

Abstract
Plant and microbial toxins are considered bioterrorism threat agents because of their extreme toxicity and/ or ease of availability. Additionally, some of these toxins are increasingly responsible for accidental food poisonings. The current study utilized an ELISA-based protein antibody microarray for the multiplexed detection of ten biothreat toxins, botulinum neurotoxins (BoNT) A, B, C, D, E, F, ricin, shiga toxins 1 and 2 (Stx), and staphylococcus enterotoxin B (SEB), in buffer and complex biological matrices. The multiplexed assay displayed a sensitivity of 1.3 pg mL−1 (BoNT/A, BoNT/B, SEB, Stx-1 and Stx-2), 3.3 pg mL−1 (BoNT/C, BoNT/E, BoNT/F) and 8.2 pg mL−1 (BoNT/D, ricin). All assays demonstrated high accuracy (75–120 percent recovery) and reproducibility (most coefficients of variation <20%). Quantification curves for the ten toxins were also evaluated in clinical samples (serum, plasma, nasal fluid, saliva, stool, and urine) and environmental samples (apple juice, milk and baby food) with overall minimal matrix effects. The multiplex assays were highly specific, with little cross-reactivity observed between the selected toxin antibodies. The results demonstrate a multiplex microarray that improves current immunoassay sensitivity for biological warfare agents in buffer, clinical, and environmental samples.
Introduction
Protein toxins, such as the botulinum neurotoxins (BoNT), ricin, shiga toxins (Stx), and staphylococcal enterotoxin B (SEB), are considered to be potential biological threat agents. These toxins pose a threat because of their extreme toxicity, wide-spread availability, and ease of use. Because of these characteristics, biothreat toxins have been stockpiled for bioweapon use and even used in previous bioterrorism events.1,2 In the event of a bioterrorist attack, it may not be obvious which agent was released, although this knowledge is critical for delivery of appropriate medical treatment. Therefore, public health officials require sensitive and specific detection systems that can identify multiple biothreat toxins early enough that appropriate care can be given.
BoNT consists of a ~100 kDa heavy chain (HC) and a ~50 kDa light chain (LC). The toxin enters the body through ingestion, inhalation, or open wounds and is thereafter transported to cholinergic synapses via circulation.3 The HC facilitates toxin entry into neurons by specific receptor mediated endocytosis and the LC functions as a metallopeptidase that cleaves soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs), thereby inhibiting acetylcholine release and resulting in flaccid muscle paralysis.4 Seven distinct serotypes (A–G) of botulinum neurotoxin are produced by the bacterium Clostridium botulinum; however, only serotypes A, B, E and F have been confirmed to cause botulism in humans. Exceptionally low doses of BoNT are sufficient for poisoning and with an LD50 of approximately 1 ng kg−1 it is the most toxic substance known to man.1 The mouse bioassay is currently the gold standard for BoNT detection and while this assay is highly sensitive, it is laborious and time-consuming.5 There are promising alternative approaches to the detection of BoNT that have been reviewed recently which include immunological methods, mass spectrometry, endopeptidase activity assays, and cell based assays.6–8
Ricin toxin is derived from the seeds produced by the castor bean plant, Ricinus communis. Because of the world-wide distribution of this plant and the ease of toxin purification, ricin is considered a major bioterrorism threat. Ricin consists of two polypeptide chains, a 30 kDa A-chain and a 32 kDa B-chain. The ricin B-chain facilitates toxin entry into cells via receptor-mediated endocytosis.2,9 Inside the cell, the ricin A-chain initiates depurination and cleavage of the 28S rRNA subunit leading to inhibition of protein synthesis and cell death.10–12 The toxicity of ricin has not been extensively studied in humans and varies depending upon the route of exposure. The lethal oral dose for humans is estimated at 1–20 mg kg−1 and the lethal inhalational dose extrapolated from extensive rodent and primate studies is predicted to be around 5 μg kg−1.13,14 Currently, the analysis of ricin relies on immunological methods,13–17 mass spectrometry analysis,18–20 or functional in vitro and in vivo assays.21‘22
Some strains of Escherichia coli produce protein toxins that are closely related to Shiga toxin (Stx) from Shigella dysenteriae. As a group, these E. coli are known as Stx-producing E. coli (STEC). STEC are responsible for many outbreaks of hemorrhagic colitis or bloody diarrhea and hemolytic uremic syndrome which can cause kidney failure. There are two major types of Shiga-like toxin, shiga-like toxin 1 (Stx1) and Shiga-like toxin 2 (Stx2). Stx1 is indistinguishable from the shiga toxin produced by Shigella dysenteriae while Stx2 is more divergent. Shiga toxins act to inhibit protein synthesis using essentially the same mechanism as ricin.12 Stx is composed of a single enzymatic A subunit and a multimer of receptor-binding B subunits, non-covalently associated with the A subunit.23 Like ricin, A chain of Stx, once it has gained entry into the cytosol of the cell, initiates depurination and cleavage of the 28S rRNA subunit, leading to inhibition of protein synthesis and cell death. The Vero cell assay is the gold standard for detection of Stx in clinical and environmental samples; however it is time-consuming and labor intensive.24 Sensitive and specific PCR-based assays are available for STX-producing organisms; however, they detect the toxin gene sequence, not the toxin itself. Several commercially available immunoassays have been evaluated25 and other more sensitive immunoassays have been developed.24,26,27
Staphylococcal enterotoxin B (SEB) is an exotoxin produced by the gram positive cocci Staphylococcus aureus. SEB is one of the toxins responsible for staphylococcal food poisoning and it has been stockpiled as a potential biological weapon by some countries. The toxin is a superantigen and intoxication leads to an excessive inflammatory response by the immune system with a release of large amounts of cytokines.28–30 SEB is extremely toxic with an inhalation exposure LD50 of 20 ng kg−1 and the ability to incapacitate 50% of the population (ED50) with just 0.4 ng kg−1.31,32 Multiple highly sensitive immunoassays, including immunosensors, point-of-care assays,33 and protein microarrays,34 have been developed for the sensitive detection of SEB.15‘35‘36
The majority of assays for the detection of toxins have targeted one to two toxins at a time in a test format; however, there is a growing need for assays that analyze multiple toxins simultaneously. Several multiplex toxin platforms have been developed including mass-spectrometry based,19‘37 suspension array technology,17‘38–40 and planar protein microarrays.41–45 Some multiplex assays have been developed to detect toxins in clinical samples39‘41‘46 or in food samples.17‘38‘47 Assay sensitivities for the most relevant multiplexed toxin assays are summarized in ESI, Table 1.† For this work we have developed a protein ELISA microarray that detects 10 toxins with high sensitivity and specificity in both clinical and food samples.
Protein microarray technology relies on a miniaturized version of the traditional ELISA whereby multiple antibodies are immobilized onto a solid surface, allowing for the simultaneous analysis of hundreds of antigens within a single experiment. We have expanded upon the ELISA-based protein antibody micro array developed by Varnum et al.41,42 to include six botulinum neurotoxins, ricin, Stx1, Stx2, and SEB. Here we employed a sensitive and specific “sandwich” protein ELISA microarray to simultaneously detect all 10 toxins in a number of biological fluids including serum, urine, and saliva and in food samples. This multiplex microarray format allowed us to monitor multiple toxins and screen hundreds of antibodies efficiently and cheaply to determine the most sensitive antibodies. This, together with a powerful yet simple biotin-tyramide amplification system, dramatically increases assay sensitivity producing assays capable of detecting toxins to pg mL−1 levels in clinical and environmental samples. The assay design can be easily adopted by other research groups, uses relatively inexpensive, commercially available reagents and equipment, while maintaining excellent sensitivity at minimal sample volumes.
Experimental
Assay reagents
BoNT holotoxins were purchased from Metabiologics (Madison, WI). Ricin toxin (Ricinus communis agglutinin II; RCA60) was purchased from Vector Laboratories (Burlingame, CA). Nontoxic recombinant SEB was acquired from BEI resources (Manassas, VA). Shiga toxin was purchased from List Biological Laboratories (Campbell, CA). Antibodies used as capture or detection antibodies were either obtained from the lab of Dr James D. Marks at the University of California-San Fran-cisco48–52 or were commercially available. In order to test all combinations of possible antibody pairs for our sandwich ELISA, an aliquot of the antibodies were labeled with biotin using EZ-Link NHS-Chromogenic-Biotin (Pierce, Rockford, IL). The manufacturer’s protocol was followed to biotinylate about 130 μg of protein to be used as detection antibody. Biotin-NHS was purchased from Peirce (Rockford, IL) and bio-tinyltyramide was prepared as previously described.53 HRP-streptavidin and Cy3-streptavidin was obtained from Jackson ImmunoResearch (West Grove, PA).
Slide preparation and microarray production
The ELISA microarray was carried out as previously described.41‘42‘54 In brief, capture antibodies specific to each toxin were prepared at concentrations of 1 mg mL−1 in phosphate buffer saline (PBS, pH 7.2) and printed as 4 replicate spots in each well on the slide. Initially, spotting was done with a NanoPlotter 2.1 (GeSim, Germany); however, current arrays are contract printed at ArrayIt (Arrayit Corp., Sunnyvale, CA). Custom made aminopropylsilane-coated glass slides (Erie Scientific, Portsmouth, NH) were used with a stamped grey hydrophobic barrier that defined 16 individual wells in an 8 × 2 grid. Following printing, slides were air-dried and blocked for one hour in 1% casein in PBS (Bio-rad). Slides were then washed with PBS containing 0.05% Tween 20 (PBS-T) before proceeding with the ELISA microarray assays or stored dry at –20 °C.
Sandwich ELISA
Each standard curve consisted of 12 points spanning the full range of the assay, including an assay blank of 0.1% casein in PBS. Toxins were diluted into the appropriate sample diluent. Twenty microliters of diluted toxin was applied to each well of the microarray and incubated for up to 16 hours on a gently rotating orbital shaker. Three washes were performed after each incubation step with PBS containing 0.05% Tween 20 (PBS-T). The slides were then incubated with the appropriate mix of biotinylated detection antibodies and incubated for 2 hours. The signal was enhanced using the biotinyltyramide amplification system.54,55 The slides were incubated with 1 μg mL−1 HRP-conjugated streptavidin for 30 minutes, followed by incubation for 10 minutes with 1 μg mL−1 biotinyltyramide. Finally, the slides were incubated with 1 μg mL−1 Cy3-conjugated streptavidin for 30–60 minutes in the dark with gentle rocking followed by a final wash and then rinsed with distilled water and dried. Experiments in the shortened assay series were incubated as indicated in the text. Cy3 fluorescence was detected by scanning slides on an LS Reloaded (Tecan, Switzerland) microarray scanner (laser: 532 nm; filter: 575 nm).
Analysis of complex clinical and environmental samples
Milk (2% reduced fat), apple juice, and baby food (Gerber Beef and Beef Gravy) were purchased from a local grocery store. Apple juice was neutralized to pH 7.0 using 1 N sodium hydroxide. Toxins were spiked directly into undiluted apple juice (after neutralization), milk, and baby food samples. Milk samples, with exogenous toxin, were centrifuged at 5000 × g for 5 minutes and the interphase layer, between the fat layer on top and the sediment, was saved for analysis. Baby food samples were incubated with toxin for the indicated time and then PBS-T was added at a 1 : 1 w/v ratio (i.e. 1 gram baby food : 1 mL PBS-T). The mixture was vortexed for 30 seconds and then centrifuged at 1000 × g for 5 minutes at room temperature. The supernatant was again centrifuged at 5000 × g for 10 minutes and the supernatant from this final spin was saved for analysis.
Urine, saliva, serum, and plasma were from an anonymous female donor (Golden West Biologicals, Inc., Temecula, CA). These samples were centrifuged at 12 000 × g for 15 minutes, aliquoted, and stored at –80 °C until used. Nasal specimens were collected by rotating a sterile swab against the anterior nasal mucosa for about 3 seconds and repeating for the other naris. The swab was immediately placed in a 1.5 mL tube with 1 mL assay buffer (0.1% casein solution in PBS) and vortexed for min, aliquoted, and stored at –80 °C. Stool was collected by an anonymous donor, mixed with equal volume to weight of gelatin phosphate buffer (0.2% bovine gelatin and 0.4% Na2PO4, pH 6.4), and centrifuged at 2500 × g for 30 minutes at 4 °C. The supernatant was stored at –80 °C until use.56,57 For experiments with clinical samples, toxins were spiked into undiluted nasal, urine and saliva samples. Centrifuged stool samples were diluted 1 : 4 in 40% fetal bovine serum in PBS-T, while serum and plasma were diluted 1 : 4 in assay buffer before toxin addition.
Assay validation
Specificity was determined by incubating individual toxin antigens, at a concentration of 1250 pg mL−1, with microarrays containing all ten toxin capture antibodies. The assays were then incubated with a complete mix of detection antibodies. Cross-reactivity was expressed in percent of the analyte concentration divided by the concentration of the specific antigen and multiplied by 100.
Assay precision for both within-(intra) and between-(inter) runs was evaluated by spiking toxins in assay buffer at two concentrations within the linear portion of each calibration curve and measuring them at least three times each day over three days with four replicates per concentration. Using the predicted concentrations the coefficient of variation (%CV) was determined as the ratio of the standard deviation to the mean for three values of a given concentration.
The limit of detection (LOD) was determined as the tested concentration of analyte that produced a mean signal intensity greater than 3 times the standard deviation of the antigen-free blank. Assay accuracy was determined by the recovery rate expressed as a percentage of the expected concentration divided by the predicted concentration calculated in ProMAT.
Data analysis
Fluorescence data were quantified using ScanArray Express software (Perkin-Elmer). Data analysis was performed using ProMAT Calibrator and the Protein Microarray Analysis Tool (ProMAT).58,59 This publicly available software normalizes data to a control protein, then fits the data to standard curves and predicts protein concentrations, in addition to performing statistical analysis specifically for ELISA microarray data. GFP protein (Millipore, Billerica, MA) was added as a calibrant at a concentration of 100 pg mL−1 to standard and spiked toxin mixtures which were made new for each assay. Biotinylated detection anti-GFP (Rockland Inc., Gilbertsville, PA) was used at a concentration of 25 ng ml−1. ProMAT Calibrator normalized spot intensities relative to GFP in order to minimize inherent variability due to slide and reagent processing within and between assays.58,60 Standard curves were generated using the adjusted values in ProMat.
Results and discussion
Optimization of antibody microarrays
In order to develop sensitive assays, over 70 antibodies were screened in a microarray format, including commercially available antibodies and high-affinity antibodies produced by the Marks lab. Initially, all possible capture antibodies specific to a single toxin were printed in a single chip. Each specific toxin chip was exposed to a serial dilution of the corresponding toxin and probed with all possible relevant detection antibodies. The capture and detection antibody pair that produced the best standard curve based on sensitivity, high reproducibility, and R2 value was chosen for further analysis in the multiplex assay. Optimal antibody pairs are listed in Table 1 and were previously established for the six BoNT.41 The optimal detection antibody concentrations were established by analyzing the signal-to-noise ratio for each assay at varying concentrations of detection antibody concentration as described previously.41,61 The detection antibodies and their corresponding optimal concentration are listed in Table 1.
Table 1.
Optimal antibody sets used for multiplex toxin protein ELISA microarraya
| Antigen | Capture antibody | Source | Detection antibody | Source | Detection antibody concentration (ng mL−1) |
|---|---|---|---|---|---|
| BoNT/A | mA b AR4 | 1 | mA b RAZl | 1 | 100 |
| BoNT/B | lgY AHcB | 2 | mA b 1B10.1 | 1 | 100 |
| BoNT/C | mA b AF5425 | 3 | mA b lCl | 1 | 100 |
| BoNT/D | mA b 8DCl | 1 | mA b 8DC2 | 1 | 25 |
| BoNT/E | mA b 3E6.2 | 1 | mA b 3E4.l | 1 | 100 |
| BoNT/F | mA b 6F8 | 1 | mA b 6F5 | 1 | 100 |
| Ricin | mA b AB-RIC-MAB2 | 4 | mA b R2031–52H | 7 | 150 |
| SEB | Sheep anti-SEB, SLB1202 | 5 | Sheep anti-SEB, SBBC202 | 5 | 150 |
| Stxl | mA b MBS311734 | 6 | mA b STX1–10D11 | 5 | 150 |
| Stx2 | mA b MBS311736 | 6 | mA b STX2-BB12 | 5 | 150 |
Antibody sources: (1) James Marks laboratory, UCSF; (2) Gallus Immunotech, Fergus, ON; (3) R& D Systems, Minneapolis, MN; (4) Critical Reagents Program, Frederick, MD; (5) Toxin Technology, Sarasota, FL; (6) My BioSource Inc., San Diego, CA; (7) US Biological, Swampscott, MA.
Calibration curves and performance of the multiplex toxin assay
Following assay optimization, the ten capture antibodies were printed on one chip for multiplexed detection of all ten toxins simultaneously within one assay. Fig. 1 shows the standard curves for all ten toxins over a concentration range from 0 to 5000 pg mL−1 in assay buffer (blue line). The LODs in assay buffer were: 1.3 pg mL−1 for BoNT/A, BoNT/B, SEB, Stx-1, and Stx-2; 3.3 pg mL−1 for BoNT/C, BoNT/E, and BoNT/F; and 8.2 pg mL−1 for BoNT/D, and ricin (Table 2). All curves had an excellent linear correlation fit (R2) of at least 0.98. For all ten toxins, the dynamic range of the assay covered between two to three orders of magnitude of concentration above the LOD (Fig. 1).
Fig. 1.
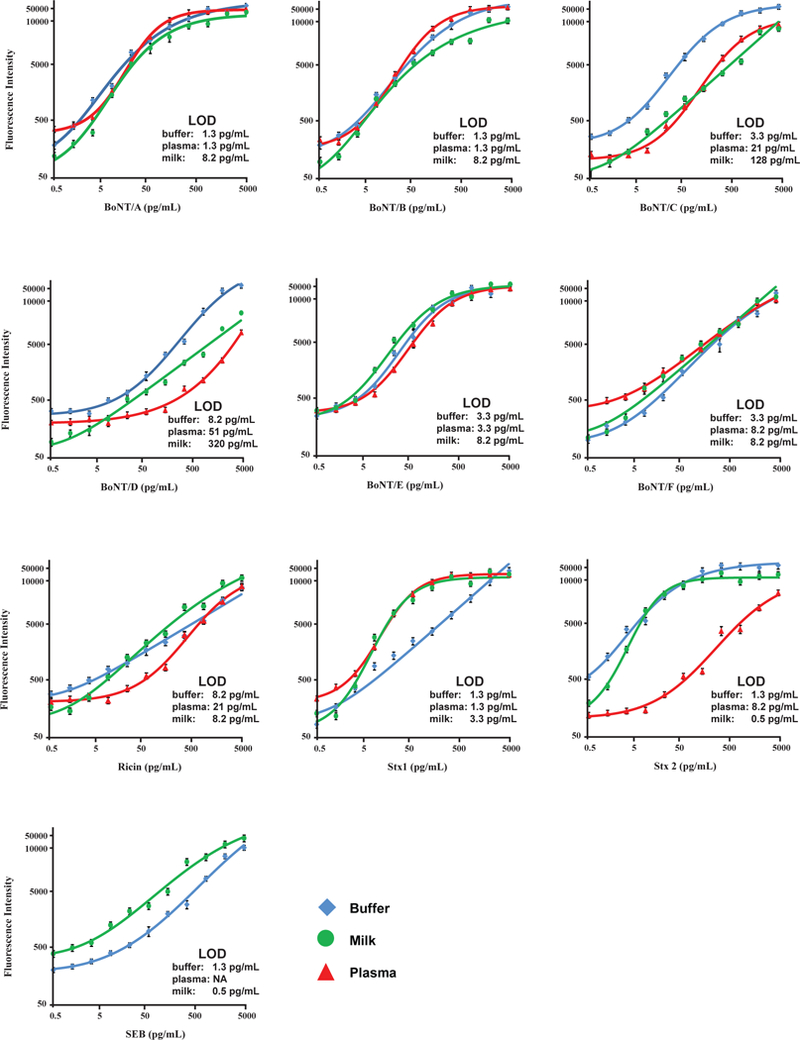
Standard curves for the simultaneous detection of the toxins in buffer, milk and plasma using an ELISA protein microarray. Error bars refer to the standard deviations of four microarray replicates.
Table 2.
Limits of detection (pg mL −1) of toxins in buffer and other complex matrices with the ELISA microarray
| LOD (pg mL−1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample | BoNT/A | BoNT/B | BoNT/C | BoNT/D | BoNT/E | BoNT/F | Ricin | SEB | Stx-1 | Stx-2 |
| Buffer | 1.3 | 1.3 | 3.3 | 8.2 | 3.3 | 3.3 | 8.2 | 1.3 | 1.3 | 1.3 |
| Nasal | 8.2 | 8.2 | 8.2 | 128 | 21 | 51 | 51 | 51 | 21 | 8.2 |
| Plasma | 1.3 | 1.3 | 21 | 51 | 3.3 | 8.2 | 21 | NA | 1.3 | 8.2 |
| Saliva | 3.3 | 1.3 | 21 | 128 | 3.3 | 320 | 320 | NA | 8.2 | 8.2 |
| Serum | 8.2 | 3.3 | 3.3 | 128 | 8.2 | 8.2 | 21 | NA | 8.2 | 1.3 |
| Stool | 51 | 51 | 51 | 800 | 51 | 21 | 800 | 8.2 | 51 | 128 |
| Urine | 3.3 | 8.2 | 3.3 | 128 | 3.3 | 3.3 | 1.3 | 3.3 | 1.3 | 1.3 |
| Milk | 8.2 | 8.2 | 128 | 320 | 8.2 | 8.2 | 8.2 | 0.5 | 3.3 | 0.5 |
| Baby food | 21 | 21 | 51 | 51 | 21 | 51 | 51 | 51 | 21 | 21 |
| Apple juice | 51 | 128 | 3.3 | 320 | 3.3 | 51 | 51 | 1.3 | 8.2 | 3.3 |
Assay accuracy was evaluated in a series of recovery studies by spiking assay buffer with three known concentrations of antigen within the linear portion of the standard curve. Toxin recovery varied from 75% to 120% (Table 3).
Table 3.
Percent recovery for toxins in buffer
| Toxin | pg mL–1 | ||
|---|---|---|---|
| 51 | 128 | 320 | |
| BoNT/A | 111 | 97 | 88 |
| BoNT/B | 108 | 99 | 96 |
| BoNT/C | 109 | 96 | 97 |
| BoNT/D | 98 | 104 | 88 |
| BoNT/E | 109 | 83 | 99 |
| BoNT/F | 105 | 103 | 95 |
| Ricin | 109 | 101 | 99 |
| SEB | 116 | 106 | 103 |
| Stx-1 | 100 | 90 | 75 |
| Stx-2 | 120 | 107 | 105 |
Assay precision was determined at two toxin concentrations by calculating intra- and inter-assay variability and is summarized in Table 4. The percent CV was calculated either for 12 test replicates run at the same time on the same day (intra-assay) or from test samples assayed on 3 separate days (inter-assay). The intra-assay variability ranged from 2% to 20% except for BoNT/E which had a higher variability of 43% at the lower concentration of 51 pg mL−1. The inter-assay variability ranged from 4% to 20% except for Stx-1 which was 29% at 320 pg mL−1. These results demonstrate that the multiplexed ELISA microarray is highly accurate, precise and has great potential for the simultaneous detection of even low concentrations of multiple bio-terrorism toxin agents.
Table 4.
Precision of the toxin microarray ELISA expressed as intraand inter-assay CVs
| Intra-assay %CVa |
Inter-assay %CVb |
|||
|---|---|---|---|---|
| Toxin | 51 | 320 | 51 | 320 |
| BoNT/A | 13 | 12 | 8 | 7 |
| BoNT/B | 3 | 19 | 17 | 14 |
| BoNT/C | 17 | 11 | 20 | 7 |
| BoNT/D | 17 | 2 | 7 | 11 |
| BoNT/E | 43 | 13 | 10 | 18 |
| BoNT/F | 16 | 13 | 8 | 4 |
| Ricin | 20 | 19 | 9 | 4 |
| SEB | 19 | 17 | 17 | 19 |
| Stx-1 | 11 | 15 | 16 | 29 |
| Stx-2 | 16 | 7 | 11 | 8 |
Intra-assay %CVs were calculated from the average values from three samples analyzed simultaneously.
Inter-assay %CVs were calculated from the average values from three samples analyzed on three consecutive days
Assessment of reagent interference between arrays
Because all ten toxins are measured simultaneously, it is important to test the specificity of each assay. To demonstrate that the assays are specific for each toxin, experiments were undertaken to determine potential cross-reactivity between assays. This involved testing each antigen singly, at a high concentration (1250 pg mL−1), on the completed multiplexed microarray consisting of all ten capture antibodies immobilized on the slides and a mixture of all detection antibodies. All of the ten toxins were specifically detected with most antigens having less than 2% cross-reactivity with their non-specific capture antibodies (Table 5). A minor cross-reactivity was detected between BoNT/C toxin and BoNT/D capture antibody. This cross-reactivity has been previously documented and shown to be specific to a cross-reaction between the BoNT/D capture antibody and BoNT/C toxin and not due to interactions between the BoNT/D capture antibody and a BoNT detection antibody.41 It has been found recently that the antibodies generated against BoNT/D were raised to a mosaic strain of BoNT/D-C.62 The D-C mosaic strain consists of BoNT/D amino acid sequence in the light chain, but with amino acid sequence similar to BoNT/C in the heavy chain. For whatever reason, it is likely that the BoNT/ D capture antibody recognizes a conserved epitope, likely within the heavy chain, shared between BoNT/C and BoNT/D. Nevertheless, the observed signal from the BoNT/D capture antibody and the BoNT/C antigen was minor (<15% of the BoNT/C assay with all 10 toxins) and does not have an impact on the assay’s ability to distinguish the BoNT serotypes C and D.
Table 5.
Percent cross-reactivity of antigens and capture antibodies
| Toxin | Capture antibody | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | Ricin | SEB | Stx-1 | Stx-2 | |
| BoNT/Aa | <2 | <2 | <6 | <2 | <2 | <2 | <5 | <2 | <2 | |
| BoNT/B | <2 | <2 | <6 | <2 | <2 | <2 | <2 | <2 | <2 | |
| BoNT/C | <2 | <2 | <15 | <5 | <2 | <2 | <2 | <2 | <2 | |
| BoNT/D | <2 | <2 | <2 | <5 | <5 | <5 | <2 | <2 | <2 | |
| BoNT/E | <2 | <2 | <2 | <6 | <2 | <2 | <2 | <2 | <2 | |
| BoNT/F | <2 | <2 | <2 | <5 | <2 | <2 | <2 | <2 | <2 | |
| Ricin | <2 | <2 | <2 | <5 | <2 | <2 | <2 | <2 | <2 | |
| SEB | <5 | <2 | <2 | <5 | <2 | <10 | <2 | <2 | <2 | |
| STX-1 | <2 | <2 | <2 | <10 | <2 | <5 | <2 | <2 | <10 | |
| STX-2 | <2 | <2 | <2 | <5 | <2 | <2 | <2 | <2 | <2 | |
Percent cross-reactivity is determined as the median (n = 3) relative fluorescence units corresponding to the non-specific capture antibody divided by the median relative fluorescence units of the specific capture antibody.
Analysis of multiplex toxin assay in complex clinical and food matrices
To assess the potential use of the toxin ELISA microarray in real-world samples, we spiked the toxins into both clinical (serum, plasma, nasal fluid, urine, saliva, and stool) and food samples (milk, apple juice and beef baby food). The detection of multiple biodefense toxins in clinical samples can provide a powerful diagnostic tool for use by medical personnel in the event of a bioweapon event. While a majority of the toxin assays only experienced a slight decrease in assay sensitivity, a few of the assays were affected more significantly (Table 2). The sensitivity of BoNT/D detection is slightly reduced in plasma samples to 51 pg Ml−1, and is further reduced to 128 pg mL−1 in nasal, saliva, urine, and serum samples and 800 pg mL−1 in stool. Similarly, the LOD of ricin was reduced when measured in specific clinical fluid matrices. However, the toxin microarray assay detects ricin in serum, plasma, nasal and saliva samples with a sensitivity of greater than 2 orders of magnitude required to detect a lethal dose in an average weight human (Table 6).
Table 6.
Established oral and inhalational LD50 values
| Oral |
Inhalation |
|||||
|---|---|---|---|---|---|---|
| Toxin | LD50
(μg kg −1) |
Relevant clinical detection levela (ng mL−1) |
Relevant detection level in 100 mL food (μg kg −1) |
LD50
(μg kg−1) |
Relevant clinical detection levela (pg mL−1) |
Reference |
| BoNT | 1 | 14 | 0.7 | 0.003 | 42 | 1 |
| Ricin | 10 000 | 140 000 | 7000 | 3 | 42 000 | 73 and 74 |
| SEBb | — | — | — | 0.02 | 280 | 32 |
| Stx-1c | 8 | 112 | 5.6 | 8 | 112 000 | 75 |
| Stx-2c | 0.29 | 4 | 0.2 | 0.29 | 4000 | 76 |
Relevant clinical detection level calculated as the minimal concentration of toxin to detect an exposure equivalent to the LOD assuming an average weight person (70 kg) with 5 L of blood in circulation.
For SEB, the oral LD50 value has not been reported.
For Stx-1 and Stx-2, the oral and inhalational LD50 values are based upon the intraperitoneal LD50.
SEB detection in serum, plasma and saliva demonstrate a considerable loss in sensitivity. Our initial experimental assays showed a high background signal and subsequent decreased LOD for SEB in these fluids. Earlier studies made similar observations43,63–65 where it was noted that the interference was attributed to pre-existing SEB antibodies in blood.66,67 We attempted, unsuccessfully, to remove antibodies using protein A/G and L columns (data not shown). Therefore, SEB detection was not performed in serum, plasma, or saliva samples. However, SEB was detected with good sensitivity in urine, nasal, and stool with LODs of 3.3, 51 and 8.2 pg mL−1 respectively (Table 2). These LODs are well below the clinically relevant detection level of 280 pg mL−1 (LD50 of 20 ng kg−1) (Table 6).32
All ten assays were also performed in stool samples. Initially, low signal was detected for all of the toxins, despite thorough centrifugation, filtration, and dilution. This drastic interference was also seen by Dezfulian et al.,68 in stool samples from infants with botulism. Further studies confirmed these interfering factors to be caused by proteolytic activity in fecal specimens which release the capture antibodies from the slide surface.69 The addition of 40% fetal bovine serum (FBS) was able to sufficiently block the interference when it was used as a fecal diluent at a 1 : 4 ratio. Therefore, stool samples were analyzed following dilution with 40% FBS. With this modification, most of the toxins had an LOD of less than 51 pgmL−1 except BoNT/D (800 pg mL−1), ricin (800 pg mL−1), and Stx2 (128 pg mL−1) (Table 2).
Because bacterial and plant toxins have been the cause of both accidental and intentional poisonings, there is a need to detect them in food matrices to assure a safe food supply. Previous studies have detected multiple toxins in food matrices with good to excellent LODs, however these studies have measured a small set of 5 or fewer toxins17‘19‘38‘39‘70‘71 or have focused on the measurement of toxins in a single food matrix, such as milk.40‘44 Therefore, we tested our toxin microarray assay for the detection of the 10 toxins spiked into milk, apple juice and baby food. As shown in Table 2, the fluorescence signal and subsequent LODs did decrease in food matrices, particularly for all ten toxins spiked into baby food samples. This is likely due to fluorescence quenching due to some component in the baby food matrix as has been shown in previous work.17 Despite the reduced LOD, the toxin assay was sufficiently sensitive to detect the 10 toxins in food samples well below that of an oral lethal dose (Table 2 and 6). For the majority of the assays, the limit of detection is at least two orders of magnitude lower than the oral LD50.
Rapid assay
In order to produce a more rapid assay, the incubation times for specific steps in the assay, including incubation with antigen and detection antibody, were reduced. Fig. 2 shows the calibration curves of the developed rapid assays with total assay times of either 2- or 4-hours. The 2 hour assay was performed with the following incubation times: antigen (1 hour); detection antibody (25 minutes); HRP-conjugated streptavidin (15 minutes); biotinyltyramide (10 min); and Cy3-conjugated streptavidin (10 minutes); whereas the 4 hours assay used these incubation times: antigen (2 hours); detection antibody (1 hour); HRP-conjugated streptavidin (20 minutes); bio-tinyltyramide (10 min); and Cy3-conjugated streptavidin (15 minutes). A comparison of the detection limits of the two rapid assays and our standard 12 hour assay is shown in Table 7. As the assay time is reduced there is a corresponding increase in the detection limit of the assay. However, most of the assays still display excellent sensitivity limits even with a 2 hours total assay time. For instance, BoNT/A, BoNT/B, Stx-1, and Stx-2 all have LOD of either 3.3 or 8.2 pg mL−1. The LOD for BoNT/C and BoNT/E are reduced (51 pg mL−1); however, these LOD are still comparable with the mouse hemidiaphragm assay. While the LOD for ricin and SEB are reduced even further, the assay sensitivity is still greater than the relevant clinical detection levels required for either oral or inhalational exposure (Table 6) by greater than two orders of magnitude for ricin and by 2-fold for SEB.
Fig. 2.
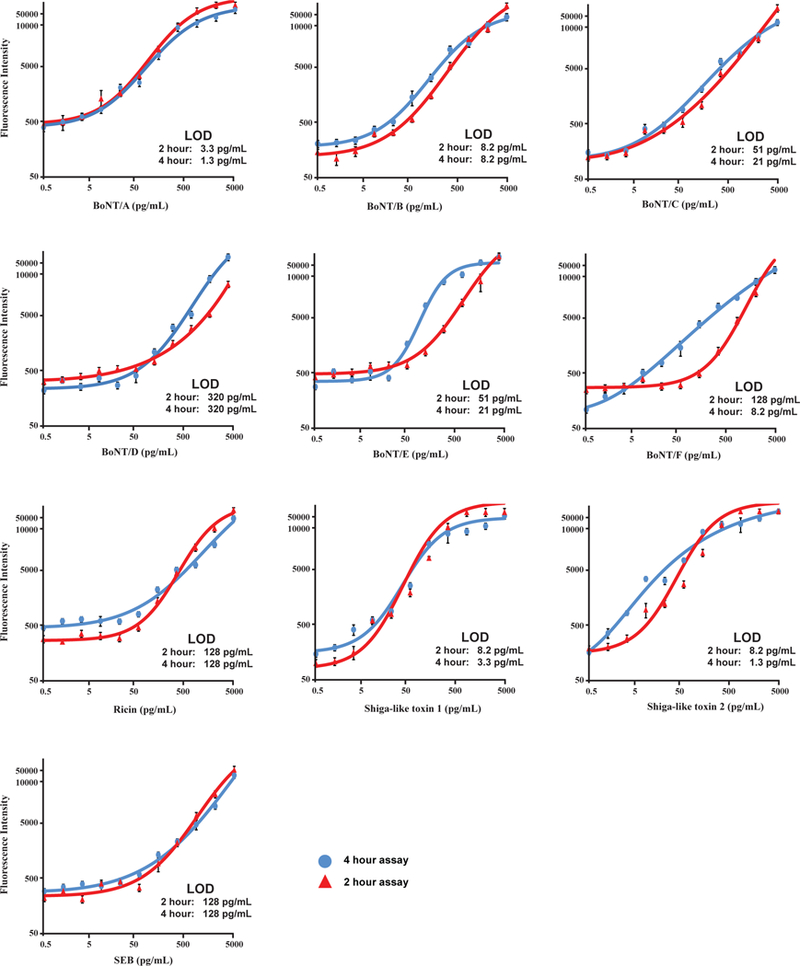
Standard curves for the simultaneous detection of select toxins in buffer using a shortened ELISA protein microarray. Error bars refer to thestandard deviations of four microarray replicates.
Table 7.
Limits of detection (pg mL−1) for toxin microarray assay with shortened assay times
| Assay | Total assay time (hours) |
||
|---|---|---|---|
| 12 | 4 | 2 | |
| BoNT/A | 1.3 | 1.3 | 3.3 |
| BoNT/B | 1.3 | 8.2 | 8.2 |
| BoNT/C | 3.3 | 21 | 51 |
| BoNT/D | 8.2 | 320 | 320 |
| BoNT/E | 3.3 | 21 | 51 |
| BoNT/F | 3.3 | 8.2 | 128 |
| Ricin | 8.2 | 128 | 128 |
| SEB | 1.3 | 128 | 128 |
| Stx-1 | 1.3 | 3.3 | 8.2 |
| Stx-2 | 1.3 | 1.3 | 8.2 |
Conclusion
Using high-affinity capture and detection antibody pairs we developed a highly sensitive ELISA protein microarray assay capable of simultaneously detecting ten biodefense toxins, BoNT/A-F, ricin, SEB, Stx1, and Stx2. These toxins were not only sensitively detected in buffer but also in complex clinical and environmental matrices at levels in the low pg mL−1 range and with a minimal sample volume of 20 μL. These LODs are among the lowest reported for the multiplexed detection of protein toxins (ESI, Table 1†) and, to our knowledge, one of only a few multiplexed toxin assays verified in both clinical and food samples. Many multiplex assays currently exist that can detect and quantify several biological toxins.15‘19‘38‘43‘44‘63‘64‘72 To date, the multiplex ELISA-based protein antibody microarray presented here demonstrates an excellent assay that is able to achieve some of the lowest detection limits and maintain sensitivity below the reported LD50 in a wide range of biological fluids. The assay uses relatively inexpensive and commercially available reagents along with the powerful biotintyramide amplification system which can be easily adopted by other laboratories. Most notably, the simple microarray format can be readily developed for high-throughput analysis of numerous biological toxins in complex clinical and environmental samples.
Supplementary Material
Acknowledgements
This research was supported by the National Institute of Allergy and Infectious Diseases (NIAID) through awards U01 AI081895 (S.M.V.), U01 A1075443 (J.D.M), PSWRCE U54AI065359 (J.D.M.), and R01AI104579 (J.D.M.). The Pacific Northwest National Laboratory (PNNL) is operated by Battelle for the U.S. Department of Energy (DOE) under contract (AC06–76RLO1830).
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c4an01270d
References
- 1.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL and Tonat K, JAMA, J. Am. Med. Assoc, 2001, 285, 1059–1070. [DOI] [PubMed] [Google Scholar]
- 2.Bigalke H and Rummel A, Toxicology, 2005, 214, 210–220. [DOI] [PubMed] [Google Scholar]
- 3.Simpson L, Toxicon, 2013, 68, 40–59. [DOI] [PubMed] [Google Scholar]
- 4.Rossetto O, Megighian A, Scorzeto M and Montecucco C, Toxicon, 2013, 67, 31–36. [DOI] [PubMed] [Google Scholar]
- 5.Lindstrom M and Korkeala H, Clin. Microbiol. Rev, 2006, 19, 298–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorner MB, Schulz KM, Kull S and Dorner BG, Curr. Top. Microbiol. Immunol, 2013, 364, 219–255. [DOI] [PubMed] [Google Scholar]
- 7.Grate JW, Ozanich RM, Warner MG, Marks JD and Bruckner-Lea CJ, TrAC, Trends Anal. Chem, 2010, 29, 1137–1156. [Google Scholar]
- 8.Singh AK, Stanker LH and Sharma SK, Clin. Microbiol. Rev, 2013, 39, 43–56. [DOI] [PubMed] [Google Scholar]
- 9.Lord JM and Spooner RA, Toxins, 2011, 3, 787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Endo Y and Tsurugi K, J. Biol. Chem, 1988, 263, 8735–8739. [PubMed] [Google Scholar]
- 11.Lord JM, Roberts LM and Robertus JD, FASEB J, 1994, 8, 201–208. [PubMed] [Google Scholar]
- 12.Walsh MJ, Dodd JE and Hautbergue GM, Virulence, 2013, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He X, McMahon S, Henderson, TD 2nd, Griffey SM and Cheng LW, PLoS One, 2010, 5, e12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffiths GD, Toxins, 2011, 3, 1373–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ler SG, Lee FK and Gopalakrishnakone P, J. Chromatogr.A, 2006, 1133, 1–12. [DOI] [PubMed] [Google Scholar]
- 16.Shriver-Lake LC, Golden J, Bracaglia L and Ligler FS, Anal. Bioanal. Chem, 2013, 405, 5611–5614. [DOI] [PubMed] [Google Scholar]
- 17.Pauly D, Kirchner S, Stoermann B, Schreiber T, Kaulfuss S, Schade R, Zbinden R, Avondet MA, Dorner MB and Dorner BG, Analyst, 2009, 134, 2028–2039. [DOI] [PubMed] [Google Scholar]
- 18.Aberg A, Bjornstad K and Hedeland M, Biosecurity and Bioterrorism: Biodefense Strategy, Practice and Science, 2013, 11, S215–S226. [DOI] [PubMed] [Google Scholar]
- 19.Kull S, Pauly D, Stormann B, Kirchner S, Stammler M, Dorner MB, Lasch P, Naumann D and Dorner BG, Anal. Chem, 2010, 82, 2916–2924. [DOI] [PubMed] [Google Scholar]
- 20.Kalb SR and Barr JR, Anal. Chem, 2009, 81, 2037–2042. [DOI] [PubMed] [Google Scholar]
- 21.Pauly D, Worbs S, Kirchner S, Shatohina O, Dorner MB and Dorner BG, PLoS One, 2012, 7, e35360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rasooly R and He X, J. Food Prot, 2012, 75, 951–954. [DOI] [PubMed] [Google Scholar]
- 23.Bergan J, Lingelem ABD, Simm R, Skotland T and Sandvig K, Toxicon, 2012, 60, 1085–1107. [DOI] [PubMed] [Google Scholar]
- 24.Bettelheim KA and Beutin L, J. Appl. Microbiol, 2003, 95, 205–217. [DOI] [PubMed] [Google Scholar]
- 25.Willford J, Mills K and Goodridge LD, J. Food Prot, 2009, 72, 741–747. [DOI] [PubMed] [Google Scholar]
- 26.Mendes-Ledesma MR, Rocha LB, Bueris V, Krause G, Beutin L, Franzolin MR, Trabulsi LR, Elias WP and Piazza RM, Microbiol. Immunol, 2008, 52, 484–491. [DOI] [PubMed] [Google Scholar]
- 27.He X, Patfield S, Hnasko R, Rasooly R and Mandrell RE, PLoS One, 2013, 8, e76368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi YW, Kotzin B, Herron L, Callahan J, Marrack P and Kappler J, Proc. Natl. Acad. Sci. U. S. A, 1989, 86, 8941–8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White J, Herman A, Pullen AM, Kubo R, Kappler JW and Marrack P, Cell, 1989, 56, 27–35. [DOI] [PubMed] [Google Scholar]
- 30.Miethke T, Wahl C, Heeg K, Echtenacher B, Krammer PH and Wagner H, J. Exp. Med, 1992, 175, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rusnak JM, Kortepeter M, Ulrich R, Poli M and Boudreau E, Emerging Infect. Dis, 2004, 10, 1544–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gill DM, Microbiol. Rev, 1982, 46, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang M, Sun S, Kostov Y and Rasooly A, Anal. Biochem, 2011, 416, 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wojciechowski J, Danley D, Cooper J, Yazvenko N and Taitt CR, Sensors, 2010, 10, 3351–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Liu F, Yan R, Xue P, Li Y, Chen L, Song C, Liu C, Jin B, Zhang Z and Yang K, Talanta, 2011, 85, 1070–1074. [DOI] [PubMed] [Google Scholar]
- 36.Han JH, Kim HJ, Sudheendra L, Gee SJ, Hammock BD and Kennedy IM, Anal. Chem, 2013, 85, 3104–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyer AE, Gallegos-Candela M, Lins RC, Kuklenyik Z, Woolfitt A, Moura H, Kalb S, Quinn CP and Barr JR, Molecules, 2011, 16, 2391–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garber EA, Venkateswaran KV and O’Brien TW, J. Agric. Food Chem, 2010, 58, 6600–6607. [DOI] [PubMed] [Google Scholar]
- 39.Rivera VR, Gamez FJ, Keener WK, White JA and Poli MA, Anal. Biochem. , 2006, 353, 248–256. [DOI] [PubMed] [Google Scholar]
- 40.Simonova MA, Valyakina TI, Petrova EE, Komaleva RL, Shoshina NS, Samokhvalova LV, Lakhtina OE, Osipov IV, Philipenko GN, Singov EK and Grishin EV, Anal. Chem, 2012, 84, 6326–6330. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Lou J, Jenko KL, Marks JD and Varnum SM, Anal. Biochem, 2012, 430, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varnum SM, Warner MG, Dockendorff B, Anheier NC Jr, Lou J, Marks JD, Smith LA, Feldhaus MJ, Grate JW and Bruckner-Lea CJ, Anal. Chim. Acta, 2006, 570, 137–143. [DOI] [PubMed] [Google Scholar]
- 43.Lian W, Wu D, Lim DV and Jin S, Anal. Biochem, 2010, 401, 271–279. [DOI] [PubMed] [Google Scholar]
- 44.Weingart OG, Gao H, Crevoisier F, Heitger F, Avondet MA and Sigrist H, Sensors, 2012, 12, 2324–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ligler FS, Sapsford KE, Golden JP, Shriver-Lake LC,Taitt CR, Dyer MA, Barone S and Myatt CJ, Anal. Sci, 2007, 23, 5–10. [DOI] [PubMed] [Google Scholar]
- 46.Kalb SR, Moura H, Boyer AE, McWilliams LG, Pirkle JL and Barr JR, Anal. Biochem, 2006, 351, 84–92. [DOI] [PubMed] [Google Scholar]
- 47.Sharma SK, Ferreira JL, Eblen BS and Whiting RC, Appl. Environ. Microbiol, 2006, 72, 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nowakowski A, Wang C, Powers DB, Amersdorfer P, Smith TJ, Montgomery VA, Sheridan R, Blake R, Smith LA and Marks JD, Proc. Natl. Acad. Sci. U. S. A, 2002, 99, 11346–11350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amersdorfer P, Wong C, Chen S, Smith T, Deshpande S, Sheridan R, Finnern R and Marks JD, Infect. Immun, 1997, 65, 3743–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Razai A, Garcia-Rodriguez C, Lou J, Geren IN,Forsyth CM, Robles Y, Tsai R, Smith TJ, Smith LA, Siegel RW, Feldhaus M and Marks JD, J. Mol. Biol, 2005, 351, 158–169. [DOI] [PubMed] [Google Scholar]
- 51.Garcia-Rodriguez C, Levy R, Arndt JW, Forsyth CM, Razai A, Lou J, Geren I, Stevens RC and Marks JD, Nat. Biotechnol, 2007, 25, 107–116. [DOI] [PubMed] [Google Scholar]
- 52.Levy R, Forsyth CM, LaPorte SL, Geren IN, Smith LA and Marks JD, J. Mol. Biol, 2007, 365, 196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hopman AH, Ramaekers FC and Speel EJ, J. Histochem. Cytochem, 1998, 46, 771–777. [DOI] [PubMed] [Google Scholar]
- 54.Varnum SM, Woodbury RL and Zangar RC, Methods Mol. Biol, 2004, 264, 161–172. [DOI] [PubMed] [Google Scholar]
- 55.Woodbury RL, Varnum SM and Zangar RC, J. Proteome Res, 2002, 1, 233–237. [DOI] [PubMed] [Google Scholar]
- 56.Wang D, Baudys J, Kalb SR and Barr JR, Anal. Biochem, 2011, 412, 67–73. [DOI] [PubMed] [Google Scholar]
- 57.Kalb SR, Moura H, Boyer AE, McWilliams LG, Pirkle JL and Barr JR,Anal. Biochem, 2006, 351, 84–92. [DOI] [PubMed] [Google Scholar]
- 58.White AM, Daly DS, Varnum SM, Anderson KK, Bollinger N and Zangar RC, Bioinformatics, 2006, 22, 1278–1279. [DOI] [PubMed] [Google Scholar]
- 59.Daly DS, White AM, Varnum SM, Anderson KK and Zangar RC, BMC Bioinf, 2005, 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zangar RC, Daly DS, White AM, Servoss SL, Tan RM and Collett JR, J. Proteome Res, 2009, 8, 3937–3943. [DOI] [PubMed] [Google Scholar]
- 61.Gonzalez RM, Seurynck-Servoss SL, Crowley SA, Brown M, Omenn GS, Hayes DF and Zangar RC, J. Proteome Res, 2008, 7, 2406–2414. [DOI] [PubMed] [Google Scholar]
- 62.Webb RP, Smith TJ, Wright PM, Montgomery VA, Meagher MM and Smith LA, Vaccine, 2007, 25, 4273–4282. [DOI] [PubMed] [Google Scholar]
- 63.Peruski AH, Johnson LH 3rd and Peruski LF Jr, J. Immunol. Methods, 2002, 263, 35–41. [DOI] [PubMed] [Google Scholar]
- 64.Liu F, Li Y, Song C, Dong B, Liu Z, Zhang K, Li H, Sun Y, Wei Y, Yang A, Yang K and Jin B, Anal. Chem, 2010, 82, 7758–7765. [DOI] [PubMed] [Google Scholar]
- 65.Kijek TM, Rossi CA, Moss D, Parker RW and Henchal EA, J. Immunol. Methods, 2000, 236, 9–17. [DOI] [PubMed] [Google Scholar]
- 66.Franz DR, Jahrling PB, Friedlander AM, McClain DJ,Hoover DL, Bryne WR, Pavlin JA, Christopher GW and Eitzen EM Jr, JAMA, J. Am. Med. Assoc, 1997, 278, 399–411. [DOI] [PubMed] [Google Scholar]
- 67.LeClaire RD and Bavari S, Antimicrob. Agents Chemother, 2001, 45, 460–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dezfulian M, Hatheway CL, Yolken RH and Bartlett JG, J. Clin. Microbiol, 1984, 20, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanvanich M, Viscidi R, Laughon BE, Bartlett JG and Yolken RH, J. Clin. Microbiol, 1985, 21, 184–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shlyapnikov YM, Shlyapnikova EA, Simonova MA, Shepelyakovskaya AO, Brovko FA, Komaleva RL, Grishin EV and Morozov VN, Anal. Chem, 2012, 84, 5596–5603. [DOI] [PubMed] [Google Scholar]
- 71.Dunning FM, Ruge DR, Piazza TM, Stanker LH, Zeytin FN and Tucker WC, Appl. Environ. Microbiol,2012, 78, 7687–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Poli MA, Rivera VR and Neal D, Toxicon, 2002, 40, 1723–1726. [DOI] [PubMed] [Google Scholar]
- 73.Audi J, Belson M, Patel M, Schier J and Osterloh J, JAMA, J. Am. Med. Assoc, 2005, 294, 2342–2351. [DOI] [PubMed] [Google Scholar]
- 74.Worbs S, Kohler K, Pauly D, Avondet MA, Schaer M, Dorner MB and Dorner BG, Toxins, 2011, 3, 1332–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith MJ, Carvalho HM, Melton-Celsa AR and O’Brien AD, Infect. Immun, 2006, 74, 6992–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cheng LW, Henderson TD, Patfield S, Stanker LH and He X, Toxins, 2013, 5, 1845–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.