

Summary
Despite abundant evidence associating CD38 overexpression and CD4 T cell depletion in HIV infection, no causal relation has been investigated. To address this issue, we propose a series of mechanisms, supported by evidence from different fields, by which CD38 overexpression could facilitate CD4 T cell depletion in HIV infection. According to our model, increased catalytic activity of CD38 may reduce CD4 T cells’ cytoplasmic nicotinamide adenine dinucleotide (NAD), leading to a chronic Warburg effect. This would reduce mitochondrial function. Simultaneously, CD38’s catalytic products ADPR and cADPR may be transported to the cytoplasm, where they can activate calcium channels and increase cytoplasmic Ca2+ concentrations, further altering mitochondrial integrity. These mechanisms would decrease the viability and regenerative capacity of CD4 T cells. These hypotheses can be tested experimentally, and might reveal novel therapeutic targets.
Keywords: HIV/AIDS, CD38, immune activation, CD4-Positive T lymphocytes, NAD, Immunometabolism, Warburg effect
Graphical Abstract
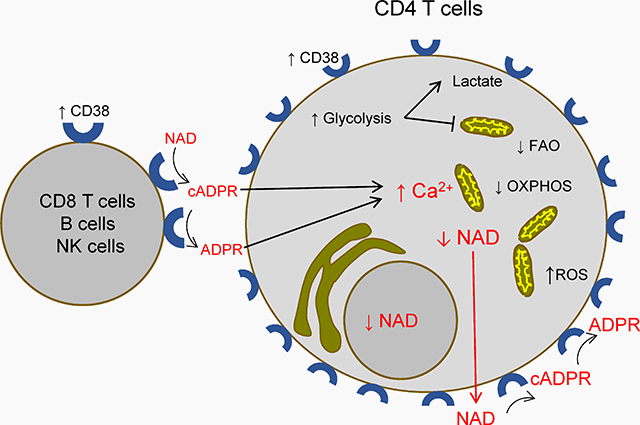
In HIV disease, CD38 expression and its catalytic activity are increased in several cell types. This may lower intracelular NAD levels in CD4 T cells, shifting their metabolism and compromising mitocondria. CD38’s products may increase cytoplasmic Ca2+. These effects may reduce CD4 T cell viability and regeneration capacity.
1. Introduction
1. 1. CD38 and Immune activation in HIV infection
An early finding in blood from patients with Acquired Immune Deficiency Syndrome was the presence of a large number of lymphocytes expressing the surface protein CD38.[1–3] Because CD38 is expressed by activated T cells, extensive research on immune activation in HIV infection ensued. Extensive evidence shows a strong correlation between the proportion of activated circulating T cells and HIV disease progression, as measured by survival time,[4,5] time to onset of opportunistic infections,[6,7] and loss of circulating CD4 T cells.[8–11] Under antiretroviral treatment, T cell activation is associated with the immune reconstitution inflammatory syndrome (IRIS)[12,13] and with insufficient recovery of CD4 T cells.[14–16] In this research area, however, CD38 has been measured as a mere activation marker,[17] leaving unanswered the question of whether CD38 overexpression has a causal relation with these outcomes.
1. 2. CD38 is a strong and independent predictor of CD4 T cell depletion
Looking back at early studies, one can see that CD38 overexpression is a very salient aspect of chronic activation in HIV infection. The number of CD38 molecules on the surface of CD8 T cells (as well as the proportion of CD8 and CD4 T cells expressing CD38) add capacity to predict the onset of AIDS to blood counts of CD4 T cells, HIV’s main target.[9,18,19] CD38 density on the surface of CD8 T cells could predict HIV disease progression and death, and was found to be independent of blood viral load (number of HIV copies per mL of blood).[20] CD38 expression on CD8 T cells was consistently predictive throughout the whole duration of untreated HIV infection, while viral load lost its predictive capacity in the long term, and CD4 T cell count had a low predictive capacity in early infection.[21] Importantly, CD38 expression on CD8 T cells was a stronger predictor than several other activation markers, including soluble mediators of inflammation, and the percentage of T cells with different combinations of HLADR and CD38 expression, as indicators of immune activation.[5] More direct indicators of T cell activation, like proliferation and cell cycle, also predict HIV disease progression.[22–24] In this regard, we have found that CD38 expression on T cells from patients with HIV correlates with lower circulating CD4 T cell counts independently of the frequency of cycling cells,[25] which further describes CD38 independence as predictor.
1.3. CD38 as predictor: implications for HIV disease pathogenesis
A possible implication of CD38’s statistical independence is that CD38 is reflecting a pathogenic mechanism independent of killing of CD4 T cells by infecting virions. An alternative implication is that CD38 itself is facilitating CD4 T cell loss. Such role requires the involvement of a particular biological function of CD38. At the same time, CD38 overexpression should increase its biological activity, which should be present in the different body compartments of CD4 T cells in the setting of HIV infection. This increased function should promote CD4 T cell loss or decreased cell renewal of CD4 T cells (which are already decimated by HIV and keep being eliminated by the cytopathic effect of cellular infection by virions). Currently, there are findings from different fields supporting that these conditions may concur in HIV infection. This scenario poses a number of hypotheses. Throughout this text we will provide an account of mechanisms supporting these hypotheses, and how they can be addressed experimentally.
2. Problem and proposed approach
It is currently unknown whether CD38 actually participates as an indirect pathogenic mechanism of HIV disease. We propose that CD38 overexpression may promote CD4 T cell depletion in the setting of HIV infection via its enzymatic functions. This would add to HIV’s direct effects on CD4 T cells. The mechanisms that can mediate this indirect pathogenic role can be compiled in the following hypotheses.
-
2.1
In HIV infection, excessive chronic CD38 expression by different cell types causes an increase in CD38’s catalytic activity in different body compartments.
-
2.2
CD38’s catalytic activity can decrease intracellular concentrations of nicotinamide-adenine dinucleotide (NAD) in CD4 T cells.
-
2.3
CD38 catalysis yields cADPR and ADPR, which are transported to the cytoplasm.
-
2.4
A decrease in cytoplasmic NAD may promote a metabolic switch favoring glycolysis over mitochondrial respiration (Warburg effect).
-
2.5
cADPR and ADPR, when transported into the cell, can increase cytoplasmic Ca2+ concentration.
-
2.6
Ca2+ increase, in addition to NAD depletion, may affect mitochondrial integrity.
-
2.7
These mechanisms can result in a reduced capacity survive and proliferate of CD4 T cells from HIV+ persons.
These hypotheses, integrating our model (Figure 1), are based in current knowledge of CD38 as an enzyme (see next) and can be addressed experimentally (Table 1).
Figure 1.-. General hypothesis outline.

HIV infection is accompanied by chronic activation of CD4 T cells and CD8 T cells (among others). This increases CD38 catalytic activity in the milieu of CD4 T cells. CD38 activity removes cytoplasmic NAD from CD4 T cells and yields the calcium-mobilizing compounds cyclic adenosine diphosphate ribose (cADPR) and adenosine diphosphate ribose (ADPR). cADPR and ADPR increase cytoplasmic Ca2+. Together, these effects reduce mitochondrial function and integrity, leading to a decreased cell viability in the long term. Dotted line indicates that less evidence is available.
Table 1.
Hypotheses conforming the proposed model of CD38-mediated indirect pathogenesis in HIV infection, and possible experimental approaches to test each.
| Hypothesis | Possible experiments |
|---|---|
| 2. 1 Catalytic activity where CD38 is present |
Comparison of catalytic activity between CD38+ and CD38− cells. Comparison of catalytic activity of CD4 T cells from HIV+ patients and healthy controls. Analysis of correlation of catalytic activity with CD38 expression. Possible methods: Degradation of NAD or an analog in culture media by colorimetry or HPLC-MS. |
| 2.2 NAD depletion by CD38 activity |
NAD measurement in lysates from CD4 T cells from patients and controls (colorimetry or HPLC-MS), and determination of its correlation with CD38 expression. |
| 2. 3 cADPR- and ADPR production by CD38 |
ADPR and cADPR concentration determination in culture media and lysates of CD4 T cells from patients and healthy controls. Correlation with CD38 expression. Possible method: HPLC-MS |
| 2. 4 Metabolic switch induced by NAD depletion |
Determination of oxygen consumption rate (OCR, a measure of mitochondrial respiration) and extracellular acidification rate (ECAR, a measure of glycolytic activity) in CD4 T cells from HIV+ patients and controls. Purified cells with the same degree of differentiation should be used*. Possible method: OCR/ECAR ratio by mitocondrial stress and glycolytic stress kinetic determinations with available instrumentation and kits. |
| 2. 5 Increased cytoplasmic Ca2+ |
Comparison of basal Ca2+ levels in CD4 T cells from HIV+ patients and healthy controls*. Analysis of correlation with CD38 expression. Comparison of Ca2+ flux after TCR-mediated stimulation in CD4 T cells from HIV+ patients and healthy controls with the same degree of differentiation. * Analysis of correlation with CD38 expression. Effect of antagonists of cADPR (8-Br-cADPR) and of ADPR in cytoplasmic Ca2+ concentrations and functionality (see below) of CD4 T cells from HIV+ patients and healthy controls*. Possible method: flow cytometry using a Ca2+ molecular probe. |
| 2. 6. Mitochondrial damage |
Mitochondrial morphology determination (e.g. mitocondrial fusion or fission and cristae integrity by confocal and electronic microspcopy)*. Mitochonrial biogenesis determination. Possible methods: microscopy and flow Cytometry with mitocondria-specific molecular probes*. |
| 2.7 Altered survival, proliferation, cytokine production, and differentiation |
Effect of pharmacological inhibition of CD38’s catalytic function in proliferation, survival, differentiation, and cytokine production after stimulation of CD4 T cells from patients and controls *. Determination of functional properties of CD4 T cell lines (for instance, Jurkat cells) expressing functional or non-catalytic variants of CD38. Possible method: CRISPR-Cas9 deletion of CD38 gen, followed by transfection with plasmids codifying for CD38 variants with or without catalytic function. Functional tests may be carried out by flow Cytometry using fluorochrome-conjugated monoclonal antibodies, cell division molecular probes, and viabiity molecular probes. |
The use of CD4 T cells from patients and controls with a same degree of differentiation is proposed to rule out confounding effects of differentiation. Variables affected by differentiation include cytokine production, viability, proliferative capacity, differentiation capacity, and metabolic profiles.
3. Evidence supporting CD38-mediated pathogenesis in HIV disease
3.1. Biological functions of CD38
3. 1. 1. CD38 enzymatic activity
The first evidence of CD38 expression on human leukocytes was obtained employing the monoclonal antibody OKT-10.[26] Thereafter, it was reported that CD38 is expressed by activated human T cells and several cancer cell types. CD38 shows a substantial similarity with an enzyme from Aplysia californica that catalyzes the conversion of nicotinamide adenine dinucleotide (NAD) to cyclic adenosine diphosphate ribose (cADPR), and the hydrolysis of cADPR into adenosine diphosphate ribose (ADPR).[27] Human CD38 gene is encoded at chromosome 4 p15.32.[28] The protein is formed by 300 amino acids, and has a molecular mass of 42–45 KDa.[29] CD38 is a type II transmembrane glycoprotein with a 21-amino acid cytoplasmic N-terminal domain, a 21-amino acid transmembrane domain, and a 258-amino acid extracellular catalytic and receptor domain.[30] CD38 belongs to the ribosyl family of NAD/ADP cyclases. Its extracellular portion possesses a catalytic pocket buried in the middle cleft of the protein,[31] where amino acid positions critical for these catalytic activities are found.[32,33] Importantly, the catalytic portion of human CD38 has been mapped to its extracellular domain.[31]
In an acid medium, CD38 may also catalyze the synthesis of nicotinic acid-adenine dinucleotide phosphate (NAADP);[34] however, at neutral pH, CD38 actually hydrolyzes NAADP.[35] This constrains the sites where NAADP is produced,[36] rendering unlikely the synthesis of NAADP by CD38 oriented to the extracellular milieu.
3. 1. 2. CD38 as a possible receptor
A possible function of CD38 as a receptor has been suggested, partly based on the effect of cross-linking CD38 with specific monoclonal antibodies. For instance, antibody IB4, initiates T cells signaling within a subset of membrane rafts[37] and recruits the signal transduction molecules TCR/CD3ζ chain, ZAP-70, phospholipase C-γ, Raf-1/mitogen-activated protein kinase, and calcium mobilization.[38–40] Previous work has proposed that CD31 and hyaluronic acid may act as natural CD38 ligands; however, this was based mainly on the binding of these molecules to CD38[41,42] and on the induction of calcium fluxes, cytokine production, and gene expression on heterogeneous cell populations or in transformed cell lines.[41,43] Agonistic monoclonal antibodies do not necessarily represent a specific ligand, and existing studies using putative ligands cannot be extrapolated to primary T cells. Evidence of a receptor function of CD38 should include a well described ligand with a physiological role on defined T cell functions. Therefore, we focus on CD38’s catalytic functions.
3.2. High CD38 activity in CD4 T cell-homing tissues in HIV infection
Our model implies that CD4 T cells are exposed to an increased CD38 catalytic activity. As mentioned above, CD4 and CD8 T cells from patients with HIV express CD38 more frequently than cells from uninfected controls, and CD38-expressing CD8 T cells from HIV+ patients show more CD38 molecules on its surface than those of uninfected people.[18,19] Moreover, CD8 T cells are expanded in HIV infection,[44–46] which further increases the number of CD38 molecules in blood. In addition to T cells, a greater proportion of NK[47,48] and B cells[49] from HIV+ persons express surface CD38, while patients’ monocytes show a tendency to greater CD38 expression.[50] These cells can further increase the amount of CD38 in contact with blood and tissues. Given that CD38 expression dictates NAD concentration,[51,52] we can expect that increased CD38 activity of CD38 in blood yields a decrease in blood NAD levels, unless NAD removal is somehow compensated. This possibility can be investigated by measuring CD38 expression and the actual catalytic activity of CD4 T cells, CD8 T cells, and other cell populations from HIV+ patients and healthy controls. Concurrently, blood NAD content can be analyzed (Table 1).
Given that CD38’s activity is determined by its expression,[27,52,53] CD4 T cells of HIV-infected patients will be exposed to a milieu with an enhanced CD38 catalytic activity.[54] In HIV infection, CD38 expression is also increased in T cells from lymph nodes,[55–58] lymphoid tissue associated with the small[59] and large[60,61] intestine, and the female genital tract.[62] This would extend the span of increased CD38 activity to most of the homing sites of CD4 T cells. Given this relative ubiquity of CD38, it can be expected that its overexpression affects other tissues and cell types. In agreement with this, CD8 T cells suffer activation-associated exhaustion in HIV infection,[63] even though they are not infected by the virus. In several tissues, age-related increased CD38 expression causes a gradual decrease of NAD levels with aging,[52] which promotes insulin resistance, mitochondrial dysfunction, and senescence.[64] Interestingly, accelerated senescence is in fact observed in patients with HIV (reviewed elsewhere[65]). Therefore, it could be very informative to determine CD38’s activity and NAD concentrations in blood and homing tissues of CD4 T cells, both in people with HIV infection, and healthy controls (Table 1).
Our model refers specifically to the setting of HIV infection. CD38 is normally expressed by several cells in lymphoid tissues, like CD8 T cells in colon-associated lymphoid tissue,[66] CD4 T cells in the mesenteric lymph nodes,[67] intraepithelial lymphocytes in the duodenum,[68] and B cell blasts in the lamina propria,[69] without being deleterious to CD4 T cells. This changes with HIV infection. We think that CD38 expression becomes pathogenic because it is overexpressed, its overexpression is chronical, and, importantly, it adds to the direct effects of HIV particles on CD4 T cells. Also delimiting our proposal, the naive subsets of CD4 AND CD8 T cells show lower activation in HIV infection (implying lower CD38 expression),[70] and their degree of activation is not predictive of HIV disease progression.[71] Moreover, they express CD38 constitutively.[72] Therefore, our proposal may be more relevant to memory CD4 T cells in HIV infection.
There is, to our knowledge, one study with results apparently opposite to our proposal. It finds a possible role of CD38 on in vitro protecting CD4 T cells from direct HIV infection. This conclusion was reached, based on differential infection of cell lines with different degrees of CD38 expression.[73] This work did not study important infectivity determinants like HIV coreceptors CCR5 and CXCR4, and it did not investigate primary CD4 T cells, precluding inferences about possible in vivo effects. In any case, this suggested role of CD38 does not seem to be relevant to HIV disease progression, given the robustness, consistency, and abundance of studies demonstrating the positive correlation of CD38 expression with CD4 T cell loss in vivo (see Introduction). The role we propose for CD38 constitutes an indirect pathogenic mechanism. There is evidence suggesting that indirect mechanisms are necessary to fully explain CD4 T cell dysfunction and loss in HIV disease.[74,75]
3.3. Enhanced CD38 catalytic activity, possible effects on CD4 T cells
3.3.1. CD38-induced reduction of NAD content of CD4 T cells in HIV infection
A possible consequence of excessive CD38 is an increased removal of its substrate, NAD, from the cytoplasm of T lymphocytes. This immediately raises the question of whether and how extracellular CD38 activity could regulate intracellular NAD concentrations.
Genetic CD38 ablation in CD4 T memory cells increases in vivo survival capacity, via an increased NAD cytoplasmic concentration.[53] Notably, this effect can also be attained by the use of an anti-CD38 antibody that blocks CD38’s NADase activity. Antibodies can only block extracellularly-oriented catalytic domain of CD38; therefore, cytoplasmic NAD reduction in this model depends at least in part on extracellular CD38 activity. Importantly, the reduction of surface CD38 on these CD4 T cells resulted in higher cytoplasmic NAD concentrations, with a shift to enhanced oxidative phosphorylation.[53] This finding is of great relevance to our model, since extracellular CD38 is the form detected in standard phenotyping[72] and it is the one that strongly correlates with HIV disease progression. Thus, increased extracellular CD38 activity could translate in reduced cytoplasmic NAD. This has important consequences in cellular metabolism and senescence.
There are particular mechanisms that may mediate reduction of cytoplasmic NAD concentration by extracellular CD38 catalytic activity. NAD can be transported from the cytoplasm to the extracellular space by Cx43 connexin in an equilibrative flow,[76] and T cells express a surface functional Cx43.[77–80] Blood NAD concentrations range from 24 to 41.4 μM,[81–83] while the intracellular concentration ranges from 200 to 500 μM, and it may increase ten-fold.[64] This could generate a NAD gradient that, in the presence of membrane Cx43, could sustain a flow of NAD from the cytoplasm to the extracellular milieu. CD38 overexpression in HIV infection could translate in reduced NAD concentration in plasma and/or in T cell cytoplasm, increasing NAD flow from cytoplasm, and surpassing the cell’s capacity to maintain its NAD levels. In consonance with a possible NAD scavenging pressure on intracellular NAD pools, we have found overexpression of nicotinamide phosphoribosyl transferase mRNA (NAMPT), the rate limiting enzyme of the salvage pathway of NAD synthesis) in central memory CD4 T cells from persons living with HIV,[84] which might be compensating NAD removal due to CD38 overexpression. In this way, increased presence of CD38 with an extracellular active site might decrease cytoplasmic NAD concentration. In agreement with this possible mechanism, cytoplasmic NAD maintenance depends more on the intracellular NAD synthesis via a salvage pathway, than in the intake of NAD,[64] while NAD can be transported outside the cell.
There is currently scant information of NAD contents of CD4 T cells from HIV+ individuals. An early study compared nucleotide intracellular concentrations in whole T cells (which include both CD4 and CD8 cells), from 8 healthy controls, 4 symptomatic patients, and 4 asymptomatic patients.[85] Interestingly, they find a failure to increase intracellular NAD in response to in vitro activation in T cells from symptomatic patients, while basal concentrations did not differ between groups. However, no distinction can be made between CD8 and CD4 T cells, and inference is limited by the small sample. Therefore, further investigation of NAD levels in plasma and cytoplasm of CD4 T cells from HIV+ patients and controls will be relevant for testing our hypotheses.
3. 3. 2. Low activity of Sirtuins and PARPs in CD4 T cells in HIV infection
If NAD overexpression is reducing intracellular NAD of CD4 T cells, as it does in other cells,[52] it may have important consequences. NAD has important functions in addition to its well-known biochemical role as an electron transporter in glycolysis, tricarboxylic acid cycle, and electron transfer chain. These functions include regulation of gene expression by sirtuins (Sirts), NAD-consuming deacetylases, and transfer of ADP-ribose groups from NAD to acceptor proteins by Poly (ADP-ribose) polymerases (PARPs), a process necessary for the regulation of cell death (reviewed elsewhere[86]). Sirtuins and PARP activities have important roles in metabolism, inflammation, DNA repair, and cell survival.[64] It is worth noting that nuclear Sirts 1 and 6 inhibit glycolysis and promote mitochondrial function, by promoting expression of PGC-1α,[87] and inhibiting HIF-1α and NFκB,[88] respectively (Figure 2). Likewise, mitochondrial Sirts 3 and 4 inhibit glycolysis and promote mitochondrial function, with Sirt 3 inhibiting generation of reactive oxygen species (Figure 2).[89] Cytoplasmic NAD is transported to mitochondria, and constitutes the main NAD source for this organelle,[90] which lends support to the possibility that a decrease in cytoplasmic NAD may reduce mitochondrial NAD content, along with decreases in Sirt 3 and Sirt 4 activity. Sirt 1, a nuclear deacetylase, promotes mitochondrial biogenesis and ROS protection.[87,91] Overall, NAD depletion affects mitochondrial integrity and function, thus reducing cell viability.
Figure 2.-. Metabolic profile of quiescent CD4 T cells.

Quiescent CD4 T cells have a metabolism dominated by oxidative phosphorylation (OXPHOS) in mitochondria. OXPHOS is fueled by pyruvate from glycolysis and by β oxidation of lipids. This metabolic program is enforced by the activity of NAD-dependent Sirtuins (Sirt). Nuclear Sirtuin 1 induces PGC-1, which increases mitochondrial biogenesis. Sirtuin 6 suppresses HIF-1 α and NFκB, decreasing glycolytic metabolism and effector functions. Mitochondrial Sirt 3 promotes OXPHOS, and Sirt 4 lowers the production of reactive oxygen species (ROS). ER, endoplasmic reticulum. NAD, nicotinamide-adenine dinucleotide. Glut1, glucose transporter 1.
3. 3. 3.-. HIV infection and Warburg effect in CD4 T cells
Quiescent T cells have a metabolism dominated by mitochondrial oxidative phosphorylation, beta oxidation of fatty acids providing the main carbon source for mitochondria. Importantly, pyruvate produced by glycolysis can be transported to the mitochondria, where it is converted to acetyl-coenzyme A, and contributes to the tricarboxylic acid cycle[92] (Figure 2). In contrast, activated T cells undergo Warburg effect.[93] In the case of T cells, this effect is described as an increase in aerobic glycolysis and a relatively reduced mitochondrial respiration after activation, which is reflected by a reduced oxygen consumption rate relative to extracellular acidification rate (OCR/ECAR ratio).[94,95] This shift may affect mitochondria fusion and cristae integrity[94] and yields an increased glucose intake and secretion of lactate.
Chronically activated CD4 T cells in HIV disease show increased aerobic glycolysis, as demonstrated by an increased frequency of cells expressing the glucose transporter Glut1, an increased glucose intake, an increased synthesis of glucose-6-phosphate from glucose, and an increased lactate secretion.[96] Importantly, the percentage of activated CD4 T cells (co-expressing CD38 and HLADR) was greater among Glut1+ cells, which showed an increased density of surface CD38.[96] This could be reflecting an activation-associated Warburg effect, or a similar metabolic change, as has been noted in the case of monocytes from HIV+ patients.[97] Since this study did not measure mitochondrial respiration, which would fully demonstrate Warburg effect, a full metabolic profiling of CD4 T cells from persons with HIV and healthy donors is necessary (Figure 3) (Table 1).
Figure 3. Activation and Warburg effect in CD4 T cells in HIV disease.

T cell activation increases CD38 expression, which increases catalytic activity on T cells, and in their homing sites. NAD removal by CD38 inactivates sirtuins, which decreases oxidative phosphorylation via the inhibition of PGC-1 and the activation of HIF-1 alfa. This blocks the entrance to mitochondria of pyruvate and products of lipid beta oxidation. NAD depletion additionally decreases the use of lipid beta oxidation products by mitochondria, via inactivation of mitochondrial Sirt3. Inactivation of Sirt 4 increases mitochondrial production of reactive oxygen species (ROS). Activation also increases CD4 T cell expression of the glucose transporter Glut 1, the entrance of glucose, and an increase in glycolysis. Pyruvate produced by glycolysis is not transported to mitochondria, but reduced to lactate, which is transported outside the cell. Warburg effect, entailing glycolysis-dominated metabolism, is thus enforced.
3. 3. 4. Low CD4 T cell viability due to activation-induced Warburg effect
Mitochondria integrity is related with the state of activation and is important for T cell viability. Loss of mitochondria integrity due to cytochrome oxidase ablation in CD4 T cells translates in a decrease in oxidative phosphorylation and a simultaneous increase in glycolysis.[98] Experimental increase in mitochondrial fission suffices to enforce an activated effector functionality in T cells, with a metabolic shift to aerobic glycolysis,[94] whereas mitochondrial biogenesis and functionality (promoted by the activity of NAD-dependent Sirt 3[99]) is associated with long term survival of T cells.[53,100,101] We think that an increase in CD38 catalytic activity may yield a decrease in cytoplasmic and mitochondrial NAD, leading to a metabolic shift to Warburg effect, which in turn would reduce mitochondrial function and biogenesis. We propose that this mechanism may be present in CD4 T cells in HIV infection (Figure 3). This hypothesis can be tested by measuring mitochondrial biogenesis in CD4 T cells from HIV+ patients and uninfected controls, and by the determination of mitochondrial respiration (Table 1). Regarding this objective, a crucial test will be to measure the oxygen consumption rate in proportion to the extracellular acidification rate (OCR/ECAR ratio[53]). In this regard, it will be useful to study the same differentiation subpopulations to rule out differences due to diverging degrees of differentiation between samples. Variables affected by differentiation include cytokine production, viability, proliferative capacity, differentiation capacity, and metabolic profiles.[72,102,103]
3. 3. 5. CD38-products ADPR and cADPR increase cytoplasmic Ca2+ in CD4 T cells
Independently of the final NAD balance outside and inside the cell, an increased turnover of NAD by CD38 would yield an increased production of cyclic adenosine diphosphate-ribose (cADPR) and adenosine diphosphate-ribose (ADPR), its two main products at neutral pH. Cyclic ADPR is an activator of the ryanodine receptor (RyR) calcium channel, able to trigger Ca2+ flow from the endoplasmic reticulum to the cytosol.[104] ADPR is the only known activator of the transient receptor potential melastatin 2 calcium channel (TRPM2).[105] TRPM2 channels are situated in the cytoplasmic membrane and transport Ca2+ from the cell exterior to the cytoplasm. ADPR and cADPR act intracellularly, which also raises the question of whether and how CD38, with its catalytic site facing outside the cell, can increase cADPR and ADPR concentrations in the cytoplasm. Such autocrine and paracrine activities of extracellular CD38 would require that cADPR and ADPR are transferred to the cytoplasm to act on ryanodine receptors (RyR) and TRPM2, correspondingly, thus triggering release of Ca2+ from endoplasmic reticulum and import of extracellular Ca2+.
An alternative to autocrine and paracrine cADPR and ADPR would be an intracellular origin of cytoplasmic cADPR, as has been shown in HL-60 cells, U937 cells, and human monocytes. In these cells, CD38 can exist as a type III protein with its catalytic domain towards the cytoplasm.[106,107] This type III CD38 produces intracellular cADPR in HEK293 cells.[106,107] Nevertheless, there is also evidence of mechanisms by which extracellular cADPR and ADPR could reach their target Ca2+ channels in the cytoplasm. Of particular relevance to our proposal, in Jurkat cells (a CD4 T cell line), extracellularly oriented CD38 can be internalized and transport cADPR to the cytoplasm.[108] In 3TC fibroblasts, endocytic vesicles containing CD38 can also transport NAD, transform it to cADPR, and liberate it to the cytosol.[109] Extracellular cADPR can also be transported to the cytoplasm by the concentrative nucleoside transporter CNT2 in 3TC fibroblasts,[110] and in HL-60 cells.[111] Connexin-43 hemichannels can also internalize cADPR.[112] As stated before, this protein is expressed by CD4 T cells,[79,80] further suggesting the likeliness of our proposal.
Regarding ADPR, its transport by the cell membrane from the outer milieu to the cytosol is possible, although less evidence is available. ADPR uptake has been found in human erythrocytes.[113] This uptake requires NAD glycohydrolase activity plus the presence of NAD, and was not observable by the mere addition of ADPR to the culture.[113] Red cell NAD glycohydrolase is likely to be CD38, since erythrocytes do not express CD157, the only other known cell surface NADase.[114] ADPR transport may involve a dual activity of CD38, synthesizing ADPR and transferring it to the cell. ADPR uptake has been verified, with the additional finding that some ADPR was incorporated to the cell membrane.[115] Interestingly, in the latter study transport was inhibited by ADPR but not by NAD, suggesting that only recently synthesized ADPR was transported by CD38. Addition of extracellular ADPR increases the intracellular Ca2+ concentration of human monocytes, also suggesting ADPR transport to the cytosol.[116] These mechanisms must be thoroughly studied in CD4 T cells.
3. 3. 6.-. Reduced CD4 T cell viability due to increased cytoplasmic Ca2+
As a final aspect of our model of CD38-promoted CD4 T cell dysfunction and depletion, chronically increased cytoplasmic Ca2+ concentration elicited by cADPR or ADPR may be detrimental CD4 T cells, even though it has physiological functions (for an example see[117]). Calcium release from the endoplasmic reticulum can in some conditions activate the mitochondrial pathway of apoptosis, mediated by mitochondrial permeability transition and liberation of Cytochrome C, which promotes apoptosis[118,119] (Figure 4). It will be necessary to compare Ca2+ concentration in CD4 T cells from HIV+ patients and healthy controls, as well as to compare calcium flux upon TCR engagement (Table 1). We would expect these variables to be increased in cells bearing more CD38 molecules. Likewise, measuring the levels of cADPR and ADPR in plasma and cell culture media from patients and controls would further evaluate this possible mechanism (Table 1). Studying CD4 T cells from patients and controls from a same differentiation subpopulation (for instance, central memory CD4 T cells) is necessary to rule out differences due to diverging degrees of differentiation between samples. Variables affected by differentiation include cytokine production, viability, proliferative capacity, differentiation capacity, and metabolic profiles.[72,102,103]
Figure 4.-. Cytoplasmic Ca2+ increase in activated CD4 T cells in HIV disease.

Concomitant to Warburg effect, NAD turnover yields cADPR and ADPR, which can be transported to the cytoplasm and activate the calcium channels ryanodine receptor (RyR) on the endoplasmic reticulum, and Transient Receptor Potential Melastatin 2 channels (TRPM2) on the cell membrane, respectively. The ensuing increase in cytoplasmic Ca2+ further stresses mitochondria, which can cause the release of cytochrome C, a programmed cell death signal.
If our hypotheses are verified in CD4 T cells in the setting of HIV infection, CD38 antagonists could be used as a therapeutic tool, adjunct to antiretroviral therapy. Such antagonists have already been proposed for other clinical applications,[120] which in principle makes such application feasible. A particularly interesting possible target tissue of CD38 inhibition is gut-associated lymphoid tissue, in which CD4 T cell recovery under antiretroviral therapy is particularly limited, coincident with a high level of immune activation (with its associated increase in CD38 expression[58,59,62]).
4. Conclusions and outlook
We provide background supporting the pertinence of asking whether CD38 overexpression facilitates CD4 T cell depletion in the setting of HIV infection. We describe existing evidence of the enzymatic function of CD38 and its effects in different cells, supporting proposed mechanisms that could mediate an indirect pathogenic role of chronic over-expression of CD38 in HIV infection. Chronic increased activity of CD38 in the milieu of memory CD4 T cells may increase NAD turnover, causing cytoplasmic NAD depletion. This in turn can promote Warburg effect, affecting mitochondria function and integrity. Additionally, CD38 catalytic products may increase cytoplasmic Ca2+ concentrations, which may further increase mitochondrial stress, ultimately leading to lower survival of CD4 T cells. This series of hypotheses is amenable to experimental testing. If verified, they may provide a new therapeutic strategy as adjunct to antiretroviral therapy.
Acknowledgements
Funding: Mexican National Council for Science and Technology (CONACYT) FC-2015-214 (E. Espinosa), The National Institutes of Health grants AI091526 (W. Jiang) and AI128864 (W. Jiang). We gratefully thank María-José Flores for language editing.
Footnotes
Conflict of interests statement
Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas has made an application for a patent based on the ideas presented this manuscript, with EE as inventor (Instituto Mexicano de la Propiedad Industrial, September 26, 2018, file MX/a/2018/011698, application number MX/E/2018/072036).
References
- [1].Murray JL, Reuben JM, Munn CG, Newell G, Mansell PW, Hersh EM, Int. J. Immunopharmacol 1985, 7, 661. [DOI] [PubMed] [Google Scholar]
- [2].Gottlieb MS, Schroff R, Schanker HM, Weisman JD, Fan PT, Wolf RA, Saxon A, N Engl J Med 1981, 305, 1425. [DOI] [PubMed] [Google Scholar]
- [3].Salazar-Gonzalez JF, Moody DJ, Giorgi JV, Martinez-Maza O, Mitsuyasu RT, Fahey JL, J Immunol 1985, 135, 1778. [PubMed] [Google Scholar]
- [4].Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP, Shih R, Lewis J, Wiley DJ, Phair JP, Wolinsky SM, Detels R, J.Infect.Dis 1999, 179, 859. [DOI] [PubMed] [Google Scholar]
- [5].Liu Z, Cumberland WG, Hultin LE, Prince HE, Detels R, Giorgi JV, J Acquir Immune Defic Syndr Hum Retrovirol 1997, 16, 83. [DOI] [PubMed] [Google Scholar]
- [6].Karim R, Mack WJ, Stiller T, Operskalski E, Frederick T, Landay A, Young MA, Tien PC, Augenbraun M, Strickler HD, Kovacs A, AIDS Lond. Engl 2013, 27, 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hazenberg MD, Otto SA, van Benthem BH, Roos MT, Coutinho RA, Lange JM, Hamann D, Prins M, Miedema F, AIDS 2003, 17, 1881. [DOI] [PubMed] [Google Scholar]
- [8].Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narvaez AB, Hunt P, Martin JN, Kahn JO, Levy J, McGrath MS, Hecht FM, Blood 2004, 104, 942. [DOI] [PubMed] [Google Scholar]
- [9].Carbone J, Gil J, Benito JM, Navarro J, Munoz-Fernandez A, Bartolome J, Zabay JM, Lopez F, Fernandez-Cruz E, AIDS 2000, 14, 2823. [DOI] [PubMed] [Google Scholar]
- [10].Sousa AE, Carneiro J, Meier-Schellersheim M, Grossman Z, Victorino RM, J Immunol 2002, 169, 3400. [DOI] [PubMed] [Google Scholar]
- [11].Resino S, Seoane E, Gutierrez MD, Leon JA, Munoz-Fernandez MA, J Acquir Immune Defic Syndr 2006, 42, 269. [DOI] [PubMed] [Google Scholar]
- [12].Antonelli LR, Mahnke Y, Hodge JN, Porter BO, Barber DL, DerSimonian R, Greenwald JH, Roby G, Mican J, Sher A, Roederer M, Sereti I, Blood 2010, 116, 3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Espinosa E, Romero-Rodríguez DP, Cantoral-Díaz M-T, Reyes-Terán G, J. Inflamm. Lond. Engl 2013, 10, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Goicoechea M, Smith DM, Liu L, May S, Tenorio AR, Ignacio CC, Landay A, Haubrich R, J Infect Dis 2006, 194, 29. [DOI] [PubMed] [Google Scholar]
- [15].Marchetti G, Gori A, Casabianca A, Magnani M, Franzetti F, Clerici M, Perno C-F, d’Arminio Monforte A, Galli M, Meroni L, AIDS Lond. Engl 2006, 20, 1727. [DOI] [PubMed] [Google Scholar]
- [16].Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D, PLoS One 2008, 3, e2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Paiardini M, Müller-Trutwin M, Immunol. Rev 2013, 254, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Giorgi JV, Liu Z, Hultin LE, Cumberland WG, Hennessey K, Detels R, J Acquir Immune Defic Syndr 1993, 6, 904. [PubMed] [Google Scholar]
- [19].Liu Z, Hultin LE, Cumberland WG, Hultin P, Schmid I, Matud JL, Detels R, Giorgi JV, Cytometry 1996, 26, 1. [DOI] [PubMed] [Google Scholar]
- [20].Liu Z, Cumberland WG, Hultin LE, Kaplan AH, Detels R, Giorgi JV, J Acquir Immune Defic Syndr Hum Retrovirol 1998, 18, 332. [DOI] [PubMed] [Google Scholar]
- [21].Giorgi JV, Lyles RH, Matud JL, Yamashita TE, Mellors JW, Hultin LE, Jamieson BD, Margolick JB, Rinaldo CR, Phair JP, Detels R, J Acquir Immune Defic Syndr 2002, 29, 346. [DOI] [PubMed] [Google Scholar]
- [22].Mohri H, Perelson AS, Tung K, Ribeiro RM, Ramratnam B, Markowitz M, Kost R, Hurley A, Weinberger L, Cesar D, Hellerstein MK, Ho DD, J.Exp.Med 2001, 194, 1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Marchetti G, Bellistrì GM, Borghi E, Tincati C, Ferramosca S, La Francesca M, Morace G, Gori A, Monforte AD, AIDS Lond. Engl 2008, 22, 2035. [DOI] [PubMed] [Google Scholar]
- [24].Hazenberg MD, Stuart JW, Otto SA, Borleffs JC, Boucher CA, de Boer RJ, Miedema F, Hamann D, Blood 2000, 95, 249. [PubMed] [Google Scholar]
- [25].Würsch D, Ormsby CE, Romero-Rodríguez DP, Olvera-García G, Zúñiga J, Jiang W, Pérez-Patrigeon S, Espinosa E, Dis. Markers 2016, 2016, 9510756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Reinherz EL, Kung PC, Goldstein G, Levey RH, Schlossman SF, Proc. Natl. Acad. Sci. U. S. A 1980, 77, 1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, Walseth TF, Lee HC, Science 1993, 262, 1056. [DOI] [PubMed] [Google Scholar]
- [28].Yates B, Braschi B, Gray KA, Seal RL, Tweedie S, Bruford EA, Nucleic Acids Res. 2017, 45, D619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rappaport N, Fishilevich S, Nudel R, Twik M, Belinky F, Plaschkes I, Stein TI, Cohen D, Oz-Levi D, Safran M, Lancet D, Biomed. Eng. Online 2017, 16, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jackson DG, Bell JI, J. Immunol. Baltim. Md 1950 1990, 144, 2811. [PubMed] [Google Scholar]
- [31].Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q, Structure 2005, 13. [DOI] [PubMed] [Google Scholar]
- [32].Tohgo A, Munakata H, Takasawa S, Nata K, Akiyama T, Hayashi N, Okamoto H, J. Biol. Chem 1997, 272, 3879. [DOI] [PubMed] [Google Scholar]
- [33].Munshi C, Aarhus R, Graeff R, Walseth TF, Levitt D, Lee HC, J. Biol. Chem 2000, 275, 21566. [DOI] [PubMed] [Google Scholar]
- [34].Aarhus R, Graeff RM, Dickey DM, Walseth TF, Lee HC, J. Biol. Chem 1995, 270, 30327. [DOI] [PubMed] [Google Scholar]
- [35].Schmid F, Bruhn S, Weber K, Mittrücker H-W, Guse AH, FEBS Lett. 2011, 585, 3544. [DOI] [PubMed] [Google Scholar]
- [36].Davis LC, Morgan AJ, Chen J-L, Snead CM, Bloor-Young D, Shenderov E, Stanton-Humphreys MN, Conway SJ, Churchill GC, Parrington J, Cerundolo V, Galione A, Curr. Biol. CB 2012, 22, 2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Muñoz P, Navarro M-C, Pavón EJ, Salmerón J, Malavasi F, Sancho J, Zubiaur M, J. Biol. Chem 2003, 278, 50791. [DOI] [PubMed] [Google Scholar]
- [38].Zubiaur M, Izquierdo M, Terhorst C, Malavasi F, Sancho J, Immunol J 1997, 159, 193. [PubMed] [Google Scholar]
- [39].Zubiaur M, Guirado M, Terhorst C, Malavasi F, Sancho J, J. Biol. Chem 1999, 274, 20633. [DOI] [PubMed] [Google Scholar]
- [40].Zubiaur M, Fernández O, Ferrero E, Salmerón J, Malissen B, Malavasi F, Sancho J, J. Biol. Chem 2002, 277, 13. [DOI] [PubMed] [Google Scholar]
- [41].Deaglio S, Morra M, Mallone R, Ausiello CM, Prager E, Garbarino G, Dianzani U, Stockinger H, Malavasi F, J. Immunol. Baltim. Md 1950 1998, 160, 395. [PubMed] [Google Scholar]
- [42].Nishina H, Inageda K, Takahashi K, Hoshino S, Ikeda K, Katada T, Biochem. Biophys. Res. Commun 1994, 203, 1318. [DOI] [PubMed] [Google Scholar]
- [43].Deaglio S, Aydin S, Grand MM, Vaisitti T, Bergui L, D’Arena G, Chiorino G, Malavasi F, Mol. Med. Camb. Mass 2010, 16, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nasi A, Chiodi F, J. Intern. Med 2018, 283, 257. [DOI] [PubMed] [Google Scholar]
- [45].Younes S-A, Freeman ML, Mudd JC, Shive CL, Reynaldi A, Panigrahi S, Estes JD, Deleage C, Lucero C, Anderson J, Schacker TW, Davenport MP, McCune JM, Hunt PW, Lee SA, Serrano-Villar S, Debernardo RL, Jacobson JM, Canaday DH, Sekaly R-P, Rodriguez B, Sieg SF, Lederman MM, J. Clin. Invest 2016, 126, 2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mudd JC, Lederman MM, Curr. Opin. HIV AIDS 2014, 9, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lichtfuss GF, Cheng W-J, Farsakoglu Y, Paukovics G, Rajasuriar R, Velayudham P, Kramski M, Hearps AC, Cameron PU, Lewin SR, Crowe SM, Jaworowski A, J. Immunol. Baltim. Md 1950 2012, 189, 1491. [DOI] [PubMed] [Google Scholar]
- [48].Kuri-Cervantes L, de Oca GS-M, Avila-Ríos S, Hernández-Juan R, Reyes-Terán G, J. Leukoc. Biol 2014, 96, 7. [DOI] [PubMed] [Google Scholar]
- [49].Tanko RF, Soares AP, Müller TL, Garrett NJ, Samsunder N, Abdool Karim Q, Abdool Karim SS, Riou C, Burgers WA, J. Immunol. Baltim. Md 1950 2017, 198, 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yong YK, Shankar EM, Westhorpe CLV, Maisa A, Spelman T, Kamarulzaman A, Crowe SM, Lewin SR, Medicine (Baltimore) 2016, 95, e4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chiang S-H, Harrington WW, Luo G, Milliken NO, Ulrich JC, Chen J, Rajpal DK, Qian Y, Carpenter T, Murray R, Geske RS, Stimpson SA, Kramer HF, Haffner CD, Becherer JD, Preugschat F, Billin AN, PloS One 2015, 10, e0134927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, Chini EN, Cell Metab. 2016, 23, 1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, Fu J, Zhang J, Nguyen H, Kang I, Toth K, Al-Homrani M, Husain M, Beeson G, Ball L, Helke K, Husain S, Garrett-Mayer E, Hardiman G, Mehrotra M, Nishimura MI, Beeson CC, Bupp MG, Wu J, Ogretmen B, Paulos CM, Rathmell J, Yu X-Z, Mehrotra S, Cell Metab. 2018, 27, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schultz MB, Sinclair DA, Cell Metab. 2016, 23, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ramzaoui S, Jouen-Beades F, Michot F, Borsa-Lebas F, Humbert G, Tron F, Clin.Exp.Immunol 1995, 99, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yang OO, Ferbas JJ, Hausner MA, Hultin LE, Hultin PM, McFadden D, Sawicki M, Detels R, Majchrowicz M, Matud JL, Giorgi JV, Jamieson BD, Acquir J Immune Defic. Syndr. 1999 2005, 39, 507. [PubMed] [Google Scholar]
- [57].Biancotto A, Grivel JC, Iglehart SJ, Vanpouille C, Lisco A, Sieg SF, Debernardo R, Garate K, Rodriguez B, Margolis LB, Lederman MM, Blood 2007, 109, 4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].d’Ettorre G, Ceccarelli G, Andreotti M, Selvaggi C, Giustini N, Serafino S, Schietroma I, Nunnari G, Antonelli G, Vullo V, Scagnolari C, Mediators Inflamm. 2015, 2015, 395484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yukl SA, Shergill AK, Girling V, Li Q, Killian M, Epling L, Li P, Kaiser P, Haase A, Havlir DV, McQuaid K, Sinclair E, Wong JK, PloS One 2015, 10, e0121290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Crowell TA, Fletcher JL, Sereti I, Pinyakorn S, Dewar R, Krebs SJ, Chomchey N, Rerknimitr R, Schuetz A, Michael NL, Phanuphak N, Chomont N, Ananworanich J, RV254/SEARCH010 Study Group, J. Int. AIDS Soc 2016, 19, 21163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cassol E, Malfeld S, Mahasha P, Bond R, Slavik T, Seebregts C, Poli G, Cassol S, van der Merwe SW, Rossouw T, J. Infect. Dis 2013, 208, 1113. [DOI] [PubMed] [Google Scholar]
- [62].Jaspan HB, Liebenberg L, Hanekom W, Burgers W, Coetzee D, Williamson A-L, Little F, Myer L, Coombs RW, Sodora D, Passmore J-A, J. Infect. Dis 2011, 204, 1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hoffmann M, Pantazis N, Martin GE, Hickling S, Hurst J, Meyerowitz J, Willberg CB, Robinson N, Brown H, Fisher M, Kinloch S, Babiker A, Weber J, Nwokolo N, Fox J, Fidler S, Phillips R, Frater J, SPARTAC and CHERUB Investigators, PLoS Pathog. 2016, 12, e1005661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cantó C, Menzies KJ, Auwerx J, Cell Metab. 2015, 22, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Deeks SG, Annu. Rev. Med 2011, 62, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Dock J, Ramirez CM, Hultin L, Hausner MA, Hultin P, Elliott J, Yang OO, Anton PA, Jamieson BD, Effros RB, PloS One 2017, 12, e0182498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].du Pré MF, van Berkel LA, Ráki M, van Leeuwen MA, de Ruiter LF, Broere F, Ter Borg MND, Lund FE, Escher JC, Lundin KEA, Sollid LM, Kraal G, Nieuwenhuis EES, Samsom JN, Am. J. Gastroenterol 2011, 106, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Eiras P, Roldán E, Camarero C, Olivares F, Bootello A, Roy G, Cytometry 1998, 34, 95. [DOI] [PubMed] [Google Scholar]
- [69].Farstad IN, Carlsen H, Morton HC, Brandtzaeg P, Immunology 2000, 101, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Srinivasula S, Lempicki RA, Adelsberger JW, Huang C-Y, Roark J, Lee PI, Rupert A, Stevens R, Sereti I, Lane HC, Di Mascio M, Kovacs JA, Blood 2011, 118, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Benito JM, Lopez M, Lozano S, Ballesteros C, Martinez P, Gonzalez-Lahoz J, Soriano V, J Acquir Immune Defic Syndr 2005, 38, 373. [DOI] [PubMed] [Google Scholar]
- [72].Chattopadhyay PK, Roederer M, Cytom. Part J. Int. Soc. Anal. Cytol 2010, 77, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Savarino A, Pugliese A, Martini C, Pich PG, Pescarmona GP, Malavasi F, J. Biol. Regul. Homeost. Agents 1996, 10, 13. [PubMed] [Google Scholar]
- [74].Douek DC, Picker LJ, Koup RA, Annu Rev Immunol 2003, 21, 265. [DOI] [PubMed] [Google Scholar]
- [75].Rodríguez B, Sethi AK, Cheruvu VK, Mackay W, Bosch RJ, Kitahata M, Boswell SL, Mathews WC, Bangsberg DR, Martin J, Whalen CC, Sieg S, Yadavalli S, Deeks SG, Lederman MM, JAMA J Am. Med. Assoc 2006, 296, 1498. [DOI] [PubMed] [Google Scholar]
- [76].De Flora A, Zocchi E, Guida L, Franco L, Bruzzone S, Ann. N. Y. Acad. Sci 2004, 1028, 176. [DOI] [PubMed] [Google Scholar]
- [77].Oviedo-Orta E, Hoy T, Evans WH, Immunology 2000, 99, 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Oviedo-Orta E, Gasque P, Evans WH, FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 2001, 15, 768. [DOI] [PubMed] [Google Scholar]
- [79].Bermudez-Fajardo A, Ylihärsilä M, Evans WH, Newby AC, Oviedo-Orta E, J. Leukoc. Biol 2007, 82, 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Oviedo-Orta E, Perreau M, Evans WH, Potolicchio I, J. Leukoc. Biol 2010, 88, 79. [DOI] [PubMed] [Google Scholar]
- [81].Creeke PI, Dibari F, Cheung E, van den Briel T, Kyroussis E, Seal AJ, J. Nutr 2007, 137, 2013. [DOI] [PubMed] [Google Scholar]
- [82].Fukuwatari T, Shibata K, Ishihara K, Fushiki T, Sugimoto E, J. Nutr. Sci. Vitaminol. (Tokyo) 2001, 47, 177. [DOI] [PubMed] [Google Scholar]
- [83].Fukuwatari T, Shibata K, J. Nutr. Sci. Vitaminol. (Tokyo) 2009, 55, 279. [DOI] [PubMed] [Google Scholar]
- [84].Olvera-García G, Aguilar-García T, Gutiérrez-Jasso F, Imaz-Rosshandler I, Rangel-Escareño C, Orozco L, Aguilar-Delfín I, Vázquez-Pérez JA, Zúñiga J, Pérez-Patrigeon S, Espinosa E, BMC Genomics 2016, 17, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bofill M, Fairbanks LD, Ruckemann K, Lipman M, Simmonds HA, J. Biol. Chem 1995, 270, 29690. [PubMed] [Google Scholar]
- [86].Schreiber V, Dantzer F, Ame J-C, de Murcia G, Nat. Rev. Mol. Cell Biol 2006, 7, 517. [DOI] [PubMed] [Google Scholar]
- [87].Zhang T, Kraus WL, Biochim. Biophys. Acta 2010, 1804, 1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Beauharnois JM, Bolívar BE, Welch JT, Mol. Biosyst 2013, 9, 1789. [DOI] [PubMed] [Google Scholar]
- [89].Guarente L, Nat. Med 2014, 20, 24. [DOI] [PubMed] [Google Scholar]
- [90].Davila A, Liu L, Chellappa K, Redpath P, Nakamaru-Ogiso E, Paolella LM, Zhang Z, Migaud ME, Rabinowitz JD, Baur JA, eLife 2018, 7, DOI 10.7554/eLife.33246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, Mottis A, Jo Y-S, Viswanathan M, Schoonjans K, Guarente L, Auwerx J, Cell 2013, 154, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Maciolek JA, Pasternak JA, Wilson HL, Curr. Opin. Immunol 2014, 27, 60. [DOI] [PubMed] [Google Scholar]
- [93].Pearce EL, Curr. Opin. Immunol 2010, 22, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang C-H, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJW, Chen Q, Huang SC-C, O’Neill CM, Edelson BT, Pearce EJ, Sesaki H, Huber TB, Rambold AS, Pearce EL, Cell 2016, 166, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR, Immunity 2011, 35, 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Palmer CS, Ostrowski M, Gouillou M, Tsai L, Yu D, Zhou J, Henstridge DC, Maisa A, Hearps AC, Lewin SR, Landay A, Jaworowski A, McCune JM, Crowe SM, AIDS Lond. Engl 2014, 28, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Aounallah M, Dagenais-Lussier X, El-Far M, Mehraj V, Jenabian M-A, Routy J-P, van Grevenynghe J, Cytokine Growth Factor Rev. 2016, 28, 1. [DOI] [PubMed] [Google Scholar]
- [98].Tarasenko TN, Pacheco SE, Koenig MK, Gomez-Rodriguez J, Kapnick SM, Diaz F, Zerfas PM, Barca E, Sudderth J, DeBerardinis RJ, Covian R, Balaban RS, DiMauro S, McGuire PJ, Cell Metab. 2017, 25, 1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Yang Y, Cimen H, Han M-J, Shi T, Deng J-H, Koc H, Palacios OM, Montier L, Bai Y, Tong Q, Koc EC, J. Biol. Chem 2010, 285, 7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, Klebanoff CA, Ji Y, Li P, Yu Z, Whitehill GD, Clever D, Eil RL, Palmer DC, Mitra S, Rao M, Keyvanfar K, Schrump DS, Wang E, Marincola FM, Gattinoni L, Leonard WJ, Muranski P, Finkel T, Restifo NP, Cell Metab. 2016, 23, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Sukumar M, Kishton RJ, Restifo NP, Curr. Opin. Immunol 2017, 46, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Appay V, Van Lier RA, Sallusto F, Roederer M, Cytom. A 2008, 73, 975. [DOI] [PubMed] [Google Scholar]
- [103].Murray PJ, Rathmell J, Pearce E, Cell Metab. 2015, 22, 190. [DOI] [PubMed] [Google Scholar]
- [104].Mészáros LG, Bak J, Chu A, Nature 1993, 364, 76. [DOI] [PubMed] [Google Scholar]
- [105].Rosenbaum T, J. Gen. Physiol 2015, 145, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Liu J, Zhao YJ, Li WH, Hou YN, Li T, Zhao ZY, Fang C, Li SL, Lee HC, Proc. Natl. Acad. Sci. U. S. A 2017, 114, 8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Zhao YJ, Lam CMC, Lee HC, Sci. Signal. 2012, 5, ra67. [DOI] [PubMed] [Google Scholar]
- [108].Franco L, Guida L, Bruzzone S, Zocchi E, Usai C, De Flora A, FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 1998, 12, 1507. [DOI] [PubMed] [Google Scholar]
- [109].Zocchi E, Usai C, Guida L, Franco L, Bruzzone S, Passalacqua M, De Flora A, FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 1999, 13, 273. [DOI] [PubMed] [Google Scholar]
- [110].Guida L, Bruzzone S, Sturla L, Franco L, Zocchi E, De Flora A, J. Biol. Chem 2002, 277, 47097. [DOI] [PubMed] [Google Scholar]
- [111].Guida L, Franco L, Bruzzone S, Sturla L, Zocchi E, Basile G, Usai C, De Flora A, J. Biol. Chem 2004, 279, 22066. [DOI] [PubMed] [Google Scholar]
- [112].Song E-K, Rah S-Y, Lee Y-R, Yoo C-H, Kim Y-R, Yeom J-H, Park K-H, Kim J-S, Kim U-H, Han M-K, J. Biol. Chem 2011, 286, 44480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kim UH, Han MK, Park BH, Kim HR, An NH, Biochim. Biophys. Acta 1993, 1178, 121. [DOI] [PubMed] [Google Scholar]
- [114].Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, Funaro A, Horenstein AL, Malavasi F, Cytometry B Clin. Cytom 2013, 84, 207. [DOI] [PubMed] [Google Scholar]
- [115].Albeniz I, Demir O, Nurten R, Bermek E, Biosci. Rep 2004, 24, 41. [DOI] [PubMed] [Google Scholar]
- [116].Gerth A, Nieber K, Oppenheimer NJ, Hauschildt S, Biochem. J. 2004, 382, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Podestà M, Zocchi E, Pitto A, Usai C, Franco L, Bruzzone S, Guida L, Bacigalupo A, Scadden DT, Walseth TF, De Flora A, Daga A, FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 2000, 14, 680. [DOI] [PubMed] [Google Scholar]
- [118].Orrenius S, Gogvadze V, Zhivotovsky B, Biochem. Biophys. Res. Commun 2015, 460, 72. [DOI] [PubMed] [Google Scholar]
- [119].Zhang D, Armstrong JS, Cell Death Differ. 2007, 14, 703. [DOI] [PubMed] [Google Scholar]
- [120].Deaton DN, Haffner CD, Henke BR, Jeune MR, Shearer BG, Stewart EL, Stuart JD, Ulrich JC, Bioorg. Med. Chem 2018, 26, 2107. [DOI] [PubMed] [Google Scholar]