

Abstract
Gulf War Illness (GWI) is a chronic multi-symptom disorder afflicting the veterans of the First Gulf War, and includes neurological symptoms characterized by depression and memory deficits. Chronic exposure to organophosphates (OPs) is considered a leading cause for GWI, yet its pathobiology is not fully understood. We recently observed chronic elevations in neuronal Ca2+ levels ([Ca2+]i) in an OP-diisopropyl fluorophosphate (DFP)-based rat model for GWI. This study was aimed at identifying mechanisms underlying elevated [Ca2+]i in this DFP model and investigating whether their therapeutic targeting could improve GWI-like neurological morbidities. Male Sprague-Dawley rats (9 weeks) were exposed to DFP (0.5 mg/kg, s.c., 1×-daily for 5 days) and at 3 months postDFP exposure, behavior was assessed and rats were euthanized for protein estimations and ratiometric Fura-2 [Ca2+]i estimations in acutely dissociated hippocampal neurons. In DFP rats, a sustained elevation in intracellular Ca2+ levels occurred, and pharmacological blockade of Ca2+-induced Ca2+-release mechanisms significantly lowered elevated [Ca2+]i in DFP neurons. Significant reductions in the protein levels of the ryanodine receptor (RyR) stabilizing protein Calstabin2 were also noted. Such a posttranslational modification would render RyR “leaky” resulting in sustained DFP [Ca2+]i elevations. Antagonism of RyR with levetiracetam significantly lower elevated [Ca2+]i in DFP neurons and improved GWI-like behavioral symptoms. Since Ca2+ is a major second messenger molecule, such chronic increases in its levels could underlie pathological synaptic plasticity that expresses itself as GWI morbidities. Our studies show that treatment with drugs targeted at blocking intracellular Ca2+ release could be effective therapies for GWI neurological morbidities.
Keywords: organophosphates, DFP, depression, anxiety, memory deficits, levetiracetam, calcium dynamics, Sprague-Dawley rats
Approximately one-third of the deployed First Gulf War soldier population suffer from a debilitating disorder called Chronic Multi-Symptom Illness, popularly known as the Gulf War Illness (GWI) (Gulf War and Health 2016; White et al., 2016).This disorder continues to affect our Veterans 25 years after the War. The Institute of Medicine defines GWI as a spectrum disorder characterized by the presence of at least 2 of the following 6 categories of symptoms in the absence of other underlying disorders, in soldiers deployed during the First Gulf War, for a period of at least 6 months: fatigue, mood and cognitive changes, pain, gastrointestinal symptoms, respiratory difficulty, and neurologic abnormalities (Gulf War and Health, 2016). Thus, GWI is a multi-symptom condition that has multi-system origins with the nervous system being particularly affected. GWI neurological impairments commonly include mood disorder, anxiety, chronic depression, and cognitive deficits (Black et al., 2004; Blore et al., 2015). These neuropsychiatric conditions are particularly difficult to treat and significantly lower the quality of life in GWI Veterans (Yee et al., 2016).
Deployment complexities of military personnel and their exposure to multiple chemicals present in the First Gulf War theater have made it difficult to identify the underlying etiology of GWI (Brimfield, 2012). Amongst numerous purported causative factors, it is now believed that chronic exposures to organophosphate (OP) compounds, including pesticides such as chlorpyrifos and malathion, and accidental exposure to OP nerve agents sarin and cyclosarin possibly underlies the development of this disorder (Chao et al., 2015; Kerr, 2015; Tuite and Haley, 2013; White et al., 2016). Investigators have mimicked these exposure conditions to develop rodent models of GWI (Abdel-Rahman et al., 2004; Abdullah et al., 2013; Hernandez et al., 2015; O’Callaghan et al., 2015; Parihar et al., 2013). One such model uses Diisopropyl fluorophosphate (DFP), an OP compound, which is used experimentally to mimic effects of the nerve agent sarin (Deshpande et al., 2010). We previously developed a model for GWI by exposing rats to repeated, low-dose DFP and observed delayed neuronal injury and chronic neurobehavioral morbidities including depression, anxiety, and cognitive deficits similar to those reported in GWI Veterans, in the absence of other deployment related confounding factors (Phillips and Deshpande, 2016).
Calcium ions (Ca2+) are signaling molecules that modulate memory, mood, and behavior functions (Bengtson and Bading, 2012). Neuronal Ca2+ homeostasis is a tightly controlled process involving a complex interplay between influx, efflux, release, and buffering mechanisms within the cytoplasm. Ca2+-induced Ca2+-release (CICR) is one such process that is mediated by ryanodine receptors (RyRs) and inositol tris-phosphate receptors (IP3R) that amplify Ca2+ signal by releasing additional Ca2+ from the endoplasmic reticulum (ER) in response to Ca2+ influx via plasmalemmal channels. Disruptions in neuronal Ca2+ homeostasis are implicated in many neurodegenerative disorders including Alzheimer’s disease (Popugaeva et al., 2017), Parkinson’s disease (Surmeier et al., 2017), acquired epilepsy (Nagarkatti et al., 2009), and traumatic brain injury (TBI) (Tehse and Taghibiglou, 2018). These disorders manifest psychiatric comorbidities like those seen in GWI Veterans. In line with these observations, we recently reported that repeated, low-dose DFP-exposed rats exhibited significantly higher hippocampal Ca2+ levels than their age-matched counterparts (Phillips and Deshpande, 2018). However, the mechanisms underlying these sustained neuronal Ca2+ elevations in GWI are unknown. In this study, we investigated a molecular basis for the development of GWI-like neurological morbidities in our DFP-based rodent model. We further investigated whether pharmacological inhibition of CICR-mediated Ca2+ elevations could also prove to be an effective therapy for GWI-like neurological conditions.
MATERIALS AND METHODS
Animals
All animal use procedures were in strict accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. The experimental protocol was approved by Virginia Commonwealth University’s Institutional Animal Care and Use Committee and by the Animal Care and Use Review Office of the U.S. Army Medical Research and Material Command. Male Sprague-Dawley rats (Envigo, Indianapolis, Indiana) weighing approximately 300 g and 9 weeks of age were used in this study. Animals were housed 2 per cage at 20°C–22°C with a 12-h light-dark cycle (lights on 06:00–18:00 h) and given free access to food and water.
Chemicals
All the chemicals were obtained from Sigma Aldrich Company (St Louis, Missouri) unless otherwise noted. Levetiracetam (LEV, Keppra) in injectable form was obtained from VCU Division of Animal Resources Pharmacy. Drugs and buffer solutions were made fresh daily.
DFP Exposure
Immediately upon arrival, DFP (Catalog no. D-0879) was inspected for discoloration that would indicate possible degradation due to oxidation. The vial containing colorless DFP was stored at −80°C. DFP has a short half-life (1 h, pH 7.5 at 25°C). On the day of the prep, DFP was removed from the vial using a Hamilton syringe and diluted with ice-cold phosphate buffered saline to a concentration of 0.5 mg/ml. Syringes filled with DFP were kept on ice and rats were injected with DFP or vehicle within 15 min of DFP preparation.
Rats were injected with DFP (0.5 mg/kg, s.c.) once daily for 5 days, while control rats received DFP vehicle injections for the same period. DFP was injected subcutaneously on the back of the rat with a volume ranging between 0.3 and 0.32 ml. Animal health including weight measurements were assessed daily during the exposure and for an additional 7 days following DFP injections. A small percentage of DFP injected rats (<5%) exhibited mild tremors around the fifth day of dosing that resolved within 24 h without the need of any pharmacological intervention. DFP-exposed rats showed no observable seizures or other hyper-cholinergic activity.
Experimental Design
At 3 months following DFP exposures, no significant differences in weights were observed between control and DFP exposed rats. At this time point postDFP exposure, rats were separated into 4 groups: group I: neuronal ratiometric intracellular Ca2+ measurements and group II: Western blot analysis. Groups III and IV rats were administered LEV (50 mg/kg, i.p.) twice daily for 4 days followed by behavioral analysis or ratiometric Ca2+ measurement on the fifth day (Figure 1). Age-matched control rats also received LEV injections and were subjected to behavioral analysis.
Figure 1.

Timeline of experimental studies. Nine-week-old male Sprague-Dawley rats (n = 45) were exposed once daily to OP agent DFP (0.5 mg/kg, s.c.). Three months later, DFP rats and age-matched vehicle control rats were divided in 4 groups. Group 1 DFP rats were euthanized, CA1 hippocampal neurons acutely isolated and immediately subjected to Ca2+ imaging studies including basal Ca2+ measurements and in vitro antagonist studies. Group 2 DFP rats were euthanized, hippocampal homogenates obtained and subjected to Western blot analysis. Group 3 DFP rats received either LEV (50 mg/kg, i.p.) or saline twice daily for 4 days. Age-matched control rats also received similar treatments. On the fifth day, these rats were subjected to behavioral tests to assess for the symptoms of depression, anxiety, and memory function. Group 4 DFP and control rats received similar treatments as group 3 but were subjected to Ca2+ imaging.
Assessment of Depression and Memory
Symptoms of depression and memory deficit were carried out using tests described below. Testing was carried out between 09:00 and 15:00 h. Normal lighting in the testing room was approximately 600 lux. For behavioral testing, the lighting was adjusted to a dimmer setting of approximately 300 lux. For elevated plus maze (EPM), open arm light levels were approximately 200 lux and closed arm light levels were approximately 150 lux. All light was reflected off photography-grade white shades and did not directly shine on the testing apparatus. The room was not sound-proof, but the interior location ensured that it remained quiet during behavioral assessments. Reviewers were blinded to treatment conditions.
Sucrose preference test
This test measures hedonia (pleasure-seeking) or lack of it (anhedonia) by monitoring a rat’s preference to sucrose-laced water (Deshpande et al., 2014; Forbes et al., 1996; Overstreet, 2012; Phillips and Deshpande, 2016). Briefly, rats were habituated to having 2 bottles in the cage lid for 4 days. The bottles were fitted with ball-bearing sipper tubes that prevented fluids from leaking. Water intake was measured daily to ascertain that rats were progressively showing lesser side-bias. Following this acclimation, on the fifth day, rats were deprived of water but not food overnight for a total of 19 h. On the sixth day, 2 bottles were again introduced in the cage-top. Rats had the free choice of either drinking a 1% sucrose solution or plain water for a total period of 1 h. After 30 min, positions of water and sucrose bottles were swapped to further control for any confounding effects produced by a side bias. Both the volume of fluid and weight of the bottle were measured before and after the test. Sucrose preference was calculated as a percentage of the sucrose intake over the total fluid intake. A spill-cage without rat was also employed using bottles and sipper tubes from the same batch as test cages. Measurement errors were ± 2 ml.
Forced swim test
Porsolt’s modified forced swim test (FST) was used to assess behavioral despair (Castagne et al., 2011; Deshpande et al., 2014; Overstreet, 2012; Phillips and Deshpande, 2016). Briefly, animals were placed in a glass cylindrical chamber (46-cm height × 30-cm depth) filled with water (30-cm height, 25°C) forced to swim for 6 min. Swimming sessions were recorded for off-line analysis. Active (swimming, climbing, and diving) and passive (immobility) behavior was evaluated by 2 reviewers blinded to the treatment conditions. Immobility (primary outcome) was defined as the period during which the animal floats in the water making only those movements necessary to keep its head above water. The tank was emptied and thoroughly cleaned for every rat to be tested in a session.
Elevated plus maze
This test assesses anxiety by considering the innate behavior of rats to prefer dark enclosed spaces over bright open spaces (Deshpande et al., 2014; Phillips and Deshpande, 2016; Walf and Frye, 2007). By using rodents’ proclivity toward dark, enclosed spaces (approach) and an unconditioned fear of heights/open spaces (avoidance), EPM allows for the measurement of anxiety state in the absence of noxious stimuli that typically produce a conditioned response (Walf and Frye, 2007). The maze (Med Associates Inc., St Albans, Vermont) was made of black polyvinyl chloride and consisted of 4 arms, 50-cm long × 10-cm wide, connected by a central square, 10 × 10 cm: 2 open without walls and 2 closed by 31-cm high walls. All arms were attached to sturdy metal legs; the maze was elevated 55 cm above the floor level and was set in a dimly lit room. A video camera was suspended above the maze to record the rat movements for analysis. A video-tracking system (Noldus Ethovision XT 11) was used to automatically collect behavioral data. The procedure consisted of placing the rats at the junction of the open and closed arms, the center of the maze, facing the open arm opposite to where the experimenter was. The video-tracking system was started after the animal was placed in the maze so that the behavior of each animal was consistently recorded for 5 min. At the end of the 5-min test session, the rat was removed from the plus maze and returned to its home cage. The maze was cleaned with 70% ethanol and air-dried to remove any scent traces and allowed to dry completely before introducing the next animal in the arena. Time spent and entries made in the various arms of EPM were calculated.
Novel object recognition test
This test assesses object recognition memory by calculating the preference of the rat to explore a novel object (Bevins and Besheer, 2006; Deshpande et al., 2014; Phillips and Deshpande, 2016). Briefly, rats were placed in black Perspex box 90 × 60 × 50 cm in a dimly illuminated and quiet animal behavior testing room. Rats were habituated individually, by allowing them to explore the empty box for 10 min per session for 2 days. The arena was cleaned with a 70% ethanol solution and dried completely in between each subject so as to eliminate any potential odor cues left by previous subjects. On the third day, in the sample phase, 2 identical objects (A, A) were placed in opposite corners of the box, 20 cm from the wall. A rat was allowed to explore for 3 min, and then it was removed from the box and returned to its home cage. In the choice phase (1 h later), one of the objects was replaced with a novel object (B), and the rat was allowed to explore for 2 min. Objects were similar in size and emotionally neutral. A video-tracking system (Noldus Ethovision XT 11) was used to automatically collect behavioral data. Direct contacts included any contact with mouth, nose or paw and did not include contacts that were accidental (backing or bumping into the object). Also, standing, sitting or leaning on the object was not scored as object interaction. A rat is exploring an object when its nose is within 2 cm of the object. Frequency and time spent exploring the new object (B) versus the familiar object (A) was calculated for each group.
Isolation of Hippocampal CA1 Neurons and Loading With Fura-2
Acute isolation of CA1 hippocampal neurons was performed by established procedures routinely used in our laboratory (Deshpande et al., 2010; Phillips and Deshpande, 2018; Raza et al., 2004). Animals were anesthetized with isoflurane and decapitated. Brains were rapidly dissected and placed in 4°C oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (aCSF) consisting of (in mM): 201.5 sucrose, 10 glucose, 1.25 NaH2PO4, 26 NaHCO3, 3 KCl, 7 MgCl2, and 0.2 CaCl2). MK-801 (1 μM) was added to all solutions to increase cell viability and was removed 15 min prior to imaging. Hippocampal slices (450 μm) were cut on a vibrating microtome (Leica Microsystems, Wetzlar, Germany) and then equilibrated for 10 min at 34°C in a piperazine-N, N'-bis[2-ethanesulfonic acid] (PIPES)-aCSF solution containing (in mM): 120 NaCl, 25 glucose, 20 PIPES, 5 KCl, 7 MgCl2, and 0.1 CaCl2. Slices were then treated with 8 mg/ml protease in PIPES-aCSF for 6 min at 34°C and rinsed. Enzyme-treated slices were visualized on a dissecting microscope to excise the CA1 hippocampal region which was then triturated with a series of Pasteur pipettes of decreasing diameter in cold (4°C) PIPES-aCSF solution containing 1 μM Fura-2 AM (Invitrogen, Carlsbad, California). The cell suspension was placed in the middle of 2-well glass-bottom chambers (Nunc, Thermo Scientific). These glass chambers were previously treated overnight with 0.05 mg/ml poly-L-lysine followed by multiple rinses with distilled water and then further treated with Cell-Tak (BD-Biosciences, San Jose, California) biocompatible cellular adhesive (3.5 μg/cm2) for 30 min, rinsed and air-dried. Neuronal suspension placed in the center of adhesive coated dishes when settled firmly adhered to the bottom. This technique simplified further manipulations on the dissociated neurons. Plates were then incubated at 37°C in a 5% CO2/95% air atmosphere for 45 min. Fura-2 was washed off with PIPES-aCSF containing no MK-801 and plates were incubated an additional 15 min to allow for complete cleavage of the AM moiety from Fura-2.
Measurement of [Ca2+]i
Fura-2 loaded cells were transferred to a 37°C heated stage (Harvard Apparatus, Hollington, Massachusetts) on an Olympus IX-70 inverted microscope coupled to a fluorescence imaging system (Olympus America, Center Valley, Pennsylvania) and subjected to [Ca2+]i measurements by procedures well established in our laboratory (Deshpande et al., 2010; Phillips and Deshpande, 2018; Raza et al., 2004). All experiments were performed using a 20×, 0.7 N.A. water immersion objective and images were recorded by an ORCA-ER high-speed digital CCD camera (Hammamatsu Photonics K.K., Japan). Fura-2 was excited with a 75 W xenon arc lamp (Olympus America). Ratio images were acquired by alternating excitation wavelengths (340/380 nm) by using a Lambda 10-2 filter wheel (Sutter Instruments Co., Novato, California) and a Fura filter cube at 510/540 emission with a dichroic at 400 nm. All image acquisition and processing was controlled by a computer connected to the camera and filter wheel using Metafluor Software ver 7.6 (MDS Analytical Technologies, Downington, Pennsylvania). Image pairs were captured every 5 s and the images at each wavelength were averaged over 10 frames. Background fluorescence was obtained by imaging a field lacking Fura-2. Hippocampal CA1 neurons were identified based on their distinct morphology. These neurons displayed pyramidal shaped cell body, long axon and dendrites and have been demonstrated to be devoid of immunoreactivity for specific protein markers for interneurons, including parvalbumin, cholecystokinin, vasoactive intestinal peptide, somatostatin, and neuropeptide Y (Deshpande et al., 2010; Phillips and Deshpande, 2018; Raza et al., 2004). The process of enzymatic treatment and mechanical trituration can add minimal stress during acute dissociation of neurons. However, we have shown previously that the neurons isolated using these procedures exhibit electrophysiological properties identical to neurons in slices or in cultures, are viable, and not apoptotic or necrotic (Sun et al., 2008).
Calcium Calibration
We performed Ca2+ calibration determinations as described previously (Deshpande et al., 2010; Phillips and Deshpande, 2018; Raza et al., 2004) to provide estimates of absolute [Ca2+]i concentrations from the 340/380 ratio values. A Ca2+ calibration curve was constructed using solutions of calibrated Ca2+ buffers ranging from 0 Ca2+ (Ca2+ free) to 39 μM Ca2+ (Invitrogen). Values from the calibration curve were used to convert fluorescent ratios to [Ca2+]i. Final [Ca2+]i were calculated from the background corrected 340/380 ratios using the Grynkiewicz equation: [Ca2+]i = (Kd × Sf2/Sb2) × (R − Rmin)/(Rmax − R); where R was the 340/380 ratio at any time; Rmax was the maximum measured ratio in saturating Ca2+ solution (39 μM free Ca2+); Rmin was the minimal measured ratio Ca2+ free solution; Sf2 was the absolute value of the corrected 380-nm signal at Rmin; Sb2 was the absolute value of the corrected 380-nm signal at Rmax; the Kd value for Fura2 was 224 nM.
Western Blot Studies
Rats were deeply anesthetized under saturated isoflurane conditions and then euthanized by decapitation. Brains were immediately removed and placed on glass surface cooled over ice. The hippocampus was dissected out and flash frozen in liquid nitrogen and stored at −80°C until use. Frozen hippocampal samples were added to 10 volumes of ice cold buffer containing: 50 mM Tris-HCL pH 7.5, 7 mM EGTA, 5 mM EDTA, 0.32 M sucrose, 0.1 mM PMSF, and protease/phosphatase inhibitor cocktails (SigmaFast/PhosSTOP, Millipore Sigma, Burlington, Massachusetts) and homogenized on ice using a motor driven glass-teflon homogenizer (Glas-Col, Terre Haute, Indiana) at 900 rpm with 15 strokes. Samples underwent low speed (900 × g, 4°C) centrifugation to pellet nuclei. An aliquot of the supernatant (homogenate fraction) was saved and the remainder of each sample underwent a second centrifugation at 10 000 × g, 4°C for 15 min to obtain a crude cytosol (supernatant) and P2 pellet fraction. Protein concentrations for all fractions were determined by the Bradford micro-assay (Bio-Rad Laboratories, Hercules, California) and read on a spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific, Waltham, Massachusetts). Homogenate and cytosolic proteins (10–15 μG/lane) were reduced in SDS sample buffer and resolved by denaturing gel electrophoresis (NuPage Novex SDS-PAGE, Invitrogen) and then transferred to PVDF membrane for Western analysis. For immunoblot analysis of medium to small proteins, samples were resolved using 4%–12% Bis-Tris gels and transferred to PVDF with 0.2-μ pore size, while analysis of large M.W. proteins were resolved using 7% Tris-Acetate gels and transferred to PVDF with 0.4-μ pore size. Following transfer, immunoblots were incubated in tris-buffered saline (TBS) pH 7.4 containing 3% blotting-grade dry milk block (Bio-Rad) for 60 min on a rocking platform at room temperature. Immunoblots were then incubated in primary antibodies in TBS + block for 1–24 h (overnight incubation was carried out at 4°C) on a rocking platform. The following antibodies were used: antiFKBP 12.6 (1:200, goat polyclonal, R&D Systems, Minneapolis, Minnesota), antiRyR2 (phosphor S2808) (1:2 K, rabbit polyclonal, Abcam, Cambridge, Massachusetts), antiRyR2 (1:1 K, rabbit polyclonal, Alomone Labs, Jerusalem, Israel), antiPKA RII (1:2 K, goat polyclonal, EMD Millipore, Billerica, Massachusetts), antiIP3-1 (1:2 K, rabbit polyclonal, EMD Millipore), antiPLC-γ1 (1:1 K, clone 10: mouse monoclonal, BD Biosciences), and antiβ-actin (1:20 K, mouse monoclonal, Millipore Sigma). Immunoblots were washed 4 × 10 min in TBS containing 0.05% Tween-20 (TBS-T) (Bio-Rad), followed by a 60-min incubation with HRP-conjugated secondary antibodies in TBS + block (1:10 K; Santa Cruz Biotechnology, Dallas, Texas). Immunoblots were washed 4 × 10 min in TBS-T followed by incubation in ECL detection reagent (Amersham ECL Prime, GE Healthcare Biosciences, Pittsburg, Pennsylvania) and exposed to x-ray film. Digital images of Western films were obtained using high-resolution scanning (Epson Expression 1680, Epson America Inc., Long Beach, California) and underwent analysis using ImageJ analysis software (ImageJ, NIH, public domain) to determine relative density values. Values were normalized against antiβ-actin for loading control.
Data Analysis
Data were analyzed, and graphs plotted using the SigmaPlot 14 software (SPSS Inc, Chicago, Illinois). Data were first examined for normality using Shapiro-Wilk’s tests. Variables not normally distributed were log transformed and normality confirmed on the transformed data prior to analyses with t tests. Normally distributed data were analyzed with independent t tests or a 1-way analysis of variance (ANOVA) followed by post hoc Tukey’s test wherever appropriate. In all cases, statistical significance was indicated by *p < .05.
For [Ca2+]i comparison between control and DFP-treated animals, a t-test was applied. For comparing the distributions of [Ca2+]i a Chi-square test was used. We used 5–9 animals for each experimental condition. Dissociating the hippocampal slices routinely yielded 20–25 healthy, phase-bright neurons that were used for the recordings. The means of each group of neurons from each animal were used to evaluate results and conduct statistical analysis. Viable neurons included in the study had a smooth surface and a pyramidal-like morphology with processes. Nonviable neurons were swollen or circular and the surface was uneven and irregular. Data from each animal were pooled together in respective groups and ultimately represented as total number of cells studied. For measuring changes in protein levels, relative densities for the specific protein bands were measured, corrected for protein loading against β-actin and then calculated as a % of control ± SEM. Student’s t test was used to compare optical densities between control and DFP rats. For behavioral studies, we used a t test or 1-way ANOVA followed by post hoc Tukey’s test wherever appropriate.
RESULTS
Chronic Hippocampal [Ca2+]i Elevation in DFP Rats
Acutely isolated CA1 hippocampal neurons (Figure 2A) harvested from DFP rats manifested mean [Ca2+]i of 348 ± 11 nM that was significantly higher than [Ca2+]i of 181 ± 8 nM in neurons harvested from age-matched controls (*p < .01, t test, n = 8 rats, Figure 2B).
Figure 2.
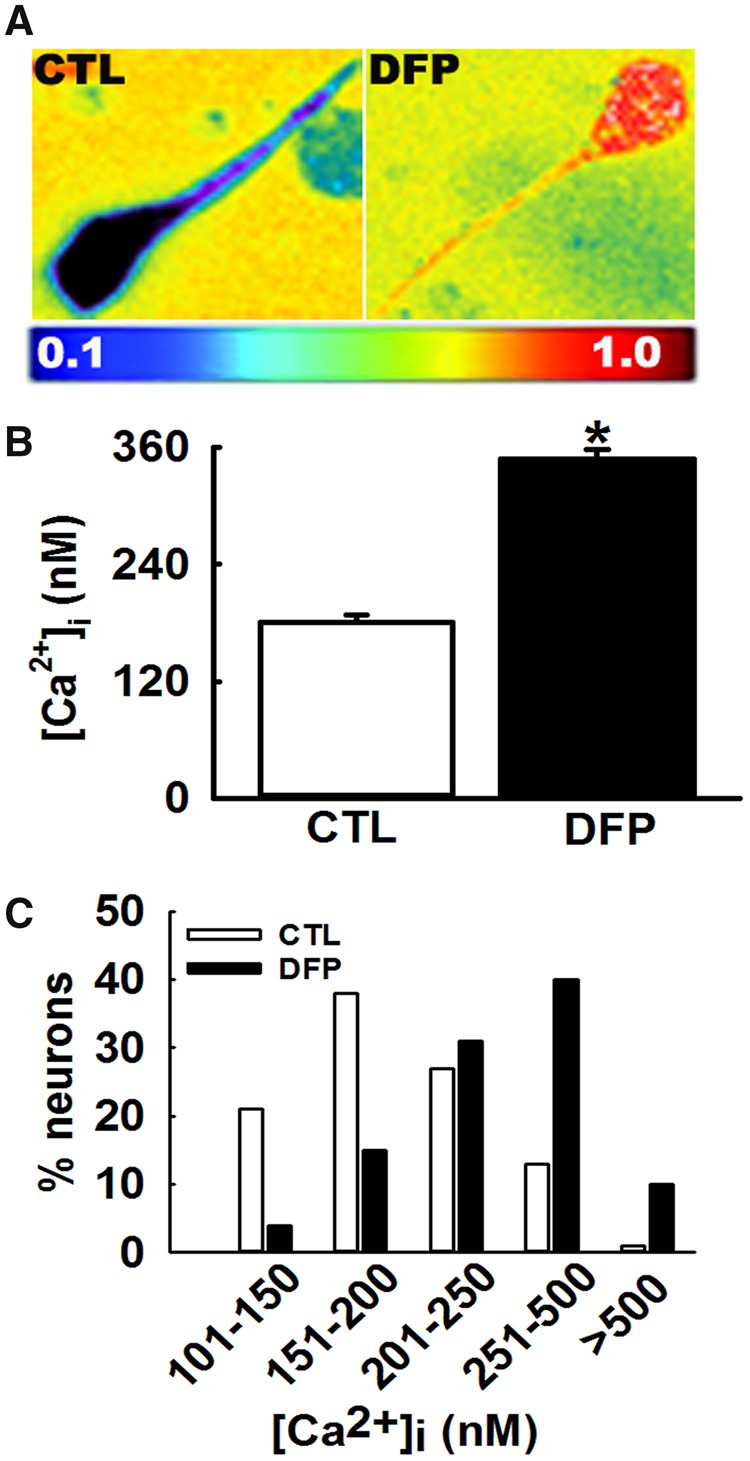
Protracted hippocampal [Ca2+]i following repeated, low-dose OP-DFP exposure in rats. A, Pseudocolor ratiometric images of representative acutely isolated hippocampal CA1 neurons from age-matched control and DFP rat. Control neurons had bluish color that corresponds to lower Fura-2 ratio. DFP neurons had orange-red color that corresponds to higher Fura-2 ratio. B, Bar graph comparing [Ca2+]i in control versus DFP neurons. CA1 hippocampal neurons acutely isolated at 3 months following repeated, low-dose DFP exposure exhibited an increased Fura-2AM 340/380 ratios indicative of [Ca2+]i that were significantly higher compared with the calibrated [Ca2+]i from age-matched control rats (data expressed as mean ± SEM, *p < .05, t test, n = 8 rats). C, Distribution of [Ca2+]i levels for control and DFP hippocampal neurons. CA1 neurons from age-matched control rats demonstrated a normal distribution for [Ca2+]i with approximately 95% of neurons exhibiting [Ca2+]i <500 nM and only 2% neurons exhibiting very high [Ca2+]i. In contrast, hippocampal neurons acutely isolated from DFP rats demonstrated a rightward shift towards higher [Ca2+]i with approximately 10% neurons exhibiting [Ca2+]i >500 nM and approximately 50% neurons exhibiting [Ca2+]i between 250 and 500 nM (p < .001, Chi-square test, n = 150 neurons).
Analysis of the population distributions of [Ca2+]i revealed only 1% of age-matched control neurons exhibited [Ca2+]i >500 nM, with the majority of neurons falling into the lower Ca2+ concentration range. In contrast, approximately 40% neurons isolated from DFP rats exhibited [Ca2+]i between 250 and 500 nM and approximately 10% neurons exhibited [Ca2+]i > 500 nM indicating a population shift towards higher Ca2+ concentration range. This rightward shift in distribution of [Ca2+]i levels in DFP neurons was significantly different from control neurons (p < .001, Chi-square test, n = 150 neurons, Figure 2C).
Mechanism for Chronic Hippocampal [Ca2+]i Elevations in DFP Rats
Ratiometric recordings in the presence of effective concentrations of inhibitors of voltage-gated Ca2+ channels (Nifedipine, 5 μM), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) AMPA/kainate channels (6,7-dinitroquinoxaline-2,3-dione (DNQX) DNQX, 10 μM) or other nonspecific cation channels Gadolinium chloride (GdCl3, 10 μM) did not produce a significant decrease in [Ca2+]i suggesting that these routes of neuronal Ca2+ entry do not mediate chronic [Ca2+]i elevations in GWI rats. Application of NMDAR antagonist (MK-801, 10 μM) produced a small but significant reduction in [Ca2+]i suggesting that NMDAR activity could be an important source of hippocampal Ca2+ dysregulation in DFP neurons (*p < .05, t test, n = 5 rats, Figure 3A).
Figure 3.
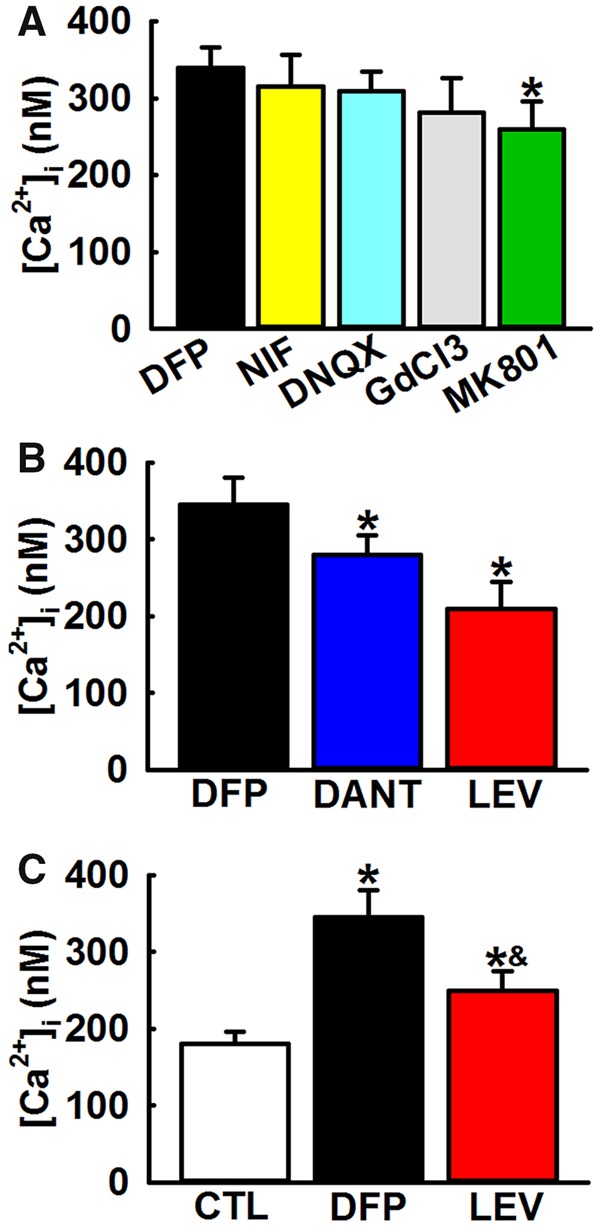
Mechanism for elevated hippocampal [Ca2+]i in DFP rats. A, Effect of inhibitors of Ca2+ influx pathways on [Ca2+]i in DFP. Bath application of inhibitors of voltage-gated Ca2+ channels (Nifedipine, 5 μM), AMPA/kainate channels (DNQX, 10 μM) or other nonspecific cation channels (GdCl3, 10 μM) did not produce a significant decrease in [Ca2+]i. Bath application of NMDA antagonist (MK-801, 10 μM) produced a small but significant reduction in [Ca2+]i (Data expressed as mean ± SEM, *p < .05, t test, n = 5 rats). B, Effect of inhibitors of intracellular Ca2+ release pathways on [Ca2+]i in DFP neurons. Bath application of IP3/RyR antagonist LEV (100 μM) or RyR antagonist dantrolene (Dant, 50 μM) produced significant reductions in [Ca2+]i. (Data expressed as mean ± SEM, *p < .05, t test, n = 6 rats). C, Effect of LEV administration on [Ca2+]i in DFP rats. Treatment with LEV (50 mg/kg, i.p.) produced a significant reduction in [Ca2+]i. compared with DFP neurons but was significantly higher than age-matched control neurons (CTL) (Data expressed as mean ± SEM, *p < .05, 1-way ANOVA, Tukey’s test, n = 5 rats).
We next investigated the contribution of intracellular Ca2+ release mechanisms towards protracted [Ca2+]i increases in DFP rats. Application of RyR antagonist dantrolene (50 μM) to hippocampal neurons isolated from DFP rats resulted in a significant drop in [Ca2+]i from 399 ± 26 (no drug) to 240 ± 11 nM (*p < .05, t test, n = 5 rats, Figure 3B). LEV has been shown to inhibit both RyR- and IP3R-activated CICR in hippocampal neurons (Angehagen et al., 2003; Cataldi et al., 2004; Fukuyama et al., 2012; Nagarkatti et al., 2008). Addition of LEV (100 μM) to hippocampal neurons isolated from DFP rats reduced [Ca2+]i to 251 ± 19 nM that was significantly lower than [Ca2+]i in neurons isolated from DFP rats (*p < .05, t test, n = 5 rats, Figure 3B).
To further confirm involvement of CICR machinery in DFP-induced Ca2+ elevations, we investigated the effect of in vivo LEV treatment in DFP rats. CA1 neurons from LEV treated DFP rats exhibited [Ca2+]i of 270 ± 21 nM that were significantly lower than [Ca2+]i in neurons isolated from DFP rats. LEV treatment, however, did not restore [Ca2+]i to baseline control levels suggesting involvement of additional mechanisms in maintenance of protracted [Ca2+]i elevations in DFP rats (*p < .05, 1-way ANOVA, n = 5 rats, Figure 3C). Overall, an almost 60% reduction in [Ca2+]i in the presence of dantrolene or LEV suggests that IP3Rs and RyRs are significantly contributing to maintaining the DFP-induced sustained Ca2+ elevations.
Estimations of Protein Levels of the Components of CICR Machinery in DFP Rats
Western blot analysis of hippocampal homogenates were carried out to evaluate possible alterations in the levels of CICR protein targets in DFP rats. No significant changes in levels of the CICR receptor proteins RyR-2 (0.82 ± 0.13, 1.25 ± 0.38) or IP3-R (1.02 ± 0.12, 0.99 ± 0.08) or in the protein levels of signaling enzymes PLCγ1 (1.00 ± 0.14, 1.13 ± 0.04) or PKA (0.99 ± 0.21, 1.03 ± 0.24) were observed in DFP rats compared with control (Figure 4).
Figure 4.

Protein expression levels of the components of GWI hippocampal CICR machinery. Western immunoblots of hippocampal protein from DFP and age-matched control (CTL) rats were stained for A, PKA RII; B, PLCγ1; C, RyR2; D, IP3R1; E, phospho-RyR2; and F, Calstabin2. To adjust for sample loading, the blots were stained with an antibody specific for the 42 kDa β-actin protein. G, Densitometric analyses of the Western blotting revealed no significant changes in proteins (A–E) but identified significant reduction in the levels of Calstabin2 (F) compared with age-matched control (Data expressed as mean optical density ± SEM, *p < .05, t test, n = 6 rats).
We then measured protein levels of phosphorylated RyR2. A small, but statistically insignificant increase in the p-RyR2 was observed in DFP rats (1.37 ± 0.35, 1.1 ± 0.15; p = .12, n = 6 rats). We next investigated expression of Calstabin2, also known as FKBP12.6, which is a RyR2 stabilizer and keeps the receptor in a closed-state. Western blot analysis with Calstabin2-specific antibody staining revealed a significant downregulation in its expression in DFP rats. Levels for the hippocampal Calstabin2 binding protein showed a 42% decrease in DFP rats when compared with control (0.87 ± 0.069, 1.51 ± 0.029). (*p < .05, t test, n = 6 rats, Figure 4G).
Effect of CICR Inhibitor on Neurological Morbidities in DFP Rats
Performance on sucrose preference test
Saline-treated control rats exhibited a strong preference for sucrose water (78.1% ± 10.1%) which was not altered by LEV therapy (75% ± 9%) suggesting that LEV treatment does not have an effect on baseline sucrose preference in age-matched control rats. In contrast, saline-treated DFP rats consumed significantly low volume (44.2% ± 8.1%) of sucrose-laced water indicative of anhedonia. LEV-treated DFP rats exhibited a greater preference for sucrose water (68.8% ± 7.1%) which was significantly higher than DFP rats but not significantly different from age-matched control rats, suggesting that LEV therapy restored sucrose preference and improved anhedonia in DFP rats (1-way ANOVA, *p < .05, n = 8 rats, Figure 5A). No differences were observed between total fluid consumption and fluid consumed on right versus left-side amongst the different groups (p > .5, n = 8, Figure 5B).
Figure 5.
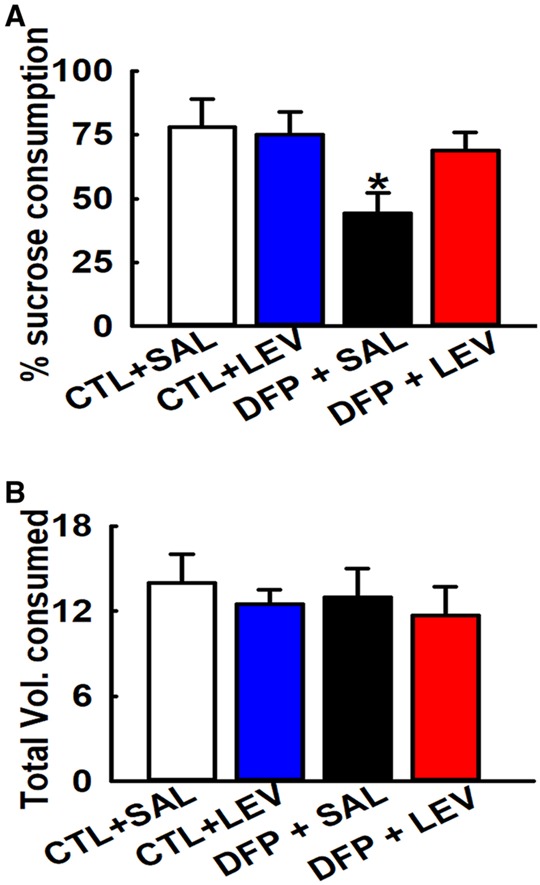
Effect of LEV therapy on sucrose preference in DFP rats. Sucrose preference test: A, Bar graph shows significantly lower sucrose consumption in DFP+SAL rats compared with age-matched CTL groups indicative of significant anhedonia. DFP+LEV rats were significantly higher than on DFP+SAL but not significantly different than CTL+SAL group indicative of restoration of sucrose preference in DFP rats following LEV therapy. CTL+SAL and CTL+LEV groups were not different from each other. B, No differences were observed in the total fluid volume consumed between the various groups. (Data expressed as mean ± SEM, *p < .05, 1-way ANOVA, Tukey’s test, n = 8 rats; *compared with CTL+SAL).
Performance on FST
The FST is an effective test in evaluating the presence of a despair; a common symptom of depression. Control rats treated with saline exhibited an immobility time of 57.12 ± 12.4 s while age-matched control rats treated with LEV exhibited an immobility time of 63.7 ± 11 s, demonstrating that LEV alone has no significant impact on mobility. DFP rats subjected to FST exhibited an increased immobility time of 127.12 ± 11.69 s indicative of a despair-like state. In the presence of LEV therapy, there was a reduction in the immobility time of DFP rats (81.08 ± 6.97 s). One-way ANOVA revealed significant differences amongst the groups. Post hoc Tukey’s test revealed that DFP rats exhibited significantly higher immobility time compared with age-matched control rats. Further, LEV therapy did not alter baseline behavior in control rats but significantly lowered immobility time in DFP rats indicative of LEV’s potential antidepressant effect. LEV therapy, however, was not able to restore DFP immobility time to control values (n = 7, *compared with CTL+SAL, #compared with DFP+SAL, p < .05, Figure 6).
Figure 6.
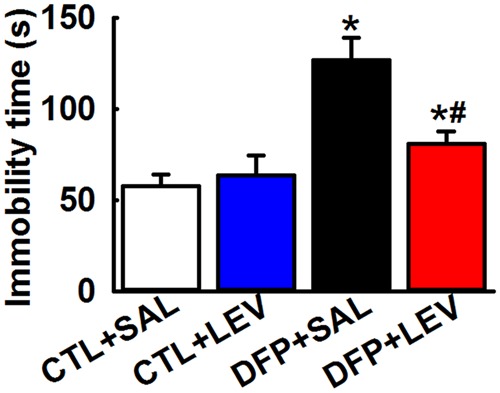
Effect of LEV therapy on immobility time in DFP rats. FST: Graph shows increased immobility time in DFP+SAL rats compared with CTL+SAL rats indicating that DFP treatment produced increased behavioral despair. A decreased immobility time in DFP+LEV compared with DFP+SAL rats during the FST, indicated improvement in behavioral despair score. DFP+LEV immobility time was significantly higher than CTL+LEV group suggesting that LEV therapy could not restore despair-like symptoms to baseline values. No significant differences in immobility times were noted between CTL+SAL and CTL+LEV groups (Data expressed as mean ± SEM, *#p < .05, 1-way ANOVA, Tukey’s test, n = 7 rats; *compared with CTL+SAL and #compared with DFP+SAL).
Performance on EPM
Measuring time spent and entries made by rats in the open versus closed arm of EPM reflects the anxiety state of rats and allows for identification of anxiolytic effects following drug treatment. Both the age-matched control rats treated with either saline and LEV spent approximately 20% of time in the open arms of EPM, which was not significantly different from each other. In contrast, DFP rats spend only 5.3% ± 1.7% of time in the open arm which was significantly different from age-matched control rats. In the presence of LEV therapy, there was a significant improvement in time spent in the open-arm of EPM of DFP rats as evidenced by significantly greater open-arm time (15.6% ± 2.7%) (*p < .05, 1-way ANOVA, n = 8 rats, Figure 7A). Improved EPM behavior in DFP+LEV rats were further evidenced by significantly higher entries in the open-arm compared with DFP+SAL rats. EPM performance by DFP rats in the presence of LEV therapy matched the open-arm behavior seen in age-matched control rats, indicative of LEV’s significant anxiolytic effect (*p < .05, 1-way ANOVA, n = 8 rats, Figure 7B). We did not observe any significant differences in total arm entries, distance moved, and speed between the various groups suggesting no involvement of motor deficits in the EPM outcomes (n = 8, 1-way ANOVA, Figs. 7C–E).
Figure 7.

Anxiolytic effects of LEV therapy in DFP rats. EPM: Bar graph shows significantly lower time spent in the open-arm (A) and significantly higher closed-arm and less open-arm entries (B) by DFP rats indicative of anxiety-like condition and a significant anxiolytic effect of LEV therapy in DFP rats. LEV alone did not alter baseline EPM exploratory behavior while DFP+LEV rats were not significantly different than age-matched control rats. Bar graph shows no significant changes between total arm entries (C), distance moved (D), and velocity (E) between the age-matched control and DFP rats treated with saline or LEV. (Data expressed as mean ± SEM, *p < .05, 1-way ANOVA, Tukey’s test, n = 8 rats; *compared with CTL+SAL).
Performance on novel object recognition
In the novel object recognition (NOR) choice phase, saline-treated control rats spent 84.3% ± 8.7% of time with the novel object which was significantly higher than time spent with familiar object (*p < .05, t test, n = 6 rats, Figure 8A). LEV-treated control rats also spent significantly higher time (80.2% ± 10%) with the novel object compared with familiar object (*p < .05, t test, n = 7 rats, Figure 8B). This indicates that LEV therapy did not alter baseline greater attraction for the novel object exhibited under control conditions. However, saline-treated DFP rats exhibited impaired recognition memory as evidenced by comparable time spent with both the familiar and the novel object. Time spent with novel object was 53.3% ± 11% which was not significantly different than time spent with familiar object (*p < .05, t test, n = 6 rats, Figure 8C). In contrast, DFP rats treated with LEV spent a significantly greater amount of time exploring the novel object (70.5% ± 14%) compared with familiar object (*p < .05, t test, n = 7 rats, Figure 8E).
Figure 8.

LEV therapy improves NOR memory in DFP rats. NOR: Bar charts compare percentages of time spent exploring the familiar object (FAM) and the novel object (NOV) in age-matched saline (SAL, A), LEV (B), DFP (C), and DFP + LEV (D) rats. Note the comparable time spent by DFP rats interacting with FAM and NOV objects (Data expressed as mean ± SEM, *p < .05, t test, n = 7 rats). E, Discrimination ratio comparisons between various groups. DFP+LEV discrimination ratio was significantly higher than DFP+SAL group but was significantly lower than CTL+LEV group suggesting that while LEV therapy improves NOR memory in DFP rats, it does not restore NOR memory to baseline values. No significant differences in discrimination ratios were noted between CTL+SAL and CTL+LEV groups (Data expressed as mean ± SEM, *#p < .05, 1-way ANOVA, Tukey’s test, n = 7 rats; *compared with CTL+SAL and #compared with DFP+SAL). Bar graphs compares the total distance traveled (F), the total object exploration time (G), and the velocity of movement (H) between the age-matched control and DFP rats treated with saline or LEV. Note that the overall motor activity was similar between the various groups.
To permit comparison across the groups, we calculated a discrimination ratio that provided an index of time spent with novel object as a fraction of total time spent exploring the 2 objects during the choice phase. As shown in Figure 8E, DFP rats exhibited a discrimination ratio of 0.47 ± 0.08 that was significantly lower compared with the discrimination ratio of 0.86 ± 0.05 obtained from age matched control rats indicative of impaired recognition memory. When treated with LEV, DFP rats exhibited a discrimination ratio of 0.72 ± 0.06 that was significantly higher than DFP discrimination ratio indicating that LEV therapy significantly improved NOR performance in DFP rats. DFP+LEV discrimination ratio was also significantly lower than age-matched control rats suggesting that LEV therapy could not completely restore the object exploration behavior to the values exhibited by control rats (*p < .05, 1-way ANOVA, n = 7 rats, Figure 8E). The distance traveled within the arena, total objects exploration time, and the velocity in the choice phase were not significantly different across groups suggesting that variable object exploration or motor deficits did not affect the outcome of NOR (p > .05, 1-way ANOVA, Figs. 8F–H).
DISCUSSION
Using an established OP DFP rat model (Phillips and Deshpande, 2016, 2018), our study has identified a unique molecular mechanism that could possibly underlie the chronic expression of neurological symptoms associated with GWI. A sustained increase in intracellular Ca2+ levels was observed in the hippocampal neurons of DFP rats that had its origins in persistent release of Ca2+ from intracellular stores. Western analysis revealed a reduction in the levels of RyR stabilizing protein Calstabin2. Further, pharmacological antagonism of RyR in isolated hippocampal CA1 neurons from DFP rats significantly lowered the observed elevated Ca2+ and in vivo LEV therapy improved GWI-like behavioral symptoms in DFP rats.
Neuronal Ca2+ influx is mediated by voltage-gated channels and neurotransmitter ligand-gated channels (Hagenston and Bading, 2011). Our studies showed that pharmacological inhibition of voltage-gated Ca2+ channels did not contribute to the sustained Ca2+ increases in DFP neurons. However, blockade of the NMDAR channel produced a small but significant drop in DFP Ca2+ elevations. Increased glutamatergic neurotransmission has been reported following repeated low-dose OP exposures (Torres-Altoro et al., 2011) and in chronic neurodegenerative disorders (Carvajal et al., 2016). Together these could lead to excitotoxic activation of NMDAR leading to neuronal death in DFP rats. Thus, NMDAR blockade could be neuroprotective and reduce neuronal injury and ongoing cell-death in GWI. Unfortunately, NMDAR antagonists have not been able to be safely translated for therapy (Williams and Schatzberg, 2016). Our observations that NMDAR blockade could not restore Ca2+ levels to baseline also suggested that there could be mechanisms beyond Ca2+ influx contributing to chronic Ca2+ dys-homeostasis in DFP rats.
Another major player in neuronal Ca2+ homeostasis is Ca2+ release from intracellular stores that occurs via RyRs and IP3Rs on the ER. Aberrant CICR has been implicated in many neurodegenerative diseases and neuropsychiatric conditions (Baker et al., 2013; Kushnir et al., 2018; Popugaeva et al., 2017; Surmeier et al., 2017; Tehse and Taghibiglou, 2018). In line with these reports, our studies here show that RyR and RyR/IP3R antagonists significantly lowered elevated Ca2+ levels in DFP neurons. The percent decrease in Ca2+ levels produced by CICR inhibition was higher than NMDAR antagonism suggesting that CICR could be a dominant source of neuronal Ca2+ dysregulation in DFP rats.
We next investigated how CICR could contribute to sustained Ca2+ elevations following DFP exposures. Studies have shown enhanced IP3R mediated Ca2+ release under conditions of hypoxia, stress, and seizures (Berridge and Taylor, 1988; Brini et al., 2014; DeLorenzo et al., 2005; Hagenston and Bading, 2011). Our Western blot studies did not reveal any significant change in the protein expression of IP3R or PLCγ in hippocampal homogenates from DFP rats. It is possible that IP3R activity could be affected via mechanisms independent of changes in expression of the receptor protein, and additional studies are needed to further decipher involvement of IP3R machinery in this model.
There are 3 isoforms of RyR in the brain, all abundantly expressed in the hippocampus (Hertle and Yeckel, 2007). RyR2 specifically is reported to be intricately involved in modulating behavior (Abu-Omar et al., 2018; Kushnir et al., 2018) and has been implicated in stress-induced cognitive dysfunction (Liu et al., 2012). Further, pharmacological blockade of RyRs afforded neuroprotection in a rat model of OP intoxication (Deshpande et al., 2016). In this study, we did not observe significant changes in the protein expression of RyR2 between age-matched control and DFP rats. Posttranslational remodeling of brain and cardiac RyR2 including phosphorylation, oxidation, and nitrosylation has been reported in animal models of PTSD, Alzheimer’s, and heart failure (Lacampagne et al., 2017; Lehnart et al., 2008; Liu et al., 2012; Marx et al., 2000). We probed for p-RYR2 expression in hippocampal homogenates from control and DFP rats and observed a small but statistically insignificant increase in the p-RYR2 expression in DFP rats. In our attempts to further probe the molecular mechanisms underlying sustained Ca2+ elevations in GWI, we investigated expression of Calstabin2, also known as FKBP12.6, in DFP rats. Calstabin2 is a critical regulatory subunit of the RyR2 macromolecular complex (Yuan et al., 2016). It selectively binds to and stabilizes RyR2 in a closed-state (Zalk et al., 2015). We observed that hippocampal homogenates from DFP rats exhibited significant depletions in Calstabin2 expression. It has been reported that under stress-induced conditions, posttranslational modification of RyR2 results in dissociation of Calstabin2 rendering RyR2 “leaky” and releasing Ca2+ from ER (Lehnart et al., 2008; Liu et al., 2012; Yuan et al., 2016). Thus, “leaky” neuronal RyR could also underlie GWI neuropathology. Additional studies are needed to experimentally confirm leaky RyRs in our dissociated neuronal preparation. Our data that hippocampal neurons isolated from LEV-treated DFP-rats display significantly lower Ca2+ levels supports the involvement of RyR in GWI-like phenotype.
We next investigated whether blockade of CICR mechanism would also provide relief from the symptoms of GWI-like neuropsychiatric abnormalities. Several studies have demonstrated that LEV (Keppra), an FDA-approved antiepileptic drug, decreases CICR (Angehagen et al., 2003; Cataldi et al., 2005; Fukuyama et al., 2012; Nagarkatti et al., 2008). LEV is also reported to produce significant memory improvement in Alzheimer’s’ patients (Musaeus et al., 2017) and improved neurobehavioral outcomes after TBI (Zou et al., 2013). These neurological conditions are also associated Ca2+ dysregulation (Surmeier et al., 2017; Tehse and Taghibiglou, 2018). We therefore investigated if LEV could be repurposed for the treatment of GWI-like neurological symptoms. In agreement with previous studies, LEV did not affect baseline behavior in naïve rats (Barker-Haliski et al., 2016; Gower et al., 2003). In contrast, LEV significantly improved symptoms of depression, anxiety, and cognitive deficits in DFP rats. However, LEV was not able to completely restore all the DFP behavioral morbidities to control baseline performance.
A critical question remains if LEV therapy is also able to reverse the underlying molecular changes that are responsible for elevated Ca2+ levels in DFP neurons. Our observation that in vitro or in vivo LEV couldn’t completely restore elevated Ca2+ levels or GWI-like behavioral symptoms to baseline suggests that there could be additional mechanisms contributing to the maintenance of elevated Ca2+ levels and GWI phenotype. Indeed, many other mechanisms have been purported for GWI-like neuropsychiatric symptoms in OP-based rodent models including neuroinflammation (Koo et al., 2018; Parihar et al., 2013), oxidative stress (Shetty et al., 2017), mitochondrial dysfunction (Shetty et al., 2017), and increased glutamatergic transmission (Torres-Altoro et al., 2011). Furthermore, LEV has several biological effects in addition to blocking RyR and IP3R activity, including inhibition of n-type calcium channels, the modulation of GABA and glycine receptors, and binding to SV2A protein (reviewed in De Smedt et al., 2007). These pleiotropic mechanisms could be contributing to the in vivo actions of LEV in addition to its effects on CICR observed here. Our study provides a possible molecular mechanism for GWI-like neurological morbidities in DFP rats and suggests that agents such as LEV could prove to be promising therapeutic option for managing GWI neurological symptoms.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Gulf War Illness Research Program under (Award No. W81XWH-14-1-0478 and W81XWH-17-1-0573). Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
REFERENCES
- Abdel-Rahman A., Abou-Donia S., El-Masry E., Shetty A., Abou-Donia M. (2004). Stress and combined exposure to low doses of pyridostigmine bromide, deet, and permethrin produce neurochemical and neuropathological alterations in cerebral cortex, hippocampus, and cerebellum. J. Toxicol. Environ. Health A 67, 163–192. [DOI] [PubMed] [Google Scholar]
- Abdullah L., Evans J. E., Montague H., Reed J. M., Moser A., Crynen G., Gonzalez A., Zakirova Z., Ross I., Mullan C. (2013). Chronic elevation of phosphocholine containing lipids in mice exposed to gulf war agents pyridostigmine bromide and permethrin. Neurotoxicol. Teratol. 40, 74–84. [DOI] [PubMed] [Google Scholar]
- Abu-Omar N., Das J., Szeto V., Feng Z. P. (2018). Neuronal ryanodine receptors in development and aging. Mol. Neurobiol. 55, 1183–1192. [DOI] [PubMed] [Google Scholar]
- Angehagen M., Margineanu D. G., Ben-Menachem E., Ronnback L., Hansson E., Klitgaard H. (2003). Levetiracetam reduces caffeine-induced Ca2+ transients and epileptiform potentials in hippocampal neurons. Neuroreport 14, 471–475. [DOI] [PubMed] [Google Scholar]
- Baker K. D., Edwards T. M., Rickard N. S. (2013). The role of intracellular calcium stores in synaptic plasticity and memory consolidation. Neurosci. Biobehav. Rev. 37, 1211–1239. [DOI] [PubMed] [Google Scholar]
- Barker-Haliski M. L., Vanegas F., Mau M. J., Underwood T. K., White H. S. (2016). Acute cognitive impact of antiseizure drugs in naive rodents and corneal-kindled mice. Epilepsia 57, 1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson C. P., Bading H. (2012). Nuclear calcium signaling. Adv. Exp. Med. Biol. 970, 377–405. [DOI] [PubMed] [Google Scholar]
- Berridge M. J., Taylor C. W. (1988). Inositol trisphosphate and calcium signaling. Cold Spring Harb. Symp. Quant. Biol. 53(Pt 2), 927–933. [DOI] [PubMed] [Google Scholar]
- Bevins R. A., Besheer J. (2006). Object recognition in rats and mice: A one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat. Protocols 1, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Black D. W., Carney C. P., Peloso P. M., Woolson R. F., Schwartz D. A., Voelker M. D., Barrett D. H., Doebbeling B. N. (2004). Gulf war veterans with anxiety: Prevalence, comorbidity, and risk factors. Epidemiology 15, 135–142. [DOI] [PubMed] [Google Scholar]
- Blore J. D., Sim M. R., Forbes A. B., Creamer M. C., Kelsall H. L. (2015). Depression in gulf war veterans: A systematic review and meta-analysis. Psychol. Med. 45, 1565–1580. [DOI] [PubMed] [Google Scholar]
- Brimfield A. A. (2012). Chemicals of military deployments: Revisiting Gulf War syndrome in light of new information. Prog. Mol. Biol. Transl. Sci. 112, 209–230. [DOI] [PubMed] [Google Scholar]
- Brini M., Cali T., Ottolini D., Carafoli E. (2014). Neuronal calcium signaling: Function and dysfunction. Cell Mol. Life Sci. 71, 2787–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal F. J., Mattison H. A., Cerpa W. (2016). Role of NMDA receptor-mediated glutamatergic signaling in chronic and acute neuropathologies. Neural Plast. 2016, 2701526.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagne V., Moser P., Roux S., Porsolt R. D. (2011). Rodent models of depression: Forced swim and tail suspension behavioral despair tests in rats and mice. Curr. Protoc. Neurosci. Chapter 8: Unit 8. 10A. [DOI] [PubMed] [Google Scholar]
- Cataldi M., Lariccia V., Secondo A., di Renzo G., Annunziato L. (2004). The antiepileptic drug levetiracetam decreases the inositol 1, 4, 5-trisphosphate-dependent [Ca2+]i increase induced by ATP and bradykinin in PC12 cells. J. Pharmacol. Exp. Ther. 313, 720–730. [DOI] [PubMed] [Google Scholar]
- Chao L. L., Zhang Y., Buckley S. (2015). Effects of low-level sarin and cyclosarin exposure on white matter integrity in gulf war veterans. Neurotoxicology 48, 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smedt T., Raedt R., Vonck K., Boon P. (2007). Levetiracetam: The profile of a novel anticonvulsant drug-part i: Preclinical data. CNS Drug Rev. 13, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorenzo R. J., Sun D. A., Deshpande L. S. (2005). Cellular mechanisms underlying acquired epilepsy: The calcium hypothesis of the induction and maintainance of epilepsy. Pharmacol. Ther. 105, 229–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande L. S., Blair R. E., Huang B. A., Phillips K. F., DeLorenzo R. J. (2016). Pharmacological blockade of the calcium plateau provides neuroprotection following organophosphate paraoxon induced status epilepticus in rats. Neurotoxicol. Teratol. 56, 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande L. S., Carter D. S., Blair R. E., DeLorenzo R. J. (2010). Development of a prolonged calcium plateau in hippocampal neurons in rats surviving status epilepticus induced by the organophosphate diisopropylfluorophosphate. Toxicol. Sci. 116, 623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande L. S., Phillips K., Huang B., DeLorenzo R. J. (2014). Chronic behavioral and cognitive deficits in a rat survival model of paraoxon toxicity. Neurotoxicology 44, 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes N. F., Stewart C. A., Matthews K., Reid I. C. (1996). Chronic mild stress and sucrose consumption: Validity as a model of depression. Physiol. Behav. 60, 1481–1484. [DOI] [PubMed] [Google Scholar]
- Fukuyama K., Tanahashi S., Nakagawa M., Yamamura S., Motomura E., Shiroyama T., Tanii H., Okada M. (2012). Levetiracetam inhibits neurotransmitter release associated with CICR. Neurosci. Lett. 518, 69–74. [DOI] [PubMed] [Google Scholar]
- Gower A. J., Falter U., Lamberty Y. (2003). Anxiolytic effects of the novel anti-epileptic drug levetiracetam in the elevated plus-maze test in the rat. Eur. J. Pharmacol. 481, 67–74. [DOI] [PubMed] [Google Scholar]
- Gulf War and Health C. 2016. Committee on Gulf War and Health, Volume 10: Update of Health Effects of Serving in the Gulf War; Board on the Health of Select Populations; Institute of Medicine; National Academies of Sciences, Engineering, and Medicine; (Cory-Slechta D. and Wedge R., Eds.), National Academies Press, Washington DC. [PubMed] [Google Scholar]
- Hagenston A. M., Bading H. (2011). Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb. Perspect. Biol. 3, a004564.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez C. M., Beck W. D., Naughton S. X., Poddar I., Adam B. L., Yanasak N., Middleton C., Terry A. V. Jr., (2015). Repeated exposure to chlorpyrifos leads to prolonged impairments of axonal transport in the living rodent brain. Neurotoxicology 47, 17–26. [DOI] [PubMed] [Google Scholar]
- Hertle D. N., Yeckel M. F. (2007). Distribution of inositol-1, 4, 5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience 150, 625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr K. J. (2015). Gulf war illness: An overview of events, most prevalent health outcomes, exposures, and clues as to pathogenesis. Rev. Environ. Health 30, 273–286. [DOI] [PubMed] [Google Scholar]
- Koo B. B., Michalovicz L. T., Calderazzo S., Kelly K. A., Sullivan K., Killiany R. J., O’Callaghan J. P. (2018). Corticosterone potentiates DFP-induced neuroinflammation and affects high-order diffusion imaging in a rat model of gulf war illness. Brain Behav. Immun. 67, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir A., Wajsberg B., Marks A. R. (2018). Ryanodine receptor dysfunction in human disorders. Biochim. Biophys. Acta Mol. Cell Res. 1865, 1687–1697. [DOI] [PubMed] [Google Scholar]
- Lacampagne A., Liu X., Reiken S., Bussiere R., Meli A. C., Lauritzen I., Teich A. F., Zalk R., Saint N., Arancio O., et al. (2017). Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer's disease-like pathologies and cognitive deficits. Acta Neuropathol. 134, 749–767. [DOI] [PubMed] [Google Scholar]
- Lehnart S. E., Mongillo M., Bellinger A., Lindegger N., Chen B. X., Hsueh W., Reiken S., Wronska A., Drew L. J., Ward C. W., et al. (2008). Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Invest. 118, 2230–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Betzenhauser M. J., Reiken S., Meli A. C., Xie W., Chen B. X., Arancio O., Marks A. R. (2012). Role of leaky neuronal ryanodine receptors in stress-induced cognitive dysfunction. Cell 150, 1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx S. O., Reiken S., Hisamatsu Y., Jayaraman T., Burkhoff D., Rosemblit N., Marks A. R. (2000). Pka phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 101, 365–376. [DOI] [PubMed] [Google Scholar]
- Musaeus C. S., Shafi M. M., Santarnecchi E., Herman S. T., Press D. Z. (2017). Levetiracetam alters oscillatory connectivity in alzheimer’s disease. J Alzheimers Dis. 58, 1065–1076. [DOI] [PubMed] [Google Scholar]
- Nagarkatti N., Deshpande L. S., DeLorenzo R. J. (2008). Levetiracetam inhibits both ryanodine and IP3 receptor activated calcium induced calcium release in hippocampal neurons in culture. Neurosci. Lett. 436, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarkatti N., Deshpande L. S., DeLorenzo R. J. (2009). Development of the calcium plateau following status epilepticus: Role of calcium in epileptogenesis. Expert Rev. Neurother. 9, 813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Callaghan J. P., Kelly K. A., Locker A. R., Miller D. B., Lasley S. M. (2015). Corticosterone primes the neuroinflammatory response to DFP in mice: Potential animal model of gulf war illness. J. Neurochem. 133, 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet D. H. (2012). Modeling depression in animal models. Methods Mol. Biol. 829, 125–144. [DOI] [PubMed] [Google Scholar]
- Parihar V. K., Hattiangady B., Shuai B., Shetty A. K. (2013). Mood and memory deficits in a model of Gulf War Illness are linked with reduced neurogenesis, partial neuron loss, and mild inflammation in the hippocampus. Neuropsychopharmacology 38, 2348–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips K. F., Deshpande L. S. (2016). Repeated low-dose organophosphate DFP exposure leads to the development of depression and cognitive impairment in a rat model of Gulf War Illness. Neurotoxicology 52, 127–133. [DOI] [PubMed] [Google Scholar]
- Phillips K. F., Deshpande L. S. (2018). Chronic neurological morbidities and elevated hippocampal calcium levels in a DFP-based rat model of Gulf War Illness. Mil. Med. 183, 552–555. [DOI] [PubMed] [Google Scholar]
- Popugaeva E., Pchitskaya E., Bezprozvanny I. (2017). Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease - A therapeutic opportunity? Biochem. Biophys. Res. Commun. 483, 998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza M., Blair R. E., Sombati S., Carter D. S., Deshpande L. S., DeLorenzo R. J. (2004). Evidence that injury-induced changes in hippocampal neuronal calcium dynamics during epileptogenesis cause acquired epilepsy. Proc. Natl. Acad. Sci. U.S.A. 101, 17522–17527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty G. A., Hattiangady B., Upadhya D., Bates A., Attaluri S., Shuai B., Kodali M., Shetty A. K. (2017). Chronic oxidative stress, mitochondrial dysfunction, nrf2 activation and inflammation in the hippocampus accompany heightened systemic inflammation and oxidative stress in an animal model of Gulf War Illness. Front. Mol. Neurosci. 10, 182.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D. A., Deshpande L. S., Sombati S., Baranova A., Wilson M. S., Hamm R. J., DeLorenzo R. J. (2008). Traumatic brain injury causes a long-lasting calcium-plateau of elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons surviving the brain injury. Eur. J. Neurosci. 27, 1659–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier D. J., Halliday G. M., Simuni T. (2017). Calcium, mitochondrial dysfunction and slowing the progression of parkinson's disease. Exp. Neurol. 298, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehse J., Taghibiglou C. (2018). The overlooked aspect of excitotoxicity: Glutamate-independent excitotoxicity in traumatic brain injuries. Eur. J. Neurosci. doi: 10.1111/ejn.14307. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Torres-Altoro M. I., Mathur B. N., Drerup J. M., Thomas R., Lovinger D. M., O’Callaghan J. P., Bibb J. A. (2011). Organophosphates dysregulate dopamine signaling, glutamatergic neurotransmission, and induce neuronal injury markers in striatum. J. Neurochem. 119, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuite J. J., Haley R. W. (2013). Meteorological and intelligence evidence of long-distance transit of chemical weapons fallout from bombing early in the 1991 Persian Gulf War. Neuroepidemiology 40, 160–177. [DOI] [PubMed] [Google Scholar]
- Walf A. A., Frye C. A. (2007). The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat. Protocols 2, 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R. F., Steele L., O'Callaghan J. P., Sullivan K., Binns J. H., Golomb B. A., Bloom F. E., Bunker J. A., Crawford F., Graves J. C., et al. (2016). Recent research on gulf war illness and other health problems in veterans of the 1991 Gulf War: Effects of toxicant exposures during deployment. Cortex 74, 449–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams N. R., Schatzberg A. F. (2016). NMDA antagonist treatment of depression. Curr. Opin. Neurobiol. 36, 112–117. [DOI] [PubMed] [Google Scholar]
- Yee M. K., Seichepine D. R., Janulewicz P. A., Sullivan K. A., Proctor S. P., Krengel M. H. (2016). Self-reported traumatic brain injury, health and rate of chronic multisymptom illness in veterans from the 1990-1991 Gulf War. J. Head Trauma Rehabil. 31, 320–328. [DOI] [PubMed] [Google Scholar]
- Yuan Q., Deng K. Y., Sun L., Chi S., Yang Z., Wang J., Xin H. B., Wang X., Ji G. (2016). Calstabin 2: An important regulator for learning and memory in mice. Sci. Rep. 6, 21087.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalk R., Clarke O. B., des Georges A., Grassucci R. A., Reiken S., Mancia F., Hendrickson W. A., Frank J., Marks A. R. (2015). Structure of a mammalian ryanodine receptor. Nature 517, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H., Brayer S. W., Hurwitz M., Niyonkuru C., Fowler L. E., Wagner A. K. (2013). Neuroprotective, neuroplastic, and neurobehavioral effects of daily treatment with levetiracetam in experimental traumatic brain injury. Neurorehabil. Neural Repair 27, 878–888. [DOI] [PubMed] [Google Scholar]