

Abstract
A method for the preparation of aryl- and heteroarylamine products by triethylphosphine-mediated deoxygenative coupling of nitroarenes and boronic acids is reported. This method provides access to an array of functionalized (hetero)arylamine products from readily available starting materials under the action of an inexpensive commercial reagent. The developed triethylphosphine-mediated transformation highlights the capability of organophosphorus compounds to carry out this useful deoxygenative transformation without the necessity of any transition metal additives.
Keywords: C-N bond formation, Nitroarene, Boronic Acid, Phosphine
1. Introduction
Aryl- and heteroaryl amines find many uses in pharmaceutical, agrichemical, and organic materials chemistry; they are also present in naturally occurring secondary metabolites and derivatives thereof [1]. Developments in intermolecular arylation of anilines, especially transition metal catalyzed C–N coupling methods (Buchwald-Hartwig [2], Chan-Lam [3], Ullmann [4]), have enabled broad access to these compounds. Recently, we have reported that intermolecular (hetero)aryl C–N bond formation is driven by deoxygenative amination of boronic acids with nitroarenes [5]. Leveraging the utility of tricoordinate phosphorus reagents as O-atom acceptors [6], this chemistry advances a line of work from Cadogan who showed that P(III)-mediated deoxygenation of nitroarenes can give nitrogen containing heterocyclic products through intramolecular C-N bond formation (Figure 1A) [7,8], Indeed, nitroarenes are attractive substrates for aryl amination reactions because they are readily available from both commercial and synthetic sources, thereby comparing favorably with respect to nitrosoarenes [9], N-alkyl hydroxylamines [10], azides [11], and tosyl triazenes [12] substrates for boronic acid amination reactions. Knochel [13], Kürti [14], and Niggemann [15] have all reported main group reagent approaches for direct intermolecular C-N bond formation with nitroarene substrates. Relatedly, Suárez‐Pantiga and Sanz reported that phosphine-mediated reductive coupling of nitroarenes and boronic acids is catalyzed by an oxomolybdenum compound [16]. Here, we show that an inexpensive commercial phosphine reagent (triethylphosphine) can drive the deoxygenative coupling of nitroarenes and boronic acids to give di(hetero)arylamine products in a direct synthetic operation without the need of any transition metal additives (Figure 1B). The method serves as an operationally simple entry into valuable C–N coupling products from readily available reaction partners.
Figure 1.
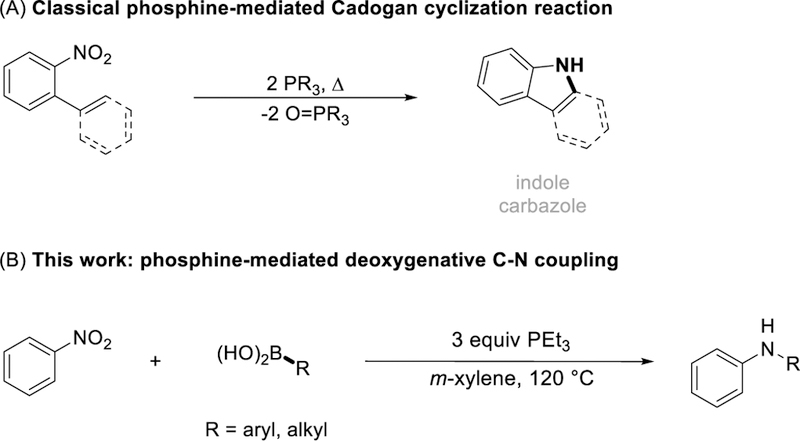
Intra- and intermolecular C-N bond forming reactions resulting from nitroarene deoxygenation. (A) Classical Cadogan cyclization reactions. (B) This work: stoichiometric phosphine-mediated deoxygenative C-N coupling of nitroarenes and boronic acids.
2. Results and Discussion
The coupling reaction of nitrobenzene (1) and phenylboronic acid (2) to give diphenylamine (3) was selected for development and optimization studies (Table 1). Commercially available phosphine and phosphite reagents (3 equiv relative to 1) were assessed for their efficacy in the reductive coupling under previously defined reaction conditions (m-xylene, 120 oC). While triphenylphosphine 4 (entry 1) was found to produce only 4% of the desired diphenylamine product 3, trialkylphosphines tri-tert-butylphosphine 5 (entry 2, 24%), tricyclohexylphosphine 6 (entry 3, 46%), and tributylphosphine 7 (entry 4, 66%) demonstrated an increase in yield from tertiary to primary alkyl substituents (Bu>Cy>tBu). Further investigation of trialkylphosphines indicated that triethylphosphine 8 (entry 5, 81%) provided desired product diphenylamine 2 with a yield comparable to our previously developed catalytic system within the same time frame of 4 hours. Trimethylphosphine 9 (entry 6, 39%) was less effective, perhaps attributed to the low boiling point of this reagent (bp 38–40 °C for PMe3 vs 127–128 °C for PEt3). Triethylphosphite 10 (entry 7, 8%) and tris(dimethylamino)phosphine (entry 8, 10%) proved to be poor reagents for the desired coupling reaction.
Table 1.
Reaction Development and Optimizationa
 | ||
|---|---|---|
| Entry | PR3 Reagent | Yield %b |
| 1 | Triphenylphosphine (PPh3, 4) | 4 |
| 2 | Tri-tert-butylphosphine (P(tBu)3, 5) | 24 |
| 3 | Tricyclohexylphosphine (PCy3, 6) | 46 |
| 4 | Tributylphosphine (PBu3, 7) | 66 |
| 5 | Triethylphosphine (PEt3, 8) | 81 |
| 6 | Trimethylphosphine (PMe3, 9) | 39c |
| 7 | Triethylphosphite (P(OEt)3, 10) | 8 |
| 8 | Tris(dimethylamino)phosphine (P(NMe)3, 11) | 10 |
0.5 mmol scale.
Yields determined via GC vs internal standard.
PMe3 was introduced as a 1 M solution in THF.
With respect to variation of the nitro coupling partner, a series of mixed diarylamine products by coupling with phenylboronic acid were prepared in good yield (Figure 2A). Halogen-(12), boryl-(13), and free aniline-containing (14) are all prepared in straightforward fashion, presenting opportunities for further functionalization, including subsequent transition metal catalyzed cross coupling if desired. It was found that electronic-deficient nitroarenes excel as substrates, providing the desired products in high yield and short reaction times. Indeed, while 3,5-bis(trifluoromethyl)aniline is considered to be a challenging substrate for Pd-catalyzed C-N coupling [17], 1-nitro-3,5-bis(trifluoromethyl)benzene is readily arylated under the deoxygenative phosphine-mediated coupling protocol to give 16. Further highlighting the variety of possible nitroarene coupling partners, heterocyclic substrate 5-nitroisoquinoline was successfully coupled with phenylboronic acid to give product heteroarylamine 17 in good yield.
Figure 2.
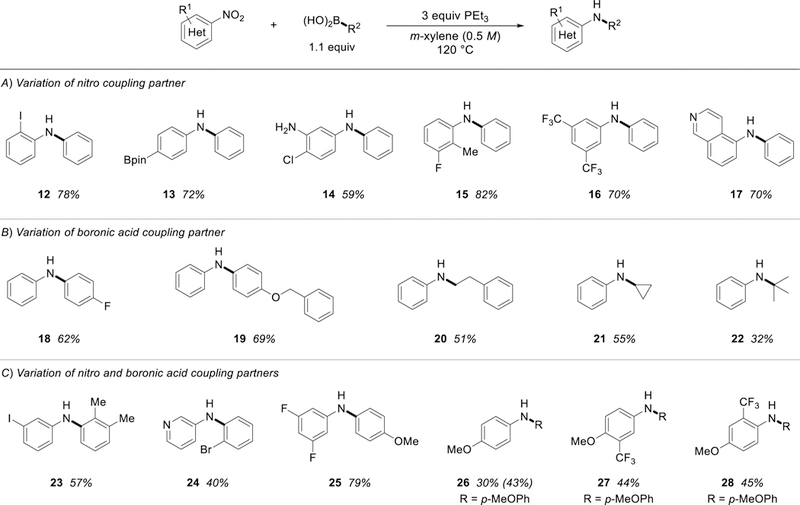
Examples of stoichiometric C-N coupling. See experimental section for specific reaction details.
The coupling reaction is similarly amenable to deoxygenative arylation of nitrobenzene by variation of the boronic acid partner (Figure 2B). Para-substituted boronic acids containing inductively withdrawing (18) and resonance donating (19) substitutents were found to be suitable partners for the coupling reaction. It was also found that primary (20), secondary (21), and even tertiary (22) carbons can transferred from alkylboronic acid partners in serviceable yields. Consequently, the deoxygenative coupling reaction can be used to access N-alkyl arylamines. That said, coupling with tert-butylboronic acid remains challenging, thus presenting a direction for future development.
To further demonstrate the synthetic utility of the developed transformation, variations were made to both the nitro and boronic acid components (Figure 2C). For instance, the deoxygenative C-N coupling 3-iodo-1-nitrobenzene with 2,3-dimethylphenylboronic acid gave diarylamine 23 in 57% yield. The method is similarly applicable to the reductive union of 3-nitropyridine with 2-bromophenylboronic acid to give heteroaryl amine product 24, albeit in modest yield. The identity of the nitroarene substrates plays a significant role in controlling the efficiency of the deoxygenative coupling with more electron deficient substrates performing at a higher level. Specifically, whereas the coupling of 3,5-difluoro-1-nitrobenzene with 4-methoxyphenylboronic acid evolves efficiently to product 25 (79%), the related coupling of 4-methoxy-1-nitrobenzene with 4-methoxyphenylboronic acid proceeded in a much reduced yield 26, 40%). To study the effects of introducing withdrawing substituents on to the core structure of challenging 4-nitroanisole, nitroanisole substrates bearing a trifluoromethyl group either meta (27) or ortho (28) to the nitro substituent were tested. In both cases, yields were slightly improved and nearly identical (44% vs 45%) – emphasizing that the introduction of withdrawing groups to the nitroarene partner can improve the overall yield in some difficult substrates.
3. Conclusion
We have demonstrated that stoichiometric reductive coupling of nitroarenes and boronic acids using triethylphosphine enables the preparation of useful di(hetero)arylamine products. While phosphines are commonly encountered as spectator ligands in transition metal-catalyzed transformations [18], we have shown that deoxygenative C-N coupling of nitroarenes and boronic acids can be mediated by a phosphine alone. The phosphine-mediated transformation showcases the ability of triethylphosphine to deoxygenate nitroarenes and expands on our previously reported catalytic C-N cross coupling by utilizing a commercially available phosphine reagent.
4. Experimental section
All reagents (including commercial phosphorus reagents used in optimization studies) were purchased from commercial vendors (Sigma-Aldrich, Alfa Aesar, Acros, TCI, or Oakwood Chemical, Combi-Blocks) and used without further purification unless otherwise indicated. Nitrobenzene was distilled over CaH prior to use. Anhydrous m-xylene was obtained from a Sigma-Aldrich and used as received. All other solvents were ACS grade or better and were used without further purification. Manipulations were conducted under an atmosphere of dry N2 gas unless otherwise noted. The phosphine-mediated deoxygenative C–N coupling reactions were carried out in threaded glass culture tubes outfitted with a phenolic screw-thread open top cap and PTFE-lined silicone septum. No special drying of the reaction vessel components was conducted prior to the reported reactions. Column chromatography was carried out on silica gel (SiliFlash® Irregular Silica Gel, P60 40–63μm). 1H, 13C, and 19F NMR were collected with a Bruker AVANCE III DRX 400 spectrometer and processed using MestReNova. 1H NMR chemical shifts are given in ppm with respect to solvent residual peak (CDCl3, δ 7.26 ppm) and 13C{1H} NMR shifts are given in ppm with respect to (CDCl3 δ 77.16 ppm). Multiplicities are described as s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, tt = triplet of triplets, m = multiplet. Coupling constants are reported in Hertz (Hz). High-resolution ESI mass spectra were obtained from the MIT Department of Chemistry Instrumentation Facility on an Agilent 6545 Q-TOF instrument.
4.1. General Procedure
A glass culture tube containing a magnetic stir bar was charged with a nitroarene substrate and boronic acid, then fitted with a PTFE-lined silicone septum within a phenolic screw-thread open-top cap. The vessel was evacuated and backfilled with nitrogen on a Schlenk line. Dry m-xylene (2 mL, 0.5 M) was added via syringe, followed by triethylphosphine (0.44 mL, 3 equivalents) and the reaction mixture was heated 120 °C with stirring. Upon completion, the reaction mixture was cooled to ambient temperature then diluted with 10 mL of distilled water. With the aid of ethyl acetate (ca. 3×10 mL), the reaction mixture was transferred to a separatory funnel. After mixing and separating the phases, the organic layer was washed with 10 mL of a 1 M NaOH aqueous solution, and 10 mL of brine. Each aqueous phase was back-extracted with one 10 mL portion of ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated by rotary evaporation. The crude residues were purified via column chromatography to yield pure coupling products. Columns were primarily slurry packed with hexanes and mobile phase polarity was increased gradually to the mixture indicated. Note: hexanes = Hex, dichloromethane = DCM, ethyl acetate = EA.
4.1.2. 2-Iodo-N-phenylaniline (12)
Following the general procedure using phenylboronic acid (134 mg, 1.10 mmol, 1.1 equiv) and 1-iodo-2-nitrobenzene (249 mg, 1.00 mmol, 1.0 equiv) for 3 h at 120 °C. The product was purified by column chromatography with a gradient from hexanes to 10% DCM/1% EA/89% Hex on silica (231 mg, 0.78 mmol, 78%). Rf (10% DCM/1% EA/89% Hex) 0.61. 1H NMR (400 MHz, Chloroform-d) δ 7.66 (d, J = 7.9 Hz, 1H), 7.21 (t, J = 7.7 Hz, 2H), 7.14 – 7.05 (m, 2H), 7.02 (d, J = 7.9 Hz, 2H), 6.93 (t, J = 7.3 Hz, 1H), 6.57 – 6.45 (m, 1H), 5.80 (br s, 1H). 13C NMR (101 MHz, Chloroform-d) δ 144.04, 142.11, 139.63, 129.57, 129.14, 122.66, 122.04, 120.11, 115.98, 88.93. HRMS (ESI) calculated for C12H10IN [M+H]+: 295.9931; Found: 295.9927.
4.1.3. N-Phenyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline (13)
Following the general procedure using phenylboronic acid (134 mg, 1.10 mmol, 1.1 equiv) and 4-nitrobenzeneboronic acid pinacol ester (249 mg, 1.00 mmol, 1.0 equiv) for 5 h at 120 °C. The product was purified by column chromatography with 10% DCM/1% EA/89% Hex on silica (212 mg, 0.72 mmol, 72%). Rf (10% DCM/1% EA/89% Hex) 0.13. 1H NMR (400 MHz, Chloroform-d) δ 7.73 (d, J = 8.5 Hz, 2H), 7.31 (t, J = 8.3, 7.4 Hz, 2H), 7.14 (d, J = 8.3 Hz, 2H), 7.06 – 6.97 (m, 3H), 5.88 (br s, 1H), 1.35 (s, 12H). 13C NMR (101 MHz, Chloroform-d) δ 145.98, 141.73, 136.11, 129.17, 121.71, 118.93, 115.32, 83.25, 24.67. HRMS (ESI) calculated for C18H22BNO2 [M+H]+: 296.1820; Found: 296.1820.
4.1.4. 4-Chloro-N1-phenylbenzene-1,3-diamine (14)
Following the general procedure using phenylboronic acid (134 mg, 1.10 mmol, 1.1 equiv) and 2-chloro-5-nitroaniline (173 mg, 1.00 mmol, 1.0 equiv) for 7 h at 120 °C. The product was purified by column chromatography with 20% EA in hexanes on silica (130 mg, 0.59 mmol, 59%). Rf (20% EA in hexanes) 0.27. 1H NMR (400 MHz, Chloroform-d) δ 7.27 (t, J = 7.8 Hz, 2H), 7.14 – 7.02 (m, 3H), 6.95 (t, J = 7.3 Hz, 1H), 6.49 (d, J = 2.4 Hz, 1H), 6.40 (dd, J = 8.5, 2.4 Hz, 1H), 5.57 (br s, 1H), 3.98 (br s, 2H). 13C NMR (101 MHz, Chloroform-d) δ 143.63, 143.14, 142.90, 130.05, 129.49, 121.48, 118.55, 111.42, 109.19, 104.49. HRMS (ESI) calculated for C12H11ClN2 [M+H]+: 219.0684; Found: 219.0684.
4.1.5. 3-Fluoro-2-methyl-N-phenylaniline (15)
Following the general procedure using phenylboronic acid (134 mg, 1.10 mmol, 1.1 equiv) and 1-fluoro-2-methyl-3-nitrobenzene (122 μL, 1.00 mmol, 1.0 equiv) for 16 h at 120 °C. The product was purified by column chromatography with 5% EA in hexanes on silica (164 mg, 0.82 mmol, 82%). Rf (5% EA in hexanes) 0.40. 1H NMR (400 MHz, Chloroform-d) δ 7.31 (t, J = 7.7 Hz, 2H), 7.15 – 6.96 (m, 5H), 6.73 (t, J = 8.6 Hz, 1H), 5.45 (br s, 1H), 2.20 (s, 3H). 13C NMR (101 MHz, Chloroform-d) δ 162.00 (d, J = 242.3 Hz), 143.42, 143.33 (d, J = 6.5 Hz), 129.48, 126.91 (d, J = 10.2 Hz), 121.36, 118.39, 113.64 (d, J = 2.7 Hz), 108.36 (d, J = 23.1 Hz), 9.21 (d, J = 6.3 Hz). 19F NMR (376 MHz, Chloroform-d) δ −115.62. HRMS (ESI) calculated for C13H12FN [M+H]+: 202.1027; Found: 202.1024
4.1.6. N-Phenyl-3,5-bis(trifluoromethyl)aniline (16)
Following an altered general procedure using phenylboronic acid (134 mg, 1.10 mmol, 1.1 equiv) 3,5-bis(trifluoromethyl)nitrobenzene (169 μL, 1.00 mmol, 1.0 equiv), and distilled water (0.16 mL, 9 mmol, 9 equiv) for 2 h at 120 °C. The product was purified by column chromatography with a gradient from hexanes to 10% DCM/2% EA in hexanes on silica (215 mg, 0.70 mmol, 70%). Rf (10% DCM/2% EA in hexanes) 0.28. 1H NMR (400 MHz, Chloroform-d) δ 7.41 – 7.35 (m, 4H), 7.33 (s, 1H), 7.20 – 7.08 (m, 3H), 5.98 (br s, 1H). 13C NMR (101 MHz, Chloroform-d) δ 145.46, 140.48, 132.85 (q, J = 33.0 Hz), 130.00, 123.99, 123.49 (q, J = 272.8 Hz), 120.57, 115.28 (d, J = 3.3 Hz), 113.11 (p, J = 3.9 Hz). 19F NMR (376 MHz, Chloroform-d) δ −63.21. HRMS (ESI) calculated for C14H9F6N [M+H]+: 306.0712; Found: 306.0706
4.1.7. N-Phenylisoquinolin-5-amine (17)
Following the general procedure using phenylboronic acid (134 mg, 1.10 mmol, 1.1 equiv) and 5-nitroisoquinoline (174 mg, 1.00 mmol, 1.0 equiv) for 3 h at 120 °C. The product was purified by column chromatography with 20% DCM/20% EA in hexanes to full EA on silica (154 mg, 0.70 mmol, 70%). Rf (EA) 0.52. 1H NMR (400 MHz, Chloroform-d) δ 9.25 (s, 1H), 8.51 (d, J = 6.0 Hz, 1H), 7.77 (d, J = 6.0 Hz, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.56 – 7.47 (m, 2H), 7.30 (t, J = 7.9 Hz, 2H), 7.05 (d, J = 7.6 Hz, 2H), 6.99 (t, J = 7.4 Hz, 1H), 6.05 (br s, 1H). 13C NMR (101 MHz, Chloroform-d) δ 153.09, 143.62, 142.89, 138.55, 129.87, 129.82, 129.64, 127.68, 121.71, 121.56, 118.42, 117.96, 114.81. HRMS (ESI) calculated for C15H12N2 [M+H]+: 221.1073, Found: 221.1066.
4.1.8. 4-Fluoro-N-phenylaniline (18)
Following the general procedure using 4-fluorophenylboronic acid (154 mg, 1.10 mmol, 1.1 equiv) and nitrobenzene (103 μL, 1.00 mmol, 1.0 equiv) for 18 h at 120 °C. The product was purified by column chromatography with a gradient from hexanes to 10% DCM in hexanes to 20% DCM/1% EA in hexanes to 20% DCM/5% EA in hexanes on silica (116 mg, 0.62 mmol, 62%). Rf (10% DCM in hexanes) 0.10. 1H NMR (400 MHz, Chloroform-d) δ 7.28 (t, J = 7.5 Hz, 2H), 7.12 – 6.97 (m, 6H), 6.93 (t, J = 7.3 Hz, 1H), 5.59 (br s, 1H). 13C NMR (101 MHz, Chloroform-d) δ 158.06 (d, J = 240.1 Hz), 143.94, 138.94 (d, J = 2.4 Hz), 129.41, 120.62, 120.55, 116.80, 115.94 (d, J = 22.5 Hz). 19F NMR (376 MHz, Chloroform-d) δ −122.00.
4.1.9. 4-(Benzyloxy)-N-phenylaniline (19)
Following the general procedure using 4-(benzyloxy)phenylboronic acid (342 mg, 1.50 mmol, 1.5 equiv) and nitrobenzene (103 μL, 1.00 mmol, 1.0 equiv) for 18 h at 120 °C. The product was purified by column chromatography with a gradient from hexanes to 8% DCM/4% EA in hexanes on silica (189 mg, 0.69 mmol, 69%). Rf (8% DCM/4% EA in hexanes) 0.37. 1H NMR (400 MHz, Chloroform-d) δ 7.51 – 7.45 (m, 2H), 7.42 (t, J = 7.3 Hz, 2H), 7.39 – 7.33 (m, 1H), 7.28 – 7.21 (m, 2H), 7.08 (d, J = 8.5 Hz, 2H), 6.99 – 6.92 (m, 4H), 6.87 (t, J = 7.3 Hz, 1H), 5.50 (br s, 1H), 5.07 (s, 2H). 13C NMR (101 MHz, Chloroform-d) δ 154.50, 145.09, 137.31, 136.16, 129.42, 128.69, 128.04, 127.61, 122.01, 119.78, 115.91, 115.87, 70.59. HRMS (ESI) calculated for C19H17NO [M+H]+: 276.1383; Found: 276.1380.
4.1.10. N-Phenethylaniline (20)
Following the general procedure on 0.5 mmol scale using phenethylboronic acid (83 mg, 0.55 mmol, 1.1 equiv), nitrobenzene (51 μL, 0.50 mmol, 1.0 equiv), and 3 equivalents of PEt3 (0.22 mL, 1.5 mmol) for 4 h at 120 °C. The product was purified by column chromatography with hexanes to 20% DCM/0.4% EA in hexanes gradient on silica (50 mg, 0.25 mmol, 51%). Rf (20% DCM/0.4% EA in hexanes) 0.73. 1H NMR (400 MHz, Chloroform-d) δ 7.35 (t, J = 7.3 Hz, 2H), 7.31 – 7.18 (m, 5H), 6.74 (t, J = 7.3 Hz, 1H), 6.65 (d, J = 7.6 Hz, 2H), 3.70 (br s, 1H), 3.43 (t, J = 7.0 Hz, 2H), 2.95 (t, J = 7.0 Hz, 2H). 13C NMR (101 MHz, Chloroform-d) δ 148.12, 139.43, 129.40, 128.91, 128.72, 126.54, 117.58, 113.11, 45.15, 35.64. HRMS (ESI) calculated for C14H15N [M+H]+: 198.1277; Found: 198.1276.
4.1.11. N-Cyclopropylaniline (21)
Following the general procedure using cyclopropylboronic acid (96 mg, 1.10 mmol, 1.1 equiv) and nitrobenzene (103 μL, 1.00 mmol, 1.0 equiv) for 9 h at 120 °C. The product was purified by column chromatography with 10% DCM/0.4% EA in hexanes on silica (73 mg, 0.55 mmol, 55%). 1H NMR (400 MHz, Chloroform-d) δ 7.22 (dd, J = 8.5, 7.4 Hz, 2H), 6.82 (d, J = 7.6 Hz, 2H), 6.76 (t, J = 7.3 Hz, 1H), 4.18 (br s, 1H), 2.50 – 2.41 (m, 1H), 0.79 – 0.72 (m, 2H), 0.57 – 0.51 (m, 2H). 13C NMR (101 MHz, Chloroform-d) δ 148.79, 129.22, 117.84, 113.26, 25.35, 7.53. HRMS (ESI) calculated for C9H11N [M+H]+: 134.0964; Found: 134.0961.
4.1.12. N-(tert-Butyl)aniline (22)
Following an altered general procedure on 0.5 mmol scale using tert-butylboronic acid (112 mg, 1.1 mmol, 2.2 equiv), nitrobenzene (51 μL, 0.50 mmol, 1.0 equiv), and 6 equivalents of PEt3 (0.44 mL, 3.0 mmol) in m-xylene (2 mL, 0.25 M) for 12 h at 120 °C. The product was purified by column chromatography with hexanes to 25% EA in hexanes gradient on silica (24 mg, 0.16 mmol, 32%). 1H NMR (400 MHz, Chloroform-d) δ 7.18 (t, J = 8.1, 7.6 Hz, 2H), 6.83 – 6.71 (m, 3H), 3.42 (br s, 1H), 1.36 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 146.99, 129.01, 118.43, 117.60, 51.60, 30.23. HRMS (ESI) calculated for C10H15N [M+H]+: 150.1277; Found: 150.1273. Tert-butylboronic acid was prepared using tert-butylmagnesium chloride and trimethylborate according to a known procedure [19 ].
4.1.13. N-(3-Iodophenyl)-2,3-dimethylaniline (23)
Following the general procedure using 2,3-dimethylphenylboronic acid (165 mg, 1.10 mmol, 1.1 equiv) and 1-iodo-3-nitrobenene (249 mg, 1.00 mmol, 1.0 equiv), for 18 h at 120 °C. The product was purified by column chromatography with hexanes to 10% EA in hexanes gradient on silica (185 mg, 0.57 mmol, 57%). Rf (10% EA in hexanes) 0.63. 1H NMR (400 MHz, Chloroform-d) δ 7.18 – 7.13 (m, 2H), 7.11 – 7.06 (m, 2H), 7.01 – 6.96 (m, 1H), 6.91 (t, J = 8.1 Hz, 1H), 6.75 (d, J = 8.1 Hz, 1H), 5.36 (br s, 1H), 2.34 (s, 3H), 2.16 (s, 3H). 13C NMR (101 MHz, Chloroform-d) δ 146.85, 139.67, 138.27, 130.80, 130.11, 128.32, 126.25, 125.95, 124.38, 120.43, 115.01, 95.12, 20.78, 13.93. HRMS (ESI) calculated for C14H14IN [M+H]+: 324.0244; Found: 324.0242.
4.1.14. N-(2-Bromophenyl)pyridin-3-amine (24)
Following an altered general procedure using 2-bromophenylboronic acid (221 mg, 1.10 mmol, 1.1 equiv) and 3-nitropyridine (124 mg, 1.00 mmol, 1.0 equiv), for 12 h at 80 °C. The product was purified by column chromatography with 75% EA in hexanes on silica (100 mg, 0.40 mmol, 40%). Rf (75% EA in hexanes) 0.34. 1H NMR (400 MHz, Chloroform-d) δ 8.47 (d, J = 2.6 Hz, 1H), 8.27 (dd, J = 4.7, 1.4 Hz, 1H), 7.55 (d, J = 7.6 Hz, 1H), 7.47 (d, J = 8.2 Hz, 1H), 7.25 – 7.18 (m, 3H), 6.81 (ddd, J = 8.0, 5.6, 3.2 Hz, 1H), 6.08 (br s, 1H). 13C NMR (101 MHz, Chloroform-d) δ 143.73, 142.30, 140.51, 138.46, 133.35, 128.40, 126.30, 123.90, 122.22, 116.32, 113.13. HRMS (ESI) calculated for C11H9BrN2 [M+H]+: 250.0052, Found: 250.0052.
4.1.15. 3,5-Difluoro-N-(4-methoxyphenyl)aniline (25)
Following the general procedure using 4-methoxyphenylboronic acid (167 mg, 1.10 mmol, 1.1 equiv) and 3,5-difluoronitrobenzene (113 μL, 1.00 mmol, 1.0 equiv) for 3 h at 120 °C. The product was purified by column chromatography with 9% EA/1% DCM in hexanes on silica (186 mg, 0.79 mmol, 79%). Rf (9% EA/1% DCM in hexanes) 0.32. 1H NMR (400 MHz, Chloroform-d) δ 7.10 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.9 Hz, 2H), 6.31 (dd, J = 9.8, 2.1 Hz, 2H), 6.22 (tt, J = 9.1, 2.3 Hz, 1H), 5.63 (br s, 1H), 3.82 (s, 3H). 13C NMR (101 MHz, Chloroform-d) δ 164.21 (dd, J = 244.5, 15.7 Hz), 156.75, 148.55 (t, J = 12.9 Hz), 133.61, 124.64, 114.95, 98.09 – 96.61 (m), 94.06 (t, J = 26.1 Hz), 55.67. 19F NMR (376 MHz, Chloroform-d) δ −110.02. HRMS (ESI) calculated for C13H11F2NO [M+H]+: 236.0881, Found: 236.0883.
4.1.16. Bis(4-methoxyphenyl)amine (26)
Following the general procedure using 4-methoxyphenylboronic acid (167 mg, 1.10 mmol, 1.1 equiv) and 4-nitroanisole (153 mg, 1.00 mmol, 1.0 equiv), for 7 h at 120 °C. The product was purified by column chromatography with 5% EA/5% DCM in hexanes on silica (69 mg, 0.30 mmol, 30%). Rf (5% EA/5% DCM in hexanes) 0.07. 1H NMR (400 MHz, Chloroform-d) δ 6.94 (d, J = 8.9 Hz, 4H), 6.83 (d, J = 8.9 Hz, 4H), 5.29 (br s, 1H), 3.79 (s, 6H). 13C NMR (101 MHz, Chloroform-d) δ 154.37, 138.07, 119.67, 114.85, 55.79. HRMS (ESI) calculated for C14H15NO2 [M+H]+: 230.1176; 230.1175.
For the catalytic reaction on the same scale (1 mmol), an altered general procedure was followed in which 1,2,2,3,4,4-hexamethylphosphetane 1-oxide (26 mg, 0.15 mmol, 15 mol%) was introduced prior to evacuation and phenylsilane (0.25 mL, 2.00 mmol, 2.0 equiv) was added via syringe instead of triethylphosphine. After heating at 120 °C for 16 h, the product was purified by column chromatography with a gradient of 5% EA/5% DCM in hexanes to 10% EA/10% DCM in hexanes on silica (98 mg, 0.43 mmol, 43%). 1H NMR (400 MHz, Chloroform-d) δ 6.95 (d, J = 8.7 Hz, 4H), 6.83 (d, J = 8.7 Hz, 4H), 5.29 (br s, 1H), 3.79 (s, 6H).13C NMR (101 MHz, Chloroform-d) δ 154.37, 138.07, 119.67, 114.85, 55.79.
4.1.17. 4-Methoxy-N-(4-methoxyphenyl)-3-(trifluoromethyl)-aniline (27)
Following the general procedure using 4-methoxyphenylboronic acid (167 mg, 1.10 mmol, 1.1 equiv) and 1-methoxy-4-nitro-2-(trifluoromethyl)benzene (221 mg, 1.00 mmol, 1.0 equiv), for 16 h at 120 °C. The product was purified by column chromatography with a gradient of 10% EA in hexanes on silica to 25% EA in hexanes (131 mg, 0.44 mmol, 44%). Rf (10% EA in hexanes) 0.17. 1H NMR (400 MHz, Chloroform-d) δ 7.17 (d, J = 2.7 Hz, 1H), 7.07 (dd, J = 8.8, 2.6 Hz, 1H), 6.97 (d, J = 8.8 Hz, 2H), 6.93 – 6.83 (m, 3H), 5.36 (br s, 1H), 3.86 (s, 3H), 3.80 (s, 3H). 13C NMR (101 MHz, Chloroform-d) δ 155.18, 151.57, 138.23, 136.59, 123.72 (q, J = 272.3 Hz), 121.50, 120.97, 116.31 (q, J = 5.3 Hz), 114.99, 113.87, 56.71, 55.75. 19F NMR (376 MHz, Chloroform-d) δ −62.30. HRMS (ESI) calculated for C15H14F3NO2 [M+H]+: 298.1049, Found: 298.1048.
4.1.18. 4-Methoxy-N-(4-methoxyphenyl)-2-(trifluoromethyl)-aniline (28)
Following the general procedure using 4-methoxyphenylboronic acid (167 mg, 1.10 mmol, 1.1 equiv) and 4-methoxy-1-nitro-2-(trifluoromethyl)benzene (221 mg, 1.00 mmol, 1.0 equiv), for 16 h at 120 °C. The product was purified by column chromatography with a 5% DCM/1% EA in hexanes (133 mg, 0.45 mmol, 45%). Rf (5% DCM/1% EA in hexanes) 0.17. 1H NMR (400 MHz, Chloroform-d) δ 7.12 – 7.05 (m, 2H), 6.99 (d, J = 8.5 Hz, 2H), 6.93 (d, J = 9.0 Hz, 1H), 6.86 (d, J = 8.6 Hz, 2H), 5.61 (br s, 1H), 3.80 (s, 6H). 13C NMR (101 MHz, Chloroform-d) δ 155.58, 153.01, 137.14, 136.17, 124.61 (q, J = 272.8 Hz), 122.33, 120.12, 119.07, 114.93, 111.79 (q, J = 5.7 Hz), 55.97, 55.73. 19F NMR (376 MHz, Chloroform-d) δ −61.81. HRMS (ESI) calculated for C15H14F3NO2 [M+H]+: 298.1049, Found: 298.1047.
Supplementary Material
Acknowledgments
Funding was provided by NIH NIGMS (GM114547) and MIT. J.Y. thanks the MIT Undergraduate Research Opportunities Program. T.V.N. thanks Dr. Jeffrey Lipshultz and Connor Gilhula for HRMS data collection.
Footnotes
Supplementary Material
Supplementary data (including 1H and 13C NMR spectra) for this article can be found online.
References and notes
- 1.(a) Ruiz-Castillo P; Buchwald SL Chem. Rev, 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lambrecht MJ; Kelly JW; Shenvi RA ACS Chem. Biol 2018, 13, 1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Hartwig JF Angew. Chem. Int. Edit 1998, 37, 2046–2067. [DOI] [PubMed] [Google Scholar]; (b) Wolfe JP; Buchwald SL Angew. Chem. Int. Ed 1999, 38, 2413–2416. [DOI] [PubMed] [Google Scholar]; (c) Jiang L; Buchwald SL Metal-Catalyzed CrossCoupling Reactions De Meijere A; Diderich F, Eds.; WileyBlackwell: Hoboken, NJ, 2008; ed. 2, pp. 699–760. [Google Scholar]; (d) Jiang Y; Ma D Catalysis without Precious Metals Bullock RM Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2010; pp 213–233. [Google Scholar]
- 3.Qiao J; Lam PYS Synthesis 2011, 2011, 829–856. [Google Scholar]; Bariwal J; Eycken E. V. der. Chem. Soc. Rev 2013, 42, 9283–9303. [DOI] [PubMed] [Google Scholar]
- 4.Sambiagio C; Marsden SP; Blacker AJ; McGowan PC Chem. Soc. Rev 2014, 43, 3525–3550. [DOI] [PubMed] [Google Scholar]
- 5.Nykaza TV; Cooper JC; Li G; Mahieu N; Ramirez A; Luzung MR; Radosevich AT J. Am. Chem. Soc 2018, 140, 15200–15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Xu S; He Z RSC Advances 2013, 3, 16885–16904. [Google Scholar]; (b) Karanam P; Reddy GM; Koppolu SR; Lin W Tetrahedron Lett 2018, 59, 59–76. [Google Scholar]
- 7.(a) Cadogan JIG; Cameron-Wood M; Mackie RK; Searle RJ G. J. Chem. Soc 1965, 0, 4831–4837. [Google Scholar]; (b) Cadogan JI G. Q. Rev. Chem. Soc 1968, 22, 222–251. [Google Scholar]
- 8.(a) For Cadogan cyclization reactions catalytic in phosphorus see:Nykaza TV; Harrison TS; Ghosh A; Putnik RA; Radosevich AT J. Am. Chem. Soc 2017, 139, 6839–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nykaza TV; Ramirez A; Harrison TS; Luzung MR; Radosevich AT J. Am. Chem. Soc 2018, 140, 3103–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Yu Y; Srogl J; Liebeskind LS Org. Lett 2004, 6, 2631–2634. [DOI] [PubMed] [Google Scholar]; (b) Roscales S; Csákÿ AG Org. Lett 2018, 20, 1667–1671. [DOI] [PubMed] [Google Scholar]
- 10.Sun H-B; Gong L; Tian Y-B; Wu J-G; Zhang X; Liu J; Fu Z; Niu D Angew. Chem. Int. Ed 2018, 57, 9456–9460. [DOI] [PubMed] [Google Scholar]
- 11.Ou L; Shao J; Zhang G; Yu Y Tetrahedron Lett 2011, 52, 1430–1431. [Google Scholar]
- 12.Sarma MJ; Phukan P Synth. Commun 2018, 48, 656–662. [Google Scholar]
- 13.(a) Sapountzis I; Knochel PJ Am. Chem. Soc 2002, 124, 9390–9391. [DOI] [PubMed] [Google Scholar]; (b) Doyle W; Staubitz A; Knochel P Chem. Eur. J 2003, 9, 5323–5331. [DOI] [PubMed] [Google Scholar]; (c) Kopp F; Sapountzis I; Knochel P Synlett, 2003, 885–887.; (d) Sapountzis I; Knochel P Synlett 2004, 955–958.; (e) Dhayalan V; Saemann C; Knochel P Chem. Commun 2015, 51, 3239–3242. [DOI] [PubMed] [Google Scholar]
- 14.Gao H; Xu Q-L; Ess DH; Kürti L Angew. Chem. Int. Ed 2014, 53, 2701–2705. [DOI] [PubMed] [Google Scholar]
- 15.Rauser M; Ascheberg C; Niggemann M Angew. Chem. Int. Ed 2017, 56, 11570–11574. [DOI] [PubMed] [Google Scholar]
- 16.Suárez‐Pantiga S; Hernández‐Ruiz R; Virumbrales C; Pedrosa MR; Sanz R Angew. Chem. Inter. Ed 2019, 58, in press, DOI: 10.1002/anie.201812806. [DOI] [PubMed] [Google Scholar]
- 17.Pompeo M; Farmer JL; Froese RDJ; Organ MG Angew. Chem. Int. Ed 2014, 53, 3223–3226. [DOI] [PubMed] [Google Scholar]
- 18.(a) Tolman CA Chem. Rev 1977, 77, 313–348. [Google Scholar]; (b) Surry DS; Buchwald SL Chem. Sci 2010, 2, 27–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srebnik M; Cole TE; Ramachandran PV; Brown HC J. Org. Chem 1989, 54, 6085–6096. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.