

Abstract
Antibodies that recognize amyloidogenic aggregates with high conformational and sequence specificity are important for detecting and potentially treating a wide range of neurodegenerative disorders, including Alzheimer's and Parkinson's diseases. However, these types of antibodies are challenging to generate because of the large size, hydrophobicity, and heterogeneity of protein aggregates. To address this challenge, we developed a method for generating antibodies specific for amyloid aggregates. First, we grafted amyloidogenic peptide segments from the target polypeptide [Alzheimer's amyloid-β (Aβ) peptide] into the complementarity-determining regions (CDRs) of a stable antibody scaffold. Next, we diversified the grafted and neighboring CDR sites using focused mutagenesis to sample each WT or grafted residue, as well as one to five of the most commonly occurring amino acids at each site in human antibodies. Finally, we displayed these antibody libraries on the surface of yeast cells and selected antibodies that strongly recognize Aβ-amyloid fibrils and only weakly recognize soluble Aβ. We found that this approach enables the generation of monovalent and bivalent antibodies with nanomolar affinity for Aβ fibrils. These antibodies display high conformational and sequence specificity as well as low levels of nonspecific binding and recognize a conformational epitope at the extreme N terminus of human Aβ. We expect that this systematic approach will be useful for generating antibodies with conformational and sequence specificity against a wide range of peptide and protein aggregates associated with neurodegenerative disorders.
Keywords: antibody engineering, monoclonal antibody, amyloid, amyloid-β (Aβ), directed evolution, fibril, neurodegeneration, Alzheimer's disease, aggregate, conformation, gammabody, scFv, yeast surface display
Introduction
Protein aggregation is the seminal event in some of the most devastating neurodegenerative disorders (1, 2). The process of amyloidogenic protein aggregation is a surprisingly complex one, as a single polypeptide can form multiple types of prefibrillar oligomers and fibrillar aggregates with unique 3D structures (3). Intriguingly, there is growing evidence that amyloidogenic aggregates with different 3D structures, for diverse polypeptides including Aβ3 and tau (associated with Alzheimer's disease) and α-synuclein (associated with Parkinson's disease), are linked to unique types of neurodegenerative diseases in a manner that is reminiscent of prion strains (4–10).
Antibodies with both conformational and sequence specificity are essential for evaluating structural differences between various types of amyloidogenic protein aggregates (11, 12). Unfortunately, generating such antibodies is challenging because of the large size and conformational heterogeneity, hydrophobicity, low solubility, and (in some cases) low kinetic stability of protein aggregates. Although some conformational antibodies against amyloidogenic aggregates have been generated using in vivo (immunization) or in vitro (phage and yeast surface display) methods (11, 13–15), it remains extremely challenging to reliably identify monoclonal antibodies with high levels of conformational and sequence specificity for diverse types of amyloidogenic aggregates.
To address this challenge, we have sought to integrate multiple aspects of our previous work (16–22) to develop robust methods for discovering antibodies specific for amyloidogenic aggregates. First, we have previously developed a strategy that involves grafting key amyloidogenic peptide motifs from amyloid-forming polypeptides (Aβ and IAPP) into the CDRs of single domain (variable heavy, VH) or multidomain (single-chain variable fragment, scFv) antibodies (19, 22). The resulting grafted amyloid-motif antibodies (gammabodies) bind to their cognate amyloid fibrils with conformational and sequence specificity. These grafted antibodies recognize amyloid fibrils via homotypic interactions between the grafted amyloidogenic peptides and the cognate peptides in fibrils.
Nevertheless, the key next steps in improving our anti-amyloid grafted antibodies are to improve their affinity and specificity. We have previously developed a mutagenesis method (Natural Diversity Mutagenesis) (17) with the goal of designing antibody libraries for affinity maturation. Given that it is not possible to sample all combinations of mutations using 20 amino acids even in a single antibody CDR (e.g. >1019 possible variants for a CDR with 15 residues), our method involved sampling the WT residue at each targeted CDR site as well as one to five mutations that are most common in human antibodies and which are compatible with degenerate codons. This approach enables testing all possible single, double, and higher-order combinations of CDR mutations in a single library by restricting the diversity at each CDR site. We previously used this library design approach for affinity maturation of single-domain (VHH) antibodies and found that it was highly effective for isolating antibody variants with significant improvements in affinity and specificity (17).
Therefore, we have sought to combine our motif-grafting (16, 18, 19, 22) and natural diversity mutagenesis (17) methods to enable robust and systematic isolation of conformational antibodies against Alzheimer's Aβ42 fibrils with high affinity and specificity (Fig. 1). We reasoned that diversifying the grafted Aβ peptide segments in antibody CDRs (e.g. C-terminal Aβ residues 33–42) would lead to the isolation of antibody mutants with either improved homotypic-like interactions (e.g. mutated Aβ C-terminal segments in antibody CDRs interacting with the Aβ C-terminal region in fibrils) or heterotypic interactions (e.g. mutated Aβ C-terminal segments in antibody CDRs interacting with the Aβ N-terminal or central regions in fibrils). These hypotheses are based on the fact that both homotypic (intermolecular) and heterotypic (intramolecular) interactions involving different Aβ peptide segments are observed in Aβ fibrils (23–28). We also reasoned that the immobilization of Aβ fibrils on magnetic beads (29) would enable reproducible and controlled antigen presentation to antibody-displaying yeast cells. Moreover, we posited that negative selections using magnetic beads coated with disaggregated Aβ42 would eliminate antibodies that possess sequence specificity but not conformational specificity. We also expected that these negative selections would be particularly effective at eliminating antibodies with poor specificity in general because antibody CDRs enriched in positively charged and hydrophobic residues, which are expected to associate with the negatively charged and extremely hydrophobic Aβ42 peptide, have been linked to low antibody specificity (16, 30–35). Finally, we reasoned the conformational specificity of monovalent antibodies that we generate against multivalent amyloid aggregates could be significantly increased by reformatting them as bivalent antibodies to enhance avidity effects (36). Here, we report integrated library design and selection methods that lead to the isolation of anti-amyloid antibodies with high affinity and specificity.
Figure 1.

Approach for designing and sorting antibody libraries to identify antibodies with conformational and sequence specificity for Aβ fibrils. A parental antibody (scFv) was generated with Aβ residues 33–42 grafted into HCDR3 (red). Next, HCDR3 was diversified using natural diversity mutagenesis at 15 CDR sites, and the antibody library was displayed on the surface of yeast (S. cerevisiae). The library was then sorted using negative selections against disaggregated Aβ and positive selections against Aβ fibrils using antigens immobilized on magnetic beads. Finally, antibody variants from the enriched library were expressed as soluble proteins, and their conformational specificity was evaluated for Aβ fibrils relative to disaggregated Aβ.
Results
Design and sorting of a motif-grafted, natural diversity antibody library against Aβ42 fibrils
Toward our goal of isolating conformation-specific antibodies against Aβ fibrils with high specificity, we first designed an antibody library using a methodology that involved several key components. First, we grafted Aβ residues 33GLMVGGVVIA42 into heavy chain CDR3 (HCDR3) of a stable single-chain variable fragment (scFv) (16). Next, we diversified a total of 15 positions within HCDR3 in a manner that sampled the WT or grafted residue as well as one to five of the most frequently occurring residues in human antibodies (Fig. 2A). The natural frequency of each amino acid at a given position in HCDR3 was obtained from the abYsis database (37). We chose to target a high level of mutagenesis, which results in sampling only two to three grafted Aβ residues per antibody variant on average, to enable the selection of antibodies with potentially large improvements in their binding properties.
Figure 2.

Antibody library design based on diversifying heavy chain CDR3 using combinations of amino acids that are most common in human antibodies. A, design of a single-chain antibody library based on sampling the WT or grafted HCDR3 residue as well as 1–5 of the most commonly occurring residues in human antibodies. Degenerate codons were designed for each position to encode the residues highlighted in black (bold) font. The corresponding coverage of natural diversity (as defined as the sum of the average observed percentages of each amino acid at each CDR position in human antibodies) is reported below each codon. Limitations in the combinations of codons that can be sampled using a single degenerate codon prevented sampling some of the most frequently occurring residues (residues that were not sampled are indicated in gray font). B, amino acid logo summary of the HCDR3 sequences for the parental (WT) antibody as well as the theoretical and observed diversity of the initial antibody libraries. The theoretical diversity is based on the expected frequencies of amino acids encoded by each degenerate codon, whereas the observed frequency was obtained by sequencing 27 members of the initial library.
Our library design methodology aimed to maximize the coverage of amino acid diversity observed in human antibodies at each CDR site in our designed libraries. We define natural diversity coverage as the sum of the average percentage occurrence of each amino acid in human antibodies at each CDR site (as given by the abYsis database) for the subset of residues sampled in our library. For example, the average occurrence at the HCDR3 site 100 (Kabat numbering) is 16% glycine and 12% serine for tens of thousands of human antibody sequences in the abYsis database (37), and the degenerate codon used in our library to sample both residues resulted in 28% coverage of natural diversity for human antibodies. At some CDR sites, we achieved >50% coverage of human antibody natural diversity, whereas at other sites the coverage was much lower (e.g. 15%) due to limitations in the combinations of amino acids that could be sampled using a single degenerate codon. For example, at HCDR3 position 100b, sampling the WT residue (methionine) along with the most frequently occurring amino acids (glycine and tyrosine) would lead to inclusion of eight additional amino acids. This is undesirable because it would greatly increase the library size as well as incorporate stop codons and other codons that are rare in human antibodies at this CDR site. Our final design (Fig. 2A) resulted in a library with 6.4 × 107 unique antibody variants, which was small enough to enable sampling of all possible variants using yeast surface display given that we are able to achieve >108 yeast transformants using standard electroporation methods (16, 20, 38).
We next synthesized the antibody DNA library and cloned it into a yeast surface display plasmid as a C-terminal fusion to a yeast cell-surface protein (Aga2). Sequencing analysis of 27 unique variants from the initial library revealed that the amino acids were generally sampled in HCDR3 as expected based on the library design (Fig. 2B and Fig. S1). Moreover, we expressed the antibody library on the surface of yeast and confirmed expression via a c-Myc tag at the antibody C terminus using flow cytometry (data not shown).
Our antibody sorting strategy (as summarized in Fig. 1) involved first performing three negative selections against disaggregated Aβ in round 1. Yeast cells were incubated with magnetic beads coated with disaggregated Aβ, and cells that failed to bind were separated and incubated with a second batch of magnetic beads coated with disaggregated Aβ. After repeating this process for a third time as part of round 1, the remaining (unbound) yeast cells were incubated with magnetic beads coated with Aβ fibrils. Yeast cells that bound to immobilized Aβ fibrils were recovered by regrowing them in fresh media. The number of isolated yeast cells in round 1 was also evaluated by plating them on selective agar plates. As a control to evaluate the performance of our negative selections, we also performed a mock-positive sort against disaggregated Aβ in the same manner as was done for Aβ fibrils. If the negative selections were effective at eliminating antibodies that recognize disaggregated Aβ, we would expect many fewer cells to be retained in these mock-positive selections against disaggregated Aβ relative to the positive selections against aggregated Aβ.
In the first three rounds of antibody sorting, we used a large (100-fold) excess of yeast cells (109 cells) relative to magnetic beads (107 beads). Our reasoning was that most of the library would lack affinity and/or specificity for Aβ fibrils, and thus we sought to enrich rare clones in these early rounds of sorting. In these first two rounds, we retained relatively few cells (Fig. 3A), and the antibody library displayed preference for disaggregated Aβ (Fig. 3B). However, in round 3, we observed a modest (2-fold) preference of the library for aggregated Aβ relative to disaggregated Aβ (Fig. 3B). At this point, we reduced the number of yeast cells (107) while maintaining the number of magnetic beads (107) to improve the stringency of the negative selections. This led to significant improvements in the library specificity for fibrils, including a 47-fold preference of the library for recognizing Aβ fibrils in round 5 (Fig. 3B).
Figure 3.
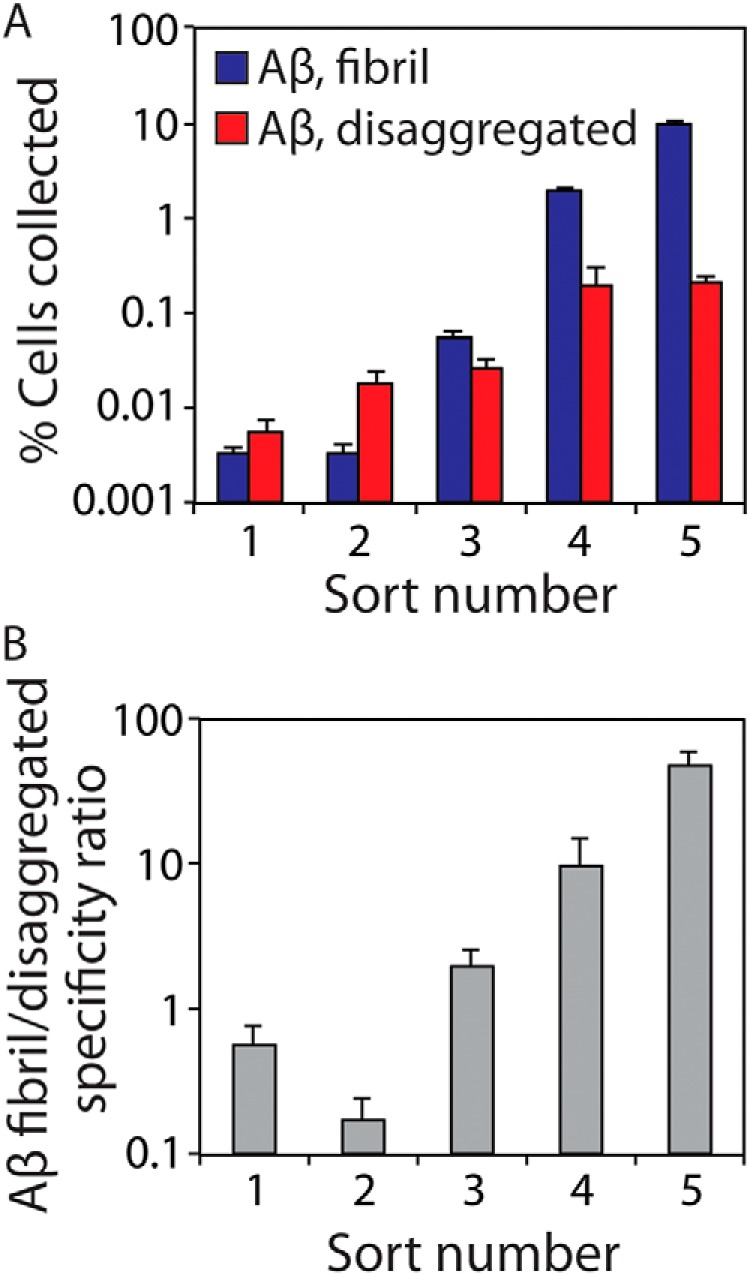
Combinations of positive and negative antibody sorts lead to strong enrichment in the fraction of antibody-displaying yeast cells that are selective for Aβ fibrils. A, percentage of yeast cells collected after each round of library sorting. Three negative selections were performed against disaggregated Aβ (immobilized on magnetic beads) for each round of sorting. Afterward, the unbound yeast cells were positively sorted against immobilized Aβ fibrils. Finally, the number of bound cells in the positive sorts was quantified via growth on selective agar plates. A total of 109 cells were used for sorts 1–3 and 107 cells for sorts 4 and 5. B, conformational specificity ratio for each sort is reported as the number of yeast cells bound to immobilized Aβ fibrils divided by the number of cells bound to disaggregated Aβ. The error bars are standard deviations for the cell counts from two dilutions of the collected cells.
These encouraging results led us to recover the enriched library and clone it a bacterial expression plasmid for sequencing analysis. Interestingly, this analysis of >10 plasmids revealed a single antibody sequence, herein referred to as AF1, that was enriched via our sorting methods. As expected, AF1 only contained mutations in HCDR3 (Fig. S2). The HCDR3 sequence (95DGYDGSYFVGYDYNDFYDY102, grafted sites are underlined, and Aβ grafted residues are italicized and in bold) has several interesting characteristics. First, it contains six tyrosine and two phenylalanine residues (out of 19 HCDR3 residues), suggesting that these aromatic residues may play a key role in selectively recognizing Aβ fibrils. Second, the HCDR3 sequence lacks positively charged residues and contains five negatively charged (aspartic acid) residues, suggesting that negative charge may also be important for selective recognition of Aβ fibrils. Finally, multiple hydrophobic grafted Aβ residues were mutated to more hydrophilic residues (G100S, V100fD, I100hN, and A100iD). The fact that only two (consecutive) grafted Aβ residues were retained (V100c and G100d), which is different from our previous results for related grafted antibodies (16, 18, 20), is likely due to the high level of HCDR3 diversification (only 2.7 grafted residues on average were expected per antibody library variant) and the extremely strong negative selections against disaggregated Aβ that we used to eliminate hydrophobic antibody variants.
Isolated AF1 antibody displays significant conformational and sequence specificity for Aβ fibrils
We next evaluated the conformational and sequence specificity of the isolated AF1 antibody in multiple soluble antibody formats. First, we expressed AF1 as monovalent (scFv) and bivalent (scFv–Fc, referred to herein as AF1–Fc) antibodies (Fig. S3). SDS-PAGE analysis revealed that the monovalent AF1 scFv runs primarily as a single band at ∼35 kDa (expected molecular mass of 33 kDa) in a manner that is similar to the scaffold (4D5) and two control anti-Aβ scFvs (A10 and B2) (16). Likewise, similar analysis of the purified AF1–Fc antibody revealed that it runs primarily as a single band at ∼120 kDa in the absence of reducing agent (expected molecular mass of 119 kDa) and ∼60 kDa in the presence of reducing agent (expected molecular mass of 59.5 kDa for the unpaired heavy chain).
To characterize the conformational and sequence specificity of AF1, we first performed immunoblotting experiments for the monovalent and bivalent AF1 antibodies (Fig. 4). These experiments were performed using disaggregated and fibrillar forms of Aβ, tau, and α-synuclein that were immobilized on nitrocellulose membranes at a range of dilutions. Notably, both monovalent and bivalent formats of AF1 show high conformational specificity for Aβ fibrils as compared with a sequence-specific anti-Aβ antibody (NAB228). The AF1 antibodies also show strong sequence specificity and fail to cross-react with tau and α-synuclein (Fig. 4). We also confirmed that the bivalent AF1–Fc antibody fails to recognize IAPP fibrils (Fig. S4). Moreover, the bivalent AF1 antibody displays excellent detection performance at lower concentrations (10 nm) than its monovalent counterpart (100 nm; Fig. 4).
Figure 4.
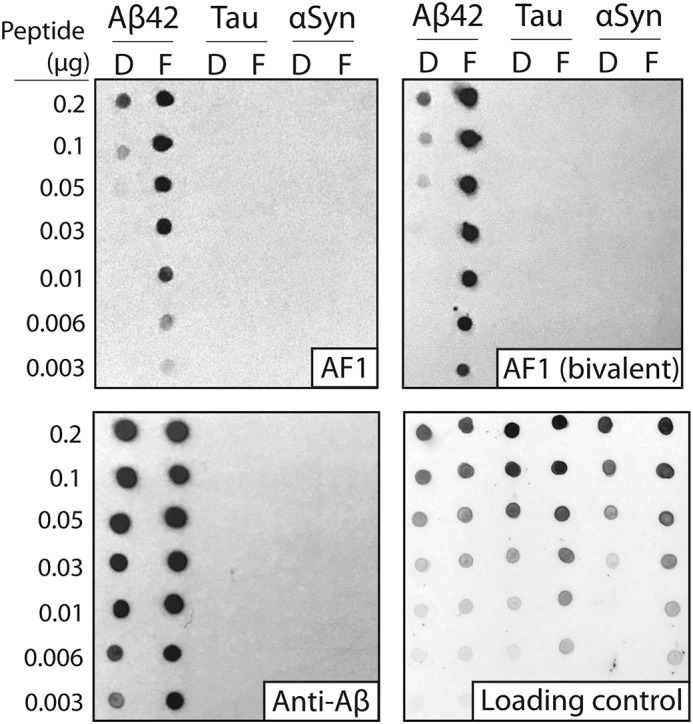
Immunoblot analysis of the conformational and sequence specificity of monovalent and bivalent AF1 antibodies. Dilutions of Aβ, tau, and α-synuclein (αSyn) (disaggregated (D) and fibril (F)) were deposited on nitrocellulose membranes. Binding of monovalent (scFv) and bivalent AF1 (scFv–Fc) antibodies were evaluated at 100 nm (scFv) and 10 nm (scFv–Fc) in 5% milk. A sequence-specific anti-Aβ antibody (NAB228, 1:1000 dilution, 5% milk) was also evaluated for comparison. Immunoblots were imaged using X-ray film (1–20 s of exposure time). The loading control blot was detected using silver colloidal stain.
Given the high conformational and sequence specificity of AF1, we next evaluated its performance using Western blotting (Fig. 5). Samples of disaggregated and fibrillar forms of Aβ, tau, and α-synuclein were first separated using SDS-PAGE and then transferred to nitrocellulose membranes for evaluation. Importantly, we find that the AF1–Fc antibody recognizes Aβ fibrils with minimal binding to disaggregated Aβ and other amyloidogenic proteins. Weak binding signals of AF1–Fc to disaggregated Aβ and intermediate size (∼80–120 kDa) Aβ aggregates were observed at long exposure times, although these signals are low relative to the strong binding to fibrils (Fig. S5). Moreover, we also performed Western blotting using brain tissues from transgenic mice that overexpress human amyloid precursor protein (APP; Fig. S6). We observe that AF1–Fc displays enhanced reactivity with large, insoluble aggregates in the forebrains of transgenic mice expressing human APP relative to the control (WT) mice. Our attempts to perform similar experiments using human brain samples were frustrated by the human Fc region of the AF1–Fc antibody, which necessitated the use of anti-human IgG detection reagents and resulted in strong cross-reactivity with human IgGs in the human brain samples (data not shown). Nevertheless, these results collectively demonstrate that AF1 displays high conformational and sequence specificity for Aβ fibrils.
Figure 5.
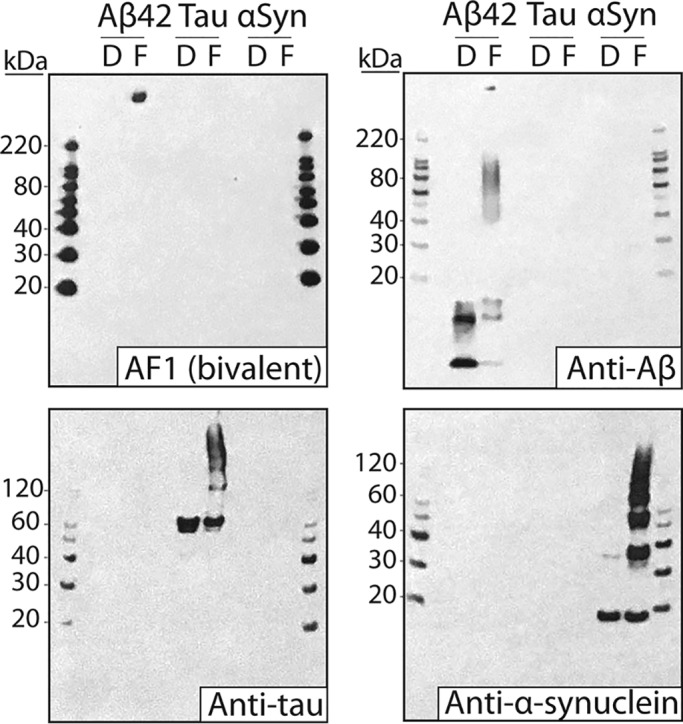
Western blot analysis of the conformational and sequence specificity of the AF1 antibody. Aβ, tau, and α-synuclein (αSyn) (disaggregated (D) and fibril (F); 0.85 μg) were separated via SDS-PAGE and transferred to nitrocellulose membranes. The AF1–Fc antibody was used at 10 nm (1% milk). Sequence-specific antibodies against Aβ (NAB228, 1:1000 dilution, 1% milk), tau (1E1/A6, 1:5000 dilution, 1% milk), and α-synuclein (5C2, 1:10,000 dilution, 1% milk) were also used as controls. Blots were imaged at short (15 s; AF1–Fc, NAB228) and long (5 min; 1E1/A6, 5C2) exposure times. The differences in the intensities of the molecular weight markers between different blots are due to the use of different secondary antibodies.
We also evaluated the specificity of the AF1 antibody by testing its propensity to interact with a panel of nontarget molecules (Fig. 6). We used an ELISA method (39, 40) that measures antibody nonspecific binding to six different types of immobilized molecules with diverse chemical properties (cardiolipin, keyhole limpet hemocyanin, lipopolysaccharide, ssDNA, dsDNA, and insulin). Importantly, we find that AF1–Fc displays low levels of nonspecific binding that are similar to its parental antibody (4D5-Fc) and multiple full-length IgGs, and displays much lower levels than other scFv–Fc antibodies that recognize Aβ (A10 and B2) (41). Given that the scFv–Fc antibodies only differ in HCDR3 sequence, these findings demonstrate that the selected mutations in AF1 HCDR3 promote specific binding to Aβ fibrils without significantly promoting nonspecific interactions.
Figure 6.
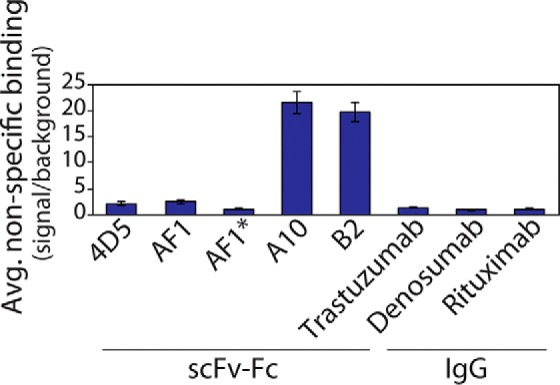
Nonspecific binding analysis for the AF1–Fc antibody relative to other scFv–Fc and IgG antibodies. The average levels of nonspecific binding (signal/background) for scFv–Fc and IgG antibodies (100 nm) were evaluated using six immobilized molecules (cardiolipin, keyhole limpet hemocyanin, lipopolysaccharide, ssDNA, dsDNA, and insulin). The 4D5, AF1, A10, and B2 antibodies are scFv–Fc antibodies with a common human IgG1 Fc fragment, and they were detected using a goat anti-human Fc antibody. AF1 and AF1* are the same scFv–Fc antibodies, but AF1* was detected using an anti-FLAG antibody instead of an anti-Fc antibody. The human IgGs (mAbs) have human IgG1 or IgG2 Fc fragments, and they were detected using a goat anti-human Fc antibody. The background values were evaluated without primary antibody and with immobilized polyspecificity molecules. The nonspecific binding values (signal/background) for each immobilized molecule were evaluated separately and then averaged together for each antibody. The data are average values for three independent experiments, and the error bars are standard deviations.
We also sought to identify the conformational epitope recognized by the AF1 antibody in Aβ fibrils. Therefore, we first generated thioflavin T (ThT)-positive aggregates for human Aβ(1–42) (control) as well as for several human Aβ peptide fragments (Fig. 7). Interestingly, AF1 recognizes both human Aβ(1–40) and Aβ(1–42) aggregates but not aggregates that lack the first residue. Moreover, we also find that AF1 fails to recognize ThT-positive aggregates for rodent Aβ(1–40). The only sequence differences between human and rodent Aβ occur at amino acid positions 5 (arginine in human and glycine in rodent), 10 (tyrosine in human and phenylalanine in rodent), and 13 (histidine in human and arginine in rodent). These results collectively suggest that the epitope of AF1 is located in the N-terminal region of Aβ. This finding is consistent with AF1 binding to Aβ fibrils via heterotypic interactions between the mutated Aβ C-terminal segment in AF1 HCDR3 and the Aβ N-terminal region in Aβ fibrils, which may be related to intramolecular interactions between these two regions in Aβ fibrils (23).
Figure 7.
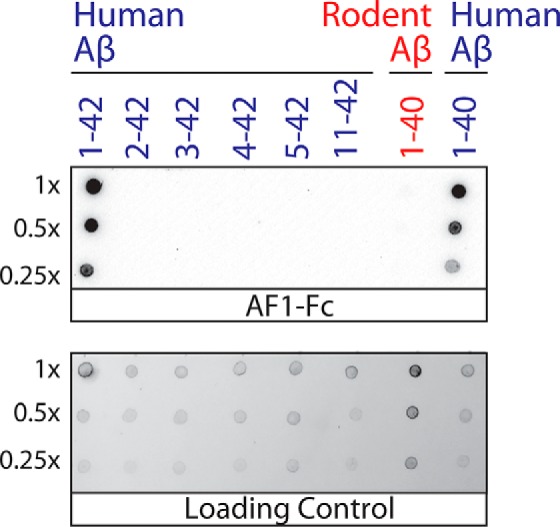
Immunoblot analysis of the conformational epitope of the AF1 antibody. Human and rodent Aβ peptides and fragments thereof were assembled into thioflavin T-positive aggregates, purified via sedimentation, and deposited on nitrocellulose membranes. The samples were loaded at equal thioflavin T values for each dilution, except for human Aβ(1–40) fibrils (which had approximately five times lower thioflavin T values). AF1–Fc immunostaining was evaluated at 10 nm (1% milk). The loading control blot was detected using silver stain.
These results next led us to evaluate the impact of bivalency on the affinity and conformational specificity of the AF1 antibody. To better understand how different bivalent formats impact antibody conformational specificity and affinity, we compared the performance of the AF1–Fc antibody with a second bivalent variant that contains two AF1 scFv antibodies connected via a 150-residue peptide linker. We reasoned that the length and flexibility of the peptide linker (30 repeats of GGSGG) may enable this novel bivalent version of AF1 to bind in unique ways relative to AF1–Fc and lead to enhanced affinity and/or conformational specificity.
To quantify the conformational specificity of the bivalent AF1 antibodies relative to their monovalent counterparts, we used a magnetic bead assay in which Aβ was immobilized on micron-sized magnetic beads (Fig. 8). The beads were then incubated with the monovalent and bivalent AF1 antibodies at different antibody concentrations, and the binding signals were quantified using flow cytometry (Fig. 8, A and B). Importantly, we find that the conformational specificity of both bivalent antibodies is greatly improved. The bivalent AF1–Fc antibody displays up to 23-fold improvement in the apparent conformational specificity for Aβ fibrils relative to monovalent AF1 (Fig. 8C). This improvement is due to greatly reduced binding of the AF1–Fc antibody to disaggregated Aβ (Fig. 8A). The EC50 value for AF1–Fc is 83 ± 6 nm for fibrils. In contrast, the bivalent AF1 antibody prepared using a peptide linker also displays up to an order of magnitude (up to 10-fold) improvement in its apparent conformational specificity for Aβ fibrils relative to the monovalent AF1 antibody (Fig. 8C), but the mechanism of this improvement is largely due to an improved apparent affinity for Aβ fibrils (EC50 of 11 ± 5 nm for bivalent AF1 relative to 146 ± 21 nm for monovalent AF1 with an N-terminal StrepTag-II tag; Fig. 8B). These findings collectively demonstrate that the valency of antibodies can be engineered in a systematic manner to greatly enhance the conformational specificity of antibodies for amyloid fibrils.
Figure 8.

Bivalent AF1 antibodies display high conformational specificity for Aβ fibrils. A and B, conformational specificities of AF1 antibodies for Aβ fibrils relative to disaggregated Aβ were evaluated for two bivalent formats of AF1 (Fc fusion (A) and 150-amino acid linker (B)) relative to monovalent AF1. The flow cytometry analysis was performed using soluble AF1 antibodies and immobilized Aβ, and antibody binding is reported in terms of relative fluorescent units (RFU). The monovalent and bivalent AF1 antibodies in B have an additional N-terminal (StrepTag-II) tag relative to the AF1 antibodies in A. C, apparent conformational specificity ratios for the AF1 antibodies. The ratios are reported as the signals for AF1 binding to Aβ fibrils relative to those for AF1 binding to disaggregated Aβ. In A and B, three independent experiments were performed, and results from a representative experiment are shown. In C, average values for 2–4 independent experiments are shown, and the error bars are standard errors.
Discussion
An interesting aspect of the conformational antibody (AF1) isolated in this study is its low levels of nonspecific interactions that are similar to those observed for antibodies identified via immunization and lower than typical antibodies isolated from in vitro libraries (based on our previous experience). It is notable that our library design method biased HCDR3 composition toward four amino acids, namely tyrosine, aspartic acid, glycine, and phenylalanine, that were each expected to be present (on average) at least twice per antibody variant. Interestingly, we observed an enrichment in the number of tyrosine residues in HCDR3 of AF1 (6) relative to the average expected number of tyrosines (3.8) for our designed library. This preference for tyrosine is consistent with the fact that tyrosine is one of the most common CDR residues in human antibodies (37, 42, 43), and increased tyrosine content in HCDR3 has been observed to be largely uncorrelated with increased levels of nonspecific interactions (44).
We also observed more aspartic acid residues in AF1 HCDR3 (five) compared with the average expected number (3.6) in the designed antibody library. It is also notable that AF1 lacks positively charged residues in HCDR3 despite the fact that arginine (average of 0.8 residues per HCDR3) and histidine (average of 0.3 residues per HCDR3) were sampled in the library. Moreover, the theoretical net charge of HCDR3 for AF1 is −5 (pH 7.4), whereas the corresponding average expected net charge of the designed library was −2.7 (assuming +1 for arginine and lysine, −1 for aspartic and glutamic acid, and a partial charge for histidine of +0.1 (pH 7.4)). We have previously reported that the enrichment in negative charge in HCDR3 is closely linked to the high specificity of AF1 (41), and this finding is also potentially consistent with the opposite finding that overenrichment in positively charged residues in the CDRs and, more generally, in the variable regions of antibodies can greatly reduce specificity (16, 30, 32, 44–49). It is also notable that the net charge of HCDR3 for AF1 is more negative than that observed for several previously reported Aβ antibodies, including those with conformational specificity (Table S1).
The other two residues in AF1 HCDR3 that were sampled at relatively high frequency (glycine and phenylalanine) were observed in AF1 at a similar frequency as expected (3 glycines observed relative to 3.3 expected and two phenylalanines observed relative to 2.3 expected). The need for glycine and phenylalanine in HCDR3 is consistent with the fact that both residues are commonly observed in the CDRs of human antibodies (37) and are expected to be important for affinity (phenylalanine) and CDR structure and flexibility (glycine). However, the lack of enrichment of these residues in AF1 is also consistent with previous observations that enrichment of these residues in antibody CDRs is linked to reduced specificity (32, 34, 44).
The conformational epitope recognized by AF1 deserves further consideration. Our finding that AF1 recognizes an N-terminal conformational epitope within Aβ fibrils is unexpected because this region is not as stably structured as the central and C-terminal regions of Aβ. However, multiple structural studies have demonstrated that some residues near the Aβ N terminus also form a β-sheet–like structure in Aβ fibrils (23, 24). Solid-state NMR analysis revealed that Aβ residues 1–14 are partially structured, and a subset of them form a β-sheet–like structure, especially Aβ residues 2–6 (24). Cryo-EM analysis also identified a similar β-sheet structure involving Aβ residues near the N terminus, especially Aβ residues 2–8 (23). This same study also found that (i) Aβ residue 1 (Asp) forms an intermolecular salt bridge with residue 28 (Lys) at the interface between Aβ protofibrils and that (ii) Aβ residues 2 (Ala) and 4 (Phe) form an intramolecular hydrophobic pocket with Aβ residues 34 (Leu) and 36 (Val). The fact that AF1 fails to recognize aggregates composed of Aβ residues 2–42 may suggest that it recognizes a conformational epitope involving the intermolecular salt bridge between Aβ residues 1 (Asp) and 28 (Lys). It could also be that AF1 recognizes a β-sheet–like structure involving residues ∼2–8 and that disrupting the salt bridge between residues 1 and 28 leads to destabilization of the N-terminal β-sheet structure and loss of AF1 binding. It will be necessary to evaluate these speculative hypotheses in the future via structural analysis of Aβ fibrils in complex with the AF1 antibody.
It is also notable that several Aβ antibodies previously or currently in clinical trials for treating Alzheimer's disease recognize epitopes involving the N terminus of Aβ, including aducanumab (Aβ residues 3–7) (50, 51), bapineuzumab (Aβ residues 1–5) (52), and gantenerumab (Aβ residues 3–11 and 18–27) (53). All three of these mAbs display high affinity for Aβ fibrils. However, aducanumab displays unusually low affinity for disaggregated Aβ (EC50 >1 μm) (50, 51), whereas bapineuzumab displays high affinity for disaggregated Aβ (EC50 of 4.4 nm; gantenerumab displays intermediate behavior) (50, 52, 53). Indeed, the ability of aducanumab to engage and clear amyloid in the brains of rodents and humans appears to be due to its high conformational specificity (50, 51). It is interesting that the AF1–Fc antibody also displays high conformational specificity due to its extremely low affinity for disaggregated Aβ (apparent KD >1 μm) in a manner that is most similar to aducanumab of the three clinical mAbs, albeit with lower affinity for Aβ fibrils. Moreover, it is important to emphasize that the conformational epitope recognized by AF1, to the best of our knowledge, is unique relative to previously reported Aβ antibodies. For example, aducanumab recognizes murine Aβ, whereas AF1 does not (50). Bapineuzumab strongly recognizes disaggregated Aβ (50, 52), whereas AF1–Fc does not, despite that both epitopes involve the first residue (Asp) of Aβ. Nevertheless, more work is needed to better define the molecular origins of the fascinating and complex diversity of antibody conformational epitopes in the immunodominant N-terminal region of Aβ.
Our encouraging findings related to the impact of bivalency on antibody conformational specificity also deserve further consideration. The bivalency of antibodies is important for efficient antibody/antigen recognition at low antibody and/or antigen concentrations. Moreover, we expected that the multivalent nature of amyloid aggregates coupled with the bivalent nature of antibodies would lead to significant increases in antibody conformational specificity relative to monovalent antibody fragments (36, 50). Indeed, our observation that bivalency results in large increases in the conformational specificity of the AF1 antibody for both bivalent constructs is consistent with avidity effects. Interestingly, the mechanism of the improved conformational specificity for the AF1–Fc antibody appears to be due to its greatly reduced affinity for disaggregated Aβ (Fig. 8). In contrast, the improvement of conformational specificity for the bivalent AF1 antibody prepared using a relatively long peptide linker is primarily due to its increased apparent affinity for Aβ fibrils relative to disaggregated Aβ. The differences in binding properties between the two bivalent antibodies are likely due to the significant differences involving the spatial positioning between the AF1 scFvs in each bivalent construct. The AF1–Fc construct has a short (three amino acid) linker between the scFvs and Fc fragment relative to the much longer (150 amino acid) linker between the scFvs in the other AF1 bivalent construct. The advantage of using the long-peptide linker for preparing the bivalent AF1 antibody is higher conformational specificity at lower antibody concentrations, whereas the advantage of using the Fc fragment is higher overall conformational specificity. We expect that these findings, coupled with our systematic approaches for designing antibody libraries and selecting conformational antibodies, will lead to robust methods for generating high-quality conformation-specific antibodies against a wide range of different types of amyloidogenic aggregates.
Experimental procedures
Aβ solubilization and fibril preparation
Aβ peptides (biotin-Aβ42 (AS-23523-05; Anaspec) and Aβ42 (AS20276; Anaspec)) were initially dissolved in hexafluoro-2-propanol (HFIP) at 1 mg/ml, aliquoted, and stored at −80 °C. After evaporation of HFIP, peptide aliquots were then dissolved in 50 mm NaOH at 1 mg/ml. The biotin–Aβ42 and Aβ42 were either used independently or mixed to give various molar fractions of biotinylated peptide. To remove any aggregated peptide, the NaOH solutions with Aβ peptide (∼250 μl) were first ultracentrifuged at 208,000 × g (Microfuge tube, polypropylene; 357448, Beckman Coulter) for 1 h (4 °C). The top 85% of the supernatant was then filtered (0.2-μm filter; SLGV004SL, Millipore). Peptide concentrations were analyzed via UV absorbance at 280 nm (extinction coefficient of 0.283 ml mg−1 cm−1) using a Nanodrop spectrophotometer, and the filtered solutions were neutralized into PBS (pH 7.4) at a final peptide concentration of 12.5 μm.
To promote Aβ fibril formation, disaggregated Aβ peptide was seeded with 5–10 volume % of preformed Aβ fibrils and incubated at 37 °C for 2–4 days without agitation. The initial amyloid seed was generated in the same manner, except that the peptide was incubated for 7 days. Amyloid formation was monitored via thioflavin T fluorescence. Aβ fibrils were purified via ultracentrifugation at 208,000 × g for 1 h (4 °C). Most of the supernatant was removed, and the fibril pellet was resuspended in the original volume of fresh PBS. The fibrils were then sonicated (FB-120 Sonic Dismembrator; Thermo Fisher Scientific) for 30 s (antibody binding analysis) or 2 min (antibody sorting) at 100% power on ice (3 or 12 cycles, each cycle with 10 s of sonication and 30 s of cooling).
Library generation
The Aβ(33–42) HCDR3 natural diversity library was generated via overlap extension PCR using the pCT-40 yeast display vector (54) that contained the 4D5 scFv sequence. This vector has a linker between Aga2 and the scFv that is longer (additional 40 residues) than the corresponding linker in the conventional pCTCON2 yeast display vector. Two PCRs were performed that encompassed the scFv gene along with 45 base pairs (bp) of homology on both ends of the vector. The first PCR used a forward primer that annealed 45 bp upstream of the scFv and a reverse degenerate codon primer that spanned HCDR3 as well as 20 bp of homology on the ends of HCDR3. The second PCR used a forward primer that encoded 20 bp of homology on the 5′ end of framework 4 and a reverse primer that provided 45 bp of homology downstream of the scFv gene.
After assembling the scFv gene with homologous ends via a second round of PCR, the resulting library was cloned into the yeast display plasmid at the C terminus of Aga2 (Aga2–scFv) via homologous recombination. The library was transformed into the EBY100 Saccharomyces cerevisiae strain via electroporation (55). After 48 h of growth in low pH SD-CAA media (20 g/liter dextrose, 6.7 g/liter yeast nitrogen base without amino acids, 5 g/liter casamino acids, 14.7 g/liter sodium citrate, and 4.3 g/liter citric acid), a total of 7 × 107 yeast transformants was obtained.
Yeast surface display and sorting
Yeast surface display of the Aβ(33–42) HCDR3 natural diversity library was performed using standard methods (38). Briefly, the library was grown in 500 ml of SD-CAA media at 30 °C and 220 rpm to an OD600 of ∼1–2. Surface display was induced by replacing the growth media with 500 ml of SG-CAA induction media (20 g/liter galactose, 6.7 g/liter yeast nitrogen base without amino acids, 5 g/liter casamino acids, 8.56 g/liter NaH2PO4·H2O, and 5.4 g/liter Na2HPO4) and incubating the cells overnight for ∼16 h at 30 °C and 220 rpm.
The library was sorted using methods similar to those described previously (29). Disaggregated Aβ (2 μm, 100 mol % biotinylated) and Aβ fibrils (6 μm, 20 mol % biotinylated) were separately immobilized on Dynabeads coated with streptavidin (11047, Thermo Fisher Scientific) via overnight incubation at 4 °C (2.5 × 107 beads/ml). A single batch of fibril-coated beads (∼7 × 107 beads, ∼2.8 ml) was prepared that was sufficient for all of the positive fibril sorts to maximize the consistency of fibrils used in each sort. The beads coated with disaggregated Aβ (∼7 × 107 beads, ∼2.8 ml) were prepared freshly for each sort to minimize the fibril content during the immobilization process. Antigen immobilization on magnetic beads was confirmed via flow cytometry analysis using an anti-Aβ antibody (∼1000× dilution; 37-4200, Thermo Fisher Scientific) and a corresponding secondary antibody (AlexaFluor 488 goat anti-mouse IgG; A11001, Thermo Fisher Scientific).
Magnetic bead sorting of the yeast-displayed scFv library was performed using both negative selections against immobilized disaggregated Aβ and positive selections against immobilized Aβ fibril. Initially, 109 yeast cells were incubated with 107 Dynabeads coated with disaggregated Aβ in a volume of 5 ml of PBS-B (PBS with 1 mg/ml BSA). After 1 h of incubation (room temperature with end–over–end mixing), the beads were collected on a Dynal magnet (12301D, Thermo Fisher Scientific). Unbound cells were subjected to a second negative sorting step (and this negative sorting process was also repeated a third time). The collected beads from each negative sort were washed with PBS-B (5 ml) and diluted onto SD-CAA agar plates for colony counting. After three negative sorts, the remaining (unbound) cells were incubated with 107 fibril-coated beads for 3–5 h in PBS-B (5 ml) at room temperature (gentle rocking). The yeast cells bound to the fibril-coated beads were isolated using the Dynal magnet, and the unbound cells were discarded. The fibril-coated beads with bound yeast cells were then released from the magnet and washed with PBS-B (5 ml). The beads were then recollected after ∼30–60 s via the Dynal magnet; the supernatant was discarded, and the beads were resuspended in SD-CAA media (5 ml). This suspension was then diluted further in SD-CAA media (50 ml final volume) and grown for 1–2 days (30 °C, 220 rpm). A small fraction of the culture was diluted onto SD-CAA agar plates for counting the number of yeast colonies.
During later rounds of sorting (after enrichment was observed), the number of cells was reduced from 109 to 107. Sorts with 107 cells were incubated and washed in PBS-B (1 ml). The yeast cells collected in the positive sorts were grown and induced in 5 ml of media when using 107 cells during the sorting process. A mock-positive sort was also performed against disaggregated Aβ (using the previous round of sorted cells that were subjected to positive sorts against immobilized Aβ fibrils) to estimate the specificity of the enriched library. The mock-positive sorting process was identical to that for the positive fibril sorting process.
Bacterial plasmid cloning, expression, and purification
The genes of the enriched scFv antibodies were first isolated by miniprepping the sorted yeast cells (D2004, Zymo Research). The isolated yeast plasmids were amplified by PCR to generate DNA fragments that contained HindIII and KpnI restriction sites on the 5′ and 3′ ends of the scFv genes, respectively. Agarose gel purification (28706, Qiagen) was used to isolate the amplified scFv product, and both the scFv genes and vector (pET-17b bacterial expression plasmid, EMD Millipore) were digested with HindIII (R3104M, New England Biolabs) and KpnI (R3142M, New England Biolabs). The digested vector was treated with a phosphatase (calf intestinal; M0290L, New England Biolabs), and then it was ligated with the digested scFv genes (T4 DNA ligase; M0202M, New England Biolabs). The ligated scFv in the pET-17b vector contained a flanking 5′ PelB leader sequence to direct periplasmic secretion as well as 3′-flanking residues that encoded 3× FLAG and 7× histidine tags for detection and purification. The ligated products were transformed into bacteria (DH5α) and selected on LB agar plates (0.1 g/liter ampicillin). Individual colonies were grown, miniprepped (27106, Qiagen), and sequenced.
Individual expression plasmids were transformed into bacteria (BL21(DE3) pLysS cells; 200132, Agilent Technologies) via heat shock. Successful transformants were selected on LB agar plates (0.1 g/liter ampicillin) after overnight growth at 37 °C. Bacterial colonies were then scraped from the LB agar plates and used to inoculate cultures of autoinduction media (56) (200 ml) supplemented with ampicillin (0.1 g/liter) and chloramphenicol (0.035 g/liter). Expression cultures were incubated for 48 h (30 °C, 220 rpm). The cultures were then centrifuged at 4000 × g, and the bacterial pellet was discarded. The supernatant (∼175 ml) was then incubated overnight (4 °C, 80 rpm) with Ni-NTA–agarose resin (1.5 ml; 30230, Qiagen).
Next, the resin was isolated and washed with 250 ml of PBS (pH 7.4). To remove any weakly bound contaminants, the washed resin was then incubated in 3 ml of PBS supplemented with 50 mm imidazole (pH 7.4) at room temperature (15 min). After removing this solution, the resin was incubated in 3 ml of PBS supplemented with 500 mm imidazole and 6 m guanidine HCl (pH 7.4) for 15 min (room temperature). The eluted antibody was incubated overnight (4 °C) and then refolded via two buffer exchanges into PBS (pH 7.4) using desalting columns (89894, Thermo Fisher Scientific). The antibody solutions were then stored at least overnight (4 °C), and then the solutions were centrifuged at 21,000 × g (5 min) and filtered using 0.2-μm filters (SLGV004SL, EMD Millipore). Antibody concentrations were determined via UV absorbance at 280 nm (extinction coefficient of 55,030 m−1 cm−1), and antibody purities were evaluated using SDS-PAGE analysis (10% BisTris; WG1203BOX, Thermo Fisher Scientific).
The AF1 scFv was also cloned into the pET28b vector (EMD Millipore) as a dimer connected via a (GGSGG)30 peptide linker. The gene was inserted between the NdeI and XhoI restriction sites. The bivalent scFv contained an N-terminal StrepTag-II and C-terminal 3× FLAG and 7× histidine tags. A monovalent AF1 scFv was also cloned in a similar manner with the same purification and detection tags. For expression of the monovalent and bivalent AF1 scFvs, plasmids were transformed into SHuffle T7 chemically competent Escherichia coli K12 (C3026J, New England Biolabs). The antibodies were expressed using two methods. For the first method, the antibodies were expressed using a similar procedure as described above for the AF1 scFv, except that kanamycin was added to the autoinduction media as the only antibiotic. Additionally, a protein A purification step was performed after the refolding step. Protein A–agarose resin (1 ml of a 50% slurry; 20334, Thermo Fisher Scientific) was added to 3 ml of the expressed protein and incubated for 1 h at room temperature. The resin was collected and washed with 250 ml of PBS (pH 7.4). The resin was then incubated with 2 ml of 0.1 m glycine (pH 3) for 15 min. After elution, the protein was neutralized by adding 100 μl of 1 m K2HPO4 and buffer exchanged into PBS (Zeba spin desalting columns; 89892, Thermo Fisher Scientific). Aggregates were removed by centrifugation (21,000 × g for 5 min) and filtration (0.22-μm filter; SLGV004SL, EMD Millipore). Antibody concentration was determined via UV absorbance measurements at 280 nm (extinction coefficient of 111,090 or 60,530 m−1 cm−1 for bivalent and monovalent AF1 scFvs, respectively), and purity was analyzed via SDS-PAGE (10% BisTris; WG1203BOX, Thermo Fisher Scientific).
In the second expression method, a 5-ml overnight culture of transformed SHuffle T7 cells was used to inoculate 1 liter of 2× YT media. The culture was grown at 30 °C until an OD of ∼0.8–1.0 (∼5–6 h). Expression was induced using isopropyl 1-thio-β-d-galactopyranoside (0.4 mm), and the temperature was reduced to 16 °C for overnight expression. Next, the cells were pelleted at 6000–8000 × g for 15 min and gently resuspended in 25 ml of lysis buffer (20 mm Tris, 500 mm NaCl, 20 mm imidazole, 5% (v/v) glycerol (pH 8.0)) supplemented with a protease inhibitor tablet (quarter of tablet; S8830, Sigma), lysozyme (0.5 mg/ml; J60701, Alfa Aesar), and benzonase (0.5 μl; E1014, Sigma). The cells were then incubated on ice for 10–15 min with intermittent mixing. A final concentration of 0.1% (w/v) sodium deoxycholate (302-95-4, Alfa Aesar) was added before sonication for a total of 6 min (3 s on, 3 s off, and 25% amplitude; Sonifier S-450, Branson Ultrasonics). The cell debris was then sedimented at 25,000 × g (30 min). The supernatant was filtered (0.45-μm filter; 6869-2504, GE Healthcare) before purification of the antibodies via immobilized metal-affinity chromatography at 4 °C. The supernatant was passed twice over a bed of 2–3 ml Ni-NTA resin (88222, Thermo Fisher Scientific) in a drip column (786-197, G-Biosciences) that was pre-equilibrated in the binding buffer (20 mm Tris, 500 mm NaCl, 20 mm imidazole, 5% v/v glycerol (pH 8.0)). The resin was then washed with 30–40 column volumes of the binding buffer. For elution, a one-column volume of elution buffer (20 mm Tris, 500 mm NaCl, 400 mm imidazole, 5% v/v glycerol (pH 8.0)) was incubated with the resin for 4–5 min. This elution step was repeated four additional times to obtain five-column volumes of the eluted protein.
The protein was next purified using StrepTrap chromatography. First, the protein was dialyzed into the binding buffer (100 mm Tris, 150 mm NaCl, 1 mm EDTA (pH 8.0)) using 3.5-kDa MWCO tubing (132-724, Spectra/Por) overnight at 4 °C. Second, the protein was loaded onto a 5-ml StrepTrap HP column (28-9075-47, GE Healthcare) on an AKTA start purification system (29022094, GE Healthcare) at a flow rate of 0.5 ml/min. The column was next washed with the binding buffer (8–10 column volumes) and followed by elution with the elution buffer (2–3 column volumes of 100 mm Tris, 150 mm NaCl, 1 mm EDTA (pH 8.0), 2.5 mm desthiobiotin). The antibodies were then concentrated to 1 ml using spin concentrators (Amicon Ultra-15, 10- kDa MWCO, Millipore Sigma). Finally, the antibodies were further purified via size-exclusion chromatography using a Superdex 75 10/300 GL column (GE Healthcare) on an AKTA Pure purification system (29018224, GE Healthcare) with a PBS running buffer (0.5 ml/min).
Mammalian expression and purification
The AF1 scFv was subcloned and expressed as an scFv–Fc fusion protein (AF1–Fc). The AF1 gene was amplified from an existing yeast display plasmid via PCR to introduce NcoI and NotI sites at the 5′ and 3′ termini of the scFv. The digested PCR product was then ligated into a digested and phosphatase-treated pBIOCAM5 mammalian expression plasmid (plasmid 39344, Addgene) (57) with the corresponding restriction sites. The expression plasmid contained the scFv fused to a human Fc IgG1 fragment along with a 7× histidine tag and a 3× FLAG tag at the C terminus of the Fc domain. Antibody expression was performed by transiently transfecting the plasmid into adherent HEK293T cells (CRL-3216, ATCC) using Lipofectamine 2000 (11668019, Thermo Fisher Scientific). After 36 h of transfection in DMEM (10569-044, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (35010CV, Corning) and 1% penicillin/streptomycin (15140122, Thermo Fisher Scientific), the cells were freshly passaged and cultured for 3 days at 37 °C in T75 tissue culture flasks (10062-860, VWR International) with 5% CO2. The growth medium containing the secreted scFv–Fc antibody was then removed and stored at 4 °C. Fresh growth medium (DMEM with 10% fetal bovine serum and 1% penicillin/streptomycin) containing the plasmid and Lipofectamine was then added, and the cells were cultured for an additional 48–72 h. The medium was then removed and combined with the original growth medium from the first transfection. This combined medium (∼120 ml) was then centrifuged at 2500 × g for 5 min to remove any cellular debris.
The secreted AF1–Fc antibody was next purified using Protein A chromatography. The medium was incubated with 1.5 ml of Protein A-agarose resin (20333, Thermo Fisher Scientific) overnight at 4 °C with end–over–end mixing. Next, the resin was collected via vacuum filtration in a 10-ml column (89898, Thermo Fisher Scientific) and subsequently washed with 250 ml of PBS. The washed resin was then incubated in 3 ml of 0.1 m glycine buffer (pH 2.5–3) for 15 min, and the AF1–Fc antibody was eluted from the resin. The eluted antibody was then neutralized by the addition of 1 m K2HPO4 buffer. Following purification, the protein was then buffer-exchanged into PBS (pH 7.4) via desalting columns (89894, Thermo Fisher Scientific). The protein solutions were then centrifuged (21,000 × g for 5 min) and filtered (0.2 μm filter; SLGV004SL, EMD Millipore). Antibody concentrations were measured using the micro BCA assay (23235, Thermo Fisher Scientific), and purity was evaluated using SDS-PAGE analysis (10% BisTris; WG1203BOX, Thermo Fisher Scientific).
Immunoblotting and Western blotting
Antibody binding was analyzed via immunoblotting and Western blotting. Aβ, tau, IAPP (AS-60254-1, Anaspec), and α-synuclein fibrils were purified via sedimentation (ultracentrifugation) at 221,000 × g (PC thick-wall tubes; 45237, Thermo Fisher Scientific). Fibril samples were resuspended in approximately one-fifth the original volume in PBS (pH 7.4 for Aβ fibrils), HEPES (20 mm HEPES, 100 mm NaCl, 1 mm EDTA (pH 7.4) for tau and α-synuclein fibrils), or Tris (20 mm Tris (pH 7.4) for IAPP fibrils) buffers. The concentrations of Aβ, tau, α-synuclein, and IAPP were measured using the BCA assay (23225, Thermo Fisher Scientific). Aβ, tau, α-synuclein, and IAPP were spotted (2 μl) onto nitrocellulose membranes (10600004, GE Healthcare) at various dilutions. The blots were then left to dry for at least 2 h. Protein loading was confirmed using colloidal silver stain. For Western blot analysis, 0.85 μg of Aβ, tau, and α-synuclein were run (unboiled) on an SDS-polyacrylamide gel (WG1203BOX, Thermo Fisher Scientific). The polypeptides were then transferred to nitrocellulose membranes using the iBlot dry blotting system and transfer stacks (IB301001, Thermo Fisher Scientific).
For both blotting procedures (immunoblots and Western blots using synthetic or recombinant polypeptides), the membranes were blocked with 10 ml of PBS with 10% (w/v) milk for 2 h. Blocked membranes were subsequently washed three times with 10 ml of PBS-T (PBS containing 0.1% Tween 20). Antibody binding was performed by incubating the blocked membranes with 10 ml of 100 nm scFv, 10 nm scFv–Fc fusion, 1:1000 dilution of NAB228 (2 mg/ml stock concentration; A8354, Sigma), 1E1/A6 (stock concentration unknown; 05-804, EMD Millipore), or 1:10,000 of 5C2 (1 mg/ml stock concentration; NBP1-04321, Novus Biologicals). All antibodies were diluted in PBS with 1 mg/ml BSA, 0.1% Tween 20, and either 1% milk (Western blots) or 5% milk (dot blots) and incubated with the blots at room temperature with gentle rocking (1 h). After binding, the membranes were again washed three times with 10 ml of PBS-T. Bound scFvs were detected using 10 ml of a 1:1000 dilution of anti-FLAG M2 antibody (1 mg/ml stock concentration; F1804, Sigma) in PBST with 1% milk (incubation for 1 h at room temperature with agitation). The membranes were then washed three times with 10 ml of PBS-T, and subsequently incubated with 10 ml of a 1:1000 dilution of goat anti-mouse IgG–HRP (0.01 mg/ml stock concentration, for detection of scFvs, NAB228, 1E1/A6, and 5C2; 32430, Invitrogen) or a 1:1000 dilution of goat anti-human IgG–HRP conjugate (0.5 mg/ml stock concentration, for detection of AF1–Fc; A18817, Invitrogen) in PBS-T with 1% milk (incubation for 1 h at room temperature with agitation). After washing three times with 10 ml of PBST, Pierce ECL Western blotting substrate (32106, Thermo Fisher Scientific) was added to the membranes. Chemiluminescence signal was detected by X-ray film using exposure times of ∼1 s to 5 min (images were converted to grayscale and auto contrasted).
For Western blotting using mouse samples, transgenic (B6SJL-Tg (APPSwFlLon, PSEN1*M146L*L286V) 6799Vas/Mmjax; The Jackson Laboratory stock no. 34840-JAX on a UM-HET3 background, aged ∼22–24 months) and WT (nontransgenic littermates) mouse brain tissue samples (courtesy of Geoffrey Murphy, University of Michigan) were homogenized in PBS with a protease inhibitor mixture (11873580001, Sigma) using a 1:3 dilution of tissue/PBS (w/v). Samples were centrifuged at 9300 RCF for 10 min at 4 °C. For the insoluble fractions, pellets were resuspended in PBS with protease inhibitor mixture (Roche Applied Science) and centrifuged at 9300 RCF for 10 min at 4 °C, and supernatants were discarded. Remaining pellet was resuspended in RIPA buffer with protease inhibitor, vortexed for 1 min, and incubated at room temperature for 1 h. Samples were water-sonicated for 5 min and centrifuged for 30 min at 16,000 RCF at 4 °C. Insoluble fractions of brain extracts containing 50 μg of total protein were loaded (without boiling) on precast NuPAGE 4–12% BisTris gels (Invitrogen) for SDS-PAGE analysis. Gels were subsequently transferred onto nitrocellulose membranes and blocked for 2 h at room temperature with 10% nonfat dry milk in TBS-T buffer. Membranes were then probed overnight at 4 °C in AF1–Fc antibody (10 nm concentration), NAB228 (1 mg/ml stock concentration, 2000× dilution), or anti-GAPDH (1 mg/ml stock concentration, 10,000× dilution; MAB374, Millipore) diluted in 5% nonfat dry milk. HRP-conjugated goat anti-human IgG (0.5 mg/ml stock concentration, 3000× dilution) or goat anti-mouse IgG (0.01 mg/ml stock concentration, 3000× dilution) were used for detection as appropriate. Western Lightning Plus ECL (ORT2755, PerkinElmer Life Sciences) was used to visualize bands. Analyses were completed in triplicate.
For the epitope mapping of AF1, ThT-positive aggregates were assembled using different Aβ peptides and fragments thereof, including human Aβ(2–42) (4036028.0500, Bachem), Aβ(3–42) (4090137.0500, Bachem), Aβ(4–42) (4090138.0500, Bachem), Aβ(5–42) (4041241.0500, Bachem), Aβ(1–40) (Anaspec, AS24236), Aβ1(1–42) (Anaspec, 63317), and rodent Aβ(1–40) (NC0609838, Thermo Fisher Scientific). The assembly and sedimentation procedures for these peptides were similar to the one described for human Aβ(1–42) except for rodent Aβ(1–40), which involved forming aggregates at 37 °C and 1000 rpm (3–4 days).
For the data in Fig. 7, the different Aβ fibril samples were normalized to the same ThT fluorescence values (normalized signals of ∼11 for 1× samples) except for human Aβ(1–40) aggregates. The normalized ThT signal for human Aβ(1–40) aggregates was ∼2.5 for the 1× sample. The antibody binding and detection procedures were similar to those described above for the immunoblots. For the antibody-binding step, the membranes were incubated with 10 ml of 10 nm AF1–Fc (Fig. 7) in PBS supplemented with 1 mg/ml BSA, 0.1% Tween 20, and 1% milk for 3–4 h at room temperature (gentle agitation). The immunoblots were detected via chemiluminescence (∼2–10 min exposure time) and quantified using a ChemiDoc XRS+ (Bio-Rad) or FluorChem M imager (ProteinSimple).
Antibody-binding analysis
Antibody binding was measured using a Dynabead-binding assay (16). The Dynabeads were prepared in the same manner as described for the antibody sorting methods and blocked (PBS supplemented with 1% milk; 1-h incubation) after antigen immobilization. Aβ-coated Dynabeads (1.25 × 105 beads) were incubated with various concentrations of bivalent AF1 scFv (scFv–Fc or scFv dimer) or monovalent scFv (respective scFv control for each bivalent format) for 3 h in PBS with 1 mg/ml BSA, 0.1% Tween 20, and 1% milk (25 °C, 350 rpm and ∼200-μl total volume). The beads were then sedimented and washed with 200 μl of ice-cold PBS-B. Antibody binding was then detected by incubating the beads with 200 μl of a 1:1000 dilution of mouse anti-FLAG M2 antibody (1 mg/ml stock; F1804, Sigma) for 30–45 min on ice. After washing with 200 μl of ice-cold PBS-B, the beads were incubated with 200 μl of a 1:1000 dilution of goat anti-mouse IgG-AlexaFluor 488 conjugate (2 mg/ml stock; A11001, Thermo Fisher Scientific) for 10 min on ice. The beads were then washed with 200 μl of ice-cold PBS-B, and the fluorescence signals were measured using a ZE5 cell analyzer (Bio-Rad). The singlet bead population was gated based on forward- and side-scatter signals, and ∼25,000–50,000 events were recorded. The mean fluorescence values across the various antibody concentrations were fit to a three-parameter sigmoidal dose-response curve using SigmaPlot to determine the EC50 values.
Nonspecific binding analysis
Antibody nonspecific binding was measured using several immobilized molecules, including cardiolipin (C0563, Sigma), lipopolysaccharide (LPS-EB, tlrl-eblps, InvivoGen), keyhole limpet hemocyanin (KLH, H8283, Sigma), ssDNA (D8899, Sigma), dsDNA (D4522, Sigma), and insulin (I9278, Sigma). Each molecule was diluted to 50 μg/ml in 95% ethanol (cardiolipin), PBS (KLH and insulin), or water (LPS, ssDNA, and dsDNA), and immobilized in 96-well plates (3369, Corning) by adding 50 μl (per well) of each solution and incubating overnight at 4 °C.
The antibody solutions for nonspecific binding analysis were prepared by measuring the concentrations of the antibody stock solutions and diluting appropriately. The concentrations of the antibodies were measured by UV absorbance using an extinction coefficient of 1.40 ml/(mg cm) for the mAbs (Trastuzumab, Denosumab, and Rituximab) and 1.52 ml/(mg cm) for the scFv–Fc fusions (4D5, A10, B2, and AF1). The antibodies were diluted to 100 nm into PBS containing 0.1% Tween 20.
The 96-well plates with immobilized molecules were next washed three times using 100 μl of PBS, and 50 μl of each antibody (100 nm) was added for 1 h. Afterward, the plates were washed six times with 100 μl of PBS, and 50 μl of goat anti-human IgG–HRP (0.5 mg/ml stock concentration, 1000× dilution; A18817, Invitrogen) was then added to each well for 1 h. A second method of detection was also used for AF1–Fc, which involved detecting AF1–Fc binding using a rabbit anti-FLAG IgG–HRP (stock concentration not given; 2044S, Cell Signaling Technologies). After secondary antibody incubation, the plates were washed six times (as described above), and 50 μl of one-step TMB Ultra (34028, Thermo Fisher Scientific) was added to the wells. The signals in the wells were developed for 1–2 min and quenched with 50 μl of 2 m H2SO4. The absorbances were read at 450 nm using a plate reader (Synergy Neo plate reader, BioTek). The absorbance values were normalized by the background from the secondary antibodies (no test antibody added) for each molecule. The normalized antibody nonspecific binding values for each molecule were averaged together to generate a single (average) value for each antibody.
Author contributions
M. C. J., L. A. R., A. A. D., A. A., J. E. G., H. L. P., R. S. K., and P. M. T. conceptualization; M. C. J., L. A. R., A. A. D., A. A., and J. E. G. investigation; M. C. J., L. A. R., A. A. D., A. A., R. S. K., and P. M. T. methodology; M. C. J., L. A. R., A. A. D., and P. M. T. writing-original draft; M. C. J., L. A. R., A. A. D., A. A., J. E. G., H. L. P., R. S. K., and P. M. T. writing-review and editing; L. A. R., A. A. D., and P. M. T. visualization.
Supplementary Material
Acknowledgments
We thank Shannon Moore and Geoffrey Murphy (University of Michigan) for the mouse brain samples and members of the Tessier laboratory for their helpful suggestions.
This work was supported by National Institutes of Health Grants R01GM104130 (to P. M. T.) and RF1AG059723 (to P. M. T. and R. S. K.,), National Science Foundation Grants CBET 1159943, 1605266, and 1813963 (to P. M. T.), Graduate Research Fellowships (to M. C. J. and L. A. R.), and the Albert M. Mattocks Chair (to P. M. T). P. M. T. has received consulting fees and/or honorariums for presentations of this and/or related research findings at Pfizer, Adimab, Roche Applied Science, Boehringer Ingelheim, Bayer, and Novo Nordisk. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6 and Table S1.
- Aβ
- amyloid β
- CDR
- complementarity-determining region
- scFv
- single-chain variable fragment
- VH
- variable domain of heavy-chain
- Fc
- fragment crystallizable
- HCDR3
- heavy chain CDR3
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- IAPP
- islet amyloid polypeptide
- RCF
- relative centrifugal force
- APP
- amyloid precursor protein
- HFIP
- hexafluoro-2-propanol
- Ni-NTA
- nickel-nitrilotriacetic acid
- DMEM
- Dulbecco's modified Eagle's medium
- KLH
- keyhole limpet hemocyanin
- ssDNA
- single-stranded DNA
- RFU
- relative fluorescence units.
References
- 1. Ross C. A., and Poirier M. A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17 10.1038/nm1066 [DOI] [PubMed] [Google Scholar]
- 2. Soto C. (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60 10.1038/nrn1007 [DOI] [PubMed] [Google Scholar]
- 3. Chiti F., and Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- 4. Narasimhan S., Guo J. L., Changolkar L., Stieber A., McBride J. D., Silva L. V., He Z., Zhang B., Gathagan R. J., Trojanowski J. Q., and Lee V. M. (2017) Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 37, 11406–11423 10.1523/JNEUROSCI.1230-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peelaerts W., and Baekelandt V. (2016) α-Synuclein strains and the variable pathologies of synucleinopathies. J. Neurochem. 139, 256–274 10.1111/jnc.13595 [DOI] [PubMed] [Google Scholar]
- 6. Peelaerts W., Bousset L., Van der Perren A., Moskalyuk A., Pulizzi R., Giugliano M., Van den Haute C., Melki R., and Baekelandt V. (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344 10.1038/nature14547 [DOI] [PubMed] [Google Scholar]
- 7. Melki R. (2015) Role of different α-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J. Parkinsons Dis. 5, 217–227 10.3233/JPD-150543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanders D. W., Kaufman S. K., DeVos S. L., Sharma A. M., Mirbaha H., Li A., Barker S. J., Foley A. C., Thorpe J. R., Serpell L. C., Miller T. M., Grinberg L. T., Seeley W. W., and Diamond M. I. (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288 10.1016/j.neuron.2014.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guo J. L., Covell D. J., Daniels J. P., Iba M., Stieber A., Zhang B., Riddle D. M., Kwong L. K., Xu Y., Trojanowski J. Q., and Lee V. M. (2013) Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117 10.1016/j.cell.2013.05.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bousset L., Pieri L., Ruiz-Arlandis G., Gath J., Jensen P. H., Habenstein B., Madiona K., Olieric V., Böckmann A., Meier B. H., and Melki R. (2013) Structural and functional characterization of two α-synuclein strains. Nat. Commun. 4, 2575 10.1038/ncomms3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kayed R., and Glabe C. G. (2006) Conformation-dependent anti-amyloid oligomer antibodies. Methods Enzymol. 413, 326–344 10.1016/S0076-6879(06)13017-7 [DOI] [PubMed] [Google Scholar]
- 12. Glabe C. G. (2004) Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem. Sci. 29, 542–547 10.1016/j.tibs.2004.08.009 [DOI] [PubMed] [Google Scholar]
- 13. Lafaye P., Achour I., England P., Duyckaerts C., and Rougeon F. (2009) Single-domain antibodies recognize selectively small oligomeric forms of amyloid β, prevent Aβ-induced neurotoxicity and inhibit fibril formation. Mol. Immunol. 46, 695–704 10.1016/j.molimm.2008.09.008 [DOI] [PubMed] [Google Scholar]
- 14. Habicht G., Haupt C., Friedrich R. P., Hortschansky P., Sachse C., Meinhardt J., Wieligmann K., Gellermann G. P., Brodhun M., Götz J., Halbhuber K. J., Röcken C., Horn U., and Fändrich M. (2007) Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Aβ protofibrils. Proc. Natl. Acad. Sci. U.S.A. 104, 19232–19237 10.1073/pnas.0703793104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., and Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 10.1126/science.1079469 [DOI] [PubMed] [Google Scholar]
- 16. Tiller K. E., Li L., Kumar S., Julian M. C., Garde S., and Tessier P. M. (2017) Arginine mutations in antibody complementarity-determining regions display context-dependent affinity/specificity trade-offs. J. Biol. Chem. 292, 16638–16652 10.1074/jbc.M117.783837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tiller K. E., Chowdhury R., Li T., Ludwig S. D., Sen S., Maranas C. D., and Tessier P. M. (2017) Facile affinity maturation of antibody variable domains using natural diversity mutagenesis. Front. Immunol. 8, 986 10.3389/fimmu.2017.00986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Julian M. C., Li L., Garde S., Wilen R., and Tessier P. M. (2017) Efficient affinity maturation of antibody variable domains requires co-selection of compensatory mutations to maintain thermodynamic stability. Sci. Rep. 7, 45259 10.1038/srep45259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee C. C., Julian M. C., Tiller K. E., Meng F., DuConge S. E., Akter R., Raleigh D. P., and Tessier P. M. (2016) Design and optimization of anti-amyloid domain antibodies specific for β-amyloid and islet amyloid polypeptide. J. Biol. Chem. 291, 2858–2873 10.1074/jbc.M115.682336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Julian M. C., Lee C. C., Tiller K. E., Rabia L. A., Day E. K., Schick A. J. 3rd., and Tessier P. M. (2015) Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. 28, 339–350 10.1093/protein/gzv050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perchiacca J. M., Ladiwala A. R., Bhattacharya M., and Tessier P. M. (2012) Aggregation-resistant domain antibodies engineered with charged mutations near the edges of the complementarity-determining regions. Protein Eng. Des. Sel. 25, 591–601 10.1093/protein/gzs042 [DOI] [PubMed] [Google Scholar]
- 22. Perchiacca J. M., Ladiwala A. R., Bhattacharya M., and Tessier P. M. (2012) Structure-based design of conformation- and sequence-specific antibodies against amyloid β. Proc. Natl. Acad. Sci. U.S.A. 109, 84–89 10.1073/pnas.1111232108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gremer L., Schölzel D., Schenk C., Reinartz E., Labahn J., Ravelli R. B. G., Tusche M., Lopez-Iglesias C., Hoyer W., Heise H., Willbold D., and Schröder G. F. (2017) Fibril structure of amyloid-β(1–42) by cryo-electron microscopy. Science 358, 116–119 10.1126/science.aao2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wälti M. A., Ravotti F., Arai H., Glabe C. G., Wall J. S., Böckmann A., Güntert P., Meier B. H., and Riek R. (2016) Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U.S.A. 113, E4976–E4984 10.1073/pnas.1600749113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paravastu A. K., Leapman R. D., Yau W. M., and Tycko R. (2008) Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 10.1073/pnas.0806270105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petkova A. T., Leapman R. D., Guo Z., Yau W. M., Mattson M. P., and Tycko R. (2005) Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science 307, 262–265 10.1126/science.1105850 [DOI] [PubMed] [Google Scholar]
- 27. Petkova A. T., Ishii Y., Balbach J. J., Antzutkin O. N., Leapman R. D., Delaglio F., and Tycko R. (2002) A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747 10.1073/pnas.262663499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B., Döbeli H., Schubert D., and Riek R. (2005) 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347 10.1073/pnas.0506723102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ackerman M., Levary D., Tobon G., Hackel B., Orcutt K. D., and Wittrup K. D. (2009) Highly avid magnetic bead capture: an efficient selection method for de novo protein engineering utilizing yeast surface display. Biotechnol. Prog. 25, 774–783 10.1002/btpr.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Datta-Mannan A., Thangaraju A., Leung D., Tang Y., Witcher D. R., Lu J., and Wroblewski V. J. (2015) Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs 7, 483–493 10.1080/19420862.2015.1016696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dobson C. L., Devine P. W., Phillips J. J., Higazi D. R., Lloyd C., Popovic B., Arnold J., Buchanan A., Lewis A., Goodman J., van der Walle C. F., Thornton P., Vinall L., Lowne D., et al. (2016) Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci. Rep. 6, 38644 10.1038/srep38644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Birtalan S., Fisher R. D., and Sidhu S. S. (2010) The functional capacity of the natural amino acids for molecular recognition. Mol. Biosyst. 6, 1186–1194 10.1039/b927393j [DOI] [PubMed] [Google Scholar]
- 33. Kehoe J. W., Whitaker B., Bethea D., Lacy E. R., Boakye K., Santulli-Marotto S., Ryan M. H., Feng Y., and Wheeler J. C. (2014) Isolation and optimization for affinity and biophysical characteristics of anti-CCL17 antibodies from the VH1–69 germline gene. Protein Eng. Des. Sel. 27, 199–206 10.1093/protein/gzu012 [DOI] [PubMed] [Google Scholar]
- 34. Kelly R. L., Le D., Zhao J., and Wittrup K. D. (2018) Reduction of nonspecificity motifs in synthetic antibody libraries. J. Mol. Biol. 430, 119–130 10.1016/j.jmb.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kelly R. L., Zhao J., Le D., and Wittrup K. D. (2017) Nonspecificity in a nonimmune human scFv repertoire. mAbs 9, 1029–1035 10.1080/19420862.2017.1356528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brännström K., Lindhagen-Persson M., Gharibyan A. L., Iakovleva I., Vestling M., Sellin M. E., Brännström T., Morozova-Roche L., Forsgren L., and Olofsson A. (2014) A generic method for design of oligomer-specific antibodies. PLoS ONE 9, e90857 10.1371/journal.pone.0090857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Swindells M. B., Porter C. T., Couch M., Hurst J., Abhinandan K. R., Nielsen J. H., Macindoe G., Hetherington J., and Martin A. C. (2017) abYsis: integrated antibody sequence and structure-management, analysis, and prediction. J. Mol. Biol. 429, 356–364 10.1016/j.jmb.2016.08.019 [DOI] [PubMed] [Google Scholar]
- 38. Chao G., Lau W. L., Hackel B. J., Sazinsky S. L., Lippow S. M., and Wittrup K. D. (2006) Isolating and engineering human antibodies using yeast surface display. Nat. Protoc 1, 755–768 10.1038/nprot.2006.94 [DOI] [PubMed] [Google Scholar]
- 39. Jain T., Sun T., Durand S., Hall A., Houston N. R., Nett J. H., Sharkey B., Bobrowicz B., Caffry I., Yu Y., Cao Y., Lynaugh H., Brown M., Baruah H., Gray L. T., et al. (2017) Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. U.S.A. 114, 944–949 10.1073/pnas.1616408114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mouquet H., Scheid J. F., Zoller M. J., Krogsgaard M., Ott R. G., Shukair S., Artyomov M. N., Pietzsch J., Connors M., Pereyra F., Walker B. D., Ho D. D., Wilson P. C., Seaman M. S., Eisen H. N., et al. (2010) Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature 467, 591–595 10.1038/nature09385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rabia L. A., Zhang Y., Ludwig S. D., Julian M. C., and Tessier P. M. (2019) Net charge of antibody complementarity-determining regions is a key predictor of specificity. Protein Eng. Des. Sel. 2019, gzz002 10.1093/protein/gzz002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zemlin M., Klinger M., Link J., Zemlin C., Bauer K., Engler J. A., Schroeder H. W. Jr, and Kirkham P. M. (2003) Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J. Mol. Biol. 334, 733–749 10.1016/j.jmb.2003.10.007 [DOI] [PubMed] [Google Scholar]
- 43. Mian I. S., Bradwell A. R., and Olson A. J. (1991) Structure, function and properties of antibody binding sites. J. Mol. Biol. 217, 133–151 10.1016/0022-2836(91)90617-F [DOI] [PubMed] [Google Scholar]
- 44. Birtalan S., Zhang Y., Fellouse F. A., Shao L., Schaefer G., and Sidhu S. S. (2008) The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J. Mol. Biol. 377, 1518–1528 10.1016/j.jmb.2008.01.093 [DOI] [PubMed] [Google Scholar]
- 45. Datta-Mannan A., Lu J., Witcher D. R., Leung D., Tang Y., and Wroblewski V. J. (2015) The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies. mAbs 7, 1084–1093 10.1080/19420862.2015.1075109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bumbaca Yadav D., Sharma V. K., Boswell C. A., Hotzel I., Tesar D., Shang Y., Ying Y., Fischer S. K., Grogan J. L., Chiang E. Y., Urban K., Ulufatu S., Khawli L. A., Prabhu S., Joseph S., and Kelley R. F. (2015) Evaluating the use of antibody variable region (Fv) charge as a risk assessment tool for predicting typical cynomolgus monkey pharmacokinetics. J. Biol. Chem. 290, 29732–29741 10.1074/jbc.M115.692434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li B., Tesar D., Boswell C. A., Cahaya H. S., Wong A., Zhang J., Meng Y. G., Eigenbrot C., Pantua H., Diao J., Kapadia S. B., Deng R., and Kelley R. F. (2014) Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs 6, 1255–1264 10.4161/mabs.29809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alam M. E., Geng S. B., Bender C., Ludwig S. D., Linden L., Hoet R., and Tessier P. M. (2018) Biophysical and sequence-based methods for identifying monovalent and bivalent antibodies with high colloidal stability. Mol. Pharm. 15, 150–163 10.1021/acs.molpharmaceut.7b00779 [DOI] [PubMed] [Google Scholar]
- 49. Starr C. G., and Tessier P. M. (2019) Selecting and engineering monoclonal antibodies with drug-like specificity. Curr. Opin Biotechnol. 60, 119–127 10.1016/j.copbio.2019.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Arndt J. W., Qian F., Smith B. A., Quan C., Kilambi K. P., Bush M. W., Walz T., Pepinsky R. B., Bussière T., Hamann S., Cameron T. O., and Weinreb P. H. (2018) Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 8, 6412 10.1038/s41598-018-24501-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sevigny J., Chiao P., Bussière T., Weinreb P. H., Williams L., Maier M., Dunstan R., Salloway S., Chen T., Ling Y., O'Gorman J., Qian F., Arastu M., Li M., Chollate S., Brennan M. S., et al. (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537, 50–56 10.1038/nature19323 [DOI] [PubMed] [Google Scholar]
- 52. Feinberg H., Saldanha J. W., Diep L., Goel A., Widom A., Veldman G. M., Weis W. I., Schenk D., and Basi G. S. (2014) Crystal structure reveals conservation of amyloid-β conformation recognized by 3D6 following humanization to bapineuzumab. Alzheimers Res. Ther. 6, 31 10.1186/alzrt261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bohrmann B., Baumann K., Benz J., Gerber F., Huber W., Knoflach F., Messer J., Oroszlan K., Rauchenberger R., Richter W. F., Rothe C., Urban M., Bardroff M., Winter M., Nordstedt C., and Loetscher H. (2012) Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimers Dis. 28, 49–69 10.3233/JAD-2011-110977 [DOI] [PubMed] [Google Scholar]
- 54. Stern L. A., Schrack I. A., Johnson S. M., Deshpande A., Bennett N. R., Harasymiw L. A., Gardner M. K., and Hackel B. J. (2016) Geometry and expression enhance enrichment of functional yeast-displayed ligands via cell panning. Biotechnol. Bioeng. 113, 2328–2341 10.1002/bit.26001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Benatuil L., Perez J. M., Belk J., and Hsieh C. M. (2010) An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng. Des. Sel. 23, 155–159 10.1093/protein/gzq002 [DOI] [PubMed] [Google Scholar]
- 56. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- 57. Falk R., Falk A., Dyson M. R., Melidoni A. N., Parthiban K., Young J. L., Roake W., and McCafferty J. (2012) Generation of anti-Notch antibodies and their application in blocking Notch signalling in neural stem cells. Methods 58, 69–78 10.1016/j.ymeth.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.