

Abstract
Functional activation of the transforming growth factor–β (TGF-β) receptors (TGFBRs) is carefully regulated through integration of post-translational modifications, spatial regulation at the cellular level, and TGFBR availability at the cell surface. Although the bulk of TGFBRs resides inside the cells, AKT Ser/Thr kinase (AKT) activation in response to insulin or other growth factors rapidly induces transport of TGFBRs to the cell surface, thereby increasing the cell's responsiveness to TGF-β. We now demonstrate that TGF-β itself induces a rapid translocation of its own receptors to the cell surface and thus amplifies its own response. This mechanism of response amplification, which hitherto has not been reported for other cell-surface receptors, depended on AKT activation and TGF-β type I receptor kinase. In addition to an increase in cell-surface TGFBR levels, TGF-β treatment promoted TGFBR internalization, suggesting an overall amplification of TGFBR cycling. The TGF-β–induced increase in receptor presentation at the cell surface amplified TGF-β–induced SMAD family member (SMAD) activation and gene expression. Furthermore, bone morphogenetic protein 4 (BMP-4), which also induces AKT activation, increased TGFBR levels at the cell surface, leading to enhanced autocrine activation of TGF-β–responsive SMADs and gene expression, providing context for the activation of TGF-β signaling in response to BMP during development. In summary, our results indicate that TGF-β– and BMP-induced activation of low levels of cell surface–associated TGFBRs rapidly mobilizes additional TGFBRs from intracellular stores to the cell surface, increasing the abundance of cell-surface TGFBRs and cells' responsiveness to TGF-β signaling.
Keywords: transcription regulation, SMAD transcription factor, Akt PKB, bone morphogenetic protein (BMP), cell-surface receptor, autocrine signaling, Smad activation
Introduction
Delicate coordination and control of signaling responses to extracellular molecules inform cell differentiation and behavior and direct tissue development and homeostasis, whereas their dysregulation underlies developmental disorders and cancers. The cell-surface levels and regulation of transmembrane kinase receptors, which are critical to transduction of extracellular signals, define the sensitivity and amplitude of the cell's response to extracellular growth and differentiation factors. Mutation or dysregulation of kinase receptors often leads to aberrant receptor function and signaling responses, as well-illustrated in studies of receptor tyrosine kinases. Changes in cell-surface expression of these receptors help define the cell's responsiveness to growth factors, whereas spatial regulation at the cell surface or cycling between the plasma membrane and endocytic vesicles also control ligand responsiveness (1–3).
In contrast to the receptor tyrosine kinases, much less is known about the control of responsiveness to the secreted TGF-β family proteins, which often act as differentiation factors yet have a plethora of additional functions in cell physiology and development (4). TGF-β and the closely related activins and bone morphogenetic protein (BMPs)2 activate dual-specificity transmembrane kinases that phosphorylate serine and threonine, as well as tyrosine (5). Genetic aberrations in TGF-β receptor signaling components have been linked to developmental deficiencies and disorders, whereas dysfunction in receptor expression and activation is implicated in the initiation and progression of fibrosis and cancer (4, 6, 7). Mostly studied using TGF-β as prototype, the dimeric TGF-β family proteins bind to cell-surface complexes of two pairs of type II and type I receptors and induce the activation of type I receptor kinases through their phosphorylation by the proximal type II receptor kinases. The activated type I receptors recruit and C-terminally phosphorylate, and thus activate, Smads (5, 8). The receptor-activated Smads dissociate from the receptors and associate with Smad4 in trimeric complexes that translocate into the nucleus where they interact with DNA-binding transcription factors and cofactors to amplify or repress gene expression (8–10). Smad2 and Smad3 are activated in response to TGF-β, with Smad3 most frequently acting as direct effector of TGF-β target gene expression, whereas Smad1, Smad5, and Smad8 act as BMP signaling effectors (8–10). TGF-β family receptors also activate non-Smad signaling pathways, including PI3K–Akt–mTOR signaling, Erk, p38, and c-Jun N-terminal kinase MAPK pathways, and Rho-like GTPase signaling (11, 12). Like the receptor tyrosine kinases, the TGF-β receptors remain catalytically active upon internalization. However, although growth factor ligand binding induces receptor tyrosine kinases to internalize in endocytic compartments, TGF-β family receptors are thought to continuously recycle and enable ligand binding; however, ligand binding may also enhance their internalization (8, 13, 14). Smad activation in response to TGF-β ligands occurs in nascent or fully formed endosomal compartments, whereas TGF-β–induced Erk MAPK and PI3K–Akt signaling associate with caveolar compartments (15–19). Differential compartmentalization enables the cells to calibrate the contributions of different signaling pathways to the cellular response (3, 13). Further control of TGF-β responsiveness at the level of cell-surface receptors is provided by the activity of the metalloprotease ADAM17, better known as TACE (tumor necrosis factor-α–converting enzyme), which proteolytically removes the ectodomain of the type I TGF-β receptors (TβRI), leading to decreased TGF-β responsiveness and nuclear functions of the TβRI cytoplasmic domains (20). Finally, cells can rapidly enhance their sensitivity to TGF-β by mobilizing TGF-β receptors from an intracellular pool toward the plasma membrane. The increased abundance of type I and type II TGF-β receptors confers increased responsiveness to autocrine TGF-β signaling (13, 21, 22). Insulin induces enhanced TGF-β receptor transport to the cell surface through activation of Akt, thus enabling enhanced autocrine TGF-β signaling (22). Akt activation may also drive the rapid increase of cell-surface TGF-β receptors from intracellular stores and increased TGF-β responsiveness in response to high glucose (21, 22).
Here, we introduce a novel mode of signal amplification that is elaborated at the level of the cell-surface TGF-β receptors. Such regulation has, to our knowledge, not been described for any other cell-surface receptors. Specifically, TGF-β ligand induces a rapid up-regulation of the levels of type I and type II TGF-β receptors, i.e. TβRII and TβRI, at the cell surface, dependent on TβRI kinase activity and TGF-β–induced Akt activation. The consequently increased TGF-β responsiveness results in ligand-induced amplification of signaling and Smad-mediated gene responses. Furthermore, BMP signaling through Akt also enhances the TGF-β receptors at the cell surface, thus increasing autocrine TGF-β signaling and allowing TGF-β responsiveness to participate in the BMP response.
Results
TGF-β induces a rapid increase in cell-surface TGF-β receptor levels
We evaluated the effect of TGF-β on cell-surface TGF-β receptor levels using two different cell lines, the human nontransformed keratinocyte cell line HaCaT, and the human alveolar adenocarcinoma cell line A549. Both cell lines are established models for studying the TGF-β response, exhibiting TGF-β–induced Smad phosphorylation and gene expression changes (23, 24). Cell-surface TGF-β receptors were detected by biotinylation of cell-surface proteins of intact, nonpermeabilized cells, followed by selective adsorption of the biotinylated proteins to NeutrAvidin beads, SDS-PAGE, and immunoblotting for TβRI or TβRII. In both cell lines, TGF-β induced within 15 min a substantial increase in the cell-surface levels of both TβRI and TβRII (Fig. 1). No effects were apparent on the overall levels of TβRI or TβRII. TGF-β treatment did not affect the cell-surface abundance of transferrin receptor (TfR), a transmembrane protein that is not regulated by TGF-β signaling (21), suggesting that the increase of TGF-β receptors at the cell surface was selective or specific. These data reveal a rapid translocation of TGF-β receptors from intracellular stores to the cell surface that, considering its rapid kinetics and the lack of change in overall receptor levels, is likely to not depend on new protein synthesis.
Figure 1.

TGF-β induces an increase in TGF-β receptor levels at the cell surface. HaCaT (A) and A549 (B) cells were stimulated with TGF-β1 for the indicated times. Cell-surface proteins were labeled by biotinylation, affinity-purified by NeutrAvidin adsorption, and visualized by SDS-PAGE and immunoblotting (IB). The upper panels show the immunoblotting of biotinylated TβRI and TβRII receptors and TfR at the cell surface, whereas the lower panels show the abundance of these proteins in whole cell lysates. GAPDH and TfR served as controls in total cell lysates.
TGF-β–induced Akt activation promotes the increase in cell-surface TGF-β receptors
Akt signaling has been shown to mediate the insulin-induced up-regulation of the TGF-β receptor levels at the cell surface by promoting the transport of the receptors from intracellular membrane compartments to the plasma membrane (22). We therefore tested whether specifically inhibiting Akt activation using the highly selective allosteric Akt inhibitor AktVIII (25, 26) affects the TGF-β–induced increase in TGF-β receptor levels at the cell surface. TGF-β treatment resulted in a rapid increase in Akt phosphorylation at serine 473 and threonine 308 in both HaCaT and A549 cells. Both basal and TGF-β–induced Akt phosphorylation on both residues were inhibited by treatment with AktVIII (Fig. 2, A and B, left panels). Another specific Akt inhibitor, MK2206 (27, 28), similarly repressed TGF-β–induced Akt activation (Fig. 2, A and B, right panel). Furthermore, the PI3K inhibitor LY294002 also blocked TGF-β–induced Akt activation in both cell lines (Fig. S1, A and B). These observations are consistent with previous reports that TGF-β activates PI3K signaling and induces Akt phosphorylation in epithelial cells (29–31).
Figure 2.

Inhibition of Akt activation attenuates the ligand-induced increase of cell-surface TβRI and TβRII. A and B, Akt activation in HaCaT (A) and A549 (B) cells stimulated with TGF-β1 for the indicated times. As shown in the left panels, TGF-β1 induces Akt phosphorylation, assessed by immunoblotting (IB) for phospho-Akt, which is prevented in the presence of AktVIII. Similarly, the Akt inhibitor MK2206 prevented basal and TGF-β–induced Akt activation, assessed at 30 min after TGF-β treatment (right panels). GAPDH serves as a loading control. C and D, cell-surface protein biotinylation analyses of HaCaT (C) and A549 (D) cells that were treated with TGF-β1 for the indicated times in the presence or absence of AktVIII (left panels) or MK2206 (right panels). As shown in the left panels, the TGF-β–induced increases of TβRI and TβRII levels at the cell surface were attenuated in the presence of AktVIII. Similarly, the Akt inhibitor MK2206 prevented TGF-β–induced up-regulation of TβRI and TβRII levels at the cell surface, assessed at 30 min after TGF-β treatment (right panels). Total TβRI and TβRII protein, GAPDH, and cell surface and total TfR were not affected by TGF-β1, AktVIII, or MK2206.
Cell-surface biotinylation of TGF-β receptors on HaCaT and A549 cells revealed that inhibition of Akt activation using AktVIII imparted a significant attenuation of TGF-β–induced cell-surface TGF-β receptor levels (Fig. 2, C and D, left panels). Similar results were obtained using the other Akt inhibitor, MK2206 (Fig. 2, C and D, right panels), or the PI3K inhibitor LY294002 (Fig. S1, C and D). These effects of the Akt inhibitors AktVIII or MK2206 or the PI3K inhibitor LY294002 appeared to be selective to the TGF-β receptors, because Akt inhibition did not affect cell-surface transferrin receptor levels (Fig. 2, C and D, and Fig. S1). These data reveal that Akt activation promotes an increase in cell-surface levels of the TGF-β receptors. However, inhibition of the TGF-β–induced increase in cell-surface TGF-β receptors by AktVIII or MK2206 or by LY294002 was incomplete, despite full inhibition of Akt activation.
The TβRI kinase is required for TGF-β–induced increase in cell-surface TGF-β receptors
We next evaluated whether TGF-β–induced TGF-β receptor up-regulation requires the kinase activity of TβRI, which is known to initiate TGF-β–induced Smad and Erk MAPK activation (5, 12, 32). SB431542, a specific inhibitor of TβRI kinase activity, prevented TGF-β–induced Smad2 and Smad3 activation (Fig. 3, A and B) and attenuated the TGF-β–induced enhancement of TβRI and TβRII, in both HaCaT and A549 cells, without affecting the cell-surface levels of transferrin receptor (Fig. 3, C and D). We also evaluated the effect of corilagin, another small molecule that inhibits the TβRI kinase and thus prevents TGF-β–induced Smad2/3 activation (33). Similarly to SB431542, pretreatment of the cells with corilagin decreased TGF-β–induced receptor presentation without affecting the total TGF-β receptor levels or the cell-surface levels of the transferrin receptor (Fig. 3, E and F).
Figure 3.

Inhibition of the TβRI kinase decreases the cell-surface TGF-β receptor response. A–D, effect of SB431542 on the TGF-β–induced increases of Smad3 and Smad2 activation, assessed by immunoblotting (IB) for phospho-Smad3 or -Smad2 (A and B), and TβRI and TβRII levels at the cell surface, assessed by cell-surface protein biotinylation and immunoblotting (C and D), in HaCaT (A and C) or A549 (B and D) cells. SB431542 blocked TGF-β–induced Smad2 and Smad3 activation (A and B) and attenuated the TGF-β–induced increase in cell-surface TβRI and TβRII levels without affecting cell-surface levels of TfR or whole-cell levels of TGF-β receptors. E and F, effect of corilagin on the TGF-β–induced increase of TβRI and TβRII levels at the cell surface, assessed as in C and D, in HaCaT (E) or A549 (F) cells. Corilagin attenuated the TGF-β–induced increase in cell-surface TβRI and TβRII levels without affecting cell-surface levels of TfR or whole-cell levels of TGF-β receptors. G and H, effects of SB431542 on TGF-β–induced Akt activation, assessed by immunoblotting for phospho-Akt or phospho-Smad2 or -Smad3, in HaCaT (G) and A549 (H) cells. Cells were pretreated and treated as in A and B. SB431542 inhibited the TGF-β–induced Akt activation (G and H). GAPDH and TfR levels serve as loading controls. Note that B presents data from the same experiment shown in D.
The observation that SB431542, the Akt inhibitors AktVIII and MK2206, and the PI3K inhibitor LY94002 attenuated TGF-β–induced increase of TβRII and TβRI levels at the cell surface led us to address the effect of SB431542 on TGF-β–induced Akt activation in HaCaT and A549 cells. TGF-β–induced Akt activation is thought to occur independently from Smad activation (11, 12). Inhibition of TβRI kinase activity was reported to prevent Akt activation in some cell lines (30, 34) yet was also shown to not affect TGF-β–induced Akt activation in a tumor cell line (31). Immunoblot analysis revealed that SB431542 attenuated or prevented TGF-β–induced Akt activation in both HaCaT and A549 cells without affecting overall Akt levels (Fig. 3, G and H). These data, together with those using the PI3K inhibitor LY294002 (Fig. S1), suggest that activation of the TβRI kinase, PI3K, and Akt in response to TGF-β are required for TGF-β–induced up-regulation of cell-surface TGF-β receptor levels and that in HaCaT and A549 cells, the TβRI kinase promotes TGF-β–induced Akt activation.
Preventing Akt activation inhibits TGF-β receptor cycling
Transmembrane receptor kinases cycle between endosomal compartments and the cell surface, and receptor cycling is an important determinant of the receptor availability at the cell surface and the signaling response (1–3). Accordingly, TGF-β receptor endocytosis and translocation to the cell surface control the TGF-β response (15–19). Complementary to evaluating the effect of AktVIII on the cell-surface levels of the TGF-β receptors, we examined its effect on internalization of the TGF-β receptors. For this purpose, we first labeled the cell-surface proteins at 0 °C using the reversible biotinylation reagent EZ Link Sulfo-NHS-SS-Biotin, which contains a reducible disulfide bond. After biotinylation, the cells were incubated at 37 °C for 20 min with or without TGF-β to allow endocytosis. Biotinylation of the proteins that remained at the cell surface was subsequently reversed using GSH to reduce and thus remove the disulfide-bonded biotin label (Fig. 4A). In contrast to the proteins at the cell surface, internalized proteins are protected against reversing the biotinylation and can be selectively adsorbed to NeutrAvidin-bound agarose (35). The adsorbed proteins were then analyzed by SDS-PAGE, followed by immunoblotting (Fig. 4, B and C). As shown in Fig. 4B, cell-surface TβRI and TβRII were labeled by cell-surface protein biotinylation at 37 °C (lane 1), and only a minimal background of receptor labeling remained in samples that were subsequently treated with reducing buffer (lane 2). After 20 min at 37 °C and in the absence of added TGF-β, low levels of internalized TβRII and TβRI receptors were apparent (lane 3). The internalized receptor levels were, however, increased in response to TGF-β (lane 4), indicating that TGF-β enhances receptor internalization. AktVIII attenuated the TGF-β–induced increase in TβRI and TβRII internalization (Fig. 4C). Internalization of the transferrin receptor was not affected by either TGF-β treatment or AktVIII (Fig. 4C), suggesting selective or specific regulation of TGF-β receptor cycling rather than an overall effect on endocytosis.
Figure 4.

TGF-β increases internalization of TβRI and TβRII in an Akt-dependent manner. TGF-β receptor internalization (int) analysis of HaCaT cells in the presence of TGF-β1 and/or AktVIII or vehicle control. A, schematic diagram of the experimental flow used to generate the results in B and C. The cells were subjected to cell-surface protein biotinylation for 20 min on ice and then switched to cell culture medium under normal growth conditions at 37 °C for 20 min, in the presence or absence of TGF-β1 and/or AktVIII. The biotin label was then removed from proteins at the cell surface using GSH, and the internalized biotinylated proteins, which were protected against removal of the biotinylation, were purified by affinity adsorption to NeutrAvidin. B, biotin-labeled proteins, purified by NeutrAvidin adsorption, were visualized by SDS-PAGE and immunoblot (IB) analysis. Lane 1 shows total cell-surface labeled protein prior to incubation at 37 °C and reversal of biotinylation (cell surface, CS). Lane 2 shows background (BG) levels of biotinylated proteins in cells kept on ice (0 min at 37 °C) prior to reversal of biotinylation, as control for those samples that were subsequently switched to 37 °C. The third and fourth lanes show internalized TβRI and TβRII receptors after 20 min at 37 °C, in the absence or presence of TGF-β, after reversal of biotinylation. TβRI and TβRII internalization was enhanced in response to TGF-β1. C, using the same treatment regimen, the presence of AktVIII during incubation at 37 °C attenuated the TGF-β-stimulated internalization of TβRI and TβRII. TfR and GAPDH levels in cell lysates serve as loading controls.
These results may explain the partial, rather than complete, repression of TGF-β–induced receptor presentation upon Akt inhibition. A scenario in which AktVIII treatment blocks the Akt-dependent increase in receptor translocation to the cell surface and also attenuates receptor internalization may explain the incomplete repression of TGF-β–induced cell-surface TGF-β receptor up-regulation by AktVIII.
Enhanced TGF-β receptor availability at the cell surface increases Smad activation
TGF-β binding to the receptors stabilizes the interaction between TβRI and TβRII, which results in activation of the TβRI kinase and enables it to C-terminally phosphorylate, and thus activate, the TβRI-associated Smads (5, 8). To address the extent to which TGF-β–induced TGF-β receptor up-regulation plays a role in the initiation of Smad signaling, we assessed TGF-β–induced stabilization of the TβRI–TβRII complex, the association of Smad3 with activated TGF-β receptors, and Smad2 and Smad3 activation in response to TGF-β.
TGF-β–induced association of TβRI and TβRII was assessed by coimmunoprecipitation of TβRII with TβRI, visualized by immunoblotting. As is apparent from Fig. 5A, TGF-β induced the association of TβRII with TβRI. The level of TβRI-associated TβRII was substantially less when Akt activation was prevented using AktVIII. This difference is consistent with the much lower level of TβRII and TβRI at the surface of cells treated with AktVIII. Association of Smad3 with the cell-surface TGF-β receptors was assessed by purifying the biotinylated proteins at the cell surface and then immunoblotting for associated Smad3. This analysis revealed a TGF-β–induced recruitment of Smad3 to cell-surface proteins (Fig. 5B). Concomitant treatment with AktVIII strongly repressed the amount of Smad3 associated with cell-surface proteins, which is consistent with AktVIII's ability to reduce the cell-surface TGF-β receptor up-regulation. The decrease in cell surface–associated Smad3 correlates with a decrease in cell-surface TGF-β receptor complexes.
Figure 5.

Enhanced TGF-β receptor availability at the cell surface confers increased TβRI–TβRII receptor association and Smad activation. A, coimmunoprecipitation of TβRI and TβRII in HaCaT cells treated with TGF-β1 for 30 min in the presence of AktVIII inhibitor or control solution. Inhibition of Akt with AktVIII decreased the ligand-induced interaction of TβRI and TβRII. B, cell-surface analysis of HaCaT cells treated with TGF-β1 in the presence or absence of AktVIII. TGF-β1 promoted association of Smad3 with cell-surface proteins, which was inhibited by AktVIII. C and D, immunoblot (IB) analysis of Smad2 and Smad3 activation. HaCaT cells were stimulated with TGF-β1 for the times indicated in the presence of AktVIII or control solution (C) or for 30 min with TGF-β1 in the presence of MK2206 or control (D). Smad2 and Smad3 activation in response to TGF-β1, assessed by anti–phospho-Smad2 or -Smad3 immunoblotting, was attenuated by AktVIII or MK2206, whereas total Smad2 and Smad3 protein levels were unaffected by TGF-β1 or AktVIII or MK2206. Note that in this figure, B presents data from the same experiment shown in Fig. 2C, and D presents data obtained from the same experiment as shown in the right panel of Fig. 2A.
Finally, the decreased level of receptor-associated Smad3 in conjunction with attenuated TGF-β receptor cell-surface presentation led us to evaluate TGF-β–induced Smad activation. In a timeframe consistent with the inhibition of receptor induction shown in Fig. 2, AktVIII treatment decreased TGF-β–induced Smad2 and Smad3 activation, detected by immunoblotting for C-terminally phosphorylated Smads, whereas overall Smad2 and Smad3 levels remained unchanged (Fig. 5C). A similar decrease in Smad2 and Smad3 activation was seen when the cells were treated with the Akt inhibitor MK2206 (Fig. 5D) or with the PI3 kinase inhibitor LY294002 (Fig. S1, A and B). These data correlate Smad activation with TβRI and TβRII receptor up-regulation at the cell surface.
Together, these results illustrate increased receptor activation, Smad recruitment, and Smad activation as a result of the TGF-β–induced increase in TβRII and TβRI availability at the cell surface. They also illustrate the role of TGF-β–induced Akt activation in these proximal TGF-β responses.
TGF-β–induced TGF-β receptor up-regulation at the cell surface amplifies Smad-mediated gene expression
Upon TGF-β binding to the receptors and subsequent C-terminal Smad phosphorylation, activated Smad2 and Smad3 combine with Smad4 to induce or repress target gene expression. Activated Smad3 complexes have been shown to directly bind DNA at Smad-binding elements (SBEs) and to induce transcription from tandem SBEs in reporter assays. We therefore performed reporter assays using SBE-Luc, a reporter plasmid containing a gene encoding firefly luciferase under direct control of tandem SBEs (36). TGF-β induced a 4-fold increase in the expression of luciferase, which was decreased by one-third when cells were concomitantly treated with AktVIII (Fig. 6A). As expected, the TβRI kinase inhibitor SB431542 fully inhibited TGF-β–induced luciferase expression from tandem SBEs (Fig. 6B).
Figure 6.

TGF-β–induced increase of TGF-β receptors at the cell surface enhances the cell response to TGF-β. A and B, Smad3 reporter assays in HaCaT cells treated or not with TGF-β1 and/or AktVIII (A) or SB431542 (B). Cells were transfected with a luciferase reporter under the control of Smad-binding elements (SBE-Luc) 24 h prior to addition of inhibitors and stimulation with TGF-β1 for the times indicated. AktVIII repressed the Smad-mediated transcription response, quantified by luminescence. C and D, relative mRNA levels of selected endogenous TGF-β–responsive genes. mRNAs of the indicated genes were quantified by qRT-PCR and normalized to expression of RPL19 mRNA (internal control) in HaCaT cells treated for 90 min with TGF-β1 and/or AktVIII (C) or SB431542 (D). AktVIII decreased the TGF-β–induced SERPINE1, SNAI2, and SMAD7 expression, whereas blocking the TβRI kinase with SB431542 prevented their expression. All data are shown as the means ± S.D. (error bars) of normalized results from three independent experiments. Significance was calculated from unpaired t tests (Mann–Whitney U method) using data from three independent experiments (E–F). *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. RLU, relative light units.
We also quantified TGF-β–induced endogenous expression of the SERPINE1, SNAI2, and SMAD7 genes, which encode PAI-1, Slug/Snail2, and Smad7, respectively. These genes are directly targeted by Smad3/4 complexes in response to TGF-β and are consequently rapidly activated in response to TGF-β (37–40). As shown in Fig. 6C, TGF-β induced within 90 min the expression of these genes, quantified by measuring the mRNA levels using qRT-PCR, and these responses were attenuated in the presence of AktVIII (Fig. 6C). As expected, SB431542 inhibited transcriptional activation in response to TGF-β (Fig. 6D). These transcription activation data support a model in which TGF-β receptor up-regulation in response to TGF-β enhances Smad activation, leading to amplification of Smad-mediated gene expression and enhancing the cell's responsiveness to TGF-β. Inhibition of the TGF-β response in the presence of AktVIII is consistent with contribution of Akt activation to the TGF-β–induced increase in receptor availability at the cell surface.
BMP-4 induces increased cell-surface levels of TGF-β receptors and autocrine TGF-β signaling
The role of Akt activation in driving increased cell-surface presentation of TGF-β receptors raised the question of whether BMP signaling, which results in Akt activation (41), promotes an increase in TGF-β receptor availability and autocrine TGF-β signaling. Such up-regulation of TGF-β receptors would then represent a proximal node of cross-talk between TGF-β and BMP signaling pathways. To address this question, HaCaT cells were treated with 5 ng/ml BMP-4, and the levels of cell-surface TGF-β receptors were assessed by cell-surface protein biotinylation followed by immunoblotting of the biotinylated proteins. Treatment with BMP-4 resulted in a rapid increase in cell-surface TβRI and TβRII (Fig. 7A). Similarly to the induction of cell-surface receptors in response to TGF-β, the BMP-4–induced increase of TβRI and TβRII at the cell surface was inhibited by AktVIII (Fig. 7A). The BMP-4–induced up-regulation of TGF-β receptors at the cell surface was lower than the up-regulation seen in response to TGF-β and was not affected by the presence of the pan–TGF-β neutralizing antibody 1D11 (Fig. 7B), which prevented TGF-β–induced activation of Akt, Smad2, and Smad3 (Fig. S2). A lack of antibodies that clearly recognize BMP receptors at endogenous levels prevented us from assessing the cell-surface levels of BMP receptors.
Figure 7.

BMP signaling promotes increased cell-surface TGF-β receptor levels, thus enhancing the autocrine TGF-β response. A, cell-surface biotinylation analysis of HaCaT cells stimulated with BMP-4 for the times indicated in the presence or absence of AktVIII. BMP-4 induced an increase of TβRI and TβRII at the cell surface, and AktVIII repressed this TGF-β receptor response to BMP-4. B, cell-surface biotinylation analysis of HaCaT cells stimulated for 30 min with TGF-β1 (lanes 2 and 3) or BMP-4 (lanes 4 and 5) in the absence or presence of the anti-TGF-β neutralizing antibody 1D11. The 1D11 antibody suppressed the TGF-β–induced increase of the TβRI and TβRII receptors at the cell surface, but not the milder induction of this cell-surface response to BMP-4. C, immunoblot (IB) analysis of Smad activation in HaCaT cells in response to BMP-4, in the presence or absence of SB431542. BMP-4 induced the activation of the TGF-β–responsive Smad3 and Smad2, assessed by immunoblotting for phospho-Smad3 and -Smad2, in addition to the BMP-responsive Smad1/5. SB431542 prevented activation of Smad3 and Smad2 but did not affect the BMP-responsive Smad1/5 activation. D, immunoblot analysis of Smad activation in HaCaT cells in response to TGF-β1 (lanes 1–8) versus BMP-4 (lanes 9–16) in the presence or absence of the neutralizing anti-TGF-β antibody 1D11. BMP-4 treatment resulted in activation of the TGF-β–responsive Smad2 and Smad3, assessed by immunoblotting for phospho-Smad3 and -Smad2, to a much lower level than TGF-β. Activation of Smad2 and Smad3 in response to either TGF-β or BMP-4 was suppressed by the 1D11, whereas the BMP-induced activation of Smad1/5 was not. E–G, expression of TGF-β- and BMP-regulated genes in response to BMP-4 stimulation. HaCaT cells were treated with BMP-4 for 90 min in the presence or absence of SB431542 (E and F) or AktVIII (G). mRNAs of the indicated genes were quantified by qRT-PCR and normalized to RPL19 mRNA (internal control). Induction of the TGF-β–responsive genes SERPINE1, SNAI2, and SMAD7 by BMP-4 was inhibited by SB431542 (E) and mildly repressed by AktVIII (G), whereas ID1 and ID3 expression, which are induced by the BMP-responsive Smad1 and Smad5, but not by TGF-β responsive Smad2 and Smad3, were unaffected by SB431542 (F). qRT-PCR results (E–G) are shown as the means ± S.D. (error bars) of normalized results from three independent experiments. Significance was calculated from unpaired t tests (Mann–Whitney U method) using data from three independent experiments (E–F). *, p ≤ 0.05; **, p ≤ 0.01; ns, nonsignificant.
Because the TGF-β–induced increase in cell-surface TβRI and TβRII levels enabled increased autocrine TGF-β signaling, we evaluated the level of Smad2 and Smad3 activation in response to BMP-4. As shown in Fig. 7C, BMP-4 induced a low level of Smad2 and Smad3 activation, assessed by immunoblotting for phospho-Smad2 and -Smad3 (Fig. 7B). This activation of Smad2 and Smad3 was inhibited by SB431542, which blocks the TβRI kinase without affecting BMP receptors (42), thus implicating TβRI kinase in BMP-induced phosphorylation of Smad2 and Smad3 (Fig. 7C). Activation of Smad2 and Smad3 in response to 5 ng/ml BMP-4 was substantially lower than the activation seen in response to TGF-β at 0.25 ng/ml, the TGF-β concentration used throughout this study (Fig. 7D). Furthermore, the neutralizing anti–TGF-β antibody 1D11 inhibited BMP-induced Smad2/3 activation, consistent with the notion that it is mediated by autocrine TGF-β signaling (Fig. 7D). These results illustrate the BMP-induced enhancement of cell-surface TGF-β receptors, resulting in increased autocrine TGF-β signaling.
We next evaluated whether the increased levels of TGF-β receptors at the cell surface enable BMP-induced autocrine expression of TGF-β/Smad3 target genes. As was done to evaluate the amplification of the TGF-β response (Fig. 6), we quantified the expression of mRNA for the TGF-β–responsive genes SERPINE1, SNAI2, and SMAD7, which are known to be transcriptionally activated by Smad3/4 complexes, but not by BMP-activated Smad1 and/or Smad5. The expression of all three genes was up-regulated in response to BMP-4 and was inhibited in the presence of the TβRI kinase inhibitor SB431542, consistent with autocrine TGF-β receptor signaling in response to BMP (Fig. 7E). In contrast, BMP-induced induction of ID1 and ID3, which are directly activated by BMP signaling through Smad1 and/or Smad5 (43), was not significantly affected by SB431542 (Fig. 7F). BMP-induced activation of SERPINE1, SNAI2, and SMAD7 was only mildly repressed by AktVIII (Fig. 7G). This is consistent with incomplete repression of TGF-β–induced activation of these genes by AktVIII (Fig. 6C) and the fact that Akt controls the up-regulation of cell-surface TGF-β receptors but not TβRI-mediated Smad2/3 activation.
Taken together, these data support a model of BMP-induced enhancement of TGF-β responsiveness as a result of TGF-β receptor up-regulation at the cell surface. BMP-induced sensitization of cells to autocrine TGF-β signaling can lead to activation of TGF-β–responsive Smad2 and/or Smad3 and gene expression changes that are associated with TGF-β signaling (Fig. 8).
Figure 8.
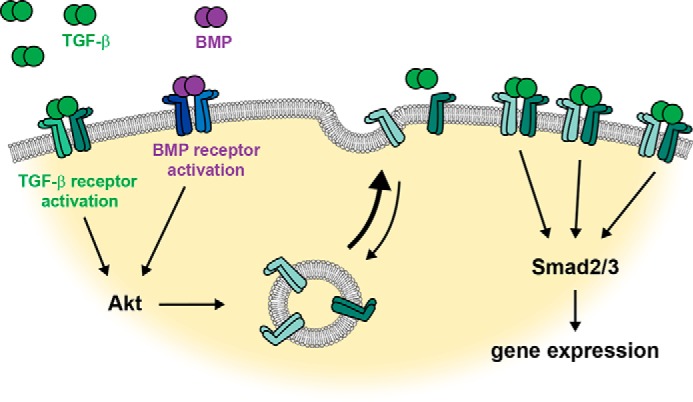
Model of cell-surface TGF-β receptor response to TGF-β and BMP. Our findings allow us to propose the following model: TGF-β or BMP binding to their type I/type II receptor complexes results in activation of Akt, which induces translocation of intracellular TGF-β type I and type II receptors to the cell surface. This increase in receptor availability increases the cell's responsiveness to exogenous or autocrine TGF-β, thus either amplifying Smad3 and Smad2 activation in response to TGF-β or inducing Smad3 and Smad2 activation by autocrine TGF-β signaling in response to BMP. The activation of TGF-β–responsive Smad3 and Smad2 results in activation of direct TGF-β/Smad target genes.
Discussion
Our results reveal a novel mechanism of ligand-induced signaling amplification. Specifically, we show that ligand-induced activation of low levels of cell-surface receptors results in a rapid mobilization of receptors from intracellular stores to the cell surface, thus increasing the abundance of cell-surface receptors and consequently the cell's responsiveness. We defined this mechanism in the context of the TGF-β receptors TβRI and TβRII in response to TGF-β. We additionally show that BMP also promotes the mobilization of intracellular TGF-β receptors to the cell surface, thus resulting in increased sensitivity to TGF-β– and BMP-induced enhancement of autocrine TGF-β signaling (Fig. 8).
TGF-β enhances TGF-β responsiveness by increasing TGF-β receptors at the cell surface
The control of receptor availability plays a major role in defining the amplitude and nature of ligand-induced cell responses, and deregulated receptor routing contributes to disease progression. This has been documented for transmembrane tyrosine kinase receptors that respond to growth factors, including epidermal growth factor, insulin, vascular endothelial growth factor, and platelet-derived growth factor receptors (3, 44–47). In general, increased cell-surface levels of receptors confer increased responsiveness yet can lead to aberrant responses. In the case of TGF-β, the receptor abundance at the cell surface is a major determinant of the cellular response, and cells control TGF-β responsiveness by modulating the availability of cell-surface receptors (13, 20, 22). Several mechanisms, including post-translational modifications, protein degradation, and subcellular compartmentalization, have been shown to control cell-surface levels of TGF-β receptors. N-Glycosylation occurs on TβRI and TβRII and promotes transport of TβRII to the cell surface and increased TGF-β responsiveness (48, 49). Polyubiquitylation resulting in degradation of TGF-β receptors at the cell surface decreases TGF-β responsiveness (5, 50), whereas ectodomain shedding of the TβRI receptor by the metalloprotease TACE (tumor necrosis factor-α-converting enzyme) also decreases cell-surface levels of TGF-β receptors in response to Erk MAPK or p38 MAPK pathway activation (20). Additionally, high glucose and insulin, which acts through a tyrosine kinase receptor, effect a rapid increase in cell-surface TGF-β receptors by promoting transport from intracellular compartments and thus induce increased TGF-β sensitivity and autocrine TGF-β signaling (21, 22). Our results now reveal that TGF-β itself, acting through a low-level population of cell-surface TβRI and TβRII, causes the cells to quickly up-regulate TGF-β receptor availability, thus amplifying TGF-β–induced Smad activation and gene responses. This novel mode of auto-induced signal amplification complements previous mechanisms of positive feedback regulation. Ligand has been shown to increase ligand expression, as first documented for TGF-α and interleukin-1 (51, 52) and subsequently also for TGF-β–induced TGF-β1 expression (53). Additionally, TGF-β ligand can induce the expression of its receptors, which is also predicted to enhance responsiveness (54). The current mode of auto-induced signal amplification is immediate, without a need for new protein synthesis, and does not rule out contributions of these other two modes of signal amplification.
Role of TGF-β–induced Akt activation
We reported that, in response to insulin, rapid increase in cell-surface TGF-β receptors results from insulin-induced Akt activation and phosphorylation of the membrane-associated RabGAP AS160 (22). Consistent with this finding, inhibition of TGF-β–induced Akt activation impaired TGF-β–induced up-regulation of TGF-β receptors at the cell surface. This result reveals a novel role of TGF-β–induced Akt activation in the TGF-β response, adding to other established responses, e.g. in TGF-β–induced gene expression, motility, and progression through EMT (4). However, inhibition of Akt did not fully repress the TGF-β–induced up-regulation of receptors, likely because of the role of Akt in TGF-β–induced internalization of TGF-β receptors. Adding our observations to previous ones, the role of Akt in TGF-β signaling may be complex. Although TGF-β induces Akt phosphorylation separately from Smad activation, Akt activation may either synergize or antagonize, depending on the response. Association of Akt with Smad3 attenuates TGF-β–induced Smad3 activation, thus decreasing TGF-β/Smad-mediated gene responses (55, 56). Akt also controls the activities of various DNA-binding transcription factors with which activated Smad complexes cooperate, thus contributing to the control of TGF-β/Smad-induced gene responses (12, 57, 58). Additionally, Akt activation promotes transport of TGF-β receptors to the cell surface and thus increases the sensitivity and responsiveness to TGF-β (22). Clearly, a complex role of Akt in TGF-β/Smad signaling emerges that may be further exacerbated by a possible role in TGF-β receptor internalization.
We also found that inhibition of TβRI kinase activity prevented TGF-β–induced Akt activation. This is consistent with some observations (30, 34), but at odds with recent observations that TGF-β induces Akt phosphorylation through PI3K activation, mediated by the E3 ligase TRAF6 and independent of TGF-β receptor kinase (31). Alternative mechanisms for the direct activation of Akt in response to TGF-β may exist, depending on cell type or cell physiological context.
BMP-induced TGF-β receptor up-regulation sensitizes cells to autocrine TGF-β
Signaling cross-talk between the BMP- and TGF-β–induced Smad pathways has been previously observed. In some cells, including endothelial cells, TGF-β induces activation of the BMP-responsive Smad1 and Smad5 (59). This “cross-activation” has been seen to result from TβRI-mediated activation of the type I BMP receptor ACVR1 and was shown to be required for cell migration or progression through EMT (60). BMPs have also been observed to stimulate low levels of TGF-β signaling, assessed by activation of TGF-β–responsive Smad2 and Smad3. BMP-induced activation of Smad2 and Smad3 was reported to occur directly through complexes comprising both TGF-β– and BMP-responsive type I receptors (61). We now show that BMP enhances autocrine TGF-β signaling by up-regulating TGF-β receptors at the cell surface, thus increasing the sensitivity to autocrine TGF-β signaling. This is apparent from the activation of TGF-β–responsive Smad2 and Smad3 and TGF-β–induced genes, in response to BMP. Inhibition of Akt activation abrogated the up-regulation of the cell-surface TGF-β receptors, whereas TβRI kinase inhibition prevented activation of Smad2 and Smad3 in response to BMP and inhibited the expression of TGF-β–responsive, but not BMP-responsive, genes.
While providing mechanistic context for activation of TGF-β response by BMP, these findings also highlight the closely intertwined nature of BMP and TGF-β signaling. BMP and TGF-β are often viewed as acting in opposition in development; however, the interaction of these signaling pathways in the spatial and temporal control of differentiation events is far more intricate (62). For example, TGF-β signaling is needed to maintain pluripotency of human embryonic stem cells in culture, whereas BMP signaling promotes their differentiation and enhances definitive endoderm formation driven by TGF-β and activin (63–66). Analysis of the induction and patterning of the definitive endoderm from human pluripotent stem cells in culture revealed that BMP, fibroblast growth factor, and Wnt signaling are needed to establish the primitive streak, whereas TGF-β and fibroblast growth factor signaling cooperatively drive formation of definitive endoderm from the primitive streak (67). In some cases, TGF-β signaling provides a permissive environment for cell fate specification, allowing BMP to initiate differentiation processes. TGF-β enhances proliferation of osteoprogenitor cells (68) and BMPs drive osteoblast differentiation (69), enabling TGF-β to greatly augment the pool of differentiated osteoblasts (70). In this context, low-level activation of TGF-β responses by BMPs may enable BMPs to provide competence for cell differentiation and drive cell fate decisions.
Experimental procedures
Cell culture and transfection
HaCaT and A549 cells were cultured at 37 °C and 5% CO2, in Dulbecco's modified Eagle's medium with 4.5 mm glucose and 10% fetal bovine serum, or RPMI 1640 medium with 10% fetal bovine serum, respectively. For assays involving stimulation with growth factor, the cells were rinsed twice in PBS and then serum-starved for 6 h prior to stimulation. HaCaT cells were starved in serum-free Dulbecco's modified Eagle's medium with 4.5 mm glucose, and A549 cells were starved in in small airway epithelial cell basal medium (Lonza). Transfections were performed in Opti-MEM minimal medium (Thermo Fisher) using TurboFect (Thermo Fisher) for A549 cells or Lipofectamine 2000 (Life Technologies) for HaCaT cells, as specified by the manufacturers.
Growth factors, inhibitors, and antibodies
TGF-β1 and BMP-4, purchased from HumanZyme, were added to culture medium at 0.25 or 5 ng/ml, respectively. The phospho-Akt inhibitor AktVIII (EMD Millipore) and the TβRI kinase inhibitor SB431542 (Sigma) were used at 5 μm. AktVIII and SB431542 were added to culture medium for 20 min and 2 h, respectively, prior to adding TGF-β. The PI3K inhibitor LY294002 (Millipore) was used at a final concentration of 100 μm. The ellagitannin corilagin (BOC Biosciences) was added to culture media at 100 nm for 6 h prior to TGF-β treatment, as described (33). The TGF-β–neutralizing antibody 1D11 (R&D Systems) was added to culture medium, at a final concentration of 0.2 μg/ml, 1 h prior to adding TGF-β or BMP-4. Immunostaining and/or coimmunoprecipitations were performed using rabbit anti-TβRII and mouse anti-transferrin receptor antibodies from Santa Cruz Biotechnology, rabbit anti-TβRI and anti-phospho-Smad3 from Abcam, and rabbit anti-Smad2, anti-phospho-Smad2, anti-Smad3, anti-phospho-Akt (Ser-473), and anti-phospho-Akt (Thr-308) from Cell Signaling. Horseradish peroxidase–conjugated secondary antibodies for visualization of Western blots were AffiniPure goat anti-mouse IgG, light chain-specific and IgG fraction monoclonal mouse anti-rabbit IgG, light chain-specific antibodies from Jackson ImmunoResearch.
Cell lysis, immunoprecipitation, and Western blotting
For immunoblotting assays without immunoprecipitation, cells were rinsed twice with PBS and lysed at in ice-cold radioimmune precipitation assay buffer, i.e. 10 mm Tris-HCl, pH 7.6, 1 mm EDTA, 1% Nonidet P-40, 0.25% sodium deoxycholate, 0.1% SDS, 150 mm NaCl, 10 mm NaF, 1 mm Na3VO4, and cOmplete Protease Inhibitor mixture (Roche). The lysates were cleared by centrifugation for 10 min at 14,000 × g and 4 °C, and protein concentration was quantified using protein assay dye reagent (Bio-Rad). Total protein was normalized between samples before denaturation with LDS sample buffer (Invitrogen) at 95 °C for 2 min and analyzed by SDS-PAGE and immunoblotting. For immunoprecipitation of endogenous TβRI, the cells were washed twice after appropriate treatment and harvested by scraping in prechilled lysis buffer containing 25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 0.75% Triton X-100, 7% glycerol, 10 mm NaF, 1 mm Na3VO4, and cOmplete Protease Inhibitor mixture (Roche) and incubated for 10 min on ice. Lysates were cleared as described above and then immunoprecipitated with anti-TβRI overnight. The antibody-lysate solution was then centrifuged for 10 min at 14,000 × g and incubated with protein G–Sepharose beads (GE Healthcare) for 2 h. The beads were washed three times for 10 min with lysis buffer, eluted with LDS sample buffer (Invitrogen), and subjected to SDS-PAGE and immunoblotting. All steps until elution were performed at 4 °C.
Cell-surface protein biotinylation
The cells were washed twice with cold PBS and incubated for 20 min with EZ Link Sulfo-NHS-LC-Biotin (Thermo Scientific) at a concentration of 0.25 mg/ml in PBS. The biotinylation reaction was quenched by washing the cells twice with PBS and incubating them for 15 min with 0.1 m glycine in PBS. Cell lysates were then harvested after cell lysis in a mild lysis buffer (MLB) containing 20 mm Tris, pH 7.6, 200 mm NaCl, 1% Nonidet P-40, 10 mm NaF, 1 mm Na3VO4, and cOmplete Protease Inhibitor mixture (Roche) for 10 min and removing the lysate by scraping. Lysates were cleared by centrifugation at 14,000 × g for 10 min and then incubated with NeutrAvidin beads (Thermo Scientific) overnight. The beads were washed three times in MLB, eluted with LDS sample buffer (Invitrogen), and subjected to SDS-PAGE and immunoblotting. All steps until elution were performed at 4 °C.
Cell-surface receptor internalization
Following serum starvation, the cells were washed twice with cold PBS and incubated for 20 min with 0.4 mg/ml EZ Link Sulfo-NHS-SS-Biotin (Thermo Scientific) in PBS on ice. The cross-linking reaction of biotin with cell-surface proteins was then quenched with 0.1 m glycine in PBS at 4 °C for 15 min. The quenching solution was removed by washing with ice cold PBS, and the cells were returned to prewarmed, serum-free medium containing growth factors and inhibitors and incubated for 20 min at 37 °C, allowing endocytosis to proceed. Endocytosis of cell-surface proteins was stopped by washing the cells twice with cold PBS on ice and immediately proceeding to removal of biotin label from proteins that remain exposed at the extracellular surface: covalently linked, disulfide-bonded biotin was removed from biotinylated proteins at the cell surface by reducing the disulfide bond, accomplished by incubating cells twice for 15 on ice with a mildly reducing buffer containing 50 mm GSH, 75 mm NaCl, 75 mm NaOH, and 10% fetal bovine serum in PBS. The disulfide bond reduction was quenched by a 30-min incubation with a solution of 50 mm sodium iodoacetamide and 1% BSA in PBS at 4 °C. The cells were then washed twice in cold PBS and lysed in prechilled MLB buffer on ice for 10 min and harvested by scraping. The lysates were cleared by centrifugation at 14,000 × g for 10 min and incubated at 4 °C overnight with NeutrAvidin beads (Thermo Scientific). The beads were washed three times in chilled MLB, eluted with LDS sample buffer (Invitrogen), and subjected to SDS-PAGE and immunoblotting.
Luciferase reporter assay
The cells were transfected with the plasmid SBE-Luc, which contains the firefly luciferase coding sequence under control of four tandem SBEs and a Renilla luciferase reporter downstream of the thymidine kinase promoter (Promega) as control. They were then serum-starved for 20 h, stimulated with the appropriate growth factors and inhibitors for defined times, rinsed with PBS, and, after aspirating liquid from the monolayer, frozen at −80 °C and thawed on ice before lysis. Relative luciferase activity was quantified using a dual luciferase assay system (Promega) according to the manufacturer's protocol. Readout was normalized against Renilla luciferase expression as an internal control. Mean and standard deviation were calculated from the Renilla-normalized readout of three independent samples.
RNA preparation and quantitative real-time PCR
RNA was purified from cells using the RNEasy mini kit (Qiagen). Total RNA (1 μg/sample) was used as a template for reverse transcription with iScript (Bio-Rad) according to the manufacturer's protocol. SERPINE1, SNAI2, SMAD7, SMAD6, ID1, and ID3 mRNA levels were quantified by RT-PCR using IQ SYBR-Green Supermix (Bio-Rad) and normalized against RPL19 mRNA for a ribosomal protein. The results of three separate experiments, each calculated from three technical replicates, were pooled using the ΔΔCT method. The primers used are shown in Table S1.
Author contributions
D. D. and R. D. conceptualization; D. D. data curation; D. D. and R. D. formal analysis; D. D. investigation; D. D. visualization; D. D. methodology; D. D. writing-original draft; D. D. and R. D. writing-review and editing; R. D. supervision; R. D. funding acquisition; R. D. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Erine Budi, whose previous observations initiated this project and whose biotinylation protocol guided this research; members of the Derynck lab and the lab of Dr. Katja Brückner for helpful discussions and feedback; and Dr. Harold Chapman for A549 cells and corilagin.
This work was supported by Grants RO1-CA63101 and RO1-CA136690 from the National Cancer Institute (to R. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1 and S2.
- BMP
- bone morphogenetic protein
- TGF-β
- transforming growth factor–β
- PI3K
- phosphatidylinositol 3-kinase
- MAPK
- mitogen-activated protein kinase
- TβR
- TGF-β receptor
- TfR
- transferrin receptor
- SBE
- Smad-binding element
- MLB
- mild lysis buffer.
References
- 1. Lemmon M. A., and Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 10.1016/j.cell.2010.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tomas A., Futter C. E., and Eden E. R. (2014) EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 24, 26–34 10.1016/j.tcb.2013.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Casaletto J. B., and McClatchey A. I. (2012) Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 12, 387–400 10.1038/nrc3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morikawa M., Derynck R., and Miyazono K. (2016) TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 8, a021873 10.1101/cshperspect.a021873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heldin C. H., and Moustakas A. (2016) Signaling receptors for TGF-β family members. Cold Spring Harb. Perspect. Biol. 8, a022053 10.1101/cshperspect.a022053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim K. K., Sheppard D., and Chapman H. A. (2018) TGF-β1 signaling and tissue fibrosis. Cold Spring Harb. Perspect. Biol. 10, a022293 10.1101/cshperspect.a022293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Massagué J. (2008) TGF-β in cancer. Cell 134, 215–230 10.1016/j.cell.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hata A., and Chen Y.-G. (2016) TGF-β signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 8, a022061 10.1101/cshperspect.a022061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feng X. H., and Derynck R. (2005) Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 10.1146/annurev.cellbio.21.022404.142018 [DOI] [PubMed] [Google Scholar]
- 10. Hill C. S. (2016) Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 8, a022079 10.1101/cshperspect.a022079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moustakas A., and Heldin C. H. (2005) Non-Smad TGF-β signals. J. Cell Sci. 118, 3573–3584 10.1242/jcs.02554 [DOI] [PubMed] [Google Scholar]
- 12. Zhang Y. E. (2017) Non-Smad signaling pathways of the TGF-β family. Cold Spring Harb. Perspect. Biol. 9, a022129 10.1101/cshperspect.a022129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Budi E. H., Duan D., and Derynck R. (2017) Transforming growth factor-β receptors and smads: regulatory complexity and functional versatility. Trends Cell Biol. 27, 658–672 10.1016/j.tcb.2017.04.005 [DOI] [PubMed] [Google Scholar]
- 14. Le Roy C., and Wrana J. L. (2005) Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat. Rev. Mol. Cell Biol. 6, 112–126 10.1038/nrm1571 [DOI] [PubMed] [Google Scholar]
- 15. Yao D., Ehrlich M., Henis Y. I., and Leof E. B. (2002) Transforming growth factor-β receptors interact with AP2 by direct binding to β2 subunit. Mol. Biol. Cell 13, 4001–4012 10.1091/mbc.02-07-0104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meyer C., Liu Y., Kaul A., Peipe I., and Dooley S. (2013) Caveolin-1 abrogates TGF-β mediated hepatocyte apoptosis. Cell Death Dis. 4, e466 10.1038/cddis.2012.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zuo W., and Chen Y. G. (2009) Specific activation of mitogen-activated protein kinase by transforming growth factor-β receptors in lipid rafts is required for epithelial cell plasticity. Mol. Biol. Cell 20, 1020–1029 10.1091/mbc.e08-09-0898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muthusamy B. P., Budi E. H., Katsuno Y., Lee M. K., Smith S. M., Mirza A. M., Akhurst R. J., and Derynck R. (2015) ShcA protects against epithelial-mesenchymal transition through compartmentalized inhibition of TGF-β–induced Smad activation. PLos Biol. 13, e1002325 10.1371/journal.pbio.1002325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Di Guglielmo G. M., Le Roy C., Goodfellow A. F., and Wrana J. L. (2003) Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 5, 410–421 10.1038/ncb975 [DOI] [PubMed] [Google Scholar]
- 20. Liu C., Xu P., Lamouille S., Xu J., and Derynck R. (2009) TACE-mediated ectodomain shedding of the type I TGF-β receptor downregulates TGF-β signaling. Mol. Cell 35, 26–36 10.1016/j.molcel.2009.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu L., and Derynck R. (2009) Essential role of TGF-β signaling in glucose-induced cell hypertrophy. Dev. Cell 17, 35–48 10.1016/j.devcel.2009.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Budi E. H., Muthusamy B. P., and Derynck R. (2015) The insulin response integrates increased TGF-β signaling through Akt-induced enhancement of cell surface delivery of TGF-β receptors. Sci. Signal. 8, ra96 10.1126/scisignal.aaa9432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zavadil J., Bitzer M., Liang D., Yang Y. C., Massimi A., Kneitz S., Piek E., and Bottinger E. P. (2001) Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc. Natl. Acad. Sci. U.S.A. 98, 6686–6691 10.1073/pnas.111614398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xi Y., Tan K., Brumwell A. N., Chen S. C., Kim Y. H., Kim T. J., Wei Y., and Chapman H. A. (2014) Inhibition of epithelial-to-mesenchymal transition and pulmonary fibrosis by methacycline. Am. J. Respir. Cell Mol. Biol. 50, 51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao Z., Leister W. H., Robinson R. G., Barnett S. F., Defeo-Jones D., Jones R. E., Hartman G. D., Huff J. R., Huber H. E., Duggan M. E., and Lindsley C. W. (2005) Discovery of 2,3,5-trisubstituted pyridine derivatives as potent Akt1 and Akt2 dual inhibitors. Bioorg. Med. Chem. Lett. 15, 905–909 10.1016/j.bmcl.2004.12.062 [DOI] [PubMed] [Google Scholar]
- 26. Calleja V., Laguerre M., Parker P. J., and Larijani B. (2009) Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 7, e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hirai H., Sootome H., Nakatsuru Y., Miyama K., Taguchi S., Tsujioka K., Ueno Y., Hatch H., Majumder P. K., Pan B. S., and Kotani H. (2010) MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 9, 1956–1967 10.1158/1535-7163.MCT-09-1012 [DOI] [PubMed] [Google Scholar]
- 28. Tan S., Ng Y., and James D. E. (2011) Next-generation Akt inhibitors provide greater specificity: effects on glucose metabolism in adipocytes. Biochem. J. 435, 539–544 10.1042/BJ20110040 [DOI] [PubMed] [Google Scholar]
- 29. Yi J. Y., Shin I., and Arteaga C. L. (2005) Type I transforming growth factor β receptor binds to and activates phosphatidylinositol 3-kinase. J. Biol. Chem. 280, 10870–10876 10.1074/jbc.M413223200 [DOI] [PubMed] [Google Scholar]
- 30. Lamouille S., and Derynck R. (2007) Cell size and invasion in TGF-β–induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178, 437–451 10.1083/jcb.200611146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hamidi A., Song J., Thakur N., Itoh S., Marcusson A., Bergh A., Heldin C. H., and Landström M. (2017) TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci. Signal. 10, eaal4186 10.1126/scisignal.aal4186 [DOI] [PubMed] [Google Scholar]
- 32. Lee M. K., Pardoux C., Hall M. C., Lee P. S., Warburton D., Qing J., Smith S. M., and Derynck R. (2007) TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 26, 3957–3967 10.1038/sj.emboj.7601818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wei Y., Kim T. J., Peng D. H., Duan D., Gibbons D. L., Yamauchi M., Jackson J. R., Le Saux C. J., Calhoun C., Peters J., Derynck R., Backes B. J., and Chapman H. A. (2017) Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J. Clin. Invest. 127, 3675–3688 10.1172/JCI94624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bakin A. V., Rinehart C., Tomlinson A. K., and Arteaga C. L. (2002) p38 mitogen-activated protein kinase is required for TGF-β-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 115, 3193–3206 [DOI] [PubMed] [Google Scholar]
- 35. Gabriel L., Stevens Z., and Melikian H. (2009) Measuring plasma membrane protein endocytic rates by reversible biotinylation. J. Vis. Exp. 34, 1669 10.3791/1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zawel L., Dai J. L., Buckhaults P., Zhou S., Kinzler K. W., Vogelstein B., and Kern S. E. (1998) Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1, 611–617 10.1016/S1097-2765(00)80061-1 [DOI] [PubMed] [Google Scholar]
- 37. Dennler S., Itoh S., Vivien D., ten Dijke P., Huet S., and Gauthier J. M. (1998) Direct binding of Smad3 and Smad4 to critical TGF-β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17, 3091–3100 10.1093/emboj/17.11.3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hua X., Liu X., Ansari D. O., and Lodish H. F. (1998) Synergistic cooperation of TFE3 and Smad proteins in TGF-β–induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev. 12, 3084–3095 10.1101/gad.12.19.3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morita T., Mayanagi T., and Sobue K. (2007) Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J. Cell Biol. 179, 1027–1042 10.1083/jcb.200708174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Denissova N. G., Pouponnot C., Long J., He D., and Liu F. (2000) Transforming growth factor β-inducible independent binding of SMAD to the Smad7 promoter. Proc. Natl. Acad. Sci. U.S.A. 97, 6397–6402 10.1073/pnas.090099297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ghosh-Choudhury N., Abboud S. L., Nishimura R., Celeste A., Mahimainathan L., and Choudhury G. G. (2002) Requirement of BMP-2-induced phosphatidylinositol 3-kinase and Akt serine/threonine kinase in osteoblast differentiation and Smad-dependent BMP-2 gene transcription. J. Biol. Chem. 277, 33361–33368 10.1074/jbc.M205053200 [DOI] [PubMed] [Google Scholar]
- 42. Laping N. J., Grygielko E., Mathur A., Butter S., Bomberger J., Tweed C., Martin W., Fornwald J., Lehr R., Harling J., Gaster L., Callahan J. F., and Olson B. A. (2002) Inhibition of transforming growth factor (TGF)-β1-induced extracellular matrix with a novel inhibitor of the TGF-β type I receptor kinase activity: SB-431542. Mol. Pharmacol. 62, 58–64 10.1124/mol.62.1.58 [DOI] [PubMed] [Google Scholar]
- 43. Miyazono K., and Miyazawa K. (2002) Id: a target of BMP signaling. Sci. STKE 2002, pe40 [DOI] [PubMed] [Google Scholar]
- 44. Maudsley S., Pierce K. L., Zamah A. M., Miller W. E., Ahn S., Daaka Y., Lefkowitz R. J., and Luttrell L. M. (2000) The β2-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J. Biol. Chem. 275, 9572–9580 10.1074/jbc.275.13.9572 [DOI] [PubMed] [Google Scholar]
- 45. Ceresa B. P., Kao A. W., Santeler S. R., and Pessin J. E. (1998) Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol. Cell Biol. 18, 3862–3870 10.1128/MCB.18.7.3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Y., Nakayama M., Pitulescu M. E., Schmidt T. S., Bochenek M. L., Sakakibara A., Adams S., Davy A., Deutsch U., Lüthi U., Barberis A., Benjamin L. E., Mäkinen T., Nobes C. D., and Adams R. H. (2010) Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486 10.1038/nature09002 [DOI] [PubMed] [Google Scholar]
- 47. Wang Y., Pennock S. D., Chen X., Kazlauskas A., and Wang Z. (2004) Platelet-derived growth factor receptor-mediated signal transduction from endosomes. J. Biol. Chem. 279, 8038–8046 10.1074/jbc.M311494200 [DOI] [PubMed] [Google Scholar]
- 48. Kim Y. W., Park J., Lee H. J., Lee S. Y., and Kim S. J. (2012) TGF-β sensitivity is determined by N-linked glycosylation of the type II TGF-β receptor. Biochem. J. 445, 403–411 10.1042/BJ20111923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Goetschy F., Letourneur O., Cerletti N., and Horisberger M. A. (1996) The unglycosylated extracellular domain of type-II receptor for transforming growth factor-β. A novel assay for characterizing ligand affinity and specificity. Eur. J. Biochem. 241, 355–362 10.1111/j.1432-1033.1996.00355.x [DOI] [PubMed] [Google Scholar]
- 50. Liu S., de Boeck M., van Dam H., and Ten Dijke P. (2016) Regulation of the TGF-β pathway by deubiquitinases in cancer. Int. J. Biochem. Cell Biol. 76, 135–145 10.1016/j.biocel.2016.05.001 [DOI] [PubMed] [Google Scholar]
- 51. Coffey R. J. Jr., Derynck R., Wilcox J. N., Bringman T. S., Goustin A. S., Moses H. L., and Pittelkow M. R. (1987) Production and auto-induction of transforming growth factor-α in human keratinocytes. Nature 328, 817–820 10.1038/328817a0 [DOI] [PubMed] [Google Scholar]
- 52. Manson J. C., Symons J. A., Di Giovine F. S., Poole S., and Duff G. W. (1989) Autoregulation of interleukin 1 production. Eur. J. Immunol. 19, 261–265 10.1002/eji.1830190207 [DOI] [PubMed] [Google Scholar]
- 53. Van Obberghen-Schilling E., Roche N. S., Flanders K. C., Sporn M. B., and Roberts A. B. (1988) Transforming growth factor β1 positively regulates its own expression in normal and transformed cells. J. Biol. Chem. 263, 7741–7746 [PubMed] [Google Scholar]
- 54. Menke A., Geerling I., Giehl K., Vogelmann R., Reinshagen M., and Adler G. (1999) Transforming growth factor-β–induced upregulation of transforming growth factor-β receptor expression in pancreatic regeneration. Biochim. Biophys. Acta 1449, 178–185 10.1016/S0167-4889(99)00011-7 [DOI] [PubMed] [Google Scholar]
- 55. Conery A. R., Cao Y., Thompson E. A., Townsend C. M. Jr., Ko T. C., and Luo K. (2004) Akt interacts directly with Smad3 to regulate the sensitivity to TGF-β–induced apoptosis. Nat. Cell Biol. 6, 366–372 10.1038/ncb1117 [DOI] [PubMed] [Google Scholar]
- 56. Remy I., Montmarquette A., and Michnick S. W. (2004) PKB/Akt modulates TGF-β signalling through a direct interaction with Smad3. Nat. Cell Biol. 6, 358–365 10.1038/ncb1113 [DOI] [PubMed] [Google Scholar]
- 57. Singh A. M., Reynolds D., Cliff T., Ohtsuka S., Mattheyses A. L., Sun Y., Menendez L., Kulik M., and Dalton S. (2012) Signaling network crosstalk in human pluripotent cells: a Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell 10, 312–326 10.1016/j.stem.2012.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen Y. G., Li Z., and Wang X. F. (2012) Where PI3K/Akt meets Smads: the crosstalk determines human embryonic stem cell fate. Cell Stem Cell 10, 231–232 10.1016/j.stem.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu I. M., Schilling S. H., Knouse K. A., Choy L., Derynck R., and Wang X. F. (2009) TGF-β-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGF-β switch. EMBO J. 28, 88–98 10.1038/emboj.2008.266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ramachandran A., Vizán P., Das D., Chakravarty P., Vogt J., Rogers K. W., Müller P., Hinck A. P., Sapkota G. P., and Hill C. S. (2018) TGF-β uses a novel mode of receptor activation to phosphorylate Smad1/5 and induce epithelial-to-mesenchymal transition. Elife 7, e31756 10.7554/eLife.31756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Holtzhausen A., Golzio C., How T., Lee Y. H., Schiemann W. P., Katsanis N., and Blobe G. C. (2014) Novel bone morphogenetic protein signaling through Smad2 and Smad3 to regulate cancer progression and development. FASEB J. 28, 1248–1267 10.1096/fj.13-239178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mullen A. C., and Wrana J. L. (2017) TGF-β family signaling in embryonic and somatic stem-cell renewal and differentiation. Cold Spring Harb. Perspect. Biol. 9, a022186 10.1101/cshperspect.a022186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. James D., Levine A. J., Besser D., and Hemmati-Brivanlou A. (2005) TGF-β/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 132, 1273–1282 10.1242/dev.01706 [DOI] [PubMed] [Google Scholar]
- 64. Xu R. H., Sampsell-Barron T. L., Gu F., Root S., Peck R. M., Pan G., Yu J., Antosiewicz-Bourget J., Tian S., Stewart R., and Thomson J. A. (2008) NANOG is a direct target of TGF-β/activin-mediated Smad signaling in human ESCs. Cell Stem Cell 3, 196–206 10.1016/j.stem.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gadue P., Huber T. L., Paddison P. J., and Keller G. M. (2006) Wnt and TGF-β signaling are required for the induction of an in vitro model of primitive streak formation using embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 103, 16806–16811 10.1073/pnas.0603916103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Teo A. K., Ali Y., Wong K. Y., Chipperfield H., Sadasivam A., Poobalan Y., Tan E. K., Wang S. T., Abraham S., Tsuneyoshi N., Stanton L. W., and Dunn N. R. (2012) Activin and BMP4 synergistically promote formation of definitive endoderm in human embryonic stem cells. Stem Cells 30, 631–642 10.1002/stem.1022 [DOI] [PubMed] [Google Scholar]
- 67. Loh K. M., Ang L. T., Zhang J., Kumar V., Ang J., Auyeong J. Q., Lee K. L., Choo S. H., Lim C. Y., Nichane M., Tan J., Noghabi M. S., Azzola L., Ng E. S., Durruthy-Durruthy J., et al. (2014) Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell 14, 237–252 10.1016/j.stem.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Erlebacher A., Filvaroff E. H., Ye J. Q., and Derynck R. (1998) Osteoblastic responses to TGF-β during bone remodeling. Mol. Biol. Cell 9, 1903–1918 10.1091/mbc.9.7.1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Grafe I., Alexander S., Peterson J. R., Snider T. N., Levi B., Lee B., and Mishina Y. (2018) TGF-β family signaling in mesenchymal differentiation. Cold Spring Harb. Perspect. Biol. 10, a022202 10.1101/cshperspect.a022202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Erlebacher A., and Derynck R. (1996) Increased expression of TGF-β2 in osteoblasts results in an osteoporosis-like phenotype. J. Cell Biol. 132, 195–210 10.1083/jcb.132.1.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.