

Summary
Neutrophils are short-lived immune cells that play important roles in a variety of diseases. The oligopotent Granulocyte Monocyte Progenitors (GMP) in the bone marrow give rise to monocytes and all granulocytes. Although several monocyte progenitors have been identified in mouse bone marrow, the unipotent neutrophil progenitors are still not well-defined. Here, we use Cytometry by Time-of-Flight (CyTOF) and Single-cell RNA-Sequencing (scRNA-Seq) methodologies to identify a committed unipotent early-stage neutrophil progenitor in adult mouse bone marrow. Importantly, we also discovered a similar unipotent, committed neutrophil progenitor (hNeP) that is present in healthy human bone marrow. Both mouse and human progenitors demonstrate unipotent neutrophil potency in vivo. Study of the identified mouse (NeP) and human (hNeP) neutrophil progenitors in cancer revealed that both NeP and hNeP significantly increased tumor growth when transferred into murine cancer models, including a humanized model. Further, we discovered that the hNeP was present in the blood of human patients recently diagnosed with melanoma, and could be readily identified by flow cytometry, suggesting that this human neutrophil progenitor could be used as a biomarker for early cancer discovery. The discovery of this early committed unipotent neutrophil progenitor in humans will allow for development of important new therapeutic targets for regulation of neutrophil levels and function in disease, particularly in cancers, where neutrophils play a significant role.
Introduction
Neutrophils are the most abundant population of circulating blood leukocytes. With many emerging studies suggesting critical roles of neutrophils in chronic inflammatory diseases, including cancer, a complete understanding of neutrophil development is imperative (Huang et al., 2016; Sagiv et al., 2015; Soehnlein et al., 2017; Summers et al., 2010). Neutrophils originate in the bone marrow (BM). In murine BM, Lin−CD117 (c-kit)+ cells (LK) cells include Lin−CD117+Ly6A/E (Sca1)+CD127−(LSK) cells that give rise to all hematopoietic cells, Lin−CD117loLy6A/E+CD127+ cells that contain the Common Lymphoid Progenitor (CLP) that give rise to all lymphoid lineages, and Lin−CD117+Ly6A/E−that selectively generate only myeloid lineages (Akashi et al., 2000). In this classic hematopoietic paradigm, the Lin−CD117+ Ly6A/E−population is divided into Common Myeloid Progenitor (CMP), Megakaryocyte-Erythroid Progenitor (MEP), and Granulocyte Monocyte Progenitor (GMP) by differential expression of surface markers CD16/32 (FcRγIII/II) and CD34 (Akashi et al., 2000). CMPs are the multi-potent progenitors for MEP and GMP whereas GMP have lost erythroid potency and thus are restricted to generate granulocyte and monocyte lineages (Manz et al., 2002).
High-dimensional mass cytometry (also known as cytometry by time-of-flight, CyTOF) has become a powerful tool to investigate the hematopoietic system (Becher et al., 2014; Bendall et al., 2011; Samusik et al., 2016). Notably, with the development of multi-channel flow cytometry, mass cytometry and scRNA-sequencing, new markers have allowed for the discovery of several new hematopoietic progenitors, including a CD41+ megakaryocyte progenitor (MkP), a Ter119+ erythroid precursor (Pro Ery) (Pronk et al., 2007), and the Ly6C+ committed Monocyte Progenitor (cMoP) (Hettinger et al., 2013). The heterogeneity of GMP has been suggested in many studies. In mouse, single-cell analysis of gene expression patterns revealed that GMP already have restricted lineage potential towards monocytes or neutrophils (Buenrostro et al., 2018; Olsson et al., 2016). Gene-expression analysis of CMP and GMP at the single-cell level showed heterogeneity in these progenitors (Paul et al., 2015), suggesting that classification of these subsets using solely CD34 and CD16/32 was not sufficient. In agreement with gene-expression analysis, additional heterogeneity has been discovered in Lin−CD117+ Ly6A/E−cells with use of additional surface markers. For example, CD105, CD150, CD41, and CD71 divide Lin−CD117+ Ly6A/E−cells into preMegE, pre CFU-E, CFU-E, MkP, preGM, GMP, and Pro Ery (Pronk et al., 2007). CX3CR1+ CD115+ CD135+ Monocyte/DC Progenitors (MDP) were also found to partially overlap with the classic CMP/GMP (Auffray et al., 2009). GMP heterogeneity has also been suggested in humans (Buenrostro et al., 2018) with similar developmental-staged transcriptional factors (such as IRF8) in mouse (Olsson et al., 2016). Comparable to GMP, myeloblasts are known to have both granulocytic and monocytic potentials (Borregaard, 2010). The use of CD64 identified a human CD34+CD64hi monocyte progenitor within human GMPs (Kawamura et al., 2017).
In mouse, several monocyte progenitors (Fogg, 2006; Hettinger et al., 2013; Liu et al., 2009; Satoh et al., 2017) and granulocyte progenitors including eosinophil progenitors (Mori et al., 2009; Zhang et al., 2004) and basophil/mast cell progenitors (Arinobu et al., 2005; Qi et al., 2013) have been identified. Several immature neutrophil precursors have also been identified (Fiedler and Brunner, 2012; Kim et al., 2017; Satake et al., 2012; Sturge et al., 2015; Yáñez et al., 2015). However, these precursors are late-stage precursors with neutrophil potency (Kim et al., 2017; Sturge et al., 2015), and several show multi- or oligopotency (Satake et al., 2012; Yáñez et al., 2015). Recently, a proliferative neutrophil precursor was identified in mouse BM that generates neutrophils after intra-BM adoptive transfer (Evrard et al., 2018). However, the long-term potency of this precursor was not tested. Thus, several gaps in understanding the complete neutrophil-lineage hierarchy from CMP to mature neutrophils remain.
In humans, the search for a unipotent neutrophil progenitor (hNeP) has been ongoing for decades (Bainton et al., 1971; Elghetany et al., 2004; Pillay et al., 2010; Terstappen and Loken, 1990). Human CMP and GMP express positive levels of CD34 and CD38 and mirror the murine CMP/GMP paradigm in myeloid cell production (Doulatov et al., 2010; Edvardsson et al., 2006; Manz et al., 2002). A missing link in human neutrophil development is the identification of neutrophil progenitors that are downstream of GMP but upstream of short-term neutrophil precursors. Recently, mass cytometry analysis of human BM neutrophils indicates that human neutrophils are heterogeneous and contain a CD117−CD34−CD49d−CD101−subset termed preNeu (Evrard et al., 2018). This subset was suggested to be a counterpart of mouse neutrophil precursors, unfortunately the neutrophil potential of this possible precursor was not evaluated.
Here, we decided to take advantage of mass cytometry and viSNE automated mapping to identify and study new neutrophil progenitors in mouse and human bone marrow.
Results
Automated single-cell analysis of Lin−CD117+ Ly6A/E−cells identifies a distinct novel neutrophil progenitor population.
We analyzed mouse bone marrow by mass cytometry with the purpose of identifying all neutrophil progenitors. We developed an antibody panel shown in Table S1 that measures 39 parameters simultaneously and used it to perform CyTOF mass cytometry on healthy mouse bone marrow. We used viSNE automated analysis to study CD45+ BM cells and found a portion of CD117(c-Kit)+ cells that had close relation to Ly6G+ cells (Figure S1A). Interestingly, CD34+ GMP clustered with different populations, including the Ly6G-enriched population and the CD115-enriched population (Figure S1B). These results agree with the reported observation that CD115+ MDP overlaps with CMP/GMP (Auffray et al., 2009), and more importantly, suggest that the GMP fraction has neutrophil progenitor (NeP) potential. We then focused exclusively on myeloid cells by examining the Lin−CD117+ Ly6A/E−fraction of LK cells which contains all myeloid cell progenitors (Figure S1C). Using viSNE automated unbiased analysis we found 5 distinct clusters of cells in Lin−CD117+ Ly6A/E−cells , which we labeled as Clusters #A-E in Figure 1A. Each of these clusters expresses distinctive biomarkers that uniquely define specific myeloid cell types. Siglec F (cluster#A) marks eosinophils, CD115 (cluster#B) marks monocytes, Ly6G (cluster#C) marks neutrophils, FcεRIα (cluster#D) marks mast cells and basophils, and CD16/32 and CD34 (cluster#E) marks both CMP and GMP. The neutrophil-specific antigen, Ly6G, is observed in a continuum from negative to high expression in Cluster#C, suggesting the presence of neutrophil progenitors and precursors within this cluster (Kim et al., 2017; Satake et al., 2012; Sturge et al., 2015; Yáñez et al., 2015). We confirmed these results using conventional flow cytometry (Figure S2A, B).
Figure 1. Automated single-cell analysis of Lin-CD117+ Ly6A/E-cells in bone marrow identifies a distinct novel neutrophil progenitor population.
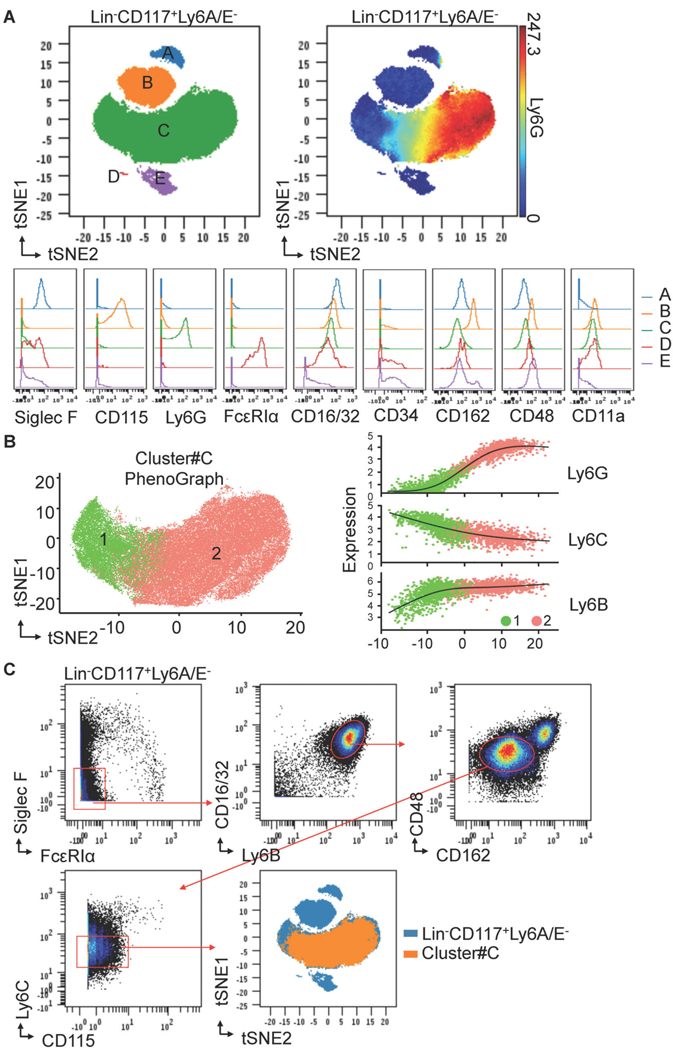
(A) ViSNE defines a largest Cluster#C of the 5 subsets in Lin−CD117+ Ly6A/E−cells from murine BM using mass cytometry (CyTOF). BM cells isolated from C57BL/6J donors were stained with the antibody panel shown in Table S1. ViSNE maps of Lin−CD117+ Ly6A/E−cells are shown as dot overlays to display the 5 automated clusters (#A-E). Ly6G expression pattern is shown on viSNE map of Lin−CD117+ Ly6A/E−cells as spectrum colored dots. The expression patterns of the indicated markers are shown as histogram overlays of each cluster. Results are representative of two independent experiments (n = 6 mice each). (B) PhenoGraph defines two subpopulations of Cluster#C using mass cytometry (CyTOF). Left, two PhenoGraph meta-clusters present two distinct populations (1, 2) in Cluster#C. Right, The expression profile of Ly6G, Ly6C, and Ly6B for randomly selected cells in each cluster is visualized on the first component of a nonlinear dimensionality reduction isomap (the regression black line estimated using the generalized linear model is added for each marker). (C) FACS gating strategy for Cluster#C using mass cytometry (CyTOF). Manually gated Cluster#C is back gated to automated viSNE map for validation.
As we were interested in identifying neutrophil progenitors, we focused our efforts on further analysis of Cluster#C, which showed a continuum of Ly6G expression. Using Phenograph, a second unbiased clustering algorithm (Chen et al., 2016; Levine et al., 2015), we found that Cluster#C consists of two major populations that display a continuum of Ly6G, Ly6C, and Ly6B expression (Figure 1B). These Ly6 proteins are highly expressed in mature neutrophils and their precursors (Kim et al., 2017; Lee et al., 2013). We developed a conventional flow cytometry gating strategy shown in Figure 1C to isolate with purity Cluster#C cells (Lin−CD117+ Ly6A/E−Siglec F−FcεRIα−CD16/32+ Ly6B+ CD162lo CD48lo Ly6Clo CD115−) from bone marrow. This cell population, when backgated onto a viSNE map fell exclusively into Cluster#C (Figure 1C).
ScRNA-Seq analysis of Cluster#C reveals two major subpopulations #C1 and #C2.
To closely investigate Cluster#C, we sorted Cluster#C cells for scRNA-Seq. We found two primary subpopulations within Cluster#C, #C1 and #C2 (Figure 2A) by scRNA-Seq. Notably #C1 shows low Ly6g expression at the mRNA level (Figure 2A), which confirms our low Ly6G protein expression in this cluster found by mass cytometry (Figure 1B).
Figure 2. ScRNA-Seq analysis of Cluster#C reveals two major subpopulations #C1 and #C2.
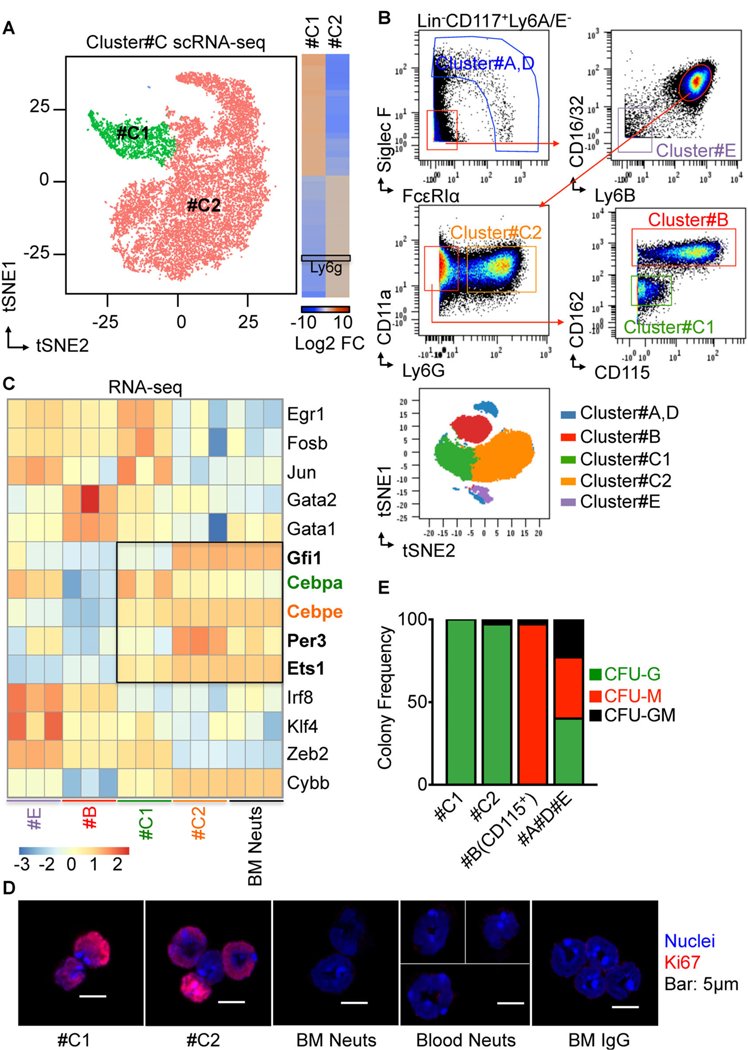
(A) Single-cell RNA Sequencing (scRNA-Seq) uncovers the heterogeneity of Cluster#C. 20,000 Cluster#C cells were sorted from healthy wild-type mouse BM for scRNA-Seq assay (3 biological triplicates, 2 technical replicates). FACS sorting strategies for Cluster#C are shown in Figure 1C using mass cytometry, and Figure S10A using flow cytometry. Left, tSNE 2D plots, obtained applying Seurat scRNA-Seq analysis R Package for the scRNA-Seq data, showing two main clusters corresponding to subsets of Cluster#C (n=16268 cells; #C1, 2149 cells (green) and #C2, 14089 cells (salmon)). Right, heatmap shows top 40 differentially expressed genes in each cluster. Black box highlights Ly6G expression. Log2 Fold Change of each gene expression is relative to the entire dataset. (B) FACS gating strategy for Cluster#A and D, #B, #C1, #C2, and #E using mass cytometry (CyTOF). Manually gated clusters are back gated to automated viSNE map for validation. (C) RNA-seq shows up-regulation of important neutrophil lineage-decision genes in #C1 and #C2. Cluster#C1, #C2, #E, and BM Neuts were sorted from healthy wild-type mice BM for RNA-seq. FACS sorting strategies for these cell types are shown in Figure 2B using mass cytometry, and Figure S10B using flow cytometry. Heatmap showing expression of important development transcriptional factors for myeloid cell development in sorted populations by RNA-seq. Black box highlights expression of important neutrophil lineage-decision genes (bold) in #C1 and #C2. Cebpa (green) expression is higher in #C1 compared to #C2. Cebpe (orange) expression is lower in #C1 compared to #C2. z-score normalization from CPM (Counts Per Million) expression level (log2 scale) was quantified from RNA-Seq. (D) Confocal microscopy detected Ki67 localization within the nuclei in Cluster#C1and #C2. #C1, #C2, BM Neuts, and Blood Neuts were sorted and stained with antibodies to Ki67 (red) and DNA was labeled with Hoechst (blue). FACS sorting strategies for these cell types are shown in Figure 2B using mass cytometry, and Figure S10B using flow cytometry. IgG stained cells served as a negative control. Bar : 5µm. (E) Cluster#C1 and #C2 cells produce only Neutrophils in vitro. Cluster#C1, #C2, #B (CD115+), #A, D, and #E cells were sorted from wild-type mice and diluted to single-cell suspensions. FACS sorting strategies for these cell types are shown in Figure 2B using mass cytometry, and Figure S10B using flow cytometry. Single cells of each cluster were cultured in methylcellulose-based medium. Numbers of colonies generated from the indicated progenitors were counted at day 10 of culture. Contingency plot shows mean value of six independent experiments (each contains 3 biological triplicates).
Using these data, we were able to design a flow cytometry panel (Figure 2B) that allowed us to isolate both #C1 and #C2 as well as other Lin−CD117+ Ly6A/E−cell fractions for further detailed study. Cluster #C1 is Lin−CD117+ Ly6A/E−Siglec F−FcεRIα−CD16/32+ Ly6B+ CD11a+ (LFA1α+) CD162lo CD48lo Ly6Clo CD115−Ly6G− and cluster #C2 is Lin−CD117+ Ly6A/E−Siglec F−FcεRIα−CD16/32+ Ly6B+ CD11a+ (LFA1α+) Ly6G+. Based on surface marker expression patterns we predict #C1 and #C2 as NeP candidates.
As identified in Figure 1A, Cluster#E is enriched with CD16/32−CD34+ CMPs and Cluster#B is enriched with CD115+ monocyte progenitors. We performed bulk RNA-Seq for transcriptome analysis on #C1 and #C2 and analyzed #E and #B cells as control groups. BM neutrophils (BM Neuts) containing immature and mature neutrophils were also sorted from the same donors for analysis. We found that #C1 expresses high levels of GMP genes including Egr1, Fosb, Jun, Gata2, Gata1 as well as genes that are shown to be critical for neutrophil development including Gfi1, Cebpa, Cebpe, Per3 and Ets1 (Avellino et al., 2016; Buenrostro et al., 2018; Evrard et al., 2018; Horman et al., 2009; Olsson et al., 2016; Radomska et al., 1998; Zhang et al., 1997). Genes that are critical for monocyte development such as Irf8 (Olsson et al., 2016; Yáñez et al., 2015), on the other hand, show low expression in #C1 and #C2. Interestingly, #C2 cells have lost expression of the GMP gene signature while the neutrophil gene signature increased in #C2 cells to levels comparable to those of BM neutrophils.
We next wanted to focus on the hierarchical structure of #C1 and #C2 within the neutrophil developmental lineage. Frequencies of #C1 are lowest in bone marrow, followed by #C2 (Figure S3B). Comparison of #C1 and #C2 by flow cytometry showed a gradient of Ly6G expression from negative in #C1 to intermediate in #C2 to high in mature BM Neuts, whereas CXCR2 is only expressed by terminally differentiated BM Neuts (Figure S3B). Reconstruction in 3-D of the nuclear architecture of #C1 and #C2 cells suggests more stem-cell like morphology than that of mature BM Neuts and Blood Neuts (Figure S3B). #C1 has more stem cell-like nuclear morphology and higher Ki67 expression and nuclear integration (Figure 2C and S3C) than does #C2, BM Neuts and Blood Neuts, suggesting an early stage of development for #C1. These data suggest that #C1 lies earlier in the neutrophil developmental hierarchy and may partially overlap with GMP from the classic myeloid progenitor paradigm. #C2, however, may represent a transitional intermediate progenitor between #C1 and terminally differentiated neutrophils in mouse BM. Thus, we then decided to focus on #C1 cells as the candidate for the early-stage committed neutrophil progenitor (NeP).
The selective neutrophil potency of #C1 cells was first tested by examining in vitro methylcellulose colony-forming unit formation (Figure 2E). All donor cell fractions were FACS sorted using the gating strategy described in Figure 2B. CD115+ CD117+ cells are monocyte progenitors and are located within Cluster#B therefore the CD115+ portion of Cluster#B was sorted as monocyte progenitors (Figure S4A). Clusters#A, D, E were collected together as a control group. As shown in Figure 2E, #C1 single cells generate colony-forming unit-granulocyte (CFU-G) in methylcellulose-based medium with 100% purity, but not colony-forming unit-macrophage (CFU-M) or colony-forming unit-granulocyte, macrophage (CFU-GM). Similar results were also observed with #C2. Cluster #B (CD115+) cells were able to generate CFU-M only, as expected. The #A#D#E control group generated all three types of colonies. These results suggest that #C1 cells have restricted granulocyte potency in vitro that lasts at least 10 days.
Cluster#C1 is the early-stage committed unipotent neutrophil progenitor (NeP) in vivo.
We next analyzed the function of #C1 in generating neutrophils in vivo using adoptive transfer approaches. The experimental scheme is shown in Figure 3A. The cell populations described in Figure 2E were FACS sorted from the same donor mice. Each of the 4 cell groups were adoptively transferred into a group of sub-lethally irradiated CD45.1 recipient mice. Blood from each group was examined at days 5, 7, 12, 14 and 28 by flow cytometry for appearance of donor-derived progeny. The flow cytometry gating for all donor cell progeny is shown in representative plots of the #A#D#E recipient group in Figure 3A right panel. Donor cells (CD45.2+) appeared in blood as early as day 5 and peaked at day 14 (Figure 3A right panel, Figure S4B left panel). Donor cells were analyzed for expression of key markers for myeloid progenies: monocytes (Mo, CD115+), neutrophils (Ne, Ly6G+), eosinophils (Eo, Siglec F+), or basophils (Ba, FcεRIα+).
Figure 3. Cluster#C1 and #C2 cells are committed hierarchical unipotent progenitors for neutrophil production in vivo.
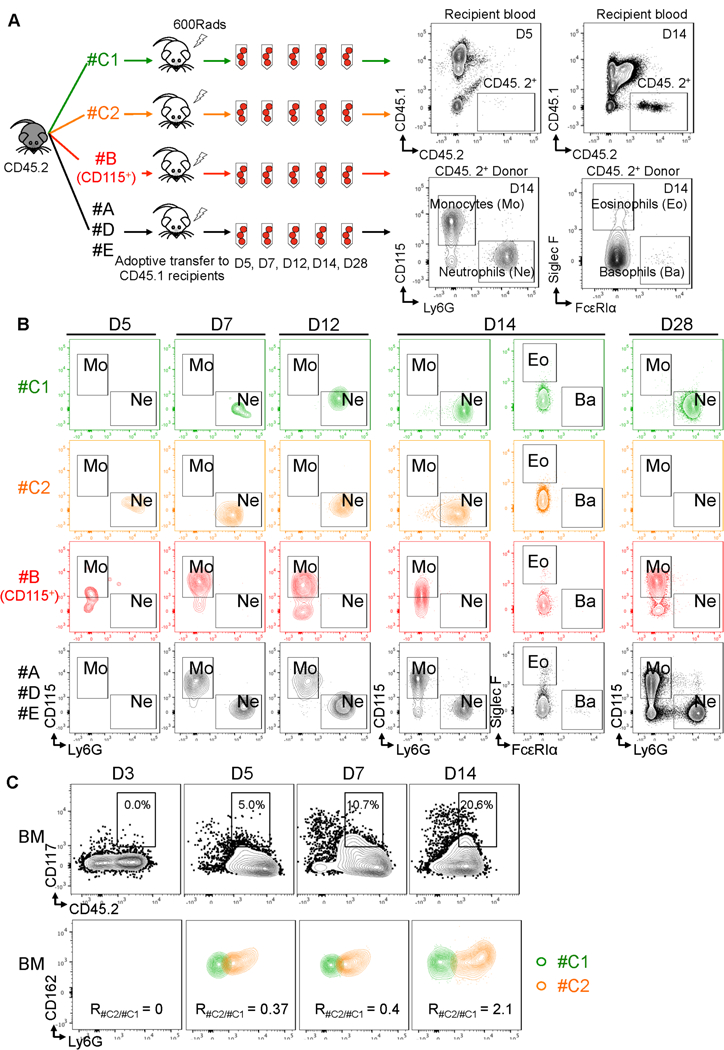
(A) Scheme showing the experimental procedure. Cluster#C1 and #C2 were sorted from CD45.2 donors and adoptively transferred into irradiated wild-type CD45.1 recipient mice. #B (CD115+), #A, D, and #E cells were sorted from the same donors for this experiment and served as controls. FACS sorting strategies for these cell types are shown in Figure 2B using mass cytometry, and Figure S10B using flow cytometry. Each recipient group includes 25 mice. Each recipient received 50,000 donor cells. After the transfer, peripheral blood was collected for flow cytometry of CD45. 2+ cells from 5 recipients of each group at days (D) 5, 7, 12, 14, 28 (D5, D7, D12, D14, D28), respectively. CD45.2+ cells were evaluated for the donor cell-derived monocytes (CD115+), neutrophils (Ly6G+), eosinophils (Siglec F+), and basophils (FcεRIα+). N = 5 mice for each time point in each group. (B) Cluster#C1 and #C2 cells produce only neutrophils in vivo. Representative plots show the appearance of neutrophils and monocytes in each recipient group at the time points indicated. Results are representative of two independent experiments. (C) Cluster#C1 produces #C2 cells in vivo. #C1 cells were sorted from CD45.2 donors and adoptively transferred into irradiated wild-type CD45.1 recipient mice. FACS sorting strategies for #C1 cells are shown in Figure 2B using mass cytometry, and Figure S10B using flow cytometry. After transfer, BM was collected for flow cytometry of CD45. 2+ cells from 3 recipients of each group at days (D) 3, 5, 7, 14 (D3, D5, D7, D14), respectively. CD117+CD45.2+ cells were evaluated for the donor #C1 homing to bone marrow and differentiation into #C2 cells. Expression in recipients of #C1 and #C2 cells are identified by the panel shown in Figure 2B and Figure S10B and overlaid for display. #C2 differentiation into #C2 cells are shown as Ratio of #C2 : #C1 (R#C2/#C1).
Donor-derived neutrophils appeared in recipient blood at Day 5 and Day 7 post-adoptive transfer in the groups reconstituted with #C1 (green) and #C2 (orange), suggesting neutrophil potency in both populations and slower kinetics of the #C1 cells in producing neutrophils (Figure 3B). Neutrophil progenies from these progenitors comprise nearly 100% of CD45.2+ donor cell-derived leukocytes in the #C1 recipients (Figure S4B middle panel). In the control groups, #B (CD115+) only produced monocytes and did not produce neutrophils and #A#D#E produced both neutrophils and monocytes (Figure 3B and S4B, red and black). These results illustrate the restricted unipotency of #C1 and #C2 progenitors to generate solely neutrophils.
Neutrophil production peaks at day 14 in #C2 recipients but at day 28, neutrophils vanished from the #C2 recipients, suggesting limited developmental potency of #C2 (Figure S4B right panel). However, in #C1 recipients, neutrophil production continued to day 28, our latest time point, indicating that the #C1 progenitors have longer-term potency. This long-term potency of #C1 is comparable to the #A#D#E fractions of Lin−CD117+ Ly6A/E−cells which contains CMP. To further determine that #C1 cells give rise to #C2 cells, we FACS-sorted only the #C1 cells (using the gating strategy shown in Figure 2B) from CD45.2 BM and adoptively transferred these CD45.2+ #C1 cells into CD45.1 recipients. We tracked the fate of the CD45.2+ #C1 donor cells by examine the the recipients’ peripheral and BM for the production of #C2 after the adoptive transfer. As shown in Figures 3C and S4C upper panel, #C1 cells infiltrated into spleen quickly by day 3 and appeared in BM 5 days after adoptive transfer. Importantly, a portion of the CD45+ #C1 donor cells started to generate #C2 very quickly after adoptive transfer while some #C1 cells seeded in the BM (Figure S4C and 3C, lower panels), again confirming that #C1 is the early-stage committed neutrophil progenitor.
Thus, by using high dimensional mass cytometry and scRNA-Seq approaches, as well as adoptive transfers in vivo, we have discovered an early-stage committed unipotent neutrophil progenitor (#C1, termed NeP) in mouse bone marrow. This progenitor can be identified as Lin−CD117+ Ly6A/E−Siglec F−FcεRIα−CD16/32+ Ly6B+ CD11a+ CD162lo CD48lo Ly6Clo CD115−Ly6G−.
Cluster#C1 cells (NePs) are increased in BM as well as in periphery with tumor and promote tumor growth in vivo.
Granulopoiesis is associated with cancer, with neutrophils having both pro-tumoral and anti-tumoral roles (Casbon et al., 2015; Hagerling and Werb, 2016; Sagiv et al., 2015). We wondered what role that #C1 NeP would have on tumorigenesis in vivo. We first asked whether #C1 or #C2 cells were increased in bone marrow in a melanoma mouse model. We injected B16F10 tumor cells SubQ into the rear flank of wild-type C57BL/6J mice (Tumor). Age-matched, gender-matched wild-type mice received D-PBS to serve as healthy controls (Healthy). At 14 days post-injection, we found a significant expansion of #C1 NeP and #C2 cells and a slight increase of #B (CD115+), but not #E (CMP) cells, in the bone marrow of tumor-bearing mice (Figure 4A), indicating that in the setting of cancer, myelopoiesis is strongly geared towards the neutrophil lineage. Interestingly, we detected minimal numbers of Cluster #C cells (less than 0.02% of all CD45+ cells in the periphery) of healthy mice, whereas #C cells are increased 10-fold in periphery of tumor-bearing mice (Figure S5A), suggesting that there is increased production and egress of these neutrophil progenitors from bone marrow to periphery in response to the tumor microenvironment. To test whether NeP can directly contribute to tumor growth, #C1 NeP cells, #C2 cells, #B (CD115+) cells, and #E cells were sorted from CD45.2 wild-type donor mice and adoptively transferred into irradiated CD45.1 recipient healthy mice. At day 1 after donor cell transfer, recipient mice were injected SubQ with B16F10 tumor cells into the rear flank. Tumor size was measured at days 12 and 22 after injection (Figure 4B, left). As shown in Figure 4B, right, mice receiving #C1 cells or #C2 cells showed increased tumor growth compared to #B (CD115+) cells or #E cells (CMP) at both timepoints. #C1 NeP promoted more potent tumor growth at the later time point compared to #C2 cells. At day 22 after tumor injection, tumors were harvested for detection of donor-derived cells. More #C1 NeP-derived cells infiltrated the tumor than did #C2 cells and over 30% of these #C1 NeP-derived cells expressed PD-L1, an inhibitory costimulatory molecule that contributes to immune suppression (Figure 4C). Further analysis revealed that tumor-infiltrated #C1 cells were able to maintain their stem cell phenotype as well as produce PD-L1+ #C2 and CD117−Ly6G+ neutrophil progenies. #C2 cells were also able to promote tumor growth via the same mechanism but to a lesser degree, whereas other cell types did not infiltrate the tumor (#B) and showed minimal PD-L1 expression (#E) (Figure S5B). Thus, #C1 NeP progenitors respond to melanoma tumor cues and have tumor-promoting functions by producing immune-suppressive progenies.
Figure 4. Cluster#C1 NeP and #C2 cells are increased in BM with tumor and promote tumor growth in vivo.
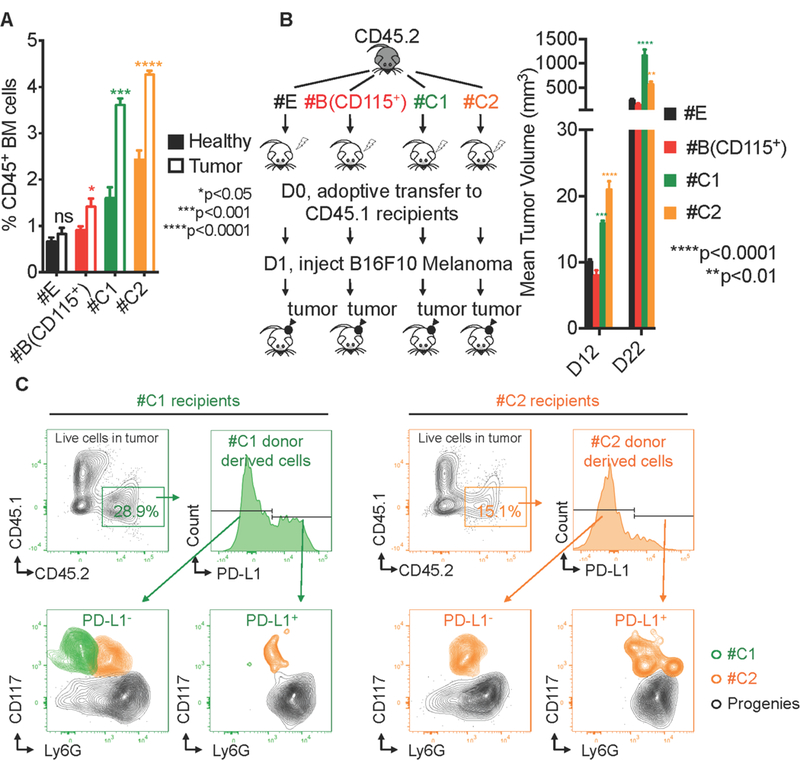
(A) Cluster#C1 NeP and #C2 cells are increased in BM of tumor-bearing mice. 5 × 105 B16F10 melanoma cells were SubQ injected into the rear flank of wild-type recipient mice for primary tumor growth. The frequency of Cluster#E, #B (CD115+), #C1 and #C2 were detected in BM from tumor-bearing mice at 14d post-injection (open bars) or their healthy counterparts (solid bars). N = 15. Error bars indicate mean (SD). (B) Left, scheme showing the experiment procedure. Cluster#E, #B (CD115+), #C1, and #C2 were sorted from the same CD45.2 wild type donors and were adoptively transferred into sub-lethally irradiated congenic CD45.1 recipients. FACS sorting strategies for these cell types are shown in Figure 2B using mass cytometry, and Figure S10B using flow cytometry. Each recipient received 5 × 104 donor cells. The next day, 3 × 105 B16F10 melanoma cancer cells were SubQ injected into each recipient mouse. Right, the tumor volume in each recipient was measured at 12d and 22d post-injection. Results are representative of 2 independent experiments. N = 5 mice in each group. Error bars indicate mean (SD). (C) #C1 and #C2 cells infiltrate to tumor and generate PD-L1 positive progenies. At D22 after the adoptive transfer, the tumors were harvested from recipients. Live singlet cells in tumor were evaluated using flow cytometry. CD45+ donor-derived cells were analyzed for PD-L1 expression. #C1, #C2, and donor-derived neutrophils were identified with the panel from Figure 2B and Figure S10B and overlaid for display.
Discovery of a heterogeneous CD66b+CD117+ CD38+ CD34+/− progenitor-like cell fraction in human BM.
Based on our cancer findings in mice showing the relevance of neutrophil progenitors to tumor growth, we next decided to look for neutrophil progenitors in human bone marrow. Human CMP and GMP express CD34, CD38, and CD117 and mirror the murine CMP/GMP paradigm in myeloid cell production (Doulatov et al., 2010; Edvardsson et al., 2006; Manz et al., 2002). CD66b is an important marker for neutrophil identification. However, it is often excluded from flow cytometry panels geared towards discovery of hematopoietic progenitors. We decided to retain this marker in our search for the early neutrophil progenitor in human bone marrow. We developed a flow cytometry panel to investigate the neutrophil lineage by focusing on CD45+ cells which excluded other HSPCs, including HSC, multipotent progenitors (MPP), CLP, multilymphoid progenitor (MLP), lymphoid-primed multipotent progenitors (LMPP), MEP, EoP, CMP, GMP, cMoP and MDP and other terminally differentiated leukocytes (Doulatov et al., 2012; Edvardsson et al., 2006; Hoebeke et al., 2007; Kawamura et al., 2017; Kohn et al., 2012; Lee et al., 2015; Manz et al., 2002; Mori et al., 2009; Notta et al., 2016; Weiskopf et al., 2016). In this panel, all gates were strictly controlled with both single color and FMO controls (Figure S6A). Indeed, we discovered that human bone marrow contains a CD66b+ population that expresses CD117 (Figure 5A), suggesting the presence of CD66b+ stem cell progenitors within human bone marrow (termed here as hNeP). This CD66b+CD117+ population expresses high levels of CD38+ (Figure S6B), an important stem cell marker that is exclusively expressed by CMP/GMP (Doulatov et al., 2010, 2012; Kohn et al., 2012; Manz et al., 2002), suggesting that this population is committed to the myeloid lineage for development. ScRNA-Seq analysis of this CD66b+CD117+ human neutrophil progenitor population revealed two major subsets which showed either positive (Subset A) or negative (Subset B) expression of CD34 (Figure 5B). Interestingly, lower CD34 gene expression in Subset B is associated with increased expression of neutrophil-specific genes such as ELANE and LYZ (Figure 5B). We then confirmed the CD34+ and CD34−subsets suggested by scRNA-seq by flow cytometry (Figure 6C). Both subsets appeared positive for Ki67 localization in the nuclei, suggesting active proliferation, with a slightly higher (about 1.3 fold) Ki67 mean fluorescence intensity value in CD34+ hNeP compared to CD34−hNeP (Figure 5D).
Figure 5. Human BM contains a CD66b+CD117+ hNeP fraction that contains CD34+ and CD34−subsets.
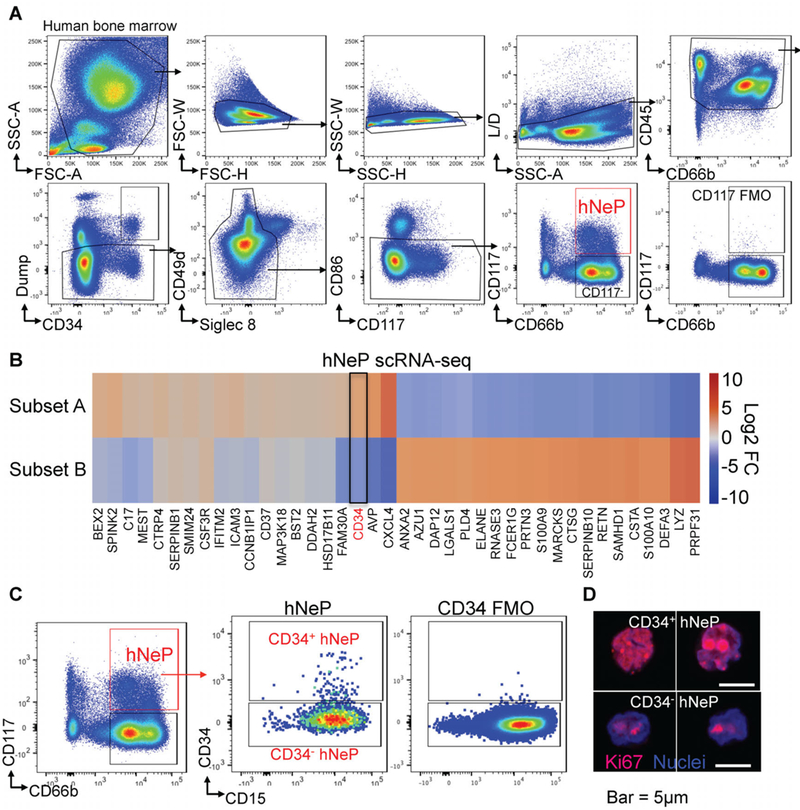
(A) Flow cytometry analysis of healthy human BM uncovers a heterogeneous Lin−CD66b+CD117+ hNeP fraction. Dump antibody cocktail contains antibodies against markers that are expressed by: HSC (CD90 (Thy1)), lymphocytes and their progenitors (CD3, CD19, CD56, CD161, CD7, CD127 (IL-7Rα)), Erythrocytes and their progenitors (CD41, CD235a (Glycophorin A)), Eosinophils/Basophils and their progenitors (Siglec 8, FcεRIα, , CD125 (IL-5Rα)), CMP/GMP and monocyte progenitors (CD123 (IL-3Rα)), DCs and macrophages (CD11c, CD169). (B) ScRNA-Seq analysis of Lin−CD66b+CD117+ hNeP cells reveals two major subpopulations, Subset A and Subset B. 20,000 hNeP cells were FACS sorted from healthy human BM for scRNA-Seq. Heatmap shows top 40 differentially expressed genes in each cluster. Log2 Fold Change of each gene expression is relative to the entire dataset. 2 biological triplicates, 2 technical replicates. (C) Lin−CD66b+CD117+ hNeP were divided into CD34+ subset and CD34−subset by flow cytometry. Confocal microscopy was used to detect Ki67 localization (red) within the nuclei (blue) in CD34+ subset and CD34−subset using antibodies to Ki67 and Hoechst. IgG stained cells served as negative control. Bar: 5µm.
Figure 6. hNeP produce only neutrophils in NSG-SGM3 (NSG-M3) mice in vivo.
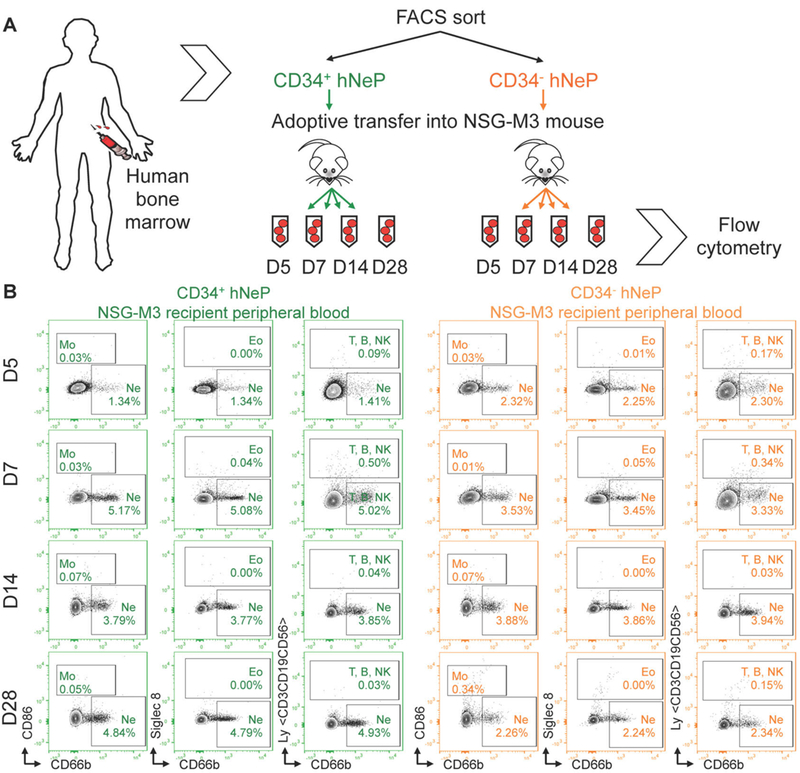
(A) Scheme showing the experimental procedure. CD34+ hNeP and CD34−hNeP subsets identified in Figure 5C were sorted from healthy human BM, and adoptively transferred into NSG-M3 recipient mice. Each recipient mouse received 25,000 donor human hNeP progenitor cells. After the transfer, peripheral blood was collected from each recipient via saphenous vein for flow cytometry on Day (D) 5, 7, 14, 28 (D5, D7, D14, D28), respectively. (B) Representative plots show the appearance of monocytes (CD86+ CD66b−), neutrophils (CD86−Siglec 8−CD66b+), eosinophils (Siglec 8+), and lymphocytes (hLy+) in each recipient group at the time points indicated. hLy antibody cocktail contains CD3, CD19, and CD56. N = 10 mice for each group.
Both hNeP subsets produce only neutrophils in NSG-SGM3 (NSG-M3) mice.
We then examined the neutrophil potency of both CD34+ and CD34−hNeP subsets in vivo by performing adoptive transfers of each subset into NSG-SGM3 (NSG-M3) mice. The triple transgenic NSG-M3 mice are immunodeficient NOD scid gamma (NSG) mice that express the human cytokines Interleukin 3 (IL-3), granulocyte/macrophage-stimulating factor (GM-CSF) and SCF, also known as KITLG. This mouse model supports stable engraftment of the human hematopoietic system, including the myeloid lineage (Billerbeck et al., 2011; Coughlan et al., 2016). The two subsets were isolated from fresh human bone marrow by FACS using the sorting panel in Figure 5 and transferred into two groups of recipient NSG-M3 mice. Peripheral blood of each NSG-M3 recipient mouse was collected at day 5, 7, 14 and 28 for flow cytometry analysis (Figure 6A). To analyze the progeny produced, we used a control group of NSG-M3 recipient mice that received all CD34+ HSPCs (that contains progenitors for all leukocyte cell types). The blood of this control group was analyzed for monocyte (Mo), neutrophils (Ne), eosinophils (Eo), and lymphocytes (Ly) including T cells, B cells and NK cells using the flow cytometry panel shown in Figure S7. This flow cytometry panel is then used for the analysis of hNeP recipient blood. After adoptive transfer, CD66b+ cells were detected in both CD34+ hNeP and CD34−hNeP recipients but no other cell types were expressed (Figure 6B), illustrating that both hNeP subsets are unipotent progenitors that produce only neutrophils. Repopulation of the neutrophil pool by either hNeP progenitor subset occurred quickly after the adoptive transfer (day 5) and lasted to day 28 (Figure 6B), indicating relatively long-term neutrophil unipotency of both progenitor subsets. These data demonstrate that the CD66b+ CD117+ CD38+ CD34+/−fraction in human BM cells contains the unipotent human neutrophil progenitor (hNeP) that occupies about 1–3% of CD45+ cells in human BM under homeostatic conditions.
hNeP increase in melanoma patient blood and promote early osteosarcoma tumor growth in humanized NSG-M3 mice.
We, of course wanted to see if hNeP played a role in tumorigenesis. First, we analyzed blood from human subjects with melanoma for the presence of hNeP (Table S2). Flow cytometry analysis of blood from melanoma versus healthy patients blood using the panel in Figure 5 revealed the presence of CD66b+CD117+ cells (about 1% of circulating CD45+ cells) in blood of healthy donors (Figure 7A). The frequency of these hNeP was significantly elevated in blood of melanoma patients, with frequencies of about 3–9% of circulating CD45+ cells (Figure 7A). We did not observe direct correlations between the hNeP frequencies and gender or age despite the small pool of donors (data not shown). Importantly, CD34+ hNeP were barely detected in healthy donor blood while elevated in blood of melanoma patients (Figure 7A). This increase of hNeP cells in human melanoma patient blood is consistent with what we have observed for mouse NeP in our mouse melanoma model (Figure S5A), suggesting that the hNeP could serve as a biomarker candidate for early cancer detection.
Figure 7. hNeP is increased in melanoma patient blood and promotes early osteosarcoma tumor growth in NSG-M3 mice.
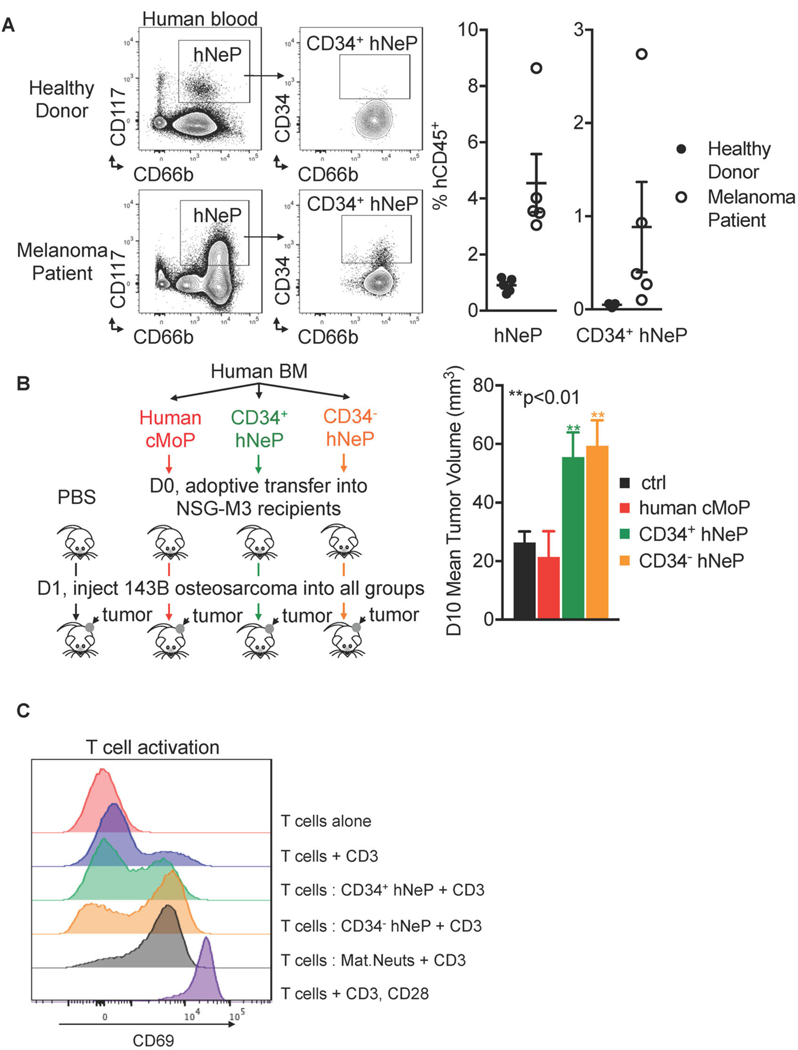
(A) hNeP is increased in melanoma patient blood. hNeP frequency was detected by flow cytometry in peripheral blood collected from healthy donors (N = 5) and melanoma patients (N = 5). Error bars indicate mean ± SEM. (B) Left, scheme showing the experiment procedure. CD34+ hNeP subset, CD34−hNeP subset, and human cMoP were FACS sorted from healthy human BM. The 3 populations were adoptively transferred into NSG-M3 recipient mice. Each recipient mouse received 25,000 donor human progenitor cells. Blank control group received only PBS for adoptive transfer. The next day, 1 × 106 143B human osteosarcoma cells were SubQ injected into each recipient mouse. Right, the tumor volume in each recipient was measured at 10d post-injection. N = 5 mice in each group. Error bars indicate mean ± SEM. (C) CD34+ hNeP cells blunt T cell activation. FACS sorting strategies for CD34+ hNeP and CD34−hNeP are shown in Figure 5A and C using using flow cytometry; FACS sorting strategies for mature neutrophils is shown in Figure S9A using flow cytometry. Histogram overlay of CD69 for CD3+ T cells cultured with CD34+ hNeP, CD34−hNeP, and mature neutrophils, in the presence of anti-CD3 for 24 hours. CD3+ T cells alone cultured without anti-CD3 served as the negative control group, and CD3+ T cells alone cultured with anti-CD3, CD28 served as the positive control group for T cell activation.
Then we examined the role of hNeP in regulating multiple solid tumor types growth to see if the tumor-promoting role of neutrophil progenitors were relevant in more than one tumor type using NSG-M3 mice. A high neutrophil to lymphocyte ratio is an indicator of worse prognosis in sarcomas (Anderson, 2017). Here we used osteosarcoma as model of solid tumor. Shown in Figure 7B, left, both CD34+ hNeP and CD34−hNeP were isolated from human BM and adoptively transferred into NSG-M3 recipient mice. Two different control groups were used in this experiment: one control group received only PBS for adoptive transfer, the other group received human cMoP as a source of human monocyte progenitors. Human cMoP were sorted from the same human BM donor using the panel described previously (Kawamura et al., 2017). One day one after adoptive transfer of progenitors, 1 × 106 human osteosarcoma cells were injected SubQ to the rear flank of mice in all 4 recipient groups. The tumor size was measured 10 days after injection. As shown in Figure 7B, right, mice receiving either CD34+ hNeP or CD34−hNeP cells showed an increase in tumor growth compared to recipient mice receiving cMoP or PBS as a control. This data is concomitant with the mouse data shown in Figure 4B, suggesting that hNeP, the counterpart of mouse NeP, also are pro-tumoral and mediate solid tumor growth.
Finally, as we observed increased tumor size with hNeP adoptive transfer, we asked whether hNeP promoted tumor growth by blunting T cell activation. CD34+ hNeP, CD34−hNeP, or mature neutrophils were FACS sorted from fresh human BM and co-cultured with purified CD3+ T cells isolated from other donor’s blood in the presence of anti-CD3. At 24h after co-culture, mature neutrophils efficiently induced CD3+ T cell activation as measured by CD69+ expression (Figure 7C). CD3+ T cells co-cultured with CD34+ hNeP expressed very low levels of CD69 compared to the mature neutrophil co-culture group, suggesting significant induction of suppression in this group. CD34−hNeP inhibited T cell activation to a lesser extent compared to CD34+ hNeP (Figure 7C). These data suggest that compared to mature neutrophils, hNeP are possibly immunosuppressive and promote tumorigenesis by attenuating tumor-destructive, pro-inflammatory T cell activation.
Discussion
In the current manuscript, we report the discovery of a new, very early-stage, committed unipotent neutrophil progenitor (NeP) that is present in mouse and human bone marrow. We found that both the mouse and human NeP promoted primary tumor growth in vivo in established cancer models. Further, we identified the presence of the human NeP (hNeP) in the blood of patients with recently diagnosed melanoma, suggesting that this hNeP is released from the bone marrow in patients with cancer, and can be readily identified in human blood.
Importantly, we found a tumor-promoting role for this new early-stage neutrophil progenitor in both mice and humans. In tumor-bearing mice, frequencies of this NeP are increased in bone marrow, suggesting aberrant myelopoiesis in response to tumor growth (Figure 4A). These results are consistent with previous studies that suggest that the tumor reprograms GMP to cause increased production of tumor-associated neutrophils (Casbon et al., 2015). Interestingly, we found that tumor-induced myelopoiesis is specific for NeP in mouse BM (Figure 4A). Further, when adoptively transferred into recipient mice, the NeP significantly promoted melanoma tumor growth compared to other myeloid progenitors and was also found in the periphery as well as in the tumor, suggesting egress from the bone marrow and infiltration to the tumor. Some of the #C1 and #C2 cells remain undifferentiated once they infiltrate into tumor and meanwhile start to express PD-L1 (Figures S5 and 4C). We also detected similar tumor-promoting effects of hNeP in human tumorigenesis using a NSG-humanized mouse model. After adoptive transfer, hNeP significantly promoted osteosarcoma tumor growth in NSG mice compared to other myeloid progenitors (Figure 7B). Importantly, we observed a 3–9 fold increase of hNeP in the blood of patients diagnosed with melanoma. This result is consistent with our observation of increased NeP in in mouse periphery in response to tumor growth (Figure S5), and suggests that this hNeP could be used in some manner as a biomarker for early cancer detection.
The earliest committed neutrophil progenitor has remained elusive for decades. Most studies have focused on murine hematopoiesis. In this regard, the classic model of hematopoiesis shows that LSK+ (Lin−CD117+Ly6A/E+CD127−) HSPCs give rise to CLP (Lin−CD117loLy6A/E+CD127+) for lymphopoiesis and to the Lin−CD117+Ly6A/E−CD127−HSPCs for myelopoiesis (Weissman et al., 2001). A higher level of heterogeneity exists within the lin−CD117+Ly6A/E−CD127−population and the committed long-term monocyte progenitor partially overlaps with this HSPC fraction (Auffray et al., 2009; Olsson et al., 2016; Paul et al., 2015; Pronk et al., 2007). Indeed, further examination of the Lin−CD117+Ly6A/E−HSPC fraction by mass cytometry showed 5 possibly committed myeloid progenitors (Figure 1A). Cluster#C in Figure 1A showed low to moderate expression of Ly6G, suggesting a neutrophil lineage potential for cells found within this cluster. This cluster was not identified in earlier hematopoiesis studies as the neutrophil marker Ly6G was routinely excluded from flow cytometry panels at that time. ScRNA-Seq analysis of this Ly6G-containing Cluster#C further revealed 2 populations: an early-stage progenitor (#C1) with stem-cell morphology and little Ly6G expression and a late-stage precursor (#C2) that expressed low levels of Ly6G with morphological features similar to transient neutrophil precursors (Figure 2D and S3B) (Evrard et al., 2018; Satake et al., 2012; Sturge et al., 2015; Yáñez et al., 2015). Recently, a late-stage neutrophil precursor was identified in bone marrow of mice (Kim et al., 2017). We located this population (termed by us as K.NeuP) on a viSNE map of Lin−CD117+Ly6A/E−HSPCs (Figure S6A). Surprisingly, we found that this K.NeuP population was highly heterogeneous and contained other myeloid progenitors. From our mass cytometry data, we were able to generate a stringent flow cytometry gating strategy (Figure S8A) that allowed us to completely purify, with no contamination from other myeloid lineages, both #C1 (NeP) and #C2 cells (late-stage precursors) (Figure 2B) in order to demonstrate their neutrophil unipotency. We also located the recently reported mouse neutrophil precursor (termed by us as Ng.preNeu) (Evrard et al., 2018) and aligned it with #C1 (NeP) and #C2 in the neutrophil developmental branch. ViSNE analysis suggested the mouse Ng.preNeu shares phenotype that shares approximately 22% similarity to #C1 NeP, while mostly similar to #C2 (Figure S8C). The signature nuclear shape of the murine Ng.preNeu also closely resembles that of #C2 (Evrard et al., 2018). In addition, we compared the human Ng.preNeu (Evrard et al., 2018) to the hNeP that we have identified. The human Ng.preNeu does not express CD117 and CD34, therefore does not overlap with the hNeP we identified (Figure S9A). Likely, the human Ng.preNeu represents a transient precursor between hNeP and terminally differentiated neutrophils. This hypothesis is supported by our analysis shown in Figure S9B that hNeP represents the lowest frequency (1–3%) of human BM CD45+ cells followed by Ng.preNeu (4–10%), immature neutrophils 20–40%, and mature neutrophils 30–40%.
The life span of human neutrophils is better studied than is neutrophil heterogeneity. It is commonly recognized that the life span of neutrophils that are isolated by gradient separation varies from hours to days (Bekkering, 2013; Pillay et al., 2010). One possible hint we can take from this notion is that gradient isolation may yield a heterogenous neutrophil population that contains neutrophil subsets and/or immature and mature neutrophils that are at different differentiation stages. Indeed, neutrophil heterogeneity has been suggested in both humans and mice (Beyrau et al., 2012; Silvestre-Roig et al., 2016). A few biological markers such as CD49 or TCRα, β variants were suggested to identify certain neutrophil subsets in addition to neutrophil markers Ly6G (mouse) and CD66b (human) (Silvestre-Roig et al., 2016). However neutrophil heterogeneity is still not well-defined. In this study, we focused on the unipotency and the functions of neutrophil progenitors rather than the heterogeneity of the produced neutrophil progeny. Although we know that the identified NePs give rise to solely neutrophils, we have not yet studied whether these progenitors give rise to specific subsets of neutrophils, particularly in the setting of cancer. Hints from our data suggest that these novel hNeP neutrophil progenitors may promote tumor growth by suppressing T cell activation. Future studies will further examine the mechanisms by which these hNeP subsets influence T cell responses within the tumor microenvironment. We also do not exclude the possibility that the NeP itself, once egressed from the bone marrow and present in the periphery, could directly participate in the tumor-promoting effects we observed (Figure 4 and 7).
In sum, using mass cytometry we have identified a novel, new, early-stage committed unipotent neutrophil progenitor that is present in both mouse and human bone marrow. This discovery may drive new therapeutic and pharmaceutical targets for neutrophil-related diseases or treatment outcomes that are associated with chronic inflammation. For example, neutropenia leads to high susceptibility to infections and is often associated as a by-product of cancer treatments (Lyman et al., 2014). Targeting hNeP could rescue patients from undesirable neutropenia. In addition, our observation of increased hNeP in blood of melanoma patients could assist in early detection for cancer diagnosis as a biomarker. As this hNeP also displays tumor-promoting effects, we suggest the possibility that this hNeP itself could be an immune-oncology target, which opens a new field of therapeutic discovery.
Methods
Mice
C57BL/6J, B6 CD45.1 congenic mice, and NSG-SGM3 mice were purchased from The Jackson Laboratory. Mice were fed a standard rodent chow diet and were housed in microisolator cages in a pathogen-free facility. Mice were euthanized by CO2 inhalation followed by cervical dislocation. All experiments followed approved guidelines of the La Jolla Institute for Allergy and Immunology Animal Care and Use Committee, and approval for use of rodents was obtained from the La Jolla Institute for Allergy and Immunology according to criteria outlined in the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health. Animals were randomly assigned to groups from available mice bred in our facility or ordered from distributor. Experiments in this study used male animals 6–10 weeks of age in good health. If animals were observed with non-experiment related health conditions (i.e. malocclusion, injuries from fighting, etc.), animals were removed from study groups. For tumor studies, B16F10 melanoma cells and 143B human osteosarcoma cells were obtained from ATCC. Cell lines were tested for being pathogen free. Cell lines were maintained in DMEM medium containing 10% heat-inactivated FBS, 2 mmol/L l-glutamine, 1 mmol/L sodium pyruvate, 50 U/mL penicillin, 50 µg/mL streptomycin. For tumor injection, the hair around the tumor injection area of the 6–10 week old mice or adoptive transfer recipients was removed before injection. For Figure 4A and S5A, 5 × 105 B16F10 cells were washed and resuspended in 100 ul D-PBS and then SubQ injected into the rear flank of the mouse, and the tumor-bearing mice were euthanized by CO2 inhalation followed by cervical dislocation at Day 14 post-tumor injection. For Figure 4B and S5B, 3 × 105 B16F10 cells were washed and resuspended in 100 ul DPBS and then SubQ injected into the rear flank of the mouse, and the tumor size were measured with a digital caliper at Day 12 and Day 22 post-tumor injection. For Figure 7B, 1 × 106 143B human osteosarcoma cells were washed and resuspended in 100 ul DPBS and then SubQ injected into the rear flank of the mouse, and the tumor size were measured with a digital caliper at Day 10 post-tumor injection. Tumor volume was calculated using the formula V (volume) = D × d2/2 (D is the largest measured tumor diameter and d is the smallest measured tumor diameter). Laboratory personnel were blinded to the identities of experimental groups during sample collection and analysis.
Human bone marrow cells
Fresh BM samples of anonymous healthy adult donors were obtained from AllCells, Inc. (Alameda, CA). The cells were stained for either flow cytometry or FACS-sorting following protocols described in the Flow Cytometry and Cell Sorting section.
Melanoma Patient Blood Collection
Blood from melanoma patients (no previous radiation, no prior chemo treatment) was collected in EDTA-tubes by the Biospecimen Repository Core Facility (BRCF) at University of Kansas Cancer Center and delivered via overnight shipping. In the meantime, blood from healthy donors were collected in EDTA-tubes in La Jolla Institute for Allergy and Immunology and stored and treated similarly as control groups for the study. All blood samples were processed at the same time and cells were stained for flow cytometry followed by the protocol described in the Flow Cytometry and Cell Sorting section.
Human Peripheral Blood Collection
EDTA-coated blood from healthy volunteers was obtained after written informed consent under the guidelines of the Institutional Review Board of the La Jolla Institute for Allergy and Immunology and in accordance with US Dept. of Health and Human Services Policy for protection of Human Research Subjects (VD-057–0217). Cells were stained for flow cytometry followed by the protocol described in the Flow Cytometry and Cell Sorting section.
Cell suspension for mass cytometry and flow cytometry
BM cells were harvested from femurs, and tibias of 6–10 week old mice. Bones were centrifuged for the collection of marrow. For the adoptive transfer experiments, donor BM cells were collected and stained under sterile conditions. Peripheral blood was obtained by cardiac puncture with an EDTA-coated syringe. For Figure 6B and S4B, a drop of blood was obtained from the saphenous vein of the adoptive transferred NSG-SGM3 mice recipients. All samples (both mouse and human) were collected in ice cold D-PBS (Gibco) with 2 mM EDTA to prevent cation-dependent cell-cell adhesion. Prior to staining cells, cells were subject to a red blood cell lysis (RBC lysis buffer, eBiosciences) at room temperature (5 min x 1 for BM cells, 10 min x 2 for blood cells). Cells were washed and filtered through a 70 µm strainer. Cell suspensions were prepared by sieving and gentle pipetting to reach final concentration of 3 × 106 cells per 100 ul buffer.
Mass Cytometry Antibodies
Metal-conjugated antibodies were purchased directly from Fluidigm for available targets. For all other targets, purified antibodies were purchased from the companies listed in Table S1. Antibody conjugations were prepared using the Maxpar Antibody Labeling Kit according to the recommended protocol provided by Fluidigm. Maxpar-conjugated antibodies were stored in PBS-based antibody stabilization solution (Candor Biosciences) supplemented with 0.05% NaN3 at 4°C. All antibodies were titrated before use.
Mass Cytometry (CyTOF)
For viability staining, cells were washed in PBS and stained with Cisplatin (Fluidigm) to a final concentration of 5 µM. Prior to surface staining, anti-CD16/32 (151Eu) antibody was added to cell suspension in ice-cold staining buffer (PBS + 2mM EDTA + 0.1% BSA + 0.05% NaN3) to stain and block the Fc receptors for 15 min. The surface antibody cocktail listed in Table S1 was then added into the cell suspension for 1h. The cells were then washed and fixed with 2% paraformaldehyde overnight at 4°C. After fixation, cells were washed in staining buffer and permeabilized using Foxp3/Transcription Factor Staining Buffer (eBioscience) for intracellular staining according to the manufacturer’s protocol. Following permeabilization, cells were washed twice with 1 ml 1X Perm Buffer (Saponin-based). The intracellular antibody cocktail listed in Table S1 were added into cell suspension for 1h. Cells were then washed in staining buffer and stained with DNA intercalator (Fluidigm) containing natural abundance Iridium (191Ir and 193Ir) prepared to a final concentration of 125 nM in 2% paraformaldehyde. Cells were washed in staining buffer, with subsequent washes in Milli-Q water (EMD Millipore) to remove buffer salts. Cells were resuspended in Milli-Q water with a 1:10 dilution of EQ Four Element Calibration beads (Fluidigm) and filtered through a 35 µm nylon mesh filter cap (Corning, Falcon). Samples were analyzed on a Helios 2 CyTOF Mass Cytometer (Fluidigm) equipped with a Super Sampler (Victorian Airship & Scientific Apparatus) at an event rate of 500 events/second or less. Mass cytometry data files were normalized using the bead-based Normalizer (Finck et al, Cytometry A 83:48) and analyzed using Cytobank analysis software (https://www.cytobank.org/). The PhenoGraph clustering(Levine et al., 2015) and isomap dimensionality reduction were done using R package cytofkit(Chen et al., 2016). Hierarchical clustering was used to determine two meta-clusters based on the median of markers’ expression from each PhenoGraph clusters.
Flow Cytometry Antibodies
Antibodies for flow cytometry were purchased from commercial sources as follows: anti-mouse CD3ε (145–2C11; BD Biosciences); anti-mouse CD19 (1D3; BD Biosciences); anti-mouse CD161 (PK136; eBiosciences); anti-mouse F4/80 (T45–2342; BD Biosciences); anti-mouse CD11c (HL3; BD Biosciences); anti-mouse CD45 (30-F11; BioLegend); anti-mouse CD45.1 (A20; BioLegend); anti-mouse CD45.2 (104; BioLegend); anti-mouse CD117 (c-kit) (2B8; BioLegend); anti-mouse Ly6A/E (Sca-1) (D7; BioLegend); anti-mouse CD16/32 (FcγRIII/II (93; BioLegend); anti-mouse/human CD11b (M1/70; BioLegend); anti-mouse CD115 (M-CSFR) (AFS98; BioLegend); anti-mouse Ly6G (1A8; BioLegend); anti-mouse Ly6C (HK1.4; BioLegend); anti-mouse Siglec F (E50–2446; BD Biosciences); anti-mouse FcεRIα (MAR-1; BioLegend); anti-mouse Ki67 (SolA15; eBiosciences); anti-human CD45 (2D1; BioLegend); anti-human CD3ε (HIT3a; BD Biosciences); anti-human CD3ε (OKT3; BD Biosciences); anti-human CD7 (MT701; BD Biosciences); anti-human CD161 (HP-3G10; BioLegend); anti-human CD56 (B159; BD Biosciences); anti-human CD19 (HIB19; BD Biosciences); anti-human CD127 (A019D5; BioLegend); anti-human Siglec 8 (7C9; MACS Miltenyi Biotec); anti-human FcεRIα (AER-37; BioLegend); anti-human CD235a (GA-R2; BD Biosciences); anti-human CD41 (HIP8; BD Biosciences); anti-human CD169 (7–239; BD Biosciences); anti-human CD69 (FN50; BioLegend); anti-human CD11c (B-ly6; BD Biosciences); anti-human CD90 (5E10; BioLegend); anti-human CD86 (IT2.2; BioLegend); anti-human CD66b (G10F5; BioLegend); anti-human CD34 (581; BD Biosciences); anti-human CD117 (YB5.B8; BD Biosciences); anti-human CD38 (HB-7; BioLegend); anti-human CD15 (W6D3; BioLegend); anti-human CD49d (9F10; BioLegend); anti-human CD10 (Hl10a; BioLegend); anti-human CD101 (BB27; BioLegend); anti-human CD16 (3G8; BioLegend). Cell viability was determined with LIVE/DEAD™ Fixable Yellow (or Blue) Dead Cell Stain Kit (ThermoFisher).
Flow Cytometry and Cell Sorting
All mouse FACS staining was performed in FACS buffer (D-PBS + 1% BSA + 0.1% sodium azide + 2mM EDTA) on ice. All human FACS staining was performed in FACS buffer (D-PBS + 1% human serum + 0.1% sodium azide + 2mM EDTA) on ice. Cells were filtered through sterile 70µm cell strainers to obtain a single cell suspension (30,000 cells per µl for flow cytometry analysis, 0.5 – 2 × 107 per ml for sorting). Prior to surface staining, anti-CD16/32 (FITC) antibody (for mouse) or human Fc receptors blocking reagent (MACS Miltenyi Biotec) was added for 15 min to stain and block the Fc receptors. Surface staining was performed for 30 minutes in a final volume of 500ul for FACS sorts and 100ul for regular flow cytometry. Cells were washed twice in at least 200ul FACS buffer before acquisition. Cells were sorted using a FACS Aria II and Aria-Fusion (BD biosciences) and conventional flow cytometry using an LSRII or a LSR Fortessa (BD Biosciences). All flow cytometry was performed on live cells. Calculations of percentages of CD45+ immune cells were based on live cells as determined by forward and side scatter and viability analysis. All analyses and sorts were repeated at least 3 times, and purity of sorted fractions was checked visually and by FACS reanalysis of the surface markers. Data were analysed using Cytobank (https://www.cytobank.org/) and FlowJo (version 10.1r5).
Confocal Microscopy
Cells were FACS-sorted by using the flow cytometry panel shown in Figure S10B and resuspended in PBS. Following fixation in 4% methanol-free formaldehyde in PBS for 10 min at room temperature, cells were washed with PBS and resuspended in 5% normal donkey serum, 0.3% Triton X-100 in PBS for one hour. Cells were then incubated with a rabbit anti-Ki67 monoclonal antibody (clone SP6, Abcam, 1:150) or negative control (normal rabbit IgG) in 1% bovine serum albumin and 0.3% Triton X-100 in PBS overnight at 4 °C. Cells were washed twice with PBS and incubated with anti-rabbit IgG (H+L) F(ab’)2 fragment conjugated to Alexa Fluor 647 (Cell Signalling, #4414, 1:500) and Hoechst (1:1000 of 10 mg/ml solution) for one hour at room temperature. After washing, cells were adhered to poly-L-lysine coated #1.5H coverslips and embedded in Prolong Gold (Thermo Fisher). Samples were imaged with a Zeiss LSM780 and Leica SP8 confocal microscopes using a 63x/1.40 NA oil-immersion objectives. Images were processed with ZEN or Leica HyVolution software and 3D reconstructions of DNA were created in Imaris software. The mean and integrated fluorescence intensity (select the one you will show) of Ki-67 within the nuclear regions were calculated in Image-Pro Premier. To reduce Z-stretching confocal images were deconvolved with Huygens Essential. Analysis of the surface area, volume and sphericity was performed in Imaris software.
Adoptive transfer
Recipient mice were housed in a barrier facility under pathogen-free conditions before and after adoptive transfer. NSG-SGM3 recipient mice were maintained in sterile conditions at all times. CD45.1 recipient mice were fed with autoclaved acidified water with antibiotics (trimethoprimsulfamethoxazole) for 3 days before the adoptive transfer. Sub-lethally irradiated recipient mice received 600 Rads. Donor BM cells were collected and FACS sorted as described in the flow cytometry section. Mouse and human progenitor cells were sorted directly into sterile FBS and kept chilled during sorting. Cells then were washed and resuspended in ice-cold D-PBS for injection. 50,000 donor progenitors in 200 ul DPBS were delivered into each recipient mouse for Figures 3 and 4. 25,000 donor progenitors in 200 ul DPBS were delivered into each recipient mouse for Figures 6 and 7. All adoptive transfer experiments were achieved via tail vein injection. After the adoptive transfer, recipient mice were provided with autoclaved food and autoclaved acidified water with antibiotics.
In vitro progenitor differentiation assay
Sorted progenitor cells were seeded into 6-well plates and cultured for 10 days with MethocultTM GF M3434 media (Stem Cell Technologies) according to the manufacturer’s protocol. The numbers of wells containing proliferated colonies were counted for colony-forming assays.
Single-cell RNA-seq. 3’ end
Mouse Cluster#C cells were FACS-sorted using the flow cytometry panel shown in Figure S10A. Human hNeP were FACS-sorted by using the flow cytometry panel shown in Figure 5A. Single cell RNA-Sequencing was performed using Chromium™ Single Cell 3’ v2 Reagent Kits (10x Genomics) following the manufacturer’s protocol(Zheng et al., 2017). Briefly, after sort collection, cells were resuspended in PBS at concentration ranging between 400 to 600 cells per µl. Between 5,000 to 10,000 cells were loaded for gel bead-in-emulsion generation and barcoding. To increase barcode diversity, samples were split in 2 technical replicates for all downstream steps: Reverse transcription, cDNA amplification, fragmentation and library preparation. Final libraries with size ranging between 200 to 1000 bp were size-selected using Ampure XP beads (Beckman Coulter). Quality and quantity of samples was controlled at multiple steps during the procedure by running small fraction (< 5%) of sample on BioAnalyzer (high sensitivity DNA chip, Agilent). Libraries were sequenced on HiSeq2500 platform to obtain 26 (read1) x 100 (read2) paired-end reads.
Single cell RNA-Seq analysis
Using Cell Ranger v1.3.0 (10x genomics), reads were aligned on the mm10 reference genome for mouse and hg19 reference genome for human and unique molecular identifier gene expression profiles were generated for every single cell reaching standard sequencing quality threshold (default parameters). On average we obtained data for 2868 cells for mouse samples and 518 cells for human samples, and on average 46,477 reads per cell for mouse and 274,080 reads per cell for human. Only confidently mapped, non-PCR duplicates with valid barcodes and UMIs were used to generate a gene-barcode matrix for further analysis. Counts were normalized to get counts per million (CPM). Unbiased clustering of single cells was performed using Seurat (version 1.4) (Rizzo, 2016; Satija et al., 2015). Principal Component Analysis (PCA) was performed using a set of top variable genes (ranging between 647 to 2142 genes) and then dimensionality reduction was performed using t-SNE algorithm with top 10 to 18 PCAs. For Figure 2A, tSNE 2D plots were obtained applying Seurat scRNA-Seq analysis R Package (using 12 first PCA, and 810 most variable genes with resolution parameter set at 0.03).
RNA-Seq
Cells were FACS-sorted by using the flow cytometry panel shown in Figure S10B. RNA-Seq was prepared on FACS sorted BM cells using Universal Plus mRNA-Seq (Nugen) and sequenced on an Illumina MiSeq instrument. Single-ended reads in FASTQ format were mapped to mouse genome (mm10) using Rsubread(Liao et al., 2013) and overlapped with UCSC mm10 transcriptome annotation from using FeatureCount R package(Liao et al., 2014). Gene expression level was then quantified as counts per million (CPM) using edgeR(Robinson et al., 2010).
Human T cell co-culture with neutrophil progenitors
To investigate the effects of neutrophil progenitor subsets on human autologous T cell activation, CD3+ T cells were negatively-selected from LJI healthy donor PBMCs according to manufacturer’s instructions (StemCell). CD34+ hNeP, CD34−hNeP, and mature neutrophils were FACS sorted from fresh human BM obtained from AllCells, Inc. (Alameda, CA). The sorted CD3+ T cells were cultured with CD34+ hNeP, CD34−hNeP, and mature neutrophils at a 2:1 ratio, in the presence or absence of 2µg/mL plate-bound anti-CD3 Ab (Biolegend; clone: OKT3) for 24 hrs at 37°C with 5% CO2. For polyclonal stimulation, purified CD3+ T cells were incubated with anti-CD3,-CD28 Dynabeads (Invitrogen). To investigate CD69 activation, cells were collected and stained with anti-human CD3, CD69 , and a fixable live/dead viability dye at 24 hours post stimulation. 75,000 events were collected on the Fortessa (BD Biosciences), and data was analyzed using FlowJo (v.10.3) (Treestar Incorporated).
Statistical analysis
Data for all experiments were analyzed with Prism software (GraphPad). For Figure 4A and S5A, unpaired t-tests were used to compare the values in tumor groups to healthy groups. P value was calculated based on two-tailed comparison with 99% confidence level. For Figure 4B and 7B, unpaired ordinary one-way ANOVA was used for multiple comparisons between experimental groups and the control group. P value was calculated based on Dunnett’s test with 95% confidence level. No statistical methods were used to predetermine sample size. No animal or sample was excluded from the analysis.
Supplementary Material
Acknowledgments
We would like to thank K. Ley, G.D. Thomas, R. Hanna, Z.C. Fan and J. Xie for helpful discussions, D. Yoakum for assistance with mouse colony management, the LJI Histology Core for assistance with cell morphology experiments, Jeremy Day and the LJI Sequencing core for assistance with RNA sequencing, and the LJI Flow Cytometry core for assistance with FACS sorting. The mass cytometry core was supported by NIH grant 1S10OD018499–01 (to C.K. and S.C.). The melanoma patient study was supported by the National Cancer Institute Cancer Center Support Grant P30 CA168524 and used the Biospecimen Shared Resource. This work was supported by NIH grants R01HL134236, P01HL136275, and R01CA202987 (all to C.C.H) and ADA7–12-MN-31 (04) (to C.C.H. and Y.P.Z).
References and Notes
- Akashi K, Traver D, Miyamoto T, and Weissman IL (2000). A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193–197. [DOI] [PubMed] [Google Scholar]
- Anderson PM (2017). Immune Therapy for Sarcomas. Adv. Exp. Med. Biol 995, 127–140. [DOI] [PubMed] [Google Scholar]
- Arinobu Y, Iwasaki H, Gurish MF, Mizuno S-I, Shigematsu H, Ozawa H, Tenen DG, Austen KF, and Akashi K (2005). Developmental checkpoints of the basophil/mast cell lineages in adult murine hematopoiesis. Proc. Natl. Acad. Sci. U. S. A 102, 18105–18110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffray C, Fogg DK, Narni-Mancinelli E, Senechal B, Trouillet C, Saederup N, Leemput J, Bigot K, Campisi L, Abitbol M, et al. (2009). CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J. Exp. Med 206, 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avellino R, Havermans M, Erpelinck C, Sanders MA, Hoogenboezem R, van de Werken HJG, Rombouts E, van Lom K, van Strien PMH, Gebhard C, et al. (2016). An autonomous CEBPA enhancer specific for myeloid-lineage priming and neutrophilic differentiation. Blood 127, 2991–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainton DF, Ullyot JL, and Farquhar MG (1971). The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J. Exp. Med 134, 907–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Schlitzer A, Chen J, Mair F, Sumatoh HR, Teng KWW, Low D, Ruedl C, Riccardi-Castagnoli P, Poidinger M, et al. (2014). High-dimensional analysis of the murine myeloid cell system. Nat. Immunol 15, 1181–1189. [DOI] [PubMed] [Google Scholar]
- Bekkering S (2013). Another look at the life of a neutrophil. World Journal of Hematology 2, 44. [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir E-AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. (2011). Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyrau M, Bodkin JV, and Nourshargh S (2012). Neutrophil heterogeneity in health and disease: a revitalized avenue in inflammation and immunity. Open Biol 2, 120134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billerbeck E, Barry WT, Mu K, Dorner M, Rice CM, and Ploss A (2011). Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rγ(null) humanized mice. Blood 117, 3076–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borregaard N (2010). Neutrophils, from marrow to microbes. Immunity 33, 657–670. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY, and Greenleaf WJ (2018). Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 173, 1535–1548.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casbon A-J, Reynaud D, Park C, Khuc E, Gan DD, Schepers K, Passegué E, and Werb Z (2015). Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. U. S. A 112, E566–E575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, and Chen J (2016a). Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline. PLoS Comput. Biol 12, e1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, and Chen J (2016b). Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline. PLoS Comput. Biol 12, e1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan AM, Harmon C, Whelan S, O’Brien EC, O’Reilly VP, Crotty P, Kelly P, Ryan M, Hickey FB, O’Farrelly C, et al. (2016). Myeloid Engraftment in Humanized Mice: Impact of Granulocyte-Colony Stimulating Factor Treatment and Transgenic Mouse Strain. Stem Cells Dev 25, 530–541. [DOI] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, and Dick JE (2010). Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat. Immunol 11, 585–593. [DOI] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Laurenti E, and Dick JE (2012). Hematopoiesis: A Human Perspective. Cell Stem Cell 10, 120–136. [DOI] [PubMed] [Google Scholar]
- Edvardsson L, Dykes J, and Olofsson T (2006). Isolation and characterization of human myeloid progenitor populations--TpoR as discriminator between common myeloid and megakaryocyte/erythroid progenitors. Exp. Hematol 34, 599–609. [DOI] [PubMed] [Google Scholar]
- Elghetany MT, Ge Y, Patel J, Martinez J, and Uhrova H (2004). Flow cytometric study of neutrophilic granulopoiesis in normal bone marrow using an expanded panel of antibodies: correlation with morphologic assessments. J. Clin. Lab. Anal 18, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrard M, Kwok IWH, Chong SZ, Teng KWW, Becht E, Chen J, Sieow JL, Penny HL, Ching GC, Devi S, et al. (2018a). Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 48, 364–379.e8. [DOI] [PubMed] [Google Scholar]
- Evrard M, Kwok IWH, Chong SZ, Teng KWW, Becht E, Chen J, Sieow JL, Penny HL, Ching GC, Devi S, et al. (2018b). Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 48, 364–379.e8. [DOI] [PubMed] [Google Scholar]
- Fiedler K, and Brunner C (2012). The role of transcription factors in the guidance of granulopoiesis. Am. J. Blood Res 2, 57–65. [PMC free article] [PubMed] [Google Scholar]
- Fogg DK (2006). A Clonogenic Bone Marrow Progenitor Specific for Macrophages and Dendritic Cells. Science 311, 83–87. [DOI] [PubMed] [Google Scholar]
- Hagerling C, and Werb Z (2016). Neutrophils: Critical components in experimental animal models of cancer. Semin. Immunol 28, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettinger J, Richards DM, Hansson J, Barra MM, Joschko A-C, Krijgsveld J, and Feuerer M (2013). Origin of monocytes and macrophages in a committed progenitor. Nat. Immunol 14, 821–830. [DOI] [PubMed] [Google Scholar]
- Hoebeke I, De Smedt M, Stolz F, Pike-Overzet K, Staal FJT, Plum J, and Leclercq G (2007). T-, B- and NK-lymphoid, but not myeloid cells arise from human CD34(+)CD38(−)CD7(+) common lymphoid progenitors expressing lymphoid-specific genes. Leukemia 21, 311–319. [DOI] [PubMed] [Google Scholar]
- Horman SR, Velu CS, Chaubey A, Bourdeau T, Zhu J, Paul WE, Gebelein B, and Grimes HL (2009). Gfi1 integrates progenitor versus granulocytic transcriptional programming. Blood 113, 5466–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Xiao Y, Xu A, and Zhou Z (2016). Neutrophils in type 1 diabetes. J. Diabetes Investig 7, 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura S, Onai N, Miya F, Sato T, Tsunoda T, Kurabayashi K, Yotsumoto S, Kuroda S, Takenaka K, Akashi K, et al. (2017). Identification of a Human Clonogenic Progenitor with Strict Monocyte Differentiation Potential: A Counterpart of Mouse cMoPs. Immunity 46, 835–848.e4. [DOI] [PubMed] [Google Scholar]
- Kim M-H, Yang D, Kim M, Kim S-Y, Kim D, and Kang S-J (2017a). A late-lineage murine neutrophil precursor population exhibits dynamic changes during demand-adapted granulopoiesis. Sci. Rep 7, 39804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M-H, Yang D, Kim M, Kim S-Y, Kim D, and Kang S-J (2017b). A late-lineage murine neutrophil precursor population exhibits dynamic changes during demand-adapted granulopoiesis. Sci. Rep 7, 39804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn LA, Hao Q-L, Sasidharan R, Parekh C, Ge S, Zhu Y, Mikkola HKA, and Crooks GM (2012). Lymphoid priming in human bone marrow begins before expression of CD10 with upregulation of L-selectin. Nat. Immunol 13, 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Breton G, Oliveira TYK, Zhou YJ, Aljoufi A, Puhr S, Cameron MJ, Sékaly R-P, Nussenzweig MC, and Liu K (2015). Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J. Exp. Med 212, 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PY, Wang J-X, Parisini E, Dascher CC, and Nigrovic PA (2013). Ly6 family proteins in neutrophil biology. J. Leukoc. Biol 94, 585–594. [DOI] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir E-AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015a). Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir E-AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015b). Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, Chu F-F, Randolph GJ, Rudensky AY, and Nussenzweig M (2009). In vivo analysis of dendritic cell development and homeostasis. Science 324, 392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman GH, Abella E, and Pettengell R (2014). Risk factors for febrile neutropenia among patients with cancer receiving chemotherapy: A systematic review. Crit. Rev. Oncol. Hematol 90, 190–199. [DOI] [PubMed] [Google Scholar]
- Manz MG, Miyamoto T, Akashi K, and Weissman IL (2002). Prospective isolation of human clonogenic common myeloid progenitors. Proc. Natl. Acad. Sci. U. S. A 99, 11872–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Iwasaki H, Kohno K, Yoshimoto G, Kikushige Y, Okeda A, Uike N, Niiro H, Takenaka K, Nagafuji K, et al. (2009). Identification of the human eosinophil lineage-committed progenitor: revision of phenotypic definition of the human common myeloid progenitor. J. Exp. Med 206, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, Kaufmann KB, McLeod J, Laurenti E, Dunant CF, et al. (2016). Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 351, aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, and Grimes HL (2016). Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 537, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul F, Arkin Y ‘ara, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, Winter D, Lara-Astiaso D, Gury M, Weiner A, et al. (2015). Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 163, 1663–1677. [DOI] [PubMed] [Google Scholar]
- Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JAM, Tesselaar K, and Koenderman L (2010). In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 116, 625–627. [DOI] [PubMed] [Google Scholar]
- Pronk CJH, Rossi DJ, Månsson R, Attema JL, Norddahl GL, Chan CKF, Sigvardsson M, Weissman IL, and Bryder D (2007). Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell 1, 428–442. [DOI] [PubMed] [Google Scholar]
- Qi X, Hong J, Chaves L, Zhuang Y, Chen Y, Wang D, Chabon J, Graham B, Ohmori K, Li Y, et al. (2013). Antagonistic regulation by the transcription factors C/EBPα and MITF specifies basophil and mast cell fates. Immunity 39, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, and Tenen DG (1998). CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol. Cell. Biol 18, 4301–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo ML (2016). Statistical Computing with R (CRC Press; ). [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, Damti P, Lumbroso D, Polyansky L, Sionov RV, et al. (2015). Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep 10, 562–573. [DOI] [PubMed] [Google Scholar]
- Samusik N, Good Z, Spitzer MH, Davis KL, and Nolan GP (2016). Automated mapping of phenotype space with single-cell data. Nat. Methods 13, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake S, Hirai H, Hayashi Y, Shime N, Tamura A, Yao H, Yoshioka S, Miura Y, Inaba T, Fujita N, et al. (2012). C/EBPβ is involved in the amplification of early granulocyte precursors during candidemia-induced “emergency” granulopoiesis. J. Immunol 189, 4546–4555. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Nakagawa K, Sugihara F, Kuwahara R, Ashihara M, Yamane F, Minowa Y, Fukushima K, Ebina I, Yoshioka Y, et al. (2017). Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 541, 96–101. [DOI] [PubMed] [Google Scholar]
- Silvestre-Roig C, Hidalgo A, and Soehnlein O (2016). Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood 127, 2173–2181. [DOI] [PubMed] [Google Scholar]
- Soehnlein O, Steffens S, Hidalgo A, and Weber C (2017). Neutrophils as protagonists and targets in chronic inflammation. Nat. Rev. Immunol 17, 248–261. [DOI] [PubMed] [Google Scholar]
- Sturge CR, Burger E, Raetz M, Hooper LV, and Yarovinsky F (2015). Cutting Edge: Developmental Regulation of IFN-γ Production by Mouse Neutrophil Precursor Cells. J. Immunol 195, 36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, and Chilvers ER (2010). Neutrophil kinetics in health and disease. Trends Immunol 31, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terstappen LW, and Loken MR (1990). Myeloid cell differentiation in normal bone marrow and acute myeloid leukemia assessed by multi-dimensional flow cytometry. Anal. Cell. Pathol 2, 229–240. [PubMed] [Google Scholar]
- Weiskopf K, Schnorr PJ, Pang WW, Chao MP, Chhabra A, Seita J, Feng M, and Weissman IL (2016). Myeloid Cell Origins, Differentiation, and Clinical Implications. Microbiol Spectr 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman IL, Anderson DJ, and Gage F (2001). Stem and progenitor cells: origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol 17, 387–403. [DOI] [PubMed] [Google Scholar]
- Yáñez A, Ng MY, Hassanzadeh-Kiabi N, and Goodridge HS (2015). IRF8 acts in lineage-committed rather than oligopotent progenitors to control neutrophil vs monocyte production. Blood 125, 1452–1459. [DOI] [PubMed] [Google Scholar]
- Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, and Tenen DG (1997). Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc. Natl. Acad. Sci. U. S. A 94, 569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JQ, Biedermann B, Nitschke L, and Crocker PR (2004). The murine inhibitory receptor mSiglec-E is expressed broadly on cells of the innate immune system whereas mSiglec-F is restricted to eosinophils. Eur. J. Immunol 34, 1175–1184. [DOI] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.