.3 Hz), 138.3, 142.7; FT-IR (neat, cm−1): 729 (m), 748 (m), 1070 (m), 1045 (m), 1377 (m), 1458 (m), 2855 (m), 2872 (m), 2928 (m), 2959 (m); Anal. Calcd for C22H40Sn: C, 62.43; H, 9.53%. Found: C, 62.30; H, 9.55%.
[1,1′-Biphenyl]-4-yltributylstannane (3e) [33]. Yield: 82% (72.7 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.93 (t, J = 7.5 Hz, 9H), 1.03–1.18 (m, JH-Sn = 43.2 Hz, 6H), 1.38 (sext, J = 7.5 Hz, 6H), 1.55–1.66 (m, 6H), 7.35–7.39 (m, 1H), 7.45–7.48 (m, 2H), 7.53–7.60 (m, 4H), 7.62–7.64 (m, 2H).
[1,1′-Biphenyl]-3-yltributylstannane (3f) [33]. Yield: 56% (49.6 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.91 (t, J = 7.5 Hz, 9H), 1.06–1.16 (m, JH-Sn = 50.8 Hz, 6H), 1.33–1.38 (m, 6H), 1.54–1.63 (m, 6H), 7.35–7.38 (m, 1H), 7.40–7.48 (m, 4H), 7.51–7.54 (m, 1H), 7.58–7.62 (m, 2H), 7.63–7.71 (d, J = 1.8 Hz, JH-Sn = 40.2 Hz, 1H).
[1,1′-Biphenyl]-2-yltributylstannane (3g) [33]. Yield: 85% (75.4 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.69–0.77 (m, JH-Sn = 51.0 Hz, 6H), 0.84 (t, J = 7.3 Hz, 9H), 1.23 (sext, J = 7.8 Hz, 6H), 1.32–1.38 (m, 6H), 7.32–7.35 (m, 3H), 7.36–7.39 (m, 3H), 7.39–7.42 (m, 2H), 7.56 (d, J = 7.3 Hz, JH-Sn = 40.8 Hz, 1H).
Tributyl(4-methoxyphenyl)stannane (3h) [31]. Yield: 64% (50.8 mg); colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 7.2 Hz, 9H), 0.96–1.09 (m, JH-Sn = 51.2 Hz, 6H), 1.33 (sext, J = 7.6 Hz, 6H), 1.48–1.59 (m, 6H), 3.81 (s, 3H), 6.91 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, JH-Sn = 37.2 Hz, 2H).
(4-Butoxyphenyl)tributylstannane (3i). Yield: 61% (53.6 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.89 (t, J = 7.2 Hz, 9H), 0.98 (t, J = 7.2 Hz, 3H), 1.01–1.04 (m, JH-Sn = 51.6 Hz, 6H), 1.33 (sext, J = 7.8 Hz, 6H), 1.47–1.58 (m, 8H), 1.75–1.80 (m, 2H), 3.96 (t, J = 6.6 Hz, 2H), 6.90 (d, J = 8.5 Hz, JH-Sn = 61.8 Hz, 2H), 7.36 (d, J = 8.5 Hz, JH-Sn = 42.6 Hz, 2H); 13C{1H} NMR (151 MHz, CDCl3) δ 9.7 (JC-Sn = 333.0 Hz), 13.9, 14.0, 19.4, 27.6 (JC-Sn = 55.5 Hz), 29.2 (JC-Sn = 20.8 Hz), 31.5, 67.4, 114.6 (JC-Sn = 43.9 Hz), 131.8, 137.6 (JC-Sn = 34.7 Hz), 159.4; FT-IR (neat, cm−1): 671 (m), 754 (m), 1072 (m), 1130 (m), 1207 (m), 1242 (m), 1273 (m), 1464 (m), 1495 (m), 1585 (m), 2855 (m), 2872 (m), 2928 (m), 2959 (m); Anal. Calcd for C22H40OSn: C, 60.15; H, 9.18%. Found: C, 60.09; H, 9.32%.
(4-(Benzyloxy)phenyl)tributylstannane (3j) [38]. Yield: 50% (47.3 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.90 (t, J = 7.2 Hz, 9H), 0.98–1.10 (m, JH-Sn = 51.0 Hz, 6H), 1.34 (sext, J = 7.2 Hz, 6H), 1.52–1.57 (m, 6H), 5.07 (s, 2H), 7.00 (d, J = 8.5 Hz, 2H), 7.32–7.36 (m, 1H), 7.37–7.43 (m, 4H), 7.46 (d, J = 7.2 Hz, 2H).
Benzo[d][1,3]dioxol-5-yltributylstannane (3k) [29]. Yield: 87% (71.5 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.90 (t, J = 7.2 Hz, 9H), 0.99–1.08 (m, JH-Sn = 49.8 Hz, 6H), 1.33 (sext, J = 7.8 Hz, 6H), 1.50–1.58 (m, 6H), 5.92 (s, 2H), 6.86 (d, J = 7.4 Hz, 1H), 6.90 (d, J = 7.5 Hz, 1H), 6.94 (s, JH-Sn = 37.2 Hz, 1H).
Tributyl(4-(trifluoromethyl)phenyl)stannane (3l) [33]. Yield: 61% (53.1 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.89 (m, J = 7.2 Hz, 9H), 1.04–1.15 (m, JH-Sn = 50.6 Hz, 6H), 1.33 (sext, J = 7.2 Hz, 6H), 1.50–1.57 (m, 6H), 7.55 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, JH-Sn = 34.8 Hz, 2H).
Tributyl(4-fluorophenyl)stannane (3m) [33]. Yield: 86% (66.2 mg); colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 7.3 Hz, 9H), 0.98–1.12 (m, JH-Sn = 51.0 Hz, 6H), 1.33 (sext, J = 8.0 Hz, 6H), 1.47–1.60 (m, 6H), 7.00–7.08 (m, 2H), 7.35–7.48 (m, 2H).
Tributyl(4′-fluoro-[1,1′-biphenyl]-4-yl)stannane (3n) [33]. Yield: 72% (66.4 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.91 (t, J = 7.2 Hz, 9H), 1.05–1.15 (m, JH-Sn = 50.1 Hz, 6H), 1.36 (sext, J = 7.2 Hz, 6H), 1.54–1.62 (m, 6H), 7.13 (dd, J = 8.7 Hz, JF-H = 8.7 Hz, 2H), 7.51–7.53 (m, 2H), 7.54–7.57 (m, 4H).
Tributyl(4-chlorophenyl)stannane (3o) [37]. Yield: 90% (72.3 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.88 (t, J = 7.5 Hz, 9H), 1.00–1.10 (m, JH-Sn = 50.1 Hz, 6H), 1.32 (sext, J = 7.2 Hz, 6H), 1.47–1.56 (m, 6H), 7.28–7.33 (m, 2H), 7.34–7.42 (m, JH-Sn = 36.0 Hz, 2H).
Tributyl(naphthalen-1-yl)stannane (3p) [33]. Yield: 51% (42.6 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.88 (t, J = 7.2 Hz, 9H), 1.19–1.22 (m, JH-Sn = 50.4 Hz, 6H), 1.35 (sext, J = 7.8 Hz, 6H), 1.54–1.60 (m, 6H), 7.42–7.51 (m, 3H), 7.63 (d, J = 6.6 Hz, JH-Sn = 46.2 Hz, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.85 (d, J = 7.2 Hz, 1H).
Tributyl(naphthalen-2-yl)stannane (3q) [33]. Yield: 79% (65.9 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.92 (m, J = 7.2 Hz, 9H), 1.09–1.21 (m, JH-Sn = 50.1 Hz, 6H), 1.38 (sext, J = 7.2 Hz, 6H), 1.56–1.65 (m, 6H), 7.44–7.51 (m, 2H), 7.59 (d, J = 8.1 Hz, JH-Sn = 33.0 Hz, 1H), 7.79–7.87 (m, 3H), 7.96 (s, JH-Sn = 44.4 Hz, 1H).
Tributyl(9H-fluoren-1-yl)stannane (3r). Yield: 82% (74.7 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.90 (t, J = 7.2 Hz, 9H), 1.11–1.23 (m, JH-Sn = 51.0 Hz, 6H), 1.36 (sext, J = 7.2 Hz, 6H), 1.52–1.62 (m, 6H), 3.85 (s, 2H), 7.30–7.42 (m, 4H), 7.57 (d, J = 7.5 Hz, 1H), 7.76 (dd, J = 7.2, 1.2 Hz, 1H), 7.80 (d, J = 7.8 Hz, 1H); 13C{1H} NMR (151 MHz, CDCl3) δ 9.8 (JC-Sn = 338.7 Hz), 13.8, 27.6 (JC-Sn = 60.1 Hz), 29.4 (JC-Sn = 19.6 Hz), 39.4, 119.9, 120.0, 125.0, 126.3, 126.6, 126.9, 135.0, 138.3, 140.3, 142.2, 143.0, 150.8; FT-IR (neat, cm−1): 731 (m), 752 (m), 1207 (s), 1220 (m), 1225 (m), 1456 (m), 1464 (m), 1695 (m), 2855 (m), 2865 (m), 2926 (m), 2959 (m); Anal. Calcd for C25H36Sn: C, 65.95; H, 7.97%. Found: C, 66.06; H, 8.20%.
Benzo[b]thiophen-2-yltributylstannane (3s) [39]. Yield: 62% (52.5 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.91 (t, J = 7.2 Hz, 9H), 1.12–1.20 (m, JH-Sn = 52.2 Hz, 6H), 1.36 (sext, J = 7.2 Hz, 6H), 1.56–1.64 (m, 6H), 7.27–7.29 (m, 1H), 7.32 (t, J = 6.6 Hz, 1H), 7.39 (s, JH-Sn = 24.0 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 7.8 Hz, 1H).
6-(Tributylstannyl)quinoline (3t) [32]. Yield: 84% (70.3 mg); colorless oil; 1H NMR (600 MHz, CDCl3) δ 0.89 (t, J = 7.5 Hz, 9H), 1.08–1.20 (m, JH-Sn = 51.0 Hz, 6H), 1.36 (sext, J = 7.8 Hz, 6H), 1.53–1.64 (m, 6H), 7.39 (dd, J = 8.4, 4.2 Hz, 1H), 7.81 (d, J = 7.8 Hz, JH-Sn = 31.5 Hz, 1H), 7.91 (s, JH-Sn = 42.0 Hz, 1H), 8.05 (d, J = 8.4 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 8.89 (dd, J = 8.4, 1.2 Hz, 1H).
6-Phenylbenzo[b]thiophene (4) [40]. Yield: 71% (30.0 mg); white solid; 1H NMR (400 MHz, CDCl3) δ 7.35–7.42 (m, 2H), 7.46–7.51 (m, 3H), 7.61 (dd, J = 8.4, 1.8 Hz, 1H), 7.66–7.90 (m, 2H), 7.95 (d, J = 8.4, 1H), 8.04 (d, J = 1.8 Hz, 1H).
4. Summary
In summary, we have developed an efficient and convenient method for the inexpensive NiCl2-catalyzed decarbonylative stannylation of a series of acyl fluorides, which is notable for being both ligand and additive-free. A one-pot decarbonylative stannylation/Migita-Kosugi-Stille reaction further demonstrated the synthetic applicability of our protocol because the isolation of toxic organotin compounds is not necessary. This study can expand the chemistry of acyl fluorides in terms of carbon-heteroatom bond formations.
Acknowledgments
We gratefully thank the SC-NMR Laboratory (Okayama University) for the NMR spectral measurements.
Supplementary Materials
The following are available online. 1H NMR, 13C{1H} NMR, 19F{1H} NMR spectra of representative starting materials and final products.
Author Contributions
X.W. developed above reactions and wrote the manuscript; X.W., Z.W. and L.L. prepared starting materials and expanded the substrates scope; Y.A. conducted the additional experiments required by reviewers; Y.N. supervised the project and revised the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 3a–3t are available from the authors.
References
- 1.Bunton C.A., Fendler J.H. The Hydrolysis of Acetyl Fluoride. J. Org. Chem. 1966;31:2307–2312. doi: 10.1021/jo01345a053. [DOI] [Google Scholar]
- 2.Carpino L.A., Beyermann M., Wenschuh H., Bienert M. Peptide Synthesis via Amino Acid Halides. Acc. Chem. Res. 1996;29:268–274. doi: 10.1021/ar950023w. [DOI] [Google Scholar]
- 3.Blanchard N., Bizet V. Acid Fluorides in Transition-Metal Catalysis: A Good Balance between Stability and Reactivity. Angew. Chem. Int. Ed. 2019;58:2–6. doi: 10.1002/anie.201900591. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y., Rovis T. A Unique Catalyst Effects the Rapid Room-Temperature Cross-Coupling of Organozinc Reagents with Carboxylic Acid Fluorides, Chlorides, Anhydrides, and Thioesters. J. Am. Chem. Soc. 2004;126:15964–15965. doi: 10.1021/ja044113k. [DOI] [PubMed] [Google Scholar]
- 5.Ogiwara Y., Maegawa Y., Sakino D., Sakai N. Palladium-Catalyzed Coupling of Benzoyl Halides with Aryltrifluorosilanes Leading to Diaryl Ketones. Chem. Lett. 2016;45:790–792. doi: 10.1246/cl.160223. [DOI] [Google Scholar]
- 6.Ogiwara Y., Sakino D., Sakurai Y., Sakai N. Acid Fluorides as Acyl Electrophiles in Suzuki-Miyaura Coupling. Eur. J. Org. Chem. 2017:4324–4327. doi: 10.1002/ejoc.201700917. [DOI] [Google Scholar]
- 7.Liu C., Szostak M. Decarbonylative cross-coupling of amides. Org. Biomol. Chem. 2018;16:7998–8010. doi: 10.1039/C8OB01832D. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Q., Szostak M. Redox-Neutral Decarbonylative Cross-Couplings Coming of Age. ChemSusChem. 2019;12 doi: 10.1002/cssc.201900408. [DOI] [PubMed] [Google Scholar]
- 9.Ogiwara Y., Sakai N. Acyl Fluorides in Late Transition-Metal Catalysis. Angew. Chem. Int. Ed. 2019 doi: 10.1002/anie.201902805. [DOI] [PubMed] [Google Scholar]
- 10.Ogiwara Y., Sakurai Y., Hattori H., Sakai N. Palladium-Catalyzed Reductive Conversion of Acyl Fluorides via Ligand-Controlled Decarbonylation. Org. Lett. 2018;20:4204–4208. doi: 10.1021/acs.orglett.8b01582. [DOI] [PubMed] [Google Scholar]
- 11.Keaveney S.T., Schoenebeck F. Palladium-Catalyzed Decarbonylative Trifluoromethylation of Acid Fluorides. Angew. Chem. Int. Ed. 2018;57:4073–4077. doi: 10.1002/anie.201800644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malapit C.A., Bour J.R., Brigham C.E., Sanford M.S. Base-free nickel-catalysed decarbonylative Suzuki-Miyaura coupling of acid fluorides. Nature. 2018;563:100–104. doi: 10.1038/s41586-018-0628-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakurai S., Yoshida T., Tobisu M. Iridium-catalyzed decarbonylative coupling of acyl fluorides with arenes and heteroarenes via C-H activation. Chem. Lett. 2019;48:94–97. doi: 10.1246/cl.180857. [DOI] [Google Scholar]
- 14.Okuda Y., Xu J., Ishida T., Wang C., Nishihara Y. Nickel-Catalyzed Decarbonylative Alkylation of Aroyl Fluorides Assisted by Lewis-Acidic Organoboranes. ACS Omega. 2018;3:13129–13140. doi: 10.1021/acsomega.8b02155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X., Wang Z., Asanuma Y., Nishihara Y. Synthesis of 2-Substituted Propenes by Bidentate Phosphine-Assisted Methylenation of Acyl Fluorides and Acyl Chlorides with AlMe3. Org. Lett. 2019 doi: 10.1021/acs.orglett.9b01059. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z., Wang X., Nishihara Y. Nickel-catalysed decarbonylative borylation of aroyl fluorides. Chem. Commun. 2018;54:13969–13972. doi: 10.1039/C8CC08504H. [DOI] [PubMed] [Google Scholar]
- 17.Liu C., Ji C.L., Hong X., Szostak M. Palladium-Catalyzed Decarbonylative Borylation of Carboxylic Acids: Tuning Reaction Selectivity by Computation. Angew. Chem. Int. Ed. 2018;57:16721–16726. doi: 10.1002/anie.201810145. [DOI] [PubMed] [Google Scholar]
- 18.Shi S., Szostak M. Decarbonylative Borylation of Amides by Palladium Catalysis. ACS Omega. 2019;4:4901–4907. doi: 10.1021/acsomega.9b00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hassan J., Sevignon M., Gozzi C., Schulz E., Lemaire M. Aryl-Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev. 2002;102:1359–1470. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 20.Johansson Seechurn C.C.C., Kitching M.O., Colacot T.J., Snieckus V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 21.Cordovilla C., Bartolom C., Martinez-Ilarduya J.M., Espinet P. The Stille Reaction, 38 Years Later. ACS Catal. 2015;5:3040–3053. doi: 10.1021/acscatal.5b00448. [DOI] [Google Scholar]
- 22.Mailhol D., Willwacher J., Kausch-Busies N., Rubitski E.E., Sobol Z., Schuler M., Lam M.H., Musto S., Loganzo F., Maderna A., et al. Synthesis, Molecular Editing, and Biological Assessment of the Potent Cytotoxin Leiodermatolide. J. Am. Chem. Soc. 2014;136:15719–15729. doi: 10.1021/ja508846g. [DOI] [PubMed] [Google Scholar]
- 23.Li J., Yang P., Yao M., Deng J., Li A. Total Synthesis of Rubriflordilactone A. J. Am. Chem. Soc. 2014;136:16477–16480. doi: 10.1021/ja5092563. [DOI] [PubMed] [Google Scholar]
- 24.Logan M.M., Toma T., Thomas-Tran R., Du Bois J. Asymmetric synthesis of batrachotoxin: Enantiomeric toxins show functional divergence against NaV. Science. 2016;354:865–869. doi: 10.1126/science.aag2981. [DOI] [PubMed] [Google Scholar]
- 25.Wursthorn K.R., Kuivila H.G. Synthesis of substituted aryltrimethylstannanes by the reaction of trimethylstannylsodium with aryl bromides. J. Organomet. Chem. 1977;140:29–33. doi: 10.1016/S0022-328X(00)84393-4. [DOI] [Google Scholar]
- 26.Knochel P., Singer R.D. Preparation and Reactions of Polyfunctional Organozinc Reagents in Organic Synthesis. Chem. Rev. 1993;93:2117–2188. doi: 10.1021/cr00022a008. [DOI] [Google Scholar]
- 27.Zhao Z.Q., Sun B., Peng L.Z., Li Y., Li Y.L. Novel Access to Organostannane Compounds under Ultrasound Irradiation. Chin. J. Chem. 2004;22:1382–1383. doi: 10.1002/cjoc.20040221133. [DOI] [Google Scholar]
- 28.Murata M., Watanabe S., Masuda Y. Palladium-Catalyzed Cross-Coupling Reaction of Tributyltin Hydride with Aryl Iodides: Formation of A Tin-Carbon Bond. Synlett. 2000;7:1043–1045. doi: 10.1002/chin.200045181. [DOI] [Google Scholar]
- 29.Komeyama K., Asakura R., Takaki K. A Sn atom-economical approach toward arylstannanes: Ni-catalysed stannylation of aryl halides using Bu3SnOMe. Org. Biomol. Chem. 2015;13:8713–8716. doi: 10.1039/C5OB01096A. [DOI] [PubMed] [Google Scholar]
- 30.Azizian H., Eaborn C., Pidcock A. Synthesis of organotrialkylstannanes. The reaction between organic halides and hexaalkyldistannanes in the presence of palladium complexes. J. Organomet. Chem. 1981;215:49–58. doi: 10.1016/S0022-328X(00)84615-X. [DOI] [Google Scholar]
- 31.Gribanov P.S., Golenko Y.D., Topchiy M.A., Minaeva L.I., Asachenko A.F., Nechaev M.S. Stannylation of Aryl Halides, Stille Cross-Coupling, and One-Pot, Two-Step Stannylation/Stille Cross-Coupling Reactions under Solvent-Free Conditions. Eur. J. Org. Chem. 2018:120–125. doi: 10.1002/ejoc.201701463. [DOI] [Google Scholar]
- 32.Gu Y., Martin R. Ni-Catalyzed Stannylation of Aryl Esters via C–O Bond Cleavage. Angew. Chem. Int. Ed. 2017;56:3187–3190. doi: 10.1002/anie.201611720. [DOI] [PubMed] [Google Scholar]
- 33.Yue H., Zhu C., Rueping M. Catalytic Ester to Stannane Functional Group Interconversion via Decarbonylative Cross-Coupling of Methyl Esters. Org. Lett. 2018;20:385–388. doi: 10.1021/acs.orglett.7b03669. [DOI] [PubMed] [Google Scholar]
- 34.Fahey D.R., Mahan J.E. Oxidative Additions of Aryl, Vinyl, and Acyl Halides to Triethylphosphinenickel(0) Complexes. J. Am. Chem. Soc. 1977;99:2501–2508. doi: 10.1021/ja00450a017. [DOI] [Google Scholar]
- 35.Lee L., Shim C.S., Chung S.Y., Kim H.Y., Lee H.W. Cross-interaction constants as a measure of the transition-state structure. Part 1. The degree of bond formation in nucleophilic substitution reactions. J. Chem. Soc. Perkin. Trans. 2. 1988;0:1919–1923. doi: 10.1039/p29880001919. [DOI] [Google Scholar]
- 36.Lal G.S., Pez G.P., Pesaresi R.J., Prozonic F.M., Cheng H.S. Bis(2-methoxyethyl)aminosulfur Trifluoride: A New Broad-Spectrum Deoxofluorinating Agent with Enhanced Thermal Stability. J. Org. Chem. 1999;64:7048–7054. doi: 10.1021/jo990566+. [DOI] [Google Scholar]
- 37.Wang D.-Y., Wang C., Uchiyama M. Stannyl-Lithium: A Facile and Efficient Synthesis Facilitating Further Applications. J. Am. Chem. Soc. 2015;137:10488–10491. doi: 10.1021/jacs.5b06587. [DOI] [PubMed] [Google Scholar]
- 38.Makaravage K.J., Brooks A.F., Mossine A.V., Sanford M.S., Scott P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016;18:5440–5443. doi: 10.1021/acs.orglett.6b02911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akram M.O., Shinde P.S., Chintawarc C.C., Patil N.T. Gold(I)-catalyzed cross-coupling reactions of aryldiazonium salts with organostannanes. Org. Biomol. Chem. 2018;16:2865–2869. doi: 10.1039/C8OB00630J. [DOI] [PubMed] [Google Scholar]
- 40.Abe M., Mori T., Osaka I., Sugimoto K., Takimiya K. Thermally, Operationally, and Environmentally Stable Organic Thin-Film Transistors Based on Bis[1]benzothieno[2,3-d:2′,3′-d′]naphtho[2,3-b:6,7-b′]dithiophene Derivatives: Effective Synthesis, Electronic Structures, and Structure–Property Relationship. Chem. Mater. 2015;27:5049–5057. doi: 10.1021/acs.chemmater.5b01608. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.