

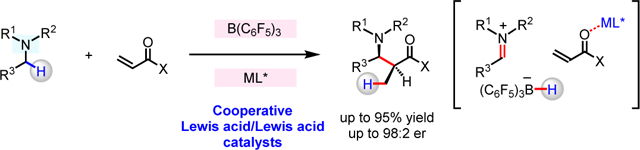
Abstract
Catalytic transformations of α-amino C–H bonds to afford valuable enantiomerically enriched α-substituted amines, entities that are prevalent in pharmaceuticals and bioactive natural products, have been developed. Typically, such processes are carried out under oxidative conditions, and require precious metal-based catalysts. Here, we disclose a strategy for enantioselective union of N-alkylamines and α,β-unsaturated compounds, performed under redox-neutral conditions, and promoted through concerted action of seemingly competitive Lewis acids, B(C6F5)3 and a chiral Mg–PyBOX complex. Thus, a wide variety of β-amino carbonyl compounds may be synthesized, with complete atom economy, through stereoselective reaction of an in situ generated enantiomerically enriched Mg-enolate and an appropriate electrophile.
Graphical Abstract
INTRODUCTION
Cooperative catalysts (e.g., Fig. 1a, A1–A4), which are capable of two distinct and simultaneous functions, can doubly activate substrate molecules.1–6 One advantage of this strategy in catalysis is the considerable rate enhancement that can become available as a result of increased proximity of the reacting substrates. Cooperative acid/base enantioselective catalysis has previously been applied to the development of reactions between in situ generated, acid-activated electrophiles and base-activated nucleophiles. The wasteful pre-activation step may thus be obviated.1–6 Nevertheless, notable shortcomings remain unaddressed. For instance, self-quenching might occur in a mixture that contains an electrophile, a nucleophile, together with an acid and a base catalyst. One way to circumvent acid–base complexation is by avoiding a combination that exhibits high affinity (i.e., hard–hard or soft–soft pairing). However, this latter approach has thus far been limited to cases in which weakly to moderately acidic and/or basic promoters are involved, where only highly acid- or base-sensitive substrates can be used. Development of potent and unquenchable cooperative two-catalyst systems that facilitate reactions between unactivated substrates is an important and largely unresolved problem in enantioselective catalysis.
Figure 1. Structures and reactivity of cooperative catalysts.

(a) Previously reported examples of cooperative acid/acid catalysts involve structurally and/or functionally disparate acidic promoters. (b) Intramolecular hydride transfer occurs within a frustrated Lewis pair complex to afford zwitterionic iminium ions. (c) Enantioselective coupling of N-alkylamines and α,β-unsaturated compounds by cooperative acid/acid catalysis. (d) A possible mechanism might involve enantio- and diastereoselective C–C bond formation between iminium ion and chiral enolate, generated in situ by cooperative functions of a chiral and an achiral Lewis acid catalyst. The reaction affords β-amino carbonyl compounds atom economically and under redox neutral conditions.
The application of frustrated Lewis pairs (FLPs), consisting of hindered and electronically disparate Lewis acids and Lewis bases, has recently emerged as an attractive strategy for overcoming mutual quenching. An unquenched acid/base pair may thus be utilized for synergistic activation of otherwise unreactive molecules such as H2 and CO2.7–9 Furthermore, FLPs that are comprised of a boron-based Lewis acid and a Lewis basic amine substrate, and contain easily accessible α-hydrogens (e.g., B; Fig. 1b), have been shown to engage in Lewis acid-mediated hydride abstraction. Such processes produce an iminium cation and a borohydride anion (C),10–13 the latter of which might then react with a carbonyl-containing species to afford D.10 A related investigation illustrates that the presence of stoichiometric amounts of B(C6F5)3 can lead to C–C bond forming reactions between N-alkylamines and α,β-unsaturated molecules to afford β-amino carbonyl compounds.13 Still, engagement of iminium ions formed by organoboron-catalyzed hydride abstraction from amines in the context of a catalytic transformation has been confined to dehydrogenation of N-containing heterocycles.14,15 To the best of our knowledge, such strategies are yet to be successfully applied to catalytic C–C bond forming transformations.16,17 One way to address this problem would be by designing a catalyst that can abstract a hydride from an amine substrate, mediate enantioselective C–C bond formation, and regenerate the active Lewis pair without significant deactivation due to the presence of a Lewis basic moiety.
While contemplating how to design a catalytic process for stereoselective coupling of N-alkylamines and carbon-based nucleophiles, we envisioned utilizing two potent Lewis acid catalysts for accomplishing separate tasks (Fig. 1c–d). We imagined that B(C6F5)3 could receive a hydride from amine 1, generating a borohydride and an iminium ion (I). Concurrently, a chiral Lewis acid co-catalyst would activate the α,β-unsaturated substrate (e.g., 2) to facilitate reduction by the aforementioned borohydride (III), furnishing the corresponding chiral enolate (IV). An ensuing stereoselective reaction between the iminium ion and the enantiomerically enriched enolate would deliver a β-amino carbonyl product (3). One key advantage of the approach would be that efficiency and stereoselectivity might be optimized through evaluation of pairs of readily accessible Lewis acids and chiral ligands. Accordingly, a central design principle is that the untethered Lewis acid catalysts must perform their separate tasks without any overlapping of their function, as otherwise, stereoselectivity would suffer (due to reaction facilitated by the achiral component).18 Enantioselective transformations promoted by the shared action of two different metal complexes,1–5,19 as well as Brønsted acid and organometallic complexes20,21 or hydrogen-bond donors22 are known (e.g., Fig. 1a, A1–A4). However, in contrast to what we aimed to accomplish, in the previous approaches, the Lewis acidic promoters were structurally and/or functionally disparate, translating to minimal overlay in their modes of action; namely, their simultaneous presence did not pose a complication. Here, we report that functionally similar B(C6F5)3 and a chiral Lewis acid co-catalyst can operate in concert to promote enantioselective coupling of N-alkylamines and various α,β-unsaturated compounds.
The plan outlined above is distinct from the traditional “direct” Mannich-type reactions, where Lewis acid and Brønsted base catalysts cooperate to generate first an enolate, and then promote its addition to an aldimine, which has already been prepared through a separate operation.4,5,23,24 There are a small number of reported examples regarding direct Mannich-type reactions that involve esters, amides or thioesters; deprotonation is more difficult in these instances, because of diminished acidity (pKa in the range of 25–30). We sought to generate the requisite electrophilic and the nucleophilic species in situ from readily available N-alkylamines and α,β-unsaturated compounds. Thus, under redox-neutral conditions and without the need for a precious metal salt,25 we would synthesize α-substituted amines 3, entities that are difficult to access by “direct” Mannich strategies. Many of the reported methods for β-amino C–H functionalization rely on transition metal-based catalysts (e.g., Fe-, Cu-, Ru-, Rh-, Pd-, Ir-, Pt-based), and, often, demand oxidative conditions.26–36 These catalyst systems may either forge C–C bonds directly,36 or first convert a substrate amine into a more active intermediate, such as an α-amino radical,26 an iminium ion,27–30 an α-halo, an α-hydroxyl or an α-metalated species.31–35 Catalytic stereoselective α-amino C–H functionalization has been investigated as well.27,29,30,34,36 However, the development of non-precious metal-based methods that complements the existing stereoselective techniques represents a compelling problem.
RESULTS AND DISCUSSION
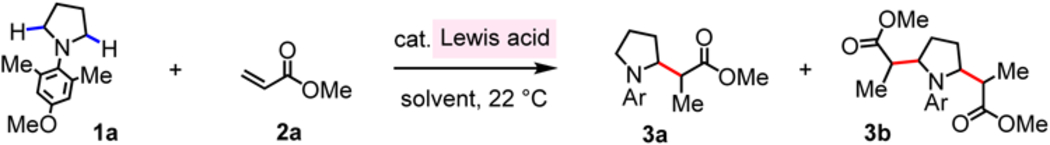
To begin, we set out to identify an appropriate combination of an acidic catalyst and an amine substrate that would allow for efficient hydride abstraction (as opposed to formation of a stable acid–base adduct). We first probed the ability of B(C6F5)3 to catalyze the reaction between N-aryl pyrrolidine (1a) and methyl acrylate (2a), affording β-amino ester 3a (Table 1; see the Supplementary Information for data regarding screening of N-substituents). In this representative process, a single Lewis acid B(C6F5)3 would be responsible for activation of the amine as well as the Michael acceptor (Fig. 1d, M–L* = B(C6F5)3). Treatment of 1a and 2a with 5.0 mol% B(C6F5)3 in CH2Cl2 at 22 °C for 12 hours afforded 3a in >95% yield as a separable mixture of diastereomers (entry 1, anti:syn = 2.3:1); there was less than 5% of the di-substituted product (3b). With Et2O and THF as the solvent, 3a was formed in 94% and 26% yield, respectively (entries 2–3). When the reaction was carried out in a relatively non-polar aromatic hydrocarbon, such as toluene and benzene, 3a was obtained in 81% and >95% yield (entries 4–5); in benzene, 3a was isolated in >95% yield in just 30 minutes (entry 6). At lower loading of B(C6F5)3 (2.5 mol%), longer reaction time was needed (48 h, 3a in 84% yield; entry 7).37 None of the desired product was formed in the absence of B(C6F5)3 (entry 8), or when the less hindered BF3•OEt2 or the less acidic BPh3 were used (entries 9–10). The latter findings support the notion that highly acidic B(C6F5)3 together with sterically demanding and electron-rich N-alkylamines represent the most effective combination.
Table 1.
 | ||||||
|---|---|---|---|---|---|---|
| entry | Lewis acid | (mol%) | solvent | reaction time (h) |
yield (%), anti:syn 3a |
yield (%) 3b |
| 1 | B(C6F5)3 | 5.0 | CH2CI2 | 12 | >95, 2.3:1 | <5 |
| 2 | B(C6F5)3 | 5.0 | Et2O | 12 | 94, 2.3:1 | <5 |
| 3 | B(C6F5)3 | 5.0 | THF | 12 | 26, 1.9:1 | <5 |
| 4 | B(C6F5)3 | 5.0 | Toluene | 12 | 81, 2.4:1 | 5 |
| 5 | B(C6F5)3 | 5.0 | Benzene | 12 | >95, 2.4:1 | <5 |
| 6 | B(C6F5)3 | 5.0 | Benzene | 0.5 | >95, 2.4:1 | <5 |
| 7 | B(C6F5)3 | 2.5 | Benzene | 48 | 84, 1.7:1 | <5 |
| 8 | none | 0 | Benzene | 12 | 0 | 0 |
| 9 | BF3•OEt2 | 5.0 | Benzene | 12 | 0 | 0 |
| 10 | BPh3 | 5.0 | Benzene | 12 | 0 | 0 |
Conditions: 1-(4-Methoxy-2,6-dimethylphenyl)pyrrolidine (0.2 mmol), methyl acrylate (0.3 mmol), Lewis acid, solvent (0.3 mL), under N2, 22 °C.
Yields were determined by 1H NMR analysis of unpurified product mixtures with mesitylene as the internal standard. See the Supporting Information for details.
A variety of α,β-unsaturated carbonyl compounds and related electrophiles may be used in reaction with N-arylpyrrolidine 1a to generate the corresponding α-substituted amines (3a–3k, Fig. 2). Reaction with methyl acrylate (2a) and benzyl acrylate (2c) afforded 3a and 3c in 97% and 91% yield, respectively. Whereas isopropyl acrylate (2d) gave 3d in 78% yield, no product was obtained with tert-butyl acrylate; this decline in efficiency probably arises from inferior ability of B(C6F5)3 in coordinating with the more sterically encumbered acrylates. Transformation with the less electrophilic thioacrylate (2e) gave 3e (53% yield), and acrylonitrile (2f) proved to be a viable starting material, furnishing 3f in 50% yield. Reaction with methyl methacrylate (2g) gave 3g, containing an α-quaternary carbon center, in 97% yield. Dimethyl acetylenedicarboxylate (2h) was converted in situ to an allenolate nucleophile, which reacted with 1a-derived iminium ion to deliver 3h in 52% yield as a separable mixture of E and Z isomers (1:1.6). Transformations with N-phenylmaleimide (2i), trans-fumaronitrile (2j), and (E)-β-nitrostyrene (2k) furnished 3i–3k (84% to >95% yield).
Figure 2. Coupling of N-alkylamines and α,β-unsaturated compounds through B(C6F5)3-catalyzed hydride abstraction.

The values correspond to yields of isolated and purified products. Diastereomeric ratio (d.r.) values were determined by analysis of the unpurified product mixture through analysis of 1H NMR spectra. *5.0 mol% of B(C6F5)3 was used. †10 mol% of B(C6F5)3 was used. See the Supplementary Information for details.
Next, we investigated reactions with cyclic and acyclic N-alkylamines. 3,3-Dimethyl-,3-((tert-butyldimethylsilyl)oxy)-substituted N-aryl pyrrolidines (1l, 1m), N-aryl piperidine (1n) and N-aryl azepane (1o) gave the corresponding products (3l–3o) in 63% to 95% yield. There was efficient hydride abstraction at the N-methyl site of various amines. 4-Methoxy-N,N,2,6-tetramethylaniline (1p) reacted with N-phenylmaleimide to afford 3p (62% yield). A series of trialkyl-substituted amines that lack the fused N-aryl group were coupled efficiently with acrylates, leading to the formation of 4a–4e (85%–95% yield). Reaction of (R)-N-methyl-1-phenyl-N-((R)-1-phenylethyl)ethan-1-amine with benzyl acrylate (2c) delivered 4d as a 1.5:1 mixture of diastereomers, which were separable through silica gel chromatography, allowing us to secure the β-amino esters in enantiomerically pure form (see the Supplementary Information for details). The less hindered N-alkylamines (e.g., N-methyl pyrrolidine) did not react with 2c, probably due to the formation of stable B(C6F5)3–amine adducts, which may compete with Lewis acid-catalyzed hydride abstraction.
Having realized the single-catalyst racemic transformation based on the hydride abstraction concept, we chose to develop an enantioselective version of the catalytic process by employing B(C6F5)3 in combination with an appropriate chiral Lewis acid. With N-arylpyrrolidine (1a) and 3-acryloyloxazolidin-2-one (2q) as model substrates, systematic evaluation of Lewis acid/chiral ligand complexes was performed (Fig. 3a). In the presence of 10 mol% of B(C6F5)3 and Mg(OTf)2, various bis-oxazoline ligands were tested (12 mol%, L1–L4). Whereas in the presence of PhBOX (L1) and DBFOX (L2) there was minimal conversion to 3q (<10% yield), with PyBOX (L3, (S,S)-2,2’-(2,6-pyridinediyl)bis(4-phenyl-2-oxazoline)) we obtained 3q in 35% yield, 1.1:1 anti:syn ratio, and 71:29 e.r. and 80:20 e.r. (syn and anti isomer, respectively). There was further improvement in efficiency (3q in >95% yield) when 2,6-bis((S)-4-(3-chlorophenyl)-4,5-dihydrooxazol-2-yl)pyridine (L4) was utilized as the chiral ligand. There was minimal conversion to the desired product and/or low enantioselectivity with Mg(OTf)2/L4, or when Cu(OTf), Ni(OTf)2, Zn(OTf)3, Sc(OTf)3, Mg(ClO4)2, and other Lewis acid co-catalysts were employed in combination with various ligands (see the Supplementary Information for details). Because B(C6F5)3 catalyzes the C–C bond forming reaction between 1a and 2q in the absence of the chiral Mg-based co-catalyst (Fig. 3b; rac-3q, 35% yield), the presence of this Lewis acid can engender considerable diminution in e.r. However, we subsequently discovered that the reaction of 1a with 3-acryloyl-4,4-dimethyloxazolidin-2-one (2r), catalyzed by the combination of B(C6F5)3 and Mg(OTf)2/L4 can deliver either diastereoisomer of 3r in high e.r. (3r-anti and 3r-syn in 97:3 and 98:2 e.r., respectively, 88% yield; Fig. 3c). The higher enantioselectivity probably arises from a preference for Mg-activated VIII over B(C6F5)3-activated VII, the latter of which might suffer from severe steric repulsion between the gem-dimethyl group of 2r and B(C6F5)3, a contention supported by the fact that 2r does not react in the absence of Mg(OTf)2/L4.
Figure 3. The effect of chiral Lewis acid co-catalysts on enantioselective coupling of an N-alkylamine and N-acryloyloxazolidinones.

(a) Initial evaluation of chiral co-catalysts revealed that Mg(OTf)2/bis(oxazoline) complexes catalyze stereoselective C–C bond formation. (b) Competing B(C6F5)3-catalyzed racemic C–C bond forming reaction could deteriorate overall enantioselectivity. (c) The use of more hindered N-acryloyl oxazolidinone suppresses the racemic process to achieve C–C bond formation with higher enantioselectivity.
With 2r as the electrophilic partner, fine-tuning of PyBOX ligands was performed (L3–L12, Fig. 4a). With derivatives that possess a halophenyl moiety (L4, L7–L10), 3r was generated in superior yield and stereoselectivity (compared to other alkyl- or aryl-substituted ligands, L3, L5, L6, L11, L12). Whereas reaction with phenyl-substituted L3 gave 3r in low yield (18%; 94:6 e.r. for anti and syn isomers), that of 3-chlorophenyl-subsituted L4 afforded 3r in 88% yield as a 4.3:1 mixture of anti:syn isomers (97:3 and 98:2 e.r., respectively). The size and position of the halogen substituent within the chiral catalyst can influence a reaction outcome. With 3-fluorophenyl- (L7) or 3-bromophenyl-substituted (L8) ligands, the transformations were moderately efficient (3r in 36% and 54% yield, respectively), but highly enantioselective (97:3 e.r. (3r-anti) and 98:2 e.r. (3r-syn)). With 2-chlorophenyl- (L9) and 4-chlorophenyl-substituted (L10) ligands, on the other hand, while 3r was isolated in higher yield 88% and 72%, respectively), there was some diminution in diastereo- and enantioselectivity (ca. 2:1 anti:syn, ca. 90:10 e.r. for 3r-anti). What is more, reactions performed in the presence of meta-tolyl-substituted L11 or electron-withdrawing 3-trifluoromethylphenyl-substituted L12 were much less efficient (<20% yield). Although the precise origin of such notable effects by the catalyst structure remains to be elucidated, the present findings suggest that non-covalent interactions38,39 (e.g., π–cation, π–π interactions) between ligand substituents and in situ-generated iminium ion and/or Mg–enolate might be critical.
Figure 4. The influence of different PyBOX ligand and oxazolidinone substituents.

(a) Notable improvements in reaction efficiency and stereoselectivity were observed with halophenyl-substituted PyBOX ligands. (b) β-Amino amides were prepared in up to 98:2 e.r. from the achiral N-acryloyloxazolidinone. With chiral oxazolidinones and Mg(OTf)2/L4, enantiomerically pure β-amino amide 3u-(R) could be obtained after separation of diastereomers by silica gel chromatography. Catalyst loading can be lowered to 5.0 mol% without deterioration in yield of isolated and purified product.
With B(C6F5)3 and Mg(OTf)2/L4 as optimal catalysts, we surveyed a series of achiral and chiral N-acryloyloxazolidinones (Fig. 4b). In contrast to dimethyl-substituted oxazolidinone 3r was obtained in 88% yield and up to 98:2 e.r. (Fig. 3c), the less hindered 3q was generated in >95% yield, but only in up to 80:20 e.r., presumably as a result of competing transformation catalyzed by B(C6F5)3. Cyclohexyl-substituted 3s was obtained in anti:syn ratio of 5.6:1 (vs. 3q (1.6:1) and 3r (4.3:1)), and enantiomerically pure N-acryloyloxazolidinones containing benzyl (2t) or phenyl (2u-(S), 2u-(R)) moieties proved to be suitable starting materials as well. (R)-3-Acryloyl-4-phenyloxazolidin-2-one (2u-(R)) represents the “matched” enantiomer with Mg(OTf)2/L4 pairing to afford 3u-(R) in >95% yield and 6.8:1:0:0 d.r.; with 2u-(S), 3u-(S) was obtained in 61% yield and 3.7:1:0:2.1 d.r. The significant impact of the chiral co-catalyst on efficiency and stereoselectivity is evident as 3u-(R) was produced in just 14% yield and 2.5:2.5:1:1 d.r. in its absence. With B(C6F5)3 and Mg(OTf)2/L4 loadings reduced to 5.0 mol%, 3u-(R) was obtained in 93% yield (6.8:1:0:0 d.r.).
The catalytic protocol is amenable to scale-up; for instance, 5.0 mmol of 1a was converted to 3u-(R) in 6.7:1:0:0 dr (>95% conv.; Fig. 5a). The diastereomers were separated readily by silica gel chromatography to afford the major isomer in 1.47 g (70% yield; minor diastereomer: 0.16 g, 8% yield). The predominant diastereomer was then converted to β-amino ester (5a), β-amino acid (5b), and β-amino alcohol (5c) in 64–91% yield and >99:1 er. Furthermore, treatment of 3u-(R) with NaOMe in methanol led to the formation of 5d in 97% yield. X-ray crystallographic analysis of 5d revealed its absolute configuration to be (R,R,R).40 The two-catalyst protocol is applicable to synthesis of a variety of N-alkylamines, affording 3v and 4f–4h as separable mixtures of diastereomers (see the Supplementary Information for details). After chromatographic separation, each of the diastereomers was converted into the corresponding benzyl ester (5), and HPLC analysis showed that 3v and 4f–4h were generated in >99:1 e.r. in either isomeric form. The presence of Mg(OTf)2/L4 was essential, as otherwise 4h was produced in only 32% and 1:1 d.r. in its absence. Reactions of various N-substituted pyrrolidines with 3-acryloyl-4,4-dimethyloxazolidin-2-one (2r) catalyzed by B(C6F5)3 and Mg(OTf)2/L4 were investigated. While no desired product was obtained with N-phenylpyrrolidine (see the Supplementary Information for details), 3w, 3x and 3y possessing the hindered and electron-donating 2,6-dimethylaryl substituents were produced in 71, 69 and 70% overall yield, respectively. An increase in enantioselectivity for both anti and syn diastereomers was observed with more hindered para-t-Bu-substituted 3y (91:9 e.r. (3y-anti) and 98:2 e.r. (3y-syn)) over para-H-substituted 3w (89:11 e.r. (3w-anti) and 93:7 e.r. (3w-syn)). Sterically encumbered N,N,N-trialkylamines could also react with 2r to afford 4i (52% yield, 88:12 er) and 4j (46% yield, 96:4 er).
Figure 5. Synthesis of chiral α-substituted amines.

(a) As demonstrated through preparation of β-amino amide 3u-(R), the catalytic reactions are amenable to gram-scale operations. (b) The versatility of 3u-(R) was demonstrated by its conversion into a β-amino ester, a β-amino alcohol and a β-amino acid. The absolute configuration of 3u-(R) was determined by X-ray crystallographic analysis of 5d. (c) A series of N-alkylamines reacted with 2 to afford the corresponding β-amino amides. For 3v and 4f–4h, diastereomers were isolated by silica gel chromatography and then converted into β-amino esters (5) that were determined to be enantiomerically pure.
We have developed a stereochemical model for reaction of the L3–Mg–enolate complex with the in situ generated iminium ion by means of density functional theory (DFT) studies (Fig. 6). These investigations suggest that the enantiomers of the minor diastereomer are probably formed via IX and X (ΔG = 20.6 and 21.1 kcal/mol respectively), whereas XI and XII generate enantiomers of the major diastereomer (ΔG = 18.1 and 18.8 kcal/mol, respectively). High enantioselectivity for the major as well as the minor diastereomer is due to effective blocking of the re face of the Mg bound enolate (IX and XI). Nonetheless, we should note that highly accurate modeling of diastereoselectivity is difficult, and depends strongly on attenuation of dispersion interactions in solution.41,42 It is possible that modes other than IX and X might similarly contribute to formation of the minor diastereomer (see the Supplementary Information).
Figure 6. Stereochemical models to account for the observed sense of stereoselectivity.

Stereochemical model for reaction of L3–Mg–enolate complex with iminium ion; DFT studies were performed at the PBE0-D3BJ/def2TZVPP//M06L/DF-def2SVP level of theory in CH2Cl2 (SMD solvation model). See the Supplementary Information for details. Abbreviations: L3, PyBOX ligand; SMD, solvation model based on density; ΔΔEdisp = ΔΔE(PBE0-D3BJ) – ΔΔE(PBE0).
CONCLUSIONS
In brief, we have designed an efficient and diastereo- and enantioselective C–C bond forming transformation by implementing the cooperative action of two non-precious metal-based Lewis acid catalysts that possess overlapping functions. We illustrate that by proper tuning of different features of structurally and electronically different Lewis acids and substrates, the ability of Lewis acid catalysts to serve as a hydride acceptor from amines, or an activator of α,β-unsaturated compounds, can be adjusted. Accordingly, enhancements in both the efficiency and stereoselectivity of C–C bond forming reactions between amines and α,β-unsaturated compounds can be attained without concomitant loss in enantioselectivity arising from any undesirable mode of catalysis by the achiral Lewis acid component. The principles outlined herein serve as a conceptual framework for the development of new processes that demand separate and independently operational Lewis acidic co-catalysts whose functions might easily overlap and the simultaneous use of which might initially seem to have a negative impact on enantioselectivity. Studies aimed at achieving highly enantioselective C–C bond forming reactions with a broader scope of hydride donors and pro-nucleophiles are in progress.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful for financial supports from the National Institutes of Health (GM-128695) and Boston College. We are thankful to Professor Amir H. Hoveyda (BC) for helpful discussions. Dr. Juan del Pozo del Valle, Dr. Filippo Romiti, Mr. Malte S. Mikus (BC), and Mr. Richard Y. Liu (MIT) are thanked for their assistance with this project. We are grateful to Professor James P. Morken for a loan of PyBOX ligands, and Dr. Bo Li for the X-ray crystallographic analysis.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Cooperative Catalysis: Designing Efficient Catalysts for Synthesis, Peters R, Eds.; Wiley–VCH: New York, 2015. [Google Scholar]
- 2.van den Beuken EK; Feringa BL Tetrahedron 1998, 54, 12985–13011. [Google Scholar]
- 3.Yamamoto H; Futatsugi K Angew. Chem., Int. Ed 2005, 44, 1924–1942. [DOI] [PubMed] [Google Scholar]
- 4.Matsunaga S; Shibasaki M Chem. Commun 2014, 50, 1044–1057. [DOI] [PubMed] [Google Scholar]
- 5.Trost BM; Bartlett MJ Acc. Chem. Res 2015, 48, 688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang MH; Scheidt KA Angew. Chem., Int. Ed 2016, 55, 14912–14922. [DOI] [PubMed] [Google Scholar]
- 7.(a) Stephan DW J. Am. Chem. Soc 2015, 137, 10018–10032. [DOI] [PubMed] [Google Scholar]; (b) Stephan DW; Erker G Angew. Chem., Int. Ed 2015, 54, 6400–6441. [DOI] [PubMed] [Google Scholar]; (c) Stephan DW Science 2016, 354, aaf7229. [DOI] [PubMed] [Google Scholar]
- 8.Keess S; Oestreich M Chem. Sci 2017, 8, 4688–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashley AE; O’Hare D Top. Curr. Chem 2013, 334, 191–218. [DOI] [PubMed] [Google Scholar]
- 10.Schwendemann S; Frohlich R; Kehr G; Erker G Chem. Sci 2011, 2, 1842–1849. [Google Scholar]
- 11.Millot N; Santini CC; Fenet B & Basset JM Eur. J. Inorg. Chem 2002, 2002, 3328–3335. [Google Scholar]
- 12.Focante F; Mercandelli P; Sironi A; Resconi L Coord. Chem. Rev 2006, 250, 170–188. [Google Scholar]
- 13.Chen G-Q; Kehr G; Daniliuc CG; Bursch M; Grimme S; Erker G Chem. Eur. J 2017, 23, 4723–4729. [DOI] [PubMed] [Google Scholar]
- 14.Maier AFG; Tussing S; Schneider T; Flörke U; Qu Z-W; Grimme S; Paradies J Angew. Chem., Int. Ed 2016, 55, 12219–12223. [DOI] [PubMed] [Google Scholar]
- 15.Kojima M; Kanai M Angew. Chem., Int. Ed 2016, 55, 12224–12227. [DOI] [PubMed] [Google Scholar]
- 16.Dureen MA; Brown CC; Stephan DW Organometallics 2010, 29, 6422–6432. [Google Scholar]
- 17.Sergej T; Zheng-Wang Q; A. SN; Ulrich F; Stefan G; Jan P Angew. Chem., Int. Ed 2016, 55, 4336–4339. [Google Scholar]
- 18.Manville N; Alite H; Haeffner F; Hoveyda AH; Snapper ML Nat. Chem 2013, 5, 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mihara H; Xu Y; Shepherd NE; Matsunaga S; Shibasaki M J. Am. Chem. Soc 2009, 131, 8384–8385. [DOI] [PubMed] [Google Scholar]
- 20.Kong J-R; Ngai M-Y; Krische MJ J. Am. Chem. Soc 2006, 128, 718–719. [DOI] [PubMed] [Google Scholar]
- 21.Inamdar SM; Shinde VS; Patil NT Org. & Biomol. Chem 2015, 13, 8116–8162. [DOI] [PubMed] [Google Scholar]
- 22.Xu H; Zuend SJ; Woll MG; Tao Y; Jacobsen EN Science 2010, 327, 986–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trost BM; Saget T; Hung C-I J. Am. Chem. Soc 2016, 138, 3659–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun B; Balaji PV; Kumagai N; Shibasaki M J. Am. Chem. Soc 2017, 139, 8295–8301. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura E; Sato K Nat. Mater 2011, 10, 158–161. [DOI] [PubMed] [Google Scholar]
- 26.Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DWC Science 2016, 352, 1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C-J Acc. Chem. Res 2009, 42, 335–344. [DOI] [PubMed] [Google Scholar]
- 28.McQuaid KM; Sames D J. Am. Chem. Soc 2009, 131, 402–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murarka S; Deb I; Zhang C; Seidel D J. Am. Chem. Soc 2009, 131, 13226–13227. [DOI] [PubMed] [Google Scholar]
- 30.DiRocco DA; Rovis T J. Am. Chem. Soc 2012, 134, 8094–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campos KR Chem. Soc. Rev 2007, 36, 1069–1084. [DOI] [PubMed] [Google Scholar]
- 32.Cordier CJ; Lundgren RJ; Fu GC J. Am. Chem. Soc 2013, 135, 10946–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osverger TJ; Rogness DC; Kohrt JT; Stepan AF; White MC Nature 2016, 537, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain P; Verma P; Xia G; Yu J-Q Nat. Chem 2017, 9, 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen W; Ma L; Paul A; Seidel D Nat. Chem 2018, 10, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies HML; Manning JR Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.As suggested by one of the reviewers, the significant difference in reaction rate observed when the loading of B(C6F5)3 is lowered from 5.0 mol% to 2.5 mol% suggests that the mechanism of the reaction proposed in Figure 1d is not the complete picture. Formations of aggregates or other preorganized states requiring multiple equivalents of B(C6F5)3 or substrate may be involved. Kinetic and spectroscopic studies carried out using N-aryl pyrrolidine (1a) and isopropyl acrylate exhibited approximately second order dependence with respect to B(C6F5)3 (see the Supplementary Information for details). Detailed mechanistic investigations are being carried out.
- 38.Phipps RJ; Hamilton GL; Toste FD Nat. Chem 2012, 4, 603–614. [DOI] [PubMed] [Google Scholar]
- 39.Brak K; Jacobsen EN Angew. Chem., Int. Ed 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.The absolute configuration of the minor diastereomer was determined as (R)-N-((R)-2-hydroxy-1-phenylethyl)-2-((S)-1-(4-methoxy-2,6-dimethylphenyl)pyrrolidin-2-yl)propanamide. See the Supplementary Information for further details.
- 41.Yang L; Adam C; Nichol GS; Cockroft SL Nat. Chem 2013, 5, 1006–1010. [DOI] [PubMed] [Google Scholar]
- 42.Pollice R; Bot M; Kobylianskii IJ; Shenderovich I; Chen P J. Am. Chem. Soc 2017, 139, 13126–13140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.