

Summary
Meiotic recombination is required for correct segregation of chromosomes to gametes and to generate genetic diversity. In mice and humans, DNA double-strand breaks (DSBs) are initiated by SPO11 at recombination hotspots activated by PRDM9-catalyzed histone modifications on open chromatin. However, the DSB-initiating and repair proteins are associated with a linear proteinaceous scaffold called the chromosome axis, the core of which is composed of cohesin proteins. STAG3 is a stromalin subunit common to all meiosis-specific cohesin complexes. Mutations of meiotic cohesin proteins, especially STAG3, perturb both axis formation and recombination in the mouse, prompting determination of how the processes are mechanistically related. Protein interaction and genetic analyses revealed that PRDM9 interacts with STAG3 and REC8 in cooperative relationships that promote normal levels of meiotic DSBs at recombination hotspots in spermatocytes. The efficacy of the Prdm9-Stag3 genetic interaction in promoting DSB formation depends on PRDM9-mediated histone methyltransferase activity. Moreover, STAG3 deficiency has a major effect on DSB number even in the absence of PRDM9, showing that its role is not restricted to canonical PRDM9-activated hotspots. STAG3 and REC8 promote axis localization of the DSB-promoting proteins HORMAD1, IHO1, and MEI4, as well as SPO11 activity. These results establish that PRDM9 and axis-associated cohesin complexes together coordinate and facilitate meiotic recombination by recruiting key proteins for initiation of DSBs, thereby associating activated hotspots with DSB-initiating complexes on the axis.
Keywords: Meiotic chromosome axis, Meiotic recombination, Cohesin, PRDM9, Recombinosome, Recombination hotspots, SPO11, Chromatin modification, Double-strand breaks, Meiosis
Graphical Abstract
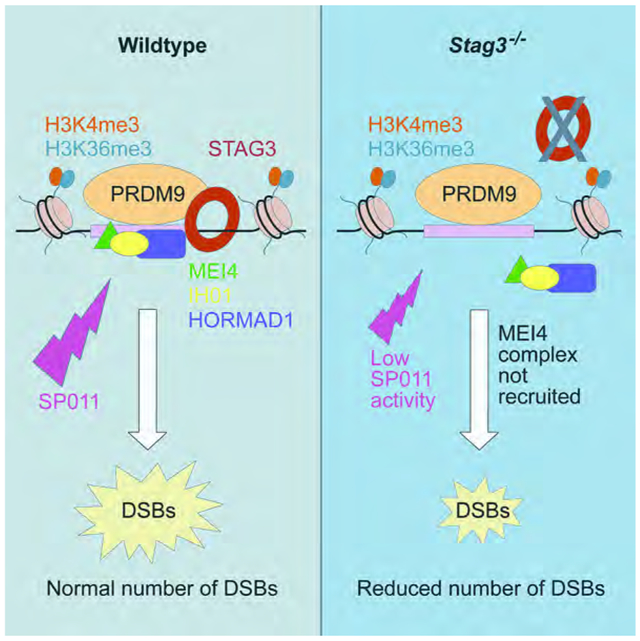
eTOC blurb
Bhattacharyya et al. investigate contribution of cohesin and recombination activator PRDM9 to DNA double-strand break (DSB) formation during mouse meiosis. They find that interactions between cohesin axis proteins and PRDM9 lead to recruitment of DSB-promoting proteins to the axis, which in turn facilitate DSB formation by the meiotic enzyme SPO11.
Introduction
During meiotic prophase, dynamic chromatin organization takes place around the proteinaceous scaffold that forms the chromosome axis. At the outset of meiosis, cohesin protein complexes establish the core of the chromosome axis [1,2] and subsequently, assemble the synaptonemal complex (SC). The cohesin complexes are ring-shaped structures comprised of two structural maintenance-of-chromosome proteins (SMC1A or B and SMC3), a kleisin subunit (RAD21, RAD21L or REC8), and a stromalin subunit (STAG1,2 or 3) [3]. Of these components, STAG3, REC8, and RAD21L are expressed specifically during meiosis, presumably with unique meiotic function(s) [2-11]. Deletion or mutation of cohesin proteins affects axis length, SC formation, DNA double-strand break (DSB) repair and chromatin organization during meiotic prophase in meiocytes [1, 3, 4, 6, 7, 11-15]. STAG3 is the only non-SMC subunit common to each of the meiosis-specific cohesin complexes (SMC3, which is also mitotic, is likewise common to all of the meiotic complexes). Its absence causes the most severe phenotype of all the meiotic cohesin single mutants [3, 6, 7, 13, 14]. Although assembly of the meiotic cohesin axis is contemporaneous with the early events of recombination activation and chromatin remodeling, the precise roles of meiotic cohesins in these processes are, as yet, incompletely understood.
In most mammalian species, during early meiotic prophase, recombination hotspots are determined by H3K4 and H3K36 trimethylation (H3K4me3 and H3K36me3) marks placed on nearby nucleosomes by a DNA binding zinc-finger protein, PR domain-containing 9 (PRDM9) [16-21]. Subsequently, meiotic DNA double-strand breaks (DSBs) are formed at hotspots by the combined action of SPO11 and TOPOVIBL [22-27]. SPO11-mediated DSB formation is facilitated by an interacting accessory protein complex, the recombinosome, comprised of MEI4 and REC114 [23, 25, 28, 29]. The temporal assembly of this pre-DSB complex at DSB sites and its relationship to hotspot activation by PRDM9 is not known. Interestingly, normal numbers of meiotic DSBs are observed in Prdm9−/− mice, but the DSBs occur at H3K4me3 enriched “default” sites, such as promoters [26]. For reasons not currently well understood, these ectopic DSBs are not repaired efficiently, and Prdm9-null spermatocytes are eliminated at a pachytene-like stage, leading to the infertility of Prdm9-null mice [26].
Emerging evidence highlights possible roles for the cohesin axis in the initiation of DSB formation, but the precise mechanistic pathways remain to be elucidated in mammals. In Saccharomyces cerevisiae (budding yeast), the association between axial element components (Hop1, Red1, and cohesins) and the pre-DSB recombinosome subunits (Mer2, Mei4, and Rec114) is required to promote DSBs [30, 31]. In Schizosaccharomyces pombe (fission yeast), the phosphorylation of axis proteins Rec8 and Rec11 (the homolog of mammalian STAG3) is required to promote DSBs [32-34]. There is also evidence for these axis-recombination relationships in mammalian spermatocytes, where most DSB-promoting and repair proteins (e.g., MEI4, REC114, RAD51, and DMC1) are localized on the chromosome axis. Moreover, mutation and deletion of meiotic cohesin proteins in mice affect not only the assembly of the axis but also DSB repair and crossover formation [4, 6, 7, 29]. Although direct evidence is lacking, this effect of axis elements might arise via formation of a pre-DSB recombinosome in mouse spermatocytes. For example, cohesin proteins are required for formation and/or stabilization of MEI4 foci [29], and axis-associated HORMAD1 and its interactor IHO1 regulates SPO11 DSB activity [35]. Taken together, these lines of evidence suggest critical roles for meiotic cohesins in DSB formation, but whether or how this is linked to recombination initiation by PRDM9 is unclear.
Our current investigations of interactions among axis-associated cohesin subunits and PRDM9 were prompted in part by a recent study revealing interactions between PRDM9 and the axis-associated cohesin protein REC8, and also SYCP3 [36]. This suggested that PRDM9-axis interaction might coordinate meiotic events from meiotic recombination initiation to DSB formation, a hypothesis we have investigated here by employing cohesin mutant mouse models, protein biochemistry, and genomic tools. We focused on the role of STAG3 in these processes because of its pivotal function in meiotic axis formation; however, we also provide evidence for the importance of REC8 in events of recombination that lead to DSBs. We found that PRDM9 interacts with STAG3 as well as REC8, suggesting the possibility that the association of hotspots with axis elements might begin at the earliest stages of recombination activation. Both STAG3 and REC8 are required for efficient DSB formation at canonical PRDM9-dependent recombination hotspots, as well as at PRDM9-independent sites. Interestingly, we found genetic evidence for a cooperative role for STAG3 and PRDM9 in the recruitment of the HORMAD1-IHO1 and MEI4-linked SPO11- protein complex that is crucial for initiation of DSBs. Additionally, there is a significant reduction in SPO11 activity in absence of STAG3, providing evidence for a mechanistic link between the cohesin-based axis elements and promotion of SPO11-catalyzed meiotic DSBs. Together, these results support a model in which axis-associated cohesin complexes coordinate events from hotspot activation to DSBs, most likely by acting as a molecular scaffold to array DSB-initiating proteins at recombination sites.
Results
STAG3-associated cohesin complexes co-express and interact with PRDM9 in mouse spermatocytes.
STAG3 is expressed in spermatocytes during the pre-leptotene to zygotene sub-stages of meiotic prophase I, concurrently with the hotspot-activating protein PRDM9 (Figure 1A); however, STAG3 is not required for PRDM9 expression or nuclear localization (Figures 1B and C, and S1A). As previously reported [6, 7, 13, 14], deletion of STAG3 leads to meiotic arrest, apparent histologically (Figure 1C). To determine the relative frequency of meiotic prophase sub-stages in Stag3−/− mutant testes, we used combinatorial antibody labeling for proteins diagnostic of specific meiotic sub-stages: STRA8 (pre-leptonema to leptonema), PRDM9 (leptonema to zygonema), the SC protein SYCP3 (exhibiting punctate to linear staining from leptonema to diplonema), and phosphorylated histone H2AFX, labeling sites of unrepaired DNA damage (refer to Materials and Methods for details). We detected an increased representation of preleptonema to zygonema-like stages in Stag3−/− mutant testes (Figures 1C and S1B-C). In concurrence with previously reported observations [6, 7, 13, 14], adult Stag3−/− mutant testes displayed a significant increase in TUNEL-positive apoptotic cells in comparison to wildtype controls (Figure S1D-E).
Figure 1. STAG3-associated cohesin complexes co-express and interact with PRDM9 in mouse spermatocytes.

(A) Immunofluorescence staining of a wildtype testis cross-section probed with PRDM9 (green) and STAG3 (red) antibodies. PRDM9 and STAG3 co-express in pre-leptotene (PL, white arrow heads), leptotene and zygotene spermatocytes (L-ZY, magenta arrow heads). No PRDM9 is detected in pachytene spermatocytes (PAC, turquoise arrow heads) or round spermatids (RS). Scale bar=100μm.
(B) Immunoblot showing expression of STAG3, PRDM9 and β-TUBULIN (loading control) in juvenile (12 d postpartum) testes from wildtype and Stags−/− males. 50 μg of protein extract was loaded per lane.
(C) Top panel: PAS-stained histological sections of wildtype (left) and Stag3−/− (right) testis at 8 weeks of age. The middle and the bottom panels are testis sections stained with different antibody combinations. Middle panel: Immunofluorescence staining of histological sections of wildtype and Stag3−/− testis stained with anti-PRDM9 (green), and anti-STRA8 (red); DNA is stained with DAPI (blue). Bottom panel: The same wildtype and Stag3−/− testis histological sections immunolabeled using anti-SYCP3 (green) and anti-STRA8 (red); DNA is stained with DAPI (blue). STRA8, SYCP3 and PRDM9 co-express in pre-leptotene and leptotene cells (yellow arrow heads), and in zygotene (-like) spermatocytes (white arrow heads) SYCP3 and PRDM9 co-express. In pachytene (-like) spermatocytes (blue arrow heads), SYCP3 is detected, but no PRDM9 is seen. Scale bars=100μm.
(D) Wildtype (two left columns) and Stag3−/− (two right columns) spermatocyte chromatin spreads immunolabeled with antibodies against the SC lateral element protein SYCP3 (blue) and hybridized with whole-chromosome FISH paint probes for Chr 1 (green) and Chr 11(red); DNA is stained with DAPI (blue). Note larger and less distinct chromosome territories, as well as separated homologs, in mutant panels (right). Scale bars=10μm.
(E&F) Co-immunoprecipitation of PRDM9 with chromosomal axis proteins STAG3, REC8, and SMC3 from wildtype 14-dpp spermatocytes (E), confirmed by reverse co-immunoprecipitation with anti-SMC3 antibody (F). Input extract, 20 μg (2.5 μg in the RAD21,SMC3 and H3 blots); lane 2, 20 μg extract coIP with nonimmune IgG; αPRDM9 and αSMC3 (F), 20 μg extract coIP with the respective antibody, either not-treated ("−") or treated ("+") with DNase. Histone H3 IP was used to detect efficiency of DNase treatment. The antibodies used to detect specific proteins are shown on the left and the relevant molecular weights are shown on right of each blot. For quantified values of protein bands please refer to Table S1 and S2.
(G) Co-immunoprecipitation of PRDM9 and cohesin proteins from Stag3−/− spermatocytes. This analysis confirmed that PRDM9 can be associated with other cohesin subunits in absence of STAG3, and demonstrates the protein-protein interaction of REC8 and RAD21L with other cohesin subunits in absence of STAG3 previously reported [7]. Input extract, 20 μg (2.5 μg in the STAG1, STAG2, RAD21 and SMC3 blots); lane 2, 20 μg extract coIP with nonimmune IgG and lane 3, 20 μg extract coIP with SMC3 antibody. The relevant molecular weights are shown on right of each blot.
As observed previously [6, 7, 13], a normal linear SC does not form in Stag3−/− spermatocytes; instead, SYCP3 protein aggregates, frequently in proximity to CREST-stained centromeric heterochromatin (Figure S2A) [6, 7, 13]. The presence of 55–80 CREST foci in Stag3−/− spermatocytes (compared to 21 foci in wildtype spermatocytes) suggests defective sister-chromatid cohesion (Figure S2A). We confirmed this using DNA fluorescence in situ hybridization (FISH) probes specific for a 200-kb region of chromosomes (Chr) 2 and 11(Figure S2B). Additionally, use of whole-chromosome paint probes to detect chromosome territories (Chr 1 and 11) revealed aberrant chromatin compaction in Stag3−/− spermatocytes; both Chr 1 and 11 occupied larger territories in Stag3−/− spermatocytes than in wildtype spermatocytes (Figure 1D). In ~75% (194 of 258 total nuclei examined) of Stag3−/− spermatocytes, we observed two chromosome territories for Chr 1,2, and 11. In comparison, only ~13% (23 of 183 total nuclei examined) of the wildtype spermatocytes have two territories for at least one of the 3 chromosomes. This observation suggests aberrant homolog pairing in Stag3−/− spermatocytes (Figure 1D and S2B). Thus, lack of a STAG3 axis element has profound effects on chromatin organization, sister chromatid cohesion, and homolog pairing during mammalian meiosis, confirming previous findings [6, 7, 13, 14].
The interaction between PRDM9 with axis-associated REC8 and SYCP3 has previously been demonstrated [36]. Because STAG3 and PRDM9 are co-expressed (Figure 1A), we tested their interaction. Co-immunoprecipitation (co-IP) assays using –-PRDM9 antibody and protein extract from juvenile mouse testes (12 days postpartum) identified interactions between PRDM9 and STAG3, REC8, and SMC3, but not with RAD21L or RAD21 (Figure 1E, Table S1). The detected interactions between cohesin proteins and PRDM9 persisted even after DNase treatment (where there is negligible IP of histone H3) (Figure 1E, Table S1), demonstrating that the detected protein interactions are independent of DNA. Co-IP with antibody to the SMC3 cohesin subunit confirmed interactions between PRDM9 and the STAG3- and REC8-associated cohesion complexes (Figure 1F, Table S2). Next, we investigated whether PRDM9 interacts with cohesin proteins in absence of STAG3, and found PRDM9 in immunoprecipitates with other cohesin protein subunits, including STAG1 and STAG2, in Stag3−/− spermatocytes (Figure 1G). Moreover, and reflecting earlier observations [7], we also detected REC8 and RAD21L with other cohesin subunits immunoprecipitated from cells lacking STAG3 (Figure 1G). These observations suggest that in the absence of STAG3, other known (STAG1 and STAG2) and perhaps unknown (stromalin-like) axis proteins may form cohesin complexes with the meiotic kleisin subunits REC8 and RAD21L, and interact with PRDM9.
Taken together, these findings raise the possibility that interactions between PRDM9 and the cohesin complexes might bring meiotic recombination hotspots into a functional relationship with the developing chromosome axis. Thus, we next investigated how these interactions might facilitate DSB formation.
Absence of key meiotic cohesin proteins and the axis influences DSB numbers at both PRDM9-dependent hotspots and PRDM9–independent sites.
To determine the importance of interactions between PRDM9 and cohesin subunit proteins for normal levels of meiotic DSBs, we assessed the number of DSBs in spermatocytes from wildtype, various single- and double-mutant (dKO) mice for cohesin subunits and/or Prdm9. We counted foci of DMC1, a protein that binds specifically to meiotic DSB sites, as a surrogate for DSB number. We scored spermatocytes at similar meiotic sub-stages [the late leptotene and zygotene (or zygotene-like) cells in wildtype and mutants were grouped together due to lack of markers that could unambiguously discriminate the stages; refer to Materials and Methods for details on staging spermatocytes]. The results are summarized in Figure 2 and tabulated in supplementary data files (Data S1).
Figure 2. Absence of key meiotic cohesin proteins and the axis influences DSB numbers in both PRDM9-dependent hotspots and PRDM9–independent sites.

(A-D) Immunofluorescence detection of DMC1 foci (green) in spermatocyte chromatin spreads also labeled with antibody against SYCP3 (red); DNA is stained with DAPI (blue). Scale bars=10μm.
(A) Wildtype, Rad21l−/−, Rec8−/− and Stag3−/− spermatocyte chromatin spreads.
(B) Spread chromatin from spermatocytes doubly-deficient for different cohesin subunits. Scale bars=10μm.
(C) Spread chromatin of spermatocytes deficient for Prdm9 or double mutants lacking Prdm9 and one of the cohesin subunits. Scale bars=10μm.
(D) Chromatin spread from spermatocytes with altered PRDM9-dependent trimethylation activity in Stag3 deficient spermatocytes. Scale bars=10μm.
(E) Dot-plots showing numbers of DMC1 foci per spermatocyte (error bars=SEM). The genotypes of mice tested are indicated below the graphs. P-values were calculated using Mann-Whitney U test with Tukey’s multiple testing corrections and are tabulated in Data S1.
See also Figure S3, S4, S5, Methods S1 and Data S1.
We first investigated the relative importance of various cohesin proteins for normal DSB levels (Figure 2E, panel 1). Deficiency of RAD21L did not significantly impact numbers of DMC1 foci (Figure 2A, E1 and Data S1). Using a KOMP allele of Rec8 (Figure S3), we found that spermatocytes deficient for REC8 exhibited a small but statistically significant reduction in DMC1 foci number, which were at ~77% of the wildtype level (Figure 2A, E1 and Data S1). Spermatocytes lacking in STAG3 exhibited ~66% of the wildtype level of DMC1 foci (Figure 2A, E1 and Data S1). Thus, the absence of either of these two cohesin proteins significantly and negatively impacts the DSB number. Most DMC1 foci in Stag3−/− spermatocytes were not associated with an α-SYCP3-stained axis (Figures 2A and S4C-E). But, we observed occasional co-occurrence of DMC1 foci and remnants of chromosomal axes in Stag3−/− spermatocytes stained for kleisin proteins REC8, RAD21, and RAD21L (Figure S4C -E; see also Figure 1G for related findings). In wildtype spermatocytes, STAG3 might influence half of all breaks, with partial compensation for its deficiency by other axis proteins. Therefore, we investigated the impact of other cohesin proteins on DSB number in Stag3−/− spermatocytes. To test the contribution of REC8 and RAD21L to DSB number in the absence of STAG3, we generated Stag3−/− Rec8−/− and Stag3−/− Rad21l−/− dKO mice (Figure 2 B). The number and sub-stage frequency of the spermatocyte population in these dKO males was similar to that in Stag3−/− mice; similarly, the spermatocytes lacked defined linear axes stained by α-SYCP3 (Figure 2B, Figure S4A-B); notably, each exhibited a significant reduction in number of DMC1 foci compared to wildtype and the respective single mutants (Figure 2E2). Although DMC1 foci in Stag3−/− Rad21l−/− spermatocytes were reduced compared to Rad21l−/− spermatocytes (~33% fewer), they were similar to Stag3−/− spermatocytes (Figures 2A-B, E1-2 and Data S1). In contrast, Stag3−/− Rec8−/− dKO spermatocytes have ~40 to 60% fewer DMC1 foci than those of either single mutant or the wildtype controls (Figures 2A-B, E1-2 and Data S1)—an additive effect that suggests interactions or compensation by other stromalins or axis-associated proteins. As anticipated by previous results suggesting redundancy of Rad21l in the presence of Rec8 [1, 29], the Rec8−/− Rad21l−/− dKO spermatocytes have a ~70 to 80% reduction in the number of DMC1 foci compared to Rec8−/−, Rad21l−/−, Stag3−/− or wildtype spermatocytes (Figures 2A-B, E1-2 and Data S1). Taken together, while these dKO models suggest the possibility of cooperative interactions between REC8 and STAG3, they also provide evidence that the REC8 kleisin may be more stringently required than RAD21L for meiotic DSB induction.
Following from our protein interaction analyses (Figure 1), we sought evidence for potentially cooperative interactions of PRDM9 with cohesin proteins promoting DSBs. Thus, we assessed DSB number in Prdm9-cohesin dKO spermatocytes (Figure 2E, panel 3). Consistent with previous studies [26, 37], we found that the number of DMC1 foci in spermatocytes lacking PRDM9 did not significantly differ from wildtype (Figure 2C and E3 and Data S1). The number of DMC1 foci in Prdm9−/− Rad21l−/− dKO spermatocytes also remained similar to wildtype, Rad21l−/− and Prdm9−/− single-mutant spermatocytes. In contrast, Prdm9−/− Rec8−/− dKO spermatocytes exhibited significantly lower numbers of DMC1 foci: ~37%, 20%, and 40% less than wildtype, Rec8−/− and Prdm9−/− spermatocytes, respectively (Figures 2A, C and E1, 3; Data S1). We also observed even more dramatic reductions (~70 to 80%) in the number of DSBs in Prdm9−/− Stag3−/− dKO spermatocytes compared to wildtype spermatocytes and the respective single mutants (Figures 2A, C and E1, 3; Data S1). To control for possible label bias, we also employed co-staining with antibody for the DSB repair proteins RAD51 and DMC1, counted the co-occurrence of both protein foci, and confirmed the decrease in DSB number in Stag3−/− and Stag3−/− Prdm9−/− dKO spermatocytes in comparison to wildtype controls (Figure S4F-G). Together these observations add to evidence that Stag3 and Rec8 promote normal numbers of DSBs, and further suggest synergistic genetic interactions with Prdm9. Moreover, because the DSBs in Prdm9-deficient spermatocytes are at PRDM9-independent sites, these results also imply that Stag3 and Rec8 influence not only recombination-initiating DSBs at PRDM9-activated hotspots, but also those at PRDM9-independent sites, which occur primarily in gene promoters and other functional elements [26]. Thus we hypothesize that axis proteins have required roles in DSB pathways that are both dependent on, and independent of, PRDM9.
Because PRDM9-dependent histone methyltransferase activity is essential for hotspot activation [27], we sought to determine whether Prdm9-Stag3 genetic interactions promoting normal DSB number depend on the histone methyltransferase activity of PRDM9. We tested this by using two genetic models exhibiting PRDM9-axis interaction, but critically low methyltransferase activity. First, functional hemizygosity for Prdm9 leads to a ~50% reduction in hotspot activation [20] and ~23% reduction in DMC1 foci (Figure 2A, D, E1,4 and Data S1). In Prdm9−/−Stag3−/− spermatocytes, we observed ~38% fewer DMC1 foci compared to Stag3−/− spermatocytes, and ~47% fewer compared to Prdm9+/− spermatocytes (Figure 2A, D, E1, 4 and Data S1). Second, we tested the requirement for PRDM9 methyltransferase activity using a new Prdm9 mutant allele with an E365K amino acid substitution within the catalytic PR/SET domain of PRDM9 (hereafter denoted as Prdm9EK, Methods S1). This amino acid substitution was induced in the endogenous Prdm9Dom2 allele, in the C57BL/6J genetic background, via CRISPR/Cas9 gene editing. Importantly, expression levels of PRDM9EK protein in the mutant were similar to wildtype (Figure S5A-C). Thus, we expect that physical interactions between PRDM9 and the chromosome axis are intact in Prdm9EK/EK spermatocytes, and indeed, Tian et al. presented evidence for such interactions in vivo, using a different mutation in the PRDM9-SET domain [38]. Similar to a recently published “methyltransferase-dead” Prdm9 allele [27], the Prdm9EK/EK homozygote phenocopied the Prdm9 null [26, 39] with respect to meiotic arrest with defective synapsis, and infertility (Figure S5A, D). The histone methyltransferase activity of the mutant PRDM9 was severely reduced, but not completely ablated; homozygous spermatocytes exhibited detectable levels of H3K4me3 at a few of the most highly trimethylated hotspots (Figure S5E-F). Similar to Prdm9-null spermatocytes, normal numbers of DMC1 foci were observed in Prdm9EK/EK spermatocytes (Figure 2D and E4; Data S1). In Prdm9EK/EK Stag3−/− double mutant spermatocytes, there was a dramatic reduction in the number of DMC1 foci, similar to that observed in Prdm9−/− Stag3−/− dKO spermatocytes (Figure 2D and E4; Data S1). These results provide evidence that the effect of the Prdm9-Stag3 genetic interaction on DSB formation depends on PRDM9-mediated histone methyltransferase activity. Therefore, genetic analyses suggest that Prdm9, Stag3 and Rec8 exhibit cooperative interactions that promote normal numbers of meiotic DSBs and/or stability of DSB repair proteins at DSB sites.
Cohesin subunit genes Stag3 and Rec8 are required for efficient DSB formation at PRDM9-activated hotspots
Although the above results revealed a reduction in DMC1 foci in Stag3- and Rec8-deficient spermatocytes, they are not informative as to whether DSBs were present at PRDM9-dependent recombination hotspots; certainly, in Prdm9−/− spermatocytes, ectopic DSBs are found at non-PRDM9-activated “default” sites, such as promoters and enhancers, also enriched in H3K4me3 [26]. We used genetic and molecular approaches to determine whether the absence of Stag3 and/or Rec8 affects the distribution and/or enrichment of meiotic DSBs at PRDM9-dependent and -independent genomic sites.
We used DMC1 single-stranded DNA (ssDNA) ChIP-seq analyses to compare the distribution of DSBs in Stag3−/− and Rec8−/−~ spermatocytes to that in wildtype. The genomic locations of hotspots depend on the identity of the PRDM9 protein variant and the genomic sequence of the strain [20, 40, 41]; therefore all mutant strains for this analysis were on a C57BL/6N (hereafter B6N) background, which is homozygous for the PRDM9Dom2 allele. As described in more detail in Materials and Methods, we used a statistical threshold of a false discovery rate (FDR) < 0.01 to call peaks utilizing MACS 2.0. We detected 11,558, 8,800, and 6,460 DMC1 peaks in wildtype, Stag3−/−, and Rec8−/− testes respectively (Figures 3A, B and D). To assess PRDM9 binding sites specifically, we included only ones that had been detected previously by two independent methods: Affinity-seq [42] and H3K4me3 ChIP-seq [19-21]. There are 13,355 such Prdm9Dom2-dependent hotspots in the C57BL/6J (B6J) genome that are detected by both of these assays (Figure 3C). Using the criteria defined above, we determined that ~75% of the DMC1 peaks in wildtype (8,625 of 11,558), ~79% of the peaks in Stag3−/− (6,949 of 8,800), and ~88% of the peaks in Rec8−/− (5,697 of 6,460) co-occur with known PRDM9-dependent hotspots in the genome (Figure 3E and F). Thus, most DSBs in cohesin-mutant spermatocytes occur at known PRDM9-dependent sites. Among the remainder of the peaks, only 3–4% of the DMC1 peaks in wildtype (402 of 11,558), Stag3−/− (293 of 8,800), and Rec8−/− (239 of 6,460) overlapped with ectopic DMC1 peaks observed in Prdm9−/− spermatocytes (25,261 DMC1 peaks) [26] (Figure 3F). In pairwise comparisons, ~92% of Stag3−/− (6,407) and ~98% of Rec8−/− (5,581) DMC1 peaks were shared with the wildtype control (Figure 3G and H). In Rec8−/− spermatocytes, ~92% of DMC1 peaks (5,193) are shared with Stag3−/− DMC1 peaks. Additionally, ~84% of peaks (7022 of 8315) in Stag3−/− Rec8−/− dKO spermatocytes are associated with known PRDM9-dependent hotspots in the genome (Figure 3D and F). Together, these data provide strong evidence that the canonical cohesin-based axis is not required for hotspot selection and activation by PRDM9.
Figure 3. Cohesin subunit genes Stag3 and Rec8 are required for efficient DSB formation at PRDM9-activated hotspots.

(A) Scatterplots showing correlation between two replicates of the B6N (wildtype control), Stag3−/− and Rec8−/− DMC1-ChIP-seq samples. Read numbers are hotspot strength for each sample expressed in fragments per million (FPM). The black line indicates correlation between autosomal hotspots, and the blue line indicates correlation between X-linked hotspots.
(B) The correlation matrix illustrating hotspot strength for each sample estimated between common hotspots derived from the same and different genotypes of wildtype (WT) and mutant mice (S3K denotes Stag3−/− and R8K denotes Rec8−/− mutants respectively).
(C) Venn diagram showing common sites between PRDM9Dom2 binding sites from an affinity-seq experiment and PRDM9Dom2-dependent H3K4me3 trimethylated sites from a ChIP-seq experiment, both in wildtype B6J mice.
(D) Coverage profiles of DMC1 peaks from a representative genomic window on chromosome 17 for chromatin with the Prdm9Dom2 allele on a wildtype B6N background (blue), Stag3−/− (pink), Rec8−/− (green) and Stag3−/− Rec8−/− (yellow) spermatocytes. PRDM9 affinity binding sites (top black bars) and H3K4me3 peaks (gray) are shown in first two panels. Recombination hotspots were defined as the sites common for PRDM9 affinity binding and H3K4me3 marks (red arrow heads); gene promoters are characterized by H3K4me3 peaks alone (blue arrow heads).
(E) Venn diagrams showing overlap between DMC1 peaks and PRDM9Dom2 activated hotspots. Numbers outside circle denote the total number of PRDM9Dom2 hotspots and total number of DMC1 peaks. Number inside the circle indicates common genomic sites.
(F) Distribution of DMC1 peaks in B6N control, Stag3−/−, Rec8−/− and Stag3−/− Rec8−/− spermatocytes among PRDM9Dom2 hotspots, Prdm9−/− DSB sites, unclear and unknown sites. The total number of observed peaks is shown on the right-hand side of the graph.
(G & H) Venn diagrams showing overlap between DSBs sites at PRDM9Dom2-activated hotspots in pair-wise comparisons among wildtype (B6N), Stag3−/− and Rec8−/− spermatocytes. Numbers outside circle denote the total number of DMC1 peaks for respective genotypes. Number inside the circle indicates common DMC1 sites.
(I) Venn diagram showing the number of DSB sites at PRDM9Dom2 activated hotspots in common among wildtype (B6N), Stag3−/− and Rec8−/− spermatocytes. Numbers outside circle denote the total number of DMC1 peaks at defined PRDM9Dom2 hotspots for respective genotypes. Number inside the circle indicates common DMC1 sites between all three genotypes.
(J & K) MA-plots of hotspot strength measured by SSDS on autosomes (grey) and the X chromosome (orange) at PRDM9Dom2 activated hotspots (n=5129) in Stag3−/− and Rec8−/− spermatocytes in comparison to B6N (wildtype) control. P-values were calculated using Analysis of Variance (ANOVA) with Tukey's correction for multiple testing to determine the significance.
(L) Hotspot strength in Stag3−/− and Rec8−/− appear weak on chromosome X (orange) and most autosomes (grey) in comparison to wildtype (B6N) control. P-values were calculated using Analysis of Variance (ANOVA) with Tukey’s correction for multiple testing to determine the significance. The statistical analysis showing significant changes is reported in Data S2.
See also Data S2.
Although DSBs are present at the expected sites, both the cytological and ChIP analyses show that they are diminished in number in the absence of the STAG3 and REC8 cohesin subunits. Therefore, an important issue to resolve is whether DSBs are initiated efficiently in the absence of cohesin proteins. To this end, we determined the hotspot strength per DSB sites (also known as DSB “heat”) calculated as the signal - background fragment count, using the method of Brick et al. 2018 on the Watson- and Crick-strand ssDNA fragment distribution [43]. We determined the fragments per million [FPM]) in mutant and control data for the 5,129 PRDM9-dependent hotspots that are in common among the mutant and control genotypes (Figure 3I). The normalized strength per hotspot in Stag3−/− and Rec8−/− spermatocytes was reduced at most hotspots in comparison to wildtype controls (Figure 3J-L; P < 0.0001; Wilcoxon rank sum test). For example, in Stag3−/− spermatocytes, ~31 % of autosomal (1515 of 4911) and ~84% of X-linked (183 of 218) DMC1 sites exhibited a significant decrease in DMC1 signal (Figure 3L and Data S2; P < 0.05; ANOVA with Tukey's correction), while in Rec8−/− spermatocytes, 40% of autosomal (1974 of 4911) and ~97% of X-linked (211 of 218) DMC1 sites showed a significant decrease in DMC1 ChIP signal (Figure 3L and Data S2; P < 0.05; ANOVA with Tukey's correction). In an interesting contrast to the majority of sites, a small minority (8%) of autosomal hotspot sites in Stag3−/− spermatocytes exhibited higher DMC1 activity (Figure 3J, Data S2). Overall, we conclude that in Stag3−/− and Rec8−/− spermatocytes DSBs at most PRDM9 hotspots either fail to activate efficiently or undergo unusually rapid repair. Rapid repair of DSBs by a sister chromatid might be possible in Rec8−/− spermatocytes, but the idea requires further investigation to explain why some sites might undergo faster repair than others. In Stag3−/− spermatocytes, with the failure of sister chromatid cohesion, such rapid repair is likely not possible. Another possible explanation is that the observed decrease in DMC1 signal could be due to the instability of DMC1 protein at DSB sites. In the following sections, we determine how deficiency of cohesins affects pre-DSB steps to address these issues.
STAG3 and REC8 influence recruitment of MEI4 to the chromosome axis
We next addressed a possible mechanism by which the absence of key meiotic cohesin proteins might diminish DSB initiation. Meiotic DSB formation is preceded by the assembly of the SPO11-accessory protein complex and the recombinosome, including MEI4 [29]. Therefore, we counted MEI4 foci in Prdm9 and cohesin single and double mutants to determine the role of the interactions in the recruitment of MEI4 to the chromosomal axis (or the rudimentary axis-like structures that occur in the absence of STAG3). There were ~50% fewer MEI4 foci in Stags’−/− (250.8 ± 124.1) and ~20% fewer in Rec8−/− (361.9± 66.72) leptotene-like spermatocytes compared to wildtype (449 ± 224.7) (Figure 4A-B and Data S3). Furthermore, Stag3−/− Rec8−/− (153.5 ± 42.22) dKO spermatocytes have fewer MEI4 foci than wildtype (~75%) or StagS−/− (~39%), and Rec8−/− (~58%) single mutant spermatocytes (Figure 4A-B and Data S3). Moreover, MEI4 protein was aggregated and aberrantly localized in both Stag3−/− mutant and Stag3−/− Rec8−/− dKO spermatocytes (Figure 4A). The number of MEI4 foci is not significantly altered in either Prdm9+/− heterozygous or Prdm9−/− homozygous mutant spermatocytes (519.3 ± 66.59 and 516.3 ± 74.72, respectively) (Figure 4A-B and Data S3), suggesting that dosage deficiencies of PRDM9 per se do not impact MEI4 localization. However, in the case of Prdm9−/− Stag3−/− dKO spermatocytes, the number of MEI4 foci was significantly reduced (139.2 ± 65.09; Figures 4A-B; Data S3) compared to wildtype (~70%), and Stag3−/− (~44%) and Prdm9−/− (~73%) single-mutant spermatocytes (Figure 4A-B and Data S3). Taken together, these results suggest that the Stag3 and Rec8 work both independently and cooperatively with each other and with Prdm9 to recruit MEI4. The cooperative function may be through the assembly of an axis, and therefore observed decreases in MEI4 foci in cohesin mutants may be a consequence of altered axis structure.
Figure 4. STAG3 and REC8 influence recruitment of MEI4 to the chromosome axis.

(A) Wildtype and mutant spermatocyte chromatin spreads labeled with antibodies against the SC protein SYCP3 (red) and the pre-DSB protein MEI4 (green); DNA is stained with DAPI (blue). For details regarding meiotic sub-staging of mutant spermatocytes see Materials and Methods. Scale bars=10μm.
(B) Dot-plots showing quantification of MEI4 foci in leptotene (and leptotene-like) spermatocytes in different mutants. Error bars=SEM. P-values were calculated using Mann-Whitney U test with Tukey’s multiple testing corrections, and are tabulated in Data S3.
See also Data S3.
STAG3 and REC8 promote localization of HORMAD1-IHO1 complexes onto the chromosome axis
We next addressed the impact of diminished cohesin and PRDM9 on other steps of DSB initiation associated with MEI4 localization. In mice, HORMAD1 and its functional partner IHO1 (interactor of HORMAD1) are required for MEI4 localization to the chromosomal axis [35]. Moreover, cohesin proteins interact with HORMAD1 [44] and IHO1 interacts with MEI4 [35]. These known interactions prompted us to assess recruitment of the HORMAD1 and IHO1 proteins to the axis or axis-like structures in cohesin-deficient spermatocytes. Both HORMAD1 and IHO1 appear in early leptotene wildtype spermatocytes, co-localizing with aggregates of SYCP3 in spermatocyte nuclei (Figures 5 and S6). By late leptonema and zygonema, HORMAD1 and IHO1 signals remained co-localized with SYCP3 along the unsynapsed chromosome axis (Figures 5 and S6). In pachytene spermatocytes, HORMAD1 and IHO1 signals disappeared from the autosomes but remained associated with the sex chromosomes (Figures 5 and S6). In Prdm9−/− zygotene-like spermatocytes, HORMAD1 and IHO1 association with SYCP3-associated axis elements was largely preserved (Figures 5 and S6). In Rec8−/− zygotene-like spermatocytes, HORMAD1 and IHO1 co-localized with the shorter SYCP3-stained axis (Figures 5 and S6). In marked contrast, HORMAD1 and IHO1 were localized only in ill-defined aggregates (Figures 5 and S6) in Stag3−/− leptotene-like and zygotene-like spermatocytes (Figures 5 and S6), consistent with lack of a defined axis. These IHO1 aggregates co-localized with MEI4, but not with SYCP3 (Figure S7). This observation suggests that while physical interaction between IHO1, HORMAD1, and MEI4 might occur independently of the cohesin axis, their role in DSB induction might depend on their association with a cohesin axis. In Stag3−/− Rec8−/− and Prdm9−/− Stag3−/− dKO spermatocytes, the observed localization of HORMAD1 and IHO1 was similar to that in Stag3−/− spermatocytes (Figures 5 and S6), indicating that Stag3 is epistatic to Prdm9 and Rec8 for this phenotype, and suggesting that formation of a proper cohesin axis is necessary for HORMAD1-IHO1 to associate with SYCP3. These results establish that both HORMAD1 and IHO1 are present in the nuclei of cohesin-mutant spermatocytes, but that STAG3 (and to a lesser extent REC8) facilitates proper localization of these proteins. Thus, the Stag3 and Rec8 genes ensure the successful formation of the cohesin axis, which in turn is necessary for proper recruitment of the HORMAD1-IHO1-MEI4 complex, leading to DSB formation by SPO11.
Figure 5. STAG3 and REC8 promote localization of IHO1 complexes onto the chromosome axis.

Wildtype (B6N) and different mutant spermatocyte chromatin spreads labeled with antibodies against the SC protein SYCP3 (red) and the axis protein IHO1 (green); chromatin is stained with DAPI (blue). For each strain, three representative patterns of staining are provided for a full depiction of the phenotype.
(A-B) Chromatin spreads from B6N (wildtype) and Prdm9−/− spermatocytes immunolabeled with IHO1 and SYCP3 antibodies. Note that IHO1 co-localizes with SYCP3 in wildtype and Prdm9−/− spermatocytes on unsynapsed axes. Scale bars=10μm.
(C) Immunostained meiotic chromatin spreads from Rec8−/− spermatocytes. Note lack of IHO1 signal on SYCP3-stained axis. Scale bars=10μm.
(D, E, & F) Immunofluorescence staining of Stag3−/−, Stag3−/− Prdm9−/− and Stag3−/− Rec8−/− spermatocyte spreads. Note that both SYCP3 and IHO1 fail to form a linear profile of axis and that there is occasional co-localization of SYCP3 and IHO1 signals. Scale bars=10μm.
(G) Chromatin spreads from Rec8−/− Rad21l−/− spermatocytes immunolabeled with IHO1 and SYCP3. IHO1 co-localizes with SYCP3 in wildtype but fail to form a linear array in Rec8−/− Rad21l−/− spermatocytes in absence of the SYCP3-stained axes. IHO1 and SYCP3 proteins form aggregates with occasional co-localization in Rec8−/− Rad21l−/− spermatocytes. Scale bars=10μm.
STAG3 facilitates SPO11 function in meiotic DSB formation
We next addressed whether the failure to recruit MEI4, HORMAD1 and IHO1 proteins in the absence of the STAG3-based cohesin axis affects SPO11 activity. We exploited the known dosage-dependent effect of the Spo11 null allele on DSB number [45] and examined SPO11 activity in Spo11+/− spermatocytes in the presence and absence of STAG3. The rationale behind this approach is that if STAG3 influences SPO11 activity, we would expect STAG3 deficiency combined with functional hemizygosity for SPO11 to exacerbate the reduction in DSB number observed with Spo11 hemizygosity alone. We counted DMC1 foci as a surrogate for DSB number. In Spo11+/− zygotene spermatocytes, we observed ~33% fewer DMC1 foci (126.2 ± 32.25) than in wildtype spermatocytes (190.2 ± 59.1) (Figure 6A and D, Data S4). A considerably more dramatic effect was observed in Stag3−/− Spo11+/− dKO spermatocytes, where the formation of DMC1 foci was completely abolished in ~77% of, compared to only ~3% in Stag3−/− and ~1% in Spo11+/− spermatocytes (Figure 6A and C). In most Stag3−/− Spo11+/− spermatocytes lacking DMC1 foci, phosphorylated histone γH2AFX was observed in a protein aggregate reminiscent of the pseudo-sex-body found in Spo11−/− spermatocytes; this was rarely observed in Stag3−/− or Spo11+/− spermatocytes (Figure 6A and B). In the ~23% of Stag3−/− Spo11+/− spermatocytes that did exhibit DMC1 foci, we observed ~50% fewer foci (63.94 ± 41.02) compared to Spo11+/− (126.2 ± 32.25) or Stag3−/− (126.2 ± 30.57) spermatocytes (Figure 6C and D, Data S4). In the spermatocytes exhibiting DMC1 foci, there was no distinct pseudo-sex-body-like structure, but the accumulation of γH2AFX at a single region within the meiotic nucleus was observed (Figure 6B). Because meiotic DSB formation generates SPO11-oligonucleotide complexes representing SPO11-activity [35, 46], we assayed 13 dpp testis extracts for SPO11-oligonucleotides. We observed ~70% and 20% reduction in SPO11-oligonucleotide complexes in Stag3−/− (N=2) and Spo11+/− (N=1) extracts, respectively, in comparison to wildtype control (N=2) extracts (Figure 6E, P=0.39, Welch's t-test). Taken together, these observations reinforce evidence that in the absence of STAG3 DSB formation is defective, most likely through formation of a chromosome axis that acts as a scaffold for loading the SPO11 accessory proteins—e.g. HORMAD1, IHO1, and MEI4— that are required for DSB initiation.
Figure 6. STAG3 facilitates SPO11 function in meiotic DSB formation.

(A & B) B6N (wildtype), Spo11+/−, Stag3−/−, Stag3−/− Spo11+/− and Spo11−/− zygotene (and zygotene-like) spermatocyte chromatin spreads immunolabeled with antibodies against the SC protein SYCP3 (red in panel A and green in panel B), DMC1 (green in panel A), DSB-associated phosphorylated histone γH2AFX (red in panel B); DNA is stained with DAPI (blue). Scale bars=10μm.
(C) Bar graph showing the distribution of zygotene and zygotene-like spermatocytes with and without DSBs in B6 (wildtype), and mutant Spo11+/−, Stag3−/− and Stag3−/− Spo11+/− spermatocytes.
(D) Dot-plots showing quantification of DMC1 foci in in zygotene and zygotene-like spermatocytes from different mouse strains. Error bars=SEM. P-values were calculated using Mann-Whitney U test with Tukey’s multiple testing corrections and are tabulated in Data S4.
(E) Blot of immunoprecipitated and radioactively labelled SPO11-oligonucleotide complexes. The left vertical line marks SPO11-specific signals, the right asterisk indicates nonspecific labelling of contaminants in 13dpp old testis from B6N (wildtype), Stag3−/− and Spo11+/− mice. Radioactive signals were quantified and corrected for background noise and normalized to wild type level. Blots were re-probed with anti-SPO11 antibody. Only one alternative isoform of SPO11 (β) protein was present in 13 dpp B6N (wildtype), Stag3−/− and Spo11+/− mice. The experiments were replicated twice. ±, plus/minus represents standard deviation. P-values were calculated using Welch's t-test.
See also Data S4.
Discussion
This study illuminates the convergence of pathways of recombination and axis formation in crucial events that follow activation of meiotic hotspots by PRDM9 and lead to the formation of DSBs by SPO11. Various endpoints indicative of these events were analyzed in detail in mutant mice deficient for PRDM9 and/or cohesin proteins STAG3 and REC8. STAG3 is viewed as a particularly pivotal protein in these processes because it is one of only two cohesin proteins (other is SMC3) present in each of the meiosis-specific cohesin complexes. Both genetic and physical interaction analyses demonstrated that this core axis protein, as well as REC8, interact with PRDM9 in a manner that cooperatively promotes normal levels of meiotic DSBs at PRDM9-activated hotspots. Interestingly, DSBs at PRDM9-independent sites (e.g., promoters and enhancers) detected in Prdm9−/− mutants depend primarily on STAG3 rather than REC8, although both cohesin subunits are required for normal numbers of DSBs at PRDM9-dependent genomic sites. We demonstrate a requirement for STAG3 for the robust localization of the pre-DSB recombinosome proteins HORMAD1, its interacting partner IHO1, and MEI4, to (leptotene/zygotene) chromatin, where together they together promote the formation of meiotic DSBs by SPO11. Moreover, STAG3 is required for normal SPO11 activity. Our analyses provide evidence that axis proteins cooperate with PRDM9 and play a mechanistic role in associating recombination sites with recombination-initiating proteins, thus poising them for DSB formation. Their cooperative roles in these early events leading to DSB formation constitute a scaffolding, axis-based, mechanism that now appears to be evolutionarily conserved from yeast to mammals.
In most mammals, recombination hotspots are activated by PRDM9-mediated trimethylation of histone H3, on lysines 4 and 36. To understand the steps leading from this event, which occurs in chromatin loops, to the formation of DSBs, which occur on the chromosomal axis, we focused on potential interactions and cooperativity between PRDM9 and cohesins, principal components of the early meiotic axis. Among the meiotic cohesins, STAG3 is of key importance, as a member of each meiosis-specific cohesin complex. In fact, a primary phenotype of mutation of Stag3 in mice is failure to assemble an immunocytologically recognizable chromosomal axis [3, 6, 7, 13, 14], and therefore these mutants, and the double-mutants we have produced, in conjunction with Prdm9 mutants, provide a critical test of the role of the meiotic axis in shepherding sites activated by PRDM9 to their DSB fate. Here we pose and answer critical questions about this process: whether the axis is required for PRDM9-mediated activation of hotspots, whether the axis is required for DSBs at both PRDM9-activated hotspots and those at PRDM9-independent sites, and how PRDM9-cohesin interactions might promote DSBs.
First, is the chromosome axis required for activation of hotspots by PRDM9? We know from previous work [36] that cohesin localization does not require PRDM9, but what about the reverse? Our findings strongly suggest that PRDM9-dependent hotspot activation does not require the organization of the substrate chromatin along a cohesin-dependent chromosome axis. First, DMC1 ChIP-seq (Figures 3 and S7) shows that almost all DSBs in Stag3−/− spermatocytes (which have no axis) and Rec8−/− spermatocytes (which have only short axis profiles) are nonetheless associated with PRDM9-dependent hotspots. Furthermore, all DSBs in Stag3−/− Rec8−/− double-deficient spermatocytes are associated with PRDM9-dependent hotspots. These data are consistent with findings that PRDM9 trimethylates canonical recombination hotspots ex vivo in HEK293 cells, even though these cells lack the meiosis-specific cohesin subunits [20]. Together, the observations suggest that, in spite of the detected impact of prior chromatin modifications on PRDM9 binding [41], PRDM9 activation of hotspots appears to be independent of meiosis-specific axis assembly. We infer that PRDM9 interactions with cohesin proteins, especially STAG3 and REC8, occur subsequent to association of PRDM9 with hotspots.
Second, is the meiotic axis required for the SPO11-mediated DSBs at PRDM9-activated hotspots? Our studies provide strong evidence that the chromosome axis is required to promote normal number of DSBs, both in default sites and at PRDM9-activated hotspots. Immunocytological analyses show that both STAG3 and REC8 are required for normal numbers of DSBs (Figure 2E); nonetheless, most DSBs in Stag3−/− and Rec8−/− mutants are at PRDM9-dependent hotspots (Figure 3E). Therefore, the absence of the axis and axis proteins does not influence the post-activation selection of sites to sustain DSBs; however, our DMC1-ChIP experiments established that mutation of Stag3 and/or Rec8 affects the number of meiotic DSBs induced at PRDM9-activated hotspots (Figure 3J-L). Thus, these cohesin axis proteins are required for normal numbers of DSBs at PRDM9-dependent sites. This finding is consistent with the evidence that PRDM9 interacts physically with STAG3 and REC8 in vivo (Figure 1E-F), and the observation that modulation of PRDM9-dependent methyltransferase activity, combined with the loss of STAG3, has a synergistic effect in diminishing meiotic DSB number at recombination hotspots. Together, these data suggest that the meiotic chromosome axis proteins and PRDM9 cooperatively influence meiotic DSB formation by SPO11. Notably, however, DSBs are not abolished in the absence of individual cohesin proteins. Indeed, the fact that DSBs are still detected at PRDM9-dependent hotspots in spermatocytes deficient for STAG3 and/or REC8 strongly suggests a compensatory role for other proteins, most likely other cohesin subunits. The fact that the number of DSBs in Rec8−/− Rad21l−/− spermatocytes is lower than in Stag3−/− spermatocytes (Figure 2E and Data S1) suggests there may contribution of STAG3-independent cohesin complexes to promoting DSBs. Indeed, substitutions among cohesin subunits are well established. In the absence of STAG3, REC8 and RAD21L can also interact with the other cohesin subunits, which may include STAG1 and STAG2 (Figure 1E) [7], suggesting the possibility of STAG3-independent axis fragments, or complexes, that could contribute to DSBs. In the absence of REC8, cohesin complexes involving RAD21L might compensate in DSB formation, as suggested by the Rec8−/− Rad21l−/− dKO, where the number of DSBs is markedly lower than in either Rec8−/− or Rad21l−/− single-mutant spermatocytes (Figure 2E and Data S1) [1, 29]. However, because there is no effect of RAD21L depletion on DSB formation (Figure 2E and Data S1), the REC8 kleisin subunit appears to be more important for DSB formation. Although STAG3 and SMC1B together form a major part of the meiotic cohesin complex, SMC1B deficiency does not affect SPO11 activity [47]. This redundancy could be explained by fact that the “mitotic” SMC1A can cover some of the functions of SMC1B during meiotic prophase I [48]. In support of these data, we have found that the number of DMC1 foci in Stag3−/− Smc1b−/− spermatocytes are similar to that in Stag3−/− spermatocytes (unpublished). In summary, these data indicate that STAG3 and REC8’s interactions with PRDM9 are important determinants of DSB number in general, and at PRDM9-dependent sites, although other cohesins also contribute to overall DSB success.
Do the meiotic cohesins influence DSBs exclusively at PRDM9-dependent sites? Although the association between DMC1 foci and the axis is preserved in Prdm9−/− spermatocytes, DSBs in the absence of PRDM9 occur ectopically at H3K4me3-enriched transcription start sites and gene enhancers, sometimes referred to as "default" sites [26]. We investigated double mutants to determine the relative effects of STAG3 and REC8 on these ectopic DSBs. In Prdm9−/− Stag3−/− and Prdm9−/− Rec8−/− spermatocytes, all meiotic DSBs detected were at PRDM9-independent sites. In Prdm9−/− Rec8−/− dKO testes, there was a minor reduction in DSB number relative to that in the Rec8−/− single-mutant, indicating that loss of REC8 has a similar effect on DSB number at both PRDM9-dependent sites and ectopic PRDM9-independent sites. However, in Prdm9−/− Stag3−/− dKO testes, loss of STAG3 was associated with a much greater reduction in DSB number relative to the Stag3−/− single-mutant (Figure 2, Data S1). Thus, there may be some specialization, with STAG3 more important than REC8 for the formation of DSBs at PRDM9-independent sites than at PRDM9-dependent sites, while REC8 does not discriminate. The mechanistic basis underlying such specialization is deserving of further investigation, which may further clarify the role of axis elements in recombination initiation.
How do cohesin-Prdm9 interactions promote DSBs at recombination hotspots? In mammals, several axis-associated proteins (e.g., IHO1, HORMAD1, and MEI4) that are required for meiotic DSB formation act by promoting normal SPO11 activity [28, 29, 35]; together, these proteins are sometimes referred to as constituting a "recombinosome." Moreover, protein-protein interactions in mammalian spermatocytes suggest that cohesins, HORMAD1, IHO1, and MEI4 interact with each other [35, 44]. Here we show that Stag3 and Rec8 genes influence quantitatively successful recruitment and linearization of recombinosome proteins along the chromosome axis (or the axis-like structures that form in their absence) and that there may be cooperation between STAG3 and PRDM9 in the localization of the HORMAD1, IHO1 and MEI4 (Figure 4-5 and S6). Consistently, Stag3-mutant spermatocytes, lacking an axis, also have reduced SPO11 activity, with diminished DSB formation (Figure 6). Altered patterns of localization of HORMAD1-IHO1-MEI4 complexes in cohesin mutant spermatocytes could result if the mutant axis was severely diminished in extent and/or not linearized, which would structurally impede interactions between cohesins and other proteins. Finally, the fact that there is localization of IHO1 and MEI4 to the axis in the absence of Prdm9, and that this is decreased in Prdm9−/− Stag3−/− dKO spermatocytes, suggests that DSB formation at PRDM9-independent sites also requires pre-DSB accessory protein localization. Together, these lines of evidence imply that recruitment of the SPO11 accessory proteins to the chromosome axis, aided by STAG3 in cooperation with PRDM9, is required to promote meiotic DSBs. These observations are consistent with the model that the meiotic cohesin axis could act as a molecular scaffold to array the pre-DSB recombinosome axis-linked complexes at DSB sites, thereby facilitating SPO11 access and function at designated sites. However, although the HORMAD1, IHO1, and MEI4 proteins localize to the chromosome axis, the full roster of proteins required for their localization is not yet known. Moreover, the exact nature of the association of recombinosome proteins with the axis (e.g. whether they simply bind to the axis or are actually incorporated into it) and the importance of that association for their pro-DSB function, are important issues for future research.
This genetic, biochemical, and cytological evidence leads to our current molecular model (Figure 7) for mechanistic interactions between PRDM9 and cohesin proteins. In early-zygotene spermatocytes, PRDM9-dependent recombination hotspots become associated with the cohesin axis via a physical interaction between PRDM9 and STAG3 and/or REC8 (probably also facilitated by other associated proteins). An important question for future work is whether this concerted interaction of PRDM9 with STAG3 and REC8 assembles the axis at recombination sites, or if it physically translocates the recombination sites to preformed axis elements [36, 49-52]. The first alternative is a biologically and intellectually satisfying possibility that ensures DSBs at marked sites, and is consistent with both our results and the known timing of these early meiotic prophase events; however, the second possibility is not excluded by available evidence. In either case, our results suggest that cohesin-PRDM9 interactions at activated hotspots promote recruitment of the MEI4-associated, pre-DSB recombinosome complex via HORMAD1 and its interacting partner IHO1 (Figure 7A). Assembly of the cohesin-HORMAD1-IHO1-MEI4 complex along with the PRDM9-cohesin interaction at activated hotspots restricts SPO11 activity to PRDM9-dependent sites (Figure 7A). In spermatocytes deficient for STAG3 (and to a certain extent REC8), there might be some compensation from partially redundant STAG3-independent axis proteins; however, most potential recombination sites in these spermatocytes fail to effectively recruit the HORMAD1-IHO1-MEI4 complex and exhibit diminished DSB formation (Figure 7B). In PRDM9-deficient spermatocytes, STAG3 and other cohesin proteins might recruit the recombinosome complex to the transcription start sites that form PRDM9-independent DSBs in these spermatocytes (Figure 7C). Alternatively, the H3K4me3-marked transcription start sites might be a substrate for alternative evolutionarily conserved recombination initiation mechanisms, such as that observed in yeast and canids [30, 50, 51, 53, 54], or DSB formation might be promoted by a recombination mechanism similar to that at Prdm9-independent sites in the pseudoautosomal region of mammalian sex chromosomes [26]. In the absence of both PRDM9 and STAG3, the HORMAD1-IHO1-MEI4 complex fails to associate with H3K4me3-marked transcription start sites, leading to a failure of SPO11-dependent DSB initiation in most sites (Figure 7D), and emphasizing the crucial role of the cohesin-complex proteins. This model will surely be bolstered in the future as our understanding of the multiple functions of cohesins evolves. Nonetheless, it brings attention to several important concepts that enhance our understanding of the highly conserved role of the meiotic chromosomal axis in the initiation of recombination. First, it emphasizes the importance of interactions between the axis elements and the machinery responsible for selecting recombination sites (PRDM9 for much of mammalian recombination). Second, it demonstrates the crucial importance of key cohesin-complex proteins in the uniquely meiotic function of assembling an axis at recombination sites. Finally, it suggests that future research to unravel these mechanisms will profit from focus on the temporal order of events at recombination sites, which may clarify the exact mechanisms by which they become associated and tethered to the axis, thus ensuring the DSBs that initiate recombination.
Figure 7. A model of pathways by which meiotic cohesin proteins promote meiotic DSB formation by localizing DSB-promoting proteins onto the chromosome axis, both in spermatocytes with functional PRDM9 (yellow background) and those lacking functional PRDM9 (blue background).

(A) In wildtype spermatocytes, PRDM9 (yellow) activates hotspots of meiotic recombination by depositing H3K4me3 (red) and H3K36me3 (orange) on adjacent nucleosomes (light blue). PRDM9 also interacts with cohesin proteins (represented by the STAG3 cohesin complex, purple) and promotes assembly of HORMAD1 (magenta), IHO1 (dark green), and MEI4 (blue), which together act to facilitate meiotic DSB formation by SPO11 (black lightning bolt). Red circles highlight DSBs.
(B) In the absence of the STAG3, PRDM9 activates recombination hotspots, but due to lack of meiotic axis, pre-DSB proteins like HORMAD1, IHO1 and MEI4 fail to assemble in an orderly array. Thus meiotic DSB formation by SPO11 is less efficient (gray lightning bolt). Red circles highlight DSBs.
(C) In the absence of PRDM9, the DSB machinery induces DSBs at PRDM9-independent H3K4me3-enriched sites such as promoters and enhancers that acquire H3K4me3 (red) marks by action of non-PRDM9 histone methyltransferases (HMT, green). Cohesin proteins promote the recruitment of HORMAD1, IHO1, and MEI4 to these sites, thus promoting meiotic DSBs by SPO11 (black lightning bolt). Red circles highlight DSBs.
(D) In the absence of both PRDM9 and STAG3, there is failure to localize the pre-DSB machinery (MEI4, HORMAD1, and IHO1). This dramatically reduces the SPO11-dependent DSB formation (gray lightning bolt). Red circles highlight DSBs.
STAR Methods
Contact for Reagents and Resource Sharing
Further information and requests for reagents should be directed to the lead contact, Mary Ann Handel (MaryAnn.Handel@iax.org).
Experimental Model and Subject Details
Mouse lines
All mice were obtained from the Jackson Laboratory (JAX) and were bred and maintained in the research colony of the authors at JAX. The protocols for their care and use at suitable ages were approved by JAX Institutional Animal Care and Use Committee (IACUC) as per NIH protocols and guidelines. This study employed the following mouse strains: C57BL/6N (B6N), C67BL/6J (B6J), B6N(Cg)-Stag3tm 1 e. 1 (KOMP)Wtsi/2J [6, 7], and B6N(Cg)-Rad21/tm1b(KOMP)Wtsi/J [6], were acquired from the Jackson Laboratory. B6N(Cg)-Rec8tm1b(KOMP)Wtsi/2J mice (Stock No:27556) were acquired from the KOMP repository at the Jackson Laboratory. This mouse carries a deletion of exon 7 of the Rec8 gene. B6;129P2-Prdm9tm1Ymat/J t (Prdm9 knock-out) [39] and B6.129X1-Spo11tm1Mjn/J [23] were acquired and backcrossed for 10 generations to C57BL/6J (B6J). The tenth generation backcrossed animals were used for experiments in this manuscript. C57BL6J-Prdm9<em3/Kpgn>/Kpgn (Prdm9EK/EK) animals were generated by introducing a point mutation creating an E365K amino acid substitution in the PRDM9 PR/SET domain that leads to almost complete loss of PRDM9’s histone methyltransferase activity. This mutation was induced via CRISPR/Cas9 gene editing by the Genetic Engineering Technology core at the Jackson Laboratory.
Method Details
Histology, immunofluorescence and TUNEL apoptosis assays on testis sections
Testes from euthanized 8-week old B6/N control or mutant mice were dissected, fixed with Bouin’s solution or 4% paraformaldehyde (PFA), embedded in paraffin wax, and 5-μm sections were prepared. Bouin’s fixed testis sections were stained with Hematoxylin and eosin (H&E) and Periodic acid–Schiff–diastase (PAS), using standard techniques. The slides were scanned by Nanozoomer -XR.
For immunofluorescence, testes fixed in 4% PFA were sectioned at 5 μm thickness, matured overnight, de-waxed, rehydrated, and heated in 10 mM sodium citrate buffer (pH 6.0) for antigen retrieval. Slides were incubated with ADB buffer (1.5% BSA, 5% donkey serum, 0.05% Triton X-100 in PBS, 0.2x cocktail of protease inhibitors) for 1 hour at room temperature. Slides were incubated overnight with diluted anti-STRA8 (ab49405, Abcam; 1:100), anti-PRDM9 (1:100), anti-STAG3 (gifts from Jessberger and Watanabe labs, 1:100) or anti-SYCP3 (D-1; #74569; Santa Cruz) in ADB buffer. Fluorescent staining was obtained by staining the sections for 2 hours at 37°C, using diluted appropriate secondary antibodies conjugated with Alexa 488/FITC, Alexa 594/CY3 and Alexa 647/CY5 (Molecular Probes/Invitrogen or Jackson Immunoresearch Laboratories 1:300). Stained sections were mounted using ProLong™ Gold Antifade Mountant with DAPI (P36935; Molecular Probes/Invitrogen) at 4°C overnight and imaged after 24 hours using a Leica SP5 confocal microscope.
Identification of apoptotic cells in control and mutant cells was done on paraffin-embedded testis sections (3 biological replicates) using the In Situ Cell Death Detection Kit (Roche, 11684795910) according to the manufacturer’s protocol.
Co-immunoprecipitation assays
The co-IP was carried out using earlier reported protocol [36]. Briefly, twenty 14 dpp testes from B6N or Stag3 −/− mice were homogenized in ice-cold 1x PBS using a dounce tissue homogenizer and filtered through a 70-μm cell strainer (Falcon BD, 352370), and centrifuged for 5 min at 3000× g at 4°C. The pellet was resuspended and incubated for 30 min in 1 ml of Pierce IP buffer (ThermoFisher Scientific, 87787) with 1mM phenylmethanesulfonyl fluoride (PMSF) and 1 × cocktail of protease inhibitors (Roche# 11836145001) with constant rotation, and then spun down at 13,200× g. For the DNase I-treated co-IP samples, the supernatant was added with 100 μl of DNase I buffer and 20 U DNase I (ThermoFisher Scientific, AM1906), and incubated for 1 hr at room temperature. After incubation, 10% of the extract was set apart as input. The co-IP was performed by incubating extract with protein A or G Dynabeads conjugated with antibodies against PRDM9 [37] or SMC3 (Abcam, ab9263) overnight at 4°C. IgG from the same animal species was used as negative control. The beads were washed three times with 1 ml of Pierce IP buffer and eluted with 200 μl of GST buffer (0.2 M glycine, 0.1% SDS, 1% Tween 20, pH 2.2) for 20 min at room temperature. The sample was then neutralized with 40 μl of 1 M Tris-HCl, pH 8. After being heated at 95°C for 5 min, 10 μg of IP and input samples (if not stated otherwise) were then subjected to standard SDS-PAGE and western blotting for detection of PRDM9 (1:1000), REC8 (1:1000, Abcam, ab38372), RAD21L (1:600, gift from Watanabe lab), SMC3 (1:1000, Abcam, ab9263), STAG3 (gift from Watanabe and Jessberger labs), RAD21 (1:1000, Abcam #ab992).
Immunostaining and DNA FISH in surface-spread chromatin preparations
The spermatocyte spreads were prepared by using the hypotonic protocol as described previously [55] with some modifications. Briefly, the testes were detunicated in PBS, and the seminiferous tubules were washed and immersed in ice-cold hypotonic extraction buffer (30 mM Tris, 50 mM sucrose, 17 mM trisodium citrate dihydrate, 5 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM dithiothreitol (DTT), and 0.5 mM phenylmethylsulphonyl fluoride (PMSF), pH 8.2) for 30 mins. Segments of the tubules were minced in 100 mM sucrose solution (pH 8.2), and the cell suspension collected was spread onto a glass slide, which was previously immersed in 1% paraformaldehyde containing 0.15% Triton X-100; the slide was then placed in a humidified box overnight at 4° C. The slides were air-dried at room temperature and washed twice for 2 min with PBS with 0.04% Photo-Flo 200 (Kodak, Rochester, NY, USA). After blocking with ADB buffer (1.5% BSA, 5% donkey serum, 0.05% Triton X-100 in PBS, 0,2x cocktail of protease inhibitors) for 1 hour at room temperature, the slides were incubated overnight with primary antibodies diluted in ADB buffer at 4°C. Primary antibodies (listed in the Key Resource Table) were used as per the indicated dilution. Secondary antibodies conjugated with Alexa 594 or 488 or 647 (Molecular Probes, Invitrogen, Carlsbad, CA, USA or Jackson Immunoresearch Laboratories 1:300) were used. The slides were mounted using ProLong™ Gold Antifade Mountant with DAPI (P36935; Molecular Probes/Invitrogen). Images were acquired with a Zeiss AxioImager.Z2 epifluorescence microscope with a Zeiss AxioCam MRm CCD camera (Carl Zeiss, USA) or a Leica SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany). The immunolabeled spermatocytes were subjected to DNA FISH using Metasystems XMP XCyting Mouse Chromosome N Whole Painting Probes for Chrs 1 and 11 or Leica TIk2 (11qE1) / Aurka (2qH3) Mouse probe as described previously [56]. The images were acquired using a Zeiss AxioImager.Z2 epifluorescence microscope. Images were processed and adjusted using Adobe Photoshop (Adobe Systems).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig anti-PRDM9 | [37] | N/A |
| Mouse anti-STAG3 | [62] | N/A |
| Rabbit anti-STAG3 | [7] | N/A |
| Rabbit anti-REC8 | [62] | N/A |
| Rabbit anti-REC8 | Abcam | Cat# ab38372; RRID: AB_882308 |
| Rabbit anti- RAD21L | [62] | N/A |
| Rabbit anti- SMC3 | Abcam | Cat# ab9263, RRID:AB_307122 |
| Rabbit anti- RAD21 | Abcam | Cat# ab992, RRID:AB_2176601 |
| Rabbit anti- H3 | Abcam | Cat# ab176842, RRID:AB_2493104 |
| Rabbit anti- SYCP3 | Novus | Cat# NB300-231; RRID: AB_10002746 |
| Mouse anti-SYCP3 | Santa Cruz | Cat# sc-74569; RRID: AB_2197353 |
| Rat anti-SYCP3 | [63] | N/A |
| Rabbit anti- STRA8 | Abcam | Cat# ab49602, RRID:AB_945678 |
| Mouse anti-γH2AX | Millipore | Cat# 05-636, RRID:AB_309864 |
| Rabbit anti-HORMAD1 | Proteintech Group | Cat# 13917-1-AP; RRID: AB_2120844 |
| Rabbit anti-HORMAD1 | [35] | N/A |
| Rabbit anti-IHO1 | [35] | N/A |
| Guinea pig anti-IHO1 | [35] | N/A |
| Rabbit anti-MEI4 | [29] | N/A |
| Goat anti-DMC1 (C-20) | Santa Cruz | Cat# sc-8973; RRID: AB_2091206 |
| Rabbit anti-DMC1 (H-100) | Santa Cruz | Cat# sc-22768, RRID:AB_2277191 |
| Rabbit anti-RAD51 | Santa Cruz | Cat# SC-8349; RRID: AB_2253533 |
| Human anti-CREST | AB Incorporated | Cat# 15–235 |
| Mouse anti-SPO11-180 | [61] | N/A |
| Goat anti-rabbit IgG-Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11034, RRID:AB_2576217 |
| Goat anti-mouse IgG-Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11029, RRID:AB_2534088 |
| Goat anti-mouse IgG Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11004, RRID:AB_2534072 |
| Goat anti-mouse IgG Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-11032, RRID:AB_2534091 |
| Goat anti-rabbit IgG Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11011, RRID:AB_143157 |
| Goat anti-rabbit IgG Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-11037, RRID:AB_2534095 |
| Goat anti-mouse IgG Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21235, RRID:AB_2535804 |
| Goat anti-rabbit IgG Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21245, RRID:AB_2535813 |
| Goat anti-guinea pig IgG-Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11073, RRID:AB_2534117 |
| Goat anti-human IgG Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21445, RRID:AB_2535862 |
| Goat anti-rat IgG-Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11006, RRID:AB_2534074 |
| Donkey anti-rabbit IgG-Alexa Fluor 488 | Jackson ImmunoResearch | Cat# 711-545-152, RRID:AB_2313584 |
| Donkey anti-mouse IgG-Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21202, RRID:AB_141607 |
| Donkey anti-mouse IgG Alexa Fluor 594 | Jackson ImmunoResearch | Cat# 715-585-151, RRID:AB_2340855 |
| Donkey anti-rabbit IgG Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-21207, RRID:AB_141637 |
| Donkey anti-mouse IgG Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31571, RRID:AB_162542 |
| Donkey anti-rabbit IgG Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31573, RRID:AB_2536183 |
| Donkey anti-guinea pig IgG-Alexa Fluor 488 | Jackson ImmunoResearch | Cat# 706-545-148, RRID:AB_2340472 |
| Donkey anti-rat IgG Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-21209, RRID:AB_2535795 |
| Rabbit anti-trimethyl-histone H3 (lys4) | EMD Millipore | Cat# 07-473, RRID:AB_1977252 |
| Donkey anti-guinea pig HRP | EMD Millipore | Cat# AP193P, RRID:AB_92662 |
| Goat anti-mouse HRP | Bio-Rad | Cat# 170-6516, RRID:AB_11125547 |
| Mouse anti-β-tubulin | Sigma | Cat # T4026, RRID:AB_477577 |
| VeriBlot for IP Detection Reagent (HRP) | Abcam | Cat# 131366 |
| Rabbit TrueBlot®: Anti-Rabbit IgG HRP | Rockland inc | Cat# 1888-16-33, RRID:AB_469529 |
| Mouse TrueBlot® ULTRA: Anti-Mouse Ig HRP (WB:1:1000) | Rockland inc | Cat# 18-0217-32, RRID:AB_2610838 |
| Normal rabbit IgG antibody | Santa Cruz | Cat# sc-2027, RRID:AB_737197 |
| Normal guinea pig IgG antibody | Santa Cruz | Cat# sc-2711, RRID:AB_737172 |
| Rabbit Normal IgG Control | Cell Signaling | Cat# 2729S, RRID:AB_1031062 |
| Native Protein A (HRP) | Abcam | Cat# ab7456 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1M Tris-Cl, pH 8.0 at 25 °C | KD Medical | Cat# RGF-3360 |
| 1M Tris-Cl, pH 6.5 at 25 °C | Boston BioProducts | Cat# BM-311 |
| Potassium hydroxide (KOH) | Sigma | Cat# P5958-250G |
| 10% SDS | KD Medical | Cat# RGF-3230 |
| Triton X100 | Sigma | Cat# T8787-250ML |
| 0.5M EDTA | KD Medical | Cat# RGF-3130 |
| EGTA | Sigma | Cat# E8145 |
| Protease Inhibitor complete | Roche | Cat# 11836 153 001 |
| 5M NaCl | Sigma | Cat# S5150-1L |
| Ultra Pure™ DNase/RNase-Free Distilled Water | Thermo Fisher Scientific | Cat# 10977-015 |
| PBS 10X, pH7.4 | KD Medical | Cat# RGF-3210 |
| 8M LiCl | Sigma | Cat# L7026-100ML |
| IGEPAL-CA630 | Sigma | Cat# 18896 |
| Sodium Deoxycholate monohydrate | Sigma | Cat# D5670-5G |
| Sodium bicarbonate | Sigma | Cat# S5761 |
| Glycine | Sigma | Cat# 50046-50G |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10004D |
| Proteinase K (20mg/ml) | Thermo Fisher Scientific | Cat# AM2546 |
| MinElute PCR Cleanup Kit | Qiagen | Cat# 28004 |
| TruSeq LT DNA Sample Library Prep. Kit, set A | Illumina | Cat# FC-121-2001 |
| TruSeq LT DNA Sample Library Prep. Kit, set B | Illumina | Cat# FC-121-2002 |
| TruSeq Nano DNA LT Library Prep Kit (24 samples) | Illumina | Cat# 15041757 |
| T4 DNA polymerase | New England Biolabs | Cat# M0203 |
| Paraformaldehyde (PFA) | Sigma | Cat# P6148 |
| DNA Polymerase I Large (Klenow) Fragment | New England Biolabs | Cat# M0210 |
| T4 Polynucleotide Kinase (PNK) | New England Biolabs | Cat# M201 |
| Klenow Fragment (3’-5’ exo−) | New England Biolabs | Cat# M0212 |
| Quick ligation™ kit | New England Biolabs | Cat# M2200 |
| 10x T4 ligation Buffer | New England Biolabs | Cat# M202 |
| dNTPs | New England Biolabs | Cat# N0447S |
| dATP | New England Biolabs | Cat# N0440S |
| Qubit kit ds DNA HS Assay kit | Thermo Fisher Scientific | Cat# Q32851 |
| 70 um Cell Strainer | Becton, Dickinson | Cat# 352350 |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat# 10001D |
| Goat Anti-Rabbit IgG Magnetic Beads | New England Biolabs | Cat# S1432S |
| Pierce IP buffer | Thermo Fisher Scientific | Cat# 87787 |
| Phenylmethylsulfonyl fluoride | Sigma | Cat# 10837091001 |
| Dithiothreitol | Sigma | Cat# 43815-1G |
| DNA-free™ DNA Removal Kit | Thermo Fisher Scientific | Cat# AM1906 |
| Mouse chromosome 1 FISH probe | MetaSystems | Cat# D-1401-050-FI |
| Mouse chromosome 11 FISH probe | MetaSystems | Cat# D-1411-050-OR |
| TIk2 (11qE1) / Aurka (2qH3) Mouse probe | Leica | Cat# KI-30501 |
| TruSeq Nano DNA LD Library Prep Kit, set A | Illumina | Cat# FC-121-4001 |
| MinElute Reaction Cleanup Kit | Qiagen | Cat# 28006 |
| QIAquick PCR Purification Kit | Qiagen | Cat# 28106 |
| Terminal Deoxynucleotidyl Transferase | Thermo Fisher scientific | Cat# EP0161 |
| Protein A-agarose | Roche/Sigma | Cat# 11134515001 |
| dCTP, [a-32P]- 6000Ci/mmol 20mCi/ml, 250 μCi | Perkin Elmer | Cat# BLU013Z250UC |
| NEBuffer™ 4 | New England Biolabs | Cat# B7004S |
| Deposited Data | ||
| Mouse genome assembly GRCm38/mm10 | http://genome.ucsc.edu | N/A |
| PRDM9 affinity seq data | [42] | GEO: GSE108259 |
| H3K4me3 data in B6/J spermatocytes (data were remapped to mm10) | [19] | GEO: GSE52628 |
| H3K4me3 data in Prdm9EK / EK spermatocytes (data were remapped to mm10) | This study | GEO: GSE112110 |
| H3K9me3 data in B6/J spermatocytes (data were remapped to mm10) | [42] | GEO: GSE61613 |
| Prdm9−/+H3K4me3 data (data were remapped to mm10) | [20] | GEO: GSE67673 |
| C57BL/6N SSDS data (data were mapped to mm10) | This study | GEO: GSE112110 |
| Prdm9−/− SSDS data (data were remapped to mm10) | [26] | GEO: GSM869781 and GSM869782 |
| Stag3−/− SSDS data (data were mapped to mm10) | This study | GEO: GSE112110 |
| Rec8−/− SSDS data (data were mapped to mm10) | This study | GEO: GSE112110 |
| Stag3−/− Rec8−/− SSDS data (data were mapped to mm10) | This study | GEO: GSE112110 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6N | The Jackson Laboratory | Stock No: 000664 |
| Mouse: B6N(Cg)-Stag3tm1e.1(KOMP)Wtsi/2J | The Jackson Laboratory [6] | Stock No: 21115 |
| Mouse: B6N(Cg)-Rec8tm1b(KOMP)Wtsi/2J | The Jackson Laboratory, This study | Stock No:27556 |
| Mouse: B6N(Cg)-Rad21l/tm1b(KOMP)Wtsi/J | The Jackson Laboratory [6] | Stock No:22102 |
| Mouse: B6;129P2-Prdm9tm1Ymat/J | The Jackson Laboratory [39] | Stock No:10719 |
| Mouse: C57BL/6J-Prdm9<em3/Kpgn>/Kpgn (Prdm9EK/EK) | The Jackson Laboratory, This study | JR#028855; Methods S1. |
| Mouse: B6.129X1-Spo11tm1Mjn/J | The Jackson Laboratory [22] | Stock No:019117 |
| Oligonucleotides | ||
| Genotyping Primers | This study | Table S3 |
| Software and Algorithms | ||
| BWA | [64] | http://bio-bwa.sourceforge.net/bwa.shtml#14 |
| Bedtools | [65] | http://bedtools.readthedocs.io/ |
| Samtools | [66] | http://samtools.sourceforge.net/ |
| MACS 2.0 | [67] | http://liulab.dfci.harvard.edu/MACS/00README.html |
| Aggregation and Correlation Toolbox | [68] | http://act.gersteinlab.org/ |
| Trimmomatic | [69] | http://www.usadellab.org/cms/?page=trimmomatic |
| R version 3.5.2 | https://www.r-project.org/ | https://www.r-project.org/ |
| Prism v7.0 | GraphPad Software | http://www.graphpad.com |
| Sequence Scanner Software v2.0 | Applied Biosystems | https://resource.thermofisher.com/page/WE28396_2/ |
| Fiji-Image J | https://fiji.sc | https://fiji.sc |
Quantification of DMC1 and MEI4 foci
Spread chromatin and immunostaining were performed as described as above on testicular cell suspensions from male mice (WT, heterozygotes and homozygous mutants), age 8-12 weeks. Because many mutant spermatocytes lack an SYCP3-stained axis, we could not stage mutant spermatocytes based solely on SYCP3-staining, and, moreover, it was not possible to use both seminiferous tubule staging and immunocytochemistry on the same cells due to different sample preparation steps for the two methods. In this combinatorial analysis, each meiotic substage has a unique signature for the expression of prophase I sub-stage-specific proteins (STRA8, PRDM9, and SYCP3). Photographs of imaged cells were processed using Adobe Photoshop. Spermatocytes were subjected to counting of DMC1, RAD51 or MEI4 foci using Fiji [57]; clearly visible DMC1, RAD51 or MEI4 foci associated with a DAPI-stained nucleus were counted across all genotypes without discrimination. To identify early leptotene spermatocytes, where MEI4 localizes to the axis, we quantified spermatocytes stained with only MEI4 and SYCP3, and lacking DMC1, among all spermatocytes with or without an SYCP3-stained axis. As previously shown [29], the maximum number of MEI4 foci appears early in leptonema and subsequently declines. Among wildtype spermatocytes, this contributes to the variation when scoring all cells that are MEI4-positive, DMC1-negative. But in mutants, the actual loading or stability of MEI4 is impaired from the earliest stage; thus, there are so few MEI4 foci at the outset that any decline does not contribute as significantly to variation. DMC1, RAD51 foci appear in late leptotene and early zygotene spermatocytes, and to determine these counts, we collected data for all spermatocytes stained with DMC1, RAD51 in mutants. In wildtype and relevant mutant spermatocytes with an SYCP3-stained axis, data collection for DMC1, RAD51 was restricted to late leptotene and zygotene spermatocytes respectively. Finer sub-categorization of spermatocytes in late-leptotene and zygotene stages was not possible due to lack of discriminating markers.
DMC1 ChIP-seq protocol
DMC1 ChIP was performed with three replicates of 8 weeks old B6N, two replicates of Stag3−/− and Rec8−/−, and single replicate for Stag3−/− Rec8−/− spermatocytes using a published method [58]. Briefly, testes from euthanized 8-week mice were removed and decapsulated and cross-linked with 1% paraformaldehyde solution for 10 min. The tissue was homogenized and filtered with a 70 um cell strainer to obtain germ cells. Cells were washed with Lysis buffer 1 (0.25% Triton X100, 10mM EDTA, 0.5mM EGTA, 10mM Tris-HCl pH 8.0), Lysis buffer 2 (0.2M NaCl, 1mM EDTA, 0.5mM EGTA, 10mM Tris-HCl pH 8.0), and resuspended in Lysis buffer 3 (1% SDS, 10mM EDTA, 50Mm Tris-HCl pH 8.0) with 1× PIC. The chromatin was then sheared to ~1000 bp by sonication, and dialyzed against ChIP buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris- HCl, pH 8.0, 167 mM NaCl) for at least 6 hrs. 10 μl of chromatin was saved as input. The rest of the chromatin was incubated with an antibody against DMC1 (Santa Cruz, sc-8973) overnight at 4°C. The mixture was then incubated with protein G Dynabeads (Thermo Fisher Scientific, 10004D) for 4 hrs at 4°C. The beads were washed with Wash buffer 1 (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.0, 150 mM NaCl), Wash buffer 2 (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mMTris-HCl, pH 8.0, 500 mM NaCl), Wash buffer 3 (0.25 M LiCl, 1% NP-40, 1mM EDTA, 10mMTris-HCl, pH 8.0, 1% Deoxycholic acid), and twice with TE buffer. The chromatin was eluted with dilution buffer (1% SDS, 0.1 M NaHCO3 pH 9.0) at 65°C for 30 min and reverse-crosslinked by adding 200 mM NaCl and incubating overnight at 65°C. ChIP and input DNA were purified by MinElute Reaction Cleanup Kit (Qiagen, 28006). For library preparation, DNA was first end-repaired by incubation with 1 × T4 DNA Ligase Reaction Buffer with 0.25 mM dNTP, 3 units of T4 DNA polymerase (NEB, M0203S), 1 unit of Klenow Enzyme (NEB, M0210S) and 10 units of T4 PNK (NEB, B0202S) at 20°C for 30 min, followed by addition of 3’-A overhangs using Klenow Fragment 3’-5’ exo- (NEB, M0212S). After denaturation of DNA at 95°C for 3 min, adapters from the TruSeq Nano DNA LD Library Prep Kit (set A, Illumina, FC-121-4001) were ligated with a Quick Ligation kit (NEB, M2200S). The libraries then were amplified using PCR Enhancer mix and primer cocktail (Illumina, FC-121-4001) for 12 cycles. For each step, DNA was purified by MinElute Reaction Cleanup Kit. Libraries were sequenced on an Illumina® HiSeq 2500, with 75 bp paired-end reads. Two B6N and Stag3−/− libraries were sequenced twice in two independent runs to get sufficient reads.
H3K4me3 ChIP-seq protocol
H3K4me3 ChIP was performed using male germ cells isolated from a 14dpp, Prdm9EK/EK mouse as previously described [19], using a commercially available polyclonal α-H3K4me3 antibody (EMD Millipore cat#07-473). DNA samples were sequenced via standard protocols on an Illumina NextSeq, with 75bp reads.
Testes extract preparation and western blotting
Crude testis extract was prepared by the following protocol: 12dpp male mice were euthanized and the testes removed and de-tunicated; testes from each mouse were placed on ice in 125ul RIPA extraction buffer (10mM Tris HCl pH=8.0, 1mM EDTA, 1mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140mM NaCl, 1X PIC, 1mM phenylmethanesulfonyl fluoride (PMSF), 1mM dithiothreitol (DTT), 1000 U Turbonuclease (Accelagen cat# N0103M)). Testes were homogenized for 10sec with an electronic pestle grinder, incubated 5min on the ice, and homogenized a second time for 10 sec. The homogenate was incubated on ice for 30min, vortexing every 5min., and then centrifuged at 10,000 × g, 4°C. The supernatant was removed to a new tube and the pellet discarded. Total protein in the supernatant was quantified using a standard Bradford assay, and stored at −20°C until electrophoresis.
50ug of each extract was electrophoresed on a pre-cast 4-15% gradient SDS-PAGE gel (Bio-Rad Cat# 4561084) at 100V. Transfer to nitrocellulose was done overnight at 22V, 4°C, in transfer buffer (25mM Tris, 192mM glycine, 20% methanol). After transfer, the blot was blocked for 1h at RT with gentle rotation in 20ml blocking buffer (Immobilon Block – CH Chemiluminescent Blocker; EMD Millipore Cat# WBAVDCH01). The blot was then incubated in the anti-PRDM9 primary antibody, diluted 1:2500 in blocking buffer, for 1h at RT with gentle rotation. The blot was then washed 3 times, 5min per wash, in 1X Tris-buffered saline with Tween (TBST; 10mM Tris HCl pH=8.0, 150mM NaCl, 0.3% Tween-20) at RT with gentle rotation. The blot was then incubated in 20ml HRP-conjugated anti-guinea pig secondary antibody (EMD Millipore Cat# AP193P), diluted 1:5000 in blocking buffer, for 1h at RT with gentle rotation. The blot was then washed twice, 15min per wash, at RT with gentle rotation. After the final wash, the blot was incubated 1min in SuperSignal West Femto chemiluminescent substrate (Thermo Scientific Cat# 34095) and imaged on a SynGene G: Box chemiluminescent imaging system.
For the loading control, the blot was stripped using Restore Western Blot Stripping Buffer (Thermo Scientific Cat# 21059) for 10min at RT with gentle rotation, then washed twice in TBST, 15min per wash, under the same conditions. The blot was then reblocked and re-probed using the procedure described above, with a 20ml mouse anti-β-tubulin primary antibody (Sigma Cat# T4026) diluted 1:10,000, and 20ml HRP-conjugated anti-mouse secondary antibody (Bio-Rad Cat# 170-6516) diluted 1:20,000.
SPO11-oligonucleotide complex estimation assay
To measure levels of SPO11-oligonucleotide complexes in wildtype and mutant spermatocytes, we carried out SPO11 immunoprecipitations and SPO11-oligonucleotide detection in 13 dpp testes as per published protocol [59-61]. This assay measures labeled oligonucleotide complexes covalently-linked with SPO11 protein generated during DSB processing. Briefly, SPO11 oligos were purified from 13-dpp juvenile mice. Testes were decapsulated and homogenized with prechilled plastic pestles in lysis buffer (1% Triton X-100, 25 mM HEPES-NaOH pH 7.4, 5 mM EDTA) containing protease inhibitors (Roche), cleared by ultracentrifugation for 25 min at 4°C, 100,000 rpm (twice), supernatant was transferred to low binding Eppendorf tubes (Fisher). Supernatants were incubated with mouse anti-SPO11-180 antibody (3 μg per immunoprecipitation (IP) sample) for 1 hr 4°C end-over-end rotation before adding protein A-agarose beads (Roche) and incubated for an additional 3 hr at 4°C. Beads containing SPO11 –oligonucleotide complexes were washed 3 times with IP buffer (1% Triton X-100, 150 mM NaCl, 15 mM Tris-HCl pH 8.0,1 Mm EDTA). SPO11 – oligonucleotide complexes were eluted in 2xSDS buffer (100 mM Tris-HCl, pH 6.8, 4%SDS, 20% glycerol, 10% beta-mercaptoethanol) for 4 min at 95°C. Washing steps were consolidated into eluates to minimize loss. Combined IP eluate and washes were diluted with 5 volumes of IP buffer and the second round of IP (IP2) was performed. 3 μg Anti-Mouse SPO11-180 antibody was added (per sample) for 1 hr at 4°C with end-over-end rotation, after which protein A-agarose beads were added and incubated at 4°C overnight for further mixing. SPO11 -oligonucleo tide complexes from IP2 were washed 3x with IP wash buffer and 2x with 1 × NEB4 before radiolabeling with terminal deoxynucleotidyl transferase (Thermo Fisher Scientific) and [α32P] CTP (PerkinElmer, Inc.) in NEB buffer 4 for 1 hr at 37°C in a thermom ixer at 300 rpm. Beads were washed 5x with IP buffer before elution with 2× with 2x SDS buffer. Proteins were separated by ready to use 7.5% SDS-PAGE gels (Biorad), 125 V for 90 min. Proteins were transferred using iBLOT Gel transfer stacks system with PVDF membrane (Invitrogen) via semi-dry transfer using iBLOT transfer system (Invitrogen). The experiment was repeated twice.
Quantification and Statistical Analysis
Statistical details for each figure can be found in the figure legends, results and/or supplementary data. Statistical tests were performed using GraphPad Prism version 7.0 and R statistical packages. Details regarding other software used are detailed in Key Resource Table.
Foci and cellular quantification and analysis.
The statistical significance of differences in DMC1, RAD51 or MEI4 counts among cells and genotypes was assessed by a Mann-Whitney U test with Tukey's correction for multiple testing. Multiple biological replicates (at least 3) of each genotype were analyzed for cellular phenotypes. For TUNEL apoptosis assays at least 15 tubules were counted for TUNEL-positive cells using Fiji-Image J software. The images were captured by a Zeiss Imager Z2 microscope. Images were processed using Adobe Photoshop (Adobe Systems). We calculated statistical significance by Mann-Whitney U test with Tukey's correction for multiple testing.
Protein and SPO11-Oligo band quantification and analysis
Western blot images were processed and quantified using Fiji-Image J software. SPO11-Oligo radiolabelled blots were detected and quantified after 48 hour exposure with Fuji phosphor screens, scanned and quantified in ImageJ. We calculated statistical significance using Welch's t-test.
DMC1 ChIP-seq analysis
Fastq files for paired-end sequenced DMC1 samples were processed with pipelines similar to Brick et al. [43]. In brief, Fastq files for paired-end sequenced DMC1 samples were trimmed using Trimmomatic (v0.32) and subsequently parsed for detection and selection of paired reads having homology at the 5' and 3' ends as established by protocols for single strand DNA enrichment. These selected paired-end reads represent the detectable single-stranded DNA reads. The micro homology sequence was removed from the read sequences and they were subsequently aligned to mm10 genome using BWA (0.5.10-tpx). Bam files from this alignment were parsed for detection and subsequent selection of single-ended reads containing true genomic sequence versus fill-in sequence at the micro-homologous region. Strandedness could be detected by the alignment flags and no biases were found between strands. Fastq files were subsequently created containing only the Watson and Crick strand sequences and subsequently processed utilizing non-paired end alignment pipelines. Peaks were called utilizing MACS 2.0 with the read extension parameter set to 800 in order to force pileups. 2,845,239, 2,938,318 and 5,108,836 aligned DMC1 reads in B6N, 8,794,169 and 6,394,126 aligned DMC1 reads in Stag3−/− spermatocytes, 5,008,222 and 6, 480,718 aligned DMC1 reads in Rec8−/− spermatocytes, 5,878,240 aligned DMC1 reads in Stag3−/− Rec8−/− spermatocytes were obtained from each library. The DMC1 activity was normalized to fragments per million (FPM). Peak calling was performed using MACS (v.2.0) with standard treatment (ChIP) and control (input) samples with an FDR value 0.01. The same Prdm9Dom2 hotspot and H3K9me3 domain locations were used as in H3K4me3 ChIP-seq analysis [19] and PRDM9 affinity-seq analysis [42]. DMC1 peaks shared by B6N, Stag3−/− Rec8−/− and Stag3−/− Rec8−/− spermatocytes, inside and outside known hotspots, or open/closed chromatin regions, were determined using bedtools (v2.22.0) intersect. Wilcoxon rank sum test was used to calculate the statistical significance. We used a generalized linear model analysis per hotspot location to assess significant differences between wildtype and mutants per hotspot. The analysis was set up to determine if the genotype is a significant (p-value < 0.05) factor in predicting read count. If genotype p.-value < 0.05, the analysis continues on to determine the least square means for each of the genotypes and the standard error. Additionally, all pairwise contrasts were performed between genotypes, and the difference and a single standard error were calculated. We used Analysis of Variance (ANOVA) with Tukey's correction for multiple testing to determine the significance.
H3K4me3 ChIP-seq analysis
Read sequence was trimmed using Trimmomatic. Sequence data were aligned to the mouse mm10 genome using BWA v. 0.5.10-tpx, and reads which failed to align to unique positions in the genome were discarded, resulting in 34,829,900 aligned reads. For comparison, peaks were also called from previously reported H3K4me3 ChIP data from a 14dpp C57BL/6J mouse [19], which was re-aligned to mm10 using the same parameters as with the Prdm9EK/EK mouse (47,576,722 aligned reads; GEO accession # GSE52628). H3K4me3 peaks were called using MACS (v 2.0), with default parameters and an FDR value of 0.01. Peaks were called from a single biological replicate for both B6 and Prdm9EK/EK, using a C57BL/6J input DNA sample as a control. Each peak was designated as a hotspot, using Bedtools (v. 2.22.0) Intersect, if it showed at least 20% overlap with one of our set of 13,355 known Prdm9Dom2 hotspots (Intersect options: -wb -f 0.20 -r).
Data and Software Availability
DMC1-ChIP and H3K4me3 ChIP seq data are available at NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) under accession number GSE112110 Software used in this paper is freely available and tabulated in Key Resource Table.
Supplementary Material
Data S1. Statistical analysis of DMC1 foci number and fold change in cohesin mutant spermatocytes in the presence, absence, and modulation of PRDM9 function. Related to Figure 2.
Significant changes are highlighted in yellow.
Data S2. Statistical analysis of DSB activity using a linear model for 5,129 DSB sites at PRDM9Dom2 hotspots common between B6 (blue columns), Stag3−/− (pink columns) and Rec8−/− (orange columns) spermatocytes. Related to Figure 3.
The analysis was set up to determine if the genotype is a significant (p-value < 0.05) factor in predicting read count. Read counts were log (value+1) transformed to achieve normality. If genotype p-value < 0.05, the analysis continues to determine the least square means for each of the genotypes and the standard error. FDR-adjusted p-values are Tukey-adjusted for multiple test correction. Significant changes are highlighted in yellow. NA=not significant.
Data S3. Statistical analysis of the number of MEI4 foci and fold change in cohesin mutant spermatocytes in the presence, absence, and modulation of PRDM9 function. Related to Figure 4.
Significant changes are highlighted in yellow.
Data S4. Statistical analysis of the number of DMC1 foci and fold change in Stag3−/− spermatocytes with modulation of SPO11 activity. Related to Figure 6.
Significant changes are highlighted in yellow.
Methods S1. Sequence information regarding the position and nature of the Prdm9EK / EK point mutation in the SET domain of the Prdm9 gene and PRDM9 protein. Related to Figures 2 and S5, and STAR Methods.
The mutation sites are highlighted in yellow.
Highlights.
PRDM9-cohesin interactions promote efficient DSB formation at recombination sites.
Cohesins promote axis localization of pre-DSB recombinosome proteins.
Cohesin proteins facilitate SPO11 function.
Cohesin deficiency affects DSBs at both PRDM9-dependent and -independent sites.
Acknowledgments
We thank three anonymous reviewers for their helpful comments. Further, we thank Drs. Ewelina Bolcun-Filas, Beth Dumont and Ken Paigen for discussion throughout this work and for their thoughtful comments on the manuscript. We also thank all members of the Handel, Paigen and Petkov labs for insightful discussions and suggestions. We are appreciative of The Jackson Laboratory Scientific Services, including the Histology core, the Microscopy core, Genome Technologies, Genetic Engineering Technology, Mouse Resources, and Computational Sciences (specifically Vivek Philip and Tim Stearns) for their expertise and help during this project. We thank the Knockout mouse project (KOMP2) at the Jackson Laboratory for providing mice. We thank Drs. Michiel Boekhout, Kevin Brick, Bernard de Massy, Yasuhiro Fujiwara, Rolf Jessberger, Scott Keeney, Attila Toth, and Yoshinori Watanabe for sharing their antibody resources and help during this project. This work was supported by grants from the NIH: P01 GM99640 (MAH) and P30 CA034196 to the Jackson Laboratory for scientific services. Dr. Tanmoy Bhattacharyya was supported by a JAX Scholar award (19042802-15-3); Dr. Natalie Powers and Alexander D Fine were supported by NICHD T32 Training Program in Developmental Genetics (T32 HD007065 to the Jackson Laboratory). The content is solely the responsibility of the authors and does not necessarily represent official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Llano E, Herran Y, Garcia-Tunon I, Gutierrez-Caballero C, de Alava E, Barbero JL, Schimenti J, de Rooij DG, Sanchez-Martin M, and Pendas AM (2012). Meiotic cohesin complexes are essential for the formation of the axial element in mice. J Cell Biol 197, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pelttari J, Hoja MR, Yuan L, Liu JG, Brundell E, Moens P, Santucci-Darmanin S, Jessberger R, Barbero JL, Heyting C, et al. (2001). A meiotic chromosomal core consisting of cohesin complex proteins recruits DNA recombination proteins and promotes synapsis in the absence of an axial element in mammalian meiotic cells. Mol Cell Biol 21, 5667–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee J, and Hirano T (2011). RAD21L, a novel cohesin subunit implicated in linking homologous chromosomes in mammalian meiosis. J Cell Biol 192, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biswas U, Hempel K, Llano E, Pendas A, and Jessberger R (2016). Distinct roles of meiosis-Specific cohesin complexes in mammalian spermatogenesis. PLoS Genet 12, e1006389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eijpe M, Offenberg H, Jessberger R, Revenkova E, and Heyting C (2003). Meiotic cohesin REC8 marks the axial elements of rat synaptonemal complexes before cohesins SMC1β and SMC3. J Cell Biol 160, 657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hopkins J, Hwang G, Jacob J, Sapp N, Bedigian R, Oka K, Overbeek P, Murray S, and Jordan PW (2014). Meiosis-specific cohesin component, Stag3 is essential for maintaining centromere chromatid cohesion, and required for DNA repair and synapsis between homologous chromosomes. PLoS Genet 10, e1004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winters T, McNicoll F, and Jessberger R (2014). Meiotic cohesin STAG3 is required for chromosome axis formation and sister chromatid cohesion. EMBO J 33, 1256–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mannini L, Cucco F, Quarantotti V, Amato C, Tinti M, Tana L, Frattini A, Delia D, Krantz ID, Jessberger R, et al. (2015). SMC1B is present in mammalian somatic cells and interacts with mitotic cohesin proteins. Sci Rep 5, 18472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Revenkova E, Eijpe M, Heyting C, Gross B, and Jessberger R (2001). Novel meiosis-specific isoform of mammalian SMC1. Mol Cell Biol 21, 6984–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Revenkova E, Eijpe M, Heyting C, Hodges CA, Hunt PA, Liebe B, Scherthan H, and Jessberger R (2004). Cohesin SMC1β is required for meiotic chromosome dynamics, sister chromatid cohesion and DNA recombination. Nat Cell Biol 6, 555–562. [DOI] [PubMed] [Google Scholar]
- 11.Xu H, Beasley MD, Warren WD, van der Horst GT, and McKay MJ (2005). Absence of mouse REC8 cohesin promotes synapsis of sister chromatids in meiosis. Dev Cell 8, 949–961. [DOI] [PubMed] [Google Scholar]
- 12.Novak I, Wang H, Revenkova E, Jessberger R, Scherthan H, and Hoog C (2008). Cohesin Smc1β determines meiotic chromatin axis loop organization. J Cell Biol 180, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukuda T, Fukuda N, Agostinho A, Hernandez-Hernandez A, Kouznetsova A, and Hoog C (2014). STAG3-mediated stabilization of REC8 cohesin complexes promotes chromosome synapsis during meiosis. EMBO J 33, 1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llano E, Gomez HL, Garcia-Tunon I, Sanchez-Martin M, Caburet S, Barbero JL, Schimenti JC, Veitia RA, and Pendas AM (2014). STAG3 is a strong candidate gene for male infertility. Hum Mol Genet 23, 3421–3431. [DOI] [PubMed] [Google Scholar]
- 15.Ward A, Hopkins J, McKay M, Murray S, and Jordan PW (2016). Genetic interactions between the meiosis-specific cohesin components, STAG3, REC8, and RAD21L. G3 (Bethesda) 6, 1713–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G, and Donnelly P (2010). Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327, 876–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, and de Massy B (2010). PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parvanov ED, Petkov PM, and Paigen K (2010). Prdm9 controls activation of mammalian recombination hotspots. Science 327, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker CL, Walker M, Kajita S, Petkov PM, and Paigen K (2014). PRDM9 binding organizes hotspot nucleosomes and limits Holliday junction migration. Genome Res 24, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker CL, Petkova P, Walker M, Flachs P, Mihola O, Trachtulec Z, Petkov PM, and Paigen K (2015). Multimer formation explains allelic suppression of PRDM9 recombination hotspots. PLoS Genet 11, e1005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powers NR, Parvanov ED, Baker CL, Walker M, Petkov PM, and Paigen K (2016). The meiotic recombination activator PRDM9 trimethylates both H3K36 and H3K4 at recombination hotspots in vivo. PLoS Genet 12, e1006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romanienko PJ, and Camerini-Otero RD (2000). The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell 6, 975–987. [DOI] [PubMed] [Google Scholar]
- 23.Baudat F, Manova K, Yuen JP, Jasin M, and Keeney S (2000). Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell 6, 989–998. [DOI] [PubMed] [Google Scholar]
- 24.Mahadevaiah SK, Turner JM, Baudat F, Rogakou EP, de Boer P, Blanco-Rodriguez J, Jasin M, Keeney S, Bonner WM, and Burgoyne PS (2001). Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet 27, 271–276. [DOI] [PubMed] [Google Scholar]
- 25.Robert T, Nore A, Brun C, Maffre C, Crimi B, Bourbon HM, and de Massy B (2016). The TopoVIB-Like protein family is required for meiotic DNA double-strand break formation. Science 351, 943–949. [DOI] [PubMed] [Google Scholar]
- 26.Brick K, Smagulova F, Khil P, Camerini-Otero RD, and Petukhova GV (2012). Genetic recombination is directed away from functional genomic elements in mice. Nature 485, 642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diagouraga B, Clement JAJ, Duret L, Kadlec J, de Massy B, and Baudat F (2018). PRDM9 methyltransferase activity Is essential for meiotic DNA double-strand break formation at its binding sites. Mol Cell 69, 853–865 e856. [DOI] [PubMed] [Google Scholar]
- 28.Kumar R, Bourbon HM, and de Massy B (2010). Functional conservation of Mei4 for meiotic DNA double-strand break formation from yeasts to mice. Genes Dev 24, 1266–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar R, Ghyselinck N, Ishiguro K, Watanabe Y, Kouznetsova A, Hoog C, Strong E, Schimenti J, Daniel K, Toth A, et al. (2015). MEI4 - a central player in the regulation of meiotic DNA double-strand break formation in the mouse. J Cell Sci 128, 1800–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panizza S, Mendoza MA, Berlinger M, Huang L, Nicolas A, Shirahige K, and Klein F (2011). Spo11 -accessory proteins link double-strand break sites to the chromosome axis in early meiotic recombination. Cell 146, 372–383. [DOI] [PubMed] [Google Scholar]
- 31.Sun X, Huang L, Markowitz TE, Blitzblau HG, Chen D, Klein F, and Hochwagen A (2015). Transcription dynamically patterns the meiotic chromosome-axis interface. Elife 4, eLife.07424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phadnis N, Cipak L, Polakova S, Hyppa RW, Cipakova I, Anrather D, Karvaiova L, Mechtler K, Smith GR, and Gregan J (2015). Casein Kinase 1 and Phosphorylation of cohesin subunit Rec11 (SA3) promote meiotic recombination through linear element formation. PLoS Genet 11, e1005225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakuno T, and Watanabe Y (2015). Phosphorylation of cohesin Rec11/SA3 by casein kinase 1 promotes homologous recombination by assembling the meiotic chromosome axis. Dev Cell 32, 220–230. [DOI] [PubMed] [Google Scholar]
- 34.Ellermeier C, and Smith GR (2005). Cohesins are required for meiotic DNA breakage and recombination in Schizosaccharomyces pombe. Proc Natl Acad Sci U S A 102, 10952–10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stanzione M, Baumann M, Papanikos F, Dereli I, Lange J, Ramlal A, Trankner D, Shibuya H, de Massy B, Watanabe Y, et al. (2016). Meiotic DNA break formation requires the unsynapsed chromosome axis-binding protein IHO1 (CCDC36) in mice. Nat Cell Biol 18, 1208–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parvanov ED, Tian H, Billings T, Saxl RL, Spruce C, Aithal R, Krejci L, Paigen K, and Petkov PM (2017). PRDM9 interactions with other proteins provide a link between recombination hotspots and the chromosomal axis in meiosis. Mol Biol Cell 28, 488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun F, Fujiwara Y, Reinholdt LG, Hu J, Saxl RL, Baker CL, Petkov PM, Paigen K, and Handel MA (2015). Nuclear localization of PRDM9 and its role in meiotic chromatin modifications and homologous synapsis. Chromosoma 124, 397–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian H, Billings T, Walker M, Petkova PM, Baker CL, and Petkov PM (2018). EWSR1 affects PRDM9-dependent histone 3 methylation and provides a link between recombination hotspots and the chromosome axis. bioRxiv 10.1101/325282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayashi K, Yoshida K, and Matsui Y (2005). A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 438, 374–378. [DOI] [PubMed] [Google Scholar]
- 40.Davies B, Hatton E, Altemose N, Hussin JG, Pratto F, Zhang G, Hinch AG, Moralli D, Biggs D, Diaz R, et al. (2016). Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smagulova F, Brick K, Pu Y, Camerini-Otero RD, and Petukhova GV (2016). The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev 30, 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker M, Billings T, Baker CL, Powers N, Tian H, Saxl RL, Choi K, Hibbs MA, Carter GW, Handel MA, et al. (2015). Affinity-seq detects genome-wide PRDM9 binding sites and reveals the impact of prior chromatin modifications on mammalian recombination hotspot usage. Epigenetics Chromatin 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brick K, Thibault-Sennett S, Smagulova F, Lam KG, Pu Y, Pratto F, Camerini-Otero RD, and Petukhova GV (2018). Extensive sex differences at the initiation of genetic recombination. Nature 561, 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukuda T, Daniel K, Wojtasz L, Toth A, and Hoog C (2010). A novel mammalian HORMA domain-containing protein, HORMAD1, preferentially associates with unsynapsed meiotic chromosomes. Exp Cell Res 316, 158–171. [DOI] [PubMed] [Google Scholar]
- 45.Bellani MA, Romanienko PJ, Cairatti DA, and Camerini-Otero RD (2005). SPO11 is required for sex-body formation, and Spo11 heterozygosity rescues the prophase arrest of Atm−/− spermatocytes. J Cell Sci 118, 3233–3245. [DOI] [PubMed] [Google Scholar]
- 46.Pan J, and Keeney S (2009). Detection of SPO11-oligonucleotide complexes from mouse testes. Methods Mol Biol 557, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biswas U, Wetzker C, Lange J, Christodoulou EG, Seifert M, Beyer A, and Jessberger R (2013). Meiotic cohesin SMC1β provides prophase I centromeric cohesion and is required for multiple synapsis-associated functions. PLoS Genet 9, e1003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biswas U, Stevense M, and Jessberger R (2018). SMC1a substitutes for many meiotic functions of SMC1β but cannot protect telomeres from damage. Curr Biol 28, 249–261 e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Imai Y, Baudat F, Taillepierre M, Stanzione M, Toth A, and de Massy B (2017). The PRDM9 KRAB domain is required for meiosis and involved in protein interactions. Chromosoma 126, 681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Massy B (2013). Spp1 links sites of meiotic DNA double-strand breaks to chromosome axes. Mol Cell 49, 3–5. [DOI] [PubMed] [Google Scholar]
- 51.Acquaviva L, Szekvolgyi L, Dichtl B, Dichtl BS, de La Roche Saint Andre C, Nicolas A, and Geli V (2013). The COMPASS subunit Spp1 links histone methylation to initiation of meiotic recombination. Science 339, 215–218. [DOI] [PubMed] [Google Scholar]
- 52.Sommermeyer V, Beneut C, Chaplais E, Serrentino ME, and Borde V (2013). Spp1, a member of the Set1 Complex, promotes meiotic DSB formation in promoters by tethering histone H3K4 methylation sites to chromosome axes. Mol Cell 49, 43–54. [DOI] [PubMed] [Google Scholar]
- 53.Munoz-Fuentes V, Di Rienzo A, and Vila C (2011). Prdm9, a major determinant of meiotic recombination hotspots, is not functional in dogs and their wild relatives, wolves and coyotes. PLoS One 6, e25498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Auton A, Rui Li Y, Kidd J, Oliveira K, Nadel J, Holloway JK, Hayward JJ, Cohen PE, Greally JM, Wang J, et al. (2013). Genetic recombination is targeted towards gene promoter regions in dogs. PLoS Genet 9, e1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhattacharyya T, Gregorova S, Mihola O, Anger M, Sebestova J, Denny P, Simecek P, and Forejt J (2012). Mechanistic basis of infertility of mouse intersubspecific hybrids. Proc Natl Acad Sci U S A 110, E468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kauppi L, Barchi M, Baudat F, Romanienko PJ, Keeney S, and Jasin M (2011). Distinct properties of the XY pseudoautosomal region crucial for male meiosis. Science 331, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khil PP, Smagulova F, Brick KM, Camerini-Otero RD, and Petukhova GV (2012). Sensitive mapping of recombination hotspots using sequencing-based detection of ssDNA. Genome Res 22, 957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neale MJ, Pan J, and Keeney S (2005). Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436, 1053–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau HG, Tischfield SE, Zhu X, Neale MJ, Jasin M, Socci ND, et al. (2011). A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 144, 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lange J, Pan J, Cole F, Thelen MP, Jasin M, and Keeney S (2011). ATM controls meiotic double-strand-break formation. Nature 479, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ishiguro K, Kim J, Fujiyama-Nakamura S, Kato S, and Watanabe Y (2011). A new meiosis-specific cohesin complex implicated in the cohesin code for homologous pairing. EMBO Rep 12, 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eaker S, Pyle A, Cobb J, and Handel MA (2001). Evidence for meiotic spindle checkpoint from analysis of spermatocytes from Robertsonian-chromosome heterozygous mice. J Cell Sci 114, 2953–2965. [DOI] [PubMed] [Google Scholar]
- 64.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jee J, Rozowsky J, Yip KY, Lochovsky L, Bjornson R, Zhong G, Zhang Z, Fu Y, Wang J, Weng Z, et al. (2011). ACT: aggregation and correlation toolbox for analyses of genome tracks. Bioinformatics 27, 1152–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Statistical analysis of DMC1 foci number and fold change in cohesin mutant spermatocytes in the presence, absence, and modulation of PRDM9 function. Related to Figure 2.
Significant changes are highlighted in yellow.
Data S2. Statistical analysis of DSB activity using a linear model for 5,129 DSB sites at PRDM9Dom2 hotspots common between B6 (blue columns), Stag3−/− (pink columns) and Rec8−/− (orange columns) spermatocytes. Related to Figure 3.
The analysis was set up to determine if the genotype is a significant (p-value < 0.05) factor in predicting read count. Read counts were log (value+1) transformed to achieve normality. If genotype p-value < 0.05, the analysis continues to determine the least square means for each of the genotypes and the standard error. FDR-adjusted p-values are Tukey-adjusted for multiple test correction. Significant changes are highlighted in yellow. NA=not significant.
Data S3. Statistical analysis of the number of MEI4 foci and fold change in cohesin mutant spermatocytes in the presence, absence, and modulation of PRDM9 function. Related to Figure 4.
Significant changes are highlighted in yellow.
Data S4. Statistical analysis of the number of DMC1 foci and fold change in Stag3−/− spermatocytes with modulation of SPO11 activity. Related to Figure 6.
Significant changes are highlighted in yellow.
Methods S1. Sequence information regarding the position and nature of the Prdm9EK / EK point mutation in the SET domain of the Prdm9 gene and PRDM9 protein. Related to Figures 2 and S5, and STAR Methods.
The mutation sites are highlighted in yellow.
Data Availability Statement
DMC1-ChIP and H3K4me3 ChIP seq data are available at NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) under accession number GSE112110 Software used in this paper is freely available and tabulated in Key Resource Table.