

Protein geranylgeranylation limits antiviral innate immunity.
Abstract
The mitochondrial antiviral signaling protein (MAVS) orchestrates host antiviral innate immune response to RNA virus infection. However, how MAVS signaling is controlled to eradicate virus while preventing self-destructive inflammation remains obscure. Here, we show that protein geranylgeranylation, a posttranslational lipid modification of proteins, limits MAVS-mediated immune signaling by targeting Rho family small guanosine triphosphatase Rac1 into the mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs) at the mitochondria-ER junction. Protein geranylgeranylation and subsequent palmitoylation promote Rac1 translocation into MAMs upon viral infection. MAM-localized Rac1 limits MAVS’ interaction with E3 ligase Trim31 and hence inhibits MAVS ubiquitination, aggregation, and activation. Rac1 also facilitates the recruitment of caspase-8 and cFLIPL to the MAVS signalosome and the subsequent cleavage of Ripk1 that terminates MAVS signaling. Consistently, mice with myeloid deficiency of protein geranylgeranylation showed improved survival upon influenza A virus infection. Our work revealed a critical role of protein geranylgeranylation in regulating antiviral innate immune response.
INTRODUCTION
In response to RNA virus infection, RIG-I–like receptors (RLRs) induce the aggregation of the mitochondrial antiviral signaling protein (MAVS) (1, 2) to initiate antiviral innate immune response (3). Aggregated MAVS further recruits signaling molecules TRADD (4), FADD, Ripk1 (5, 6), cFLIPL (7), and caspase-8 (8, 9) to form a large signaling complex—the MAVS signalosome (10), which eventually leads to the activation of two cytosolic kinases, TRAF family member-associated NFκB activator (TANK)–binding kinase 1 (TBK1) and IκB kinase (IKK), and their downstream transcription factors interferon regulatory factor 3 (IRF3) and nuclear factor κB (NFκB) (11). RNA virus infection induces the K63-linked ubiquitination of Ripk1 within the MAVS signalosome (8, 12). Ubiquitinated Ripk1 recruits the NFκB essential modulator (NEMO), a component required for the activation of TBK1 and IKK (12, 13). MAVS-mediated signaling is self-limiting. K63-linked ubiquitination of Ripk1 also renders Ripk1 susceptible to cleavage by the caspase-8/cFLIPL heterodimer within the signaling complex. The resultant 37KD Ripk1 fragment from this cleavage inhibits MAVS signaling (8).
The mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs) are the ER membranes at the mitochondria-ER contact sites (MERCs). MAM has attracted increased attention owing to its essential function in phospholipid biosynthesis, calcium and other ions transferring between ER and mitochondria, the mitochondria bioenergetics, and the initiation of autophagosome formation (14). In the event of RNA virus infection, the MAVS signalosome forms at the junction between MAM and mitochondria by recruiting RLRs and other signaling components into this subcellular location to form a signaling synapse (15). However, the detailed molecular mechanisms by which MAVS signaling is regulated at the innate immune synapse remain to be elucidated.
Protein geranylgeranylation is a posttranslational modification of proteins using the mevalonate pathway metabolite geranylgeranyl pyrophosphate (GGPP) as a ligand (16). Protein geranylgeranylation is catalyzed by protein geranylgeranyl transferase–I (GGTase-I). Using a mouse strain with myeloid deficiency of GGTase-I β subunit (Pggt1b), we report here that protein geranylgeranylation limits the MAVS-mediated antiviral innate immune response by targeting the Rho family small guanosine triphosphatase (GTPase) Rac1 to MAM. MAM-localized Rac1 engages directly with the MAVS signalosome and restricts the interaction between Trim31 and MAVS and thereby limits MAVS K63-linked ubiquitination and subsequent aggregation and activation. Furthermore, Rac1 also promotes the recruitment of caspase-8 and cFLIPL to the MAVS signalosome and hence facilitates the cleavage of ubiquitinated Ripk1 to terminate MAVS signaling.
RESULTS
Protein geranylgeranylation negatively regulates RLR-mediated antiviral innate immune response
To study whether protein geranylgeranylation regulates innate immune responses mediated by intracellular nucleic acid sensors, bone marrow–derived macrophages (BMDMs) from Pggt1bfl/fl Lyz2-Cre or Pggt1b+/+Lyz2-Cre mice were transfected with ligands to RIG-I, Sting, and cGAS. The transcription of type I interferons Ifnb1 and Ifna4, as well as proinflammatory cytokines Il6 and Tnf, was increased in Pggt1b-deficient cells compared to wild-type controls in response to stimulation with RIG-I ligands 5′ppp-dsRNA and polyinosinic:polycytidylic acid low molecular weight [poly(I:C) LMW], but not with Sting ligand 2′3′-cGAMP (cyclic guanosine monophosphate–adenosine monophosphate) or cGAS (cGAMP synthase) ligand ISD (interferon-stimulating DNA) (Fig. 1, A and B). Sendai virus (SeV, an RNA virus) infection–induced transcription of Ifnb1, Ifna4, Il6, and Tnf in Pggt1b-deficient BMDMs was also enhanced compared to wild-type controls (Fig. 1C) but not that induced by mouse cytomegalovirus (MCMV, a DNA virus) infection (fig. S1, A and B). The augmented production of cytokines was not caused by higher replication of SeV since the amount of the SeV-P gene was lower in Pggt1b-deficient BMDMs (Fig. 1C). The augmented transcription was consistent with elevated levels of cytokines in the supernatant of Pggt1b-deficient BMDMs infected with SeV (Fig. 1D). Together, these results indicate that protein geranylgeranylation suppresses MAVS but not Sting-mediated innate immune signaling pathways.
Fig. 1. Protein geranylgeranylation negatively regulates RLR-mediated antiviral innate immune response.
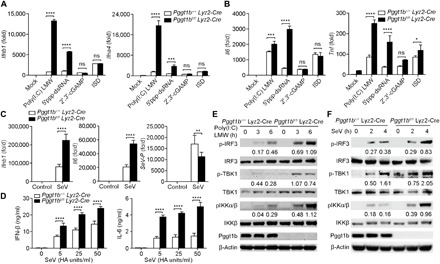
(A to C) Quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis of cytokine genes or SeV-P gene transcript abundance from Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre BMDMs after transfection of mock or RIG-I ligands [poly(I:C) LMW and 5′ppp-dsRNA (1 μg/ml)], Sting ligand 2′3′-cGAMP (8 μg/ml), and cGAS ligand ISD (1 μg/ml) with Lipofectamine for 4 hours (A and B), or after infection with SeV (50 HA (Hemagglutinin) units/ml in all succeeding experiments) for 4 hours (C); fold, fold change relative to wild-type negative control throughout. (D) Enzyme-linked immunosorbent assay (ELISA) measurement of interferon-β (IFN-β) and interleukin-6 (IL-6) in supernatants of Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre BMDMs infected with SeV for 24 hours. (E and F) Immunoblot analysis of phosphorylated (p-) or total IRF3, TBK1, IKK, and Pggt1b or β-actin (loading control throughout) in Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre BMDMs transfected with poly(I:C) LMW (E) or infected with SeV (F). Numbers below lanes indicate densitometry of phosphorylated IRF3, TBK1, or IKK relative to that of total IRF3, TBK1, or IKK, respectively. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 [two-way analysis of variance (ANOVA)]. ns, not significant. Data are representative of three independent experiments with three biological replicates.
Protein geranylgeranylation inhibits RLR downstream signaling
Consistent with augmented production of type I interferons and other cytokines, phosphorylation of IRF3, TBK1, and IKK was enhanced in Pggt1b-deficient BMDMs transfected with poly(I:C) LMW (Fig. 1E) or infected with SeV (Fig. 1F) compared to wild-type controls. However, phosphorylation of IRF3 and IKK was not enhanced in response to cGAS ligand ISD (fig. S1C) or infection with DNA virus MCMV (fig. S1D). We have previously shown that activation of the phosphoinositide 3-kinase (PI3Kinase)–Akt–Gsk3β signaling axis by Toll-like receptors was compromised in Pggt1b-deficient BMDMs (17). However, poly(I:C) LMW– or ISD-induced activation of Akt and Gsk3β was not substantially changed in Pggt1b-deficient BMDMs compared to wild-type controls (fig. S1, E and F). Together, these data suggest that protein geranylgeranylation inhibition of RLR-mediated innate immune signaling is independent of the PI3Kinase-Akt signaling axis.
Rho GTPase Rac1 inhibition of MAVS-mediated signaling depends on protein geranylgeranylation
To study the molecular mechanisms by which protein geranylgeranylation regulates MAVS signaling, we focused on GGTase-I substrates. Rho family small GTPases Rac1 and Rac2 are typical GGTase-I substrates. When wild-type, Rac1−/−, or Rac2−/− immortalized BMDMs (iBMDMs) were infected with SeV, only Rac1−/− but not Rac2−/− iBMDMs showed increased transcription of Ifnb1 and Il6 compared to wild-type controls, mimicking the phenotype of Pggt1b-deficient macrophages (Fig. 2, A to C). Phosphorylation of IRF3 and IKK in Rac1−/− iBMDM was also enhanced compared to that in wild-type controls (Fig. 2D). These results suggest that Rac1 but not Rac2 is an inhibitor of RLR signaling. To further evaluate the role of protein geranylgeranylation in Rac1 regulation of RLR signaling, Rac1−/− iBMDMs were reconstituted with various mutant forms of Rac1. Reconstitution with wild-type and the constitutively active G12V Rac1 inhibited SeV infection–induced Ifnb1 and Il6 transcription that correlated with more viral propagation, whereas the non-geranylgeranylated C189S (18) did not (Fig. 2, E to G) although the expression level of C189S Rac1 was much higher than its wild-type or G12V counterpart (Fig. 2H). The G12V/C189S double mutant failed to inhibit cytokine production in reconstituted cells (Fig. 3, E and F), indicating that protein geranylgeranylation is required for the inhibitory effects of Rac1. The wild-type, G12V, and C189S Rac1 mutants were also stably expressed in mouse embryonic fibroblasts (fig. S2A). Stable expression of wild-type and G12V Rac1 inhibited RIG-I ligand poly(I:C) LMW transfection–induced transcription of Ifnb1 and Il6, whereas the non-geranylgeranylated C189S mutant Rac1 failed to do so (fig. S2, B and C). Together, these data suggest that Rac1 inhibits MAVS signaling in a protein geranylgeranylation–dependent manner.
Fig. 2. MAM-localized Rac1 inhibits RLR signaling in a protein geranylgeranylation–dependent manner.
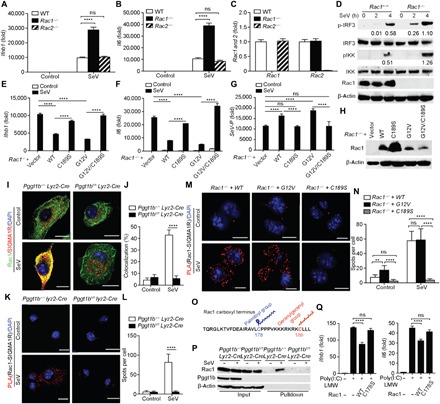
(A to C) Quantitative RT-PCR analysis of transcript abundance of Ifnb1, Il6, Rac1, and Rac2 in wild-type (WT), Rac1−/−, or Rac2−/− iBMDMs 4 hours after SeV infection. (D) Immunoblot of phospho-IRF3 and phospho-IKK in cell lysates of wild-type or Rac1−/− iBMDMs following SeV infection. Numbers below lanes indicate densitometry of phosphorylated IRF3 or IKK relative to that of total IRF3 or IKK, respectively. (E to H) Quantitative RT-PCR analysis of transcript abundance of Ifnb1, Il6, and SeV-P (E to G) and immunoblot of Rac1 (H) in Rac1−/− iBMDMs reconstituted with wild-type, C189S, G12V, or G12V/C189S of Rac1 4 hours after SeV infection. (I and J) Confocal microscopy of immunofluorescent staining of Rac1 (green) and SIGMA1R (red) in Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre BMDMs 4 hours after SeV infection. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bars, 10 μm (I). See each single channel in fig. S3J. The percentage of colocalization between Rac1 and SIGMA1R was calculated using the ImageJ software from at least 50 cells randomly selected from the immunostaining slides (J). (K to N) Representative PLA images of Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre BMDMs (K) or Rac1−/− iBMDMs reconstituted with wild-type, G12V, or C189S of Rac1 (M) 4 hours after SeV infection. The cells were probed with Rac1 and SIGMA1R antibodies. Red spots indicate the association of Rac1 with SIGMA1R. (K and M) Cells were counterstained with DAPI (blue). Scale bars, 10 μm. (L and N) Quantitation of PLA spots per cell in (K) and (M) (n = 20 cells). (O) Schematic display of the C-terminal amino acid sequence of Rac1 highlighting the C178 for palmitoylation and C189 for geranylgeranylation sites. (P) Immunoblots of Rac1 after S-palmitoylated protein assay (acyl-RAC; see Materials and Methods) of captured proteins (Pulldown) and inputs from Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre BMDM cell lysate following infection with or without SeV for 4 hours. (Q) Quantitative RT-PCR of Ifnb1 and Il6 abundance in MEF cells stably expressing wild-type and C178S mutant forms of Rac1 after transfection with poly(I:C) LMW. **P < 0.01, ****P < 0.0001 (two-way ANOVA). Data are representative of three independent experiments.
Fig. 3. Rac1 directly engages MAVS signalosome and inhibits MAVS signaling in a protein geranylgeranylation–dependent manner.
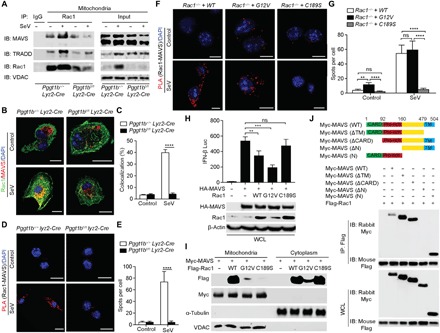
(A) Immunoblots (IB) of MAVS and TRADD in anti-Rac1 immunoprecipitates (IP) from the mitochondrial fraction of BMDMs infected with or without SeV. IgG, immunoglobulin G; VDAC, voltage-dependent anion channel. (B and C) Confocal microscopy of immunofluorescent staining of Rac1 (green) and MAVS (red) in BMDMs 4 hours after infection with SeV. Scale bars, 10 μm (B). See each single channel in fig. S5A. The percentage of colocalization of Rac1 and MAVS was calculated using ImageJ (C). (D to G) Representative PLA confocal images for Rac1 and MAVS in Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre BMDMs (D) or Rac1−/− iBMDMs reconstituted with wild-type, G12V, or C189S of Rac1 (F) 4 hours after SeV infection. Cells were counterstained with DAPI (blue). Scale bars, 10 μm (D and F). (E and G) Quantification of PLA spots per cell in (D) and (F) (n = 20 cells). (H) Luciferase activity in 293T cell lysate 24 hours after transfection with an IFN-β luciferase reporter, HA-MAVS together with wild-type, G12V, or C189S Rac1 (top). Immunoblots of HA-MAVS and Rac1 in whole-cell lysates (WCL) of transfected 293T cells (bottom). (I) Immunoblot of crude mitochondrial and cytoplasm fractions from 293T cells cotransfected with Myc-MAVS and Flag-Rac1 wild-type, G12V, or C189S mutants with indicated antibodies. (J) Schematic of MAVS (wild-type) and its truncation mutants (top), and coimmunoprecipitation analysis of the interaction of Flag-Rac1 with Myc-MAVS or its truncation mutants in 293T cells (bottom). **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). Data are representative of three independent experiments.
Deficiency of another Rho family member, RhoA, in BMDMs did not significantly increase the innate immune response to SeV infection (fig. S2, D to F). Expression of RhoA was not completely abrogated in RhoAfl/fl Lyz2-Cre BMDMs presumably owing to counterselection of RhoA-deficient cells (fig. S2G). When wild-type BMDMs were treated with the clostridial glucosylating lethal toxin TcsL from Clostridium sordellii that inactivates Rac and Cdc42 (19), production of type I interferons in response to SeV infection was enhanced (fig. S2H). Treatment of wild-type BMDMs with a Rac-specific inhibitor, EHT1864 (20), enhanced type I interferon transcription (fig. S2I), whereas treatment of BMDMs with a Cdc42-specific inhibitor, ML141 (21), did not affect macrophages’ innate immune response to viral infection (fig. S2J), suggesting that Cdc42 is not involved in the regulation of innate immune response to viral infection. Together, Rac1 likely plays a dominant role in limiting RLR signaling.
Viral infection–induced MAM translocation of Rac1 depends on protein geranylgeranylation and subsequent palmitoylation
Mitochondrial anchorage of MAVS is essential for its innate immune function (2). Rho GTPases are conventionally thought to be either membrane-associated or cytosolic when bound to Rho-GDI (Rho GDP-dissociation inhibitor) (22). Since MAVS-mediated innate immune response happens at the MAM-mitochondria junction (15), we speculated that Rac1 might be recruited to MAM to engage with the MAVS signalosome. To test this hypothesis, Pggt1bfl/fl Lyz2-Cre or Pggt1b+/+Lyz2-Cre BMDMs infected with or without SeV were fractionated into crude mitochondria or cytosolic fractions and subjected to Western blot. Two MAM markers, FACL4 and SIGMA1R (23, 24), were readily detected in the crude mitochondrial fraction (fig. S3A), indicating that the crude mitochondrial fraction contains MAM protein components as reported earlier (15). Rac1 was also recovered in the crude mitochondrial fraction from wild-type, but to a much lower level than that from Pggt1b-deficient BMDMs (fig. S3A). The amount of Rac1 in the crude mitochondrial fraction was further increased upon SeV infection only in wild-type but not mutant BMDMs, suggesting that SeV infection–induced enrichment of Rac1 in the crude mitochondrial fraction depends on protein geranylgeranylation (fig. S3, A and B). Nevertheless, RIG-I enrichment in the mitochondrial crude extract was not affected by Pggt1b deficiency, suggesting that protein geranylgeranylation regulates RLR signaling downstream of RIG-I (fig. S3, A to C). It is notable that when cells were fractionated, the total amount of Rac1 in both the crude mitochondrial and cytosolic fractions was less in Pggt1b-deficient macrophages compared to wild-type controls. This raised the possibility that Rac1 expression or stability is regulated by protein geranylgeranylation. Western blot of total cell lysate with an anti-Rac1 antibody indicated that there was equal amount of Rac1 in Pggt1b-deficient cells and wild-type control cells (fig. S3, D and E). We believe that the phenomenon of lower recovery of Rac1 from Pggt1b-deficient crude mitochondrial and cytosolic fractions was due to fractionation of part of the non-geranylgeranylated Rac1 into the nuclear fraction. RIG-I ligand poly(I:C) LMW transfection induced similar effects of Rac1 enrichment in the crude mitochondrial fraction from wild-type BMDMs, and such enrichment happened in a protein geranylgeranylation–dependent manner (fig. S3, F and G). In line with the biochemical results, confocal microscopy analysis showed that Rac1 was brought to the proximity of mitochondria in a Pggt1b-dependent manner (fig. S3, H and I). To further verify whether Rac1 in the crude mitochondrial fraction is actually on MAMs, we used confocal microscopy to determine whether Rac1 colocalizes with MAM markers SIGMA1R and FACL4. Confocal microscopy analysis indicates that Rac1 was randomly distributed in the unprovoked cells. SeV infection induced colocalization of Rac1 with MAM markers SIGMA1R and FACL4. Such colocalization was abrogated in the absence of Pggt1b (Fig. 2, I and J, and fig. S3, J to L). These observations indicate that Rac1 translocates into MAMs upon SeV infection in a protein geranylgeranylation–dependent manner.
The chaperon protein SIGMA1R is a MAM marker that has been shown to interact with Rac1 (25). We speculated that SIGMA1R might serve as an anchor to recruit Rac1 into MAM upon viral infection. We used in situ proximity ligation assay (PLA) to assess the interaction between Rac1 and SIGMA1R. Rac1 and SIGMA1R were inducibly associated with each other upon SeV infection in wild-type but not Pggt1b-deficient BMDMs (Fig. 2, K and L). PLA analysis of Rac1−/− iBMDMs reconstituted with various mutant forms of Rac1 showed that only wild-type and G12V but not the non-geranylgeranylated C189S mutant Rac1 could interact with SIGMA1R in macrophages upon viral infection (Fig. 2, M and N), further supporting the notion that viral infection–induced Rac1 interaction with SIGMA1R depends on protein geranylgeranylation of Rac1. Together, Rac1 is recruited into MAM upon viral infection where it is associated with MAM-resident protein SIGMA1R in a protein geranylgeranylation–dependent manner.
SIGMA1R receptor agonists such as N,N-dimethyltryptamine (NN-DMT) can activate SIGMA1R signaling and modulate innate immune response (26). NN-DMT treatment did not alter SeV-induced cytokine production (fig. S4, A to C). However, RNAi knockdown of SIGMA1R in primary BMDMs led to enhanced innate immune response to SeV infection (fig. S4, D to G). Together, it is highly likely that the chaperon but not the signaling function of SIGMA1R is important for anchoring Rac1 to MAM to execute the immune suppressive activity.
The MAMs are enriched with cholesterols (27). Protein palmitoylation facilitates modified protein translocation to the cholesterol-enriched microdomains within MAMs (28). Viral infection causes ER stress and increases protein palmitoylation (29). It has been reported that Rac1 can be palmitoylated at Cys178 (Fig. 2O) that facilitates Rac1 translocation into cholesterol-enriched lipid microdomains and that Rac1 palmitoylation depends on prior geranylgeranylation (30). On the basis of these previous findings, we speculated that viral infection might induce the palmitoylation and subsequent translocation of Rac1 to the cholesterol-rich MAM. SeV infection induced palmitoylation of Rac1 in wild-type but not Pggt1b-deficient BMDMs (Fig. 2P). When stably expressed in mouse embryonic fibroblast (MEF) cells, C178S mutant Rac1 failed to inhibit transfected poly(I:C) LMW–induced Ifnb1 and Il6 transcription (Fig. 2Q). Consistent with these findings, confocal microscopy showed that C178S mutant Rac1 failed to colocalize with MitoTracker or the MAM marker FACL4 upon SeV infection (fig. S4, H and I). Together, these data indicate that protein geranylgeranylation and subsequent palmitoylation target Rac1 into MAM upon viral infection.
MAM-localized Rac1 directly engages MAVS signalosome and inhibits MAVS signaling in a protein geranylgeranylation–dependent manner
When activated, MAVS forms a signaling complex on the mitochondria that includes TRADD, termed TRADDosome (4). We reasoned that MAM-localized Rac1 might directly interact with the MAVS signaling complex on the mitochondria given the close proximity between MAM and mitochondria (14). Crude mitochondrial fractions were immunoprecipitated with an anti-Rac1 antibody. Western blot analyses showed that Rac1 was coimmunoprecipitated with both MAVS and TRADD in wild-type BMDMs. This association was further enhanced following SeV infection (Fig. 3A). However, the interaction was significantly decreased in Pggt1b-deficient BMDMs, presumably because less Rac1 was present in the MAM in the absence of protein geranylgeranylation (Fig. 3A). Confocal microscopy corroborated the biochemical results that, upon SeV infection, Rac1 colocalized with MAVS in wild-type but not Pggt1b-deficient macrophages (Fig. 3, B and C, and fig. S5A). Consistently, PLA analysis indicated that the association of Rac1 with MAVS was abrogated in the absence of Pggt1b (Fig. 3, D and E). Additional PLA analysis of the Rac1-deficient iBMDMs reconstituted with various mutant forms of Rac1 showed that SeV infection induced the association between endogenous MAVS with wild-type and G12V Rac1 but not the non-geranylgeranylated C189S mutant Rac1 (Fig. 3, F and G). Together, these data indicate that Rac1 engagement with MAVS signalosome depends on protein geranylgeranylation.
Overexpression of MAVS in the absence of viral infection can activate TBK1 and IKK and induce transcription of type I interferons (2). When ectopically expressed in 293T cells together with MAVS and an interferon-β (IFN-β) luciferase reporter, wild-type and the constitutively active G12V but not the C189S non-geranylgeranylated Rac1 mutant inhibited MAVS-induced IFN-β luciferase reporter activity (Fig. 3H). Unlike the wild-type and G12V Rac1, the non- geranylgeranylated C189S mutant Rac1 was not associated with MAM-containing crude mitochondrial fractions when ectopically expressed in 293T cells (Fig. 3I). In addition to MAVS, overexpression of the MAVS downstream signaling components TBK1 or IRF3 can also activate IFN-β reporters. When coexpressed, Rac1 inhibited MAVS- but not TBK1- or IRF3-induced IFN-β luciferase reporter activity (fig. S5B). These results imply that Rac1 acts on MAVS, but not its downstream signaling components, to inhibit the RLR-mediated innate immune response.
It is of note that the constitutively active G12V Rac1 seemed to associate more with MAVS than the wild-type Rac1 in PLA (Fig. 3, F and G). When ectopically expressed, G12V Rac1 more potently inhibited MAVS-induced IFN-β reporter activity than the wild-type Rac1 (Fig. 3H), suggesting that the Rac1 effector function likely plays a role in its MAVS-inhibiting activity. To further explore this, MAVS−/− iBMDMs or those reconstituted with wild-type MAVS (MAVS−/− + MAVS) were pretreated with the clostridial glucosylation toxin TcsL that specifically inactivates Rac and Cdc42 effector function by modifying their switch region (31). Since Rac2 and Cdc42 are not involved in MAVS signaling (Fig. 2, A and B, and fig. S2J), the outcome of TcsL treatment in this experimental setting was most likely attributed to the inactivation of Rac1. TcsL treatment led to increased Ifnb1 and Il6 transcription upon SeV infection and enhanced activation of IRF3 and IKK only in the MAVS-reconstituted, but not the MAVS−/− parental iBMDM cell line (fig. S5, C to E). These observations further demonstrate that Rac1 effector function is required for inhibiting MAVS signaling. Using different truncated mutant forms of MAVS, the portion of MAVS interacting with Rac1 has been mapped to the proline-rich region (Fig. 3J). Together, these data indicate that MAM-localized Rac1 directly engages with the MAVS signalosome and that Rac1 inhibition of MAVS signaling depends on protein geranylgeranylation and its effector function.
MAM-localized Rac1 inhibits MAVS aggregation and activation by limiting MAVS K63 ubiquitination
When activated, MAVS forms functional, prion-like aggregates to activate downstream signaling kinases such as TBK1 and IKK (3). We reasoned that augmented MAVS signaling in the absence of protein geranylgeranylation might be caused by increased MAVS aggregation. MAVS aggregates are resistant to detergent in radioimmunoprecipitation assay (RIPA) buffer and therefore would not be recovered in RIPA-soluble extracts (3). Western blot detected less MAVS in RIPA buffer extracts of Pggt1b-deficient BMDMs compared to that of wild-type control at 3 hours following SeV infection (Fig. 4, A and B). This observation suggests that MAVS aggregates form with a faster kinetics in Pggt1b-deficient BMDMs than wild-type control cells. To further inspect MAVS aggregation, mitochondrial crude extracts were resolved by semidenaturing detergent agarose gel electrophoresis (SDD-AGE) (3). Before SeV infection, there was a substantial amount of aggregated MAVS in the Pggt1b-deficient BMDMs compared to almost none in wild-type control BMDMs (Fig. 4C). Viral infection increased the amount of MAVS aggregates in both genotypes. However, there were substantially more viral infection–induced MAVS aggregates in the Pggt1b-deficient BMDMs than in wild-type controls (Fig. 4C). Inactivation of Rac1 effector function with the clostridial toxin TcsL also led to increased MAVS aggregation in MAVS-deficient iBMDMs reconstituted with a hemagglutinin (HA)–tagged wild-type MAVS (fig. S6A). SDD-AGE analysis of wild-type and Rac1−/− iBMDMs indicated that Rac1 deficiency promotes SeV-induced MAVS aggregation. When Rac1−/− iBMDMs were reconstituted with various forms of Rac1, only wild-type or G12V but not the non-geranylgeranylated C189S Rac1 inhibited SeV-induced MAVS aggregation (fig. S6B). Together, we conclude that Rac1 inhibits SeV-induced MAVS aggregation in a protein geranylgeranylation–dependent manner.
Fig. 4. MAM-localized Rac1 inhibits Trim31-MAVS interaction and subsequent MAVS Lys63-linked ubiquitination, aggregation, and activation upon RNA virus infection.
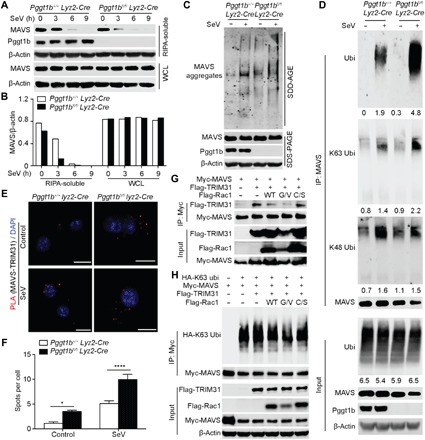
(A) Immunoblot of MAVS and Pggt1b in the RIPA-soluble fractions and WCL of BMDMs infected with SeV. (B) Densitometry of MAVS relative to that of β-actin in the RIPA-soluble fractions and WCL of (A). (C) Anti-MAVS immunoblot of crude mitochondrial extracts of BMDMs infected with SeV for 4 hours and resolved by SDD-AGE (top) or total cell lysate of BMDMs resolved by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (bottom). (D) Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre BMDMs infected with SeV for 4 hours were lysed, boiled, and sheared by sonication. After centrifugation, supernatants were immunoprecipitated with an anti-MAVS antibody and probed with pan-ubiquitin, K63-, or K48-ubiquitin–specific antibodies. Numbers below lanes indicate densitometry relative to β-actin. (E and F) PLA confocal images for MAVS and TRIM31 (red) in BMDMs 4 hours after SeV infection. Cells were counterstained with DAPI (blue). Scale bars, 10 μm (E); (F) quantification of PLA foci per cell in (E) (n = 20 cells), *P < 0.05, ****P < 0.0001 (two-way ANOVA). (G) Immunoblot of total cell lysate (bottom) or anti-Myc immunoprecipitates (top) of 293T cells transfected with Myc-tagged Mavs and Flag-tagged Trim31 together with the wild-type, G12V, or C189S mutant form of Rac1. (H) Immunoblot of total cell lysate (bottom) or anti-Myc immunoprecipitates (top) of 293T cells transfected with Myc-tagged Mavs, Flag-tagged Trim31, and HA-tagged K63-ubiqutin together with the wild-type, G12V, or C189S mutant form of Rac1. Data are representative of three independent biological experiments.
It has been shown recently that K63-linked ubiquitination of MAVS plays an important role in promoting MAVS aggregation (32). We speculated that enhanced MAVS aggregation might be due to increased K63-linked MAVS ubiquitination in the absence of Pggt1b. To test this hypothesis, MAVS immunoprecipitates from total cell lysate of SeV- or mock-infected BMDMs were probed with an anti-ubiquitin antibody. SeV-induced MAVS ubiquitination was substantially enhanced in Pggt1b-deficient BMDMs compared to wild-type controls. Only K63-linked, but not K48-linked, ubiquitination of MAVS was enhanced in Pggt1b-deficient cells (Fig. 4D). These observations indicate that protein geranylgeranylation inhibits MAVS aggregation, at least in part, by suppressing MAVS K63-linked ubiquitination.
To explore whether protein geranylgeranylation inhibits MAVS ubiquitination through Rac1 effector function, MAVS was immunoprecipitated from wild-type primary BMDMs pretreated with or without TcsL followed by infection with mock or SeV. Treatment with TcsL, which presumably inactivates Rac1, caused increased ubiquitination of MAVS in comparison to untreated cells. More specifically, the increased MAVS ubiquitination was Lys63- but not Lys48-linked (fig. S6C). Together, these data further support the idea that Rac1 inhibits SeV-induced MAVS K63-linked ubiquitination and aggregation.
MAM-localized Rac1 limits the interaction between MAVS and the E3 ligase Trim31
Trim31, a ring domain–containing E3 ligase that belongs to the tripartite motif family proteins, has recently been shown to catalyze MAVS K63 ubiquitination upon RNA virus infection (32). The increased ubiquitination of MAVS in Pggt1b-deficient cells prompted us to investigate whether MAM-localized Rac1 affects the interaction between Trim31 and MAVS in a protein geranylgeranylation–dependent manner. Before viral infection, there was a stronger association between endogenous Trim31 and MAVS in Pggt1b-deficient cells compared to almost none in wild-type controls (Fig. 4, E and F). SeV infection induced a substantially higher amount of Trim31-MAVS foci in Pggt1b-deficient cells compared to wild-type controls (Fig. 4, E and F). When coexpressed in 293T cells, a robust interaction between Trim31 and MAVS was detected by Western blot. This interaction was inhibited by wild-type and, more profoundly, the G12V mutant Rac1, but not by the non-geranylgeranylated C189S Rac1 (Fig. 4G). To further investigate the role of Rac1 in the regulation of Trim31-MAVS interaction, MAVS-deficient iBMDMs reconstituted with Myc-tagged MAVS (Myc-MAVS iBMDMs) were pretreated with TcsL to inactivate Rac1 effector function, followed by infection with mock or SeV. MAVS was immunoprecipitated with anti-Myc antibody. Western blot analysis showed that inactivation of Rac1 by TcsL substantially enhanced the interaction between Trim31 and MAVS (fig. S6D), suggesting that inhibition of the interaction between Trim31 and MAVS by Rac1 depends on its effector function. PLA analysis of Rac1−/− iBMDMs reconstituted with various mutant forms of Rac1 indicated that only wild-type and G12V Rac1 efficiently inhibited MAVS-Trim31 association, whereas C189S Rac1 failed to do so (fig. S6, E and F). Consistently, when K63-ubiquitin was included in the ectopic expression combination in 293T cells, Trim31-mediated K63-linked ubiquitination of MAVS was inhibited by wild-type and G12V Rac1, but not by the non-geranylgeranylated C189S Rac1 (Fig. 4H). Together, we conclude that protein geranylgeranylation enables Rac1 inhibition of MAVS ubiquitination, aggregation, and activation by limiting the MAVS-Trim31 interaction.
Rac1 facilitates the recruitment of caspase-8 and cFLIPL to the MAVS signalosome and promotes Ripk1 cleavage
Within the MAVS signalosome, Ripk1 is K63-ubiquitinated upon RNA virus infection. Ubiquitinated Ripk1 promotes the MAVS-mediated innate immune response (5). Ubiquitination also renders Ripk1 susceptible to cleavage by the caspase-8–cFLIPL heterodimer into a 37KD fragment that inhibits MAVS signaling (8). SeV infection–induced Ripk1 cleavage into the 37KD fragment was impaired in Pggt1b-deficient macrophages compared to wild-type controls (Fig. 5A). Consistently, inactivation of Rac1 with the bacterial toxin TcsL also compromised Ripk1 cleavage upon SeV infection (fig. S7, A and B). These observations imply that protein geranylgeranylation and Rac1 restrict MAVS signaling by promoting Ripk1 cleavage.
Fig. 5. MAM-localized Rac1 facilitates the recruitment of caspase-8 and cFLIPL to MAVS signalosome and promotes the cleavage of Ripk1.
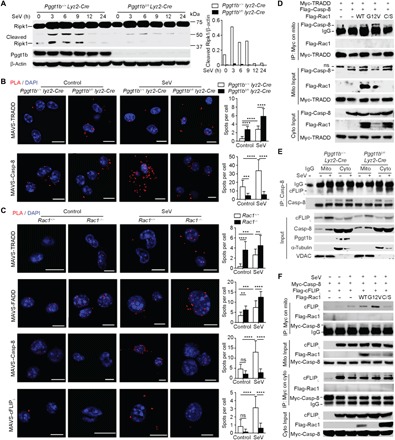
(A) Immunoblot with anti-Ripk1 of cell lysate from BMDMs at different time points after SeV infection and the densitometry of cleaved 37KD Ripk1 fragment relative to that of β-actin in the WCL. (B and C) Representative PLA confocal images for MAVS and TRADD and Casp-8 in Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre BMDMs (B) or MAVS and TRADD, FADD, Casp-8, and cFLIPL in wild-type (Rac1+/+) and Rac1−/− iBMDMs (C) 4 hours after SeV infection. Cells were counterstained with DAPI (blue). Scale bars, 10 μm. Quantification of PLA spots per cell is shown in the right panels of (B) and (C) (n = 20 cells). (D) Immunoblot of input (middle and bottom) or anti-Myc immunoprecipitates (top) in mitochondrial fractions of 293T cells transfected with Myc-tagged TRADD and Flag-tagged Casp-8 together with the wild-type, G12V, or C189S mutant form of Rac1. (E) Immunoblot of anti–caspase-8 immunoprecipitates and input from mitochondrial (mito) or cytosolic (cyto) fractions of BMDMs 4 hours after SeV infection. (F) Immunoblot of input (bottom) or anti-Myc immunoprecipitates (top) in mitochondrial (left) or cytosolic (right) fractions of 293T cells transfected with Myc-tagged caspase-8 and Flag-tagged cFLIPL together with the wild-type, G12V, or C189S mutant form of Rac1. **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). Data are representative of three independent experiments.
When activated, aggregated MAVS recruits Ripk1, TRADD, FADD, caspase-8, and cFLIPL to form the MAVS signaling complex—the TRADDosome. Consistent with the observation that Pggt1b deficiency enhances MAVS aggregation and activation, the recruitment of TRADD to MAVS complex was increased in the absence of Pggt1b (Fig. 5B). Nevertheless, the recruitment of Ripk1 to MAVS was similar between wild-type and Pggt1b-deficient cells (fig. S7, C and D). Contrary to TRADD, the recruitment of caspase-8 to the MAVS complex upon SeV infection was almost abrogated in the absence of protein geranylgeranylation (Fig. 5B). Similar to Pggt1b-deficient cells, the recruitment of TRADD and FADD to the MAVS complex was also enhanced in Rac1-deficient iBMDMs. However, the recruitment of caspase-8 and cFLIPL was almost abrogated in the absence of Rac1 upon SeV infection (Fig. 5C). Consistently, when ectopically expressed in 293T cells, the wild-type and G12V but not C189S mutant Rac1 promoted the association between TRADD and caspase-8 on mitochondria (Fig. 5D). These data suggest that though hyperactivation of MAVS in the absence of Pggt1b or Rac1 leads to increased recruitment of TRADD and FADD into the MAVS signalosome, the recruitment of negative regulators, caspase-8 and cFLIPL, to the TRADDosome depends on Rac1 and protein geranylgeranylation.
When caspase-8 was immunoprecipitated from mitochondrial extracts, the interaction of caspase-8 with cFLIPL was abrogated in the absence of Pggt1b (Fig. 5E). It is notable that the association between caspase-8 and cFLIPL in the cytosol was not affected by Pggt1b deficiency (Fig. 5E). These observations prompted us to investigate whether Rac1 also promotes the association of caspase-8 with cFLIPL on mitochondria. Coexpression of wild-type or the constitutively active form of G12V Rac1 substantially increased the interaction between ectopically expressed caspase-8 and cFLIPL on mitochondria but not that in the cytoplasm, whereas the non-geranylgeranylated C189S mutant Rac1 had no such effect (Fig. 5F). This result indicates that Rac1 inhibits MAVS signaling by promoting the association of caspase-8 with cFLIPL on mitochondria. Consistent with the decreased Ripk1 cleavage (Fig. 5A), the MAVS downstream signaling complex immunoprecipitated with the anti-TANK antibody (8) was associated with more high–molecular weight species of Ripk1 (presumably ubiquitinated forms) and NEMO in Pggt1b-deficient BMDMs (fig. S7E), leading to sustained MAVS signaling to TBK1 and IKK (8). Together, we conclude that protein geranylgeranylation licenses Rac1 inhibition of MAVS signaling by facilitating the recruitment of caspase-8 and cFLIPL to the MAVS complex and by promoting the association of caspase-8 with cFLIPL on mitochondria to enable Ripk1 cleavage.
Myeloid deficiency of Pggt1b improved mice survival upon lethal influenza A virus infection
Pandemic, endemic, and seasonal influenza A virus infection cause severe morbidity and mortality (33). Alveolar macrophages are among the first line of innate immune cells counteracting viral infection in the respiratory tract. Lung alveolar macrophages (mAMs) from Pggt1bfl/fl Lyz2-Cre or Pggt1b+/+Lyz2-Cre mice were infected with influenza A/Puerto Rico/8/1934 H1N1 (PR8) virus. The transcription of type I interferons and proinflammatory cytokines was significantly augmented in Pggt1b-deficient mAMs infected with PR8 virus compared to wild-type control cells (Fig. 6, A and B). Pggt1b-deficient BMDMs infected with PR8 also produced a higher amount of type I interferons and proinflammatory cytokines (fig. S8, A and B). When infected with PR8-mNeon influenza A virus (34) at a low multiplicity of infection (MOI), the propagation of virus [assessed by the number of mNeon fluorescence-positive mAMs (Fig. 6, C and D) or by reverse transcription polymerase chain reaction (RT-PCR) quantitation of viral M1 and NS1 genes (fig. S8C)] was impaired in Pggt1b-deficient BMDMs compared to wild-type controls. In alignment with these in vitro observations, Pggt1bfl/flLyz2-Cre mice showed improved survival upon intranasal infection with a lethal dose of PR8 compared to wild-type control mice (Fig. 6E). At the early phase (days 2 to 5) after infection, there was significantly more body weight loss in Pggt1bfl/fl Lyz2-Cre versus control mice (Fig. 6F, days 1 to 5), suggesting a more severe inflammatory response in these mice. Lung virus titers were lower in Pggt1bfl/fl Lyz2-Cre mice than in wild-type control mice on day 3 after infection (Fig. 6G). Consistent with that, IFN-β and interleukin-6 (IL-6) levels in the lung homogenates were higher on day 3 following infection in Pggt1bfl/fl Lyz2-Cre mice compared to wild-type control mice (Fig. 6, H and I). Together, these data indicate that myeloid deficiency of protein geranylgeranylation enhances innate immunity and improves survival upon lethal dose of influenza A virus challenge.
Fig. 6. Myeloid Pggt1b deficiency better protects mice from lethal PR8 challenge.
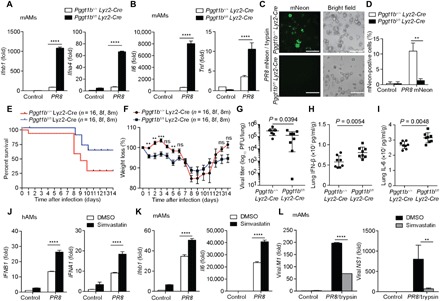
(A and B) Quantitative RT-PCR analysis of Ifnb1, Ifna4, Il6, and Tnf in murine alveolar macrophages (mAMs) 9 hours after acute infection with influenza A virus (PR8, 1 MOI). (C and D) Fluorescence microscopy of mAMs 24 hours after infection with a PR8-mNeon strain of (0.01 MOI) containing TPCK-trypsin (1 μg/ml) (C) and percentage of mNeon-positive cells (D). (E and F) Survival curves (E) and body weights (F) of Pggt1b+/+Lyz2-Cre or Pggt1bfl/f Lyz2-Cre mice infected with 50 plaque-forming units (PFU) PR8 (n = 16); results are pooled together from two independent biological experiments. (G to I) Viral titer (G), IFN-β (H), and IL-6 (I) levels in lung homogenates from mice 3 days after infection with 50 PFU of PR8 (n = 8 per group). (J) Quantitative RT-PCR analysis of IFNB1 and IFNA1 in mock or 10 μM simvastatin-treated human lung alveolar macrophages (hAMs) 9 hours after infection with 1 MOI PR8. DMSO, dimethyl sulfoxide. (K) Quantitative RT-PCR of Ifnb1 and Il6 in mouse lung alveolar macrophages (mAMs) pretreated with 10 μM simvastatin overnight followed by PR8 (1 MOI) infection for 9 hours. (L) Quantitative RT-PCR of IAV viral genes M1 and NS1 in mAMs pretreated with simvastatin overnight followed by infection with PR8 (0.01 MOI) containing TPCK-trypsin (1 μg/ml) for 24 hours. **P <0.01, ***P <0.001, ****P <0.0001 (two-way ANOVA). Data are representative of three independent biological experiments.
The protein geranylgeranylation ligand GGPP is a metabolite of the mevalonate pathway. Inhibition of the mevalonate pathway with statins may decrease the abundance of GGPP in the cell and impede protein geranylgeranylation. Simvastatin-treated mouse BMDMs showed enhanced transcription of Infb1 and Il6 upon PR8 infection (fig. S8D). Similarly, when infected with PR8, human and mouse lung alveolar macrophages (hAMs and mAMs) displayed a higher level of transcription of type I interferons and IL-6 following treatment with simvastatin compared to nontreated control cells (Fig. 6, J and K). The increase of cytokine production following simvastatin treatment was not as robust as in Pggt1b-deficient cells. This is likely due to the fact that Pggt1b expression is almost abrogated in Pggt1b-deficient cells, whereas simvastatin may not completely deplete GGPP in treated cells. PR8 expansion in mouse alveolar macrophages was also inhibited by simvastatin treatment (Fig. 6L). It is known that statins have antiviral activity through various mechanisms (35). Our results indicate that part of the antiviral effect of statins may be attributed to its inhibition of GGPP production, impairment of protein geranylgeranylation, and hyperinduction of cytokines.
DISCUSSION
The molecular mechanisms underlying the activation of MAVS-mediated antiviral innate immunity have been extensively studied. However, how such innate immune response is optimally controlled to prevent rampant inflammation remains obscure. Here, we identified protein geranylgeranylation as a crucial molecular mechanism in limiting the antiviral innate immune response. Protein geranylgeranylation and subsequent palmitoylation target Rac1 into the MAMs. MAM localization brings Rac1 to the proximity of MAVS on mitochondria. MAM-associated Rac1 is incorporated into the MAVS signaling complex and inhibits MAVS K63-linked ubiquitination, aggregation, and activation by inhibiting the interaction between MAVS and the E3 ligase Trim31. MAM-associated Rac1 also facilitates the recruitment of caspase-8 and cFLIPL to the MAVS signaling complex and promotes the association between caspase-8 and cFLIPL that cleaves Ripk1 to terminate MAVS signaling. A graphic summary is shown in fig.S9.
MAVS aggregation plays a central role in RLR-mediated antiviral immune response (3). For many years since the discovery of MAVS, the molecular mechanisms that prevent spontaneous MAVS aggregation on mitochondria in unprovoked cells remain enigmatic. We have observed that, in the absence of Pggt1b or Rac1, MAVS forms aggregates spontaneously without viral infection. Consistent with this observation, elevated levels of activated TBK1 were detected in unprovoked Pggt1b-deficient macrophages but not in wild-type control cells. Although this elevated level of TBK1 activities in Pggt1b-deficient cells is not sufficient to drive detectable transcription of type I interferons and proinflammatory cytokines, it might greatly diminish the threshold to initiate an antiviral innate immune response. Our results support the notion that protein geranylgeranylation, together with the GGTase-I substrate Rac1, constitutes a key molecular mechanism in keeping MAVS from spontaneous aggregation and activation on mitochondria.
MAVS-orchestrated signaling is self-limiting through the function of caspase-8 (8) and cFLIPL (7). MAVS signaling resembles that of death receptor in which death domain–containing proteins TRADD, FADD, Ripk1, caspase-8, and cFLIPL are recruited to MAVS, forming a complex termed TRADDosome (4). Upon activation, TRADD and FADD are recruited into the MAVS signalosome (1, 4). FADD further recruits caspase-8 (8) and cFLIPL through the death effector domain homophilic interaction to form a heterodimer that cleaves Ripk1 and terminates MAVS signaling. Consistently, cells deficient for caspase-8 or cFLIPL showed an enhanced innate immune response to SeV infection or transfected RNA (7, 8). We showed here that Rac1 is an integral component of the MAVS signalosome. MAM-localized Rac1 is required for the recruitment of caspase-8 and cFLIPL to TRADDosome. Rac1 promotes the heterodimerization between caspase-8 and cFLIPL within the TRADDosome and facilitates subsequent Ripk1 cleavage. Our results indicated that the self-limiting capacity of MAVS signalosome depends on Rac1 and protein geranylgeranylation.
Our data elucidated the molecular mechanisms by which protein geranylgeranylation suppresses innate immunity to RNA virus infection. Protein geranylgeranylation uses GGPP, a mevalonate pathway metabolite, as a ligand. Treatment of human and mouse alveolar macrophages with statins enhances innate immunity to influenza A virus infection. Statins have been shown with antiviral activities through various mechanisms (35). Our results indicate that at least part of the statins’ antiviral effects can be attributed to inhibition of protein geranylgeranylation.
MATERIALS AND METHODS
Mice
Pggt1bfl/fl Lyz2-Cre mice were described previously (17). Rhoafl/fl Lyz2Cre mouse bones were provided by M. Kloc of Houston Methodist Hospital. Littermate controls were used in the experiments. Mouse strains were maintained under specific pathogen–free conditions in the animal facility at Duke University, and the animal protocols were carried out in accordance with the guidelines set forth by the Institutional Animal Care and Use Committee of Duke University.
Transfections of BMDMs
Bone marrow cells from wild-type and various knockout mice were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) and 20% L929 supernatants. For transfection, Lipofectamine 2000 reagents (Invitrogen) were used. Before transfection, DMEM was replaced with serum-free media and 5′ppp-dsRNA (InvivoGen, catalog no. tlrl-3prna), poly(I:C) LMW (InvivoGen, catalog no. tlrl-picw), 2′3′-cGAMP (InvivoGen, catalog no. tlrl-nacga23), ISD (InvivoGen, catalog no. tlrl-isdn), or other stimulators-lipid complex was added to cells. After 4-hour incubation, cells were collected for immunoblot assay or RT-PCR. For Sigma1r knockdown, BMDMs were transfected with scramble small interfering RNA (siRNA) or siGENOME mouse Sigma1r siRNA (Dharmacon, SMARTpool, catalog no. 18391). After 48-hour incubation, transfected cells were infected with SeV (50 HA units/ml) for 4 hours and cells were lysed for RT-PCR.
Isolation of human and mouse alveolar macrophages
Human bronchoalveolar lavage (BAL) was collected according to a protocol approved by the Duke Institutional Review Board committee as described previously (36). Mouse BAL was collected as described previously (37). Bronchoalveolar cells were separated from around 10 ml of lavage fluid by centrifugation at 250g for 7 min. The cell pellet was washed twice with phosphate-buffered saline (PBS) and resuspended in RPMI 1640 medium [10% heat-inactivated FBS, penicillin (100 U/ml), and streptomycin (100 mg/ml), supplemented with 2 mM glutamine]. Human and murine alveolar macrophages were purified by adhering for 90 min at 37°C under 5% CO2 in 150-cm2 culture flasks. The majority of the adhesive cells were alveolar macrophages.
BMDM and MEF cell immortalization and reconstitution
Rac1−/−, Rac2−/−, or MAVS−/− iBMDMs were established according to a protocol described previously (17). For reconstitution experiments, iBMDMs were infected with retrovirus (pMSCV, Clontech) harboring complementary DNAs (cDNAs) encoding Myc- or Flag-tagged mouse MAVS or Flag-tagged mouse Rac1 wild-type, constitutively active (G12V), and C189S mutant. The iBMDM culture was selected in BMDM medium with puromycin (5 μg/ml). For MEF cells stably expressing various Rac1 mutants, wild-type C57BL/6 MEF cells were infected with retrovirus harboring cDNAs encoding wild-type, or G12V and C189S mutant of Rac1. The MEF culture was selected in BMDM medium with puromycin (5 μg/ml). Viable cells were used for subsequent experiments.
Immunoblot analysis and immunoprecipitation assay
Cells were lysed with RIPA buffer [25 mM tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Triton X-100] supplemented with protease and phosphatase inhibitors. Immunoprecipitation was performed using a previously described protocol (38). Antibodies against RhoA (catalog no. 2117), p-IRF3 (catalog no. 29047), p-IKKα/β (catalog no. 2697S), p-AKT S473 (catalog no. 4060), p-GSK-3β Ser9 (catalog no. 9322), IKKβ (catalog no. 2684P), pan-AKT (catalog no. 4691), GSK-3β (catalog no. 9315S), RIG-I (catalog no. 3743), MAVS (catalog no. 4983), TRADD (catalog no. 3694), cFLIP (catalog no. 56343), Ubi (catalog no. 3936), K48 Ubi (catalog no. 4289), K63 Ubi (catalog no. 5621), NEMO (catalog no. 2685), SIGMA1R (catalog no. 61994), Calnexin (catalog no. 2679), VDAC (catalog no. 4866), Myc (catalog no. 2278P), and HA-HRP (catalog no. 2999) were from Cell Signaling Technology. Antibodies against IRF3 (catalog no. sc-9082), MAVS (catalog no. sc-365334), TANK (catalog no. sc-166643), α-tubulin (catalog no. sc-23948), and Myc (catalog no. sc-40) were from Santa Cruz. Antibodies against Pggt1b (catalog no. HPA030646), TRIM31 (catalog no. AV34717), β-actin–HRP (catalog no. A3854), and Flag (catalog no. F4042) were from Sigma-Aldrich. Antibodies against FACL4 (catalog no. PA5-27137), Alexa Fluor 488 donkey anti-mouse (catalog no. A21202), and Alexa Fluor 594 goat anti-rabbit (catalog no. A-11037) were from Thermo Fisher Scientific. Anti-Rac1 (catalog no. 05-389) was from EMD Millipore Corporation. Anti-Ripk1 (catalog no. 610458) was from BD Transduction Laboratories. Anti–caspase-8 (catalog no. ALX-804-447) was from Enzo Life Sciences. Anti–influenza A nucleoprotein (catalog no. EMS 010) was from Kerafast.
Enzyme-linked immunosorbent assay and quantitative RT-PCR
Cell culture supernatants or sera were analyzed by enzyme-linked immunosorbent assay (ELISA) for IFN-β (BioLegend, catalog no. 439407) and IL-6 (eBiosciences, catalog no. 555240). A sandwich ELISA for mouse IFN-β was performed as previously described (39). Transcript abundance of cytokines was determined by RT-PCR using primers listed in table S1.
Detection of S-palmitoylated Rac1 by acyl-RAC
This procedure was performed with the CAPTUREome S-Palmitoylated Protein Kit (Badrilla). Each sample of 2 mg of total cellular protein was collected and subjected to acyl-RAC. After blocking free thiols with a thiol blocking agent, 30 μl of supernatant was saved as the “total input.” A palmitoylate thioester linkage–specific reagent is then used to cleave the thioester bond and to release the palmitoylate group only. Newly liberated thiols are captured with CAPTUREome capture resin. After thorough washing, captured proteins from the resin were eluted with a reductant provided in the kit, resolved by SDS–polyacrylamide gel electrophoresis, and analyzed by immunoblotting for Rac1.
Ubiquitination assay
The analysis of the ubiquitination of endogenous MAVS in BMDMs or overexpressed MAVS in 293T cells (American Type Culture Collection) transfected with plasmids encoding mouse Myc-MAVS or that together with HA-K63 ubiquitin, mouse Flag-Trim31, and Rac1 and its mutant forms was performed according to a protocol described previously (40). Briefly, cells were collected with complete cell lysis buffer [2% SDS, 150 mM NaCl, and 10 mM tris-HCl (pH 8.0)] with 2 mM sodium orthovanadate, 50 mM sodium fluoride, and protease inhibitors and then boiled for 10 min. Cells were immediately sheared with sonication, diluted with dilution buffer [10 mM tris-HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, and 1% Triton], and incubated at 4°C for 30 min with rotation. After centrifugation for 30 min at 20,000g, supernatants were immunoprecipitated with the anti-MAVS or anti-Myc–specific antibodies and analyzed by immunoblot with anti-ubiquitin (total), anti-ubiquitin (K48), anti-ubiquitin (K63), or anti-HA antibodies.
Semidenaturing detergent agarose gel electrophoresis
SDD-AGE was performed as described previously (3).
Viral infection and plaque assay
For in vitro infection, SeV (Charles River Laboratories, catalog no. 10100816) and MCMV were diluted to indicated concentrations with DMEM serum-free media. After washing with Dulbecco’s PBS, cultured BMDMs were infected with virus for 1 hour and then changed with DMEM with 10% FBS. For H1N1 influenza A virus Puerto Rico/08/1934 (IAV PR8) infection, BMDMs were infected with IAV PR8 in Opti-MEM supplemented with 1% bovine serum albumin (BSA). Single-round acute infections were performed at an MOI of 1.0, while multicycle spreading infections were performed at an MOI of 0.01. Cells were incubated with infectious medium for 1 hour before this was replaced with DMEM supplemented with 1% BSA and antibiotics. For multicycle spreading infections, postinfection media were also supplemented with TPCK-treated trypsin (1 μg/ml; Sigma-Aldrich, catalog no. T8802).
Animal infections and plaque assays were performed as described previously (34). Briefly, age- and sex-matched Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre mice were anesthetized and infected with influenza virus A/PR/8/34 (IAV, 50 PFU per mouse) by intranasal administration. Mice were weighed daily and euthanized once their body weight reached 80% of starting weight. Mice were euthanized 3 days after infection, and lung homogenates were generated for plaque assay and cytokine analyses on day 3.
Lung viral titer was determined via standard plaque assay procedures on Madin-Darby canine kidney cells. Cells were incubated with diluted virus suspension for 1 hour before removing the virus and applying the agar overlay. Cells were then incubated for 72 hours before being fixed in 4% paraformaldehyde (PFA) for at least 4 hours. After aspirating off PFA and removing the agar layer, cells were incubated with an anti–influenza A nucleoprotein antibody overnight. After washing and incubating with a secondary antibody, cells were then washed and incubated in a TrueBlue reagent (SeraCare, catalog no. 5510-0050) for 30 min to allow for the staining of plaques.
Luciferase assays
Luciferase activity was measured with the Dual-Luciferase Reporter Assay system according to the manufacturer’s instructions (Promega, catalog no. E1910). Data were normalized for transfection efficiency by calculating the ratio between firefly luciferase activity and Renilla luciferase activity.
Deconvolution of confocal microscopy and colocalization analysis
Cells grown on glass coverslips were infected with or without SeV for 4 hours and incubated with 200 nM MitoTracker Red CMXRos (Thermo Fisher Scientific, catalog no. M7512) for 20 min at 37°C. The cells were then fixed in ice-cold methanol for 15 min at −20°C, permeabilized with 0.3% Triton X-100, and blocked with 10% FBS. The cells were stained with the indicated primary antibodies, followed by incubation with fluorescent dye–conjugated secondary antibodies. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich, catalog no. D9542). Imaging of the cells was carried out using a Leica confocal microscope (Leica SP8). Nine images of stacks were collected in 0.15-μm Z-steps. The stacks were further subjected to three-dimensional deconvolution using the Huygens Essential software (Scientific Volume Imaging). Deconvolved images were exported as single-channel tiff images. Colocalization analysis was performed using Fiji software [National Institutes of Health (NIH), USA].
Proximity ligation assay
Primary Pggt1b+/+ Lyz2-Cre and Pggt1bfl/fl Lyz2-Cre BMDMs, Rac1−/− iBMDM, or that reconstituted with various mutant forms of Rac1 were fixed with ice-cold 100% methanol, permeabilized with 0.3% Triton X-100, and blocked with 5% goat serum. PLA was performed according to the manufacturer’s instructions (Duolink, Sigma-Aldrich). Fluorescent red spots were detected by using an upright confocal microscope, Leica SP8 (Leica). Quantification of interaction spots per cell was performed using the ImageJ software.
Quantification and statistical analyses
Two-way ANOVA multiple-factor comparison was performed using Prism software (GraphPad Software Inc.).
Supplementary Material
Acknowledgments
We thank E. W. St.Clair and M. Krangle for critically reading our manuscript as well as M. P. Hamilton and E. Park for technical support. Funding: This work was supported in part by funds from the NIH (National Institute of Allergy and Infectious Diseases 1R01AI110695-01A1 to D.W.). A.T.H. was supported by NIH Virology and Oncology Training grant T32-CA009111. Author contributions: D.W. conceived the study. D.W. and S.Y. designed the experiments. S.Y. performed most of the experiments. A.T.H. and N.S.H. provided viruses and helped with in vivo infection. K.A.F. and Z.J. provided MCMV. C.Su. and M.G. provided Rac1 and Rac2 knockout mice bones. G.S., C.Sw., C.Z., and D.M. provided technical support for gene cloning and in vivo experiments. H.K.K. and D.B.L. provided TcsL. G.H., and M.O.B. provided scientific advice. L.G.Q. helped with alveolar macrophage isolation. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The mNeon PR8 virus was constructed by N.S.H. and can be requested from N.S.H. pending scientific review and a complete material transfer agreement.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav7999/DC1
Fig. S1. Protein geranylgeranylation has no effect on DNA virus–induced antiviral innate immune response.
Fig. S2. Rac1 but not RhoA or Cdc42 suppresses MAVS signaling in a protein geranylgeranylation–dependent manner.
Fig. S3. Protein geranylgeranylation targets Rac1 to MERC ER membranes upon RLR engagement.
Fig. S4. Chaperon but not the signaling function of SIGMA1R and Rac1 palmitoylation is important for anchoring Rac1 to MERC ER membranes.
Fig. S5. Rac1 directly engages MAVS signalosome and inhibits MAVS signaling in a protein geranylgeranylation–dependent manner.
Fig. S6. MERC-localized Rac1 inhibits Trim31-MAVS interaction and subsequent MAVS Lys63-linked ubiquitination, aggregation, and activation upon RNA virus infection.
Fig. S7. MERC-localized Rac1 promotes cleavage of Ripk1 and the association of TANK with ubiquitinated Ripk1.
Fig. S8. Pggt1b deficiency and Statin pretreatment in BMDMs enhance antiviral innate immune response after influenza A virus PR8 challenge.
Fig. S9. Graphic summary.
Table S1. Primers for quantitative RT-PCR.
REFERENCES AND NOTES
- 1.Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K. J., Takeuchi O., Akira S., IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Seth R. B., Sun L., Ea C.-K., Chen Z. J., Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122, 669–682 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Hou F., Sun L., Zheng H., Skaug B., Jiang Q.-X., Chen Z. J., MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michallet M.-C., Meylan E., Ermolaeva M. A., Vazquez J., Rebsamen M., Curran J., Poeck H., Bscheider M., Hartmann G., König M., Kalinke U., Pasparakis M., Tschopp J., TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity 28, 651–661 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Balachandran S., Thomas E., Barber G. N., A FADD-dependent innate immune mechanism in mammalian cells. Nature 432, 401–405 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Takahashi K., Kawai T., Kumar H., Sato S., Yonehara S., Akira S., Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J. Immunol. 176, 4520–4524 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Handa P., Tupper J. C., Jordan K. C., Harlan J. M., FLIP (Flice-like inhibitory protein) suppresses cytoplasmic double-stranded-RNA-induced apoptosis and NF-κB and IRF3-mediated signaling. Cell Commun. Signal 9, 16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajput A., Kovalenko A., Bogdanov K., Yang S.-H., Kang T.-B., Kim J.-C., Du J., Wallach D., RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity 34, 340–351 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Buskiewicz I. A., Koenig A., Huber S. A., Budd R. C., Caspase-8 and FLIP regulate RIG-I/MDA5-induced innate immune host responses to picornaviruses. Future Virol. 7, 1221–1236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blander J. M., A long-awaited merger of the pathways mediating host defence and programmed cell death. Nat. Rev. Immunol. 14, 601–618 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Wu J., Chen Z. J., Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 32, 461–488 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Zhao T., Yang L., Sun Q., Arguello M., Ballard D. W., Hiscott J., Lin R., The NEMO adaptor bridges the nuclear factor-κB and interferon regulatory factor signaling pathways. Nat. Immunol. 8, 592–600 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Zeng W., Xu M., Liu S., Sun L., Chen Z. J., Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol. Cell 36, 315–325 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giacomello M., Pellegrini L., The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 23, 1417–1427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horner S. M., Liu H. M., Park H. S., Briley J., Gale M. Jr., Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 108, 14590–14595 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang F. L., Casey P. J., Protein prenylation: Molecular mechanisms and functional consequences. Annu. Rev. Biochem. 65, 241–269 (1996). [DOI] [PubMed] [Google Scholar]
- 17.Akula M. K., Shi M., Jiang Z., Foster C. E., Miao D., Li A. S., Zhang X., Gavin R. M., Forde S. D., Germain G., Carpenter S., Rosadini C. V., Gritsman K., Chae J. J., Hampton R., Silverman N., Gravallese E. M., Kagan J. C., Fitzgerald K. A., Kastner D. L., Golenbock D. T., Bergo M. O., Wang D., Control of the innate immune response by the mevalonate pathway. Nat. Immunol. 17, 922–929 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan O. M., Ibrahim M. X., Jonsson I.-M., Karlsson C., Liu M., Sjogren A.-K. M., Olofsson F. J., Brisslert M., Andersson S., Ohlsson C., Hultén L. M., Bokarewa M., Bergo M. O., Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J. Clin. Invest. 121, 628–639 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voth D. E., Ballard J. D., Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 18, 247–263 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onesto C., Shutes A., Picard V., Schweighoffer F., Der C. J., Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. Methods Enzymol. 439, 111–129 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Chen H.-Y., Yang Y. M., Stevens B. M., Noble M., Inhibition of redox/Fyn/c-Cbl pathway function by Cdc42 controls tumour initiation capacity and tamoxifen sensitivity in basal-like breast cancer cells. EMBO Mol. Med. 5, 723–736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaffe A. B., Hall A., Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Hayashi T., Rizzuto R., Hajnoczky G., Su T.-P., MAM: More than just a housekeeper. Trends Cell Biol. 19, 81–88 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bozidis P., Williamson C. D., Colberg-Poley A. M., Isolation of endoplasmic reticulum, mitochondria, and mitochondria-associated membrane fractions from transfected cells and from human cytomegalovirus-infected primary fibroblasts. Curr. Protoc. Cell Biol. Chapter 3, Unit 3.27 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Natsvlishvili N., Goguadze N., Zhuravliova E., Mikeladze D., Sigma-1 receptor directly interacts with Rac1-GTPase in the brain mitochondria. BMC Biochem. 16, 11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szabo A., Kovacs A., Frecska E., Rajnavolgyi E., Psychedelic N,N-dimethyltryptamine and 5-methoxy-N,N-dimethyltryptamine modulate innate and adaptive inflammatory responses through the sigma-1 receptor of human monocyte-derived dendritic cells. PLOS ONE 9, e106533 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujimoto M., Hayashi T., Su T. P., The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem. Biophys. Res. Commun. 417, 635–639 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lynes E. M., Bui M., Yap M. C., Benson M. D., Schneider B., Ellgaard L., Berthiaume L. G., Simmen T., Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 31, 457–470 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lynes E. M., Raturi A., Shenkman M., Sandoval C. O., Yap M. C., Wu J., Janowicz A., Myhill N., Benson M. D., Campbell R. E., Berthiaume L. G., Lederkremer G. Z., Simmen T., Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. J. Cell Sci. 126, 3893–3903 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Navarro-Lérida I., Sánchez-Perales S., Calvo M., Rentero C., Zheng Y., Enrich C., Del Pozo M. A., A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J. 31, 534–551 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Craven R., Lacy D. B., Clostridium sordellii lethal-toxin autoprocessing and membrane localization activities drive GTPase glucosylation profiles in endothelial cells. mSphere 1, e00012-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu B., Zhang M., Chu H., Zhang H., Wu H., Song G., Wang P., Zhao K., Hou J., Wang X., Zhang L., Gao C., The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 18, 214–224 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Uyeki T. M., Katz J. M., Jernigan D. B., Novel influenza A viruses and pandemic threats. Lancet 389, 2172–2174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harding A. T., Heaton B. E., Dumm R. E., Heaton N. S., Rationally designed influenza virus vaccines that are antigenically stable during growth in eggs. MBio 8, e00669-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mehrbod P., Hair-Bejo M., Ibrahim T. A. T., Omar A. R., El Zowalaty M., Ajdari Z., Ideris A., Simvastatin modulates cellular components in influenza A virus-infected cells. Int. J. Mol. Med. 34, 61–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collins A. M., Rylance J., Wootton D. G., Wright A. D., Wright A. K. A., Fullerton D. G., Gordon S. B., Bronchoalveolar lavage (BAL) for research; obtaining adequate sample yield. J. Vis. Exp., e4345 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Hoecke L., Job E. R., Saelens X., Roose K., Bronchoalveolar lavage of murine lungs to analyze inflammatory cell infiltration. J. Vis. Exp., e55398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonham K. S., Orzalli M. H., Hayashi K., Wolf A. I., Glanemann C., Weninger W., Iwasaki A., Knipe D. M., Kagan J. C., A promiscuous lipid-binding protein diversifies the subcellular sites of Toll-like receptor signal transduction. Cell 156, 705–716 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts Z. J., Goutagny N., Perera P. Y., Kato H., Kumar H., Kawai T., Akira S., Savan R., van Echo D., Fitzgerald K. A., Young H. A., Ching L.-M., Vogel S. N., The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J. Exp. Med. 204, 1559–1569 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choo Y. S., Zhang Z., Detection of protein ubiquitination. J. Vis. Exp., e1293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav7999/DC1
Fig. S1. Protein geranylgeranylation has no effect on DNA virus–induced antiviral innate immune response.
Fig. S2. Rac1 but not RhoA or Cdc42 suppresses MAVS signaling in a protein geranylgeranylation–dependent manner.
Fig. S3. Protein geranylgeranylation targets Rac1 to MERC ER membranes upon RLR engagement.
Fig. S4. Chaperon but not the signaling function of SIGMA1R and Rac1 palmitoylation is important for anchoring Rac1 to MERC ER membranes.
Fig. S5. Rac1 directly engages MAVS signalosome and inhibits MAVS signaling in a protein geranylgeranylation–dependent manner.
Fig. S6. MERC-localized Rac1 inhibits Trim31-MAVS interaction and subsequent MAVS Lys63-linked ubiquitination, aggregation, and activation upon RNA virus infection.
Fig. S7. MERC-localized Rac1 promotes cleavage of Ripk1 and the association of TANK with ubiquitinated Ripk1.
Fig. S8. Pggt1b deficiency and Statin pretreatment in BMDMs enhance antiviral innate immune response after influenza A virus PR8 challenge.
Fig. S9. Graphic summary.
Table S1. Primers for quantitative RT-PCR.