

Abstract
Virtual molecular catalogs have limited utility if member compounds are (1) difficult to synthesize or (2) unlikely to have biological activity. The Distributed Drug Discovery (D3) program addresses the synthesis challenge by providing scientists with a free virtual D3 catalog of 73,024 easy-to-synthesize N-acyl unnatural α-amino acids, their methyl esters, and primary amides. The remaining challenge is to document and exploit the bioactivity potential of these compounds. In the current work, a search process is described that retrospectively identifies all virtual D3 compounds classified as bioactive hits in PubChem-cataloged experimental assays. The results provide insight into the broad range of drug-target classes amenable to inhibition and/or agonism by D3-accessible molecules. To encourage computer-aided drug discovery centered on these compounds, a publicly available virtual database of D3 molecules prepared for use with popular computer docking programs is also presented.
Keywords: unnatural amino acid derivatives, peptidomimetic, structure-based drug design, small molecule diversity, molecular modeling, virtual screening, in silico chemoinformatics, synthetic methods
Graphical Abstract
The Distributed Drug Discovery (D3) program provides scientists with a free virtual catalog of 73,024 easy-to-synthesize N-acyl unnatural α-amino acids, their methyl esters, and primary amides. In the current work, we identify all virtual D3 compounds classified as bioactive hits in PubChem-cataloged experimental assays. The results 1) provide insight into the broad range of drug-target classes amenable to modulation by D3-accessible molecules and 2) suggest future avenues for virtual-screening/ drug-discovery projects.
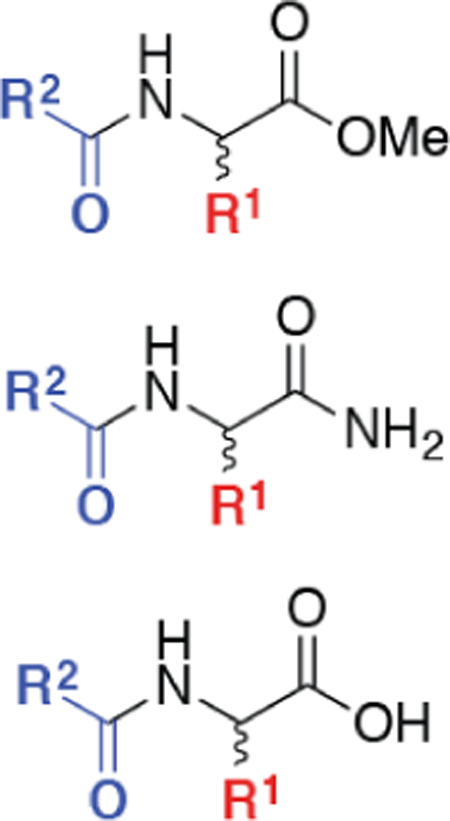
Virtual compound catalogs [1–4]/libraries [5–9] can be useful sources of candidate molecules for drug discovery. However, two major factors often limit their utility: (1) member compounds and analogs may be difficult to synthesize [10] or (2) there is no inherent probability of biological activity. To the first issue, we have created large virtual catalogs of N-acyl α-amino acids and their methyl ester and primary amide derivatives based on reproducible procedures and demonstrated student syntheses of individual catalog members 1 [2, 4] 2 [3], and 3 [11] (presented here for the first time, Figure 1).
Figure 1.
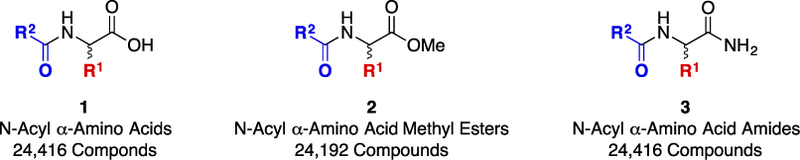
Generic structures of the 73,024 unnatural and natural N-acyl α-amino acid derivatives in the D3 virtual catalog.
The purpose of the current work is to address the second issue by assessing the existing or potential biological activity of these compounds through a chemoinformatics analysis, and providing an example of D3-centered computer-aided drug discovery. The pharmacological activity of virtual D3 molecules can be demonstrated in two ways: prospectively by synthesizing and testing catalog members in biological assays, or retrospectively by searching the literature and other databases for previously documented activity (Figure SI-1).
The prospective approach was first pursued as an initial proof of concept (Figure SI-1, left arrow). A small number of D3 compounds 1 were synthesized and submitted to the NIH Molecular Libraries Initiative for testing in established high-throughput screens [12], resulting in hits in disparate NIH bioassays. Further investigation of one of these hits (CID 971933, AID 132162) identified a published analog, also present in the D3 virtual catalog, with documented biologically activity. That analog had been key to the development of nateglinide, a marketed drug to treat diabetes mellitus type 2 [13, 14].
Inspired by this suggestive finding, a retrospective search (Figure SI-1, right arrow) was undertaken to identify other examples of virtual catalog members with documented biological activity. In this article, we identify all enumerated D3 unnatural and natural N-acyl α-amino acids (1), N-acyl α-amino acid methyl esters (2), and N-acyl α-amino acid amides (3) that are listed among the active compounds in the PubChem database of >1 million bioassays (AID, Assay ID). By unnatural, we mean compounds with D (normally R) stereochemistry and/or a non-proteinogenic side chain, vs. natural compounds with L (normally S) stereochemistry and/or a proteinogenic side chain. Members of this list are termed “D3/PubChem actives.” The hits from this search give credibility to the prospective drug-discovery potential of the D3 catalog and suggest areas for future discovery research. Within the D3/PubChem actives list, relevant PubChem screens that specifically target single proteins are also categorized (Figure SI-2). These protein targets then offer the opportunity to construct structure-based computational models for predictive analysis of D3 virtual catalogs.
In addition to this chemoinformatics analysis, a new publicly available database of three-dimensional D3 structures for use in virtual-screening projects, available free of charge from http://durrantlab.com/liglib/iupui/d3_docking/ (accessed February 27, 2017), is described. To demonstrate its utility, this database was used in a virtual screen against peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1), a potential target for future cancer chemotherapies [15]. This analysis predicted the biological activity of known compounds and suggested molecules with the potential for even greater activity. The methodology presented should assist others in the field who similarly wish to harness the power of enumerated compound catalogs in drug discovery.
1 |. Materials and Methods
1.1 |. Enumeration protocol
Virtual catalogs of 73,024 unnatural or natural N-acyl α-amino acids (1; 24,416 compounds), N-acyl α-amino acid esters (2; 24,192 compounds), and N-acyl α-amino acid amides (3; 24,416 compounds), called the “D3 N-Acyl α-Amino Acid,” “D3 N-Acyl α-Amino Acid Ester,” and “D3 α-N-Acyl Amino Acid Amide” sets, respectively, were enumerated from diversity sets of commercially available electrophiles and carboxylic acids (100 members each) [2] using CombiChem for Excel [16] or, more recently, ChemAxon [17]. Issues of stereochemistry were addressed in this enumeration to yield compounds with two to eight stereoisomers (see Supporting Information, Figure SI-4, and associated text for a detailed discussion).
1.2 |. PubChem/D3 searching protocol
A series of Boolean substructure searches was used to identify all D3-like N-acyl α-amino acid derivatives (4, 5, or 6) and N-acyl dipeptide derivatives (7, 8, or 9) present in PubChem (i.e., “PC-CID” compounds in Supporting Information Figures SI-3, SI-4, SI-5, and SI-6). See the Supporting Information for full Boolean-search details.
1.3 |. Calculating molecular properties
Schrodinger’s Maestro Suite [18] was used to calculate the chemical properties of the compounds in the D3 N-Acyl α-Amino Acid set, NCI Diversity Set III [19], and ChemBridge Diversity CombiSet [20, 21] libraries, which contain 24,416, 1,597, and 30,000 compounds, respectively. One electrically neutral, three-dimensional structure of each compound was created using Schrodinger’s LigPrep module [22], as required to calculate molecular properties. Molecular geometries were optimized using the OPLS_2005 force field [23], hydrogen atoms were added or removed as required to achieve electrical neutrality (e.g., carboxylates were protonated, protonated amines were deprotonated, etc.), salts were removed, only the most probable tautomer was considered, and one low-energy conformation was generated for each ring. The D3 input files explicitly specify the stereochemistry at each chiral center; for compounds from other virtual catalogs that lack specified chiralities, a single chiral molecule was created per input structure, chosen from among the many possible enantiomers that could have been enumerated. Once these 3D structures had been created, Schrodinger’s QikProp module [24] was used to calculate the molecular weight, predicted LogP, and number of hydrogen-bond donors and acceptors for each compound.
Occasionally, the Schrodinger software was unable to process a given molecule. In the end, molecular properties were generated for 92%, 96%, and 99% of the D3 N-Acyl α-Amino Acid Set, the NCI Diversity Set III, and ChemBridge Diversity CombiSet libraries, respectively. The relevant figures and statistics were generated using these compounds.
1.4 |. Preparing a virtual catalog of three-dimensional D3 structures for docking
Schrodinger’s LigPrep module was used to generate 3D structures of all D3 compounds for use in docking studies. Unlike the virtual library of 3D compounds generated to calculate molecular properties, the docking library should account for the multiple ionic, tautomeric, and conformational states potentially associated with each compound under physiological conditions. To this end, Schrodinger’s Epik algorithm [25, 26] was used to determine all protonation states at pH values ranging from 5.0 to 9.0. All tautomers were enumerated, and one low-energy ring conformation was considered per compound. After salt molecules had been removed, the 3D geometry of each compound was optimized using the OPLS_2005 force field [23]. The chiralities explicitly specified in the D3 database were retained. When alternate protonation and tautomeric states were considered, there were ultimately 27,220, 26,794, and 27,036 N-acyl α-amino acid (1), N-acyl α-amino acid ester (2), and N-acyl α-amino acid amide (3) derivatives, respectively. These structures were saved in the SDF format, which is compatible with computer docking programs like Schrodinger’s Glide [27, 28].
To facilitate docking with programs such as AutoDock [29] and Vina [30], each of the structures was also saved in the PDBQT format. The SDF to PDB conversions were performed using Open Babel [31]. The PDB to PDBQT conversions, which involved computing Gasteiger partial charges for each atom [32], assigning AutoDock atom types, and merging non-polar hydrogen atoms with their parent heteroatoms, were performed using AutoDockTools [29]. Compound torsions were assigned using AutoDock’s AutoTors utility to allow full molecular flexibility during docking.
1.5 |. Benchmark virtual screen
AutoDock Vina was used to dock the entire 24,416-member D3 N-acyl α-amino acid set into the 3KAI Pin1 structure [33]. The ligand PDBQT files were taken from the database described in the previous paragraph. The protein was processed using Schrödinger’s Protein Preparation Wizard [34] to add hydrogen atoms, optimize hydrogen-bond networks, relax the structure, etc. The processed structure was then converted to the PDBQT format using AutoDockTools [29]. Compounds were docked into a cube (20 Å x 20 Å x 20 Å) centered on the Pin1 active site, using the Rocce cluster provided by the National Biomedical Computation Resource. Default Vina parameters were used.
1.6 |. Combinatorial chemistry and the D3 initiative
In theory, the number of drug-like compounds exceeds 1060 [35]. Any hope of exploring all of “drug space,” whether by computational or experimental means, must be abandoned from the outset. Given that researchers are restricted to a limited subspace, it is prudent to focus on small molecules that can be readily synthesized using simple and robust synthetic procedures [1]. These requirements can be met by designing drug-discovery projects around readily accessible small molecules derived using carefully designed synthetic methodologies. Enumerated virtual combinatorial catalogs can potentially include far more compounds than even the largest pre-existing physical collections used in traditional screening, and subsequent drug optimization is simpler because analogs of any initial hits can often be easily prepared using the same chemistry.
In this spirit, a subset of the chemistry employed in the D3 initiative focuses on chemical reactions that produce unnatural and natural N-acyl α-amino acids (1) [2, 4, 36, 37], N-acyl α-amino acid esters (2) [3], and N-acyl α-amino acid amides (3) [11], as well as their dipeptide counterparts. These compounds are derived from combinatorial scaffolds 1 - 3 (Scheme 1) with “sites of diversity” to which new chemical groups can be added (e.g., new α-side chains, N-terminal substituents, and/or various carboxylic acid derivatives: -CO2H, -CO2Me, and -CONH2). The simple solid-phase reactions employed in D3 can also be readily adapted to an educational setting with undergraduate or graduate students [1–4].
Scheme 1.
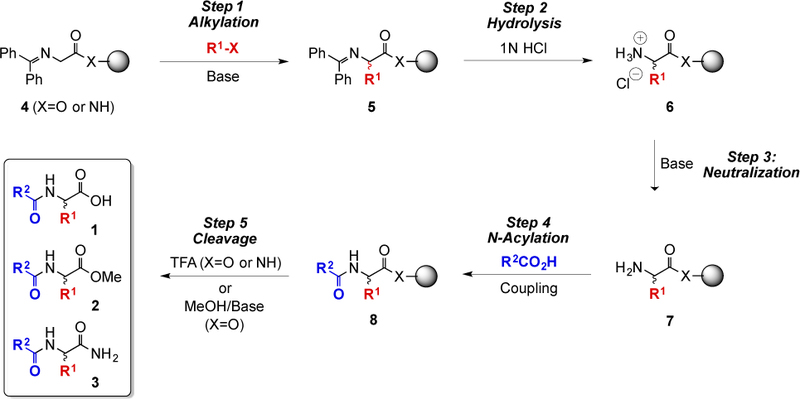
Synthesis of D3 Catalog Members 1, 2, and 3.
The synthetic chemistry for the five-step preparation of D3-catalog compounds 1 [2, 4], 2 [3], and 3 [11] is outlined below (Scheme 1). The starting benzophenone imine (Schiff base) of glycine bound to either Wang (X = O) or Rink (X = NH) resin (4) is deprotonated with base. The intermediate carbanion then reacts with an alkyl halide or Michael acceptor (both represented as the generic red electrophile R1X) to form racemic 5, which contains the first variable input, an R1 group. Hydrolysis to salt 6 is followed by neutralization to the free amine 7. The second variable input, R2 in blue, is then added with an N-acylation by coupling 7 with a carboxylic acid R2CO2H to give 8 (when an N-acyl or other protected α-amino acid is used as the acylating agent R2CO2H, and the D3 products are protected dipeptide derivatives). Finally, the resin-bound product is cleaved from the resin to yield an unnatural N-acyl α-amino acid 1 (using trifluoroacetic acid, TFA), an unnatural N-acyl methyl ester 2 (MeOH/Base) from Wang resin, or an unnatural N-acyl primary amide 3 from Rink amide resin (TFA).
This protocol produces a racemic mixture of both (S)- and (R)-N-acyl α-amino acid derivatives, allowing both epimers to be simultaneously tested in any biological assay. When activity is detected, an optically enriched sample of the mixture can be produced by enantioselective solution- or solid-phase synthesis [36, 37], or chiral chromatography [38, 39].
A virtual catalog of 73,024 synthesizable compounds accessible by D3 solid-phase reaction sequences was enumerated from a diversity set of 100 and 100 commercially available electrophiles and carboxylic acids, respectively [2]. This catalog contains 12,208 each of the (S)- and (R)-N-acyl α-amino acids 1; 12,096 each of the (S)- and (R)-N-acyl α-amino acid methyl esters 2; and 12,208 each of the (S)- and (R)-N-acyl α-amino acid primary amides 3. See the Supporting Information (pp. SI-4 & SI-5) for a more complete explanation of the chemical enumeration and library sizes.
2 |. Results and Discussion
2.1 |. α-Amino acid derivatives and biological activity
Many endogenous α-amino acids have known biological effects beyond the fundamental role they play as protein building blocks. Since D3 compounds are based on the α-amino acid scaffold, it is reasonable to suppose that they may be enriched with bioactive compounds. A collection of 40 approved drugs containing the α-amino acid structural framework (nitrogen to alpha-carbon to carbonyl unit, highlighted in red), is shown in Table SI-1 (see Supporting Information for full structures, references, and other data). Of these compounds, 35% (14/40) are N-acyl α-amino acids, N-acyl dipeptides, or their ester or amide derivatives: alvimopan, bortezomid, ceftaroline, doripenem, folic acid, lacosamide, lenalidomide, methotrexate, pemetrexed, penicillin V, pralatrexate, raltitrexed, tacrolimus, and valsartan.
2.2 |. Identifying known drug targets of D3 compounds
To verify the pharmacological utility of the D3 libraries, we explored whether or not any virtual D3 molecules had previously reported biological activity. Among the >51 million molecules with unique chemical structures (CIDs) deposited in the PubChem database (Figure SI-2) [40], approximately 3,000 compounds were also serendipitously present in D3 catalogs (Figure 2A). These molecules included 76 compounds (Figure 2B) that were active in 95 different PubChem bioassays (AIDs).
Figure 2.
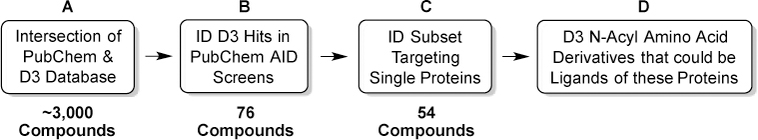
The chemoinformatics analysis. Common D3 virtual and bioactive PubChem compounds.
Many PubChem assays detect phenotypic responses or changes to entire biochemical pathways. These types of screens are useful for drug discovery, but structure-based drug design (“SBDD,” e.g., computer docking) requires a specific macromolecular target. By manually examining the PubChem assay descriptions of the 95 assays noted above, we identified 45 that are likely to involve binding to the orthosteric pockets [41] of 32 distinct single-protein drug targets (Table SI-2). Given that D3 compounds are α-amino acid derivatives, the majority of these protein targets (25 of 32) had endogenous or natural substrates that were free α-amino acids, short peptides, specific protein residues, or α-amino acid metabolites. However, targets with nucleic acid and steroidal endogenous ligands are also amenable to D3 binding, suggesting even broader utility.
Given that so many of these drug targets bind to natural α-amino acids, it is not surprising that there are many N-acyl α-amino acids with natural side chains among the D3/PubChem actives. The strength of the D3 protocol lies in its ability to produce combinatorially diverse sets of both unnatural and natural α-amino acid derivatives. The 45 SBDD-amenable PubChem assays contained 54 unique D3 active compounds (Figure 2C; Figure 3; Table SI-3). Insight into the structural features governing the activity of these 54 compounds is provided in the section entitled “Comparing the D3 virtual-catalog members with D3/PubChem actives,” below.
Figure 3.
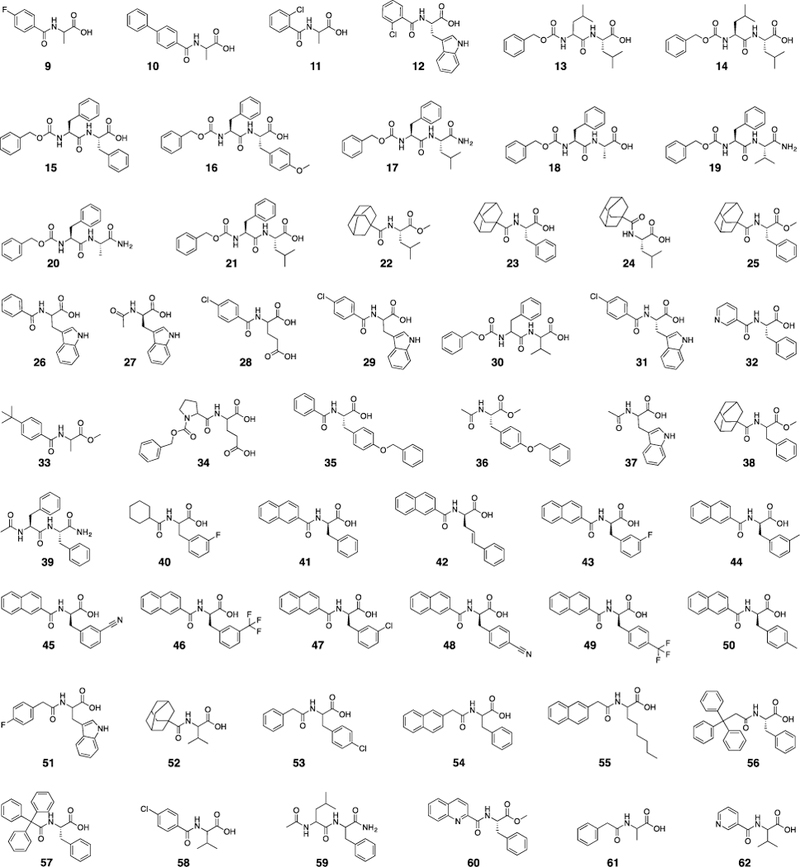
Fifty-four D3/PubChem hits in SBDD-amenable assays (i.e., assays judged likely to involve binding to orthosteric pockets of single-protein targets). See Table SI-3 for further details.
2.3 |. A computer-aided drug-discovery example: Pin1 inhibitors
To further demonstrate the utility of the D3 virtual database, attention was next focused on PubChem SBDD-amenable assays that include active compounds also present in D3 catalogs. One of these was a PubChem assay against Pin1 reported in 2010 (AID: 474989, Table SI-2, entry 14). Also known as “peptidyl-prolyl cis-trans isomerase (PPIase) NIMA-interacting 1,” Pin1 catalyzes the cis-trans isomerization of prolyl amide bonds in select substrate proteins. Because it is thought to play a role in cancer pathogenesis, Pin1 has been studied as a potential drug target for novel cancer chemotherapies. The PubChem assay AID 474989 detected specific binding (leading to inhibition) to the primary Pin1 active site [15] and reported compounds that have a Ki less than 50 μM.
Thirteen of the 32 molecules tested for Pin1 activity in 2010 [15] had been previously disclosed in the 2008 virtual D3 N-acyl α-amino acid set [2]. Of these thirteen compounds, ten were reported as active in PubChem (Ki < 50 μM), and three with lower affinities were reported as inactive. Twelve of the 13 D3 compounds were monosubstituted N-acyl phenylalanines (63, G = H, 2- or 3-F, 2- or 3- or 4-CF3, 2- or 3- or 4-CN, 3- or 4-Me, 3-Cl), and one was a phenylalanine vinylog 42 (Figure 4).
Figure 4.
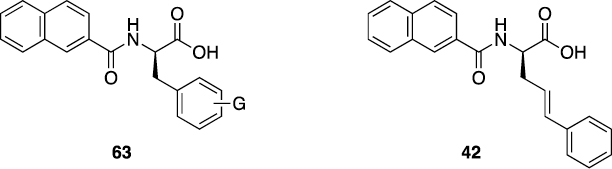
Known Pin1 inhibitors [15] present in the D3 N-acyl α-amino acid set [2].
It is notable that these Pin1 inhibitors have the (R)- or D-stereochemistry not normally present in proteinogenic α-amino acids and their derivatives. This highlights a strength of the D3 approach. D3-based chemistry enables the synthesis and testing of both α-amino acid stereochemistries (usually in the racemic form, and then separately after racemic activity is observed). In contrast, medicinal chemistry is often biased in favor of α-amino acids (and their derivatives) with the naturally occurring stereochemistry, perhaps due to availability of starting materials. Without access to the unnatural stereoisomer, important drugs or drug leads could be missed. Examples of drugs that are unnatural α-amino acids or their derivatives include nateglinide [13], lacosamide [42], penicillamine [43], penicillin V [44], and ximelagatran [45] (see Table SI-1 and the Supporting Information text for more examples).
To further demonstrate the utility of the D3 catalogs, D3-compound binding to Pin1 was evaluated in an in silico benchmark screen. A crystal structure of Pin1 (PDB ID: 3KAI) was used for docking [33]. To validate this target, the co-crystalized compound 64 (Figure 5) was removed from the 3KAI pocket, and a known inhibitor and member of the D3 virtual catalog (42, Figures 3 and 4, Table SI-3) was docked using AutoDock Vina [30].
Figure 5.
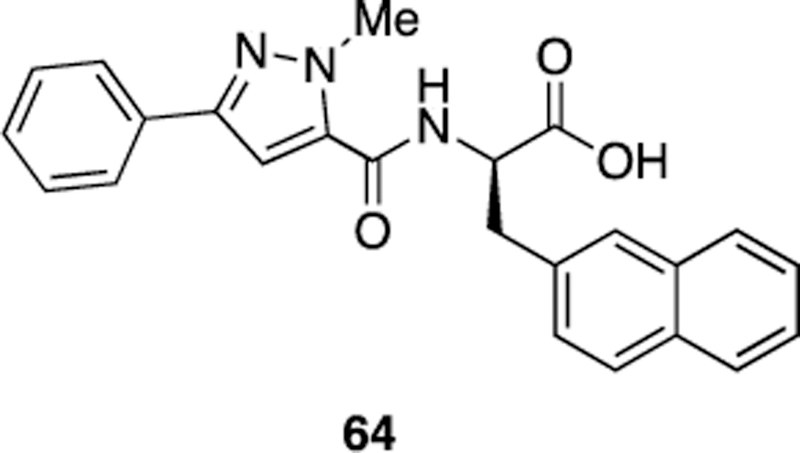
The co-crystallized compound removed from the 3KAI Pin1 structure prior to docking.
The resulting docked pose of bound 42 (Figure 6) closely approximated the crystallographic pose captured in the 3JYJ Pin1 structure [15]. Given that AutoDock Vina was able to reproduce the experimental pose, the entire 24,416-member D3 N-acyl α-amino acid set (including the identified Pin1 inhibitors) was next docked into 3KAI. Since the co-crystallized compound 64 was not present in the D3 database, the results of the virtual screen were not biased in favor of any D3 compound.
Figure 6.
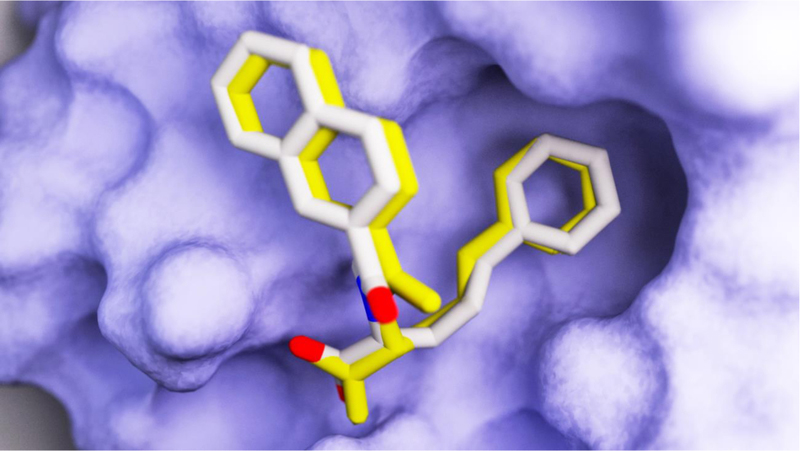
A comparison of the docked and crystallographic poses of 42 (CID 45100499, a known Pin1 inhibitor and also a member of the D3 virtual database). The docked and crystallographic poses are shown in white and yellow, respectively. The compound was docked into the 3KAI structure [33]. The actual crystallographic pose was taken from the 3JYJ structure [15].
This benchmark virtual screen was particularly adept at predicting the activity of the ten known Pin1 inhibitors present in the 24,000-member D3 virtual catalog of N-acyl α-amino acids (1, Table 1). Six of the ten compounds listed as active in PubChem were contained in the top 5% (roughly 1,200) of Vina-ranked D3 compounds. A receiver operating characteristic (ROC) curve was next generated, with the known inhibitors and remaining compounds serving as the true positives and negatives, respectively. The area under this curve was 0.93, suggesting that, given a randomly selected pair of compounds comprised of one known inhibitor and one uncharacterized compound, there is a 93% chance that the known inhibitor has the better Vina score [30]. Random ranking would put this probability at only 50%.
Table 1.
The Vina scores and percentile rankings of the ten positive controls (i.e., known inhibitors) included in the benchmark Pin1 screen.
| ID | Vina Score | Percentile Rank |
|---|---|---|
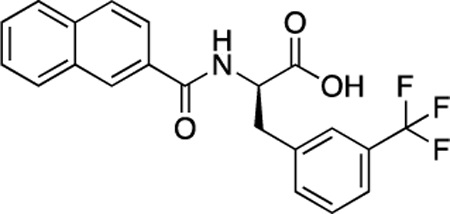 (R)-52 (DDD-000012382) |
−7.3 | 1.5 |
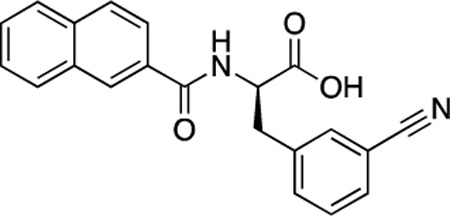 (R)-51 (DDD-000012386) |
−7.3 | 1.5 |
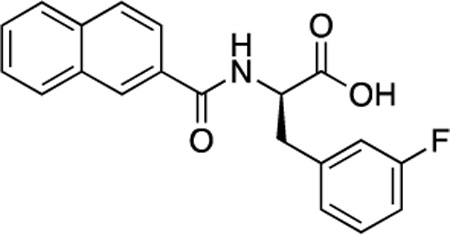 (R)-49 (DDD-000012361) |
−7.2 | 2.4 |
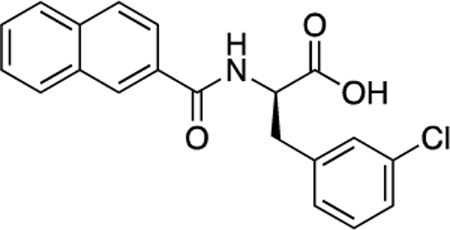 (R)-53 (DDD-000012365) |
−7.2 | 2.4 |
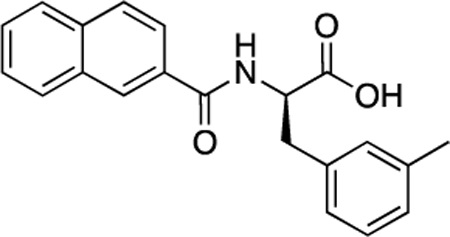 (R)-50 (DDD-000012379) |
−7.2 | 2.4 |
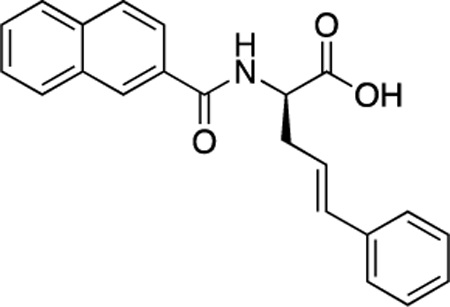 (R)-48 (DDD-000012394) |
−7.2 | 2.4 |
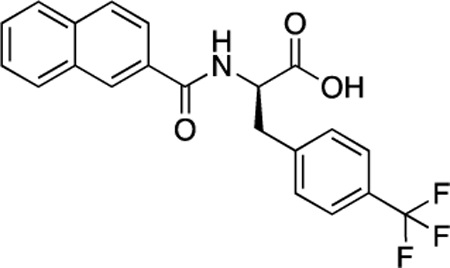 (R)-55 (DDD-000012383) |
−6.9 | 7.5 |
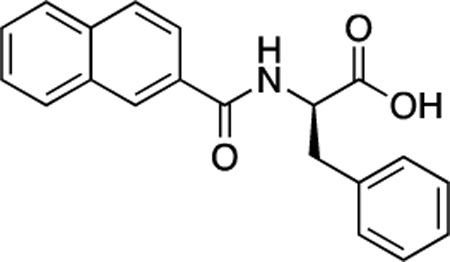 (R)-47 (DDD-000012350) |
−6.8 | 10.5 |
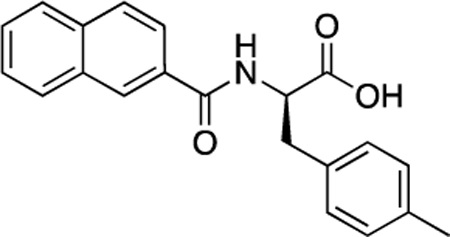 (R)-56 (DDD-000012380) |
−6.8 | 10.5 |
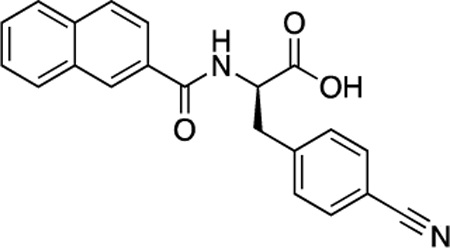 (R)-54 (DDD-000012387) |
−6.6 | 18.8 |
Given the performance of this virtual screen, some top-ranking but untested D3 compounds are also likely Pin1 inhibitors. Though it is beyond the scope of this paper, future directions include experimentally synthesizing and testing a number of these top-ranked compounds in order to identify small molecules with improved activity.
2.4 |. Comparing the D3 virtual-catalog members with D3/PubChem actives
We next compared the structural characteristics of the 73,024-member D3 virtual catalog with the 54-member D3/PubChem active set (Figure 7). These two sets both contain all three D3 structural classes [N-acyl α-amino acids (1) and their methyl ester (2) and amide (3) derivatives], but in differing proportions. In the larger D3 set (Figure 7, left column), the numbers of compounds in each class are nearly equal because the D3 molecules were derived combinatorially from the same set of 100 electrophile (R1X) and 100 carboxylic acid (R2CO2H) inputs, differing only in the method of resin cleavage (Scheme 1). The varying numbers of 2 are a result of the different cleavage condition used, which caused different side-chain side reactions. For example, if R1 in compounds 1 – 3 is CH2CO2tBu, cleavage of 1 or 3 with TFA converts the protected t-butyl ester to the carboxylic acid (CH2CO2tBu -> CH2CO2H). In contrast, cleavage of 2 with Et3N leaves the side chain unchanged (CH2CO2tBu -> CH2CO2tBu).
Figure 7.
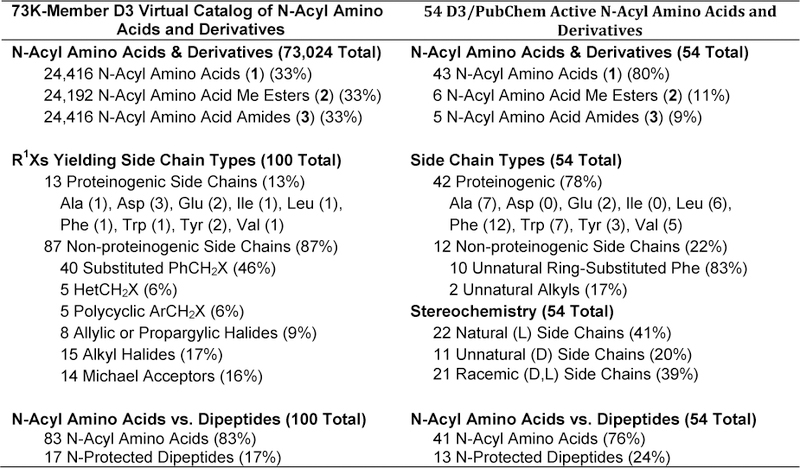
Comparing the 73,024 D3 compounds and the 54 D3/PubChem actives.
As might be expected, the proportions of structural classes in the D3 virtual catalog set is quite different from the D3/PubChem active set (Figure 7, right column). A lower percentage of a particular structural class in the active set could imply that it is not fertile territory for drug discovery and is overrepresented in the D3 virtual catalog. On the other hand, perhaps that structural class is simply underrepresented among the PubChem molecules made and tested. Without going through a complete analysis of the structural classes present in PubChem, both tested and untested, we are confident that this latter hypothesis is the case, especially when comparing the structural categories of “proteinogenic/non-proteinogenic side chains” and “natural/unnatural side chain stereochemistry.” The introduction, via a racemic alkylation reaction, of side chains usually not found in proteinogenic amino acids is at the core of D3 chemistry. This process gives rise to many unnatural-side-chain residues with equal proportions of the natural (e.g. L- or S-) and unnatural D- or R- stereoisomers. To account for the presence, potential activity, and computational accessibility of all enantiomers, each is explicitly enumerated in the D3 virtual catalog. In cases where D3 compounds contain two or three stereogenic centers, all four or eight stereoisomers, respectively, are included in the catalog.
The preponderance of the natural stereochemistry and side chains in the PubChem active set is not surprising given that amino-acid-based compounds submitted to PubChem for testing are likely biased in favor of derivatives of the more readily available natural (L)-α-amino acid stereoisomer. What is noteworthy is the significant number of D3/PubChem active compounds (20%) with the unnatural (D)-stereochemistry. These would not have been present in the D3/PubChem intersection if the D3 project had considered only enantiomerically pure molecules with the (L)-stereochemistry. The observed activity of some racemic PubChem actives (39%) may also be explained in part by the D3 methodology, which generates the unnatural and natural stereoisomers in equal proportions. In any case, the presence of these “unnatural” amino-acid derivatives in both the D3 virtual catalog and PubChem active set demonstrates the value of the D3 virtual catalog as a resource for identifying biologically active compounds from an underrepresented structural class.
Finally, when an N-acyl or other protected α-amino acid is used as the acylating agent R2CO2H, the D3 products are protected dipeptide derivatives (see Figure SI-5 and associated text). In total, such dipeptide derivatives comprise 17% of the 73K D3 virtual catalog and 24% of the 54-member D3/PubChem set (Figures 3 and 7), pointing to the ready availability of such derivatives via the D3 methodology.
2.5 |. The D3 N-acyl α-amino acid virtual catalog
The Pin1 virtual screen of the above 24,416-member “D3 N-Acyl α-Amino Acid set” illustrates how the D3 chemical library can be particularly useful for drug discovery projects. Computed chemical descriptors suggest that the compounds in this catalog are chemically similar to those of other libraries commonly used in virtual and experimental screens (Figure 8). For comparison, consider the NCI Diversity Set III (NCI III) and the ChemBridge Diversity CombiSet (ChemBridge). The average calculated molecular properties of the D3 N-acyl α-amino acid set are intermediate between those of these two popular libraries, with the exception of the hydrogen-bond-donor count, where the ChemBridge set is anomalous (Table 2, Figure 7). The overwhelming majority of the catalog compounds (97.2%, 98.6%, and 99.8% of the D3 N-Acyl α-Amino Acid, NCI III, and ChemBridge compounds, respectively) satisfy Lipinski’s “Rule of Five” for druglikeness [46].
Figure 8.
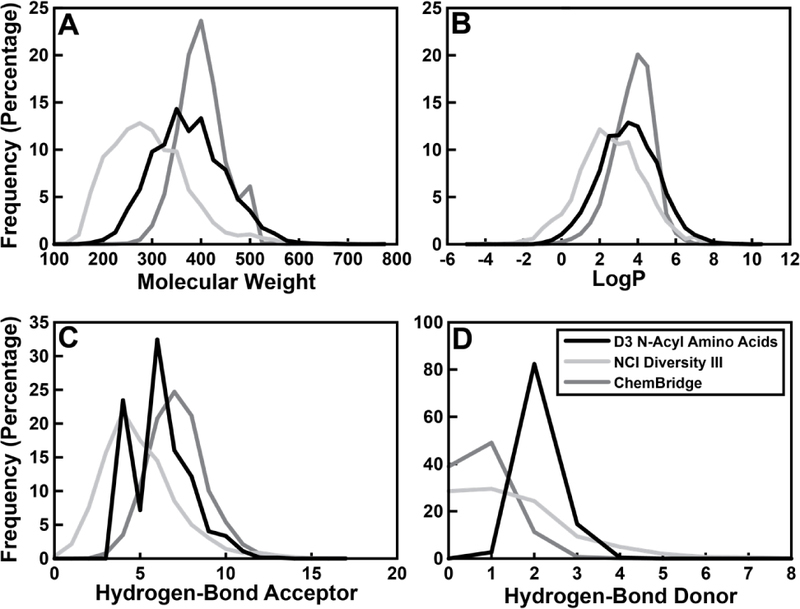
Histograms showing the molecular weights, calculated LogP values, and hydrogen-bond acceptor/donor counts for the D3 N-Acyl α-Amino Acid set, the NCI Diversity set III, and the ChemBridge Diversity CombiSet catalogs.
Table 2.
The average molecular properties of compounds in the D3 N-Acyl α-Amino Acid Set, the NCI Diversity Set III, and the ChemBridge Diversity CombiSet. Standard deviations are shown between parentheses.
| D3 | NCI III | ChemBridge | |
|---|---|---|---|
| Molecular Weight (Daltons) | 361.8 (73.4) | 280.5 (80.6) | 388.6 (47.3) |
| logP | 3.3 (1.5) | 2.3 (1.7) | 3.5 (1.1) |
| Hydrogen-Bond Acceptors | 5.8 (1.6) | 4.7 (2.2) | 6.8 (1.6) |
| Hydrogen-Bond Donors | 1.5 (0.4) | 1.4 (1.3) | 0.7 (0.7) |
To encourage broad use of the D3 virtual catalog, structures of the D3 N-acyl α-amino acids, N-acyl α-amino acid esters, and N-acyl α-amino acid amides have been prepared in formats that are compatible with several computer docking programs, including AutoDock [29], Vina [30], and Schrodinger’s Glide [27, 28]. Various possible tautomeric, protonation, and ring conformational states of each compound were carefully considered. These virtual catalogs may be downloaded free of charge from http://durrantlab.com/liglib/iupui/d3_docking/ (accessed February 27, 2017).
Many researchers may wish to prepare D3 structures according to their own protocols. The SMILES string of each D3 compound has also been published on both the D3 (http://durrantlab.com/liglib/iupui/d3_docking/, accessed February 27, 2017) and Collaborative Drug Discovery (CDD) (https://www.collaborativedrug.com, accessed February 27, 2017) websites [47, 48]. CDD also allows researchers to search and download subsets of the entire D3 virtual catalog.
2.6 |. Conclusions
Two complementary strengths of D3 virtual catalogs are their bioactivity potential and synthetic accessibility. By demonstrating the retrospective and prospective biological utility of these catalogs, this manuscript encourages computational chemists to probe D3 molecules with their own tools and hypotheses. Compounds enumerated in the virtual catalog are based on well-documented D3 procedures, so synthesizing and testing computationally selected molecules is entirely feasible. The D3 catalogs thus enable collaborations between computationalists, synthetic chemists, and biologists.
In summary, to demonstrate the utility of the D3 virtual catalog, we identified a number of D3 molecules with known biological activity that were present in the PubChem database. These results will help guide future drug-discovery efforts. Using computational techniques such as docking, students and researchers can identify analogs of known modulators that are likely to bind to the same target. These compounds can then be synthesized and distributed to collaborators, who evaluate the pharmacological, enzymatic, or signaling effect to confirm or refute the computational predictions. Others who wish to identify additional D3 drug targets via virtual screening can download a carefully prepared catalog of D3 compounds from http://durrantlab.com/liglib/iupui/d3_docking/.
Supplementary Material
Acknowledgements
The National Science Foundation (NSF/DUE-1140602, NSF/MRI-CHE-0619254, and NSF/MRI-DBI-0821661), National Institutes of Health (RO1-GM28193), IUPUI International Development Fund, Indiana University Purdue University Indianapolis (IUPUI) STEM Summer Scholars Institute, and Analytical Technologies at Eli Lilly and Company are acknowledged for their generous support. We thank Collaborative Drug Discovery Inc. for generously hosting D3 molecular data on their servers, ChemAxon for granting access to their chemical-enumeration software, Guillermo Morales for helpful discussions, and the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov, accessed February 27, 2017) for providing compound structures for download.
Footnotes
Conflict of Interest
The authors declare that they have no financial or commercial conflicts of interest.
References
- 1.Scott WL, O’Donnell MJ, J. Comb. Chem 2009, 11, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scott WL, Alsina J, Audu CO, Babaev E, Cook L, Dage JL, et al. , J. Comb. Chem 2009, 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott WL, Audu CO, Dage JL, Goodwin LA, Martynow JG, Platt LK, et al. , J. Comb. Chem 2009, 11, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott WL, Denton RE, Marrs KA, Durrant JD, Samaritoni JG, Abraham MM, et al. , J. Chem. Educ 2015, 92, 819. [Google Scholar]
- 5.Leach AR, Hann MM, Drug Discov. Today 2000, 5, 326. [DOI] [PubMed] [Google Scholar]
- 6.Barnard JM, Downs GM, von Scholley-Pfab A, Brown RD, J. Mol. Graph. Model 2000, 18, 452. [PubMed] [Google Scholar]
- 7.Lobanov VS, Agrafiotis DK, J. Mol. Graph. Model 2001, 19, 571. [DOI] [PubMed] [Google Scholar]
- 8.Lobanov VS, Agrafiotis DK, Comb. Chem. High Throughput Screening 2002, 5, 167. [DOI] [PubMed] [Google Scholar]
- 9.Green DVS, Pickett SD, Mini. Rev. Med. Chem 2004, 4, 1067. [DOI] [PubMed] [Google Scholar]
- 10.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, et al. , J. Med. Chem 2014, 57, 4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samaritoni JG, Copes AT, Crews DK, Glos C, Thompson AL, Wilson C, et al. , J. Org. Chem 2014, 79, 3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Austin CP, Brady LS, Insel TR, Collins FS, Science 2004, 306, 1138. [DOI] [PubMed] [Google Scholar]
- 13.Shinkai H, Nishikawa M, Sato Y, Toi K, Kumashiro I, Seto Y, et al. , J. Med. Chem 1989, 32, 1436. [DOI] [PubMed] [Google Scholar]
- 14.Swinney DC, Anthony J, Nat. Rev. Drug Discov 2011, 10, 507. [DOI] [PubMed] [Google Scholar]
- 15.Dong LM, Marakovits J, Hou X, Guo C, Greasley S, Dagostino E, et al. , Bioorg. Med. Chem. Lett 2010, 20, 2210. [DOI] [PubMed] [Google Scholar]
- 16.CombiChem for Excel from CambridgeSoft, PirkinElmer, 2015.
- 17.ChemAxon, ChemAxon Kft, 2015.
- 18.Maestro, Schrödinger, LLC, 2015.
- 19.Holbeck SL, Eur. J. Cancer 2004, 40, 785. [DOI] [PubMed] [Google Scholar]
- 20.Desai PV, Patny A, Sabnis Y, Tekwani B, Gut J, Rosenthal P, et al. , J. Med. Chem 2004, 47, 6609. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Fourches D, Sedykh A, Zhu H, Golbraikh A, Ekins S, et al. , J. Chem. Inf. Model 2013, 53, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LigPrep, Schrödinger, LLC, 2015.
- 23.Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, et al. , J. Comput. Chem 2005, 26, 1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.QikProp, Schrödinger, LLC, 2015.
- 25.Greenwood JR, Calkins D, Sullivan AP, Shelley JC, Comput J .-Aided Mol. Des 2010, 24, 591. [DOI] [PubMed] [Google Scholar]
- 26.Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M, J. Comput. Aid. Mol. Des 2007, 21, 681. [DOI] [PubMed] [Google Scholar]
- 27.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. , J. Med. Chem 2004, 47, 1739. [DOI] [PubMed] [Google Scholar]
- 28.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. , J. Med. Chem 2004, 47, 1750. [DOI] [PubMed] [Google Scholar]
- 29.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. , J. Comput. Chem 2009, 30, 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trott O, Olson AJ, J. Comput. Chem 2009, 31, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR, J. Cheminf 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasteiger J, Marsili M, Tetrahedron 1980, 36, 3219. [Google Scholar]
- 33.Potter AJ, Ray S, Gueritz L, Nunns CL, Bryant CJ, Scrace SF, et al. , Bioorg. Med. Chem. Lett 2010, 20, 586. [DOI] [PubMed] [Google Scholar]
- 34.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W, Comput J .-Aided Mol. Des 2013, 27, 221. [DOI] [PubMed] [Google Scholar]
- 35.Ohlmeyer M, Zhou MM, Mt. Sinai J. Med 2010, 77, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Donnell MJ, Aldrichimica Acta 2001, 34, 3. [Google Scholar]
- 37.O’Donnell MJ, Acc. Chem. Res 2004, 37, 506. [DOI] [PubMed] [Google Scholar]
- 38.Maier NM, Franco P, Lindner W, J. Chromatogr. A 2001, 906, 3. [DOI] [PubMed] [Google Scholar]
- 39.Kennedy JH, Belvo MD, Sharp VS, Williams JD, J. Chromatogr. A 2004, 1046, 55. [DOI] [PubMed] [Google Scholar]
- 40.Cheng TJ, Pan YM, Hao M, Wang Y, Bryant SH, Drug Discov. Today 2014, 19, 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Smet F, Christopoulos A, Carmeliet P, Nat. Biotechnol 2014, 32, 1113. [DOI] [PubMed] [Google Scholar]
- 42.de Biase S, Gigli GL, Valente M, Merlino G, Expert Opin. Drug Metab. Toxicol 2014, 10, 459. [DOI] [PubMed] [Google Scholar]
- 43.Wadhwa S, Mumper RJ, Cancer Lett 2013, 337, 8. [DOI] [PubMed] [Google Scholar]
- 44.Brown K, Penicillin Man: Alexander Fleming and the Antibiotic Revolution, Sutton Publishing Limited, London, 2005. [Google Scholar]
- 45.Testa L, Bhindi R, Agostoni P, Abbate A, Zoccai GGLB, van Gaal WJ, Expert Opin. Drug Saf 2007, 6, 397. [DOI] [PubMed] [Google Scholar]
- 46.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Adv. Drug. Deliv. Rev 2001, 46, 3. [DOI] [PubMed] [Google Scholar]
- 47.Gupta RR, Gifford EM, Liston T, Waller CL, Hohman M, Bunin BA, et al. , Drug Metab. Dispos 2010, 38, 2083. [DOI] [PubMed] [Google Scholar]
- 48.Ekins S, Bunin BA, Computational Approaches and Collaborative Drug Discovery for Trypanosomal Diseases, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.