

Jiang et al. show that Ccr4–Not controls the ubiquitylation and turnover of Rpb1, the largest subunit of RNAPII, during transcription arrest.
Keywords: RNAPII, Ccr4–Not, Rsp5, ubiquitylation, transcription elongation
Abstract
The Ccr4–Not complex regulates essentially every aspect of gene expression, from mRNA synthesis to protein destruction. The Not4 subunit of the complex contains an E3 RING domain and targets proteins for ubiquitin-dependent proteolysis. Ccr4–Not associates with elongating RNA polymerase II (RNAPII), which raises the possibility that it controls the degradation of elongation complex components. Here, we demonstrate that Ccr4–Not controls the ubiquitylation and turnover of Rpb1, the largest subunit of RNAPII, during transcription arrest. Deleting NOT4 or mutating its RING domain strongly reduced the DNA damage-dependent ubiquitylation and destruction of Rpb1. Surprisingly, in vitro ubiquitylation assays indicate that Ccr4–Not does not directly ubiquitylate Rpb1 but instead promotes Rpb1 ubiquitylation by the HECT domain-containing ligase Rsp5. Genetic analyses suggest that Ccr4–Not acts upstream of RSP5, where it acts to initiate the destruction process. Ccr4–Not binds Rsp5 and forms a ternary complex with it and the RNAPII elongation complex. Analysis of mutant Ccr4–Not lacking the RING domain of Not4 suggests that it both recruits Rsp5 and delivers the E2 Ubc4/5 to RNAPII. Our work reveals a previously unknown function of Ccr4–Not and identifies an essential new regulator of RNAPII turnover during genotoxic stress.
Transcription elongation is a highly dynamic and regulated process (Jonkers and Lis 2015). RNA polymerase II (RNAPII) undergoes cycles of productive elongation, pausing, arrest, and reactivation. Prolonged stalling of RNAPII prevents normal transcription, and arrested RNAPII must be resolved to allow transcription to resume. Starvation of nucleotides, physical barriers, or DNA lesions arrest RNAPII (Wilson et al. 2013a). RNAPII arrested at DNA lesions initiates transcription-coupled repair (TCR), which depends on Rad26 in yeast and CSB in humans, to repair the damage and recover transcription (Hanawalt and Spivak 2008). However, if the Rad26-TCR mechanism does not restore transcription, RNAPII is removed by proteasome-mediated degradation as a last resort (Wilson et al. 2013a).
Removal of arrested RNAPII is achieved by polyubiquitylation and proteasome-dependent degradation of its largest subunit, Rpb1. A series of ubiquitin-conjugating enzymes ubiquitylates Rpb1. In yeast, the HECT domain E3 ligase Rsp5 (Nedd4 family in humans) recruits the E2 ubiquitin-conjugating enzyme Ubc4/5 to build Lys63-linked ubiquitin chains onto Rpb1 (Huibregtse et al. 1997; Somesh et al. 2005, 2007; Anindya et al. 2007) that are subsequently trimmed by the deubiquitinase Ubp2 to a monoubiquitylated species (Harreman et al. 2009). Def1 binds monoubiquitylated Rpb1 and then recruits a second E3 ubiquitin ligase complex containing Elc1–Cul3, which adds Lys48-linked ubiquitin chains to Rpb1 (Ribar et al. 2006, 2007; Yasukawa et al. 2008; Harreman et al. 2009; Wilson et al. 2013b). The proteasome is then recruited to polyubiquitylated Rpb1 by a Cdc48-containing complex (Verma et al. 2011). The regulation of RNAPII destruction has turned out to be more elaborate than once anticipated. Many factors regulating its removal from genes remain to be identified (Daulny et al. 2008; Karakasili et al. 2014). Significant unresolved questions include what determines whether RNAPII is salvaged or destroyed and what triggers the recruitment of Rsp5 to initiate the destruction program.
Ccr4–Not is a multisubunit complex that is highly conserved throughout the eukaryotic kingdom (Miller and Reese 2012; Collart 2016) and has been implicated in the control of gene expression from transcription to mRNA decay and, finally, protein destruction. The complex can be broken down into two physical and functional “modules” linked via the Not1 scaffold protein: the Ccr4–Caf1 deadenylase module and the Not module (Collart 2016). Contained within the “Not module” is Not4, a RING domain-containing protein that partners with the E2 enzymes Ubc4/5 to ubiquitylate proteins (Mulder et al. 2007b). In the cytoplasm, Not4 ubiquitylates proteins involved in translation, such as the ribosomal protein Rps7A (Panasenko and Collart 2012) and the nascent polypeptide-associated complex (Panasenko et al. 2006). In the nucleus, Not4 targets the histone demethylase Jhd2 (Mersman et al. 2009), the transcription factor Yap1 (Gulshan et al. 2012), and a subunit of the mediator (Cooper et al. 2012). However, many Not4 targets have not been identified.
Deleting genes encoding subunits of the Ccr4–Not complex causes sensitivity to DNA damage agents, and a not5Δ mutant is defective in TCR (Gaillard et al. 2009). The precise role of Ccr4–Not in DNA repair is unknown, and, given its many functions in both the cytoplasm and nucleus, it is not clear whether Ccr4–Not is directly involved in the repair process. For example, Ccr4 imparts resistance to the replication inhibitor hydroxyurea (HU) by controlling the stability of the mRNA encoding for a regulator of the ribonucleotide reductase genes (Woolstencroft et al. 2006). In the nucleus, Ccr4–Not associates with the RNAPII elongation complex (EC) (Kruk et al. 2011; Reese 2013; Babbarwal et al. 2014); thus, it may play a direct role in the repair process. Interestingly, Not4 binds Ubc4/5, the E2 involved in Rpb1 degradation (Mulder et al. 2007b; Somesh et al. 2007), but there is no evidence that Ccr4–Not participates in the turnover of Rpb1 by the proteasome.
Here, we provide important insights into how Ccr4–Not maintains genomic integrity and transcription fidelity. Ccr4–Not controls the DNA damage-dependent destruction of RNAPII by promoting the ubiquitylation of the largest subunit of RNAPII, Rpb1. Surprisingly, Ccr4–Not does not directly ubiquitylate Rpb1 but instead initiates the cascade of RNAPII removal by promoting Rsp5-dependent ubiquitylation. Here we reveal a novel function for the fascinating Ccr4–Not complex and identify a new mechanism for resolving arrested RNAPII throughout the genome.
Results
Degradation of RNAPII requires Not4
Ccr4–Not controls multiple stress responses, including that caused by genotoxic stress. Ccr4–Not mutants are ultraviolet (UV) radiation-sensitive, and a not5Δ strain has DNA repair defects (Gaillard et al. 2009). However, it is not clear whether Ccr4–Not plays a direct role in, or which of its many activities is important for, DNA repair. Ccr4–Not associates with elongating RNAPII, and Not4 mediates the destruction of proteins; thus, we speculated that it might play a role in resolving arrested RNAPII by targeting Rpb1 for destruction. Strains containing a deletion of nonessential Ccr4–Not genes were treated with the UV-mimetic drug 4-NQO to induce damage and with cycloheximide to inhibit new protein synthesis using published protocols (Verma et al. 2011). Western blotting showed that Rpb1 was rapidly degraded in wild-type cells, reaching a level of ∼20% within 90 min of 4-NQO treatment (Fig. 1A,B). Rpb1 degradation was severely reduced in the “Not group” mutants not2Δ, not4Δ, and not5Δ (Fig. 1A; Supplemental Fig. S1). The degradation defect was similar to that of a def1Δ mutant, a factor required for Rpb1 turnover after DNA damage (Fig. 1A; Woudstra et al. 2002; Wilson et al. 2013b). In contrast, deleting other subunits of the complex had little to no effect on Rpb1 degradation. The caf1Δ mutant displayed a small reduction in Rpb1 degradation, but the magnitude was not nearly as great as that observed in the NOT2, NOT4, and NOT5 mutants. Ccr4 is essential for the mRNA decay function of the complex (Tucker et al. 2001; Miller and Reese 2012; Collart 2016). Because Rpb1 was degraded normally in the ccr4Δ mutant, the Rpb1 degradation defect cannot be explained by changes in mRNA decay rates or global mRNA metabolism.
Figure 1.
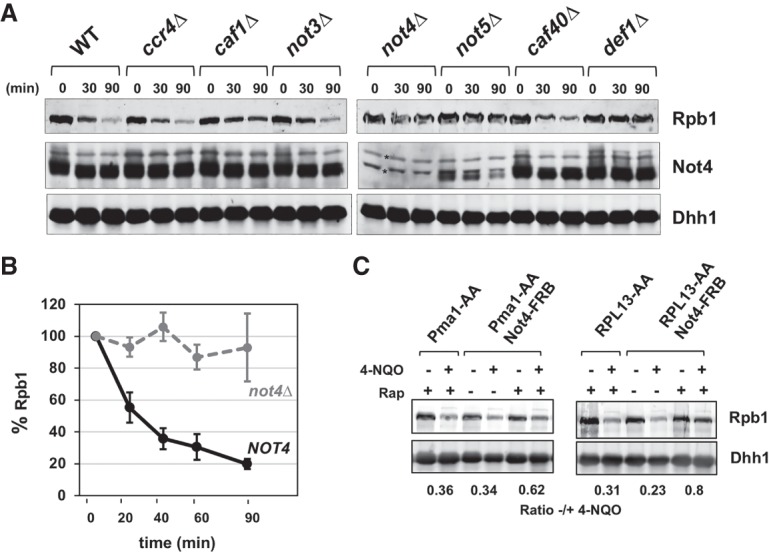
Not4 is crucial for Rpb1 degradation. (A) Western blot of Rpb1 levels in cell extracts from wild-type and Ccr4–Not mutant strains. Cells were treated with 5 µg/mL 4-NQO together with 100 µg/mL cycloheximide to block new protein synthesis for the times indicated. Rpb1 was detected using 8WG16, and Not4 and Dhh1 were detected using polyclonal antiserum. Dhh1 was used as the loading control. Asterisks indicate proteins cross-reacting with the Not4 antisera. (B) Quantification of Rpb1 turnover in wild-type and not4Δ cells. The percentage of Rpb1 remaining was calculated, setting the untreated value (t = 0) at 100%. The Rpb1 signal was normalized to the loading control. Each data point represents the mean and standard deviation. N = 4. (C) Analysis of Rpb1 levels in Not4 anchor-away (AA) strains. Not4 was tagged in-frame with FRB in the PMA1-AA (PMA1-2xFKBP) or RPL13-AA (RPL13B-2xFKBP) strain. Cells were treated with 1 µg/mL rapamycin for 30 min and then with 5 µg/mL 4-NQO and 100 µg/mL cycloheximide for 60 min. Blots were normalized and quantified as described in B.
Not2, Not4, and Not5 are required for Rpb1 degradation (this study) and H3K4 trimethylation (H3K4me3) (Laribee et al. 2007; Mulder et al. 2007a), suggesting that Not2 and Not5 affect Not4 activity. One possibility is that Not2 and Not5 are important for the stability of Not4. Probing the extracts with Not4 antiserum revealed that Not4 protein levels were significantly reduced in both the not2Δ and not5Δ mutants (Fig. 1A; Supplemental Fig. S1A). In contrast, Not2 and Not5 levels are unaffected in a not4Δ mutant (Supplemental Fig. S1B). The stability of the Ccr4–Not complex is dependent on Not2 and Not5, and this may explain why Not4 fails to accumulate in these mutants (Bai et al. 1999; Azzouz et al. 2009). The reduction of Not4 protein in the not2Δ and not5Δ mutants is likely caused by a substantial reduction in Not1 protein levels (Supplemental Fig. S2). We attempted to suppress the Rpb1 degradation defect in the not2Δ and not5Δ mutants by overexpressing NOT4 from a high-copy vector. We found that even though Not4 levels were elevated, Rpb1 degradation was not restored in the not2Δ or not5Δ mutants (Supplemental Fig. S2). These results suggest that the “free” pool of overexpressed Not4 cannot carry out Rpb1 degradation and that it must be in the complex to function. We performed structure-guided mutagenesis of the interface between Not4 and Not1 to directly address whether Not4 can function outside of the Ccr4–Not complex but found that mutations that disrupted the interaction between Not4 and Not1 strongly reduced Not4 protein levels in cells (data not shown).
Ccr4–Not is present in both the nucleus and cytoplasm (Collart 2016). We expected that if Ccr4–Not is directly involved in the destruction of Rpb1, it must reside in the nucleus to perform this function. We conditionally depleted Not4 from the nucleus using the anchor-away (AA) technique (Haruki et al. 2008). AA strains were constructed to relocate Not4-FRB to the plasma membrane (PMA1-AA) or the ribosome (RPL13A-AA). Although these cells had no obvious phenotype when plated on medium lacking rapamycin, they displayed slow growth and sensitivity to HU in the presence of rapamycin (Supplemental Fig. S3A). Thus, relocating Not4 to the cytoplasm caused phenotypes similar to deleting its gene. Rapamycin was added to cells for 30 min to deplete Not4 from the nucleus, and then 4-NQO/cycloheximide was added for an additional 60 min. Retaining Not4 in the cytoplasm significantly reduced the degradation of Rpb1 (Fig. 1C; Supplemental Fig. S3B). The RPL13A-AA strain displayed more severe growth and Rpb1 turnover defects than the PMA1-AA strain. This is not surprising because RPL13A shuttles between the nucleus and the cytoplasm, whereas Pma1 localizes to the plasma membrane. RPL13A would remove Not4 present in the nucleus before rapamycin treatment or retrieve any protein that escaped retention in the cytoplasm (Haruki et al. 2008). Yeast can adapt to the loss of a gene product over time (Stern et al. 2007; Moore et al. 2014); however, the AA experiment suggests that long-term adaption to the loss of NOT4 does not explain the Rpb1 degradation defect because the phenotype was evident within 90 min.
The Not4 RING domain is essential for polymerase turnover
The RING domain of Not4 binds to the E2 enzyme Ubc4/5 and is required for Not4's activity (Albert et al. 2000, 2002; Mulder et al. 2007b; Mersman et al. 2009; Panasenko and Collart 2012). We constructed three RING domain mutants, each predicted to impair Not4–Ubc4/5 interactions progressively. A deletion of the entire RING domain was constructed, as well as single (L35A) and double (L35A,I37A) point mutations in amino acids that contact residues in Ubc4 (Bhaskar et al. 2015). As reported previously, deleting the RING domain (ΔRING) caused slow growth and temperature sensitivity (Panasenko and Collart 2011; Halter et al. 2014). We screened these mutants for DNA damage sensitivity and Rpb1 destruction. Deleting the RING domain rendered the cells sensitive to DNA damage (Fig. 2A). The single point mutation caused no detectable phenotypes in these assays, while the double point mutation resulted in an intermediate phenotype. Western blotting revealed that deleting the RING domain phenocopied the Rpb1 destruction defect of the null allele, and the double point mutant displayed an intermediate phenotype. Expression of the mutants was similar to the wild-type protein (Fig. 2B), and we found that the RING domain is dispensable for Not4 to associate with the scaffold of the Ccr4–Not complex, Not1 (Supplemental Fig. S4 and see Fig. 6A below).
Figure 2.

The RING domain of Not4 is required for stress tolerance and normal Rpb1 degradation. (A) Spot growth assays. Strains were spotted on YPD plates with the treatments indicated. The images of growth at 37°C with 60 J/m2 UV and 100 mM HU were taken after 2 d. (B) Western blot of extracts from wild-type and mutant strains. The experiment was conducted as described in the legend for Figure 1A, except that cells were treated for 60 min. Asterisks mark nonspecific bands reacting with the Not4 antiserum. (C) Spot growth assays, as in A. (D) Western blotting of extracts from ΔRING and ΔRRM (RNA recognition motif) mutant strains.
Not4 has an RNA recognition motif (RRM), but its function is unclear (Albert et al. 2000). The RRM has been reported to be dispensable for Jhd2 turnover and histone H3K4me3 (Mersman et al. 2009). However, a recent study showed that mutating residues in both the RRM and RING domains caused stronger proteostasis defects than only introducing mutations in the RING domain alone (Chen et al. 2018), suggesting the RRM may have specialized functions in the cell. The RRMΔ mutant did not show an apparent defect in cell growth, resistance to HU or UV, or Rpb1 turnover (Fig. 2C,D); therefore, the RRM is not essential for Rpb1 destruction or DNA damage resistance.
Not4 is required for Rpb1 ubiquitylation
The turnover of Rpb1 is a multistep process that is initiated by the ubiquitylation of Rpb1 followed by the recruitment of downstream factors, including a Cdc48-containing complex and the proteasome (Verma et al. 2011; Wilson et al. 2013a). Since Not4 is an E3 ubiquitin ligase and associates with RNAPII, an attractive model is that Ccr4–Not is required for the ubiquitylation of Rpb1. However, Ccr4–Not could function at any of the steps leading up to the destruction of Rpb1. For example, Ccr4–Not binds the proteasome and Cdc48 (Supplemental Fig. S5; Laribee et al. 2007; Panasenko and Collart 2011), and Ccr4–Not could recruit one or more of these factors to the EC. Cells (pdr5Δ) were pretreated with the proteasome inhibitor MG132 for 30 min followed by the addition of 4-NQO for 60 min. Ubiquitylated proteins were enriched from cell extracts using immobilized GST-Dsk2, which captures both monoubiquitylated and polyubiquitylated proteins (Anindya et al. 2007; Harreman et al. 2009), and the bound fraction was analyzed by Western blotting for Rpb1 and ubiquitin. As expected, a substantial increase in ubiquitylated Rpb1 was detected in 4-NQO-treated wild-type cells (Fig. 3A). The ubiquitylation in untreated cells is likely to be caused by DNA damage-independent RNAPII arrest (Somesh et al. 2005; Karakasili et al. 2014). The amount of polyubiquitylated Rpb1 was significantly reduced in both the treated and untreated not4Δ cells, indicating a Rpb1 ubiquitylation defect. Some Rpb1 was pulled down by GST-Dsk2 in the not4Δ cell extracts that migrated near the unubiquitylated protein, which was not 4-NQO-dependent (Fig. 3A). This fraction of Rpb1 could be the monoubiquitylated species, which had been observed in Dsk2-enriched fractions of polymerase (Harreman et al. 2009), or unmodified Rpb1 that was bound by Dsk2 through ubiquitin attached to another subunit of RNAPII. Large-scale proteomic analysis of posttranslational modifications in yeast identified ubiquitin marks on Rpb2, Rpb3, Rpb5, Rpb6, Rpb7, and Rpb10 (Daulny et al. 2008; Beltrao et al. 2012). Importantly, probing the Dsk2-enriched fractions with antiubiquitin antibodies revealed that a comparable amount of total ubiquitylated proteins was observed in both NOT4 and not4Δ cells, suggesting that there was no global defect in ubiquitylation in the mutant and that the loss of Rpb1 ubiquitylation is selective.
Figure 3.
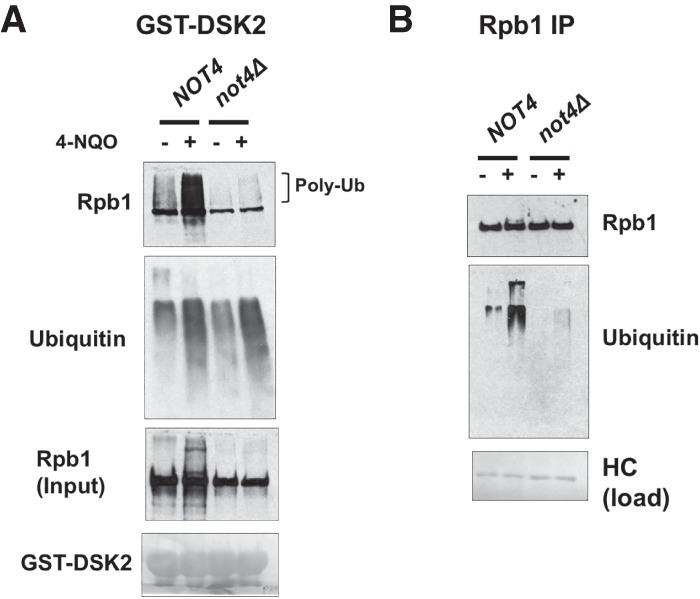
DNA damage-induced polyubiquitylation of Rpb1 is reduced in not4Δ cells. (A) Western blot of ubiquitylated proteins bound to GST-DSK2 beads. Cells were treated with 50 µM MG132 and 6 µg/mL 4-NQO for 60 min. Rpb1 was probed using 4H8 antibody. The blot was also probed with antiubiquitin antibody (P4G7-H11). The Ponceau S stain of the membrane displays the amount of GST-DSK2, which was used as the loading control. (B) Blotting for ubiquitylated proteins in Rpb1 immunoprecipitates. RNAPII was immunoprecipitated using 4H8 antibody. Rpb1 levels were detected using an antibody that recognizes the N terminus of Rpb1 (Santa Cruz Biotechnology, Y-80). Ubiquitin levels were probed with P4G7-H11. A Ponceau S stain of the heavy chain (HC) was used as a recovery control.
We further confirmed Not4-dependent Rpb1 ubiquitylation by probing immunoprecipitated Rpb1 with antiubiquitin antibodies. The results in Figure 3B show significant accumulation of polyubiquitylated proteins after 4-NQO treatment. Consistent with the results shown in Figure 3A, ubiquitylation of Rpb1 was strongly reduced, but not eliminated, in not4Δ cells. In sum, all of these results suggest that the ubiquitylation of Rpb1 requires Not4, which is likely responsible for the reduced turnover of Rpb1 in NOT4 mutants.
Ccr4–Not promotes Rpb1 ubiquitylation by a novel mechanism
The most straightforward explanation for Ccr4–Not's role in polymerase destruction is that Not4 directly ubiquitylates Rpb1. To test this, we established a Rpb1 ubiquitylation assay using highly purified components, replicating conditions that lead to robust ubiquitylation of Rpb1 by Rsp5 (Supplemental Fig. S6A,B; Somesh et al. 2005). We observed robust ubiquitylation of Rpb1 and confirmed that the change in mobility in gels was caused by ubiquitylation by adding the deubiquitinating enzyme Usp2 (Supplemental Fig. S6C).
Like many E3s, Not4 displays autoubiquitylation activity (Mulder et al. 2007b). We confirmed that our purified Ccr4–Not complex (Fig. 4A) has robust autoubiquitylation activity in the presence of E1 and E2 enzymes. Almost all of Not4 was converted to polyubiquitylated species within 5 min, and Not4 was progressively modified up to 30 min (Fig. 4B). The Not4 signal diminished significantly at later time points (Fig. 4B, lanes 5,6), most likely because the more extensively ubiquitylated species failed to migrate into the gel or transfer out of the gel onto the membrane. Next, we examined whether Ccr4–Not ubiquitylates free RNAPII. In the absence of either E2 (Ubc5) or E3 (Ccr4–Not), no ubiquitylation of Rpb1 was observed, as expected (Fig. 4C, lanes 1–4). However, Rpb1 was not ubiquitylated when Ccr4–Not was used as a source of E3 even though the complex displayed robust autoubiquitylation activity under the same conditions.
Figure 4.

Ccr4–Not does not directly ubiquitylate RNAPII. (A) Coomassie blue stain of purified Ccr4–Not complex. (B) Not4 autoubiquitylation assay using the Ccr4–Not complex. The Ccr4–Not complex (1.3 µg) was incubated with 50 ng of GST-Ube1 (E1), 50 ng of UbcH5c (E2), and 8 µg of ubiquitin. Reactions were terminated after the times indicated, and Not4 was detected by Western blotting. The minus E2 and E3 samples were incubated for 30 min. (C) Ubiquitylation assay using RNAPII as a substrate. Three-hundred nanograms of RNAPII, 0.6 µg of Ccr4–Not, 50 ng of GST-Ube1, 50 ng of 6xHis-Ubc4, and 8 µg of ubiquitin were used in the assay. Rpb1 was detected by Western blotting using 8WG16. (D) Schematic of the procedure producing RNAPII ECs. Details are described in the Materials and Methods. (E) Ubiquitylation reactions comparing free polymerase and ECs. (Top panel) Free RNAPII and that incorporated into immobilized ECs were incubated with E1 (50 ng of GST-Ube1), E2 (50 ng of UbcH5c), and either 1.8 µg of Ccr4–Not (E3) or, as a control, 75 ng of recombinant GST-Rsp5 (E3). Two of the reactions contained either full-length Def1 or the truncated active form (amino acids 1–500). Rpb1 was detected by Western blotting using 8WG16.
The EC is a better substrate for Rsp5 than free RNAPII (Somesh et al. 2005), and RNAPII incorporated into ECs is a more relevant substrate of the ubiquitylation machinery. Ccr4–Not associates with ECs (Kruk et al. 2011; Babbarwal et al. 2014); therefore, we tested whether Ccr4–Not can ubiquitylate Rpb1 incorporated into ECs. Arrested ECs were prepared on biotinylated templates as described in the Materials and Methods. Trapping RNAPII over a DNA lesion initiates Rpb1 destruction, but ECs arrested by nucleotide omission also stimulated Rsp5 activity in vitro. Furthermore, RNAPII arrested in vivo by nucleotide starvation induced Rpb1 ubiquitylation and destruction (Somesh et al. 2005; Karakasili et al. 2014). Thus, an EC prepared using the method described here produces a suitable substrate. Ubiquitylation assays were conducted on ECs purified on streptavidin beads and, in parallel, with equivalent amounts of free RNAPII. As described above, Ccr4–Not did not ubiquitylate free RNAPII, although Rsp5 could do so in the same assay (Fig. 4E, top panel, cf. lanes 4 and 5). However, Ccr4–Not did not ubiquitylate Rpb1 incorporated into ECs (Fig. 4E, bottom panel, lane 5).
Def1 is necessary for Rpb1 polyubiquitylation and destruction by enhancing Rsp5 activity and recruiting the Elc1–Cul3 complex to RNAPII (Reid and Svejstrup 2004; Somesh et al. 2005; Wilson et al. 2013b). A high-throughput study indicated that Def1 binds to Ccr4–Not components, and our coimmunoprecipitation (co-IP) experiments confirm this interaction (data not shown). We considered the possibility that Def1 stimulates Ccr4–Not ubiquitylation activity, so assays were repeated in the presence of recombinant Def1. Def1 is cleaved into an active ∼500-amino-acid form that enters the nucleus (Wilson et al. 2013a), so we added full-length Def1FL or its activated N-terminal version, Def11–500, to the ubiquitylation reaction. However, adding either Def1FL or Def11–500 did not stimulate Ccr4–Not activity (Fig. 4E, lanes 6,7). Together, the ubiquitylation assays suggest that Not4 does not directly ubiquitylate Rpb1 under the conditions used here and that it promotes Rpb1 ubiquitylation by a novel mechanism.
Ccr4–Not promotes Rsp5-dependent ubiquitylation
In light of our failure to demonstrate Ccr4–Not-dependent ubiquitylation of Rpb1, we considered the possibility that Ccr4–Not may promote the activity of one of the two E3 proteins that ubiquitylate Rpb1, Rsp5, or Elc1–Cul3. Deleting NOT4 greatly impaired the destruction and ubiquitylation of Rpb1, and one might predict that if Ccr4–Not were in an alternative pathway, Rsp5 and/or Elc1–Cul3 would carry out ubiquitylation in the absence of Not4. The minimal amount of Rpb1 ubiquitylation in the not4Δ mutant suggests that Ccr4–Not lies upstream of Rsp5. One possibility is that it may recruit Rsp5 to RNAPII or stimulate its activity. We tested this by conducting ubiquitylation assays in the presence or absence of Ccr4–Not while titrating in Rsp5. Consistent with the hypothesis that Ccr4–Not enhances Rsp5 activity, adding Ccr4–Not increased the ubiquitylation of Rpb1 (Fig. 5A, lanes 3,8,4,9). This result strongly suggests that Ccr4–Not regulates RNAPII destruction by promoting Rsp5-dependent ubiquitylation of Rpb1. This conclusion is consistent with the result showing that ubiquitylation of Rpb1 is significantly reduced, but not eliminated, in not4Δ cells (Fig. 3A).
Figure 5.

Ccr4–Not enhances Rsp5-dependent ubiquitylation and forms a ternary complex with the EC and Rsp5. (A) Ubiquitylation assay in the presence or absence of Ccr4–Not. Recombinant Rsp5 was titrated into the reaction (0, 12.5, 25, 50, and 100 ng) containing 300 ng of RNAPII with or without 1.5 µg of Ccr4–Not. After 60 min, Rpb1 was detected by Western blotting. (B) Immunoprecipitation of Rsp5 with Ccr4–Not. Ccr4–Not was immunoprecipitated from an extract of a Rsp5-13MYC strain using Not4 antiserum. Western blotting was performed using antimyc (9E10) or anti-Not4 antibody. The control is protein A beads without antibody. (C) Pull-down assay using GST-Rsp5 and purified Ccr4–Not. Ccr4–Not was incubated with GST-Rsp5 or GST in solution and then captured onto glutathione sepharose (GSH) beads. The eluted proteins were analyzed by Western blotting using anti-Not1 and anti-Caf1 antibody. Images of gels containing proteins used in the assays and GST and GST-Rsp5 loading are displayed in Supplemental Figure S6. (D) Electrophoretic mobility shift assay (EMSA) of ECs. ECs were labeled by incorporating [a-32P]UTP into the transcript. GST-Rsp5 was titrated into the binding assay (0, 50, 100, or 300 ng) with or without 1 µg of Ccr4–Not complex. (E) Quantification of the intensity of the EC band with (black) or without (gray) Ccr4–Not. The amount of EC in the absence of Rsp5 was set to 1.0. The data points represent averages and standard deviation of multiple experiments. N = 3.
We considered that the binding of Rsp5 to Ccr4–Not could stimulate Not4's activity and enhance the ubiquitylation of Rpb1. This possibility was tested by conducting assays with a Rsp5 catalytic mutant that has a cysteine mutated to an alanine (C777A). No ubiquitylation of Rpb1 was observed when the mutant was used, even in the presence of Ccr4–Not, indicating that Rsp5 is attaching ubiquitin to Rpb1 in the assay (Supplemental Fig. S7A). Rsp5 and Elc1–Cul3 act in a sequential manner. Rsp5 places the initial ubiquitin on Rpb1, which is then used to build a K48 polyubiquitin chain by the Elc1–Cul3 complex (Harreman et al. 2009). We tested whether the enhanced ubiquitylation of Rbp1 is caused by Ccr4–Not extending K48 chains from a monoubiquitin mark laid down by Rsp5 by conducting assays with ubiquitin mutants that block chain elongation (K63R and K48R). As reported previously (Harreman et al. 2009), Rsp5 only catalyzed monoubiquitylation of Rpb1 when K63R ubiquitin was used (Supplemental Fig. S7B). Moreover, adding Ccr4–Not did not cause further modification of Rpb1 (Supplemental Fig. S7B), indicating that Ccr4–Not was not extending a K48 chain from the Rsp5-dependent monoubiquitin modification. These results suggest that Ccr4–Not is enhancing the ubiquitylation activity of Rsp5.
A potential mechanism for the Ccr4–Not-dependent enhanced ubiquitylation is that it binds to Rsp5 and recruits it to RNAPII. We first investigated whether Ccr4–Not and Rsp5 associate in cells by co-IP. Ccr4–Not was immunoprecipitated with Not4 antiserum from extracts of a Rsp5-13MYC strain, and Western blotting indeed revealed that Rsp5 binds to Ccr4–Not (Fig. 5B). However, this interaction was not dependent on DNA damage (Supplemental Fig. S5B). GST pull-down assays were performed using purified Ccr4–Not and GST-Rsp5 to provide additional support for the interaction and to test whether the binding is direct. The binding of Ccr4–Not to Rsp5 was detected by Western blotting for Not1 and Caf1, which revealed that Rsp5 directly interacts with Ccr4–Not (Fig. 5C).
Rsp5 binds to the C-terminal domain (CTD) of Rpb1, while Ccr4–Not binds to the Rpb4/7 module and the transcript (Wang et al. 1999; Somesh et al. 2007; Babbarwal et al. 2014). Since each factor recognizes distinct features of RNAPII, it is possible that Ccr4–Not and Rsp5 form a ternary complex with, and enhance the binding of each other to, the EC. Radiolabeled ECs were incubated with increasing amounts of Rsp5 with or without Ccr4–Not, and the complexes were resolved on native gels. An amount of Ccr4–Not was added to the assay to cause a shift of a fraction of the ECs so that additive or cooperative effects on complex formation when titrating in Rsp5 could be observed (see below). In the absence of Ccr4–Not, Rsp5 shifted a small amount of EC when added at a stoichiometry of 2:1 to polymerase, and nearly a complete shift of the EC was observed when the highest amount of Rsp5 was added (Fig. 5D, lanes 3,4). The Ccr4–Not–EC complex migrated to almost the same position in the gel as the Rsp5–EC band (Fig. 5D, lane 5) even though Ccr4–Not (∼1 MDa) is ∼10 times the mass of Rsp5 (92 kDa). Ccr4–Not binds to the structured portion of RNAPII, while Rsp5 binds as a dimer to the flexible extended CTD (Somesh et al. 2007; Babbarwal et al. 2014); thus, it is not surprising that the migration of these two binary complexes in native gels does not correspond to the mass of protein incorporated into the EC. The Ccr4–Not–EC binary complex was shifted to a new position by even the smallest amount of Rsp5 (Fig. 5D, lane 6), suggesting the formation of a larger complex containing Rsp5. Adding the maximum amount of Rsp5 caused a further shift of the band upward in the gel (Fig. 5D, lane 7 vs. 8), which may be a ternary complex binding two Rsp5 rather than one (Somesh et al. 2007).
Quantification of large complexes in native gels is a challenge because they do not migrate as sharp, distinct species. In an attempt to measure the effects of adding both Rsp5 and Ccr4–Not on their binding to the EC, we generated semiquantitative numbers by measuring the fraction of the EC complex remaining as a function of Rsp5 concentration. The graph produced from multiple experiments indicates that including Ccr4–Not in the binding reaction depleted the EC significantly more effectively than occurred in its absence. Taken together, the data above indicate that Ccr4–Not and Rsp5 form a ternary complex with, and influence the binding of, each other to the EC. These results suggest that Ccr4–Not can recruit Rsp5 to the EC.
The RING domain of Not4 is required for Rpb1 degradation in cells (Fig. 2). The stimulation of Rsp5 ubiquitylation by Ccr4–Not suggests that either Rsp5 binds to the RING domain or the RING domain brings the E2 Ubc4/5 to Rsp5. To discriminate between these two possibilities, we purified a version of the Ccr4–Not complex that contains a deletion of the RING domain of Not4 and analyzed its biochemical activities. The composition of the “ring-less” complex is shown in Figure 6A. Western blotting for Not4 revealed that an equal amount of Not4 was present in the wild-type and Δring mutant complexes (data not shown). A pull-down assay revealed that Rsp5 bound to the wild-type and mutant complexes equally well (Fig. 6B). Next, we performed a Rpb1 ubiquitylation assay using the wild-type and RING-less complexes. Representative gels from the assay (Fig. 6C) and the quantification of three experiments (Fig. 6D) are shown. The wild-type complex stimulated ubiquitylation twofold to threefold, depending on the amount of Rsp5. However, the RING-less complex failed to enhance ubiquitylation significantly. There was a slight but reproducible enhancement at the highest amount of Rsp5. Given that the role of the RING domain of Not4 is to recruit Ubc4/5 to substrates (Mulder et al. 2007b; Bhaskar et al. 2015), these results suggest that in addition to binding Rsp5, Ccr4–Not also delivers the E2 enzyme that it uses to ubiquitylate Rpb1.
Figure 6.

The RING domain of Not4 is required for Ccr4–Not to promote ubiquitylation. (A) Coomassie blue-stained gel of the ΔRING Ccr4–Not complex. Two quantities of the complex were loaded. The band migrating near 30 kDa is TEV protease that was used in the purification. (B) Pull-down assay using GST-Rsp5 and purified Ccr4–Not. Wild-type or a ΔRING version of Ccr4–Not was incubated in solution with GST-Rsp5 and then captured onto GSH beads. After washing, the beads were eluted with glutathione, and the bound proteins were analyzed by Western blotting using anti-Not1 and anti-Caf1 antibody. The membrane was cut to probe for Not1 and Caf1 independently and then reprobed with antibody recognizing GST. The splice in the membrane is indicated by a line. (C) Representative ubiquitylation assay gels comparing wild-type and a ΔRING mutant of Ccr4–Not. The conditions of the assay are described in the legend for Figure 5A. (D) Ubiquitylation assay comparing wild-type and a ΔRING mutant of Ccr4–Not. The conditions of the assay are described in the legend for Figure 5A. The Western signals were detected using a CY5-labeled secondary antibody. The signal intensities of modified Rpb1 were quantified, and the relative amounts were compared with the “no E3” lane (−), which was set to 1.0. The bars display the mean and standard deviation of three experiments. N = 3.
Not4 has functions distinct from Def1 and Rad26
Two pathways resolve RNAPII arrested over DNA lesions: the DEF1-dependent ubiquitin–proteasome pathway and the RAD26–TCR pathway. Rad26 belongs to the SWI/SNF2 helicase superfamily and plays an important role in TC-NER (Vangool et al. 1994). It is believed that Rad26 protects Rpb1 from degradation by moving it away from the lesion to allow access of the TC-NER machinery and by preventing Def1 from initiating ubiquitylation (Woudstra et al. 2002; Xu et al. 2017). We conducted genetic analyses to explore the relationship between NOT4 and the two pathways used to resolve arrested polymerase.
First, we attempted to determine whether NOT4 and DEF1 are epistatic for DNA damage resistance. However, we could not produce a double mutant, suggesting synthetic lethality between the two mutations. We confirmed synthetic lethality between the two genes using the plasmid shuffle technique; double mutants could not lose the URA3-marked plasmid expressing wild-type NOT4 in a not4Δ/Δdef1 background (Fig. 7A). RING domain mutants I64A and ΔRING could not rescue the lethality of the null mutation, indicating that the protein destruction function of Not4 is required to prevent lethality in combination with the def1Δ mutation. Therefore, even though Rpb1 degradation requires DEF1 and NOT4 and they reside in the same pathway, the genetic analysis described above suggests that they have other nonredundant functions, such as elongation (see the Discussion).
Figure 7.
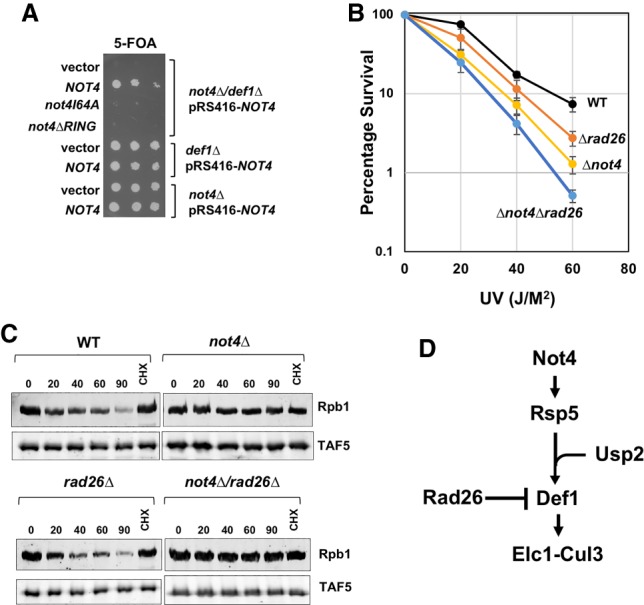
Genetic analysis of NOT4 in the Rpb1 destruction pathway. (A) Plasmid shuffle assay. Single or double DEF1 and/or NOT4 deletion strains containing pRS416-NOT4 (URA3 marked plasmid) were transformed with a LEU2-marked empty vector, one containing wild-type NOT4 or RING domain mutants. Strains were plated onto 5-FOA medium. (B) Epistasis analysis between NOT4 and RAD26. Each point represents the average and standard deviations of three biological replicates. (C) Rpb1 degradation in double mutants. The experiment was performed as described in Figure 1A. The lane labeled “CHX” indicates cells treated with cycloheximide only for 90 min. (D) Proposed role of Not4 in Rpb1 ubiquitylation.
Next, we looked for genetic interactions between RAD26 and NOT4. Deleting DEF1 enhances the UV sensitivity of rad26Δ mutants, presumably because both pathways used to resolve arrested polymerase are impaired (Woudstra et al. 2002). Both the rad26Δ and not4Δ single mutants displayed modest UV sensitivity, as reported previously (Vangool et al. 1994; Gaillard et al. 2009), and the double mutant exhibited lower resistance than the single mutants (Fig. 7B). This result provides additional genetic evidence that NOT4 plays a role in mediating the destruction of Rpb1. Rad26 antagonizes Def1-dependent Rpb1 degradation because deleting RAD26 suppressed the Rpb1 turnover defect in def1Δ cells (Woudstra et al. 2002). We next determined whether deleting RAD26 could likewise reverse the degradation defect in the not4Δ mutant. Deleting RAD26 increased the rate of Rpb1 turnover slightly compared with wild-type cells, as reported by others (Woudstra et al. 2002). However, deleting RAD26 did not suppress the Rpb1 degradation defect in the not4Δ cells (Fig. 7C). Therefore, even though both NOT4 and DEF1 are required for Rpb1 ubiquitylation and degradation, each acts at a different point in the Rpb1 destruction pathway. It is likely that NOT4 functions upstream of DEF1, which is consistent with our biochemical assays indicating that Ccr4–Not acts to promote Rsp5-dependent ubiquitylation of Rpb1 (Fig. 7D).
Discussion
Removal of “terminally” arrested RNAPII from DNA is essential for gene expression and genomic integrity. Failure to clear arrested polymerase prevents transcription and ultimately results in collisions between polymerase and the DNA replication machinery. RNAPII is removed by the ubiquitin–proteasome system, which is initiated by the conjugation of ubiquitin to Rpb1. Rsp5/NEDD4 was the first ubiquitin ligase identified to target Rpb1 for destruction >20 yr ago (Huibregtse et al. 1997). We now know that two E3 enzymes act in series (Rsp5/NEDD4 and Elc1–Cul3) to ubiquitylate Rpb1 (Harreman et al. 2009). The process is more complicated than anticipated, and many factors involved in degrading RNA polymerase have not been identified. For example, there is evidence that Asr1 and an unidentified ubiquitin ligase modify Rpb1 at telomeres and during DNA damage-independent arrest, respectively (Daulny et al. 2008; Karakasili et al. 2014; McCann et al. 2016). Moreover, what distinguishes terminally arrested from paused RNAPII and what triggers the binding of the ubiquitin conjugation machinery are not known. We make a case below that Ccr4–Not initiates the RNAPII destruction program by recruiting Rsp5 and helping to bring Ubc4/5 to the EC.
Ccr4–Not regulates Rpb1 degradation by an unexpected mechanism
Genetic studies have implicated Ccr4–Not in DNA damage responses. For example, mutating subunits of the complex causes sensitivity to DNA-damaging agents, altered checkpoint recovery, and, in the case of a not5Δ mutant, defective repair of DNA (Westmoreland et al. 2004; Traven et al. 2005; Manukyan et al. 2008; Gaillard et al. 2009). However, considering that Ccr4–Not controls so many steps in gene regulation, it was unclear whether it is directly involved in sensing damage or repair or whether it regulates the DNA damage response indirectly by controlling the production and turnover of mRNAs encoding repair factors. Moreover, Not4 has been implicated in multiple aspects of proteostasis, including regulating the assembly of the proteasome (Panasenko and Collart 2011). Given the known function of the proteasome in DNA damage responses, the action of Ccr4–Not could have been more distant and indirect.
We provide strong evidence that Ccr4–Not (specifically, the Not4 subunit) is required for DNA damage-dependent Rpb1 ubiquitylation. If the mutation of Not4 affected a “downstream” step such as proteasome activity or targeting, this would result in the accumulation of ubiquitylated Rpb1. On the contrary, not4Δ cells displayed reduced Rpb1 ubiquitylation. The most straightforward explanation for the function of Ccr4–Not in Rpb1 destruction would be that Not4 directly ubiquitylates Rpb1; however, this was not seen under the conditions used here. Instead, Ccr4–Not promoted Rsp5-dependent Rpb1 ubiquitylation, one of the earliest steps in the pathway. Genetically, this mechanism makes more sense. If Not4 directly ubiquitylated Rpb1 as part of a separate pathway, it would be hard to reconcile why Rpb1 ubiquitylation is abolished in Rsp5 or Elc1–Cul3 mutants and, likewise, why Rsp5/Elc1–Cul3 would not ubiquitylate Rpb1 in not4Δ cells. Finally, since deleting RAD26 did not suppress the Rpb1 degradation defect in not4Δ cells, as it does in def1Δ cells, this suggests that NOT4 acts upstream of DEF1 and Elc1–Cul3, precisely where Rsp5 lies in the pathway. Collectively, our data support a model in which Ccr4–Not acts early in the pathway, promoting the initial ubiquitylation of Rpb1 by Rsp5 (Fig. 7D). Although def1Δ and not4Δ mutations are synthetically lethal in combination, the lethality is probably caused by their redundant roles in promoting elongation. DEF1 and NOT4 mutants have common phenotypes, such as synthetic enhanced growth phenotypes in combination with a dst1Δ mutation (Woudstra et al. 2002; Dutta et al. 2015). Recently, Def1 was shown to “restart” RNAPII elongation after arrest (Damodaren et al. 2017). Therefore, both Ccr4–Not and Def1 reactivate arrested RNAPII, and synthetic lethality is most likely caused by inactivating two elongation pathways. If RNAPII is terminally arrested due to unrepairable damage, they then promote Rpb1 degradation.
One function of Ccr4–Not is to promote Rsp5 association with the EC. A recruitment mechanism is consistent with how these two factors recognize RNAPII. Ccr4–Not binds the EC by contacting Rpb4/7 and the transcript (Kruk et al. 2011; Babbarwal et al. 2014). On the other hand, Rsp5 binds to the CTD of Rpb1 (Somesh et al. 2007). This presents a scenario in which Ccr4–Not and Rsp5 associate with the EC simultaneously, which we observed in electrophoretic mobility shift assays (EMSAs) (Fig. 5). The direct interaction of Ccr4–Not with ECs and the requirement for nuclear accumulation of Not4 for Rpb1 degradation further support the idea that Ccr4–Not must bind to ECs to promote Rpb1 degradation. A tantalizing possibility is that Ccr4–Not could be part of the sensor that coordinates the activity of the degradation machinery at arrested RNAPII. Ccr4–Not binds to the nascent transcript in the EC (Kruk et al. 2011). One might predict that the transcript passes through Ccr4–Not during elongation. The arrest of RNAPII stops transcript movement, which could cause Ccr4–Not to engage or “clamp down” onto the RNA, causing a conformation change that signals the ubiquitylation of Rpb1. We did not observe a DNA damage-dependent association of Rsp5 and Not4 (Supplemental Fig. S5B), but both factors associate with RNAPII in the absence of damage (Huibregtse et al. 1997; Somesh et al. 2007; Kruk et al. 2011). Furthermore, as we show below, Not4 and Rsp5 likely collaborate to target additional substrates in the cell; thus, only a fraction of the pool of these proteins is involved in targeting Rpb1 under damage-inducing conditions.
Our results suggest that the mechanism is more involved than recruiting or stabilizing the association of Rsp5 with the EC. A RING-less mutant of the complex binds Rsp5 equally well yet does not stimulate ubiquitylation. The function of the RING domain of Not4 is to recruit the E2 Ubc4 (and Ubc5) to substrates, and mutations in this domain disrupt E2 binding (Albert et al. 2000, 2002; Mulder et al. 2007b). Ubc4/5 is the very same E2 used by Rsp5 to ubiquitylate Rpb1 (Somesh et al. 2005; Yasukawa et al. 2008; Harreman et al. 2009). The best explanation for these results is that the second function of Ccr4–Not is “delivery” of the E2 to Rsp5 (Fig. 8). Somesh et al. (2007) proposed that arresting RNAPII leads to a conformational change that positions Rsp5 and Ubc4/5 over the sites of ubiquitylation in Rpb1. Ccr4–Not likewise could direct the E2 to the correct location on polymerase, enhancing the ability of Rsp5 to target the ubiquitylation sites. Thus, Ccr4–Not plays multiple roles in mediating Rpb1 degradation, and there may be more yet to be discovered.
Figure 8.

Model of how Ccr4–Not promotes Rpb1 ubiquitylation. Ccr4–Not promotes the recruitment of Rsp5 (E3) and Ubc4 (E2) through its RING domain to the core of RNAPII to position the E2–E3 pair and direct the ubiquitylation of Rpb1.
The collaboration between Not4 and Rsp5 extends to other substrates and may represent a paradigm for how two ubiquitin ligase complexes function together. Other reported substrates of Not4 are the nascent polypeptide-associated complex subunits Egd1 and Egd2 (Panasenko et al. 2006). However, another group reported that the stress-dependent ubiquitylation and destruction of NAC are dependent on Rsp5 (Hiraishi et al. 2009). Both reports agree that NAC ubiquitylation requires Ubc4. This controversy can be explained if Not4 and Rsp5 work together to degrade NAC, similar to what we observed for Rpb1.
Ccr4–Not as a sensor and regulator of RNAPII elongation, TCR, and RNAPII destruction
As described above, deleting NOT5 and reducing Not4 protein levels reduces TCR (Gaillard et al. 2009). Preventing Rpb1 degradation in itself does not result in TCR defects. For example, a def1Δ mutant fails to destroy RNAPII yet carries out TCR. Therefore, the reduced ubiquitylation and destruction of RNAPII in NOT4 and NOT5 mutants does not account for the TCR defect in these cells; thus, Ccr4–Not does more than regulate Rpb1 degradation during the DNA damage response. One possibility is that Not4 ubiquitylates other proteins involved in resolving arrested polymerase or TCR. Such modifications do not have to trigger proteolysis but could serve as a signal for protein recruitment. Interestingly, proteomics of TFIIH-associated proteins identified subunits of the Ccr4–Not complex (Damodaren et al. 2017), suggesting that Ccr4–Not could affect TCR by regulating TFIIH activity. Finally, Ccr4–Not associates with the 19S proteasome (Supplemental Fig. S5; Laribee et al. 2007; Panasenko and Collart 2011). A function of the proteasome, which is independent of proteolysis, is promoting elongation and TCR (Russell et al. 1999; Ferdous et al. 2001; Gillette et al. 2001). Ccr4–Not's role in TCR may be to recruit the proteasome.
Ccr4–Not has multiple functions in elongation and DNA repair. It is reasonable to propose that Ccr4–Not may act as a sensor of the status of elongating RNAPII and serves as a platform or “hub” to recruit different factors to coordinate how arrested RNAPII is corrected. Ccr4–Not would use its antiarrest activity to resolve transient pausing of RNAPII under nonstress conditions and recruit TFIIS if polymerase backtracks (Kruk et al. 2011; Dutta et al. 2015). When polymerase encounters a bulky lesion, it can recruit the proteasome and TCR factors to repair the damage. If all else fails, it initiates the proteasome-dependent destruction program. Under this scenario, Ccr4–Not acts as a direct player and a coordinator of the pathways that resolve arrested RNAPII throughout the genome.
Materials and methods
Strains and media
The strains used in this study are listed in Supplemental Table S1. Cells were grown in YP medium (1% yeast extract, 2% peptone) supplemented with 0.02 mg/mL adenine sulfate and 2% dextrose. Yeast synthetic dropout medium supplemented with the appropriate amino acid/nucleotide mix and 2% dextrose was used to maintain selection when necessary. Gene deletion and epitope tagging were carried out by homologous recombination using PCR-generated cassettes (Longtine et al. 1998). NOT4 mutants were constructed using CRISPR–Cas9 (Laughery et al. 2015). Double mutants were isolated by mating single mutants of opposite mating types, sporulating, and tetrad analysis. The plasmid shuffle strain containing a double def1Δ1/not4Δ was constructed by transforming the heterologous diploid with pRS416-NOT4 followed by sporulation and tetrad dissection. The double mutant supported by the plasmid copy was then transformed with LEU2-based plasmids containing NOT4 and its mutant derivatives. Cells were grown in the absence of uracil and then plated onto 5-FOA plates. UV sensitivity was measured by plating cells onto YPD plates in triplicate and then treating with the indicated doses of UV. The percentage of viability was calculated after growth in the dark at 30°C.
Rpb1 degradation assay and analysis of Rpb1 ubiquitylation in vivo
Cells were grown in YPD at 30°C until an OD600 of 0.8–1.0 and then treated with cyclohexidine and 4-NQO at concentrations of 100 µg/mL and 5 µg/mL, respectively. Cells were harvested, and cell extracts were prepared by the TCA method (Miller et al. 2018). In the AA studies, cells were treated with 1 µg/mL rapamycin for 30 min prior to treatment with 4-NQO. Measurement of Rpb1 ubiquitylation using GST-Dsk2 beads was performed as described by others (Wilson et al. 2012). Details are in the Supplemental Material. Western signals were detected using CY5-labeled secondary antibodies, and images were captured on a Typhoon (GE Lifesciences). Quantification was performed using ImageJ software.
Protein purification
Twelve-subunit RNAPII was purified from 12 L of Rpb4-TAP strain using a modification to two procedures (Suh et al. 2005; Kaplan et al. 2008; Crickard and Reese 2019). The Ccr4–Not complex was purified from either a Not4-TAP or Not1-TAP strain with DST1 deleted, as described in a previous publication and detailed in the Supplemental Material (Kruk et al. 2011). The RING-less Ccr4–Not complex was purified through a TAP tag on the Not1 subunit in a mutant constructed by a CRISPR–Cas9 strategy. Recombinant Ubc5, Def1, GST-Rsp5, and GST-Dsk2 were expressed in Rosetta2 cells and purified. Details on protein purification are in the Supplemental Material.
In vitro ubiquitylation assay
The procedure was modified based on a previous study (Somesh et al. 2005). In a typical ubiquitylation reaction, 300 ng of purified RNAPII was incubated with 50 ng of GST-Ube1 (Boston Biochem, E-300), 50 ng of UbcH5c (Boston Biochem, E2-627) or recombinant yeast Ubc5, 100 ng of GST-Rsp5 or 1–2 µg of purified Ccr4–Not complex, 8 µg of ubiquitin (Boston Biochem), and 2.5 mM ATP in reaction buffer (25 mM Tris-HCl at pH 8.0, 125 mM NaCl, 3 mM MgCl2, 0.25 mM TCEP) for 1 h at 30°C. ECs were prepared as described (Kruk et al. 2011), with modifications. ECs were formed onto 100 ng of biotin-labeled EC70 template using 300 ng of purified RNAPII in 15 µL of transcription buffer (50 mM HEPES at pH 7.4, 100 mM KCl, 1 mM MnCl2, 0.3 mM UpG, 12% glycerol, 0.5 mM TCEP, 100 ng/µL BSA) for 5 min at room temperature. Transcription was initiated by adding an NTP mix without GTP to reach the final concentration of 0.5 mM ATP, 0.5 mM CTP, and 0.5 mM UTP for 30 min. One-hundred nanograms of denatured salmon sperm DNA was added to remove loosely bound RNAPII, and then the ECs were incubated with streptavidin agarose beads for 1 h. The beads were washed three times in wash buffer (20 mM HEPES at pH 7.5, 100 mM KCl, 100 ng/µL BSA, 0.02% NP-40, 0.5 mM TCEP) and three times in reaction buffer (25 mM Tris-HCl at pH 8.0, 125 mM NaCl, 3 mM MgCl2, 0.25 mM TCEP). Ubiquitylation reactions were fractionated on 5.5% SDS-PAGE gels or 6%–7% Tris-acetate gels. Blots were probed with 8WG16 monoclonal antibody (BioLegend) and Not4 antiserum.
EMSA of ECs
EMSA was carried out as described previously (Kruk et al. 2011; Babbarwal et al. 2014). Radiolabeled ECs were formed from 300 ng of purified RNAPII and 100 ng of EC70 DNA template, and 1.2 µg of Ccr4–Not complex was added and incubated for 5 min at room temperature. Two-hundred nanograms of yeast total RNA was added as the competitor. GST-Rsp5 (50, 100, or 300 ng) was added to the mixture and incubated for 5 min. Complexes were resolved on 4% native gel (acrylamide:bis-acrylamide = 39:1) for 6–9 h at 4°C with 200 V. The gel was dried and exposed to phosphoimager screen overnight.
GST pull-down assays
Five micrograms GST-Rsp5 or GST was incubated with 2.6 µg of purified Ccr4–Not complex in 30 µL of transcription buffer + 0.05% NP-40 (50 mM HEPES at pH 7.5, 100 mM KCl, 1 mM MnCl2, 0.5 mM DTT, 10% glycerol, 0.1 µg/µL BSA) for 1 h at room temperature. The mixture was incubated with 15 µL of glutathione sepharose (GSH) beads for 90 min at 4°C. GSH beads were washed three times in cold transcription buffer + 0.01% NP40 and eluted in SDS-loading buffer of with glutathione. Blots were probed with Not1 and Caf1 antiserum.
Immunoprecipitation
Log-phase cells were harvested, washed, and stored at −80°C. Cells were disrupted by bead beating using a BeadBlaster 24 (Benchmark Scientific) in lysis buffer (0.2 M Tris base, 0.39 M ammonium sulfate, 10 mM MgSO4, 1.0 mM EDTA, 20% glycerol, 1× Protease inhibitor cocktail at pH 7.9). The clarified lysate was dialyzed in dialysis buffer (20 mM HEPES, 10 mM MgOAc, 200 mM KOAc, 2 mM EGTA, 20% glycerol, 1× Protease inhibitor cocktail at pH 7.9) for 3–4 h at 4°C. Protein (0.75 mg) was diluted to 2 mg/mL using dialysis buffer and incubated with Not4 antiserum and protein-A sepharose overnight at 4°C. Beads were washed three times with immunoprecipitation wash buffer (dialysis buffer containing 0.03% NP-40) and once with NaCl immunoprecipitation wash buffer (25 mM Tris-HCL at pH 7.5 , 0.10 M NaCl, 2 mM EDTA, 10% glycerol, 0.03% NP-40). Samples were loaded onto 7.5% SDS-PAGE gel and transferred to membranes. Blots were probed with antimyc (BioLegend, 9E10) and Not4 antiserum. One-fifteenth of the input sample was loaded to the gel.
Supplementary Material
Acknowledgments
We thank Jesper Svejstrup, Martine Collart, and Thibault Mayor for plasmids used in this study. Diane Libert and Abhinaya Srikanth are acknowledged for pilot studies in the early phases of the work. Luke Creamer and Jeffrey Pfannenstein assisted in the supporting work and purified the GST-Rsp5 (C777A) mutant. The members of the Center for Eukaryotic Gene Regulation are recognized for their feedback and comments. This research was supported by funds from the National Institutes of Health (GM58672 to J.C.R.).
Author contributions: All authors conducted experiments and analyzed the data. H.J. and J.C.R. designed the experiments, interpreted the results, and wrote the paper.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.322453.118.
References
- Albert TK, Lemaire M, van Berkum NL, Gentz R, Collart MA, Timmers HTM. 2000. Isolation and characterization of human orthologs of yeast Ccr4–Not complex subunits. Nucleic Acids Res 28: 809–817. 10.1093/nar/28.3.809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert TK, Hanzawa H, Legtenberg YIA, de Ruwe MJ, van den Heuvel FAJ, Collart MA, Boelens R, Timmers HTM. 2002. Identification of a ubiquitin–protein ligase subunit within the CCR4–NOT transcription repressor complex. EMBO J 21: 355–364. 10.1093/emboj/21.3.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anindya R, Aygün O, Svejstrup JQ. 2007. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol Cell 28: 386–397. 10.1016/j.molcel.2007.10.008 [DOI] [PubMed] [Google Scholar]
- Azzouz N, Panasenko OO, Deluen C, Hsieh J, Theiler G, Collart MA. 2009. Specific roles for the Ccr4–Not complex subunits in expression of the genome. RNA 15: 377–383. 10.1261/rna.1348209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbarwal V, Fu J, Reese JC. 2014. The Rpb4/7 module of RNA polymerase II is required for carbon catabolite repressor protein 4-negative on TATA (Ccr4–Not) complex to promote elongation. J Biol Chem 289: 33125–33130. 10.1074/jbc.C114.601088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai YL, Salvadore C, Chiang YC, Collart MA, Liu HY, Denis CL. 1999. The CCR4 and CAF1 proteins of the CCR4–NOT complex are physically and functionally separated from NOT2, NOT4, and NOT5. Mol Cell Biol 19: 6642–6651. 10.1128/MCB.19.10.6642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J, Lim WA, Fraser JS, Frydman J, Krogan NJ. 2012. Systematic functional prioritization of protein posttranslational modifications. Cell 150: 413–425. 10.1016/j.cell.2012.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar V, Basquin J, Conti E. 2015. Architecture of the ubiquitylation module of the yeast Ccr4–Not complex. Structure 23: 921–928. 10.1016/j.str.2015.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HF, Sirupangi T, Wu ZH, Johnson DL, Laribee RN. 2018. The conserved RNA recognition motif and C3H1 domain of the Not4 ubiquitin ligase regulate in vivo ligase function. Sci Rep 8: 8163 10.1038/s41598-018-26576-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart MA. 2016. The Ccr4–Not complex is a key regulator of eukaryotic gene expression. Wiley Interdiscip Rev RNA 7: 438–454. 10.1002/wrna.1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KF, Scarnati MS, Krasley E, Mallory MJ, Jin CY, Law MJ, Strich R. 2012. Oxidative-stress-induced nuclear to cytoplasmic relocalization is required for Not4-dependent cyclin C destruction. J Cell Sci 125: 1015–1026. 10.1242/jcs.096479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crickard JB, Reese JC. 2019. Biochemical methods to characterize RNA polymerase II elongation complexes. Methods 10.1016/j.ymeth.2019.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damodaren N, Van Eeuwen T, Zamel J, Lin-Shiao E, Kalisman N, Murakami K. 2017. Def1 interacts with TFIIH and modulates RNA polymerase II transcription. Proc Natl Acad Sci 114: 13230–13235. 10.1073/pnas.1707955114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daulny A, Geng FQ, Muratani M, Geisinger JM, Salghetti SE, Tansey WP. 2008. Modulation of RNA polymerase II subunit composition by ubiquitylation. Proc Natl Acad Sci 105: 19649–19654. 10.1073/pnas.0809372105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Babbarwal V, Fu JH, Brunke-Reese D, Libert DM, Willis J, Reese JC. 2015. Ccr4–Not and TFIIS function cooperatively to rescue arrested RNA polymerase II. Mol Cell Biol 35: 1915–1925. 10.1128/MCB.00044-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdous A, Gonzalez F, Sun LP, Kodadek T, Johnston SA. 2001. The 19S regulatory particle of the proteasome is required for efficient transcription elongation by RNA polymerase II. Mol Cell 7: 981–991. 10.1016/S1097-2765(01)00250-7 [DOI] [PubMed] [Google Scholar]
- Gaillard H, Tous C, Botet J, González-Aguilera C, Quintero MJ, Viladevall L, García-Rubio ML, Rodríguez-Gil A, Marín A, Ariño J, et al. 2009. Genome-wide analysis of factors affecting transcription elongation and DNA repair: a new role for PAF and Ccr4–Not in transcription-coupled repair. PLoS Genet 5: e1000364 10.1371/journal.pgen.1000364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette TG, Huang W, Russell SJ, Reed SH, Johnston SA, Friedberg EC. 2001. The 19S complex of the proteasome regulates nucleotide excision repair in yeast. Genes Dev 15: 1528–1539. 10.1101/gad.869601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulshan K, Thommandru B, Moye-Rowley WS. 2012. Proteolytic degradation of the Yap1 transcription factor is regulated by subcellular localization and the E3 ubiquitin ligase Not4. J Biol Chem 287: 26796–26805. 10.1074/jbc.M112.384719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halter D, Collart MA, Panasenko OO. 2014. The Not4 E3 ligase and CCR4 deadenylase play distinct roles in protein quality control. PLos One 9: e86218 10.1371/journal.pone.0086218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G. 2008. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9: 958–970. 10.1038/nrm2549 [DOI] [PubMed] [Google Scholar]
- Harreman M, Taschner M, Sigurdsson S, Anindya R, Reid J, Somesh B, Kong SE, Banks CAS, Conaway RC, Conaway JW, et al. 2009. Distinct ubiquitin ligases act sequentially for RNA polymerase II polyubiquitylation. Proc Natl Acad Sci 106: 20705–20710. 10.1073/pnas.0907052106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. 2008. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31: 925–932. 10.1016/j.molcel.2008.07.020 [DOI] [PubMed] [Google Scholar]
- Hiraishi H, Shimada T, Ohtsu I, Sato TA, Takagi H. 2009. The yeast ubiquitin ligase Rsp5 downregulates the α subunit of nascent polypeptide-associated complex Egd2 under stress conditions. FEBS J 276: 5287–5297. 10.1111/j.1742-4658.2009.07226.x [DOI] [PubMed] [Google Scholar]
- Huibregtse JM, Yang JC, Beaudenon SL. 1997. The large subunit of RNA polymerase II is a substrate of the Rsp5 ubiquitin-protein ligase. Proc Natl Acad Sci 94: 3656–3661. 10.1073/pnas.94.8.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Lis JT. 2015. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 16: 167–177. 10.1038/nrm3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CD, Larsson KM, Kornberg RD. 2008. The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by α-amanitin. Mol Cell 30: 547–556. 10.1016/j.molcel.2008.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakasili E, Burkert-Kautzsch C, Kieser A, Sträßer K. 2014. Degradation of DNA damage-independently stalled RNA polymerase II is independent of the E3 ligase Elc1. Nucleic Acids Res 42: 10503–10515. 10.1093/nar/gku731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruk JA, Dutta A, Fu J, Gilmour DS, Reese JC. 2011. The multifunctional Ccr4–Not complex directly promotes transcription elongation. Genes Dev 25: 581–593. 10.1101/gad.2020911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laribee RN, Shibata Y, Mersman DP, Collins SR, Kemmeren P, Roguev A, Weissman JS, Briggs SD, Krogan NJ, Strahl BD. 2007. CCR4/NOT complex associates with the proteasome and regulates histone methylation. Proc Natl Acad Sci 104: 5836–5841. 10.1073/pnas.0607996104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughery MF, Hunter T, Brown A, Hoopes J, Ostbye T, Shumaker T, Wyrick JJ. 2015. New vectors for simple and streamlined CRISPR–Cas9 genome editing in Saccharomyces cerevisiae. Yeast 32: 711–720. 10.1002/yea.3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- Manukyan A, Zhang J, Thippeswamy U, Yang JY, Zavala N, Mudannayake MP, Asmussen M, Schneider C, Schneider BL. 2008. Ccr4 alters cell size in yeast by modulating the timing of CLN1 and CLN2 expression. Genetics 179: 345–357. 10.1534/genetics.108.086744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann TS, Guo Y, McDonald WH, Tansey WP. 2016. Antagonistic roles for the ubiquitin ligase Asr1 and the ubiquitin-specific protease Ubp3 in subtelomeric gene silencing. Proc Natl Acad Sci 113: 1309–1314. 10.1073/pnas.1518375113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersman DP, Du HN, Fingerman IM, South PF, Briggs SD. 2009. Polyubiquitination of the demethylase Jhd2 controls histone methylation and gene expression. Genes Dev 23: 951–962. 10.1101/gad.1769209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JE, Reese JC. 2012. Ccr4–Not complex: the control freak of eukaryotic cells. Crit Rev Biochem Mol Biol 47: 315–333. 10.3109/10409238.2012.667214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JE, Zhang LY, Jiang HY, Li YF, Pugh BF, Reese JC. 2018. Genome-wide mapping of decay factor-mRNA interactions in yeast identifies nutrient-responsive transcripts as targets of the deadenylase Ccr4. G3 8: 315–330. 10.1534/g3.117.300415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LS, Wei W, Stolovicki E, Benbenishty T, Wilkening S, Steinmetz LM, Braun E, David L. 2014. Induced mutations in yeast cell populations adapting to an unforeseen challenge. PLos One 9: e111133 10.1371/journal.pone.0111133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder KW, Brenkman AB, Inagaki A, van den Broek NJF, Timmers HTM. 2007a. Regulation of histone H3K4 tri-methylation and PAF complex recruitment by the Ccr4–Not complex. Nucleic Acids Res 35: 2428–2439. 10.1093/nar/gkm175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder KW, Inagaki A, Cameroni E, Mousson F, Winkler GS, De Virgilio C, Collart MA, Timmers HTM. 2007b. Modulation of Ubc4p/Ubc5p-mediated stress responses by the RING-finger-dependent ubiquitin-protein ligase Not4p in Saccharomyces cerevisiae. Genetics 176: 181–192. 10.1534/genetics.106.060640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panasenko OO, Collart MA. 2011. Not4 E3 ligase contributes to proteasome assembly and functional integrity in part through Ecm29. Mol Cell Biol 31: 1610–1623. 10.1128/MCB.01210-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panasenko OO, Collart MA. 2012. Presence of Not5 and ubiquitinated Rps7A in polysome fractions depends upon the Not4 E3 ligase. Mol Microbiol 83: 640–653. 10.1111/j.1365-2958.2011.07957.x [DOI] [PubMed] [Google Scholar]
- Panasenko O, Landrieux E, Feuermann M, Finka A, Paquet N, Collart MA. 2006. The yeast Ccr4–Not complex controls ubiquitination of the nascent-associated polypeptide (NAC–EGD) complex. J Biol Chem 281: 31389–31398. 10.1074/jbc.M604986200 [DOI] [PubMed] [Google Scholar]
- Reese JC. 2013. The control of elongation by the yeast Ccr4–Not complex. Biochim Biophys Acta 1829: 127–133. 10.1016/j.bbagrm.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid J, Svejstrup JQ. 2004. DNA damage-induced Def1-RNA polymerase II interaction and Def1 requirement for polymerase ubiquitylation in vitro. J Biol Chem 279: 29875–29878. 10.1074/jbc.C400185200 [DOI] [PubMed] [Google Scholar]
- Ribar B, Prakash L, Prakash S. 2006. Requirement of ELC1 for RNA polymerase II polyubiquitylation and degradation in response to DNA damage in Saccharomyces cerevisiae. Mol Cell Biol 26: 3999–4005. 10.1128/MCB.00293-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribar B, Prakash L, Prakash S. 2007. ELA1 and CUL3 are required along with ELC1 for RNA polymerase II polyubiquitylation and degradation in DNA-damaged yeast cells. Mol Cell Biol 27: 3211–3216. 10.1128/MCB.00091-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SJ, Reed SH, Huang WY, Friedberg EC, Johnston SA. 1999. The 19S regulatory complex of the proteasome functions independently of proteolysis in nucleotide excision repair. Mol Cell 3: 687–695. 10.1016/S1097-2765(01)80001-0 [DOI] [PubMed] [Google Scholar]
- Somesh BP, Reid J, Liu W-F, Søgaard TMM, Erdjument-Bromage H, Tempst P, Svejstrup JQ. 2005. Multiple mechanisms confining RNA polymerase II ubiquitylation to polymerases undergoing transcriptional arrest. Cell 121: 913–923. 10.1016/j.cell.2005.04.010 [DOI] [PubMed] [Google Scholar]
- Somesh BP, Sigurdsson S, Saeki H, Erdjument-Bromage H, Tempst P, Svejstrup JQ. 2007. Communication between distant sites in RNA polymerase II through ubiquitylation factors and the polymerase CTD. Cell 129: 57–68. 10.1016/j.cell.2007.01.046 [DOI] [PubMed] [Google Scholar]
- Stern S, Dror T, Stolovicki E, Brenner N, Braun E. 2007. Genome-wide transcriptional plasticity underlies cellular adaptation to novel challenge. Mol Syst Biol 3: 106 10.1038/msb4100147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh MH, Ye P, Zhang MC, Hausmann S, Shuman S, Gnatt AL, Fu JH. 2005. Fcp1 directly recognizes the C-terminal domain (CTD) and interacts with a site on RNA polymerase II distinct from the CTD. Proc Natl Acad Sci 102: 17314–17319. 10.1073/pnas.0507987102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traven A, Hammet A, Tenis N, Denis CL, Heierhorst J. 2005. Ccr4–Not complex mRNA deadenylase activity contributes to DNA damage responses in Saccharomyces cerevisiae. Genetics 169: 65–75. 10.1534/genetics.104.030940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M, Valencia-Sanchez MA, Staples RR, Chen JJ, Denis CL, Parker R. 2001. The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell 104: 377–386. 10.1016/S0092-8674(01)00225-2 [DOI] [PubMed] [Google Scholar]
- Vangool AJ, Verhage R, Swagemakers SMA, Vandeputte P, Brouwer J, Troelstra C, Bootsma D, Hoeijmakers JHJ. 1994. Rad26, the functional Saccharomyces-cerevisiae homolog of the Cockayne-syndrome-B gene Ercc6. EMBO J 13: 5361–5369. 10.1002/j.1460-2075.1994.tb06871.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Oania R, Fang RH, Smith GT, Deshaies RJ. 2011. Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol Cell 41: 82–92. 10.1016/j.molcel.2010.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Yang J, Huibregtse JM. 1999. Functional domains of the Rsp5 ubiquitin-protein ligase. Mol Cell Biol 19: 342–352. 10.1128/MCB.19.1.342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmoreland TJ, Marks JR, Olson JA, Thompson EM, Resnick MA, Bennett CB. 2004. Cell cycle progression in G1 and S phases is CCR4 dependent following ionizing radiation or replication stress in Saccharomyces cerevisiae. Eukaryot Cell 3: 430–446. 10.1128/EC.3.2.430-446.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Saponaro M, Leidl MA, Svejstrup JQ. 2012. MultiDsk: a ubiquitin-specific affinity resin. PLos One 7: e46398 10.1371/journal.pone.0046398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Harreman M, Svejstrup JQ. 2013a. Ubiquitylation and degradation of elongating RNA polymerase II: the last resort. Biochim Biophys Acta 1829: 151–157. 10.1016/j.bbagrm.2012.08.002 [DOI] [PubMed] [Google Scholar]
- Wilson MD, Harreman M, Taschner M, Reid J, Walker J, Erdjument-Bromage H, Tempst P, Svejstrup JQ. 2013b. Proteasome-mediated processing of Def1, a critical step in the cellular response to transcription stress. Cell 154: 983–995. 10.1016/j.cell.2013.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolstencroft RN, Beilharz TH, Cook MA, Preiss T, Durocher D, Tyers M. 2006. Ccr4 contributes to tolerance of replication stress through control of CRT1 mRNA poly(A) tail length. J Cell Sci 119: 5178–5192. 10.1242/jcs.03221 [DOI] [PubMed] [Google Scholar]
- Woudstra EC, Gilbert C, Fellows J, Jansen L, Brouwer J, Erdjument-Bromage H, Tempst P, Svejstrup JQ. 2002. A Rad26–Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature 415: 929–933. 10.1038/415929a [DOI] [PubMed] [Google Scholar]
- Xu J, Lahiri I, Wang W, Wier A, Cianfrocco MA, Chong J, Hare AA, Dervan PB, DiMaio F, Leschziner AE, et al. 2017. Structural basis for the initiation of eukaryotic transcription-coupled DNA repair. Nature 551: 653–657. 10.1038/nature24658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa T, Kamura T, Kitajima S, Conaway RC, Conaway JW, Aso T. 2008. Mammalian Elongin A complex mediates DNA-damage-induced ubiquitylation and degradation of Rpb1. EMBO J 27: 3256–3266. 10.1038/emboj.2008.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.