

Abstract
Congenital myopathies are typically characterised by early onset hypotonia, weakness and hallmark features on biopsy. Despite the rapid pace of gene discovery, ∼50% of patients with a congenital myopathy remain without a genetic diagnosis following screening of known disease genes. We performed exome sequencing on two consanguineous probands diagnosed with a congenital myopathy and muscle biopsy showing selective atrophy/hypotrophy or absence of type II myofibres. We identified variants in the gene (MYL1) encoding the skeletal muscle fast-twitch specific myosin essential light chain (ELC) in both probands. A homozygous essential splice acceptor variant (c.479-2A > G, predicted to result in skipping of exon 5 was identified in Proband 1, and a homozygous missense substitution (c.488T>G, p.(Met163Arg)) was identified in Proband 2. Protein modelling of the p.(Met163Arg) substitution predicted it might impede intermolecular interactions that facilitate binding to the IQ domain of myosin heavy chain, thus likely impacting on the structure and functioning of the myosin motor. MYL1 was markedly reduced in skeletal muscle from both probands, suggesting that the missense substitution likely results in an unstable protein. Knock down of myl1 in zebrafish resulted in abnormal morphology, disrupted muscle structure and impaired touch-evoked escape responses, thus confirming that skeletal muscle fast-twitch specific myosin ELC is critical for myofibre development and function. Our data implicate MYL1 as a crucial protein for adequate skeletal muscle function and that MYL1 deficiency is associated with severe congenital myopathy.
Introduction
Congenital myopathies are a clinically and genetically heterogeneous group of disorders characterised by muscle dysfunction, typically hypotonia and muscle weakness that are present at birth (1). Some forms can present with decreased intrauterine movements (2). Congenital myopathies are increasingly considered a spectrum of diseases, however they are usually sub-classified by the presence of distinct histopathological features on muscle biopsy which include: cores or core-like regions, internal nuclei, nemaline bodies and/or fibre-type disproportion. Mutations in over 20 genes are now known to cause congenital myopathies (1,3,4). Typically, congenital fibre type disproportion (CFTD) describes the pathological feature of a myopathy in which the type I myofibres are smaller than type II myofibres by 25%, in the absence of other pathological features (OMIM 255310). Mutations of five different genes (ACTA1, MYH7, RYR1, SEPN1 and TPM3) have been associated with CFTD; all of which are also associated with various structural features (5–9). Congenital myopathies characterised by a relative loss or hypotrophy of type II myofibres are much less frequent. Dominant and recessive mutations of MYH2 (OMIM 160740), encoding myosin heavy chain (MHC) type IIA, cause proximal myopathy, external ophthalmoplegia and loss or hypotrophy of type IIA myofibres (OMIM 605637) (10,11). Recently, bi-allelic mutation of TNNT3, encoding the skeletal muscle troponin-Tfast was identified in a patient presenting with arthrogryposis, nemaline myopathy and marked atrophy of type II myofibres (12). Small type II myofibres are also a non-specific feature in congenital myasthenic syndromes [CMSs; (13)], in ageing of muscle, critical illness myopathy and following steroid therapy, where histochemical IIB myofibres are affected (14). Here we present two isolated cases presenting with a severe congenital myopathy with absent or very small type II myofibres and bi-allelic mutations in the gene (MYL1; OMIM 160780) encoding the skeletal muscle fast-twitch specific myosin essential light chain (ELC).
Results
Clinical features and muscle pathology
The proband in Family 1 (Turkish origin) was the first child of second-degree consanguineous parents. There was no family history of neuromuscular disease. The pregnancy was complicated by polyhydramnios, with normal fetal movements. The boy was delivered at 36 weeks gestation by Caesarean section; he was floppy with no respiratory efforts and required ventilation via endotracheal tube.
Examination in the neonatal period demonstrated mild facial weakness, normal eye movements and no ptosis, a thin muscle build with severe weakness affecting axial and proximal muscles of both the upper, and to a lesser extent, lower limbs. He required nasogastric tube feeding. He had normal cardiac examination, electrocardiography and echocardiogram.
Deep tendon reflexes were absent, and there were mild flexion contractures in the elbows, knees and hips. EMG was myopathic and repetitive nerve stimulation showed a decrement at 3 Hz, increased after tetanic stimulation. Creatine kinase levels and brain MRI were normal. In view of the differential diagnosis between a congenital myopathy and a CMS, he was empirically started on pyridostigmine.
In the first few months of life some partial antigravity movements in the upper and lower limbs appeared. At the age of 3 months he was extubated into Continuous positive airway pressure but after a few weeks he required re-intubation, and gastrostomy with Nissen’s fundoplication. A stimulated single fibre EMG at 5 months showed minimal increased jitter not affected by the administration of tensilon, interpreted as a non-specific finding in the context of a myopathy. The pyridostigmine was therefore stopped, with no change in clinical status. The patient eventually died of respiratory failure at the age of 7 months.
Haematoxylin- and eosin-stained sections of the muscle biopsy (Fig. 1A) showed marked myopathic features, with severe fatty infiltration and diffuse perimysial and endomysial fibrosis. There was marked variation in myofibre size (<5–80 μm; normal range for age: 15–20 μm). There was a distinctive pattern of ‘florets’ of small slightly granular basophilic myofibres surrounding larger myofibres resembling a flower with a central area surrounded by petals. The small myofibres expressed fast (Fig. 1B) and fetal myosin and some also expressed developmental myosin and cardiac actin, whilst the hypertrophic myofibres expressed only slow myosin (Fig. 1C). Nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR) staining (Fig. 1D) was indistinct but uniform staining of larger myofibres and a mixed pattern of dark and light smaller myofibres was just apparent. Endomysial connective tissue was increased but there were very few internal nuclei. Gomori trichrome staining showed some focal circular red stained areas that were not reducing bodies (negative for menadione-linked glycerophosphate dehydrogenase without substrate) but may have been cytoplasmic bodies. There was no apparent necrosis, although acid phosphatase activity was elevated. Immunolabelling of sarcolemmal proteins was normal. In addition to fast and fetal myosin, the small myofibres also expressed high levels of cardiac α-actin, desmin and NCAM, suggesting immaturity and/or non-innervation.
Figure 1.
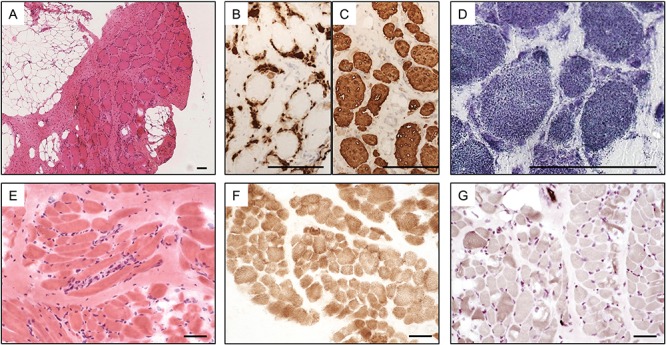
Pathological features of MYL1-myopathy. Staining of the skeletal muscle biopsy taken from the proband of Family 1 aged 5 months: haematoxylin and eosin (A), pan fast MHC (B) and slow MHC (C) showing many small myofibres with fast myosin surrounding larger myofibres with slow myosin and NADH-TR (D) showing indistinct fibre typing but uniform larger myofibres and some darker smaller myofibres; and muscle biopsy from the proband of Family 2 aged 8 months: haematoxylin and eosin (E) COX showing indistinct fibre typing (F), and ATPase pH 9.5 showing almost uniform type 1 myofibres (G). Scale bars represent 100 μm (A–D) or 50 μm (E–G).
Family 2 is Turkish; the parents are second cousins. There is no family history. The female proband was born at 37 weeks gestation by Caesarean section with a birth weight of 2400 g. Intrauterine movements were reduced. She was mechanically ventilated for the first 37 days of life; dependent on ventilatory support at the age of 3 months when she presented with sudden apnea. When evaluated at the age of 8 months (Supplementary Material, Video 1); she was an alert baby with a myopathic face, high-arched palate, facial weakness, normal conjugated eye movements, hypotonia and generalised weakness with some effort to move using mainly proximal muscles and head lag. There was markedly increased respiratory effort with lower costal recession, poor abdominal wall movements, asynchrony between the respiratory effort and mechanical ventilation. Serum creatine kinase level was normal. Cardiac examination (ECG and echocardiogram) was normal. Differential diagnosis included congenital myopathies.
At the age of 18 months the patient developed respiratory arrest due to a tracheostomy cannula obstruction and stayed in the hospital for 6 weeks. She gradually acquired head control and the ability to sit with support at ∼18 months of age, sit unaided at 24 months of age and walk with support at 3 years of age.
She is hypotonic with marked axial weakness, especially involving the neck flexors, and requires night-time ventilation without swallowing and feeding difficulties.
Muscle biopsy revealed non-specific myopathic features with mild to moderate variation in myofibre size, randomly scattered small myofibres and rare myofibres undergoing degeneration. A mild increase in endomysial and perimysial fibrous tissue was present (Fig. 1E). The typical checkerboard pattern of fibre types was not observed with staining for oxidative enzymes [NADH-TR, succinate dehydrogenase, cytochrome C oxidase (COX; Fig. 1F)]. ATPase stain (pH 9.5) revealed pronounced predominance of type I myofibres, with only a few small type II myofibres (Fig. 1G). No sample for electron microscopy was available.
Identification of mutations in MYL1
Exome sequencing of DNA from the proband of Family 1, identified a homozygous essential splice acceptor variant (intron 4; c.479-2A > G) in the gene encoding myosin, light chain 1, alkali, skeletal, fast (MYL1, OMIM 160780), also known as the fast skeletal muscle-specific ELC. Evaluation in Alamut revealed that this variant is likely to result in the formation of an acceptor site 2 bps downstream (NNSPLICE, MaxEntScan and Human Splice Finder) and in-frame skipping of exon 5. This likely removes 13% of the protein sequence, including the end of the second EF-hand motif, a region conserved to Baker’s yeast (Alamut). Muscle was not available for complementary DNA analysis. Bi-directional Sanger sequencing confirmed this variant and showed that as expected, each parent was a carrier. This variant is novel and has not previously been found in any control databases (1000 genomes, ExAC/GnomAD browsers).
Neurogenetic Sub-Exomic Sequencing (NSES) gene panel analysis of Proband 2 identified no mutations. The exome data showed 816 filtered rare coding variants. Overlaying these with the FANTOM5 52 highly enriched candidate muscle disease genes, identified a homozygous missense variant (c.488T > G) in exon 5 of MYL1 (Supplementary Material, Fig. S1A) as the most likely candidate. Skeletal muscle enrichment of MYL1 within FANTOM5 (15) and GTEx (16) is shown in Figure 2A. Sanger sequencing confirmed the variant and showed each parent was a carrier (Supplementary Material, Fig. S1B). The variant occurs within the same EF-hand domain that is likely deleted in Proband 1. It results in substitution of a highly-conserved amino-acid residue (p.(Met163Arg); Fig. 2B). This variant is novel and not in any control databases (1000 genomes, ExAC/GnomAD browsers). The substitution is suggested to be disease causing by all in-silico predictors (MutationTaster, SIFT [damaging, 0.000], PolyPhen-2 [probably damaging, 0.997], Provean [deleterious, −5.333], MutationAssessor [FI score 3.925]). In addition, the variant occurs 10 bp from the intron/exon boundary and is suggested to affect splicing (MutationTaster) and to introduce a SF2 binding site (Alamut). However, patient muscle was unavailable for cDNA studies.
Figure 2.
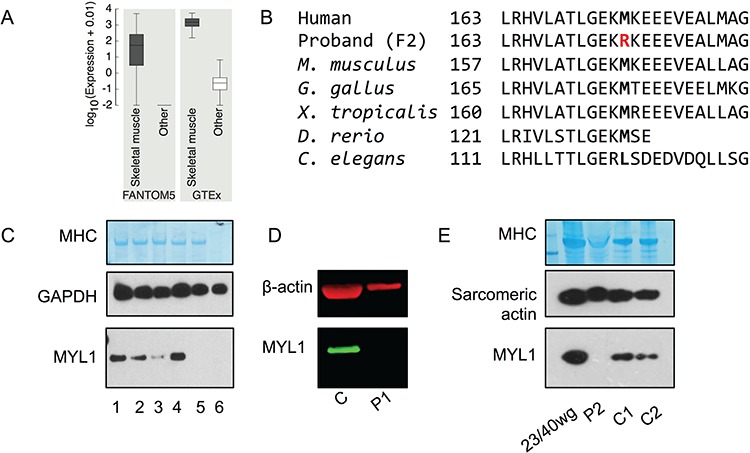
MYL1 expression and protein abundance. (A) Expression of MYL1 in skeletal muscle samples compared to other samples from FANTOM5 (tissue and primary cell) and GTEx databases. Units of expression are TPM (tags per million) for FANTOM5 and RPKM (reads per kilobase per million mapped reads) for GTEx. Number of samples in each comparison for FANTOM5 (skeletal muscle = 76, other = 1753), GTEx (skeletal muscle = 430, other = 8,125). (B) Evolutionary conservation of p.Met163. (C) Western blotting of lysates of mouse tissues (pooled from three 8-week-old C57BL6 mice) revealed that MYL1 (antibody SAB1409338) was similarly abundant in mouse skeletal muscles (diaphragm, EDL, quadriceps and soleus). Loading: quadriceps (1), EDL (2), soleus (3), diaphragm (4), heart (5) and brain (6). Western blotting for GAPDH and Coomassie staining of the MHC band are shown to demonstrate similar total protein loading. (D) Western blotting for MYL1 (green) and β-actin for control muscle and muscle from the proband (P1) of Family 1. (E) Western blotting of the muscle biopsy from the proband (P2) of Family 2, compared with human fetal (23/40 weeks gestation) and adult skeletal muscle (C1, C2) showed a relative loss of MYL1. Coomassie staining of the MHC band and western blotting for sarcomeric actin (5C5 antibody) are shown to demonstrate similar loading of sarcomeric proteins between samples.
No known or likely pathogenic variants were identified in any known neuromuscular disease genes in the exome data for either proband.
MYL1 protein studies
An antibody raised against full-length human MYL1 gave a single band at ∼21 kDa in mouse skeletal muscle (diaphragm, EDL, quadriceps and soleus) but not the heart or brain (Fig. 2C). This band corresponds to the full-length MLC1F isoform. No band corresponding to the truncated MLC3F isoform was observed. Analysis of MYL1 by immunoblotting of the muscle biopsies from the probands revealed an apparent total loss of MYL1 relative to fetal and adult human healthy control muscles (Fig. 2D–E).
MYL1 protein modelling
MYL1 encodes the fast myofibre non-regulatory myosin alkali/ELC isoforms MLC1F and MLC3F, with the N-terminus truncated MLC3 isoform being 44 residues shorter than MLC1. Two ELCs, two myosin phosphorylatable, or regulatory light chains (RLCs) and two MHCs make up each myosin multi-protein complex unit. Both ELCs and RLCs bind to the lever arm neck of the myosin cross-bridge/head, to the first two IQ motifs, which are 22–25 amino acid regions characterised by the conserved consensus sequence IQxxxRGxxxR. Although, from a structural point of view, the myosin ELC belong to the EF-superfamily of Ca2+-binding proteins, they have lost the possibility to bind Ca2+ throughout evolution (17,18). Nevertheless, the ELC still displays the helix–loop–helix fold typical of EF-hand domains, which is presented four times in the mature protein, which is formed by 194 amino acids (MLC1F; MLC3F). Despite their lost ability of actively acting as Ca2+-controlled regulators, myosin ELCs are still central to the proper functioning of myosin. The ELC plays an important role in regulating both cardiac and skeletal muscle contraction, it stabilises the neck lever arm structure to produce steps of a definite size (19) and also plays a role in fine-tuning of the molecular motor (20).
MLC1F and MLC3F, have discrete promoters within MYL1 and result from alternative splicing. To date, no high-resolution structure of human MLC1F or MLC3F is available. Nevertheless, ELC structures, invariably in their bound conformation to MHC, have been obtained for other organisms (e.g. chicken, scallop and yeast). Due to the extremely high degrees of sequence identity, these can be used as templates to safely predict the three-dimensional structure of their Homo sapiens counterparts by homology modelling. Using the Protein DataBank structure 2W4A as template, we built a model for MLC3F bound to its target site on the MHC, the first IQ motif of myosin II.
In the model, p.Met163 is in close contact with p.Leu802 and p.Cys798 of the myosin IQ domain suggesting it may be involved in a hydrophobic interaction with the leucine side chain, playing a role in IQ recognition and binding (Fig. 3A). Such an interaction would be lost upon substitution to an Arg, likely impacting the ELC interaction with MHC. Although the p.Met163 hydrophobic interaction with p.Leu802 on MHC could facilitate the binding of the ELC to the heavy chain, electrostatic interactions may play a major role. IQ domains have been reported to rely on electrostatics for acquiring the proper orientation and/or for correct binding to partners. The modelled IQ portion of MHC has a high density of surface-exposed arginine side chains and therefore a dense region of positive electrostatic potential, especially in the portion which is thought to interact directly with MLC3. The ELC exposes a wide region of negative electrostatic potential (Fig. 3B). Electrostatic interactions therefore likely play a crucial role in the binding of ELC to MHC. p.Met163 is located on the edge of the negative potential groove. Substitution to Arg should introduce repulsion towards the positive potential region of IQ, affecting the correct binding of the C-terminal lobe of ELC and its stabilisation of the heavy chain structure and thus impacting the proper structure and functioning of the myosin motor.
Figure 3.
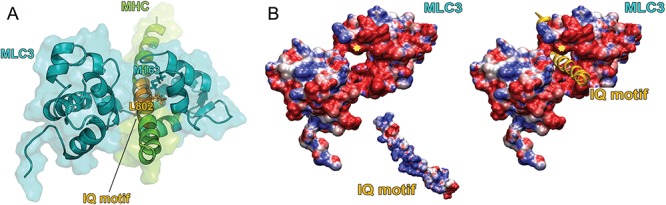
Protein modelling of the p.(Met163Arg) substitution. (A) Cartoon and surface representations of the modelled structure of MLC3 (cyan) bound to the IQ motif of the myosin heavy chain MHC (green). The MHC region contacting the MLC3 is depicted in orange. Met163 on MLC3 is represented as sticks and colored in cyan; Leu802, located on the IQ domain and interacting with Met163, is colored in orange. Met163 on MLC3 is represented as sticks and coloured in cyan; Leu802, located on the IQ domain and interacting with Met163, is coloured in orange. (B) Left: Electrostatic potential calculated separately for MLC3 and for the IQ motif of MHC and mapped on their molecular surfaces. Potentials less than −5 kT/e are coloured in red, and those greater than +5 kT/e are depicted in blue. The position of Met163 is indicated by a yellow star. Right: same as in left, but with the IQ motif represented as a cartoon and shown in the position it occupies when interacting with MLC3.
Morpholino knockdown of myl1 in zebrafish demonstrates the essential role of myl1 for muscle development and function
To study the impact of myl1 loss-of-function during muscle development, we generated morpholino (MO) knockdown myl1 zebrafish models. We used two MOs, one targeting the myl1 AUG start codon (MO AUG) and the other targeting the splice site of exon 2 (MO Spl), thus creating two independent myl1 MO zebrafish models. In un-injected control (UIC) embryos, a band corresponding to normal splicing of myl1 exon1–exon4 (440 bp) was present while no band was detected when the primer pair in exon1–intron1 was used (Fig. 4E). RT-PCR analysis of the MO Spl morphants demonstrated the normal exon1–exon4 band and an additional larger band (449 bp) due to the retention of intron 1. Sanger sequencing of the intronic inclusion revealed an in-frame stop codon at intronic position 42. The results demonstrate that the MO Spl is targeting the exon 2 splice site and is causing aberrant myl1 splicing.
Figure 4.
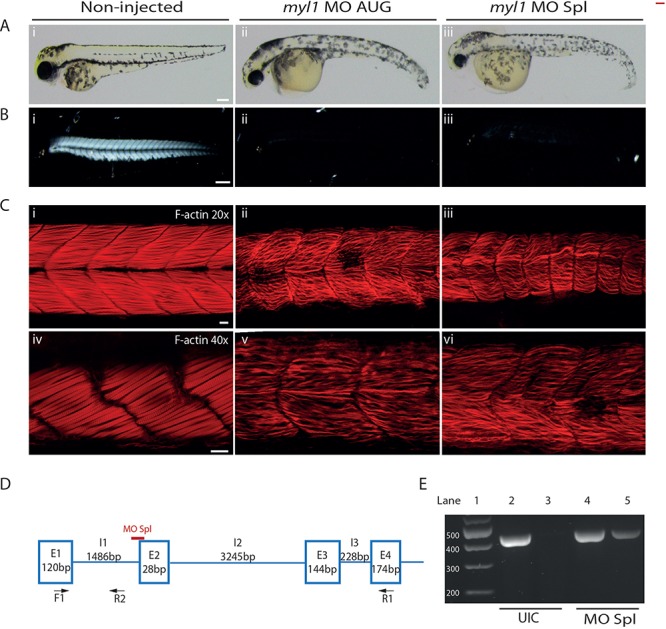
Knockdown of myl1 in zebrafish causes abnormal muscle development assessed at 48 hpf. (A) General morphology of uninjected control embryos (i), embryos injected with 1 ng of the MO targeting the myl1 AUG start codon (ii) and embryos injected with 3 ng of the myl1 splice blocking MO (iii). Scale bar represents 400 μm. (B) Birefringence of uninjected control embryos (i) and myl1 morphant embryos (ii, iii). Scale bar represents 200 μm. (C) Phalloidin staining of F-actin (red) of myl1 morphant embryos (ii, iii at 20× magnification; v, vi at 40× magnification) and uninjected control embryos (i at 20× magnification; iv at 40× magnification). Scale bar represents 20 μm (20× magnification) and 40 μm (40× magnification). (D) Schematic representation of the myl1 splice blocking MO target site shown in red. Presented are the exons and introns with the corresponding size. Arrows present the positions of the primers used to confirm the splicing defect in the myl1 splice blocking morphants. (E) RT-PCR validation of the splicing defect caused by the myl1 splice blocking MO. The primer pair F1-R1 produced a band for the endogenous myl1 product at the expected size (440 bp) in the uninjected controls (UIC, lane 2) and in myl1 splice blocking MO morphants (MO Spl, lane 4). No product was detected using primer pair F1-R2 in the uninjected controls (UIC, lane 3). In myl1 splice blocking MO morphants a band with the expected size of 449 bp corresponding to inclusion of intron 1 was detected (MO Spl, lane 5).
Consistent phenotypes were observed between the myl1 MO AUG and MO Spl morphants. At 48 h post fertilisation (hpf), the morphants had curved bodies, bent tails (Fig. 4A) and a marked reduction in touch-evoked escape response (Supplementary Materials, Videos 1, 2, 3). Muscle structure measured by the birefringence assay resulted in a bright signal indicating highly-organised myofibres in the UICs (Fig. 4Bi), whereas in the myl1 morphants a significant reduction in birefringence demonstrated gross impairment of the skeletal muscle structure (Fig. 4Bii and iii). Phalloidin labelling in the UIC embryos revealed densely packed and organised myofibres (Fig. 4 Ci, iv). In the myl1 morphants, the myofibres appeared ‘wavy’, sparse and disordered (Fig. 4Cii, iii, v and vi). Taken together, these data indicate that myl1 is required for the normal formation of myofibres and/or myofibre maintenance and thus appropriate muscle function.
To evaluate the effect of the c.488T>G MYL1 variant, we attempted a complementation assay but the injected mRNA exacerbated the phenotype (data not shown). We then injected human WT or c.488T>G MYL1 mRNA in wild-type (WT) zebrafish embryos and the resulting phenotype was compared to UIC fish. At 48 hpf, in WT mRNA injected zebrafish, 50% of the embryos showed a mild phenotype, 14% were moderately affected and 36% of the fish showed a severely affected phenotype (Supplementary Material, Fig. S2). In zebrafish injected with c.488T>G MYL1 mRNA all embryos demonstrated a mild phenotype (Supplementary Material, Fig. 2). Thus, while overexpression of WT MYL1 mRNA induced an aberrant phenotype in the injected zebrafish, the lack of a severe phenotype in the c.488T>G MYL1 mRNA injected fish demonstrates that the mutant mRNA is relatively non-functional and supports the pathogenicity of the c.488T>G MYL1 variant.
Discussion
Here we describe the identification of recessive mutations in MYL1 in two unrelated Turkish individuals with severe congenital myopathy and selective involvement of type II myofibres: small type II myofibres in Proband 1 and almost absence of type II myofibres in Proband 2. These features are highly unusual; small type I myofibres are typical in congenital myopathies, whilst small type II fibres are very rarely seen. It was not possible to determine if the type II/fast myofibres were atrophic or hypotrophic, or if they failed to develop. In Proband 1 myosin isoforms associated with immaturity were co-expressed with fast myosin; co-expression of immature proteins can occur in both atrophic and hypotrophic myofibres. In addition, some of the very small fast myofibres expressed cardiac actin suggesting that they were immature. Electron microscopy to look for redundant basal lamina associated with atrophy was not possible due to lack of tissue.
The MYL1 gene encodes two splice isoforms of skeletal muscle fast ELC (designated MLC1F and MLC3F) (OMIM 160780) (21), which are well studied in model organisms. They interact with fast MHC as either homodimers or heterodimers.
It has long been known that the ‘essential’ myosin light chains are necessary for the proper contraction of muscle. Removal of the ELC from chicken pectoralis myosin reduced the velocity of actin filament sliding to around a sixth of normal speed (22). Another study showed that selective removal of myosin ELCs from chicken skeletal myosin resulted in ∼50% reduction in isometric force, whereas removal of the RLC had no effect (23).
There are several isoforms of myosin ELCs and RLCs, each encoded by a different gene. MYL2 encodes the cardiac and slow-twitch muscle fibre specific regulatory myosin light chain (OMIM 160781), MYL3 the ventricular and slow-twitch skeletal myofibre ELC, MYL4 an ELC isoform present in embryonic cardiac muscle and adult atria (OMIM 160770) and MYL6 a smooth-muscle and non-muscle specific regulatory light chain (OMIM 609931). Human genetic diseases have previously been associated with MYL2, MYL3 and MYL4.
Dominant mutations of MYL2 (the cardiac and slow-twitch specific regulatory light chain, OMIM 160781) and MYL3 (OMIM 160790) cause hypertrophic cardiomyopathy. Recessive mutations in MYL2 cause cardiomyopathy in combination with a type I fibre myopathy (24). Affected individuals had normal prenatal and perinatal periods, but developed rapidly progressive muscle weakness from a few weeks of age and all died through heart failure by 6 months. Skeletal muscle from affected individuals showed increased variation in myofibre size with small type I myofibres and reduced abundance of MYL2 (24). Dominant mutations in MYL2 are missense mutations, whereas recessive MYL2 mutations are null mutations, including splice-site and frameshift mutations. Both dominant (25) and recessive (26) missense mutations in MYL3 cause cardiomyopathy. Missense mutation of MYL4 causes dominant atrial fibrillation (27), homozygosity for a frameshift mutation causes recessive atrial fibrillation (28).
Myl1 is an early marker of differentiating fast muscle in zebrafish (29). By in situ hybridisation, myl1 was detected in developing embryos from the 8-somite stage onwards, whereas other markers of fast-twitch muscle were only observed from the 10-somite stage. Myl1 transcript levels declined with development in post-somitogenic stages. In fish knocked-down for both myf5 and myod by MO injection, myl1 was reduced or absent, suggesting that myl1 expression is downstream of myf5/myod. Zammit et al. (30) showed that in mice, the Mlc1f promoter was active in embryonic, fetal and adult fast-twitch skeletal muscle, whereas the Mlc3f promoter was up-regulated during fetal development and remained active in adult fast-twitch skeletal muscle. Normal development of myotubes and myofibrillogenesis is impaired in chicken myoblast/myotube cultures by reduction of MLC1F, mediated by antisense knock down (31). This may relate to the atrophy/hypotrophy of type II myofibres seen on patient biopsy.
There was a marked reduction in type II myofibres in muscle biopsies from both probands. The muscle biopsy from Proband 1 was striking in having a ‘floret’ appearance of small fast myofibres surrounding slow myofibres. The larger type I fibres were, from the patient phenotype, insufficient to maintain muscle function. Stuart et al. recently studied the MHC and MLC composition of individual human skeletal myofibres by laser-capture micro-dissection followed by immunoblotting using the MYL1 antibody (SAB1409338) and mass spectrometry. Their data suggested MYL1 was not restricted to fast myofibres, but was in fact the most abundant MLC species in all myofibres (I, IIA, IIX), though with higher abundance in type IIA/IIX compared with type I myofibres (32). Thus, while MYL1 is considered the skeletal muscle fast-specific ELC, it may be critical to the function of all skeletal myofibre types. That type I fibres were not small may result from the presence of slow twitch ELC MYL3.
The severe atrophy/hypotrophy of type II myofibres also raised the possibility of a CMS. This was initially considered because of a modest electrodecrement on repetitive nerve stimulation and a borderline jitter in the proband from Family 1. However, modest secondary abnormalities of the neuromuscular junction are well known to occur in certain congenital myopathies (3), and this patient did not appear to benefit from pyridostigmine therapy. In addition, involvement of CMS genes was ruled out by the analysis of the relevant genes.
By western blotting, we showed absence of MYL1 protein from the muscle biopsies of both patients. The protein modelling suggested that the p.(Met163Arg) substitution could alter binding of MYL1 to the MHC and thereby impact myosin function. However, our western blots suggest the variant results in reduced stability of the mutant MYL1.
The patients had profound muscle weakness. This suggests that MYL1 is critical for muscle function or that compensation by other ELCs is not able to replace the function of MYL1, indicating a lack of redundancy between ELC isoforms. Relevant to this, no compensatory upregulation of other ELCs was seen by Nawrotzki et al. following experimental knock down of MLC1f (31). In this study we demonstrated that knock down of myl1 in zebrafish led to aberrant muscle development evidenced by the curved body, significantly reduced motility and gross impairment of myofibre organisation; supporting the crucial role of myl1 for muscle development and function. Over-expression of the c.488T>G MYL1 variant in WT zebrafish embryos only produced a mild phenotype. Over-expression of other skeletal muscle proteins are known to be deleterious (33,34). Whereas over-expression of WT MYL1 produced a moderate to severe phenotype in 50% of fish. This assay suggests that the c.448T>G variant is relatively non-functional and confirms the pathogenicity of this substitution.
Identification of the MYL1 mutation in Family 2 was aided by a gene prioritisation approach based on gene enrichment and abundance within the FANTOM5 datasets. This represents a novel approach to identify candidate genes. New approaches are much needed, in order to improve the rate of accurate gene discovery in small families or isolated probands with rare monogenetic diseases (4). Interestingly, Neto et al. (35) through data mining to highlight candidate genes for monogenic myopathies, had previously identified MYL1 as a top candidate for a range of muscle diseases, including congenital myopathies.
Here we present comprehensive evidence that bi-allelic loss-of-function MYL1 mutations represent a novel, albeit rare, cause of severe congenital myopathy. The unusual muscle pathology of type II myofibre absence or hypotrophy appears the most characteristic feature of MYL1 myopathy. Patients with a congenital myopathy and small or absent type II myofibres should be screened for mutations in MYL1.
Materials and Methods
Ethics approvals
All individuals were enrolled under appropriate procedures in accordance with the ethical standards of the responsible committees on human experimentation (UWA Human Research Ethics Committee RA/4/1/4403 and Great Ormond Street Hospital Research Ethics Committees GOSH 00/5802).
Exome sequencing
Family 1
Exome sequencing was performed on the proband of Family 1 within the UK10K project. Sequencing, alignment and variant calling were as previously (36).
Family 2
DNA extracted from peripheral blood of the proband from Family 2, was sequenced using version 1 of the NSES gene panel, as previously described (37), 93.7% of the targeted 336 genes were covered to >20-fold. Exome sequencing was performed using the Ion Torrent platform and data was mapped, annotated and filtered as previously (38). Average coverage was 128-fold with 90.7% of the exome covered to >20-fold.
Using the functional annotation of the mammalian genome (FANTOM5) promoter level expression atlas (15) we generated a ranked list of skeletal muscle enriched genes. Source file with precomputed relative expression was downloaded from http://fantom.gsc.riken.jp/5/tet/data/hg19.cage_peak_phase1and2combined_rel_expr.txt.gz. For each gene, we identified the promoter with the most enriched expression in skeletal muscle samples compared to median across the entire FANTOM5 collection [log10(max expression in any skeletal muscle sample + 1) – log10(median expression in the FANTOM5 collection +1)]. The 76 skeletal muscle sample tissue and primary cell expression profiles used are listed in Supplementary Material, Table 1. This identified 83 annotated protein-coding genes with a promoter showing more than 1000-fold enriched expression in skeletal muscle (Supplementary Material, Table 2). Of these, 31 had already been associated with human genetic muscle disease. The remaining 52 were prioritised as candidate myopathy genes. Many of these highly enriched genes are known striated muscle genes but many are not, thus this approach does not rely on a priori knowledge of gene function for prioritisation.
Histological analysis
An open muscle biopsy, from each proband, was frozen in isopentane cooled in liquid nitrogen, stained and immunolabelled according to standard procedures (14). The amount of muscle obtained from both cases was too small for electron microscopy to be performed. The biopsy from Proband 1 was taken from the quadriceps muscle at 5 months of age. A biopsy of the vastus lateralis was obtained from Proband 2 at 8 months of age.
Western blotting
Skeletal muscle from Proband 1 and a control was homogenised manually in lysis buffer containing 75 mm Tris-HCl (pH 6.8), 1% Sodium dodecyl sulfate and protease inhibitor. Protein concentration was assessed using BCA assay. A total of 40 μg of protein was loaded on NuPAGE 4–12% Bis-Tris protein gels, separated by electrophoresis for 1.5 h and then transferred to nitrocellulose membrane for 1.5 h. The membrane was blocked in Odyssey blocking buffer for 1 h at room temperature followed by incubation with primary antibodies against MYL1 (Sigma-Aldrich, SAB1409338) and β-Actin (Cell Signalling, 4967) overnight at 4°C. The membrane was washed three times for 10 min with 0.1 M PBS and incubated with IRDye® 800CW Donkey anti-Mouse IgG and IRDye® 680RD Donkey anti-Rabbit IgG for 1 h at room temperature. Following three washes for 10 min in 0.1 M PBS, the blot was imaged using an Odyssey Infrared Imaging System.
Muscle lysates for Proband 2, human controls (healthy human adult excess tissue from in vitro contracture testing and from a 23/40 gestation fetus) and mouse tissues were prepared as outlined below. Cryostat cut sections were lysed in Laemmli lysis buffer supplemented with protease inhibitor and DTT, boiled for 5 min at 94°C, centrifuged and the supernatant retained. The total protein was quantified using a Pierce™ BCA Protein quantification kit. NuPAGE™ Bis-Tris 4–12% gradient gels were loaded with 10 μg of protein and run at 150 V for 1 h. Gels were transferred onto PVDF, blocked for 1 h at room temperature in blocking solution and then incubated with primary antibodies overnight. Membranes were rinsed three times and then incubated with HRP-conjugated secondary antibodies. Membranes were rinsed again and detection was performed using Pierce ECL Plus Western Blotting Substrate. Primary antibodies: MYL1 (SAB1409338; 1:2000); sarcomeric actin (clone 5C5; 1:200 000) and GAPDH (clone GAPDH-71.1; 1:40 000). The Coomassie stained MHC band was also used to demonstrate approximate loading between muscle samples.
Protein modelling
The model of myosin ELC 3 (MLC3) in complex with MHC, was built by homology modelling using MODELLER 9.16 (39). We used as template the structure of the MLC3-MHC complex from Gallus Gallus (PDB entry 2W4A) (40) that displays a high degree of sequence identity (more than 80%) with the human MLC3 and myosin II. The models obtained were evaluated by DOPE score (41) and VADAR web server (42). The electrostatic properties were investigated using the APBS software. The pqr input file required to run APBS was prepared using PDB2PQR (43). The electrostatic potential was obtained by solving the linearised Poisson–Boltzmann equation at 298.5 K, using dielectric constant values of 2 and 78.54 for protein (solute) and solvent, respectively.
myl1 zebrafish analysis
Antisense MO oligonucleotides (Gene Tools, LLC) were designed against the myl1 start codon (TCTTAGCGTCCTTCTTAGGTGCCAT) and against the myl1 intron1-exon2 splice site (TCGAGCTGCAGGAAAACAGAGTTAT). Knockdown of myl1 was achieved by MO injections at doses of 1 ng for the MO against the start codon (MO AUG) and at 3 ng for the MO against the intron1-exon2 splice site (MO Spl). MOs were injected into zebrafish embryos at the 1–2 cell stage. The injected embryos were incubated at 28.5°C and dechorionated at 24 hpf.
Embryos at 48 hpf were mounted in 3% methylcellulose and the structure of the zebrafish muscle assessed using birefringence microscopy.
Immunofluorescent phalloidin (Molecular Probes, Australia) staining was performed on whole mounted zebrafish at 48 hpf. Confocal microscopy imaging was performed using a Leica confocal microscope and images were analysed using ImageJ software.
The specificity of the MO Spl was confirmed using RNA extracted from morphant and control zebrafish (n = 20 in each group) at 48 hpf. cDNA was synthetised using High Capacity RNA-to-cDNA™ Kit (ThermoFisher Scientific, Melbourne, Australia).
Clones containing human WT MYL1 or c.488T>G MYL1 variant were purchased from Piscataway, NJ. The clones were grown in NEB® 10-beta Competent E. coli cells and DNA was extracted using EndoFree Plasmid Kit (Qiagen, Melbourne, Australia). Linearisation of the plasmids was done by digestion with NotI enzyme at 37°C for 2 h. Capped WT and c.488T>G MYL1 mRNA was generated using the mMessage mMachine SP6 Transcription kit (Thermo Fisher Scientific). In total, 100 pg of WT or c.488T>G MYL1 mRNA were injected into 1-cell stage zebrafish embryos.
Electronic Database Information
1000genomes: http://www.1000genomes.org;
ExAC: http://exac.broadinstitute.org;
Exome variant server: http://evs.gs.washington.edu;
FANTOM5: http://fantom.gsc.riken.jp/5/;
gnomADbrower: http://gnomad.broadinstitute.org;
Mutation Assessor: http://mutationtaster.org;
MutationTaster: http://mutationtaster.org;
OMIM: http://omim.org;
PolyPhen-2: http://genetics.bwh.harvard.edu/pph2/;
Provean: http://provean.jcvi.org/index.php;
SIFT: http://sift.jcvi.org.
Supplementary Material
Acknowledgements
We are grateful to the families for their participation in this research. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. We are grateful to Dr Alison Testa for her contributions to this manuscript.
Conflict of Interest statement. None declared.
Funding
National Health and Medical Research Council of Australia (Early Career Researcher Fellowship, APP1035955 to G.R.); Research Fellowships (APP1002147 and APP1117510 to N.G.L.); Project Grant (APP1022707) and Project Grant (APP1146321 to ARRF, GR, NL); EU Collaborative grant (APP1055295); the Association Francaise contre les Myopathies, a UWA Collaborative Research Award (#15734); the European Community’s Seventh Framework Programme (FP7/2007–2013) funded grant “Integrated European –omics research project for diagnosis and therapy in rare neuromuscular and neurodegenerative diseases (NEUROMICS)” (grant agreement n° 2012–305121); the Muscular Dystrophy UK Grant on Gene Identification (18GRO-PG24-0271 to F.M.); the Wellcome Trust support to the UK10K consortium; the NIHR Great Ormond Street Hospital Biomedical Research Centre; National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London (to F.M.); Muscular Dystrophy UK to the Dubowitz Neuromuscular Centre; the MRC Neuromuscular Centre Biobank; Muscular Dystrophy UK to F.M. (18GRO-PG24-0271); The National Specialised Commissioned Team (NSCT) funding for the Congenital Muscular Dystrophies and Congenital Myopathy service in London.
References
- 1. Ravenscroft G., Laing N.G. and Clarke N.F. (2014) In Hilton-Jones D. (ed), Oxford Textbook of Neuromuscular Disorders. Oxford University Press, Oxford. [Google Scholar]
- 2. Ravenscroft G., Sollis E., Charles A.K., North K.N., Baynam G. and Laing N.G. (2011) Fetal akinesia: review of the genetics of the neuromuscular causes. J. Med. Genet., 48, 793–801. [DOI] [PubMed] [Google Scholar]
- 3. Ravenscroft G., Laing N.G. and Bonnemann C.G. (2015) Pathophysiological concepts in the congenital myopathies: blurring the boundaries, sharpening the focus. Brain, 138, 246–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ravenscroft G., Davis M.R., Lamont P., Forrest A. and Laing N.G. (2017) New era in genetics of early-onset muscle disease: Breakthroughs and challenges. Semin. Cell Dev. Biol., 64, 160–170. [DOI] [PubMed] [Google Scholar]
- 5. Ortolano S., Tarrio R., Blanco-Arias P., Teijeira S., Rodriguez-Trelles F., Garcia-Murias M., Delague V., Levy N., Fernandez J.M., Quintans B. et al. (2011) A novel MYH7 mutation links congenital fiber type disproportion and myosin storage myopathy. Neuromuscul. Disord., 21, 254–262. [DOI] [PubMed] [Google Scholar]
- 6. Clarke N.F., Waddell L.B., Cooper S.T., Perry M., Smith R.L., Kornberg A.J., Muntoni F., Lillis S., Straub V., Bushby K. et al. (2010) Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum. Mutat., 31, E1544–E1550. [DOI] [PubMed] [Google Scholar]
- 7. Clarke N.F., Kolski H., Dye D.E., Lim E., Smith R.L., Patel R., Fahey M.C., Bellance R., Romero N.B., Johnson E.S. et al. (2008) Mutations in TPM3 are a common cause of congenital fiber type disproportion. Ann. Neurol., 63, 329–337. [DOI] [PubMed] [Google Scholar]
- 8. Clarke N.F., Ilkovski B., Cooper S., Valova V.A., Robinson P.J., Nonaka I., Feng J.J., Marston S. and North K. (2007) The pathogenesis of ACTA1-related congenital fiber type disproportion. Ann. Neurol., 61, 552–561. [DOI] [PubMed] [Google Scholar]
- 9. Clarke N.F., Kidson W., Quijano-Roy S., Estournet B., Ferreiro A., Guicheney P., Manson J.I., Kornberg A.J., Shield L.K. and North K.N. (2006) SEPN1: associated with congenital fiber-type disproportion and insulin resistance. Ann. Neurol., 59, 546–552. [DOI] [PubMed] [Google Scholar]
- 10. Martinsson T., Oldfors A., Darin N., Berg K., Tajsharghi H., Kyllerman M. and Wahlstrom J. (2000) Autosomal dominant myopathy: missense mutation (Glu-706 --> Lys) in the myosin heavy chain IIa gene. Proc. Natl. Acad. Sci. USA, 97, 14614–14619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tajsharghi H., Hammans S., Lindberg C., Lossos A., Clarke N.F., Mazanti I., Waddell L.B., Fellig Y., Foulds N., Katifi H. et al. (2014) Recessive myosin myopathy with external ophthalmoplegia associated with MYH2 mutations. Eur. J. Hum. Genet., 22, 801–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sandaradura S.A., Bournazos A., Mallawaarachchi A., Cummings B.B., Waddell L.B., Jones K.J., Troedson C., Sudarsanam A., Nash B.M., Peters G.B. et al. (2017) Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum. Mutat., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brooke M.H. and Engel W.K. (1969) The histographic analysis of human muscle biopsies with regard to fiber types. 1. Adult male and female. Neurology, 19, 221–233. [DOI] [PubMed] [Google Scholar]
- 14. Dubowitz V., Sewry C.A. and Oldfors A. (2013) Muscle Biopsy—A Practical Approach, 4th edn. Elsevier Limited, Philadelphia. [Google Scholar]
- 15. Forrest A.R., Kawaji H., Rehli M., Baillie J.K., Hoon M.J., Haberle V., Lassmann T., Kulakovskiy I.V., Lizio M., Itoh M. et al. (2014) A promoter-level mammalian expression atlas. Nature, 507, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mele M., Ferreira P.G., Reverter F., DeLuca D.S., Monlong J., Sammeth M., Young T.R., Goldmann J.M., Pervouchine D.D., Sullivan T.J. et al. (2015) Human genomics. The human transcriptome across tissues and individuals. Science, 348, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schaub M.C., Hefti M.A., Zuellig R.A. and Morano I. (1998) Modulation of contractility in human cardiac hypertrophy by myosin essential light chain isoforms. Cardiovasc. Res., 37, 381–404. [DOI] [PubMed] [Google Scholar]
- 18. Harris S.P., Lyons R.G. and Bezold K.L. (2011) In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res., 108, 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Terrak M., Wu G., Stafford W.F., Lu R.C. and Dominguez R. (2003) Two distinct myosin light chain structures are induced by specific variations within the bound IQ motifs-functional implications. Embo J., 22, 362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hernandez O.M., Jones M., Guzman G. and Szczesna-Cordary D. (2007) Myosin essential light chain in health and disease. Am. J. Physiol. Heart Circ. Physiol., 292, H1643–H1654. [DOI] [PubMed] [Google Scholar]
- 21. Seidel U. and Arnold H.H. (1989) Identification of the functional promoter regions in the human gene encoding the myosin alkali light chains MLC1 and MLC3 of fast skeletal muscle. J. Biol. Chem., 264, 16109–16117. [PubMed] [Google Scholar]
- 22. Lowey S., Waller G.S. and Trybus K.M. (1993) Skeletal muscle myosin light chains are essential for physiological speeds of shortening. Nature, 365, 454–456. [DOI] [PubMed] [Google Scholar]
- 23. VanBuren P., Waller G.S., Harris D.E., Trybus K.M., Warshaw D.M. and Lowey S. (1994) The essential light chain is required for full force production by skeletal muscle myosin. Proc. Natl. Acad. Sci. USA, 91, 12403–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weterman M.A., Barth P.G., Spaendonck-Zwarts K.Y., Aronica E., Poll-The B.T., Brouwer O.F., Tintelen J.P., Qahar Z., Bradley E.J., Wissel M. et al. (2013) Recessive MYL2 mutations cause infantile type I muscle fibre disease and cardiomyopathy. Brain, 136, 282–293. [DOI] [PubMed] [Google Scholar]
- 25. Poetter K., Jiang H., Hassanzadeh S., Master S.R., Chang A., Dalakas M.C., Rayment I., Sellers J.R., Fananapazir L. and Epstein N.D. (1996) Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat. Genet., 13, 63–69. [DOI] [PubMed] [Google Scholar]
- 26. Olson T.M., Karst M.L., Whitby F.G. and Driscoll D.J. (2002) Myosin light chain mutation causes autosomal recessive cardiomyopathy with mid-cavitary hypertrophy and restrictive physiology. Circulation, 105, 2337–2340. [DOI] [PubMed] [Google Scholar]
- 27. Orr N., Arnaout R., Gula L.J., Spears D.A., Leong-Sit P., Li Q., Tarhuni W., Reischauer S., Chauhan V.S., Borkovich M. et al. (2016) A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat. Commun., 7, 11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gudbjartsson D.F., Helgason H., Gudjonsson S.A., Zink F., Oddson A., Gylfason A., Besenbacher S., Magnusson G., Halldorsson B.V., Hjartarson E. et al. (2015) Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet., 47, 435–444. [DOI] [PubMed] [Google Scholar]
- 29. Burguiere A.C., Nord H. and Hofsten J. (2011) Alkali-like myosin light chain-1 (myl1) is an early marker for differentiating fast muscle cells in zebrafish. Dev. Dyn., 240, 1856–1863. [DOI] [PubMed] [Google Scholar]
- 30. Zammit P.S., Cohen A., Buckingham M.E. and Kelly R.G. (2008) Integration of embryonic and fetal skeletal myogenic programs at the myosin light chain 1f/3f locus. Dev. Biol., 313, 420–433. [DOI] [PubMed] [Google Scholar]
- 31. Nawrotzki R., Fischman D.A. and Mikawa T. (1995) Antisense suppression of skeletal muscle myosin light chain-1 biosynthesis impairs myofibrillogenesis in cultured myotubes. J. Muscle. Res. Cell Motil., 16, 45–56. [DOI] [PubMed] [Google Scholar]
- 32. Stuart C.A., Stone W.L., Howell M.E., Brannon M.F., Hall H.K., Gibson A.L. and Stone M.H. (2016) Myosin content of individual human muscle fibers isolated by laser capture microdissection. Am. J. Physiol. Cell Physiol., 310, C381–C389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Glover L.E., Newton K., Krishnan G., Bronson R., Boyle A., Krivickas L.S. and Brown R.H. Jr. (2010) Dysferlin overexpression in skeletal muscle produces a progressive myopathy. Ann. Neurol., 67, 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu X., Hadhazy M., Groh M.E., Wheeler M.T., Wollmann R. and McNally E.M. (2001) Overexpression of gamma-sarcoglycan induces severe muscular dystrophy. Implications for the regulation of Sarcoglycan assembly. J. Biol. Chem., 276, 21785–21790. [DOI] [PubMed] [Google Scholar]
- 35. Abath Neto O., Tassy O., Biancalana V., Zanoteli E., Pourquie O. and Laporte J. (2014) Integrative data mining highlights candidate genes for monogenic myopathies. PLoS ONE, 9, e110888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cirak S., Foley A.R., Herrmann R., Willer T., Yau S., Stevens E., Torelli S., Brodd L., Kamynina A., Vondracek P. et al. (2013) ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain, 136, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cabrera-Serrano M., Ghaoui R., Ravenscroft G., Johnsen R.D., Davis M.R., Corbett A., Reddel S., Sue C.M., Liang C., Waddell L.B. et al. (2015) Expanding the phenotype of GMPPB mutations. Brain, 138, 836–844. [DOI] [PubMed] [Google Scholar]
- 38. Ravenscroft G., Nolent F., Rajagopalan S., Meireles A.M., Paavola K.J., Gaillard D., Alanio E., Buckland M., Arbuckle S., Krivanek M. et al. (2015) Mutations of GPR126 are responsible for severe arthrogryposis multiplex congenita. Am. J. Hum. Genet., 96, 955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sali A. and Blundell T.L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol., 234, 779–815. [DOI] [PubMed] [Google Scholar]
- 40. Wu S., Liu J., Reedy M.C., Tregear R.T., Winkler H., Franzini-Armstrong C., Sasaki H., Lucaveche C., Goldman Y.E., Reedy M.K. et al. (2010) Electron tomography of cryofixed, isometrically contracting insect flight muscle reveals novel actin-myosin interactions, 5, e12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen M.Y. and Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci., 15, 2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Willard L., Ranjan A., Zhang H., Monzavi H., Boyko R.F., Sykes B.D. and Wishart D.S. (2003) VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res., 31, 3316–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dolinsky T.J., Czodrowski P., Li H., Nielsen J.E., Jensen J.H., Klebe G. and Baker N.A. (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res., 35, W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.