

Abstract
Our recent study revealed that fibrin and the VLDL receptor (VLDLR) interact with each other through a pair of fibrin βN-domains and CR domains of the receptor and this interaction promotes transendothelial migration of leukocytes and thereby inflammation. The major objectives of the present study were to further clarify the molecular mechanism of fibrin-VLDLR interaction and to identify amino acid residues in the βN-domains involved in this interaction. Our binding experiments with the (β15–66)2 fragment, which corresponds to a pair of fibrin βN-domains, and the VLDLR(1–8) fragment consisting of eight CR domains of VLDLR, revealed that interaction between them strongly depends on ionic strength and chemical modification of all Lys or Arg residues in (β15–66)2 results in abrogation of this interaction. To identify which of these residues are involved in the interaction, we mutated all Lys or Arg in each of the three positively charged Lys/Arg clusters of the (β15–66)2 fragment, as well as single Arg17 and Arg30, and tested the affinity of the mutants obtained to VLDLR(1–8) by ELISA and SPR. The experiments revealed that the second and third Lys/Arg clusters make the major contribution to this interaction while the contribution of the first cluster is moderate. The results obtained suggest that interaction between fibrin and the VLDL receptor employs the “double-Lys/Arg” recognition mode previously proposed for the interaction of the LDL receptor family members with their ligands. They also provide valuable information for the development of highly specific peptide-based inhibitors of fibrin-VLDLR interaction.
Graphical Abstract
Activation of blood coagulation cascade upon vascular injury or other events results in generation of active proteolytic enzyme thrombin, which converts plasma protein fibrinogen into an insoluble fibrin polymer. Fibrin polymerization results in formation of a blood clot, which seals damaged vasculature thereby preventing blood loss and subsequently serves as a provisional matrix to which various cell types adhere, migrate, and proliferate during wound healing.1 Thus, fibrinogen is a multifunctional protein that plays an important role in hemostasis and wound healing. Numerous data indicate the involvement of fibrin(ogen) in inflammation, which is an integral part of wound healing and an attribute of many pathological processes.1 Such involvement occurs through the interaction of fibrin(ogen) with various leukocyte and endothelial cell receptors. Specifically, it has been proposed that fibrinogen promotes transendothelial migration of leukocytes and thereby inflammation by bridging them to the endothelium through the interaction with leukocyte integrin receptor Mac-1 and endothelial cell receptor ICAM-1.2,3 It has also been shown that fibrin interacts with endothelial cell receptor VE-cadherin4,5 and suggested that interaction of fibrin degradation products with VE-cadherin and leukocyte integrin CD11c promotes leukocyte transmigration.6,7 Finally, we found that interaction of fibrin with the very low-density lipoprotein (VLDL) receptor also promotes leukocyte transmigration and thereby inflammation.8
The VLDL receptor (VLDLR) is a member of the low-density lipoprotein (LDL) receptor family.9 VLDLR is a multifunctional cellular receptor present in different tissues including vascular endothelium.10,11 It was shown that this receptor interacts with triglyceride-rich lipoproteins,11 several proteinase-serpin complexes,12–14 thrombospondin-1,15 reelin,16 and fibrin.8 These interactions play an important role in lipid metabolism,17,18 reelin signaling and neuronal migration,19,20 angiogenesis and tumor grows,11,21 and inflammation.8 The VLDL receptor consists of eight complement-type repeats (CR repeats or domains), three EGF-like domains, β-propeller and O-linked sugar domains, and transmembrane and cytoplasmic domains.9,11 Among them, the extracellular CR domains are involved in ligand binding. We have recently demonstrated that the recombinant VLDLR(1–8) fragment containing all eight CR domains interacts with fibrin with practically the same high affinity as the entire extracellular portion of the VLDL receptor.22 Further, we localized the fibrin-binding site to the second and third CR domains of VLDLR.22 We also found that the presence of the fourth CR domain of VLDLR is required for high-affinity interaction of this receptor with fibrin and Trp residue of its third CR domain is involved in this interaction.22 Less is known about the complementary VLDLR-binding site in fibrin.
Fibrinogen is a chemical dimer consisting of two identical subunits each of which is formed by three non-identical polypeptide chains, Aα, Bβ, and γ.23 These chains form a number of structural and/or functional domains24,25 that are involved in multiple interactions. Among them, a pair of fibrin βN-domains (Figure 1A) was shown to participate in fibrin polymerization and interact with a number of ligands including heparin and proteoglycans, VE-cadherin, and the VLDL receptor.4,5,8,26,27 Each of the fibrinogen BβN-domains is formed by the N-terminal portion of the β chain including amino acid residues 15–64 and contains fibrinopeptide B at the N-terminus (amino acid residues 1–14). This fibrinopeptide is removed by proteolytic cleavage with thrombin upon conversion of fibrinogen into fibrin and only after its removal the βN-domains of fibrin exhibit high affinity to their ligands. Our recent study revealed that these domains are the only structures in fibrin that interact with the VLDL receptor with a very high affinity and make the major contribution to the fibrin-VLDLR-dependent transendothelial migration of leukocytes.8 Thus, the VLDLR-binding site was localized to the βN-domains of fibrin; however, the precise location of this site within these domains and its structure remain unclear. Therefore, the major objectives of the present study were to further characterize fibrin-VLDLR interaction, to localize the VLDL receptor-binding site within fibrin βN-domains, and to identify amino acid residues in these domains that are critical for their interaction with the VLDL receptor.
Figure 1.
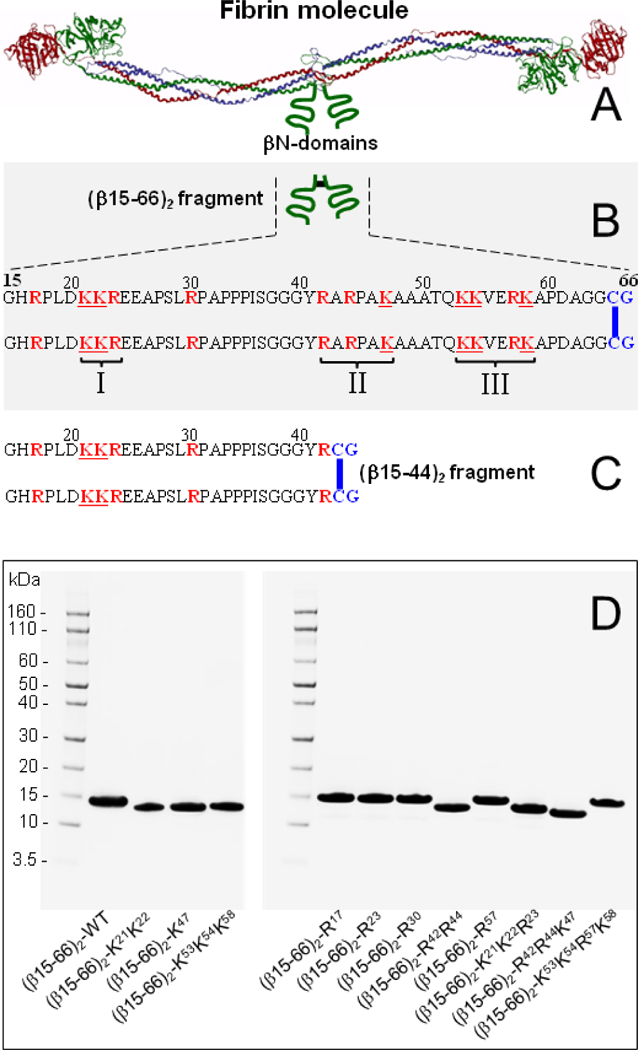
Schematic representation of fibrin, its βN-domains, and the dimeric βN-domain fragments and their mutants prepared for the present study. (A) Ribbon diagram of the fibrinogen molecule based on its crystal structure;25 the individual fibrinogen chains, Aα, Bβ, and γ, are colored in blue, green and red, respectively. The βN-domains of fibrin, whose structure have not been identified, are shown schematically as two curved green lines. (B) The recombinant dimeric (β15–66)2 fragment corresponding to the βN-domains is shown schematically on the top and its amino acid sequence is presented on the bottom; the Cys-Gly residues added to the natural sequence of the βN-domain to enable its dimerization are in blue and the disulfide bond formed by Cys65-Cys65 is shown by vertical blue bar. Roman numerals indicate three positively charged clusters of amino acid residues that are shown in red. (C) Amino acid sequence of the synthetic dimeric βN-domain fragment (β15–44)2 corresponding to the N-terminal halves of a pair of fibrin βN-domains. (D) SDS-polyacrylamide gel electrophoresis analysis of the (β15–66)2 fragment (wild type, WT) and its mutants. The left outer lanes contain Novex® Sharp Pre-stained protein markers of the indicated molecular masses.
MATERIALS AND METHODS
Antibodies and Reagents.
Mouse anti-hVLDLR monoclonal antibody mAb 5F328 was purified as described earlier.29 Goat secondary anti-mouse polyclonal antibodies conjugated with HRP and HRP substrate SureBlue TMB were purchased from KPL (Gaithersburg, MD). Thrombin CleanCleave kit was obtained from Sigma (St. Louis, MO), Blocker BSA in TBS, sulfo-NHS-acetate, and p-hydroxyphenylglyoxal were from Thermo Scientific (Rockford, IL).
Recombinant and Synthetic Fibrin and VLDLR Fragments.
The recombinant monomeric β15–64 and dimeric (β15–66)2 fragments (Figure 1B) corresponding to monomeric and dimeric fibrin βN-domain, respectively, were prepared as described earlier.30,31 The truncated variant of the (β15–66)2 fragment, β15–44 (Figure 1C), was synthesized by Bachem (Torrance, CA, USA) and dimerized through its Cys43 as described previously32 to produce dimeric (β15–44)2 fragment. The recombinant VLDLR(1–8) fragment of the VLDL receptor, which contains all eight ligand-binding CR domains, was prepared as described earlier.22
Preparation of (β15–66)2 Fragment Mutants.
The recombinant (β15–66)2-R17 mutant with Arg17Glu mutation was produced in E. coli and purified as previously described.30 The Lys/Ala and Arg/Ala mutants of the fibrinogen-related Bβ1–66 fragments (“B” indicates the presence of fibrinopeptide B), (Bβ1–66)-K21K22, (Bβ1–66)-R23, (Bβ1–66)-R30, (Bβ1–66)-R42R44, (Bβ1–66)-K47, (Bβ1–66)-K53K54K58, and (Bβ1–66)-R57 were generated by site-directed mutagenesis using QuikChange Multi Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). The pCAL-n construct containing DNA encoding the fibrinogen Bβ1–66 region30 was modified by using the mutagenic primers shown in Table 1. The desired mutations were confirmed by sequencing of mutant plasmids. Plasmids pCAL-n-(Bβ1–66)-K21K22, pCAL-n-(Bβ1–66)-K47, and pCAL-n-(Bβ1–66)-K53K54K58 were used as templates in mutant strand synthesis reactions to generate fibrinogen-related Lys-Arg mutants, (Bβ1–66)-K21K22R23, (Bβ1–66)-R42R44K47, and (Bβ1–66)-K53K54R57K58, respectively. All monomeric mutant fragments were produced in E. coli strain BL21(DE3), purified, and dimerized using procedures described earlier for the wild-type dimeric (β15–66)2 fragment.30 All mutants were treated with thrombin-agarose from Thrombin CleanCleave kit using conditions described before31 to remove their fibrinopeptides B (Bβ chain residues 1–14) and produce fibrin-related (β15–66)2-K21K22, (β15–66)2-R23, (β15–66)2-R30, (β15–66)2-R42R44, (β15–66)2-K47, (β15–66)2-K53K54K58, (β15–66)2-R57, (β15–66)2-K21K22R23, (β15–66)2-R42R44 K47, and (β15–66)2-K53K54R57K58 mutant fragments. The purity of the resultant mutant fragments was verified by SDS-PAGE analysis (Figure 1D).
Table 1.
Primers Used to Produce Mutant Fibrinogen Bβ1-66 Fragments.
| Mutation target | Mutagenic Primer sequence |
|---|---|
| K21K22 | 5’-CTGGGAGCCTCTTCTCTCGCCGCGTCAAGGGGTCGATGACC-3’ |
| K47 | 5’-GAGTGGCAGCTGCTGCGGCTGGACGAGCCC-3’ |
| K53K54K58 | 5’-CCAGCCAAAGCAGCTGCCACTCAAGCGGCAGTAGAAAGAGCAGCC CCTGATGCTGGA-3’ |
| R23 | 5’-ACCCCTTGACAAGAAGGCAGAAGAGGCTCCCAGC-3’ |
| R30 | 5’-GCTCCCAGCCTGGCGCCTGCCCCACC-3’ |
| R42R44 | 5’-CCCATCAGTGGAGGTGGCTATGCGGCTGCTCCAGCCAAAG-3’ |
| R57 | 5’-CCACTCAAAAGAAAGTAGAAGCAAAAGCCCCTGATGCTGGA-3’ |
| K21K22R2 | 5’-CCTTGACGCGGCGGCAGAAGAGGCTCCC-3’ |
| R42R44K47 | 5’-CATCAGTGGAGGTGGCTATGCGGCTGCTCCAGCCGCA-3’ |
| K53K54R57K58 | 5’-GCCACTCAAGCGGCAGTAGAAGCAGCAGCCCCTG-3’ |
Introduced Ala mutation sequences are underlined
Chemical Modification of Lys and Arg Residues in the Recombinant (β15–66)2 Fragment.
Chemical modification of (β15–66)2 to block the primary amines in lysine side chains was performed using Sulfo-NHS-acetate (Thermo Scientific) according to manufacturer’s protocol. The modified (β15–66)2 was dialyzed into HBS (HEPES-buffered saline with 10 mM HEPES, pH 7.4, and 150 mM NaCl) containing 1 mM CaCl2 to remove the excess of Sulfo-NHS-acetate. Chemical modification of arginine residues of (β15–66)2 was performed using p-Hydroxyphenylglyoxal (Thermo Scientific) according to the previously described protocol.33
Protein Concentration Determination.
Concentrations of the β15–64, (β15–66)2, (β15–44)2, (β15–66)2-R17, and VLDLR(1–8) fragments were determined as previously described.22,30,32 Concentrations of the expressed (β15–66)2 fragment mutants were determined spectrophotometrically using theoretical extinction coefficients (E280,1%) estimated from fragments’ sequences by the ProtParam online tool (http://www.expasy.ch/tools/protparam.html). Molecular masses of these fragments were also estimated using ProtParam. The following molecular masses and E280,1% values were obtained: (β15–66)2-K21K22, 10.6 kDa and 2.9; (β15–66)2-K47, 10.7 kDa and 2.9; (β15–66)2-K52K53K58, 10.4 kDa and 3.0; (β15–66)2-R23, 10.6 kDa and 2.9; (β15–66)2-R30, 10.6 kDa and 2.9; (β15–66)2-R42R44, 10.4 kDa and 3.0; (β15–66)2-R57, 10.6 kDa and 2.9; (β15–66)2-K21K22R23, 10.4 kDa and 3.0; (β15–66)2-R42R44K47, 10.3 kDa and 3.0; (β15–66)2-K52K53R57K58, 10.3 kDa and 3.0.
Solid-Phase Binding Assay.
Wells of Immulon 2HB microtiter plates were coated overnight at 4 oC with the β15–64, (β15–44)2, (β15–66)2, or mutant fragments, all at 2 μg/mL in 0.1 M Na2CO3, pH 9.5. The wells were then blocked with Blocker BSA in TBS (Thermo Scientific) for 1 hour at room temperature. Following washing TBS (Tris-buffered saline with 20 mM Tris, pH 7.4, and 150 mM NaCl) containing 0.05% Tween 20 and 1 mM CaCl2 (ELISA-binding buffer), the indicated concentrations of VLDLR(1–8) fragment in this buffer were added to the wells and also to control wells coated just with Blocker BSA in TBS and incubated for 1 hour at 37 oC. Bound fragments were detected by reaction with anti-VLDLR mAb 5F3, which does not block interaction of VLDLR with fibrin,29 for 1 h at 37 oC followed by incubation with the HRP-conjugated donkey anti-mouse polyclonal antibodies for 1 h at 37 oC. The peroxidase substrate, SureBlue TMB (KPL, Gaithersburg, MD), was added to the wells and the amount of bound ligand was measured spectrophotometrically at 450 nm. Data were analyzed by nonlinear regression analysis using equation 1:
where A represents the absorbance of the oxidized substrate, which is assumed to be proportional to the amount of ligand bound, Amax is the absorbance at saturation, [L] is the molar concentration of the ligand, and Kd is the equilibrium dissociation constant.
Surface Plasmon Resonance Analysis.
Interaction of the β15–64, (β15–44)2, and (β15–66)2 wild-type and mutant fragments with the immobilized VLDLR(1–8) fragment was studied by surface plasmon resonance (SPR) using the BIAcore 3000 biosensor (GE Healthcare), which measures the association/dissociation of proteins in real time as described earlier.22 Binding experiments were performed in HBS-P (10 mM HEPES buffer, pH 7.4, 150 mM NaCl, and 0.005% Surfactant P20) containing 1 mM CaCl2 at 20 μl/min flow rate. To regenerate the chip surface, complete dissociation of the complex was achieved by adding 100 mM H3PO4 for 30 seconds followed by re-equilibration with the binding buffer. Experimental data were analyzed using BIAevaluation 4.1 software supplied with the instrument. The dissociation equilibrium constant, Kd, was calculated as Kd = kdiss/kass, where kass and kdiss represent kinetic constants that were estimated by global analysis of the association/dissociation data using the 1:1 Langmurian interaction model (kinetic analysis). To confirm the kinetic analysis, Kd was also estimated by analysis of the association data using the steady-state affinity model (equilibrium analysis).
RESULTS
Further Localization of the VLDL Receptor-Binding Site within Fibrin βN-Domains.
We previously found that the recombinant dimeric (β15–66)2 fragment, which corresponds to a pair of fibrin βN-domains disulfide-linked through Cys65 (Figure 1B), contains the VLDLR-binding site and has practically the same high affinity to the VLDL receptor as fibrin.8 Therefore, this dimeric fragment has been used as a simple soluble mimetic of fibrin in our previous studies with the VLDL receptor.8,22 However, the question arises whether the monomeric β15–64 fragment, which corresponds to a single βN-domain of fibrin,30 may represent even a simpler soluble fibrin mimetic. To address this question, we compared the affinities of the dimeric (β15–66)2 fragment and monomeric β15–64 fragment to the recombinant VLDLR(1–8) fragment, which contains all eight CR domains of the VLDL receptor and preserves fibrin-binding properties of its parent molecule,22 i.e. represents a simple fibrin-binding mimetic of VLDLR. In ELISA experiments, when (β15–66)2 and β15–64 were immobilized on the surface of microtiter plates and incubated with increasing concentrations (up to 250 nM) of VLDLR(1–8), a prominent binding of the latter was observed only to the dimeric (β15–66)2 fragment while its binding to monomeric β15–64 was very low (Figure 2A). The equilibrium dissociation constant (Kd) value for (β15–66)2 determined by fitting its binding curve was found to be 3.6 nM (Table 2), i.e. very similar to that determined earlier22 while the Kd value for the β15–64 fragment was impossible to determine due to the low binding signal. At the same time, in SPR experiments, in which increasing concentrations of the β15–64 fragment (up to 10 μM) were added to immobilized VLDLR(1–8), this fragment exhibited a dose-dependent binding (Figure 2B). The Kd value for this binding determined by global analysis of the association/dissociation data was found to be 6.1 μM, i.e. three orders of magnitude higher than that determined from SPR data for (β15–66)2-VLDLR(1–8) interaction in this (Table 2) and our previous22 studies. These results indicate that the high affinity interaction of fibrin with the VLDL receptor requires dimerization of its βN-domains and only the dimeric (β15–66)2 fragment represents the simplest VLDLR-binding mimetic of fibrin. Therefore, in the present study the (β15–66)2 fragment has been used as soluble fibrin mimetic.
Figure 2.

Analysis of interaction of the fibrin-binding VLDLR(1–8) fragment of the VLDL receptor with the monomeric fibrin β15–64 fragment and the dimeric fibrin (β15–66)2 fragment and its truncated variant (β15–44)2 by ELISA (A) and SPR (B and C). (A) Increasing concentrations of VLDLR(1–8) were incubated with microtiter wells coated with the (β15–66)2 (●), β15–64 (Δ), or (β15–44)2 (○) fragments and bound VLDLR(1–8) was detected as described in Materials and Methods. The curves demonstrate the best fit of the data to eq. 1 and are representative of 3 independent experiments; error bars represent the standard deviation of triplicate determinations. (B and C) The β15–64 fragment (B) at increasing concentrations, 0.25, 0.5, 1.0, 2.5, 5, 7.5, and 10 μM, or the (β15–44)2 fragment (C) at 1.0, 2.5, 5, 10, 20, and 30 μM was added to the immobilized VLDLR(1–8) fragment and its association/dissociation was monitored in real time while registering the resonance signal (response) using BIAcore biosensor. The dotted curves, which essentially coincide with the experimental curves, represent the best fit of the binding data using global fitting analysis (see Materials and Methods). The Kd values determined from both ELISA and SPR binding data are presented in Table 2.
Table 2.
ELISA- and SPR-Determined Equilibrium Dissociation Constants (Kd) for the Interaction of the Fibrin-Binding VLDLR(1–8) Fragment with the Recombinant and Synthetic Fragments of Fibrin βN-Domains.
| β N-fragments | Kd (nM)a | |
|---|---|---|
| ELISA | SPR | |
| (β 15–66)2 fragment | 3.6 ± 0.8 | 3.1 ± 0.2 |
| β 15–64 fragment | n.d.b | 6.1 ± 1.6 μ M |
| (β 15–44)2 fragment | n.d.b | 67 ± 9 μ M |
| Ionic Strength Dependence | ||
| (β 15–66)2 + 0.15 M NaCl | 3.0 ± 0.1 | 3.1 ± 0.2 |
| (β 15–66)2 + 0.2 M NaCl | 6.0 ± 0.3 | 5.8 ± 0.2 |
| (β 15–66)2 + 0.25 M NaCl | 13.5 ± 1.3 | 11.1 ± 0.1 |
| (β 15–66)2 + 0.3 M NaCl | 34.1 ± 1.9 | 20.5 ± 2.7 |
All Kd values are in nM except those for the β15–64 and (β15–44)2 fragment, which are presented in μM.
Not detected due to low binding signal.
To further localize the VLDLR-binding site within the βN-domains, we prepared a synthetic truncated variant of (β15–66)2, a dimeric (β15–44)2 fragment, which contains the N-terminal halves of the βN-domains disulfide-linked through Cys43 (Figure 1C), and compared its affinity to the VLDLR(1–8) fragment with that of (β15–66)2. In ELISA experiments, when (β15–44)2 was immobilized on the surface of microtiter plates and then incubated with increasing concentrations (up to 250 nM) of VLDLR(1–8), the binding of the latter to (β15–44)2 was extremely low (Figure 2A). At the same time, in SPR experiments, in which increasing concentrations of the (β15–44)2 fragment (up to 30 μM) were added to immobilized VLDLR(1–8), this fragment exhibited a dose-dependent binding with the Kd value of 67 μM (Figure 2C and Table 2), i.e. much higher than that determined for (β15–66)2-VLDLR(1–8) interaction. These results clearly indicate that the affinity of the (β15–44)2 fragment to VLDLR(1–8) is extremely low. Altogether, these results suggest that the C-terminal portions of fibrin βN-domains are critical for high affinity interaction with the VLDL receptor, i.e. the VLDLR-binding site is located mainly in these portions. At the same time, they also suggest that some amino acid residues involved in the interaction with VLDLR may be located in the N-terminal portions of the βN-domains.
Ionic Strength-Dependence of the Interaction Between the (β15–66)2 and VLDLR(1–8) fragments.
It should be noted that all ELISA and SPR binding experiments described above were performed in 20 mM Tris or 10 mM HEPES buffers both containing 0.15 M NaCl, i.e. at near physiological ionic strength. When ELISA experiments were performed at higher ionic strength, in the buffer containing 1 M NaCl, the (β15–66)2 fragment exhibited no binding to VLDLR(1–8), suggesting involvement of electrostatic interactions in binding of fibrin to the VLDL receptor. To test this suggestion, we performed systematic study of the interaction between the (β15–66)2 and VLDLR(1–8) fragments at different concentrations of NaCl (ionic strengths). The results of ELISA experiments presented in Figure 3A clearly indicate that the affinity of VLDLR(1–8) to immobilized (β15–66)2 strongly depends on ionic strength, in agreement with the above suggestion. The determined Kd values for this interaction at different NaCl concentrations are presented in Table 2 and the analysis of the binding data using a Debye-Hȕckel plot is shown in Figure 3B. A Debye-Hȕckel plot of log10Kd versus I½ for the Kd values obtained between 0.15 and 0.3 M NaCl concentration (Table 2) gave us a straight line with a slope of 6.7 ± 0.5 (Figure 3B), suggesting the involvement of roughly 6–7 charged amino acid residues of the (β15–66)2 fragment in the interaction with the VLDLR(1–8) fragment. Such strong dependence of the affinity of (β15–66)2 to VLDLR(1–8) from ionic strength was confirmed in SPR experiments (Figure 4). As in the case with the ELISA experiments, the Kd values for the (β15–66)2-VLDLR(1–8) interaction increased with the increasing of NaCl concentration (Table 2) although the affinity decrease was less pronounced than that observed in ELISA experiments (Table 2) and the slope of the Debye-Hȕckel plot of 5.2 ± 0.2 was slightly lower (Figure 4E). Altogether, these results provide direct evidence for the involvement of electrostatic interactions in binding of fibrin to the VLDL receptor. Furthermore, they suggest that such interactions play a significant role in this binding. They also suggest that roughly six (5.2 to 6.7) charged amino acid residues of the (β15–66)2 fragment are involved in fibrin-VLDLR interaction.
Figure 3.
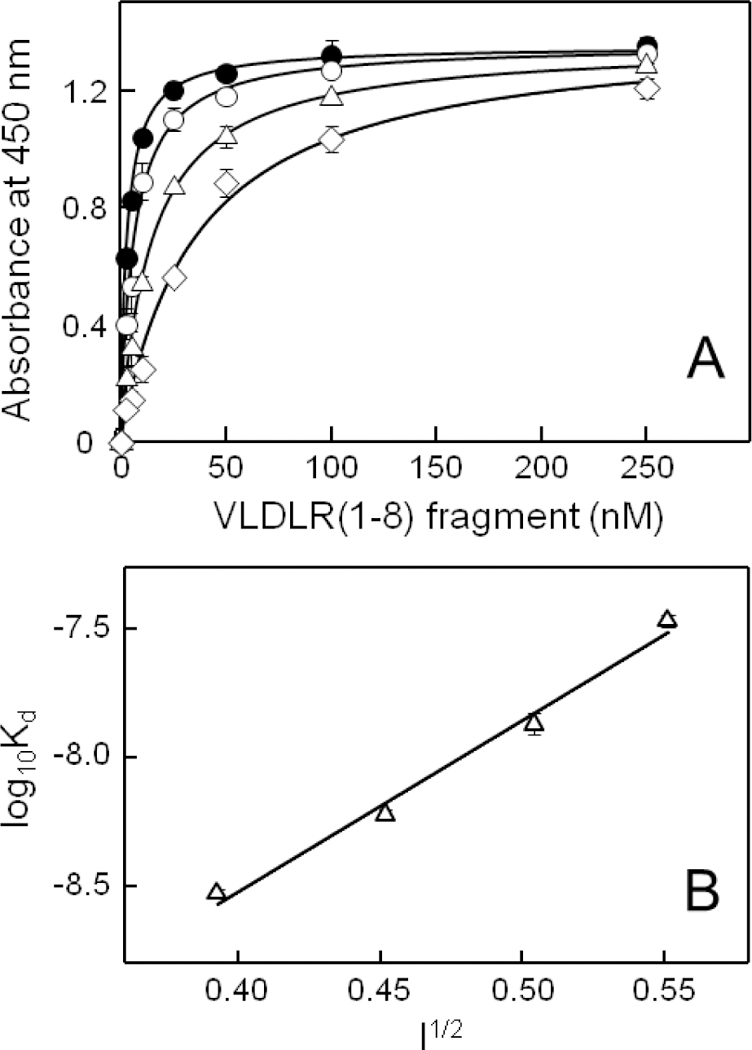
ELISA-detected interaction between the (β15–66)2 and VLDLR(1–8) fragments at different NaCl concentrations. (A) Increasing concentrations of VLDLR(1–8) were incubated with immobilized (β15–66)2 at increasing concentrations of NaCl, 0.15 M (●), 0.2 M (○), 0.25 (Δ), and 0.3 M (◊). The curves demonstrate the best fit of the data to eq. 1 and are representative of 3 independent experiments; error bars represent the standard deviation of triplicate determinations; the determined Kd values are presented in Table 2. (B) Debye-Huckel plots for binding of VLDLR(1–8) to (β15–66)2 at different ionic strengths. The Kd values were taken from ELISA data presented in panel A and Table 2, and the log10Kd values represent an average of at least three independent experiments ± SD; a slope of 6.7 ± 0.5 was determined by linear regression analysis.
Figure 4.

SPR-detected interaction between the (β15–66)2 and VLDLR(1–8) fragments at different concentrations of NaCl, 0.15 M (A), 0.2 M (B), 0.25 (C), and 0.3 M (D). The (β15–66)2 fragment at increasing concentrations, 0.25, 0.5, 1, 2.5 and 5 nM (A), 0.5, 1, 2.5, 5 and 10 nM (B), and 1, 2.5, 5, 10, and 25 (C and D), was added to the immobilized VLDLR(1–8) fragment and its association/dissociation was monitored in real time while registering the resonance signal (response) using BIAcore biosensor. The dotted curves represent the best fit of the binding data using global fitting analysis (see Materials and Methods); the determined Kd values are presented in Table 2. (E) Debye-Huckel plots for binding of VLDLR(1–8) to (β15–66)2 at different ionic strengths. The Kd values were taken from SPR data presented in panels A-D and Table 2, and the log10Kd values represent an average of at least three independent experiments ± SD; a slope of 5.2 ± 0.2 was determined by linear regression analysis.
Chemical Modification of Lys Residues in the Recombinant Fibrin βN-Domain Fragment Abrogates its Interaction with the VLDLR(1–8) Fragment.
Previous studies revealed crucial role of positively charged Lys residues of ligands in their interaction with some LDL receptor family members.34–36 Since our experiments described above revealed the involvement of charged residues in fibrin-VLDLR interaction and since each fibrin βN-domain contains 6 Lys residues (Figure 1B), we hypothesized that some of these residues may be involved in the interaction of fibrin with the VLDL receptor. To test this hypothesis, we first chemically modified Lys residues in the (β15–66)2 fragment. The modification was performed with Sulfo-NHS acetate, which was previously used to block Lys residues in factor VIII.37 In SPR experiments, when non-modified (β15–66)2 fragment at 10 nM was passed over a chip with immobilized VLDLR(1–8), it exhibited a well expressed binding (Figure 5A). At the same time, Lys-modified (β15–66)2 exhibited no binding at this concentration; even at 5 μM Lys-modified (β15–66)2 no binding was observed (Figure 5A). Thus, the results obtained with Lys-modified (β15–66)2 confirm the above hypothesis suggesting the involvement of Lys residues of fibrin βN-domains in the interaction with the VLDL receptor.
Figure 5.

Effect of modification of Lys and Arg residues in the (β15–66)2 on its interaction with the VLDLR(1–8) fragment. (A) The Lys-modified (β15–66)2 fragment at 5 μM was added to the immobilized VLDLR(1–8) fragment and its association/dissociation was monitored in real time. (B) The Arg-modified (β15–66)2 fragment was added at 10 nM (dotted curve) and 5 μM (solid curve) to the immobilized VLDLR(1–8) fragment and its association/dissociation was monitored in real time. Binding of the non-modified (β15–66)2 fragment added at 10 nM is shown in both panels for comparison.
Effect of Mutations of Lys Residues in the Recombinant Fibrin βN-Domain Fragment on its Interaction with the VLDLR(1–8) Fragment.
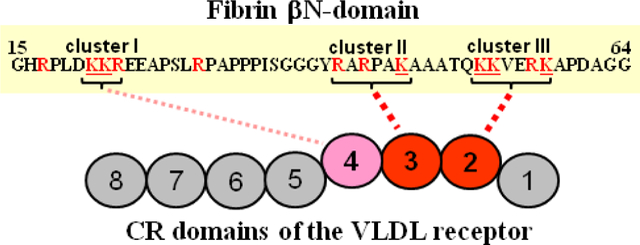
There are six Lys residues in each fibrin βN-domain; they are located in three positively charged clusters of amino acid residues (Figure 1B). Among these clusters, clusters I and II contain two and one Lys residues, respectively, and cluster III contains the remaining three Lys residues. To test the involvement of these Lys residues in the interaction with the VLDL receptor, we prepared three mutants of the (β15–66)2 fragment. They included (β15–66)2-K21K22, (β15–66)2-K47, and (β15–66)2-K53K54K58 Lys-mutants in which Lys residues 21 and 22 of the first cluster, Lys47 of the second cluster, and Lys53, 54 and 58 of the third cluster, respectively, were mutated to Ala.
In ELISA experiments, when the wild type (β15–66)2 fragment or each of the three (β15–66)2 Lys-mutants was immobilized on the surface of microtiter plates and increasing concentrations of the VLDLR(1–8) fragment were added, all species exhibited prominent bindings (Figure 6A). However, while the binding of VLDLR(1–8) to the (β15–66)2-K21K22 and (β15–66)2-K47 Lys-mutants differed only slightly from that to the wild type (β15–66)2 fragment, its binding to the (β15–66)2-K53K54K58 Lys-mutant was significantly lower. Comparison of the Kd values determined for these interactions (Table 3) revealed that the (β15–66)2-K53K54K58 Lys-mutant has more than 30-fold lower affinity to the VLDLR(1–8) fragment than the wild type fragment (Kd value of 124 nM vs 3.6 nM) while the affinities of the other two Lys-mutants, (β15–66)2-K47 and (β15–66)2-K21K22, to VLDLR(1–8) were only ∼3–6-fold lower (Kd values of 11 and 21 nM vs 3.6 nM). Similar trend in the affinities of Lys-mutants to VLDLR(1–8) was observed in SPR experiments in which Kd values for (β15–66)2-K47, (β15–66)2-K21K22, and (β15–66)2-K53K54K58 were found to be 7, 37, and 250 nM, respectively (Table 3 and Table S1). Thus, both methods revealed a significant drop in the affinity of the (β15–66)2-K53K54K58 Lys-mutant to VLDLR(1–8) while the changes in the affinities of the (β15–66)2-K47 and (β15–66)2-K21K22 Lys-mutant were much less dramatic. Altogether, these experiments suggest that Lys residues of positively charged cluster III make the major contribution to the fibrin-VLDLR interaction while the contributions of Lys residues of clusters I and II are less pronounced.
Figure 6.

Analysis of interaction of the fibrin-binding VLDLR(1–8) fragment with the (β15–66)2 fragment and its Lys-mutants (A) and Arg-mutants (B) by ELISA. Increasing concentrations of the VLDLR(1–8) fragment were incubated with microtiter wells coated with the (β15–66)2 fragment (●), and its Lys-mutants, (β15–66)2-K21K22 (○), (β15–66)2-K47 (Δ), and (β15–66)2-K53K54K58 (□), or Arg-mutants, (β15–66)2-R17 (○), (β15–66)2-R23 (Δ), (β15–66)2-R30 (∇), (β15–66)2-R42R44 (□), and (β15–66)2-R57 (◊), and bound VLDLR(1–8) was detected as described in Materials and Methods. The curves in both panels demonstrate the best fit of the data to eq. 1 and are representative of 3 independent experiments; error bars represent the standard deviation of triplicate determinations. The determined Kd values are presented in Table 3.
Table 3.
ELISA- and SPR-Determined Equilibrium Dissociation Constants (Kd) for the Interaction of the Fibrin-Binding VLDLR(1–8) Fragment with the Recombinant Fibrin (β15–66)2 Fragment and its Mutants.
| β N-fragment/mutants | Kd (nM)a | |
|---|---|---|
| ELISA | SPRb | |
| (β 15–66)2 fragmentc | 3.6 ± 0.8 | 3.1 ± 0.2 |
| Lys-mutants | ||
| (β 15–66)2-K21K22 | 21 ± 2 | 37 ± 4 |
| (β 15–66)2-K47 | 11 ± 2 | 7 ± 3 |
| (β 15–66)2-K53K54K58 | 124 ± 19 | 250 ± 23 |
| Arg-mutants | ||
| (β 15–66)2-R17 | 7.3 ± 2.3 | 5.5 ± 0.3 |
| (β 15–66)2-R23 | 12 ± 2 | 6.3 ± 2 |
| (β 15–66)2-R30 | 6.3 ± 1.5 | 7.4 ± 0.6 |
| (β 15–66)2-R42R44 | 50 ± 6 | 204 ± 8.0 |
| (β 15–66)2-R57 | 6.4 ± 1.7 | 3.6 ± 0.3 |
| Arg-Lys-mutants | ||
| (β 15–66)2-K21K22R23 | 77 ± 7 | 160 ± 2 |
| (β 15–66)2-R42R44K47 | n.d.d | 6.1 ± 0.5 μ M |
| (β 15–66)2-K53K54R57K58 | n.d.d | 11.7 ± 1.6 μ M |
Evidence for the Involvement of Arg Residues of Fibrin βN-Domains in the Interaction with the VLDL Receptor.
Since Arg residue of PAI-1, a ligand for another LDL-receptor family member, LRP1, was shown to be involved in the interaction with CR5 domain of LRP1,38 we also tested the involvement of Arg residues of fibrin βN-domains in the interaction with the VLDL receptor. There are six Arg residues in each fibrin βN-domain. Among them, one Arg is located in the first positively charged cluster, two Arg in the second, and one Arg in the third cluster; two more single Arg residues, Arg17 and Arg30, are located around cluster I. To test the involvement of these residues in the interaction with VLDLR, we first chemically modified in the (β15–66)2 fragment guanidine groups of Arg side chains with p-Hydroxyphenylglyoxal and studied the interaction of Arg-modified (β15–66)2 with the VLDLR(1–8) fragment by SPR. As in the case with Lys-modified (β15–66)2, no binding was observed when Arg-modified (β15–66)2 at 10 nM was added to immobilized VLDLR(1–8) and only very little binding was detected when much higher concentration (5 μM) of Arg-modified (β15–66)2 was added (Figure 5B). Such lack of binding suggests that one or more Arg residues of fibrin βN-domains are involved in the interaction with the VLDL receptor. However, there are two Arg residues in each βN-domain located next to Lys residues, one in the first positively charged cluster and another one in the third cluster (Figure 1B). Thus, alternatively, if these Lys residues are involved in the binding, chemical modification of neighboring Arg residues may reduce their reactivity due to steric hindrance.
To select between the above mentioned alternatives, we mutated individual Arg residues in the (β15–66)2 fragment. Namely, we produced the following single Arg-mutants, (β15–66)2-R17, (β15–66)2-R23, (β15–66)2-R30, and (β15–66)2-R57, as well as a double mutant (β15–66)2-R42R44. The interaction of these mutants with the VLDLR(1–8) fragment was tested by ELISA and SPR. In ELISA experiments, VLDLR(1–8) exhibited prominent binding to all immobilized single mutants that was comparable to its binding to the wild type (β15–66)2 fragment, while its binding to the double mutant, (β15–66)2-R42R44, was significantly lower (Fig. 6B). This trend was confirmed in SPR experiments in which all Arg-mutants exhibited dose-dependent binding. The Kd values derived from the analysis of binding curves obtained by both techniques are presented in Table 3 and Table S1. They indicate that mutation of single Arg residues, Arg17, Arg23, Arg30, and Arg57 in (β15–66)2 does not change significantly its affinity to the VLDLR(1–8) fragment while simultaneous mutation of Arg42 and Arg44 resulted in a significant (up to 66-fold) drop in the affinity of this double mutant to VLDLR(1–8). These results suggest that only Arg residues located in the second positively charged cluster of fibrin βN-domains significantly contribute to the interaction of fibrin with the VLDL receptor.
Effect of Simultaneous Mutation of Lys and Arg Residues in the Positively Charged Clusters of the Recombinant Fibrin βN-Domain Fragment on its Affinity to the VLDLR(1–8) Fragment.
To further test the involvement of Lys and Arg residues of the βN-domains in the interaction with the VLDL receptor, we then mutated Arg residues in the (β15–66)2 Lys-mutants described above in order to eliminate all charged residues in each of the three positively charged clusters. Namely, we produced the following Arg-mutants of Lys-mutants: (β15–66)2-K21K22R23, (β15–66)2-R42R44K47, and (β15–66)2-K53K54R57K58. The interaction of these mutants with the VLDLR(1–8) fragment was tested by both ELISA and SPR.
In ELISA experiments, when increasing concentrations of the VLDLR(1–8) fragment (up to 250 nM) were added to the immobilized Lys-Arg-mutants, this fragment exhibited significant binding to the (β15–66)2-K21K22R23 mutant with the Kd value of 77 nM while its binding to (β15–66)2-R42R44K47 or (β15–66)2-K53K54R57K58 was very low precluding reliable determination of Kds (Figure 7 and Table 3). At the same time, in SPR experiments, in which increasing concentrations of the (β15–66)2-R42R44K47 or (β15–66)2-K53K54R57K58 mutants (up to 10 or 20 μM, respectively) were added to immobilized VLDLR(1–8), they both exhibited a dose-dependent binding (Figure 8). The Kd values determined by global analysis of the association/dissociation data for the interaction of (β15–66)2-K21K22R23, (β15–66)2-R42R44K47, and (β15–66)2-K53K54R57K58 with VLDLR(1–8) were found to be 175 nM, 6.1 μM, and 11.7 μM, respectively (Table 3). Altogether, these experiments revealed that simultaneous mutations of Lys and Arg residues in the second or third clusters of positively charged residues of the recombinant βN-domain result in a significant drop of its affinity to the VLDLR receptor while such mutations in the first clusters had much lower effect.
Figure 7.

Analysis of interaction of the fibrin-binding VLDLR(1–8) fragment with the (β15–66)2 fragment and its Lys-Arg-mutants by ELISA. Increasing concentrations of the VLDLR(1–8) fragment were incubated with microtiter wells coated with the (β15–66)2 fragment (●), or the (β15–66)2-K21K22R23 (○), (β15–66)2-R42R44K47 (Δ), and (β15–66)2-K53K54R57K58 (□) Lys-Arg-mutants, and bound VLDLR(1–8) was detected as described in Materials and Methods. The curves demonstrate the best fit of the data to eq. 1 and are representative of 3 independent experiments; error bars represent the standard deviation of triplicate determinations. The determined Kd values are presented in Table 3.
Figure 8.
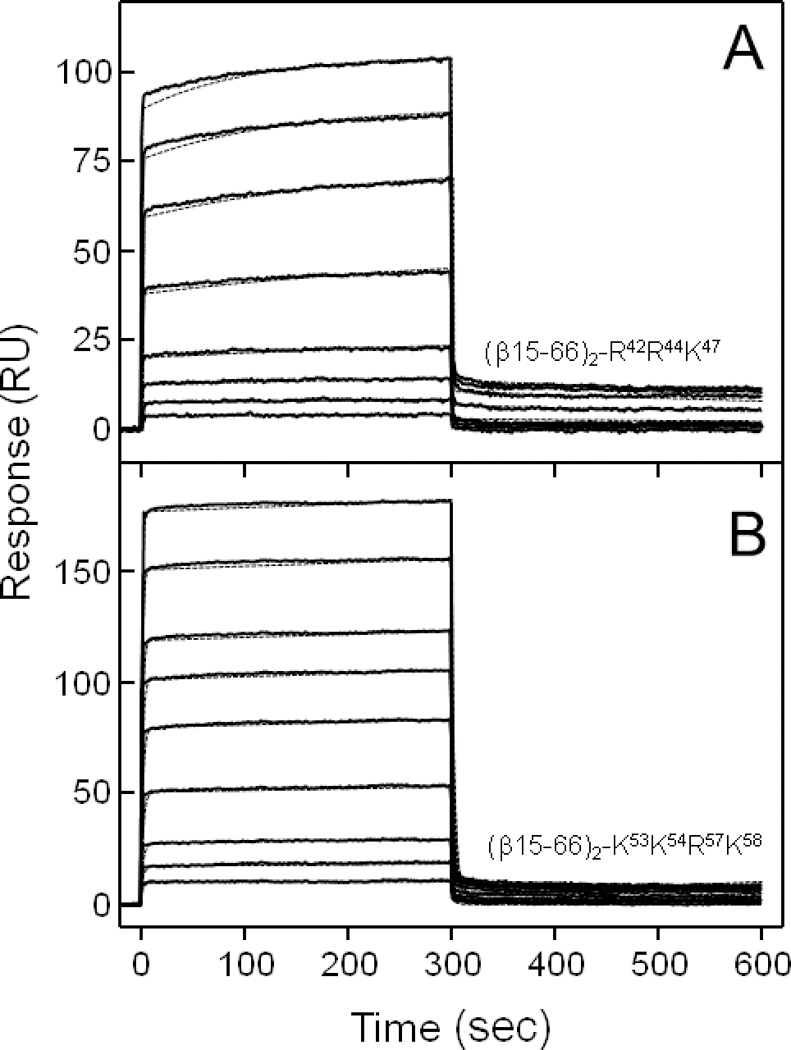
Analysis of interaction of the (β15–66)2 Lys-Arg-mutants with the fibrin-binding VLDLR(1–8) fragment by SPR. The (β15–66)2-R42R44K47 mutant at increasing concentrations, 0.1, 0.25, 0.5, 1.0, 2.5, 5, 7.5, and 10 μM (A), or the (β15–66)2-K53K54R57K58 mutant at 0.25, 0.5, 1.0, 2.5, 5, 7.5, 10, 15, and 20 μM (B) was added to the immobilized VLDLR(1–8) fragment and its association/dissociation was monitored in real time while registering the resonance signal (response) using BIAcore biosensor. The dotted curves in both panels, which essentially coincide with the experimental curves, represent the best fit of the binding data using global fitting analysis (see Experimental Procedures). The Kd values determined from SPR binding data are presented in Table 3.
DISCUSSION
Our previous study established that interaction of fibrin with the VLDL receptor promotes transendothelial migration of leukocytes and thereby inflammation.8 Furthermore, our experiments using mouse model of peritonitis revealed that fibrin-dependent leukocyte transmigration accounts for at least half of the total infiltration of leukocytes into the peritoneum.8,32 Because of the prominent role of fibrin-VLDLR interaction in promoting leukocyte transmigration, this interaction is a potential therapeutic target for controlling fibrin-dependent inflammation. Thus, one of the major goals of our current studies is to develop potent peptide-based antagonists of this interaction. The development of such antagonists requires detailed knowledge of the molecular mechanism of fibrin-VLDLR interaction. As the first step towards establishing this mechanism, we localized the fibrin-bindings site within CR domains of the VLDL receptor22 and revealed that the complementary binding site is located in fibrin βN-domains.8 In the present study, we have made our next step, namely, we clarified the nature of fibrin-VLDLR interaction, further localized the VLDLR-binding sites within the βN-domains, and identified clusters of positively charged amino acid residues critical for their interaction with the VLDL receptor.
Since our initial experiments revealed that the affinity of fibrin-VLDLR interaction strongly depends on ionic strength and chemical modification of Lys or Arg residues in the recombinant βN-domain fragment of fibrin results in abrogation of this interaction, we focused our subsequent study on identification of Lys and Arg residues involved in fibrin-VLDLR interaction by site directed mutagenesis. Specifically, we mutated Lys and Arg residues in each of the three positively charged clusters of fibrin βN-domains, as well as single Arg17 and Arg30 residues not belonging to these clusters, and tested the effect of such mutations on the affinity of the mutants produced to the fibrin-binding VLDLR mimetic, the VLDLR(1–8) fragment. It should be noted that all mutagenesis experiments were performed with the dimeric (β15–66)2 fragment that, according to our initial experiments (Figure 2), represents the simplest soluble VLDLR-binding fibrin mimetic.
To test the affinities of the (β15–66)2 mutants to VLDLR(1–8), we used two independent methods, ELISA and SPR. While the Kd values determined by these two methods for wild type (β15–66)2 and most of the mutants were comparable, those for some mutants differed by 2–4-fold (Table 3). Such a discrepancy could be explained by the fact that the state of binding partners in our SPR and ELISA experiments was different. In ELISA, all βN-domain mutants were immobilized on the surface of microtiter plates and, therefore, had fixed conformation. In contrast, in SPR experiments, all these mutants were in solution and, thus, preserved their solution conformation and flexibility. Alternatively, ELISA-detected Kd values in some cases may be less accurate than those determined by SPR since our analysis of ELISA data is based on the assumption that the absorbance of the oxidized substrate is proportional to the amount of ligand bound, as mentioned in Materials and Methods, and this assumption has not been validated for each interaction studied. Whatever the reason for such a discrepancy is, the trend in the loss of affinities by all mutants obtained by both methods was the same. Namely, both methods revealed a very significant drop (by 3 orders of magnitude) in the affinities of the (β15–66)2-R42R44K47 (cluster II) and (β15–66)2-K53K54R57K58 (cluster III) mutants to the VLDLR(1–8) fragment while the drop in the affinity of the (β15–66)2-K21K22R23 (cluster I) mutant was much less dramatic, from 20- to 50-fold (Table 3). In contrast, mutation of individual Lys or Arg residues reduced the affinities of the corresponding mutants by no more then 3-fold. These results clearly indicate that mutation of all charged residues in the second or third clusters, both located in the C-terminal half of the βN-domains, results in a very significant loss of affinity to VLDLR. This is in agreement with the results of our experiments with the truncated (β15–44)2 fragment, which suggest that the C-terminal portions of the βN-domains are critical for the interaction. The results also suggest that the two single Arg residues, Arg17 and Arg30, do not contribute noticeably to the binding.
Altogether the results obtained with the mutants indicate that Lys and Arg residues of the positively charged clusters II and III of fibrin βN-domains make the major contribution to the interaction of fibrin with the VLDL receptor. As to Lys and Arg residues of the first cluster, their contribution to the interaction is much less prominent since the (β15–66)2-K21K22R23 mutant, in which this cluster was mutated to Ala-Ala-Ala, still had quite high affinity to VLDLR (Kd of 77 and 160 nM determined by ELISA and SPR, respectively, Table 3). Furthermore, the affinity of the (β15–44)2 fragment containing only this cluster was found to be extremely low (Kd = 67 μM, Table 2). In this regard, our previous study revealed that the (β15–44)2 fragment interacts with a relatively high affinity with VE-cadherin,32 another fibrin receptor proposed to be involved in fibrin-dependent leukocyte transmigration.6 This implies that the VE-cadherin- and VLDLR-binding sites seems to be located in different portions of fibrin βN-domains. This also suggests that (β15–44)2 may be considered as a more or less specific inhibitor of interaction of fibrin with VE-cadherin. The results obtained in the present study provide valuable information for further identification of critical amino acid residues involved in fibrin-VE-cadherin and fibrin-VLDLR interactions and designing their highly specific peptide-based inhibitors.
The involvement of Lys residues of fibrin in the interaction with the VLDL receptor was not unexpected since previous X-ray study of a complex between CR domains 2–3 of VLDLR and its ligand, the minor group human rhinovirus HRV2, revealed that two Lys residues of the ligand are involved in the interaction with the third CR domain.34 Subsequent X-ray study of a complex between CR domains 3–4 of another LDL receptor family member, the LDL receptor, and its ligand, D3 domain of RAP, confirmed the involvement of Lys residues in this interaction.35 Based on this study, a general mode for ligand recognition by the LDL receptor family members, which includes electrostatic interaction of a positively charged Lys residue of a ligand with the “acidic pocket” on a receptor CR domain, has been proposed.35,36 In addition, it was shown that the aliphatic portion of this Lys forms multiple van der Waals contacts with a Trp residue, which is highly conserved in CR domains, and a second Lys residue of a ligand also contributes to the interaction by enhancing the electrostatic potential created by the first Lys residue and by formation of contacts with CR domain residues.35,39 Such “double-Lys” recognition mode39 was confirmed in subsequent studies.38,40,41 Furthermore, it was shown that not only Lys/Lys pair, but also Lys/Arg pair of a ligand may be involved in the interaction with CR domains.35,38–40 This is in a good agreement with our finding that Arg residues of fibrin βN-domains make a significant contribution to their interaction with the VLDL receptor. Thus, the results of the present study further confirm the importance of Lys and Arg residues in the interaction of LDL receptor family members with their ligands. Moreover, together with our previous finding that mutation of the highly conserved Trp residue in the third CR domain of VLDLR resulted in a significant loss of the affinity of VLDLR(1–8) to (β15–66)2,22 these results suggest that interaction of fibrin with CR domains of the VLDL receptor may occur through the proposed “double Lys/Arg” recognition mode.39
Our previous study revealed that interaction of fibrin βN-domains with the VLDL receptor occurs mainly through the second and third CR domains of the latter and the presence of the fourth CR domain increases the affinity of this interaction.22 To explain the role of the fourth CR domain in the interaction with fibrin, we assumed that this domain either has low affinity to fibrin or may stabilize the structure of the second and third CR domains in a conformation that is most favorable for high affinity binding.22 The results of the present study allow us to select between these two assumptions. Namely, our finding that all three positively charged clusters of the βN-domains contribute to the interaction with VLDLR suggests that all three CR domains of VLDLR are involved in this interaction. Furthermore, since contribution of the first cluster is the least pronounced, it most probably interacts weakly with the fourth CR domain while cluster II and III are involved in strong interaction with CR domains 3 and 2, respectively. The question which residues of the positively charged clusters of the βN-domains form interacting Lys/Lys or Lys/Arg pairs remains to be addressed.
It should be noted that our ionic strength-dependence experiments revealed the involvement of roughly 6 ionic pairs in the interaction of (β15–66)2 with the VLDLR(1–8) fragment. Assuming that this interaction occurs through the “double-Lys/Arg” recognition mode, as mentioned above, one can expect that only one Lys residue from each positively charged cluster should be involved in electrostatic interaction with the “acidic pocket” on a CR domain since another Lys or Arg contributes to the binding mainly through non-electrostatic interactions.39 This suggests that each of the two βN-domains of the dimeric (β15–66)2 fragment, which mimics the dimeric arrangement of the βN-domains in fibrin, forms three ionic pairs with the VLDLR(1–8) fragment, i.e. one molecule of (β15–66)2 should interact with two molecules of VLDLR(1–8) in the (β15–66)2-VLDLR(1–8) complex. This also implies that two closely spaced βN-domains in fibrin molecule should interact with two VLDL receptor molecules. This speculation is in a good agreement with the previous studies suggesting that VLDLR and ApoER2, another member of the VLDL receptor family members, undergo clustering upon binding to their dimeric ligands and such clustering induces activation of intracellular signaling cascade.42,43 Further structure/function studies are required to test this speculation.
In summary, the major findings of the present study are that electrostatic interactions play the major role in the interaction of fibrin with the VLDL receptor and the second and third clusters of the positively charged residues of fibrin βN-domains make the major contribution to this interaction while the contribution of the first cluster is moderate. Our study also revealed that both Lys and Arg residues of these clusters are involved in the interaction. Furthermore, the results of our present and previous studies22 suggest that the interaction between fibrin and the VLDL receptor employs the “double-Lys/Arg” recognition mode previously proposed for the interaction of the LDL receptor family members with their ligands.35,39 Thus, the results of the present study provide further insight into the molecular mechanism underlying the interaction of ligands with the LDL receptor family members. They also provide a solid basis for the development of highly specific peptide-based inhibitors of fibrin-VLDL receptor interaction.
Supplementary Material
Acknowledgments
Funding Source Statement: This work was supported by National Institutes of Health Grant HL056051 to L.M.
ABBREVIATIONS AND TEXTUAL FOOTNOTES
- VLDLR
very low density lipoprotein (VLDL) receptor
- ELISA
enzyme-linked immunosorbent assay
- SPR
surface plasmon resonance
- TBS
Tris-buffered saline containing 20 mM Tris, pH 7.4, and 150 mM NaCl
- HBS
HEPES-buffered saline containing 10 mM HEPES, pH 7.4, and 150 mM NaCl
Footnotes
ASSOCIATED CONTENT
Supporting Information
The supporting information is available free of charge on the ACS Publication website.
REFERENCES
- (1).Clark RA (2001) Fibrin and wound healing, Ann. N. Y. Acad. Sci. 936, 355–367. [DOI] [PubMed] [Google Scholar]
- (2).Languino LR, Plescia J, Duperray A, Brian AA, Plow EF, Geltosky JE, and Altieri DC (1993) Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway, Cell 71, 1423–1434. [DOI] [PubMed] [Google Scholar]
- (3).Altieri DC (1999) Regulation of leukocyte-endothelium interaction by fibrinogen, Thromb. Haemost. 82, 781–786. [PubMed] [Google Scholar]
- (4).Bach TL, Barsigian C, Yaen CH, and Martinez J (1998) Endothelial cell VE-cadherin functions as a receptor for the β15–42 sequence of fibrin, J. Biol. Chem. 273, 30719–30728. [DOI] [PubMed] [Google Scholar]
- (5).Martinez J, Ferber A, Bach TL, and Yaen CH (2001) Interaction of fibrin with VE-cadherin, Ann. N. Y. Acad. Sci. 936, 386–405. [DOI] [PubMed] [Google Scholar]
- (6).Petzelbauer P, Zacharowski PA, Miyazaki Y, Friedl P, Wickenhauser G, Castellino FJ, Gröger M, Wolff K, and Zacharowski K (2005) The fibrin-derived peptide Bβ15–42 protects the myocardium against ischemia-reperfusion injury, Nat. Med. 11, 298–304. [DOI] [PubMed] [Google Scholar]
- (7).Zacharowski K, Zacharowski P, Reingruber S, and Petzelbauer P (2006) Fibrin(ogen) and its fragments in the pathophysiology and treatment of myocardial infarction, J. Mol. Med. 84, 469–477. [DOI] [PubMed] [Google Scholar]
- (8).Yakovlev S, Mikhailenko I, Cao C, Zhang L, Strickland DK, and Medved L (2012) Identification of VLDLR as a novel endothelial cell receptor for fibrin that modulates fibrin-dependent transendothelial migration of leukocytes, Blood 119, 637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lillis AP, Van Duyn LB, Murphy-Ullrich JE, and Strickland DK (2008) LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies, Physiol. Rev. 88, 887–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wyne KL, Pathak K, Seabra MC, and Hobbs HH (1996) Expression of the VLDL receptor in endothelial cells, Arterioscler. Thromb. Vasc. Biol. 16, 407–415. [DOI] [PubMed] [Google Scholar]
- (11).Takahashi S, Sakai J, Fujino T, Hattori H, Zenimaru Y, Suzuki J, Miyamori I, and Yamamoto TT (2004) The very low-density lipoprotein (VLDL) receptor: characterization and functions as a peripheral lipoprotein receptor, J. Atheroscler. Thromb. 11, 200–208. [DOI] [PubMed] [Google Scholar]
- (12).Argraves KM, Battey FD, MacCalman CD, McCrae KR, Gåfvels M, Kozarsky KF, Chappell DA, Strauss JF III, and Strickland DK (1995) The very low density lipoprotein receptor mediates the cellular catabolism of lipoprotein lipase and urokinase-plasminogen activator inhibitor type I complexes, J. Biol. Chem. 270, 26550–26557. [DOI] [PubMed] [Google Scholar]
- (13).Heegaard CW, Simonsen AC, Oka K, Kjøller L, Christensen A, Madsen B, Ellgaard L, Chan L, and Andreasen PA (1995) Very low density lipoprotein receptor binds and mediates endocytosis of urokinase-type plasminogen activator-type-1 plasminogen activator inhibitor complex, J. Biol. Chem. 270, 20855–20861. [DOI] [PubMed] [Google Scholar]
- (14).Kasza A, Petersen HH, Heegaard CW, Oka K, Christensen A, Dubin A, Chan L, and Andreasen PA (1997) Specificity of serine proteinase/serpin complex binding to very-low-density lipoprotein receptor and α2-macroglobulin receptor/low-density-lipoprotein-receptor-related protein, Eur. J. Biochem. 248, 270–281. [DOI] [PubMed] [Google Scholar]
- (15).Mikhailenko I, Krylov D, Argraves KM, Roberts DD, Liau G, and Strickland DK (1997) Cellular internalization and degradation of thrombospondin-1 is mediated by the amino-terminal heparin binding domain (HBD). High affinity interaction of dimeric HBD with the low density lipoprotein receptor-related protein, J. Biol. Chem. 272, 6784–6791. [DOI] [PubMed] [Google Scholar]
- (16).D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, and Curran T (1999) Reelin is a ligand for lipoprotein receptors, Neuron 24, 471–479. [DOI] [PubMed] [Google Scholar]
- (17).Tacken PJ, Teusink B, Jong MC, Harats D, Havekes LM, van Dijk KW, and Hofker MH (2000) LDL receptor deficiency unmasks altered VLDL triglyceride metabolism in VLDL receptor transgenic and knockout mice, J. Lipid Res. 41, 2055–2062. [PubMed] [Google Scholar]
- (18).Takahashi S, Sakai J, Fujino T, Miyamori I, and Yamamoto TT (2003) The very low density lipoprotein (VLDL) receptor - a peripheral lipoprotein receptor for remnant lipoproteins into fatty acid active tissues, Mol. Cell. Biochem. 248, 121–127. [DOI] [PubMed] [Google Scholar]
- (19).Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, and Herz J (1999) Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2, Cell 97, 689–701. [DOI] [PubMed] [Google Scholar]
- (20).Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, and Herz J (1999) Direct binding of reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation, Neuron 24, 481–489. [DOI] [PubMed] [Google Scholar]
- (21).Hembrough TA, Ruiz JF, Swerdlow BM, Swartz GM, Hammers HJ, Zhang L, Plum SM, Williams MS, Strickland DK, and Pribluda VS (2004) Identification and characterization of a very low density lipoprotein receptor-binding peptide from tissue factor pathway inhibitor that has antitumor and antiangiogenic activity, Blood 103, 3374–3380. [DOI] [PubMed] [Google Scholar]
- (22).Yakovlev S, and Medved L (2015) Interaction of fibrin with the very low density lipoprotein receptor: Further characterization and localization of the fibrin-binding site, Biochemistry 54, 4751–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Henschen A, and McDonagh J (1986) Fibrinogen, fibrin and factor XIII, in Blood Coagulation (Zwaal RFA, Hemker HC, Eds.), pp 171–241, Elsevier Science Publishers, Amsterdam. [Google Scholar]
- (24).Medved L, and Weisel JW, on behalf of Fibrinogen and Factor XIII Subcommittee of Scientific Standardization Committee of International Society on Thrombosis and Haemostasis (2009) Recommendations for nomenclature on fibrinogen and fibrin, J. Thromb. Haemost. 7, 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kollman JM, Pandi L, Sawaya MR, Riley M, and Doolittle RF (2009) Crystal structure of human fibrinogen, Biochemistry 48, 3877–3886. [DOI] [PubMed] [Google Scholar]
- (26).Odrljin TM, Francis CW, Sporn LA, Bunce LA, Marder VJ, and Simpson-Haidaris PJ (1996) Heparin-binding domain of fibrin mediates its binding to endothelial cells, Arterioscler. Thromb. Vasc. Biol. 16, 1544–1551. [DOI] [PubMed] [Google Scholar]
- (27).Yakovlev S, Gorlatov S, Ingham K, and Medved L (2003) Interaction of fibrin(ogen) with heparin: further characterization and localization of the heparin-binding site, Biochemistry 42, 7709–7716. [DOI] [PubMed] [Google Scholar]
- (28).Ruiz J, Kouiavskaia D, Migliorini M, Robinson S, Saenko EL, Gorlatova N, Li D, Lawrence D, Hyman BT, Weisgraber KH, and Strickland DK (2005) The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor, J. Lipid Res. 46, 1721–1731. [DOI] [PubMed] [Google Scholar]
- (29).Yakovlev S, Belkin AM, Chen L, Cao C, Zhang L, Strickland DK, and Medved L (2016) Anti-VLDL receptor monoclonal antibodies inhibit fibrin-VLDL receptor interaction and reduce fibrin-dependent leukocyte transmigration, Thromb. Haemost. 116, 1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gorlatov S, and Medved L (2002) Interaction of fibrin(ogen) with the endothelial cell receptor VE-cadherin: mapping of the receptor-binding site in the NH2-terminal portions of the fibrin β chains, Biochemistry 41, 4107–4116. [DOI] [PubMed] [Google Scholar]
- (31).Yakovlev S, and Medved L (2009) Interaction of fibrin(ogen) with the endothelial cell receptor VE-cadherin: localization of the fibrin-binding site within the third extracellular VE-cadherin domain, Biochemistry 48, 5171–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yakovlev S, Gao Y, Cao C, Chen L, Strickland DK, Zhang L, and Medved L (2011) Interaction of fibrin with VE-cadherin and anti-inflammatory effect of fibrin-derived fragments, J. Thromb. Haemost. 9, 1847–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tawfik DS (2009) Modification of arginine side chains with p-hydroxyphenylglyoxal, in The Protein Protocols Handbook (Walker JM, Ed.) 3 rd ed, pp 863–864, Humana Press, New York. [Google Scholar]
- (34).Verdaguer N, Fita I, Reithmayer M, Moser R, and Blaas D (2004) X-ray structure of a minor group human rhinovirus bound to a fragment of its cellular receptor protein, Nat. Struct. Mol. Biol. 11, 429–434. [DOI] [PubMed] [Google Scholar]
- (35).Fisher C, Beglova N, and Blacklow SC (2006) Structure of an LDLR-RAP complex reveals a general mode for ligand recognition by lipoprotein receptors, Mol. Cell. 22, 277–283. [DOI] [PubMed] [Google Scholar]
- (36).Blacklow SC (2007) Versatility in ligand recognition by LDL receptor family proteins: advances and frontiers, Curr. Opin. Struct. Biol. 17, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).van den Biggelaar M, Madsen JJ, Faber JH, Zuurveld MG, van der Zwaan C, Olsen OH, Stennicke HR, Mertens K, and Meijer AB (2015) Factor VIII interacts with the endocytic receptor low-density lipoprotein receptor-related protein 1 via an extended surface comprising “hot-spot” lysine residues, J. Biol. Chem. 290, 16463–16476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gettins PG, and Dolmer K (2016) The high affinity binding site on plasminogen activator inhibitor-1 (PAI-1) for the low density lipoprotein receptor-related protein (LRP1) is composed of four basic residues, J. Biol. Chem. 291, 800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Yasui N, Nogi T, and Takagi J (2010) Structural basis for specific recognition of reelin by its receptors, Structure 18, 320–331. [DOI] [PubMed] [Google Scholar]
- (40).Gettins PG, and Dolmer K (2012) A proximal pair of positive charges provides the dominant ligand-binding contribution to complement-like domains from the LRP (low-density lipoprotein receptor-related protein), Biochem. J. 443, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Dolmer K, Campos A, and Gettins PG (2013) Quantitative dissection of the binding contributions of ligand lysines of the receptor-associated protein (RAP) to the low density lipoprotein receptor-related protein (LRP1), J. Biol. Chem. 288, 24081–24090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Strasser V, Fasching D, Hauser C, Mayer H, Bock HH, Hiesberger T, Herz J, Weeber EJ, Sweatt JD, Pramatarova A, Howell B, Schneider WJ, and Nimpf J (2004) Receptor clustering is involved in reelin signaling, Mol. Cell. Biol. 24, 1378–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Divekar SD, Burrell TC, Lee JE, Weeber EJ, and Rebeck GW (2014) Ligand-induced homotypic and heterotypic clustering of apolipoprotein E receptor 2, J. Biol. Chem. 289, 15894–15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.