

SUMMARY
During corticogenesis, ventricular zone progenitors sequentially generate distinct subtypes of neurons, accounting for the diversity of neocortical cells and the circuits they form. While activity-dependent processes are critical for the differentiation and circuit assembly of postmitotic neurons, how bioelectrical processes affect non-excitable cells such as progenitors remains largely unknown. Here, we reveal that, in the developing mouse neocortex, ventricular zone progenitors become more hyperpolarized as they generate successive subtypes of neurons. Experimental in vivo hyperpolarization shifted the transcriptional programs and division modes of these progenitors to a later developmental status, with precocious generation of intermediate progenitors and a forward shift in the laminar, molecular, morphological and circuit features of their neuronal progeny. These effects occurred through inhibition of the Wnt-beta-catenin signaling pathway by hyperpolarization. Thus, during corticogenesis, bioelectric membrane properties are permissive for specific molecular pathways to coordinate the temporal progression of progenitor developmental programs and hence neocortical neuron diversity.
Graphical Abstract
In Brief:
Progressive changes in the membrane potential of neural progenitors during corticogenesis coordinates the temporal progression of neuronal subtype specification in neocortex.
INTRODUCTION
The building of the mammalian brain involves coordinated interactions between cell-intrinsic genetic programs and input/activity-dependent processes. Activity-dependent processes are involved at several sequential steps of neuronal development, such as migration (Bortone and Polleux, 2009; De Marco García et al., 2011), axonal elongation (Mire et al., 2012), neuronal specification (Chou et al., 2013; Pouchelon et al., 2014), synaptogenesis and regulation of excitatory/inhibitory balance (Bloodgood et al., 2013; Lin et al., 2008; Wang and Kriegstein, 2008), dendritogenesis (Frangeul et al., 2017; Ewald et al., 2008) and even neurotransmitter specification (Spitzer, 2012). In contrast, the role of electrical activity on non-excitable cells has been less well studied, and neurogenic events are not classically thought to rely on bioelectrical status. However, bioelectric membrane properties, including the resting membrane potential (Vm), also determine the behavior of non-excitable cells across organs and systems (Bauer and Schwarz, 2001; Blackiston et al., 2009; Cone and Tongier, 1971; LoTurco et al., 1995; Sundelacruz et al., 2008; Urrego et al., 2014; Wang et al., 2003). In fact, across cell types, membrane potential oscillations are necessary for progression through the cell cycle (Blackiston et al., 2009). In the neocortex, progenitor depolarization inhibits DNA synthesis (LoTurco et al., 1995) and calcium waves increase in VZ progenitors during corticogenesis (Weissman et al., 2004). Similarly, membrane depolarization promotes neural development by driving proneural gene expression during adult neurogenesis in the hippocampus, and changes in potassium conductances regulate cycling behavior in cortex-derived precursors in neurospheres (Tozuka et al., 2005, Yasuda et al., 2008). Together, these findings point towards a critical role for bioelectrical processes in regulating progenitor cell properties.
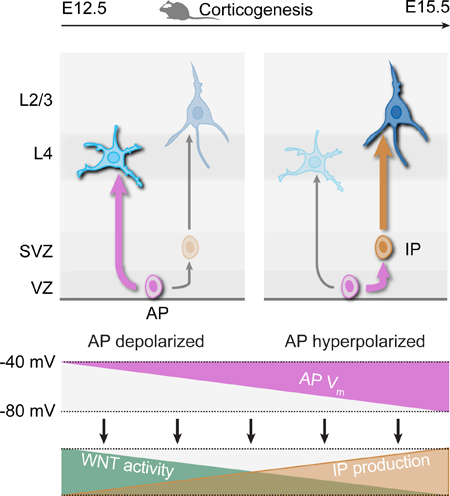
In the neocortex, distinct subtypes of excitatory glutamatergic neurons are sequentially generated from apical progenitors (APs) located in the dorsal ventricular zone (VZ). In the mouse, each embryonic day (E) sees the peak of birth of one laminar subtype of neurons. Layer (L) 6 neurons are born first, at E12.5, followed by L5 neurons (E13.5), L4 neurons (E14.5) and finally L3 (E15.5) and L2 (E16.5) neurons (reviewed in Jabaudon, 2017). While the cell-intrinsic molecular mechanisms controlling AP divisions are increasingly understood (Dehay and Kennedy, 2007; Govindan and Jabaudon, 2017; Taverna et al., 2014), the mechanisms which drive the progression in AP neurogenic competence are not, since molecular distinction across APs are not readily apparent (Toma et al., 2016, Okamoto et al., 2016). Here, we reveal that bioelectrical membrane properties regulate the temporal progression of AP competence.
Building from an initial observation that hyperpolarization affects post-mitotic neuron identity in the developing neocortex, we report that the progressive AP hyperpolarization drives progression in neurogenic fate. This progressive hyperpolarization is due to an increase in Ba2+-sensitive K+ conductances, which allows the progression in progenitor division modes by inhibiting canonical WNT signaling. Thus, as corticogenesis proceeds, bioelectric membrane properties act via specific molecular pathways to coordinate the temporal progression of progenitor identity and associated neuronal diversity.
RESULTS
Kir2.1 electroporation at E14.5 leads to premature presence of L2/3-type neurons
L4 neurons of primary sensory cortices require input from the periphery in order to complete their differentiation (Chou et al., 2013; Pouchelon et al., 2014). To directly investigate how manipulation of activity affects L4 neuron differentiation, we used in utero electroporation (IUE) of Kir2.1, an inward-rectifying potassium channel which lowers Vm (Arcangeli et al., 1995; Urrego et al., 2014). Consistent with previous findings demonstrating a critical role of activity in barrel formation (Belford and Killackey, 1980; Iwasato et al., 2000; 1997; Van der Loos and Woolsey, 1973), Kir2.1 overexpression disrupted the tangential distribution of E14.5-born L4 neurons within the somatosensory cortex, resulting in an absence of somatotopic mapping of whiskers as barrels on the cortical surface at postnatal day (P) 7 (Figure S1).
Unexpectedly, however, the radial positioning of E14.5-born neurons was also altered. In contrast to the control condition, in which E14.5-born neurons were essentially confined to L4, E14.5-born neurons following Kir2.1 IUE were located more superficially, including within L2/3 (Figure 1A). Since Kir2.1 is first electroporated into apical progenitors (APs) and then present in the progeny of these cells, three scenarios may account for this finding (Figure 1B): (1) Kir2.1 cell-autonomously affects the migration of L4 neurons; (2) Kir2.1 cell-autonomously affects the post-mitotic differentiation of E14.5-born neurons, i.e. these cells are respecified into normally later-developing L2/3-type neurons; (3) Kir2.1 non cell-autonomously affects neurons: instead, it acts on APs, leading to an anticipated production of L2/3-type neuron instead of L4-type neurons.
Figure 1.

Kir2.1 electroporation at E14.5 leads to premature presence of L2/3-type neurons.
(A) The radial location of neurons born in the Kir2.1-electroporation condition shifts superficially towards that of normally later born neurons.
(B) Possible scenarios explaining the radial shift.
(C) Left: RORB and BRN2 are expressed along opposing radial gradients in L4 and L2/3. Middle and right: Molecular expression is congruent with laminar location in single neurons in Kir2.1-electroporation condition.
(D) Fluorescently-labeled neurons collected at P3 for transcriptomic analysis.
(E) Neurons repress L4 neuron-type and induce L2/3 neuron-type genes in the Kir2.1-electroporation condition.
(F) Left: unbiased SVM classification reveals a shift of the transcriptional identity of E14.5-born neurons in the Kir2.1-electroporation. Middle: Top 20 genes used to build the model. Right: Expression of selected genes; in situ hybridizations from Allen Brain Atlas.
Data are represented as mean ± SEM, except for scatter plots represented as means ± SD. (A), two-way ANOVA for bin distribution analysis (significantly different bins indicated in darker shades); Student’s t test for scatter plot (comparing average cell position). Individual biological replicates are distinguished by color and aligned from left to right. (C), Student’s t test. (F), one-way ANOVA. *: P<0.05; **: P<10−2; ***: P<10−3; ****: P<10−4.
Ctl: Control; Epor: electroporated cells; VZ: ventricular zone. See also Figure S1.
We first differentiated between a mismigration (scenario 1) and a misdifferentiation (scenario 2), by assessing whether in addition to their L2/3 laminar positioning, E14.5-born neurons in the E14.5 Kir2.1 condition showed additional features of L2/3-type neurons, in terms of molecular identity, morphology, and input-output connectivity.
Molecular identity
L4-type neurons express high levels of RORB and lower levels of BRN2, while the converse is true for L2/3 type neurons (Fig. 1C, left) (Jabaudon et al., 2012; Oishi et al., 2016). Consistent with a graded acquisition of a L2/3-type molecular identity, E14.5-born neurons in the Kir2.1 condition repressed RORB expression, and induced BRN2 expression (Figure 1C, middle and right). Combinatorial expression in single neurons corresponded with radial location: more superficially located neurons were more likely to express the L2/3 marker BRN2 than RORB, while the converse was true for more deeply located neurons, as it is normally the case (Figure 1C). Thus, E14.5-born neurons in the Kir2.1 condition coordinately shift their radial location and molecular expression, suggesting an overall misdifferentiation rather than an isolated mismigration.
We next compared the transcriptional programs of L4-type neurons, L2/3-type neurons, and E14.5-born neurons in the Kir2.1 condition at P3, a time at which they have reached their target layers (Figures 1D–F). This approach revealed an overall repression of L4-type neuronal genes and a premature induction of L2/3-type neuronal genes (Figure 1E and Table S1). To unbiasedly assess the transcriptional identity of neurons in the E14.5 Kir2.1 condition, we trained a support-vector machine (SVM) model with L4- and L2/3-type neuron transcriptional data, and used this as a model to determine the identity of neurons in the E14.5 Kir2.1 condition (Frangeul et al., 2016). This analysis revealed that the aggregate identity of P3 neurons in the Kir2.1 condition is intermediate between that of L4- and L2/3-type neurons (Fig. 1F). Together, these results indicate that the abnormal L2/3 radial positioning of E14.5-born neurons following Kir2.1 IUE is associated with a corresponding L4-to-L2/3 transcriptional shift in their transcriptional programs.
Morphology and input-output connectivity
In the primary somatosensory cortex, the dendritic tree of L4-type neurons is normally polarized, and these neurons only rarely display an apical dendrite by P7 (De la Rossa et al., 2013; Mizuno et al., 2014; Pouchelon et al., 2014; Callaway and Borrell, 2011). In contrast, L2/3-type neurons have symmetrical dendritic arbors and a prominent apical dendrite. Consistent with a L4-to-L2/3 shift in their morphology, in the Kir2.1-electroporation condition, neurons lost their dendritic polarization (Figure 2A) and displayed an apical dendrite (Figure 2B).
Figure 2.

Kir2.1 in utero electroporation leads to a forward shift in neuronal morphology.
(A) In the Kir2.1-electroporation condition, neurons display a non-polarized dendritic arbor, as E15.5-born L2/3-type neurons do. Right: cumulative radial distribution of primary dendrites for each analyzed cell.
(B) E14.5-born neurons mostly display a stellate morphology whereas Kir2.1-electroporated neurons often display an apical dendrite, as do E15.5-born L2/3-type neurons (red arrowheads). Horizontal bars in (A) indicate median values. Data in (B) are represented as means ± SEM. (A), Kruskal-Wallis test; (B), one-way ANOVA. *: P<0.05; **: P<10−2; ***: P<10−3.
This directed change in neuronal morphology suggests a corresponding change in the circuit properties of these cells, which we examined next. Thalamocortical terminals normally represent a strong source of input to L4-type neurons but not to L2/3-type neurons (De la Rossa et al., 2013; Lefort et al., 2009) (Figure 3A). Using VGLUT2 to identify thalamocortical synapses (Jabaudon et al., 2012; Pouchelon et al., 2014), we found that in contrast to the control condition, where VGLUT2+ terminals overlapped GFP-expressing neurons in L4, thalamocortical synapses no longer overlapped these cells in the E14.5 Kir2.1 condition, suggesting loss of this key circuit feature of L4 neuron identity (Figure S2).
Figure 3.

Kir2.1 in utero electroporation leads to a L4-to-L2/3 shift in input-output circuit properties.
(A) Schematic representation of input-output connectivity in S1. Letters refer to panels.
(B) In the Kir2.1-electroporation condition, neurons acquire L2/3 neuron-type intracortical input.
(C) Kir2.1-electroporated neurons acquire L2/3 neuron-type intracortical output. Left: axons of electroporated neurons are visible in the corpus callosum. Dashed line corresponds to midline. Data are represented as mean ± SEM. (B) Fisher’s exact test; (C) one-way ANOVA. *: P<0.05; **: P<10−2.
IUE: in utero electroporation; ChR2: channelrhodopsin; CTB: cholera toxin B.
See also Figure S2.
An additional distinguishing circuit feature of L4- and L2/3-type neurons is a unidirectional L4-to-L2/3 connectivity. Thus, while L2/3-type neurons receive strong input from other L2/3 neurons, L4-type neurons do not (Johns et al., 1999; De la Rossa et al., 2013) (Figure 3A). To examine whether E14.5-born neurons acquire L2/3 neuron-type input in the Kir2.1 condition, we targeted channelrhodopsin 2 (ChR2) expression into L2 neurons via IUE at E16.5 (Figure 3B, left), and recorded evoked synaptic responses in P15 acute cortical slices. In contrast to control E14.5-born L4 neurons, following Kir2.1 IUE, E14.5-born neurons received L2 input, as was the case for genuine, E15.5-born L2/3-type neurons (Figure 3B, right). Thus, E14.5-born neurons repress L4-type and acquire L2/3-type input properties following Kir2.1 IUE.
Finally, we investigated a L4-to-L2/3 reassignment in the output properties of E14.5-born neurons following Kir2.1 IUE. L4-type neurons normally exclusively project locally, while L2/3-type neurons instead send long-range intracortical projections, including to the contralateral hemisphere (De la Rossa et al., 2013; Lefort et al., 2009). Axons of electroporated neurons were clearly visible in the corpus callosum in the E15.5 control and E14.5 Kir2.1 conditions, but less so in the E14.5 control condition (Figure 3C, left). To investigate whether E14.5-born neurons acquire long-range interhemispheric projections upon Kir2.1 IUE, we performed retrograde labelling from the contralateral hemisphere, which revealed a striking increase in callosally-projecting neurons in the E14.5 Kir2.1-electroporation condition (Figure 3C). Together, these findings are consistent with the premature emergence of neurons with L2/3 neuron-type input-output features upon IUE of Kir2.1 at E14.5.
Our findings so far reveal a congruent reassignment in the laminar, molecular, morphological and input-output circuit features of E14.5-born neurons following Kir2.1 IUE, with untimely, precocious presence of L2/3-type neurons at the time when L4 neurons are normally born.
APs precociously generate next-born neuronal subtypes following Kir2.1-induced hyperpolarization
With an isolated neuronal migration defect now excluded, the untimely presence of L2/3-type neurons could reflect either a L4-to-L2/3-type respecification of newborn neurons (scenario 2, Figure 1B), or a pre-mitotic forward shift in the competence of APs to sequentially generate specific neuronal subtypes (scenario 3, Figure 1B). Indeed, since Kir2.1 expression persists in the neuronal progeny of these cells, our observations above could reflect a pre- or post-mitotic effect of Kir2.1 on neuronal differentiation. To distinguish between these two possibilities, we targeted hyperpolarization to postmitotic neurons or to APs (Figure 4A).
Figure 4.

Apical progenitors precociously generate next-born neuronal subtypes following Kir2.1-induced hyperpolarization.
(A) Possible scenarios explaining the laminar shift. Reported from Figure 1B. Letters refer to panels.
(B) Kir2.1 overexpression lowers the Vm of E14.5 APs. Inset: Fluorescent dye-filled patched AP (arrowhead).
(C) Left: Gene expression dynamics for NeuroD1 (from Telley et al., 2016, see http://genebrowser.unige.ch/science2016). Right: Postmitotic expression of Kir2.1 does not affect the radial positioning of E14.5-born neurons.
(D) Premitotical hyperpolarization by early CNO pulse-injection in hM4D-expressing APs shifts the laminar location of E14.5-born neurons towards that of normally later-born neurons.
(E) Premitotical depolarization by early CNO pulse-injection in hM3D-expressing APs shifts the radial location of E15.5-born neurons towards that of normally earlier-born neurons.
(F) The radial distribution of E12.5-born neurons following Kir2.1 hyperpolarization is shifted towards that of later-born neurons. This shift is restricted to deep-layer neurons, a time at which the Kir2.1-expression plasmid is most concentrated.
(G) Molecular expression of the L5B marker CTIP2 is congruent with radial shift in location following Kir2.1 hyperpolarization. The percentage of cells expressing CTIP2 in L5B is unchanged in control and Kir conditions.
Data are represented as mean ± SEM, except scatter plots represented as means ± SD. (B), one-way ANOVA; (C-F), two-way ANOVA for bin distribution analysis (significantly different bins indicated in darker shades); Student’s t test for scatter plot (comparing average cell position). Individual biological replicates are distinguished by color and aligned from left to right. (G), Student’s t test. ***: P<10−3; ****: P<10−4.
SVZ: subventricular zone.
See also Figure S3.
Using whole cell patch clamp recordings of APs at E14.5, we established that Kir2.1 overexpression indeed decreases Vm in these cells (Figure 4B). We first restricted hyperpolarization to postmitotic neurons by using a NeuroD1-promoter Kir2.1 plasmid (Figure 4C, left, and Figure S3A) (Guerrier et al., 2009). In this condition, the radial positioning of E14.5-born neurons was unaffected (Figure 4C, right), indicating that postmitotic hyperpolarization alone is not sufficient to affect this defining feature of L4 neuron identity, and suggesting that hyperpolarization acts pre-mitotically on APs.
To test this possibility, we restricted hyperpolarization to APs by overexpressing hM4D, a DREADD which leads to hyperpolarization upon binding by CNO (Armbruster et al., 2007). Consistent with a critical role of membrane potential in regulating AP behavior, early and transient chemogenetic hyperpolarization of HM4D-expressing APs with 3 CNO pulse injections in the first 24 hours reassigned radial positioning to more superficial locations, although this effect was weaker than that of Kir2.1 (Figure 4D). Therefore, the L4-to-L2/3 shift in radial positioning following Kir2.1-induced hyperpolarization reflects a change in AP competence rather than a direct postmitotic effect on neuronal specification.
Confirming the critical role of Vm on the progression of AP competence, we depolarized E15.5 APs using a different DREADD, hM3D (Armbruster et al., 2007), which led to a shift in the laminar fate of their neuronal progeny to that of normally earlier-born neurons, i.e. to deeper radial positions (Figure 4E; Figure S3A), although this effect was distinctly weaker than that elicited by hyperpolarization. CNO application induced AP depolarization as early as 12 hours following IUE, demonstrating the rapid onset of HM3D expression (Fig. S3A). These neurons acquired E14.5-born neuron morphological features (Figure S3B) and RORB expression in L4 was similar to that of the control condition (data not shown), consistent with an overall reassignment of neuronal identity. Finally, we examined whether hyperpolarization APs at E12.5, a time at which deep-layer neurons are normally born, also led to the generation of normally later-born neurons. As previously reported (Telley et al., 2016; Jabaudon, 2017), early born neurons have an broader radial distribution than later born neurons, yet neurons born from Kir2.1-hyperpolarized APs at E12.5 were located more superficially within deep layers than their control counterparts, and showed a corresponding change in their molecular identity when using CTIP2 (BCL11B) as a marker of L5B neurons (Figure 4F,G) (Arlotta et al., 2005). In contrast to deep-layer neurons, the position of superficial layer neurons was unaltered, consistent with progressive plasmid dilution and a dose-dependent effect of hyperpolarization. The process described here is thus not limited to the L4-to-L2/3 transition, but instead is at play across the neurogenic period of cortical development. Together, these findings suggest that progressive hyperpolarization regulates the competence of APs to generate successive subtypes of neurons.
AP hyperpolarization regulates the progression from direct to indirect neurogenic divisions
Since upon experimental membrane hyperpolarization APs give rise to normally later-born neuronal subtypes (Figures 1A, 4D and 4F), and upon depolarization they give rise to normally earlier-born neuronal subtypes (Figure 4E), we hypothesized that our manipulations might be emulating a physiological progression in AP Vm during corticogenesis. Confirming this possibility, whole cell patch clamp recordings of APs in E12.5 to E15.5 cortical slices revealed a sharp decrease in Vm during this period (Figure 5A). In fact the average voltage-drop induced by Kir2.1 overexpression was similar to the hyperpolarization occurring between E14.5 and E15.5 (22 mV vs. 16 mV, P > 0.05), consistent with an emulation of a natural hyperpolarization of these cells.
Figure 5.

Apical progenitor hyperpolarization regulates the progression from direct to indirect neurogenic divisions.
(A) APs hyperpolarize in the course of corticogenesis. Kir2.1 overexpression and CNO activation of hM3D-expressing APs induce hyperpolarization and depolarization respectively. E14.5 Kir2.1 and E15.5 hM3D data reported from Figures 4B and S3A, respectively.
(B) Schematic representation of the shift from direct to indirect neurogenesis during corticogenesis. Note that indirect neurogenesis is also present at lower levels at early stages.
(C) Direct neurogenesis decreases during corticogenesis. Directly-born neurons are Epor+BrdU−. Kir2.1-hyperpolarization decreases direct neurogenesis in E14.5 APs. Note that while this approach identifies directly-born neurons as GFP+BrdU− cells, GFP+BrdU+ cells cannot be assumed to be born indirectly, since they may be born directly from another round of progenitor divisions (Telley et al., 2016).
(D) Summary of the findings: Kir2.1 overexpression hyperpolarizes APs and leads to a decrease in direct neurogenesis.
(E) Depolarization of hM3D-expressing APs by CNO pulse-injection increases direct neurogenesis.
(F) Kir2.1-hyperpolarization decreases direct neurogenesis in E12.5 APs.
(G) At E14.5, medial cortical areas produce L5 neurons, while lateral areas produce L4 neurons. P7 brains following FT injection at E14.5, which labels neurons directly produced by APs at that time.
(H) Laterally-located APs are more hyperpolarized than medial ones at E14.5, corresponding to the type of neuron being produced. Arrowheads point to the tip of the recording pipette.
Data are represented as mean ± SEM. (A), (C), (D) and (F): one-way ANOVA; (E) and (H): Student’s t-test. *: P<0.05; **: P<10−2; ***: P<10−3.
CP: cortical plate; FT: FlashTag; V: ventricle; AP: apical progenitor.
See also Figure S4.
Which cellular process might this progressive hyperpolarization be associated with? As corticogenesis proceeds, neurons are thought to be increasingly born from intermediate progenitors (IPs) as opposed to directly from APs (Figure 5B) (Noctor et al., 2004; Noctor et al., 2008; Kowalczyk et al., 2009; Taverna et al., 2014; Jabaudon, 2016). Supporting this possibility, using a recent single-cell transcriptomic resource in which cell types were identified by their transcriptional signatures (Yuzwa et al., 2017), we found that IP-to-AP number ratios increases 3-fold between E11.5 and E15.5 (data not shown).
As a first step to investigate whether AP hyperpolarization might regulate a progression in neurogenic modes, we quantified the developmental progression from direct to indirect neurogenesis. For this purpose, we used IUE of a control-GFP reporter plasmid to birthdate-label APs in E12.5, E13.5, E14.5 and E15.5 embryos (Figures 5C, left, and S4A). At each experimental age, GFP birthdate-labeling was immediately followed by chronic 72-hour administration of bromodeoxyuridine (BrdU) to identify intervening cell divisions between time of IUE and P7. Using this strategy, neurons born through direct neurogenesis could be identified as GFP+ BrdU− cells (Telley et al., 2016), allowing us to confirm and quantify the developmental decrease in direct neurogenic divisions of APs as corticogenesis proceeds (Martínez-Cerdeño et al., 2006; Reillo et al., 2011; Vasistha et al., 2015) (Figures 5C, left, and S4A). Importantly, while this approach identifies directly-born neurons as GFP+ BrdU− cells, GFP+ BrdU+ cells cannot be assumed to be born indirectly, since they may be born directly during a later round of progenitor division (Govindan et al., 2018; Telley et al., 2016). As shown in Figure 5D, the dynamics of this progressive decrease in direct neurogenesis corresponds to progressive AP hyperpolarization.
To investigate a causal link between AP hyperpolarization and decreased direct neurogenic divisions, we focused on the transition between L4- and L2/3-type neuron production occurring between E14.5 and E15.5. We hyperpolarized E14.5 APs by IUE of Kir2.1 (Figures 5C, right, and S4B), which led to a decrease in direct neurogenesis to values normally observed at E15.5 (Figures 5C, right, and 5D). Conversely, depolarization of hM3D-expressing E15.5 APs with CNO application reverted direct neurogenesis to levels normally found at earlier time points of corticogenesis (Figures 5E and S4C), although this effect was weaker than the one elicited by hyperpolarization. Finally, hyperpolarization E12.5 APs also precociously decreased direct neurogenesis (Figures 5F and S4D). Thus, AP hyperpolarization leads to a forward developmental decrease in direct neurogenic divisions.
Neurogenesis does not occur synchronously across cortical areas (Polleux et al., 1997a). Instead, lateral regions of the neocortex develop ahead of medial regions, such that at a given time point, APs in distinct areas are not generating the same subtypes of neurons (Figure 5G). If progressive hyperpolarization drives the progression in AP neurogenic competence, then corresponding differences in the Vm of these cells should be detected. Consistent with this hypothesis, laterally-located APs were significantly more hyperpolarized than medially-located APs, with values consistent with those corresponding to the type of neuron being produced (Figure 5H). Thus, different cortical regions contain APs with distinct Vm, whose absolute value is predictive of the type of neurons that is being produced.
Hyperpolarization leads to a forward shift in AP cycling behavior
We next examined the cellular consequences of hyperpolarization on AP division modes. To this end, we first quantified the number of neurons and IPs 24 hours after IUE at E14.5, E15.5, and following E14.5 Kir2.1-hyperpolarization, using NEUROD2 and TBR2 to distinguish these two cell types (Telley et al., 2016). Consistent with the decrease in direct neurogenic divisions described above, we observed a decrease in NEUROD2+GFP+ cells (i.e. newborn neurons) and an increase in TBR2+GFP+ cells (i.e. presumptive IPs) between E14.5 and E15.5, which was emulated by Kir2.1 overexpression at E14.5 (Figure 6A, left and middle). Supporting a genuine increase in IPs (TBR2 is also expressed in a fraction of newborn neurons, see Telley et al., 2016), Ki67+TBR2+ cells in the VZ showed similar dynamics (Figure 6A). To directly investigate the effect of Kir2.1-hyperpolarization on the division mode of single APs, we performed single-cell clonal analysis in vitro following Kir2.1 overexpression to identify the progeny of single progenitors (Pilaz et al., 2016). Kir2.1 overexpression increased the ratio of IP to neurons in the progeny, suggesting that hyperpolarization acts cell-autonomously to regulate neurogenic modes (Figures 6B and S5A–D). Thus, AP hyperpolarization leads to a cell-intrinsic increase in daughter IP production at the expense of neurons.
Figure 6.

Hyperpolarization leads to a forward shift in apical progenitor cycling behavior and transcriptional identity.
(A) Kir2.1-hyperpolarization of APs decreases neuronal production (NEUROD2+) and increases IP numbers (TBR2+) in vivo. TBR2/Ki67 double labeling is used to highlight newborn IPs in the VZ.
(B) In vitro single cell clonal analysis shows increased IP production relative to neurons in the Kir2.1-hyperpolarization condition. Dotted lines connect biological replicates.
(C) The proportion of cells that remain in the VZ after E14.5 Kir2.1-hyperpolarization is increased towards E15.5 levels.
(D) In contrast to control cells, E14.5 Kir2.1-hyperpolarized cells remain susceptible to targeting by a second IUE 12 hours after the first IUE, suggesting a prolonged pre-mitotic period (white arrowheads show double labeled neurons).
(E) E14.5 Kir2.1 hyperpolarization increases the proportion of progenitor cells (Ki67+ and SOX2+).
(F) Kir2.1-hyperpolarized APs show decreased cell cycle exit as demonstrated by double labeling for BrdU and Ki67 at 24h. Cells that remain in the cell cycle are BrdU+Ki67+ while neurons are BrdU+Ki67−.
(G) Left: E14.5 Kir2.1-hyperpolarized APs repress E14.5-type and induce E15.5-type genes 12h after IUE. Right: unbiased SVM classification reveals that the transcriptional identity of E14.5 Kir2.1-hyperpolarized APs is shifted towards E15.5 APs.
(H) Differentially-enriched genes include several cell cycle- and Wnt signaling-related transcripts.
Data are represented as mean ± SEM. (A), (C), (E-G): one-way ANOVA; (B), (D): Student’s test. IP: intermediate progenitor; N: neuron; IZ: intermediate zone.
*: P<0.05; **: P<10−2; ***: P<10−3. See also Figures S5–7.
Newborn neurons and IPs normally exit the VZ within 24 hours of their birth, while cycling APs remain within this compartment (Telley et al., 2016; Toma et al., 2016). Following E14.5 Kir2.1-hyperpolarization, GFP+ cells were retained in the VZ, up to levels normally observed at E15.5 (Figure 6C). These cells remained susceptible to plasmid DNA incorporation by a second IUE performed with a 12 hour-delay, suggesting that they were progenitors rather than hypokinetic postmitotic neurons (Figure 6D). Confirming these findings, Ki67+GFP+ and SOX2+GFP+ cell numbers were increased to E15.5 levels following E14.5 Kir2.1-hyperpolarization (Figure 6E). Dynamic cellular changes were also found following hyperpolarization of E12.5 APs, suggesting that hyperpolarization acts consistently across cortical neurogenesis (Figure S5E). Together, these findings suggest a progressive decrease in cell cycle exit of APs during corticogenesis, which is emulated by Kir2.1-hyperpolarization. To directly measure the cell cycle exit rate of APs, we performed a BrdU pulse-chase experiment (Figure S5F), which revealed that the cell cycle exit rate of hyperpolarized E14.5 APs was decreased to values found in E15.5 APs (Figures 6F and S5G). Similarly, FlashTag pulse-labeling to follow the fate of neurons born directly from APs (Telley et al., 2016) revealed a forward shift in the radial positioning of these cells following E14.5 Kir2.1-hyperpolarization (Figure S5H). Together, these results indicate that hyperpolarization leads to a forward temporal shift in AP division mode and behavior.
Membrane hyperpolarization drives the progression of AP molecular identity
We next investigated the molecular mechanisms underlying progressive AP hyperpolarization and associated changes in neurogenic division modes by using single-cell RNA sequencing. We isolated isochronic cohorts of E14.5, E15.5 and Kir2.1-hyperpolarized E14.5 cells 12 hours after mitosis using microdissection, fluorescence-activated cell sorting (FACS) and microfluidic isolation of single cells. At this time point, neurons essentially consist of directly-born cells (i.e. indirectly-born neurons are not yet born), since only 7.5% of the total neuronal population was labeled by chronic BrdU perfusion (7.5 ± 0.4 % of BrdU+NEUROD2+ cells, n = 3 mice, data not shown and see Telley et al., 2016). APs, daughter IPs and daughter neurons were distinguished based on their single-cell transcriptional signatures at E14.5 (Telley et al., 2016), and the identity of E15.5 and E14.5 Kir2.1-hyperpolarized cells was determined by using this data as a training set (Figure S6A).
Focusing on transcriptionally identified APs from the single-cell RNA sequencing data (Figure S6A), we first identified developmentally-regulated genes in APs at E14.5 and E15.5 (Figure 6G). Hyperpolarization of E14.5 APs by Kir2.1 overexpression repressed a battery of E14.5-type AP genes and precociously induced E15.5-type genes (Figure 6G), in line with the forward shift in the neurogenic features and in daughter neuron identity reported earlier. When the analysis was restricted to 12h-old postmitotic neurons instead of APs, acquisition of a L2/3-type transcriptional signature in the E14.5 condition was already detectable, revealing an early shift in postmitotic developmental programs (Figure S6B and Table S3). Unbiased training of an SVM on E14.5 APs and E15.5 APs transcriptional data and on 12h-old neurons at the same stages (Frangeul et al., 2016) revealed that hyperpolarized APs and their 12-hour old neuronal progeny shift their transcriptional identity towards normally later-born cells (Figure 6H and Fig. S6C).
Ontology analyses highlighted two classes of differentially-expressed genes: genes involved in cell-cycle regulation, consistent with our findings above, and genes involved in WNT signaling, whose functional relevance will be described in the next section (Fig. 6H and Table S2). In addition, this analysis revealed that transcripts for K+ channels which are key regulators of Vm (Arcangeli et al., 1995; Urrego et al., 2014), are specifically upregulated between E14.5 and E15.5 (Figure S7A). Supporting a critical functional role for Kir channels in developmental hyperpolarization, in vitro blockade of these channels with BaCl2 (Yasuda and Adams, 2010) at E14.5 and E15.5 led to a dose-dependent depolarization of APs, which occluded the Vm difference between these two developmental time points (Figure S7B). Furthermore, E14.5 in vivo blockade of Kir2.1 channels with BaCl2 increased net neuronal production, as determined by a decrease in TBR2+ cells (IPs) and an increase in NEUROD2+ cells (neurons) (Figure S7C). Pharmacological blockade of voltage-gated K+ channels with tetraethylammonium (TEA) and 4-aminopyridine (4-AP), which can be blocked by BaCl2, did not affect the membrane potential of APs (data not shown), effectively eliminating this subtype of channels as the source of the developmental progression in Vm. Non-voltage-gated K+ channels (and particularly Kir channels) thus remain as the most likely effectors of membrane hyperpolarization.
AP membrane hyperpolarization represses Wnt signaling to drive developmental progression in daughter neuron identity
Canonical WNT signaling is dynamically regulated and has multiple effects on progenitor function. At early stages of corticogenesis, downregulation of WNT signaling primarily decreases precursor proliferation (Woodhead et al., 2006), while at later stages it also inhibits neuronal production (Munji et al, 2011). Given the presence of a dynamically regulated cell cycle- and WNT-associated transcripts between E14.5 and E15.5 and following experimental hyperpolarization (Fig. 6H), we hypothesized that the WNT / β-catenin signaling pathway might mediate the effects of hyperpolarization on cell cycling behavior and daughter neuron fate.
We first used a reporter construct to monitor β-catenin-dependent WNT activity (Boitard et al., 2015), which confirmed that WNT signaling decreases as corticogenesis proceeds. Supporting an action of the Vm in this process, E14.5 AP hyperpolarization with Kir2.1 decreased canonical WNT signaling to levels normally found at E15.5 (Figure 7A). Hyperpolarization of E14.5 APs decreases neuronal production to levels normally found at E15.5 (Figure 6A); using a dominant-negative TCF construct (DN-TCF) to repress WNT signalling, we observed a similar effect, in line with previous findings (Munji et al, 2011) (Figure 7B). Furthermore, constitutive WNT activation using β-catenin overexpression (Boitard et al., 2015) mitigated the effect of hyperpolarization (Figure 7B), demonstrating that WNT acts as a molecular link between Vm and neurogenesis.
Figure 7.

Membrane hyperpolarization represses Wnt signaling and drives the developmental progression in daughter neuron identity.
(A) AP hyperpolarization decreases canonical Wnt pathway activity. Arrowheads show example of 2 cells with active Wnt signaling as revealed by IUE of a Wnt reporter construct.
(B) Blocking canonical Wnt signaling by overexpressing the dominant negative form of TCF4 replicates the effect of Kir2.1-mediated hyperpolarization on neuronal output. Inducing canonical Wnt signaling by β-catenin overexpression rescues hyperpolarization-mediated reduction in neuronal output. Control and Kir2.1 values have been reported from Figure 6A for comparison.
(C) E14.5 AP-targeted repression of Wnt signaling using an inducible DN-TCF4 construct causes a forward shift in their laminar identity. Compare with Fig. 1A.
(D) Molecular expression is congruent with laminar location in single neurons following E14.5 AP Wnt signaling repression.
Data are represented as mean ± SEM, except scatter plots represented as means ± SD. (A), (B): one-way ANOVA; (C), two-way ANOVA for bin distribution analysis (statistically significantly different bins indicated in darker shades); Student’s t test for scatter plot (comparing average cell position). Individual biological replicates are distinguished by color and aligned from left to right. (D), Student’s t test. *: P<0.05; **: P<10−2; ***: P<10−3; ****: P<10−4.
DN-TCF4: dominant negative TCF4; ND2: NEUROD2.
To directly test the role of this molecular link not only in the generation of neurons, but also in the progression of fate of daughter neurons, we examined whether selective repression of WNT in APs replicates the effects of Kir2.1 on daughter neuron identity. We used an inducible DN-TCF4 construct to down-regulate WNT signaling specifically in progenitors and assessed the effect of this repression on the laminar identity of daughter postmitotic neurons at P7. Consistent with a WNT-mediated effect on the neurogenic competence of progenitors, neurons born from APs with repressed WNT signaling were located more superficially than their control counterparts, replicating the effects of hyperpolarization alone (Figure 7C). In addition, their molecular identity, as assessed using RORB and BRN2 expression, was consistent with their new radial position (Figure 7D). Together these findings demonstrate that membrane hyperpolarization regulates the progression of AP neurogenic behavior and corresponding daughter neuron fate by repression of WNT β-catenin signaling.
DISCUSSION
Our results reveal that progressive membrane hyperpolarization drives the temporal progression in AP identity during corticogenesis. Activity-dependent modulation of canonical WNT signaling at later stages of differentiation, during dendritic arborization and synaptic plasticity (Ataman et al., 2008; Wayman et al., 2006; Chen et al., 2006; Yu and Malenka, 2003). Our findings show that changes in Vm also modulate this pathway in progenitors.
Vm values also determine the ability of cells to respond to electrogenic signals in the extracellular environment. Classical ligand-receptor-driven molecular pathways are triggered by the binding specific compounds. Since ionotropic receptors require some level of polarization to respond to such ligands, the dynamic progression in Vm identified here may additionally act to regulate the sensitivity of APs to extracellular signaling molecules, including neurotransmitters (Dehay et al., 2001; LoTurco et al., 1995; Polleux et al., 1997b), and play a permissive role in cell fate progression.
Given the area-specific differences in Vm identified here, our findings, by setting the sensitivity to extracellular signals, provide a potential mechanism through which specific inputs could impart activity-dependent cellular features to cortical areas during development. Other extracellular parameters such as ionic composition, metabolic supplies, or even the three-dimensional structure of the neurogenic niche may also affect Vm and hence progenitor behavior.
Chemogenetic AP depolarization reversed the normal course of neurogenesis and allow re-emergence of normally earlier-born neuronal subtypes. Although depolarization was less efficient than hyperpolarization in controlling cell fate, this result reveals an unexpected degree of plasticity in APs. In Drosophila, late neuroblasts can again produce early-born neurons when provided with appropriate extrinsic cues (Kohwi and Doe, 2013) and cortical progenitors can under certain circumstances change their competence when transplanted at different developmental time points (McConnell and Kaznowski, 1991). Vm values may determine the ability of these cells to respond to fate-determining extracellular signals in these conditions.
We used early pulse-applications of CNO soon after IUE of a DREADD construct to target APs and distinguish the pre- and post-mitotic effects of hyperpolarization on cell fate. Although APs will preferentially be targeted by this procedure, postmitotic neurons born during the period of CNO pulse application will also be affected. A significant postmitotic effect on neuronal identity was excluded by the use of an early-onset NEUROD-promoter construct, yet newborn neuron fate may also be modified by early manipulations of their Vm.
Mesenchymal stem cells progressively hyperpolarize during development and experimental depolarization keeps them in a stem state, while hyperpolarization accelerates their differentiation into postmitotic cells (Sundelacruz et al., 2008). Our findings may thus be generalizable to progenitor cells whose competence progresses with time. Since Vm is a relatively straightforward cellular parameter to record, our observations may also prove useful to monitor progression of progenitor competence.
Altogether, the bioelectrical regulation over the progression of progenitor competence identified here provides a parsimonious mechanism to coordinate and pattern progenitor behavior across space and time to generate context-specific cellular types. If bioelectrical effects are also at play in species with prolonged corticogenesis such as primates, large effects on neuronal fate and circuit assembly are to be expected.
STAR ★ METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Denis Jabaudon (denis.jabaudon@unige.ch).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experimental procedures were approved by the Geneva Cantonal Veterinary Authority. Embryonic day (E) 0.5 was established as the day of vaginal plug. Wild-type CD1 mice were provided by Charles River Laboratories. Male and female embryos between E12.5 and E15.5 were used for the in utero electroporations, and pups between postnatal day (P) 0 and P21 for the postnatal experiments. Pregnant dams were kept in single cages and pups were kept with their mothers until P21, in the institutional animal facility under standard 12 : 12 h light / dark cycles.
METHODS DETAILS
In utero electroporation
Timed pregnant CD1 mice were anaesthetized with isoflurane (5% induction, 2,5% during the surgery) and treated with the analgesic Temgesic (Reckitt Benckiser, Switzerland). Embryos were injected unilaterally with 700 nl of DNA plasmid solution (diluted in endo-free TE buffer and 0.002% Fast Green FCF (Sigma)) into the lateral ventricle. Embryos were then electroporated by holding each head between circular tweezer-electrodes (5 mm diameter, Sonidel Limited, UK) across the uterine wall while 5 electric pulses (35 V for E12.5, 40 V for E13.5, 45 V for E14.5, 50 V for E15.5, 55 V for E17.5 and E18.5, 50 ms at 1 Hz) were delivered with a square-wave electroporator (Nepa Gene, Sonidel Limited, UK). Where described, 12 hours after the first surgery the uterine horns were exposed again and embryos injected intra-ventricularly with FlashTag (488 CellTraceTM CFSE, Life Technologies, #C34554; 405 CytoTellTM Blue, AAT Bioquest, #22252) (Telley et al., 2016).
Plasmids
Injected plasmids were: pCAG-IRES-GFP (0,75 µg/µl) and pCAG-hKir2.1-IRES-GFP (2 µg/µl) (gift from Guillermina Lopez-Bendito); pCAG-IRES-mCherry (1,25 µg/µl; subcloned from pMSCV-IRES-mCherry, Addgene #52114); pCAG-ChR2-Venus (Addgene #15753); pNeuroD-IRES-GFP and pNeuroD-hKir2.1-ires-GFP (2 µg/µl; subcloned from pNeuroD-CRE-IRES-GFP, gift from Dayer lab); pCAG-hM4D-IRES-GFP (2 µg/µl; subcloned from pcDNA5-HA-hM4D, Addgene #45548); pGR-hM3D-pGK-GFP (2µg/µl; Addgene #45547); pGR-IRES-tdTOM (0,75 µg/µl; gift from Alexandre Dayer); pGR-hKir2.1-HA (2 µg/µl); pGR-M38-TOPdGFP (2 µg/µl, Addgene #17114); pGR-dnTCF4-pGK-GFP (2 µg/µl); pGR-∆45-βcatenin-pGK-GFP (2 µg/µl), pCWX-pTF-dnTCF4-pGK-GFP-E2A-rtTA (2 µg/µl).
Injections (BrdU, CNO, Doxycycline)
A 16.25 mg/ml solution of BrdU (Sigma) was prepared in 1 : 1, DMSO : water. An osmotic pump (Alzet, #1003D) was filled with this solution and placed in the peritoneal cavity at the end of the surgery (Telley et al., 2016). This procedure was repeated after 72 hours in the E12.5 conditions. For single-pulse labeling, a single dose of 50 mg/kg of animal weight of BrdU (10 mg/ml in water) was administered intra-peritoneally. CNO (Clozapine N-oxide, Sigma) was dissolved in 0.9% saline to a final concentration of 0.1 mg/ml and administered via intra-peritoneal injections (1 mg/kg animal weight) every 8 hours for the first 24 hours after surgery. Doxycycline (Sigma) was dissolved in water at the final concentration of 1 mg/ml. It was administered in drinking water immediately after in utero electroporation for 24 hours, in order to activate transgene expression from the inducible construct (pCWX-pTF-dnTCF4-pGK-GFP-E2A-rtTA).
Retrograde labeling
Focal retrograde labeling from the contralateral hemisphere was performed on hypothermic P5 mice placed on a newborn mouse-adapted stereotaxic apparatus (Stoelting, USA). Three microinjections of 92 nl Alexa Fluor 555-conjugated cholera toxin subunit B (Invitrogen) were executed in S1 (coordinates from Lambda: AP: + 1.4, ML: ± 1.8, DV: − 0.1; AP: + 1.6, ML: ± 1.8, DV: − 0.1; AP: + 1.6, ML: ± 2.0, DV: − 0.1) to retrogradely label contralateral callosal projecting neurons.
Immunohistochemistry and imaging
Embryos were collected 24 h or 48 h following in utero electroporation and post-fixed overnight in 4% paraformaldehyde (PFA, Sigma) at 4°C. Postnat al mice from P0 were perfused with 4% PFA and post-fixed overnight in 4% PFA at 4°C. Fift y to 70 µm coronal sections were performed using a vibrating microtome (Leica, #VT100S). Immunofluorescence staining was performed as followed: sections were incubated for one hour at room temperature in blocking solution (3% Bovine Serum Albumine, 0,3% Triton X-100, diluted in PBS 1X), then overnight at 4°C with primary antibodies. Treatment with HCl 2 N at 37°C for 40’ was performed after incubation with standard blocking solution for BrdU immunohistochemistry. Sections were rinsed three times in PBS 1X and incubated for 1 hour at room temperature with corresponding secondary antibodies (1:500, Life Technologies). Three washes in PBS 1X were performed, the second one using Hoechst staining solution (1:10000 in PBS 1X, Life Technologies) to label nuclei, before dry mounting on slides with Fluoromount (Sigma). For imaging, the putative primary somatosensory cortex (S1) was used as region of study for all the experiments. Images were acquired on Eclipse 90i epifluorescence microscope (Nikon) or on LSM 700 confocal laser scanning microscope (Carl Zeiss).
Antibodies
Rat anti-BrdU (1:250; Abcam, #AB6326), goat anti-BRN2 (1:50; SC Biotech, #SC6029); goat anti-BRN2 (1:500; Thermo Scientific, #PA5–1904; rabbit anti-KI67 (1:250; Abcam, #AB15580); rabbit anti-NEUROD2 (1:1000; Abcam, #AB104430); rabbit anti-RORB (1:500; Diagenode, #C15410001); mouse anti RORB (1:200; Perseus Proteomics, #PP-N7927–00); goat anti-SOX2 (1:500; SC Biotech, #SC17320); chicken anti-TBR2 (1:500; Millipore, #AB15894); rat anti-TBR2 (1:500; Invitrogen, #14–4875-82); mouse anti-guinea pig anti-VGLUT2 (1:2000; Millipore. #AB2251).
Live clonal analysis in vitro
Figure 6B and S5A–D: E14.5 dorsal cortices were microdissected from C57BL/6J mice and pooled. Primary cultures were prepared as previously described in Pilaz et al., 2016 (TBR2 immunostaining was used in the current protocol instead of Tbr2-EGFP knock-in mice). Two sets of experiments were done with 3 biological replicates (= litter) plated into several wells for replicate 1. Cortices were chemically dissociated using 0.25% Trypsin + EDTA (Gibco) for 10 min at 37°C. Trypsin inhibitor (Sigma) was added and cells were mechanically dissociated by pipetting 6–8 times with a fire polished Pasteur pipette. 1×106 cells were nucleofected with either pCAG-IRES-GFP alone (CtlGFP) or pCAG-IRES-GFP + pCAG-Kir2.1-IRES-GFP (Kir2.1GFP) using an Amaxa 4D nucleofector (Lonza) with P3 reagent. Cells were seeded in a 12 well glass bottom plate (MatTek) coated with poly-d-lysine in neural progenitor media (DMEM + Glucose, N2, B-27, bFGF, N-Acetyl Cysteine, Sodium Pyruvate, Glutamine). Cells were maintained at 37°C with 5%CO2 for 16 hours prior to live imaging. For live imaging cells were transferred to a Zeiss Axio Observer Z.1 with a XL multi S1 incubation chamber, CO2 module S, temperature module S, and humidity control and held at 37°C with 5% CO2. DIC and GFP images were captured every 1 hour for a total of 20 hrs. Following live imaging, cells were fixed in 4% PFA for 10 min and immunostaining was performed. Cells were treated with 0.25% Triton X-100 for 10 min and blocked in 10% NGS for 30 min at room temperature. Primary antibody incubations were performed for 2 hours at room temperature. Primaries used were mouse anti-TUJ1 (1:500, Covance), to identify neurons, and rabbit anti-TBR2 (1:250, Abcam), to identify IPs, when expressed in isolation. Secondary antibodies were used at 1:300 (Life Technologies). A total of 510 cells were produced from 255 divisions in the CtlGFP condition; 588 cells were produced from 558 divisions in the Kir2.1GFP condition. 90 neurons were produced in the CtlGFP condition, 123 in the Kir2.1GFP condition. Three IPs were produced in the CtlGFP condition vs. 16 in the Kir2.1GFP condition. IP/N ratio in the CtlGFP condition: 0.033 vs. 0.131 in the Kir2.1GFP condition. A χ2 test was used for statistical comparison.
Bulk and single-cell RNA sequencing
Figure 1E,F: Three brains per condition were electroporated with either pCAG-IRES-mCherry or pCAG-Kir2.1-IRES-GFP plasmid at E14.5 and with pCAG-IRES-mCherry plasmid at E15.5 and were dissected in frozen aCSF and cut coronally at P3 at a thickness of 600 µm in ice-cold and oxygenated aCSF with 3 mM kynurenic acid (Sigma) using a vibrating microtome. The presumptive somatosensory cortex was dissected under stereomicroscopic control and collected samples were mechanically dissociated on ice and chemically digested with 0.5 mg/ml pronase (Sigma), and washed in aCFS / kynurenic acid. Fluorescent cells were FAC-sorted using a MoFlo Astrios (Beckman) device and collected in RNAlater (Sigma) at 4°C. RNA extraction was performed using RNeasy Micro kit (Qiagen).
Figures 6G,H S6A–C: For embryonic collections, 3 pups were electroporated with pCAG-Kir2.1-IRES-GFP at E14.5 or with pCAG-IRES-GFP at E15.5. Since electroporated cells are affected in the S-phase and VZ progenitors take about 12 hours to move from the basal VZ (where they undergo S-phase) to the apical VZ (where they undergo M-phase and are susceptible to FlashTag labeling), FlashTag (FT) was injected in these pups after 12h to compare isochronic cohorts of labeled cells within single animals (see Telley et al., 2016 for details). Labeled populations of interest thus consisted in E14.5 FT+ cells (FT+ Kir2.1GFP–, referred to as E14.5 Ctl), E15.5 FT+ cells (Kir2.1GFP–, referred to as E15.5 Ctl) and E14.5 FT+ Kir2.1+ cells (FT+ Kir2.1GFP+, referred to as E14.5 Kir2.1). The presumptive somatosensory cortex was collected 24 hours after the electroporation (i.e. 12h after FT) and FAC-sorted as previously described (Telley et al., 2016). Two cell suspensions (E14.5 Ctl and E14.5 Kir2.1GFP, and E15.5 Ctl) were then loaded into the 2 inlets of a C1 Single-Cell AutoPrep integrated fluidic circuit (IFC) designed for 10- to 15-µm cells (HT-800, Fluidigm). The plate was processed following the manufacturer’s protocol (C1 system, Fluidigm) and then transferred to an inverted microscope to image and assign wells to conditions.
RNA library preparation
Figure 1E,F: Reverse transcription and pre-amplification of cDNA was achieved using SMARTer Ultra Low RNA kit (Clontech). RNA-sequencing libraries of the harvested cDNA were prepared using Nextera XT DNA library prep kit (Illumina). Libraries were multiplexed and sequenced with 50bp paired-end reads using HiSeq2000 platform (Illumina) with an expected depth of 60M reads.
Figures 6G,H S6A–C: Reverse transcription and pre-amplification of cDNA was achieved on the chip using manufacter protocol (Fluidigm). RNA-sequencing libraries of the harvested cDNA were prepared using Nextera XT DNA library prep kit (Illumina). Libraries were multiplexed and sequenced according to the manufacturer’s recommendations with 70bp/30bp paired-end reads using HiSeq2000 platform (Illumina) with an expected depth of 1M reads per single cell.
All preparations were done by the iGE3 Genomics Platform of the University of Geneva. The sequenced reads were aligned to the latest reference assembly for mouse genome (GRCm38) using the read-mapping algorithm TopHat. The number of reads per transcript was calculated with the open-source HTSeq Python library. All the analyses were computed on the UG Vital-It cluster administered by the Swiss Institute of Bioinformatics.
Electrophysiology
Four-hundred µm thick coronal slices were prepared 24 hours after in utero electroporation of E12.5 / E13.5 / E14.5 / E15.5 embryos with pCAG-IRES-GFP (Figure 5A AND S7B), pCAG-Kir2.1-IRES-GFP (Figure 4B), pGR-hM3D-pGK-GFP (Figure S3A), pCAG-hM4D-IRES-GFP (Figure S3A), and pNeuroD-Kir2.1-IRES-GFP (Figure S3A) or 12 hours after FlashTag injection of E15 or E16 embryos (Figures 5H). Two-hundred-fifty µm thick slices from P15 brains were used for the experiments described in Figure 3B. Slices were kept for at least 30 minutes in artificial cerebrospinal fluid (aCSF) at 32°C (119 mM NaCl, 2.5 mM KCl, 1.3 mM MgCl2, 2.5 mM CaCl2, 1.0 mM NaH2PO4, 26.2 mM NaHCO3 and 11 mM glucose, bubbled with 95% O2 and 5% CO2) before recording. The slices were then transferred in the recording chamber, submerged and continuously perfused with aCSF at 32–34°C. The internal solution used for the experiments contained 140 mM potassium gluconate, 2 mM MgCl2, 5 mM KCl, 0.2 mM EGTA, 10 mM HEPES, 4 mM Na2ATP, 0.3 mM Na3GTP and 10 mM creatine phosphate (pH 7.2, 300 mOsm). Immediately after the whole-cell configuration, resting membrane potential was measured in current-clamp mode.
For resting membrane potential recordings (Figures 4B, 5A,H, S3A and S7B), membrane potential was monitored every 10 s and averaged for 6 consecutive acquisitions, within the first 2 minutes after the whole-cell configuration establishment. In n = 9 E14.5 cells and n = 15 E15.5 cells with at least 2 minutes of recording following the current clamp configuration, Vm remained stable within conditions and significantly different across conditions throughout the duration of the recording, indicating that Vm recordings are not influenced by cytoplasmic dilution with the patch pipette solution.
For DREADD in vitro validation (Figure S3A), a stable resting membrane potential baseline was measured for 4 minutes. CNO at 100 nM concentration was bath-applied for 6–10 minutes and then washed.
For pharmacological experiments (Figure S7B and Results), a stable resting membrane potential baseline was measured for 4 minutes. Two concentrations of BaCl2 were bath applied for 5 minutes each (1 mM and 3 mM), and then washed. For voltage gated potassium channel blockage, tetraethylammonium (TEA) and 4-aminopyridine (4-AP) were bath apply for 10 minutes at 1mM each and then washed.
For optogenetic experiments (Figure 3B), EPSC were evoked in presence of PTX by a 4 msec blue-light pulse at 0.1 Hz delivered through the 40X objective (100 µM; Tocris). Currents were amplified, filtered at 5 kHz and digitized at 20 kHz. Access resistance was monitored by a hyperpolarizing step of −14 mV at each sweep, every 10 s. The illustration provided in Figure 4B was acquired by filling a patched VZ cell with a fluorescent dye using a 2-photon microscope (courtesy of Sandrine Lefort).
QUANTIFICATION AND STATISTICAL ANALYSES
Histological analyses
Zen and ImageJ softwares were used to analyse images. All results are shown as mean ± SEM, except when indicated otherwise. For statistical analyses, the following convention was used: *: P < 0.05, **: P < 0.01, ***: P < 0.001. “Student’s t-test” refers to the unpaired test. For embryonic analyses, differently electroporated embryos from the same mother were used to reduce plug timing variability. Experiments in Figure 1A, 4C–F and 7C were cross-quantified blindly (i.e. the investigator was unaware of which of the experimental conditions the sections were collected from).
Figures 1A, 4C–F and 7C: Two to 3 sections for each electroporated brain were used to define the laminar position (Y) of electroporated cells at P7. The analyses have been carried blindly. The Y value was normalized as percentage of distance from IZ. The cortex was divided in 20 bins and the mean value of frequency distribution was plotted in a bar graph. The dark-colored bins are significantly different for the considered conditions. The Y values have been plotted grouped by brain and the mean value and standard deviation are represented. Density plot and cumulative distribution have been used to additionally display the Y values. Figure 1A: IUE at E14.5 with pCAG-IRES-GFP at E14.5 (number of brains, n = 3) or with pCAG-Kir2.1-IRES-GFP (n = 3). Mean value for Y position in E14.5 control: 64.6 ± 5.644, E14.5 Kir2.1: 73.03 ± 7.085. Figure 4C: IUE at E14.5 with pNeuroD-IRES-GFP at E14.5 (number of brains, n = 5) or with pNeuroD-Kir2.1-IRES-GFP (n = 3). ). Mean value for Y position in E14.5 pNeuroD-control: 70.81 ± 4.913, E14.5 pNeuroD-Kir2.1: 70.09 ± 6.397. Figure 4D: IUE at E14.5 with pCAG-hM4D-IRES-GFP at E14.5 and injected with NaCl (number of brains, n = 5) or CNO (n = 8). Mean value for Y position in E14.5 hM4D + NaCl: 67.83 ± 4.511, E14.5 hM4D + CNO: 77.65 ± 6.432. Figure 4E: IUE at E14.5 with pGR-hM3D-IRES-GFP at E15.5 and injected with NaCl (number of brains, n = 4) or CNO (n = 3). Mean value for Y position in E15.5 hM3D + NaCl: 79.09 ± 12.23, E15.5 hM3D + CNO: 70.53 ± 10.53. Figure 4F: IUE at E12.5 with pCAG-IRES-GFP at E14.5 (number of brains, n = 3) or with pCAG-Kir2.1-IRES-GFP (n = 3). Mean value for Y position in E12.5 control: 32.67 ± 21.18, E12.5 Kir2.1: 43.2 ± 18.51. Figure 7C: IUE at E14.5 with pCAG-IRES-mCherry (number of brains, n = 3) or with pCWX-pTF-dnTCF4-pGK-GFP-E2A-rtTA (n = 3). Mean value for Y position in E14.5 control: 71.99 ± 5.537, E14.5 dnTCF4: 78.3 ± 5.018. For the experiments with hM4D and hM3D, in the same pregnant mice, some embryos were injected with pCAG-hM4D-IRES-GFP or pGR-hM3D-IRES-GFP plasmids and other with pCAG-IRES-mCherry or pGR-IRES-TdTom, respectively and then injected with either NaCl 0.9% or CNO. P7 mice electroporated with pCAG-IRES-mCherry or pGR-IRES-TdTom were used as internal controls and were statistically comparable to controls used for the analyses (hM4D or hM3D injected with NaCl; data not shown). A two-way ANOVA with Bonferroni post-hoc test was used for bin analysis; a Student’s t-test was used for mean Y position.
Figures 1C, 4G, 7D: Three sections for each brain electroporated at E14.5 with pCAG-IRES-GFP (number of brains, n > 3) or pCAG-Kir2.1-IRES-GFP (n > 3) were used to quantify the number of RORβ+ and BRN2+ cells among the fraction of electroporated cells in L2/3 at P7. Percentage of RORB+ in L2/3 in E14.5 control: 6.87 ± 2.47, E14.5 Kir2.1: 4.47 ± 1.36, E15.5 control: 2.27 ± 1.16. Percentage of BRN2+ in L2/3 in E14.5 control: 93.13 ± 0.82, E14.5 Kir2.1: 93.47 ± 3.29, E15.5 control: 82.23 ± 8.50. Two to three sections for each brain electroporated at E14.5 with pCAG-IRES-mCherry (n = 3) or pCWX-pTF-dnTCF4-pGK-GFP-E2A-rtTA (n = 3) were used to quantify the number of RORB+ and BRN2+ cells among the fraction of electroporated cells at P7 in L2/3. Percentage of RORB+ in E14.5 control: 7.915 ± 4.79, E14.5 Kir2.1: 12.6 ± 7.35. Percentage of BRN2+ in E14.5 control: 95.42 ± 3.56, E14.5 Kir2.1: 91.38 ± 4.38. Two to three sections for each brain electroporated at E12.5 with pCAG-IRES-GFP (number of brains, n > 3) or pCAG-Kir2.1-IRES-GFP (n > 3) were used to quantify the number of Ctip2+ cells among the fraction of electroporated cells in L5 at P7. Percentage of Ctip2+ in E12.5 control: 30.68 ± 14.78, E14.5 Kir2.1: 30.37 ± 0.92. A Student’s t-test was used. The fluorescence intensity (0–255 scale of 8-bit images) for RORB+, BRN2+ and Ctip2+ of the counted cells has been plotted to show the expression of these markers.
Figure 2A: Coronal sections from at least 4 different brains electroporated with pCAG-IRES-GFP at E14.5 (n = 12 neurons), at E15.5 (n = 13 neurons) or with pCAG-Kir2.1-IRES-GFP (n = 16 neurons) were used to quantify the cumulative dendritic radial location. The angle between adjacent primary dendrites was measured and plotted for each analysed cell in a cumulative way and angles were aligned based on the position of the axon. Median angle value in E14.5 control: 90.05°, E15.5 control: 153.60°, E14.5 Kir2.1: 157.80°. A Kruskal-Wallis test with Dunn post-hoc test was performed.
Figures 2B and S3B: Coronal sections from at least 3 different brains electroporated with pCAG-IRES-GFP at E14.5 (total of 240 neurons), at E15.5 (total of 140 neurons), with pCAG-Kir2.1-IRES-GFP (total of 330 neurons), or following CNO application in hM3D-expressing cells (total of 57 neurons and 87 controls with hM3D only) were used to quantify the percentage of electroporated cells displaying an apical dendrite at P7. Percentage of cells with apical dendrite in E14.5 control: 21.05 ± 4.68, E15.5 control: 82.68 ± 2.62, E14.5 Kir2.1: 41.00 ± 2.93, E15.5 hM3D control: 80.26 ± 0.79, E15.5 hM3D CNO: 23.08 ± 4.67. A one way-ANOVA with Tukey post-hoc test was used.
Figure S2: Three coronal sections for each brain electroporated at E14.5 with pCAG-IRES-GFP (number of brains, n = 5) or with pCAG-Kir2.1-IRES-GFP (n = 6) were used to quantify the intensity level of VGLUT2 immunostaining along a vertical line with ImageJ software. The length of the line was then normalized as percentage distance from the IZ and averaged values were represented.
Figure S3A: Two sections from at least 4 embryonic brains electroporated with pGR-hM3D-IRES-GFP at E15.5, pCAG-hM4D-IRES-GFP or pNeuroD-Kir2.1-IRES-GFP were used for patch-clamp experiments 12 or 24 hours after. Recorded Vm in apical progenitors in the VZ at E15.5 hM3D Ctl at 24 h: −70.13 ± 2.56 mV; E15.5 hM3D + CNO at 24 h : −43 ± 10.55 mV; n = 5 cells. E15.5 hM3D Ctl at 12 h: −70.46 ± 1.97 mV; E15.5 hM3D + CNO at 12 h : −60.94 ± 5.12, n = 10 cells. E14.5 hM4D Ctl at 24 h: −60.64 ± 2.28 mV; E14.5 hM4D + CNO at 24 h : −67.95 ± 2.45 mV, n = 7 cells. Recorded Vm in apical progenitors in the CP at E14.5 pNeuroD-control at 48 h: −55.26 ± 3.16 mV; E14.5 pNeuroD-control at 24 h : −71.38 ± 4.30 mV, n = 10 cells. A Student’s t-test was used.
Figure 3B: Two to four sections from at least four P15 brains electroporated with pCAG-IRES-GFP at E14.5 (n = 7 neurons), E15.5 (n = 8 neurons) or with pCAG-Kir2.1-IRES-GFP at E14.5 (n = 14 neurons) and with pCAG-Chr2Venus/pCAG-IRES-mCherry at E16.5 (to label L2 cells) were used to measure the connectivity. Connectivity rate of E14.5 control: 0 / 7, E15.5 control: 7 / 8, E14.5 Kir2.1: 8 / 14. A Fisher’s exact test was used.
Figure 3C: Three sections for each brain electroporated with pCAG-IRES-GFP at E14.5 (number of brains, n = 3), E15.5 (n = 3) or with pCAG-Kir2.1-IRES-GFP (n = 4) and injected with CTB at P5, were used to quantify the number of CTB+ cells among the fraction of GFP+ cells at P7. % CTB + in E14.5 control: 12.00 ± 3.60, E15.5 control: 70.43 ± 6.72, E14.5 Kir2.1: 40.85 ± 6.80. A one-way ANOVA with Tukey post-hoc test was used.
Figures 4B and 5A: Two sections from at least 4 embryonic brains electroporated with pCAG-IRES-GFP at E12.5 (number of recorded cells, n = 13), E13.5 (n = 7), E14.5 (n = 23), E15.5 (n = 21) or with pCAG-Kir2.1-IRES-GFP at E14.5 (n = 10) were used for patch-clamp experiments 24 hours after IUE. Recorded Vm at E12.5: −41.87 ± 3.00 mV, n = 13 cells; E13.5 −51.00 ± 4.23 mV, n = 7 cells; E14.5: −59.71 ± 2.13 mV, n = 23 cells; E15.5: −75.69 ± 1.88 mV, n = 21 cells; E14.5 Kir2.1: −82.36 ± 1.55 mV, n = 10 cells. A one way-ANOVA with Tukey post-hoc test, including the Kir2.1 data presented in 4B, was used.
Figures 5C,D,F and S4A,B,D: Three sections for each brain electroporated with pCAG-IRES-GFP at E12.5 (number of brains, n = 3), E13.5 (n = 3), E14.5 (n = 5), or E15.5 (n = 5) and with pCAG-Kir2.1-IRES-GFP at E14.5 (n = 6) or at E12.5 (n = 3) and chronically-delivered BrdU were used to quantify the number of BrdU− cells among the total amount of GFP+ cells at P7. Percentage of BrdU− GFP+ at E12.5: 39.74 ± 0.38, E13.5: 37.15 ± 0.44, E14.5: 24.47 ± 2.40, E15.5: 9.94 ± 2.32, E14.5 Kir2.1: 8.02 ± 2.00, E12.5 Kir2.1: 17.12 ± 1.00. A one way-ANOVA with Tukey post-hoc test was used.
Figures 5E and S4C: Three sections for each brain electroporated at E15.5 with pGR-hM3D-GFP and injected for 24 hours with NaCl 0.9% (number of brains, n = 4) or CNO (n = 3) and chronically delivered with BrdU were used to quantify the number of BrdU− GFP+ cells within the total amount of GFP+ cells at P7. Percentage of BrdU− GFP+ in NaCl-injected mice: 14.79 ± 0.50 and in CNO-injected mice: 33.28 ± 1.68. E15.5 Ctl : −70.13 ± 2.56 mV; E15.5 CNO : −43 ± 10.55 mV, n = 5 cells. A Student’s t-test was used.
Figure S5H: Three sections for each brain electroporated at E14.5 with pCAG-IRES-GFP (n = 5) or with pCAG-Kir2.1-IRES-GFP (n = 5) and injected 12 hours after with FT were used to quantify the number of FT+ cells among the total amount of GFP+ cells at P7. FT+ cells are defined as the top 10% of the brightest cells (< 10% of these cells are labeled by chronic BrdU administration, see Telley et al., 2016). The radial position of each neuron was measured with respect to the pia. Mean position of (GFP+FT+) neurons: Ctl: 263.5 ± 5.8 µm, 60 cells; Kir2.1: 220.4 ± 6.3 µm, 45 cells. A Student’s t-test was used.
Figure 5H: Two sections from at least 4 embryonic brains injected with FlashTag at E15 (which corresponds to an E14.5 electroporation, see Telley et al., 2016) were used for patch-clamp experiments 12 hours after. Recorded Vm in medial cells (n = 11): −58.80 ± 2.44 mV; lateral cells (n = 11): −73.93 ± 1.79 mV. A Student’s t-test was used.
Figure S7B: Two sections from at least 4 embryonic brains electroporated with pCAG-IRES-GFP at E14.5 (number of recorded cells, n = 9) and E15.5 (n = 8) were used for patch-clamp experiments 24 hours after electroporation. Recorded Vm after E14.5 electroporation: Ctl: −66.00 ± 2.64 mV; BaCl2 1 mM: −56.77 ± 2.12 mV; BaCl2 3 mM: −48.17 ± 2.20 mV. Recorded Vm after E15.5 electroporation: Ctl: −74.76 ± 1.20 mV; BaCl2 1 mM: −60.38 ± 2.88 mV; BaCl2 3 mM: −51.67 ± 1.61 mV. A two-way ANOVA with Tukey post-hoc test was used.
Figure S7C: Three sections for each brain injected with FT (number of brains, n = 3) or FT and BaCl2 1 mM at E15 (number of brains, n = 4) were used to quantify the number of TBR2+ and NEUROD2+ cells among the fraction of GFP+ cells 12 hours after injection. Percentage of TBR2+ in E15 control: 75.70 ± 1.87, E15 BaCl2: 67.78 ± 1.68. Percentage of NEUROD2+ in E15 control: 62.90 ± 2.35, E15 BaCl2: 70.10 ± 1.67. A Student’s t-test was used.
Figure 6A,E: Three sections for each brain electroporated with pCAG-IRES-GFP at E12.5 (n = 3), E14.5 (number of brains, n > 3), E15.5 (n > 3) or with pCAG-Kir2.1-IRES-GFP at E12.5 (n = 5), E14.5 (n > 4) were used to quantify the number of NEUROD2+, TBR2+, Ki67+ and SOX2+, and TBR2+Ki67+ cells among the fraction of GFP+ cells 24 hours after IUE. Figure 6A: Percentage of NEUROD2+ in E14.5 control: 55.43 ± 2.24, E15.5 control: 42.20 ± 3.81, E14.5 Kir2.1: 35.50 ± 2.01. Percentage of TBR2+ in E14.5 control: 51.62 ± 6.41, E15.5 control: 62.80 ± 9.71, E14.5 Kir2.1: 59.60 ± 3.89. Percentage of TBR2+/Ki67+ in the VZ in E14.5 control: 28.35 ± 3.86, E15.5 control: 52.06 ± 2.51, E14.5 Kir2.1: 53.5 ± 4.79. Figure 6E: Percentage of Ki67+ in E14.5 control: 41.84 ± 1.88, E15.5 control: 56.65 ± 4.88, E14.5 Kir2.1: 62.46 ± 2.19. Percentage of Sox2+ in E14.5 control: 49.96 ± 2.02, E15.5 control: 67.83 ± 1.50, E14.5 Kir2.1: 62.47 ± 2.56. A one way-ANOVA with Tukey post-hoc test was used.
Figure S7C: Three sections for each brain injected with FT (number of brains, n = 3) or FT and BaCl2 1 mM at E15 (number of brains, n = 4) were used to quantify the number of TBR2+ and NEUROD2+ cells among the fraction of GFP+ cells 12 hours after injections. % TBR2+ in E15 control: 75.70 ± 1.87, E15 Ba2+: 67.78 ± 1.68. % NEUROD2+ in E15 control: 62.90 ± 2.35, E15 Ba2+: 70.10 ± 1.67. A Student’s t-test was used.
Figure 6C and S5E: Three sections for each brain electroporated with pCAG-IRES-GFP at E14.5 (number of brains, n = 14), E15.5 (n = 12), E12.5 (n = 3) or with pCAG-Kir2.1-IRES-GFP at E14.5 (n = 13) or E12.5 (n = 4) were used to quantify the number of GFP+ cells in the ventricular zone among the total amount of GFP+ cells 24 hours after IUE. Percentage of GFP+ in VZ at E14.5: 38.72 ± 2.63, E15.5: 48.76 ± 2.36, E14.5 Kir2.1: 51.77 ± 2.17, E12.5: 47.23 ±, E12.5 Kir2.1: 56.22 ± 1.01. A one way-ANOVA with Tukey post-hoc test was used.
Figure 6D: Three sections for each brain electroporated at E14.5 with pCAG-IRES-GFP (number of brains, n = 4) or with pCAG-Kir2.1-IRES-GFP (n = 3) and electroporated 12 hours later with pCAG-IRES-mCherry were used to quantify the number of mCherry+GFP+ cells among the fraction of GFP+ cells at P7. Percentage of mCherry+GFP+ in control: 29.28 ± 3.14 and Kir2.1: 69.67 ± 3.38. A Student’s t-test was used.
Figures 6F and S5F,G: Three sections for each brain electroporated with pCAG-IRES-GFP at E14.5 (number of brains, n = 3), E15.5 (n = 3) or with pCAG-Kir2.1-IRES-GFP at E14.5 (n = 4) and labeled with a single pulse-injection of BrdU 24 hours after IUE (to allow time for the plasmid to be expressed) were used to quantify the number of Ki67− BrdU+ GFP+ cells 48 hours after IUE (i.e. 24 hours after the BrdU pulse). Cell cycle exit rate was determined by dividing this number by the total number of BrdU+ GFP+ cells. Percentage of Ki67− BrdU+ GFP+ at E14.5: 41.88 ± 0.65, E15.5: 17.35 ± 3.74, E14.5 Kir2.1: 17.26 ± 2.27. A one way-ANOVA with Tukey post-hoc test was used.
Figure 7A: Three sections for each brain electroporated with pGR-IRES-TdTOM and pGR-M38-TOPdGFP at E14.5 (number of brains, n = 7), E15.5 (n = 6), or with pGR-IRES-TdTOM, pGR-M38-TOPdGFP and pGR-Kir2.1-HA at E14.5 (n = 6) were used to quantify the number of GFP+ cells among the fraction of TdTOM+ cells 24 hours after IUE. Percentage of GFP+ in E14.5 control: 64.47 ± 4.00, E15.5 control: 47.88 ± 9.05, E14.5 Kir2.1: 37.96 ± 5.28. A one way-ANOVA with Tukey post-hoc test was used.
Figure 7B: Three sections for each brain electroporated with pGR-dnTCF4-pGK-GFP (number of brains, n = 5) or pGR-Kir2.1-HA and pGR-∆45-βcatenin-pGK-GFP (n = 5) at E14.5 were used to quantify the number of NEUROD2+ cells among the fraction of GFP+ cells 24 hours after IUE. % NEUROD2+ in E14.5 dnTCF4: 32.40 ± 3.70, Kir2.1 + βcat: 51.5 ± 1.88. E14.5. Control and Kir2.1 values have been reported from Figure 6A for comparison. A one way-ANOVA with Tukey post-hoc test was used.
Transcriptomic analyses
Figure 1E,F: Obtained read counts were normalized by library size using DESeq, an R/Bioconductor package. The molecular identities of L4- and L2/3-type neurons was defined using differential pairwise expression analysis. Differentially-expressed genes were defined by the amplitude (FC >1.7) and by the statistical significance of the difference in their expression level (adjusted p-value < 0.05). Genes from control E14.5 and E15.5 showing the smallest p-value in both conditions were defined as top L4 and L2/3 specific genes. Relative normalized expressions across conditions were plotted using variance-stabilizing transformation from DESeq R Package. In order to compare expression levels across all genes, for each gene, the dispersion value from the center of mean expression level of all samples was divided by the highest expression level value. The heatmap representation displays the corrected dispersion mean values of all differentially expressed genes (602 genes) (Figure 1E, left). In the scatter plot representations, the relative normalized expression levels of the significantly differentially expressed 267 genes of L4 and 335 genes of L2/3 in Kir2.1E14.5 neurons was determined (Figure 1F, right). A Fisher’s exact test was used (P < 10−4).
Figures 6G,H, S6A–C: Read counts were normalized as previously described (Telley et al., 2016). Principal component analysis (PCA) was performed on E14.5 FT+ cells using a reduced set of genes of the top 100 apical progenitors, intermediate progenitors or neuronal genes identified in ref. 12. Cells were then assigned to three groups using a hierarchical clustering and the identity of each group was assigned based on the level of expression of classical markers such as Sox2, Tbr2 and NeuroD2 (Figure S6A). E15.5 Ctl and E14.5 Kir2.1 single cells were then projected into this PCA and a k-nearest neighbour approach were used to assign each cell to each of the three E14.5 cell clusters (i.e. apical progenitors, intermediate progenitors and neurons). Single-Cell Differential Expression (SCDE) was used to identify differentially expressed genes across E14.5 and E15.5 progenitors (Figures 6G,H S6A,B). Gene expression changes are displayed as Maximum Likelihood Estimate (MLE) of the fold-change in gene expression for genes with a significant Z score. The same approach was used to determine differentially-expressed genes across E14.5 Ctl and E14.5 Kir2.1 in apical progenitors (Figures 6G) and neurons (Figure S6B,C). Gene ontologies and their statistical significance were defined using the Genego website (portal.genego.com, Figure 6H).
Figures 1F, 6G, S6C: Support Vector Machine (SVM) training: control E14.5 and E15.5 samples/single-cells were used as training sets to build the best model distinguishing the two groups using R package bmrm v3.9, with lamda = 100 and N = 50 genes per condition. The classification of Kir2.1 condition sample/cells in this model was then determined.
A total of 242 ion channel genes was identified based on their ontology (http://www.genenames.org/genefamilies/VGIC#KCA) and classified into 4 types of conductances (sodium / cations, potassium, chloride or calcium, Figure S7A). The mean expression value of the transcripts of these genes was calculated for E14.5 and E15.5 APs and normalized to E14.5 values. Statistical significance was determined using a Student’s t-test.
Supplementary Material
Table S1. Differentially expressed genes in L4-type vs. L2/3-type, and L4-type vs. L4-Kir2.1E14.5 P3 neurons, Related to Figure 1.
Figure S5. In vitro clonal analysis, cycling behavior of APs, and fate of directly-born neurons, Related to Figure 6.
(A) Schematic summary of the experimental setup for in vitro single-cell clonal analysis.
(B) In vitro hyperpolarization with Kir2.1 leads to an increase in IP-producing divisions compared to the control condition. Dashed lines link experiments from corresponding biological replicates.
(C,D) Representative photomicrographs for an IP-generating division (C) and a neuron-generating division (D). 1a and 1b refer to the daughter cells.
(E) E12.5-born Kir2.1-overexpressing cells remain longer in the VZ following in utero electroporation, as is the case following E14.5 hyperpolarization (compare with Figure 6C).
(F) Schematic representation of the cell cycle exit assay in Figure 6F.
(G) Representative photomicrographs showing cells in E14.5 Ctl, E14.5 Kir2.1 and E15.5 Ctl conditions 24 hours after BrdU pulse injection, as quantified in Figure 6F. Progenitors are GFP+ BrdU+ Ki67+ (empty arrowheads); Neurons are GFP+ BrdU+ Ki67-(full arrowheads).
(H) Left: schematic representation of the FlashTag labeling procedure (see also Telley et al., 2016 and https://www.biorxiv.org/content/early/2018/03/22/286831). Right: Directly-born neurons shift superficially following Kir2.1-hyperpolarization of APs.
Data are represented as means ± SEM. (B), Student’s t test; (E), one-way ANOVA; (H) Student’s t test. ***: P<10–3.
Figure S6. Hyperpolarization of progenitors leads to a forward shift in the molecular identity of 12h-old daughter neurons, Related to Figure 6.
(A) Apical progenitors (AP), daughter intermediate progenitors (IP), and daughter neurons (N) can be distinguished by unbiased clustering.
(B) Neurons born from E14.5 hyperpolarized APs repress L4-type and induce L2/3-type genes programs already 12 hours after their birth. Analyses were performed on the neuronal population shown in Figure S6A.
(C) Unbiased SVM classication using 12-hour old L4- and L2/3-type neurons as a training set reveals that the transcriptional identity of E14.5 Kir2.1-hyperpolarized AP daughter neurons is shifted towards that of E15.5-born neurons (i.e. L2/3 neurons).
Data are represented as means ± SEM. (B), Fisher’s exact test; (C), one-way ANOVA. ***: P<10–3.
Figure S7. Ba2+-sensitive K+ channels drive apical progenitor hyperpolarization, Related to Figure 6.
(A) Transcripts coding for K+ channels specifically increase between E14.5 and E15.5.
(B) In vitro blockade of Kir channels with BaCl2 causes embryonic age-specific depolarization of APs. (C) Blockade of Kir channels with BaCl2 results in decreased neurogenesis and increased generation of intermediate progenitors (TBR2+ cells).
Data are represented as means ± SEM. (A) and (C), Student’s t test; (B) two-way ANOVA. *: P < 0.05.
Table S2. Differentially expressed genes in E14.5 vs. E15.5, and E14.5 vs. Kir2.1-expressing apical progenitors, Related to Figure 6.
Table S3. Differentially expressed genes in L4-type vs. L2/3-type, and L4-type vs. L4-Kir2.1E14.5 neurons 24 hours after electroporation, Related to Figure 6.
Figure S1. Kir2.1 in utero electroporation leads to a disruption in the tangential distribution of E14.5-born neurons of the somatosensory cortex at P7, Related to Figure 1.
Flattened cortical preparation showing disruption of the somatotopic mapping of whiskers asbarrels.
Figure S2. E14.5-born neurons no longer overlap with thalamocortical terminals following Kir2.1 in utero electroporation, Related to Figure 3.
VGLUT2 labels presynaptic thalamocortical terminals. Data are represented as means ± SEM. Ctl: control; Epor: electroporation.
Figure S3. Technical validation of the constructs used in Fig. 5 and effect morphological effect of hM3D, Related to Figure 4.
(A) Panel 1: Neurons in the basal VZ are hyperperpolarized after in utero electroporation of a pNeuroD-Kir2.1 construct. Panel 2: CNO application hyperpolarizes hM4D-expressing APs. Panels 3 and 4: CNO application reversibly depolarizes hM3D-expressing APs. Panel 5: APs express hM4D-GFP.
(B) The fraction of neurons with an apical dendrite is decreased in the progeny of E15.5, hM3Dexpressing APs following early CNO pulse-injections.
Data are represented as means ± SEM. (A,B) Student’s t-test (paired in panels 2 and 4). *: P < 0.05; **: P < 10–2; ***: P<10–3.
Figure S4. Apical progenitor membrane potential regulates the balance between direct and indirect neurogenesis, related to Figure 5.
(A) Photomicrographs and schematic representations corresponding to the quantifications of direct neurogenesis during corticogenesis shown in Figure 5C left.
(B) Photomicrograph corresponding to the schematic and quantification of direct neurogenesis in the Kir2.1 condition at E14.5 shown in Figure 5C right.
(C) Photomicrographs corresponding to the schematics and quantifications of direct neurogenesis in the depolarizing hM3D condition shown in Figure 5E.
(D) Photomicrographs corresponding to the schematics and quantifications of direct neurogenesis in the Kir2.1 condition at E12.5 shown in Figure 5F.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rat anti-BrdU | Abcam | Cat.N: #AB6326; RRID: AB_305426 |
| goat anti-BRN2 | SC Biotech | Cat.N: #SC6029; RRID: AB_2167385 |
| rabbit anti-KI67 | Abcam | Cat.N: #AB15580; RRID: AB_443209 |
| rabbit anti-NEUROD2 | Abcam | Cat.N: #1B104430; RRID: AB_10975628 |
| mouse anti-RORB | Perseus Proteomics | Cat.N: #PP-N7927–00; RRID: N/A |
| goat anti-SOX2 | SC Biotech | Cat.N: #SC17320; RRID: AB_2286684 |
| chicken anti-TBR2 | Millipore | Cat.N: #AB15894; RRID: AB_10615604 |
| rat anti-TBR2 | Thermo Fisher Scientific | Cat.N: #14–4875-82; RRID: AB_11042577 |
| guinea pig anti-VGLUT2 | Millipore | Cat.N: #AB2251-I; RRID: AB_2665454 |
| mouse anti-TUJ1 | Covance | Cat.N: #MMS-435P; RRID: AB_2313773 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 488 CellTraceTM CFSE | Life Technologies | Cat.N: #C34554 |
| Clozapine N-oxide, CNO | Sigma | Cat.N: #C0832 |
| Barium chloride (BaCl2) | Sigma | Cat.N: #449644 |
| 5-Bromo-2’-deoxyuridine (BrdU) | Sigma | Cat.N: #B5002 |
| Doxycycline | Sigma | Cat.N: #24390–14-5 |
| Cholera Toxin Subunit B (CTB), AlexaFluor™ 555-Conjugated | TermoFischer Scientific | Cat.N: #C43776 |
| Hoechst | TermoFischer Scientific | Cat.N: #H1399 |
| Deposited Data | ||
| Postnatal bulk RNA sequencing at P3 after IUE of Ctl at E14.5, Kir2.1 at E14.5 and Ctl at E15.5 | This paper | GEO: GSE115627 |
| Embryonic single-cell RNA sequencing at 24 h after IUE of Ctl at E14.5, Kir2.1 at E14.5 and Ctl at E15.5 | This paper | GEO: GSE115628 |
| Embryonic single-cell RNA sequencing | Yuzwa et al., 2017 | GEO: GSE107122 |
| Recombinant DNA | ||
| pCAG-IRES-GFP | Laboratory of Lopez-Bendito; Mire et al., 2012 | N/A |
| pCAG-hKir2.1-IRES-GFP | Laboratory of Lopez-Bendito; Mire et al., 2012 | N/A |
| pCAG-IRES-mCherry | This paper | N/A |
| pCAG-ChR2-Venus | Petreanu et al., 2007 | Addgene #15753 |
| pNeuroD-IRES-GFP | Laboratory of Dayer; Jacobshagen et al., 2014 | N/A |
| pNeuroD-hKir2.1-ires-GFP | This paper | N/A |
| pCAG-hM4D-IRES-GFP | This paper | N/A |
| pGR-IRES-tdTOM | Laboratory of Dayer; Jacobshagen et al., 2014 | N/A |
| pGR-hKir2.1-HA | This paper | N/A |
| pGR-M38-TOPdGFP | Laboratory of Kiss; Boitard et al. 2015 | N/A |
| pGR-dnTCF4-pGK-GFP | Laboratory of Kiss; Bocchi et al., 2017 | N/A |
| pGR-D45-bcatenin-pGK-GFP | Laboratory of Kiss; Boitard et al., 2015 | N/A |
| pCWX-pTF-dnTCF4-pGK-GFP-E2A-rtTA | Laboratory of Kiss; Bocchi et al., 2017 | N/A |
| Software and Algorithms | ||
| R | N/A | https://www.r-preoject.org/ |
| GraphPad Prism | Graphpad software | https://www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Photoshop | Adobe | https://www.adobe.com/en/products/photoshop.html |
| Adobe Illustrator | Adobe | https://www.adobe.com/en/products/illustrator.html |
Highlights:
Apical progenitor (AP) resting membrane potential decreases during corticogenesis.
AP hyperpolarization allows progression from direct to indirect neurogenesis.
AP hyperpolarization drives neuronal diversity via repression of Wnt signaling.
ACKNOWLEDGMENTS
We thank Alexandre Dayer and Guillermina Lopez-Bendito for providing plasmids, and Julien Prados for his contribution to the single cell analyses. We are thankful to Audrey Benoit for technical assistance, to Didier Chollet and Mylène Docquier of the Genomics Platform of the University of Geneva. We thank Andrea Lopes for sharing the photomicrograph in Fig. 5G and Fernando Alsina for help with experiments. We thank Eiman Azim and members of the laboratory for suggestions on the manuscript. Support: Work in the Jabaudon laboratory is supported by the Swiss National Science Foundation, the Brain and Behavior Foundation, and the Synapsis Foundation. Work in the Silver laboratory is supported by the NIH (Grant number R01NS083897). EK is supported by the Machaon Foundation. Supplement contains additional data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
No competing interests to declare.
DATA AND SOFTWARE AVAILABILITY
The raw data for the postnatal bulk RNA sequencing at P3 following IUE of E14.5 Ctl, E14.5 Kir2.1 and E15.5 Ctl have been deposited in the GEO database under ID code GSE115627. The raw data for the embryonic single cell sequencing 24h after IUE of E14.5 Ctl, E14.5 Kir2.1 and E15.5 Ctl have been deposited in the GEO database under ID code GSE115628. Access links are available also in the Key Resource Table. Further information and requests for resource data should be directed to and will be fulfilled by the Lead Contact, Denis Jabaudon (denis.jabaudon@unige.ch).
REFERENCES
- Arcangeli A, Bianchi L, Becchetti A, Faravelli L, Coronnello M, Mini E, Olivotto M, and Wanke E (1995). A novel inward-rectifying K+ current with a cell-cycle dependence governs the resting potential of mammalian neuroblastoma cells. J. Physiol. (Lond.) 489 (Pt 2), 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD (2005). Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45, 207–21. [DOI] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, and Roth BL (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U.S.a 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataman B, Ashley J, Gorczyca M, Ramachandran P, Fouquet W, Sigrist SJ, and Budnik V (2008). Rapid activity-dependent modifications in synaptic structure and function require bidirectional WNT signaling. Neuron 57, 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CK, and Schwarz JR (2001). Physiology of EAG K+ channels. J. Membr. Biol 182, 1–15. [DOI] [PubMed] [Google Scholar]
- Belford GR, and Killackey HP (1980). The sensitive period in the development of the trigeminal system of the neonatal rat. J. Comp. Neurol 193, 335–350. [DOI] [PubMed] [Google Scholar]
- Blackiston DJ, McLaughlin KA, and Levin M (2009). Bioelectric controls of cell proliferation: ion channels, membrane voltage and the cell cycle. Cell Cycle 8, 3527–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood BL, Sharma N, Browne HA, Trepman AZ, and Greenberg ME (2013). The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 503, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchi R, Egervari K, Carol-Perdiguer L, Viale B, Quairiaux C, De Roo M, Boitard M, Oskouie S, Salmon P, and Kiss JZ (2017). Perturbed WNT signaling leads to neuronal migration delay, altered interhemispheric connections and impaired social behavior. Nat Commun 8, 1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitard M, Bocchi R, Egervari K, Petrenko V, Viale B, Gremaud S, Zgraggen E, Salmon P, and Kiss JZ (2015). WNT signaling regulates multipolar-to-bipolar transition of migrating neurons in the cerebral cortex. Cell Rep 10, 1349–1361. [DOI] [PubMed] [Google Scholar]
- Bortone D, and Polleux F (2009). KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron 62, 53–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway EM, and Borrell V (2011). Developmental sculpting of dendritic morphology of layer 4 neurons in visual cortex: influence of retinal input. J Neurosci 31, 7456–7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Park CS, and Tang S-J (2006). Activity-dependent synaptic WNT release regulates hippocampal long term potentiation. J. Biol. Chem 281, 11910–11916. [DOI] [PubMed] [Google Scholar]
- Chou S-J, Babot Z, Leingärtner A, Studer M, Nakagawa Y, and O’Leary DDM (2013). Geniculocortical input drives genetic distinctions between primary and higher-order visual areas. Science 340, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone CD, and Tongier M (1971). Control of somatic cell mitosis by simulated changes in the transmembrane potential level. Oncology 25, 168–182. [DOI] [PubMed] [Google Scholar]
- De la Rossa De, A., Bellone C, Golding B, Vitali I, Moss J, Toni N, Lüscher C, and Jabaudon D (2013). In vivo reprogramming of circuit connectivity in postmitotic neocortical neurons. Nat Neurosci 16, 193–200. [DOI] [PubMed] [Google Scholar]
- De Marco García NV, Karayannis T, and Fishell G (2011). Neuronal activity is required for the development of specific cortical interneuron subtypes. Nature 472, 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay C, Savatier P, Cortay V, and Kennedy H (2001). Cell-cycle kinetics of neocortical precursors are influenced by embryonic thalamic axons. J Neurosci 21, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay C, and Kennedy H (2007). Cell-cycle control and cortical development. Nat Rev Neurosci 8, 438–450. [DOI] [PubMed] [Google Scholar]
- Ewald RC, Van Keuren-Jensen KR, Aizenman CD, and Cline HT (2008). Roles of NR2A and NR2B in the development of dendritic arbor morphology in vivo. J Neurosci 28, 850–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangeul L, Pouchelon G, Telley L, Lefort S, Luscher C, Jabaudon D (2016). A cross-modal genetic framework for the development and plasticity of sensory pathways. Nature 538, 96–98. [DOI] [PubMed] [Google Scholar]
- Frangeul L, Kehayas V, Sanchez-Mut JV, Fièvre S, Krishna-K K, Pouchelon G, Telley L, Bellone C, Holtmaat A, Gräff J, Mackliss JD, and Jabaudon D (2017) Input-dependent regulation of excitability controls dendritic maturation in somatosensry thalamocortical neurons. Nat Comm, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindan S, and Jabaudon D (2017). Coupling progenitor and neuronal diversity in the developing neocortex. FEBS Lett 18, 379. [DOI] [PubMed] [Google Scholar]
- Govindan S, Oberst P, and Jabaudon D (2018). In vivo pulse-labeling of isochronic cohorts of cells in the central nervous system using FlashTag. BioRxiv 286831. [DOI] [PubMed]
- Guerrier S, Coutinho-Budd J, Sassa T, Gresset A, Jordan NV, Chen K, Jin W-L, Frost A, and Polleux F (2009). The F-BAR domain of srGAP2 induces membrane protrusions required for neuronal migration and morphogenesis. Cell 138, 990–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knöpfel T, Erzurumlu RS, and Itohara S (2000). Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406, 726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Erzurumlu RS, Huerta PT, Chen DF, Sasaoka T, Ulupinar E, and Tonegawa S (1997). NMDA receptor-dependent refinement of somatotopic maps. Neuron 19, 1201–1210. [DOI] [PubMed] [Google Scholar]
- Jabaudon D (2017). Fate and freedom in developing neocortical circuits. Nat Commun 8, 16042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabaudon D, Shnider SJ, Tischfield DJ, Galazo MJ, and Macklis JD (2012). RORβ induces barrel-like neuronal clusters in the developing neocortex. Cereb. Cortex 22, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobshagen M, Niquille M, Chaumont-Dubel S, Marin P, and Dayer A (2014). The serotonin 6 receptor controls neuronal migration during corticogenesis via a ligand-independent Cdk5-dependent mechanism. Development 141, 3370–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns DC, Marx R, Mains RE, O’Rourke B, and Marbán E (1999). Inducible genetic suppression of neuronal excitability. J Neurosci 19, 1691–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohwi M, and Doe CQ (2013). Temporal fate specification and neural progenitor competence during development. Nat Rev Neurosci 14, 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk T, Pontious A, Englund C, Daza RA, Bedogni F, Hodge R, Attardo A, Bell C, Huttner WB, Hevner RF Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. (2009). Cereb Cortex 19, 2439–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort S, Tomm C, Floyd Sarria J-C, and Petersen CCH (2009). The excitatory neuronal network of the C2 barrel column in mouse primary somatosensory cortex. Neuron 61, 301–316. [DOI] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim T-K, Hu LS, Malik AN, and Greenberg ME (2008). Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoTurco JJ, Owens DF, Heath MJ, Davis MB, and Kriegstein AR (1995). GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15, 1287–1298. [DOI] [PubMed] [Google Scholar]
- Martínez-Cerdeño V, Noctor SC, and Kriegstein AR (2006). The role of intermediate progenitor cells in the evolutionary expansion of the cerebral cortex. Cereb. Cortex 16 Suppl 1, i152–i161. [DOI] [PubMed] [Google Scholar]
- McConnell SK, and Kaznowski CE (1991). Cell cycle dependence of laminar determination in developing neocortex. Science 254, 282s–285. [DOI] [PubMed] [Google Scholar]
- Mire E, Mezzera C, Leyva-Díaz E, Paternain AV, Squarzoni P, Bluy L, Castillo-Paterna M, López MJ, Peregrín S, Tessier-Lavigne M, et al. (2012). Spontaneous activity regulates Robo1 transcription to mediate a switch in thalamocortical axon growth. Nat Neurosci 15, 1134–1143. [DOI] [PubMed] [Google Scholar]
- Mizuno H, Luo W, Tarusawa E, Saito YM, Sato T, Yoshimura Y, Itohara S, and Iwasato T (2014). NMDAR-regulated dynamics of layer 4 neuronal dendrites during thalamocortical reorganization in neonates. Neuron 82, 365–379. [DOI] [PubMed] [Google Scholar]
- Munji RN, Choe Y, Li G, Siegenthaler JA, and Pleasure SJW(2011). Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci 31,1676–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, and Kriegstein AR (2004). Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7, 136–144. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, and Kriegstein AR Distinct behaviors of neural stem and progenitor cells underlie cortical neurogenesis. (2008). J Comp Neurol 508,28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi K, Aramaki M, and Nakajima K (2016). Mutually repressive interaction between Brn½ and Rorb contributes to the establishment of neocortical layer 2/3 and layer 4. Proc. Natl. Acad. Sci. U.S.a 113, 3371–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto M, Miyata T, Konno D, Ueda HR, Kasukawa T, Hashimoto M, Matsuzaki F, and Kawaguchi A (2016). Cell-cycle-independent transitions in temporal identity of mammalian neural progenitor cells. Nat Commun 7, 11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Huber D, Sobczyk A, and Svoboda K (2007). Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat Neurosci 10, 668. [DOI] [PubMed] [Google Scholar]
- Pilaz L-J, McMahon JJ, Miller EE, Lennox AL, Suzuki A, Salmon E, and Silver DL (2016). Prolonged mitosis of neural progenitors alters cell fate in the developing brain. Neuron 89, 83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Dehay C, and Kennedy H (1997a). The timetable of laminar neurogenesis contributes to the specification of cortical areas in mouse isocortex. J. Comp. Neurol 385, 95–116. [PubMed] [Google Scholar]
- Polleux F, Dehay C, Moraillon B, and Kennedy H (1997b). Regulation of neuroblast cell-cycle kinetics plays a crucial role in the generation of unique features of neocortical areas. J Neurosci 17, 7763–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouchelon G, and Jabaudon D (2014). Nurturing the cortex’s thalamic nature. Curr. Opin. Neurol 27, 142–148. [DOI] [PubMed] [Google Scholar]
- Pouchelon G, Gambino F, Bellone C, Telley L, Vitali I, Lüscher C, Holtmaat A, and Jabaudon D (2014). Modality-specific thalamocortical inputs instruct the identity of postsynaptic L4 neurons. Nature 511, 471–474. [DOI] [PubMed] [Google Scholar]
- Reillo I, de Juan Romero C, García-Cabezas MÁ, and Borrell V (2011). A role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb. Cortex 21, 1674–1694. [DOI] [PubMed] [Google Scholar]
- Spitzer NC (2012). Activity-dependent neurotransmitter respecification. Nat Rev Neurosci 13, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundelacruz S, Levin M, and Kaplan DL (2008). Membrane potential controls adipogenic and osteogenic differentiation of mesenchymal stem cells. PLoS ONE 3, e3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna E, Götz M, and Huttner WB (2014). The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu. Rev. Cell Dev. Biol 30, 465–502. [DOI] [PubMed] [Google Scholar]
- Telley L, Govindan S, Prados J, Stevant I, Nef S, Dermitzakis E, Dayer A, and Jabaudon D (2016). Sequential transcriptional waves direct the differentiation of newborn neurons in the mouse neocortex. Science 351, 1443–1446. [DOI] [PubMed] [Google Scholar]
- Toma K, Wang T-C, and Hanashima C (2016). Encoding and decoding time in neural development. Dev. Growth Differ 58, 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozuka Y, Fukuda S, Namba T, Seki T, and Hisatsune T (2005). GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron 47, 803–815. [DOI] [PubMed] [Google Scholar]
- Urrego D, Tomczak AP, Zahed F, Stühmer W, and Pardo LA (2014). Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. Lond., B, Biol. Sci 369, 20130094–20130094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Loos H, and Woolsey TA (1973). Somatosensory cortex: structural alterations following early injury to sense organs. Science 179, 395–398. [DOI] [PubMed] [Google Scholar]
- Vasistha NA, Garcia-Moreno F, Arora S, Cheung AFP, Arnold SJ, Robertson EJ, and Molnár Z (2015). Cortical and clonal contribution of Tbr2 expressing progenitors in the developing mouse brain. Cereb. Cortex 25, 3290–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, and Kriegstein AR (2008). GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J Neurosci 28, 5547–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Yin Y, Zhao M, Forrester JV, and McCaig CD (2003). Physiological electric fields control the G1/S phase cell cycle checkpoint to inhibit endothelial cell proliferation. Faseb J 17, 458–460. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, and Soderling TR (2006). Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of WNT-2. Neuron 50, 897–909. [DOI] [PubMed] [Google Scholar]
- Weissman TA, Riquelme PA, Ivic L, Flint AC, and Kriegstein AR (2004). Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron 43, 647–661. [DOI] [PubMed] [Google Scholar]
- Woodhead GJ, Mutch CA, Olson EC, and Chenn A (2006). Cell-autonomous beta-catenin signaling regulates cortical precursor proliferation. J Neurosci 26, 12620–12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Bartlett PF, and Adams DJK K(ir) and K(v) channels regulate electrical properties and proliferation of adult neural precursor cells (2008). Mol Cell Neurosci 37, 284–97. [DOI] [PubMed] [Google Scholar]
- Yasuda T, and Adams DJ (2010). Physiological roles of ion channels in adult neural stem cells and their progeny. J. Neurochem 114, 946–959. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Borrett MJ, Innes BT, Voronova A, Ketela T, Kaplan DR, Bader GD, Miller FD Developmental emergence of adult neural stem cells as revealed by single-cell transcriptional profiling. (2017). Cell Rep 21, 3970–3986. [DOI] [PubMed] [Google Scholar]
- Yu X, and Malenka RC (2003). Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci 6, 1169–1177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed genes in L4-type vs. L2/3-type, and L4-type vs. L4-Kir2.1E14.5 P3 neurons, Related to Figure 1.
Figure S5. In vitro clonal analysis, cycling behavior of APs, and fate of directly-born neurons, Related to Figure 6.
(A) Schematic summary of the experimental setup for in vitro single-cell clonal analysis.
(B) In vitro hyperpolarization with Kir2.1 leads to an increase in IP-producing divisions compared to the control condition. Dashed lines link experiments from corresponding biological replicates.
(C,D) Representative photomicrographs for an IP-generating division (C) and a neuron-generating division (D). 1a and 1b refer to the daughter cells.
(E) E12.5-born Kir2.1-overexpressing cells remain longer in the VZ following in utero electroporation, as is the case following E14.5 hyperpolarization (compare with Figure 6C).
(F) Schematic representation of the cell cycle exit assay in Figure 6F.
(G) Representative photomicrographs showing cells in E14.5 Ctl, E14.5 Kir2.1 and E15.5 Ctl conditions 24 hours after BrdU pulse injection, as quantified in Figure 6F. Progenitors are GFP+ BrdU+ Ki67+ (empty arrowheads); Neurons are GFP+ BrdU+ Ki67-(full arrowheads).
(H) Left: schematic representation of the FlashTag labeling procedure (see also Telley et al., 2016 and https://www.biorxiv.org/content/early/2018/03/22/286831). Right: Directly-born neurons shift superficially following Kir2.1-hyperpolarization of APs.
Data are represented as means ± SEM. (B), Student’s t test; (E), one-way ANOVA; (H) Student’s t test. ***: P<10–3.
Figure S6. Hyperpolarization of progenitors leads to a forward shift in the molecular identity of 12h-old daughter neurons, Related to Figure 6.
(A) Apical progenitors (AP), daughter intermediate progenitors (IP), and daughter neurons (N) can be distinguished by unbiased clustering.
(B) Neurons born from E14.5 hyperpolarized APs repress L4-type and induce L2/3-type genes programs already 12 hours after their birth. Analyses were performed on the neuronal population shown in Figure S6A.
(C) Unbiased SVM classication using 12-hour old L4- and L2/3-type neurons as a training set reveals that the transcriptional identity of E14.5 Kir2.1-hyperpolarized AP daughter neurons is shifted towards that of E15.5-born neurons (i.e. L2/3 neurons).
Data are represented as means ± SEM. (B), Fisher’s exact test; (C), one-way ANOVA. ***: P<10–3.
Figure S7. Ba2+-sensitive K+ channels drive apical progenitor hyperpolarization, Related to Figure 6.
(A) Transcripts coding for K+ channels specifically increase between E14.5 and E15.5.
(B) In vitro blockade of Kir channels with BaCl2 causes embryonic age-specific depolarization of APs. (C) Blockade of Kir channels with BaCl2 results in decreased neurogenesis and increased generation of intermediate progenitors (TBR2+ cells).
Data are represented as means ± SEM. (A) and (C), Student’s t test; (B) two-way ANOVA. *: P < 0.05.
Table S2. Differentially expressed genes in E14.5 vs. E15.5, and E14.5 vs. Kir2.1-expressing apical progenitors, Related to Figure 6.
Table S3. Differentially expressed genes in L4-type vs. L2/3-type, and L4-type vs. L4-Kir2.1E14.5 neurons 24 hours after electroporation, Related to Figure 6.
Figure S1. Kir2.1 in utero electroporation leads to a disruption in the tangential distribution of E14.5-born neurons of the somatosensory cortex at P7, Related to Figure 1.
Flattened cortical preparation showing disruption of the somatotopic mapping of whiskers asbarrels.
Figure S2. E14.5-born neurons no longer overlap with thalamocortical terminals following Kir2.1 in utero electroporation, Related to Figure 3.
VGLUT2 labels presynaptic thalamocortical terminals. Data are represented as means ± SEM. Ctl: control; Epor: electroporation.
Figure S3. Technical validation of the constructs used in Fig. 5 and effect morphological effect of hM3D, Related to Figure 4.
(A) Panel 1: Neurons in the basal VZ are hyperperpolarized after in utero electroporation of a pNeuroD-Kir2.1 construct. Panel 2: CNO application hyperpolarizes hM4D-expressing APs. Panels 3 and 4: CNO application reversibly depolarizes hM3D-expressing APs. Panel 5: APs express hM4D-GFP.
(B) The fraction of neurons with an apical dendrite is decreased in the progeny of E15.5, hM3Dexpressing APs following early CNO pulse-injections.
Data are represented as means ± SEM. (A,B) Student’s t-test (paired in panels 2 and 4). *: P < 0.05; **: P < 10–2; ***: P<10–3.
Figure S4. Apical progenitor membrane potential regulates the balance between direct and indirect neurogenesis, related to Figure 5.
(A) Photomicrographs and schematic representations corresponding to the quantifications of direct neurogenesis during corticogenesis shown in Figure 5C left.
(B) Photomicrograph corresponding to the schematic and quantification of direct neurogenesis in the Kir2.1 condition at E14.5 shown in Figure 5C right.
(C) Photomicrographs corresponding to the schematics and quantifications of direct neurogenesis in the depolarizing hM3D condition shown in Figure 5E.
(D) Photomicrographs corresponding to the schematics and quantifications of direct neurogenesis in the Kir2.1 condition at E12.5 shown in Figure 5F.