

Abstract
Drug-induced compensatory signaling and subsequent rewiring of the signaling pathways that support cell proliferation and survival promotes the development of acquired drug resistance in tumors. Here, we sought to analyze the adaptive kinase response in cancer cells after distinct treatment with agents targeting human epidermal growth factor receptor 2 (HER2), specifically those which induce only temporary cell cycle arrest or apoptosis in HER2-overexpressing cancers. We compared trastuzumab, ARRY-380, the combination thereof, and a biparatopic HER2-targeted designed ankyrin repeat protein (DARPin; specifically 61LG) and quantified the phosphoproteome by isobaric tagging using tandem mass tag liquid chromatography/mass spectrometry (TMT LC-MS/MS). We found a specific signature of persistently phosphorylated tyrosine peptides after the non-apoptotic treatments, which we used to distinguish between different treatment-induced cancer cell fates. Next, we analyzed the activation of serine/threonine and tyrosine kinases after treatment using a bait peptide chip array and predicted the corresponding active kinases. Through a combined system-wide analysis, we identified a common adaptive kinase-response program that involved the activation of focal adhesion kinase (FAK1), protein kinase C-δ (PKCδ), and Ephrin (EPH) family receptors. The findings reveal potential targets to prevent adaptive resistance to HER2-targeted therapies.
INTRODUCTION
Mutations leading to increased phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway signaling represent the most frequent mode of acquired cancer drug resistance in late-stage human epidermal growth factor 2 (HER2, ERBB2)-positive cancers (1–3). On the other hand, a PI3K wild-type, HER2-positive cancer (HER2-dependent) relies similarly on the integrity of an active HER2/HER3/PI3K/AKT pathway (4), and HER2-targeted therapeutics (such as trastuzumab, pertuzumab, lapatinib or ARRY-380) block this signaling cascade at least transiently (5–9). This transient nature of signaling inhibition revealed the control function of negative feedbacks, which result in the reactivation of either the same pathway (10, 11) or of alternate signaling pathways that compensate for the perturbation by the inhibitor (12–15). The resulting adaptive kinase response, constituted by these stabilizing signaling relays, can be activated rapidly and reversibly (8, 16), which indicates that they are potentially suppressed by the primary oncogene in the cancer host network. Thus the inhibition of the primary oncogene by the targeted inhibitor inevitably relieves negative feedbacks and consequently causes activation of other kinases (17), which may prevent the induction of apoptosis and, thus, facilitate the development of acquired cancer drug resistance.
Previously, we developed a new class of biparatopic HER2-binding agents, called biparatopic DARPins (such as DARPin 6L1G), which blocked productive HER2 receptor homodimer and heterodimer interactions and, consequently, induced apoptosis in various HER2-dependent breast cancer models (7, 18).Similar to the trastuzumab-plus-lapatinib or trastuzumab-plus-ARRY-380 combination treatments, these biparatopic agents can overcome adaptive kinase-response signaling and effectively induce apoptosis in HER2-dependent cancer cells, which confirmed the existence of HER2 oncogene addiction and revealed the need to overcome it through simultaneous blockade of signaling from both HER2 and HER3 (7).
Through comparative analysis of distinct HER2-targeted (hereafter, anti-HER2) treatments with different effects on the cancer cell fate, one can assess the specific modes of the adaptive kinase response signaling in a time-dependent manner. Recent large-scale studies have illuminated the vulnerabilities and the adaptive responses across various HER2-positive cancer models (13, 19–22). Here, however, we focused on the analysis of a single HER2-dependent model after the mechanistically different anti-HER2 treatments, which induce either only transient cell-cycle arrest in G1 or apoptosis (5, 7, 23). This enabled us to directly compare the phosphostatus of the Tyr kinome in cancer cells undergoing treatment leading to two different outcomes: temporary cell cycle arrest and likely acquired drug resistance, or induction of apoptosis, representing effective cancer treatment. Using quantitative phospho-proteomics and kinase activity profiling, we generated a reference network of all HER2-dependent Tyr kinase signaling events after complete HER2 inhibition (HER2 “off-state” network). Next, we compared the adaptive Tyr and Ser/Thr kinase activation in a system-wide functional protein association network upon the different treatments and identified a group of kinases that followed a common activation program after the different anti-HER2 treatments. Finally, we identified focal adhesion kinase 1 (FAK1, PTK2) as a major signaling hub in this compensatory kinase signaling network upon HER2 inhibition, and we found that specific combination treatments blocked its feedback activation and, consequently, led to substantially increased induction of cancer cell death.
RESULTS
Experimental setup for quantitative pathway profiling
To dynamically monitor pathway activity after applying the anti-HER2 treatments trastuzumab (TZB), ARRY-380 (ARRY), the combination of both agents (ARRY-380 + trastuzumab, A+T) and the biparatopic DARPin (6L1G) in a HER2-dependent breast cancer cell model (the BT474 cell line in 2-dimensional culture), we performed two orthogonal and complementary quantitative large-scale studies (Fig. 1A). First, we measured peptide tryrosine (Tyr) phosphorylation after enrichment by triple antibody pulldown, immobilized metal affinity chromatography and multiplexed TMT LC-MS/MS (24). We manually verified 471 unique phospho-Tyr (p-Tyr) peptides from two biological replicates (table S1). Next, we used the commercial kinase activity-profiling array from PamGene to monitor the phosphorylation of 121 Tyr (table S2) bait peptides and 77 Ser/Thr (table S3) bait peptides in biological quadruplicates of ATP-spiked cell extracts in the absence of phosphatase activity (25). The corresponding kinases were subsequently predicted by the group-based prediction system. Finally, cognate kinase activities were combined with the measured target phospho-status from the TMT dataset in a high-confidence functional protein-protein interaction network to track potential resistance driving pathways (Fig. 1A).
Fig. 1. Quantitative effects and patterns in the phospho-Tyr kinome in BT474 cells after anti-HER2 treatments by TMT LC-MS/MS.
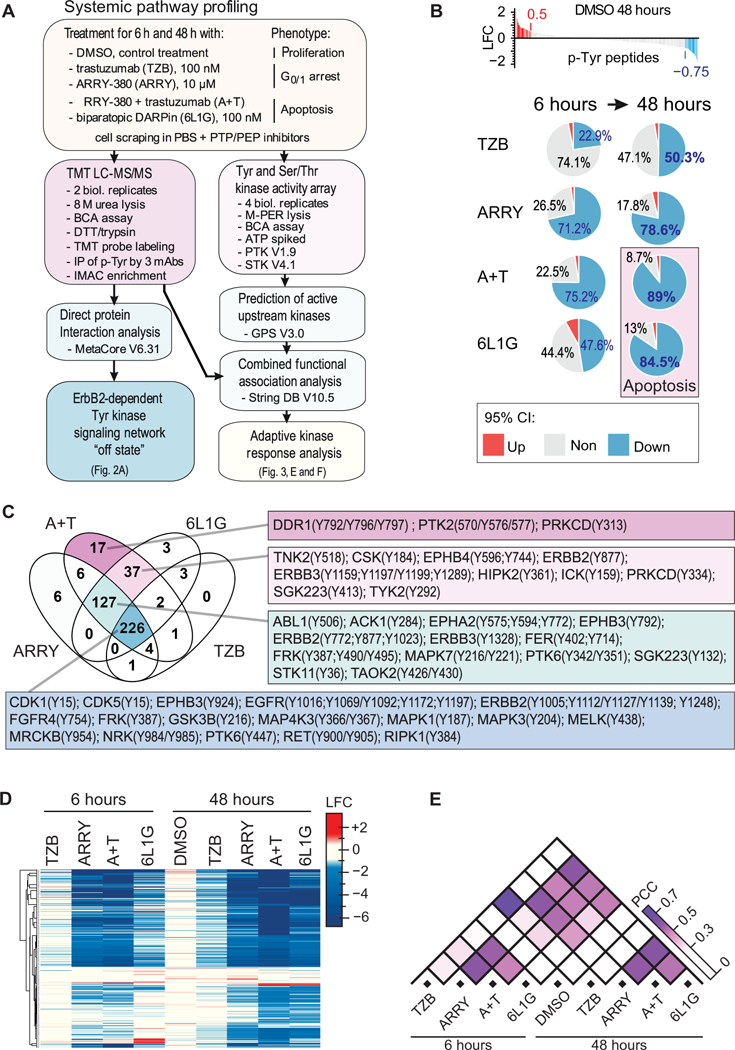
(A) Study design, sample preparation, and dataset analysis. (B) Global changes of Tyr peptide phosphorylation in BT474 cells after 6 and 48 hours of continuous treatment with 100 nM trastuzumab (TZB), 10 μM ARRY-380 (ARRY), the combination of ARRY-380 (10 μM) and trastuzumab (100 nM) (A+T), or 100 nM biparatopic DARPin (6L1G), as determined by TMT LC-MS/MS. 95% confidence interval (CI) of increase, decrease, or no change was based on the two-fold standard deviation of the data from the 48-hour DMSO treatment and used to determine upper and lower log2 fold-change ratio (LFC) thresholds. Plots are based on mean values of two biological replicates. (C) Venn diagram of the fractions of p-Tyr peptides identified in (B) of which the abundance was reduced below the threshold (95 % CI) after 48 hours of the indicated treatment. Linked tables list the associated kinases and specific Tyr (Y) phosphosites. (D and E) Pearson clustering of 471 unique p-Tyr peptides from the TMT LC-MS/MS dataset (D) and pairwise comparison of LFC values computed by absolute Pearson correlation coefficient (PCC) (E). Data are analyzed from the 2 biological replicates described in (B).
Inhibition of peptide Tyr phosphorylation after anti-HER2 treatments
The mean signal intensity of each p-Tyr peptide was normalized to the corresponding mean signal intensity after 6 hours’ exposure to DMSO control treatment and expressed as log2 fold-change ratio (LFC). To determine an upper and lower threshold for treatment-induced changes in Tyr phosphorylation, we introduced a 95% confidence interval (CI), which was based on the distribution of LFC values in the DMSO control treatment after 48 hours (Fig. 1B). We observed a globally progressive reduction in peptide tyrosine phosphorylation below the lower threshold (LFC of −0.75) from 6 to 48 hours in each of the anti-HER2 treatments. Treatments that were apoptotic, inferred from the detection of cleaved poly-ADP-ribose polymerase (PARP; A+T and 6L1G; fig. S1A) reduced the abundance of p-Tyr peptides after 48 hours more than the non-apoptotic ARRY-380 or trastuzumab single-agent treatments (fig. S1A). Thus, the antiproliferative effect of these anti-HER2 treatments is mirrored by an overall inhibition of Tyr kinase activity.
Next, we compared the fractions of p-Tyr peptides with reduced abundance between the anti-HER2 treatments after 48 hours by Venn diagram analysis (Fig. 1C) and observed that the A+T combination treatment includes quantitatively all of the reduced p-Tyr peptides of the other anti-HER2 treatments. In fact, the different anti-HER2 treatments blocked the Tyr phosphorylation after 48 hours according to the onion principle: A+T included the 6L1G inhibition pattern, which included the ARRY inhibition pattern, which finally included the TZB inhibition pattern quantitatively. Thus, our data suggest that the strength of the anti-proliferative effect of these different anti-HER2 treatments is a function of inactivating incrementally more Tyr kinases after 48 hours, and both apoptotic treatments (A+T and 6L1G) blocked activity in an apparently sufficiently large fraction of Tyr kinases to suppress compensatory signaling.
Treatment-specific patterns in Tyr-phosphorylation
To identify treatment-specific patterns in the p-Tyr signaling, we performed Pearson clustering and pairwise cross-correlation on the LFC values of the 471 unique p-Tyr peptides found by MS analysis (Fig. 1, D and E). ARRY, A+T and 6L1G induced overall strong and very similar inhibition of the Tyr phosphorylation after 48 hours with a high positive Pearson correlation coefficient (PCC). Furthermore, the responses to the ARRY and A+T treatments were highly similar at the 6-hour treatment timepoint, which mostly reflects the rapid and sustained inhibition of HER2 by the small molecule kinase inhibitor. However, we have previously shown that the same concentration of ARRY induces only a cell cycle arrest in BT474 cells, whereas A+T and 6L1G induces apoptosis (7). Here, we analyzed PARP cleavage by western blot as a measure of apoptosis in BT474 cells after all treatments (fig. S1A) and observed specific PARP cleavage only after 48-hours’ exposure to A+T and 6L1G.
Next, we observed that the overall inhibition pattern of the Tyr phosphorylation is highly indicative of the mechanism of the drug but not of the drug-induced cancer cell fate — meaning cell cycle arrest vs. apoptosis. On the other hand, trastuzumab showed only a moderate inhibition profile and a relatively weak positive PPC with the other anti-HER2 treatments, which reflects the fact that it only blocked the phosphorylation of HER3, but did not affect the phosphorylation of HER2, which can continue signaling (5, 7). Furthermore, whereas TZB and 6L1G both decreased Tyr phosphorylation over 6 to 48 hours of treatment, signaling inhibition after 48 hours in 6L1G was ultimately greater than that after TZB and almost identical to that after the combination treatment (A+T) at the same 48-hour timepoint. Therefore, we conclude that after 48 hours the quantitative inhibition after the A+T and 6L1G treatment of the HER2/HER3 receptor signaling induced the signaling “off-state” in these HER2-dependent cancer cells, which consequently led to the induction of PARP cleavage and, presumably, apoptosis (fig. S1A). Because we observed rather small quantitative differences in Tyr kinase inhibition between the ARRY and both apoptosis-inducing treatments after 48 hours’ exposure, we propose that only a small fraction of active Tyr kinases may potentially mediate escape from apoptosis after the ARRY treatment.
HER2-dependent p-Tyr signaling network
The relative importance of a particular kinase to drive cancer cell signaling depends on its biochemical wiring (17), and kinases with the highest degree of connectivity are thus potentially the most effective drug targets (26). We hypothesized that after treatment-induced cancer remodeling, active kinases with the highest degree of connectivity are probably also the most relevant mediators of the adaptive cancer drug resistance. Thus, in addition to simple intensity-based kinase activity grading, we implemented a network topology-based approach for the analysis of the adaptive kinome response.
For this purpose, we assessed the degree of connectivity based on a “direct protein interaction” network, which was obtained by feeding the corresponding search algorithm from the MetaCore database (27) with the p-Tyr peptides with significantly reduced abundance, observed after the apoptotic combination treatment (A+T) after 48 hours. Thus, this network represents the “off-state” of a HER2-dependent cell (Fig. 2A). Based on the export of this direct protein interaction network, we analyzed only those protein pairs involved in binding and phosphorylation events in the subsequent exploratory analysis in Cytoscape software. For interpretation of this HER2-dependent signaling network in the “off-state”, we analyzed the overall network topology by community clustering using ClusterMaker2.0 and the Glay algorithm in Cytoscape (28). We identified 5 major communities, which contained the following main signaling hubs: (i) the epidermal growth factor receptors 1, 2 and 3 (EGFR, HER2 and HER3); (ii) phosphatidylinositol 3-kinase regulatory subunit alpha and beta (PIK3R½); tyrosine-protein phosphatase non-receptor type 11 (PTPN11), GRB2-associated-binding protein 1 (GAB1); (iii) Abelson murine leukemia viral oncogene homolog 1 (ABL1), glycogen synthase kinase-3 beta (GSK3B), and cyclin-dependent-like kinase 5 (CDK5); (iv) mitogen-activated protein kinase 1 and 3 (MAPK1/3), PKCδ (PRKCD), and heat shock protein alpha (HSP90), (v) PTK2 (FAK1), integrin beta-1 (ITGB1) and cyclin-dependent-like kinase 1 (CDK1) (Fig. 2, A and B). These signaling hubs have been previously identified by various experimental approaches (13, 15, 19, 20, 22, 29); however, here we performed a combined data-driven analysis of their relative importance in our HER2-dependent model.
Fig. 2. HER2-dependent “off-state” network and feedback activation of Tyr kinases in BT474 cells by TMT LC-MS/MS.
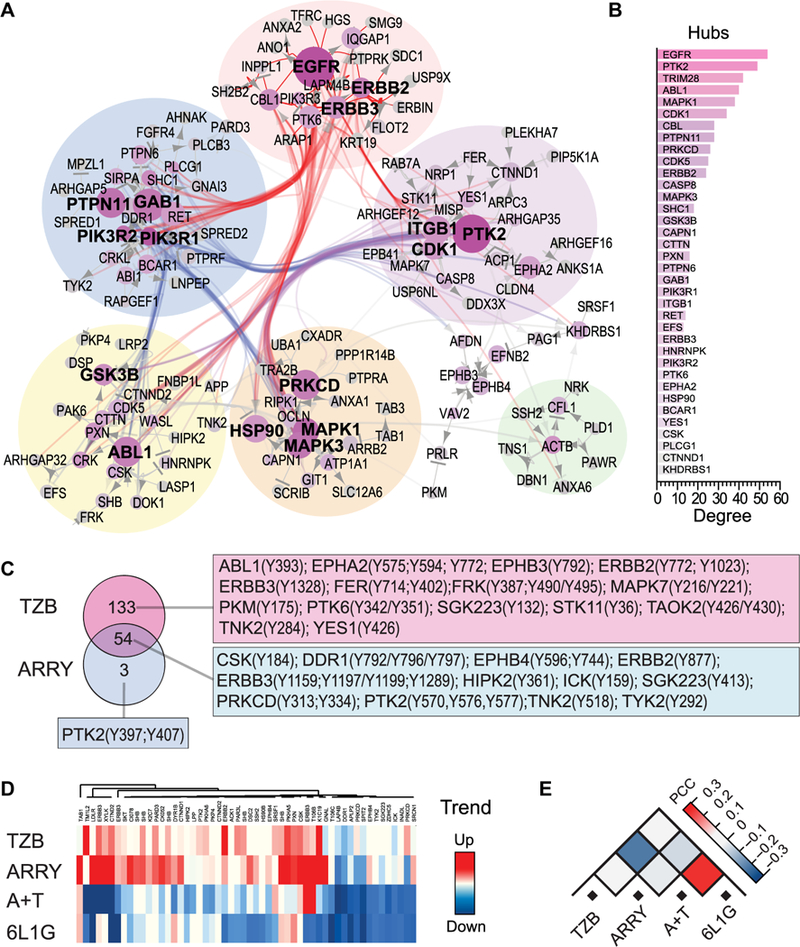
(A and B) Direct protein interaction network derived from a search of the MetaCore database in which p-Tyr peptide phosphorylation was significantly inhibited (below 95 % CI) after the 48-hour A+T treatment (Fig. 1B). Node (“hub”) size indicates number of connections (edges) to other nodes (degree) or proteins, as determined by the MetaCore algorithm (n=2 biological replicates) and edges represent known binding, phosphorylation or dephosphorylation events between both proteins. (C) Comparison of the fractions of persistently phosphorylated Tyr peptides (all which are not downregulated) after 48 hours of treatment, arranged by Venn diagram and tables of the associated kinases. A consensus of 54 p-Tyr peptides was identified after TZB_48h and ARRY_48h treatment (n=2 biological replicates). (D) Heat map of time-dependent Tyr phosphorylation trends of the 54 consensus peptides from Fig. 2C. Trends were calculated from the slope of a simple linear regression on LFC values from 6 to 48 hours of treatment based on the mean of two biological replicates. (E) Pairwise comparison of Tyr phosphorylation trends computed by absolute PCC (n=2 biological replicates).
In terms of network topology and a rational combination treatment (26), we would expect the removal or complete inhibition of the two largest hubs, EGFR and PTK2 (FAK1), to result in the highest probability of a system failure, i.e., a complete tyrosine signaling inhibition. However, the available representation of this Tyr kinase network is currently limited by the number of curated protein-protein interactions from the database, which results, for example, in the higher number of connections of EGFR over HER2 in this particular HER2-overexpressing model and therefore may not exactly reflect the underlying cancer signaling network (26). Thus, for high-accuracy prediction of the adaptive kinase response, we decided to further refine our model by implementing a second orthogonal large-scale profiling of kinase activities (tested below).
Tyr kinase phosphorylation after anti-HER2 treatments
Increased Tyr phosphorylation upon inhibition of the main driving oncogene may either originate from increased autocatalytic activity or from increased upstream kinase activity and, henceforth, indicates release of a negative feedback loop (an adaptive response) (12, 16, 17). Therefore, we compared the fraction of the p-Tyr peptides whose phosphorylation level increases or at least stays constant after 48 hours of treatment between the non-apoptotic single-agent treatments ARRY and TZB by Venn diagram analysis (Fig. 2C). The fraction of persistent phosphorylated Tyr peptides after the TZB treatment comprised almost all persistent phosphorylated Tyr peptides from the ARRY treatment after 48 hours. We identified a consensus of 54 persistent phosphorylated Tyr peptides from 46 different proteins between the two non-apoptotic treatments. This p-Tyr peptide signature included peptides from PTK2 (FAK1), paxillin (PXN), PRKCD, C-terminal Src kinase (CSK), TGF-beta-activated kinase 1- binding protein 1 (TAB1), SH2 domain-containing adapter protein B (SHB), Catenin delta-1 (CTNND1), Ephrin type-B receptor 4 (EPHB4), homeodomain-interacting protein kinase 2 (HIPK2) and EGFR family members (fig. S1C) and we analyzed their interaction (fig. S1D) and phosphorylation over time (trends) from 6 to 48 hours (Fig. 2D). Most of these p-Tyr peptides showed a positive trend of increasing abundance after the non-apoptotic treatments and, in contrast, a negative trend of decreasing phosphorylation abundance after the apoptotic treatments.
Therefore, we found that the overall time-dependent phosphorylation trend of this 54 p-Tyr-peptide signature is indicative of the treatment-induced cancer cell fate in our model, meaning mere cell cycle arrest or apoptosis. Consequently, the trends showed also a negative PCC between the treatments TZB and A+T, as well as between ARRY and A+T (Fig. 2E), which was previously hidden in the overall positive PCC of the global analysis of the 471 p-Tyr peptides (Fig. 1E). Thus, the phosphorylation trend of this fraction of p-Tyr peptides, which are apparently associated with the adaptive kinase response signaling, allowed us to distinguish with higher accuracy between apoptotic and non-apoptotic cell fate. In other words, the increased phosphorylation of this p-Tyr peptide signature appears to be associated with the adaptive cancer drug resistance.
Kinase activity profiling by peptide chip array
To measure the Tyr- and Ser/Thr-kinase activity in an orthogonal and complementary manner to the TMT approach, we used the peptide chip array from PamGene with a common peptide setup of specific phosphorylation motifs (30). Again, the mean signal intensity of each Tyr-bait peptides (PTK) and Ser/Thr-bait peptides (STK) was normalized to the corresponding DMSO treatment and expressed as log2 fold-ratio. We performed Pearson clustering with all LFC values (Fig. 3A) and determined the individual P-values versus the corresponding DMSO treatment by ANOVA and post-hoc Dunnett’s test in the proprietary Bionavigator software (Fig. 3B and fig. S2, A and B). Note that phosphatase activity was blocked by the addition of a PTP inhibitor cocktail during the assay.
Fig. 3. Peptide chip array, cognate kinase prediction, and combined adaptive kinase response analysis in BT474 cells.
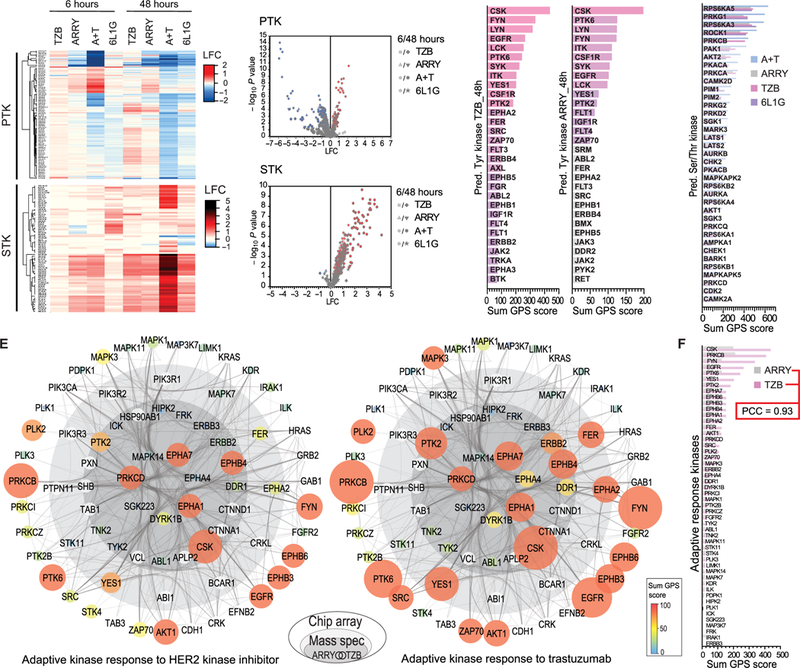
(A) Heat map of up- and down-regulated Tyr (PTK, top) and Ser/Thr (STK, bottom) bait peptide phosphorylation versus DMSO treatment by peptide chip array (n=4 biological replicates). BT474 cells were treated the same as in Fig. 1B. (B) Volcano plots of the bait peptide phosphorylation from PTK (top) and STK (bottom) chip arrays in (A), which shows significantly up- and downregulated peptide phosphorylation as defined by ANOVA and post-hoc Dunnett’s test versus DMSO treatment (p≤ 0.05). (C-D) Sum of scores from group-based prediction system (GPS) based on significantly upregulated phosphorylation of Tyr- (C) and Ser/Thr- (D) bait peptides (as determined in B). (E) Combined adaptive kinase response analysis in onion diagram representation, depicting adaptive signaling proteins (as nodes) based on co-appearance in TMT data and in the set of predicted kinases from peptide chip array data after 48 hours’ treatment with ARRY (left) or TZB (right) in a high-confidence interaction network (edges) from String DB. Inner core (dark grey) represents proteins from the consensus of the 54 persistently phosphorylated Tyr peptides after ARRY and TZB treatment from the TMT data (Fig. 2C). Middle layer (light grey) represents remaining hits form same TZB treatment (Fig. 2C). Node size and color indicates sum of GPS scores (predicted active) from indicated treatment (from C and D). Outer layer (white) represents String DB interactions to additional predicted kinases (from C and D) after TZB treatment. (F) Comparison of the upregulated Tyr- and Ser/Thr- kinases from (E).
Similar to the p-Tyr peptides found by TMT, the overall Tyr phosphorylation after 48 hours’ exposure to A+T and 6L1G was significantly reduced over DMSO treatment in the PTK array and both apoptotic treatments showed a high positive PCC (fig. S2C). We found a common group of p-Tyr peptides with increased phosphorylation over DMSO treatment after the 6-hour exposure with A+T and ARRY, which were associated with increased activity of tyrosine- protein kinase Mer (MERTK), FAK1, Janus kinase 2 (JAK2), FMS-related tyrosine kinase 4 (FLT4) and several ephrin receptor family members (EPHA4, EPHB1, EPHA8, EPHA3 and EPHA2) (fig. S2D). In the STK array, however, we observed an overall significant increase of bait peptide phosphorylation after each anti-HER2 treatment, in particular the 48-hour A+T treatment (Fig. 3, A and B), which may relate to the caspase-mediated hyper-activation of the apoptotic Ser/Thr kinases, such as PKCδ (31).
Next, significantly more highly phosphorylated peptides over the DMSO control treatment (P ≤ 0.05) in the peptide array were used for cognate kinase prediction in a group-based prediction system (http://gps.biocuckoo.org/). The sum of scores was calculated for each kinase from the individual peptide scores using a high-threshold prediction on all Tyr- or Ser/Thr-kinases, respectively (32), from which we determined the top 30 Tyr-kinases after the 48-hour ARRY and 48-hour TZB treatment (Fig. 3C) and the top 37 Ser/Thr-kinases after each 48-hour treatment (Fig. 3D). Unexpectedly, we scored almost the same active Tyr- and Ser/Thr- kinases in both non-apoptotic treatments. In fact, the group of active Tyr kinases after 48 hours’ exposure to TZB included all active Tyr kinases found after the ARRY 48-hour treatment and, thus, the Tyr kinase activation pattern followed again the onion principle (fig. S2D). We identified CSK, Tyr protein kinases Fyn and Lyn (FYN and LYN), EGFR, lymphocyte-specific protein Tyr kinase (LCK), protein Tyr kinase 6 (PTK6), Tyr kinase Yes (YES1), macrophage colony-stimulating factor 1 receptor (CSF1R) and PKT2 (FAK1) among the most active Tyr kinases after both non-apoptotic treatments (Fig. 3C). In the STK array, we scored ribosomal protein S6 kinase alpha-3 and 5 (RPS6KA3 and RPS6KA5), cGMP-dependent protein kinase 1 (PRKG1), Rho-associated protein kinase 1 (ROCK1) and protein kinase C beta (PRKCB) among the top five most activated Ser/Thr-kinases after all anti-HER2 treatments (Fig. 3D). Furthermore, we scored the activity of a number of other cancer associated Ser/Thr- kinases such as e.g. RAC-alpha and beta Ser/Thr- protein kinase (AKT½), cyclin-dependent kinase 2 (CDK2) and PKCδ (PRKCD). This suggests that the Ser/Thr- driver kinases of the adaptive response are to be found among this set and that a combined analysis of both datasets (TMT and peptide chip array) may identify the most relevant targets (this hypothesis was explored and is presented below). In summary, similar Tyr and Ser/Thr kinases were revealed after both non-apoptotic single agent treatments after 48 hours (ARRY and TZB) from the peptide chip array (Fig. 3, C and D) and from the TMT dataset (fig. S2F), respectively.
Adaptive kinase response and cancer cell signaling plasticity
To combine the phospho-status from the TMT dataset with the scored kinase activities from the peptide chip arrays into one biologically relevant system-wide analysis, we constructed a functional association network, based on high-confidence protein-protein interactions from the String database (33). Scored kinase activities (labeled nodes in the network diagram) were grouped according to their appearance in the fraction of persistent phosphorylated Tyr peptides in the TMT dataset (Fig. 2C) and placed in the center of the onion plot (Fig. 3E). Thus, active kinases shown in the core of the onion plot (dark grey inner core and light grey middle layer) were identified by two orthogonal large-scale measurements, and the inner core (darker grey) shows the consensus of ARRY (left) and TZB (right) treatment after 48 hours from the TMT dataset (Fig. 2C). Furthermore, we kept the strongly connected non-kinase Tyr peptide hits identified in the TMT dataset in this network (white circles/nodes in the network plot) and extended the protein associations by one layer of String DB interactions to additional hits from the PTK and STK peptide chip array after the 48-hour TZB treatment (Fig. 3, C and D).
The onion plot shows that CSK, EPHA1/7, EPHB4, PRKCD and PTK2 were activated after both non-apoptotic treatments, ARRY and TZB, and hence, activation of these kinases was a common process after 48 hours and apparently independent of the nature of the anti-HER2 treatment. We found increased phosphorylation and increased kinase activity of HER2 specifically after 48 hours exposure to the HER2-blocking antibody (TZB) but, as expected, not after the same duration exposure to the kinase inhibitor (ARRY), which represents the main differences between the two non-apoptotic treatments. In the middle layer, which shows the consensus hits from the TMT and the peptide chip array after the 48-hour TZB treatment, we found activation of YES1, EPHA2 and the Tyr-kinase Fer (FER) in response to both non-apoptotic treatments. In the outer layer, activation of AKT1, EGFR, EPHB3/6, FYN, MAPK3, Ser/Thr- protein kinase PLK2 (PLK2), PRKCB and PTK6 was found after both non-apoptotic treatments. Consequently, the overall kinase activation pattern of both non-apoptotic anti- HER2 treatments showed a high positive PCC (Fig. 3F), which suggested that adaptive kinase activation follows a common response program.
FAK1 inhibition in HER2-overexpressing breast cancer cells
The kinase FAK1 (also known as PTK2) was the second largest signaling hub in our HER2- dependent network (Fig. 2, A and B) and both non-apoptotic treatments caused phosphorylation of FAK1 at Tyr570/576/577 in the TMT dataset (Fig. 2C) as well as increased activity of FAK1 kinase in the peptide chip array (Fig. 3C). Therefore, FAK1 became a central player in our combined adaptive response analysis (Fig. 3E) and, thus, may be a rational target for combination treatments. The abundance of FAK1 is often increased in breast cancers and is associated with cancer cell motility and migration, survival, and cancer stem-like cell proliferation (22, 34). A series of small molecule kinase inhibitors have been developed to block FAK1 activation, such as defactinib or PF562271, and these have proceeded to clinical trials (34). Furthermore, FAK1 reportedly also participates in the adaptive response after lapatinib treatment in HER2-positive breast cancer cells (13), but FAK1 phosphorylation appears suppressed upon increased phosphorylation of HER2 (15).
To analyze the effects of FAK1 inhibition in HER2-overexpressing breast cancer with and without a PI3K-activating mutation, we compared BT474, SKBR3 and HCC1419 cells (trastuzumab-responsive group) with MDA-MB361, MDA-MB453 and UACC893 cells (trastuzumab-resistant group). Constant activation of the PI3K/AKT-pathway by a PIK3CA- activating point mutation confers strong resistance against all the anti-HER2 treatments (Fig. 4A). Furthermore, we included the allosteric small molecule inhibitor of AKT, MK2206 (35), to block AKT activation independently of both PIK3CA mutation status and HER2/HER3 signaling (Fig. 4, A and B). In comparison to the high basal abundance of phosphorylated AKT [p- AKT(Ser473)], we observed low basal abundance of p-FAK1(Tyr397) at the unperturbed steady state in the HER2-overexpressing breast cancer cells (Fig. 4B). However, upon inhibition of p-AKT, either directly by MK2206 or upstream by an anti-HER2 treatments in BT474, SKBR3 and HCC1419 cells, the p-FAK1(Tyr397) abundance was subsequently increased in all cancer cell lines, which is in agreement with previous findings (13, 15). Conversely, FAK1 inhibition by PF562271 induced decreased abundance of p-FAK1(Tyr397) in all cancer cell lines but increased amounts of p-AKT(Ser473) and reduced abundance of HER3 after 48 hours in BT474, SKBR3, HCC1419 and UACC893 cells (Fig. 4B), presumably through the AKT-FOXO-HER3 negative feedback loop (12).
Fig. 4. FAK1 activation in response to AKT inhibition and effects of combination treatments.
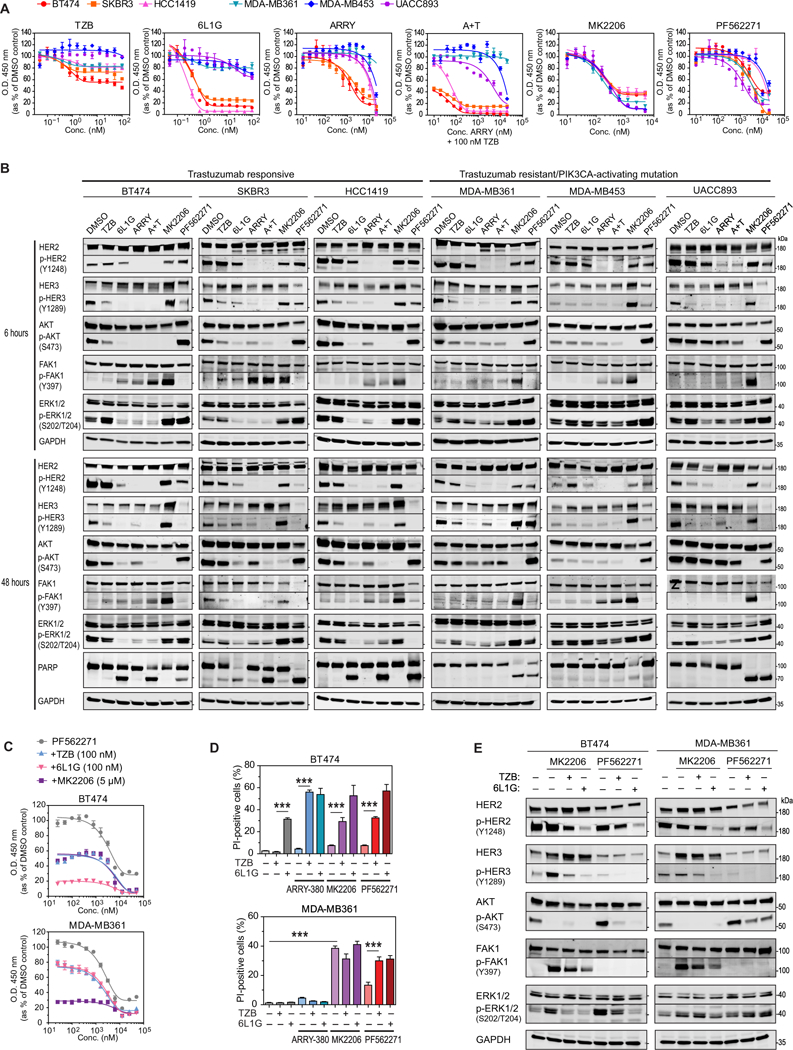
(A) XTT cell proliferation assays of HER2-positive breast cancer cell lines with or without PIK3CA point mutations [BT474 (K111N), SKBR3 (WT), HCC1419 (WT), MDA-MB361 (E345K), MDAMB453 (H1047R) and UACCC893 (H1047R)] treated for 4 days with the indicated concentration of HER2-targeted therapy (TZB, 6L1G, ARRY, A+T), AKT inhibitor (MK2206), or FAK inhibitor (PF562271). Data are mean ± SD of n=3 experiments. (B) Western blot analysis of a HER2-dependent signaling cascade after 6 (top) and 48 hours (bottom) of treatment with the indicated drug (DMSO, 100 nM TZB, 100 nM 6L1G, 10 μM ARRY380, 10 μM PF562271, or 5 μM MK2206) or combination (A+T, 10 μM ARRY380 and 100 nM TZB). Blots are representative of 2–4 independent experiments. (C) XTT cell proliferation assays of BT474 and MDAMB361 cells after 4 days of continuous treatment with titration of the FAK1-inhibitor PF562271, and subsequent addition at day 0 (less than 5 min) of the indicated anti-HER2 agents TZB (100 nM) and 6L1G (100 nM), or the AKT-inhibitor MK2206 (5 μM). Data are mean ± SD of n=3 experiments. (D) High-throughput microscopy analysis of breast cancer cells continuously treated for 3 days 10 μM ARRY-380 or 10 μM PF562271 in combination with 100 nM TZB or 6L1G, then stained with propidium iodide (PI) and Hoechst-33342 to detect the number of membrane-permeable (PI-positive, inferred as dead) cells in the population. Data are mean ± SD of n=5 experiments. ***P ≤ 0.005 by one-sided pairwise t-test. (E) Western blot analysis for the indicated proteins in BT474 and MDA-MB361 cells treated for 2 days with 10 μM PF562271 or 5 μM MK2206, either alone or in combination with either 100 nM TZB or 6L1G. Blots are representative of 2 independent experiments.
Therefore, the AKT and FAK1 kinase pathways displayed a counter-regulation in these cancer cell lines; meaning when one is inhibited, the other is activated. Next, we analyzed the time- dependent phosphorylation of FAK1 after MK2206 treatment in BT474 cells and observed progressively increasing p-FAK1 (Tyr397) abundance from 6 to 24 hours (fig. S3A). On the other hand, treatment with PF562271 induced upregulation of p-AKT(Ser473) in the same period.
Notably, MK2206 and PF562271 treatment induced marked PARP cleavage in MDA-MB361 and UAC893 cells after 48 hours (Fig. 4B). Thus, both trastuzumab-resistant cell lines showed greater vulnerability to AKT and FAK1 inhibitors in comparison to BT474 cells, which may indicate that a PI3K-activating mutation increases the dependency on PI3K/AKT and FAK1 pathway signaling.
Synergy by combined inhibition of FAK1 and HER2
As described above, we had found that FAK1 was a key player in the adaptive kinase signaling response to anti-HER2 treatments (Fig. 3E), and FAK1 showed counter-regulatory phosphorylation with AKT (Fig. 4B). Therefore, we combined the anti-HER2 treatments as well as the AKT inhibitor MK2206 with the FAK1 inhibitor PF562271 to test for potential combination benefits on the inhibition of cell proliferation (Fig. 4C and fig. S3B), induction of cell death (Fig. 4D and fig. S3C), and blockage of AKT-FAK1 counter phosphorylation (Fig. 4E). We indeed observed a strong anti-proliferative effect of the FAK1 inhibitor as single-agent treatment and a clearly additive effect of FAK1 inhibition with anti- HER2 or AKT combination treatments in both cell lines (Fig. 4C). The combination of TZB or 6L1G with PF562271 induced strong reduction of p-FAK(Tyr397), quantitatively blocked FAK1-AKT counter-regulatory phosphorylation and showed significantly increased induction of cell death (Fig. 4D and fig. S3C). Thus, the combination of an anti-HER2 treatment with a small molecule inhibitor against FAK1 blocked the activation of the adaptive kinase response and significantly enhanced cell death in both HER2-positive breast cancer cell lines.
DISCUSSION
In this study, we analyzed the early kinome changes of a HER2-positive breast cancer cell line after different anti-HER2 treatments by TMT LC-MS/MS to identify critical signaling events that are associated with the development of cancer drug resistance. As hypothesized, we found that the anti-proliferative effect of a particular treatment was proportional to the number of p-Tyr peptides with reduced abundance and, thus, inhibition of cancer cell proliferation correlated with progressively reduced Tyr kinase activity after more potent combination treatments.
In contrast, and against our initial hypothesis, the overall pattern in the p-Tyr peptide abundance did not directly correlate with the ultimate cancer cell fate after the different treatments, but it showed high correlation with the mechanism of the particular drug. In fact, the overall pattern of the Tyr peptide phosphorylation after the different treatments with, for example, the HER2 kinase inhibitor ARRY-380 was highly similar to the combination treatment of ARRY-380 plus trastuzumab, but only the latter combination treatment induced apoptosis, and did so relatively robustly. We conclude that the induced cancer cell fate and, thus, the potential of developing cancer drug resistance depends on a rather small number of residually active or compensatory active kinases that maintain cancer cell survival after single inhibitor treatment. Assuming that cancer cell signaling plasticity is proportional to the number of persistently active kinases, we conclude that the potential for cancer plasticity was reduced by the combination treatment and by the biparatopic DARPin in a similar manner.
Through comparative analysis of different non-apoptotic treatments, we identified a common persistently phosphorylated Tyr peptide signature. Analyzing the time-dependent phosphorylation trends of this particular subset of p-Tyr peptides between treatments, we could distinguish, with high confidence, between apoptotic and non-apoptotic cell fates. This finding led us to the hypothesis that a common adaptive signaling program was executed upon different non-apoptotic anti-HER2 treatments, which may have maintained the survival of the cancer cells after single inhibitor treatment.
To test this hypothesis, we analyzed the residual kinase activities with a peptide chip array after these treatments. By a combined system-wide analysis, we characterized the common adaptive kinase-response program that essentially involved the activation of FAK1, PKCδ and several Eph family receptors. Thus, we conclude that the adaptive kinase response is a specific backup program of the given cancer host network and that it operates similarly and predictably after the different treatments against the same target. Therefore, we speculate that previously reported heterogeneity in the adaptive kinase response (13, 36) may have originated mainly from the different genetic backgrounds of the various cancer cell lines. However, we do not exclude that additional heterogeneity in the adaptive kinase response may arise later from changes in kinase expression after prolonged drug exposure (6–8, 11, 12, 16).
Finally, we tested the effect of FAK1 inhibition in various HER2-overexpressing cancer cell lines with and without PI3KCA activating point mutations, which resemble a common mode of acquired cancer drug resistance to anti-HER2 therapies. By small-molecule inhibitor studies, we observed that FAK1 and AKT signaling are counter-regulated, such that inhibition of FAK1 causes activation of AKT and vice versa. The exact mechanism is currently unknown; however, we speculate that Pten 3-phosphoinositide phosphatase alpha (PTEN) may play a key role in the counter-regulation of both pathways, as previously suggested in the context of leukemia (37). Furthermore, FAK1 inhibitors showed an additive effect when combined with anti-HER2 treatments, such as trastuzumab or biparatopic DARPin, in trastuzumab-resistant cell lines.
In summary, we used a network topology-based approach to identify a common adaptive kinase-response program and its major signaling hubs after various HER2-targeted treatments in breast cancer cells. Our findings not only provide a larger resource to study HER2-dependent signaling, but also a novel approach to predict effective combination treatments against a dynamically adapting cancer signaling network.
Materials and Methods
Cell lines and reagents
The human mammary carcinoma cell lines BT474, SKBR3, HCC1419, MDA-MB453, UACC893 and MDA-MB361 were obtained from the American Type Culture Collection (ATCC; www.atcc.org) and cultured in RPMI-1640 medium from Life Technologies with 1 % penicillin streptomycin from Sigma Aldrich and 10 % fetal calf serum from PAA. Cells were seeded at a density of 10,000 cells/cm2 24 hours prior to treatment. Protease inhibitors pefabloc from Merck, leupeptin and pepstatin from Serva and marimastat from Calbiochem and the phosphatase inhibitors sodium orthovanadate, sodium metavanadate, sodium molybdate, sodium β-glycerol phosphate and sodium fluoride from Sigma Aldrich were used for western blot analysis. Antibodies against ErbB3 (D22C5) (#12708) (1:1000), p-ErbB3 (Y1289) (#4791) (1:500), Erk½ (#9102) (1:1000), p-ERK½ (T202/Y204) (#4370) (1:1000), pan-AKT (C67E7) (1:2000) ,p-AKT (S473) (#4060) (1:2000), PARP (#9542) (1:1000), FAK (D5O7U) XP (1:500), p-FAK Y397 (D20B1) (1:500) were used from Cell Signaling Technology. Anti-ErbB2 (3B5) (OP15) (1:5000) and p-ErbB2 (1248) (06–229) (1:10,000) were from Calbiochem. GAPDH (sc-365062) (1:2000) was used from Santa Cruz. Secondary anti-mouse IgG IRDye800 conjugate from Rockland (610–732-124) and anti-rabbit IgG Alexa680 (A-21109) from Invitrogen was used (1:10,000) for IR-western blot detection on an Odyssey system from Li-Cor. ARRY-380, MK-2206, PF-562271 were obtained from Selleckchem. DARPin expression and purification has been described previously (38). Trastuzumab (Herceptin®) was obtained from Kantonsapotheke Zürich.
Sample preparation for LC-MS/MS
BT474 cells were treated in biological duplicates with 100 nM trastuzumab, 100 nM DARPin 6L1G, 10 μM HER2 inhibitor ARRY-380 and the combination of 100 nM trastuzumab and 10 μM ARRY-380, each treatment for 6 and 48 hours. Cell pellets were lysed in 8 M urea supplemented with phosphatase inhibitors on ice and cell extract concentrations were determined by BCA assays (Pierce). Cell lysates were reduced with 1 M DTT in ammonium acetate at pH 8.9 and 55 mM iodoacetamide was added to quench reformation of disulfide bridges. Afterwards, ~400 μg of sample was digested with trypsin overnight at room temperature and the reaction was stopped by adding acetic acid to a final concentration of approximately 90 %. Samples were loaded on a Sep-Pak columns, washed with 90 % acetonitrile/ 0.1 % acetic acid and eluted in 25 % acetonitrile/0.1 % acetic acid.
TMT peptide labeling and p-Tyr peptide IP
Peptides were labeled with TMT10plex Mass Tag Labeling Kits (Thermo) according to the following scheme: DMSO_6h (126), TZB_6h (127C), 6L1G_6h (127N), ARRY_6h (128C), A + T_6h (128N), DMSO_48h (129C), TZB_48h (129N), 6L1G_48h (130C), ARRY_48h (130N), A + T_48h (131). Samples were resuspended in 100 μL of 70% (v/v) ethanol, 30% (v/v) 0.5 M triethylammonium bicarbonate at pH 8.5 and incubated with TMT reagent resuspended in 40 μL anhydrous acetonitrile at room temperature for 1 hour. Dried samples were resuspended in 400 μL IP buffer (100 mM Tris-HCl, 1% Nonidet P-40 at pH 7.4) and added to 60 μL protein G agarose beads, which were loaded with anti-phosphotyrosine antibodies (12 μg 4G10 (Millipore), 12 μg PY-100 (Cell Signaling Technologies), and 12 μg PT-66 (Sigma)) overnight at 4 °C. Beads were centrifuged for 60 s at 4,000 × g for supernatant collection, washed once with 400 μL IP buffer, and washed three times with 400 μL wash buffer (100 mM Tris-HCl at pH 7.4). Antibody-peptide complexes were eluted with 70 μL 100 mM glycine at pH 2.5 for 30 min at room temperature.
Immobilized metal affinity chromatography (IMAC)
After antibody-peptide complex elution, peptides were purified by IMAC as follows. A 10-cm self-packed IMAC (Poros 20MC; Applied Biosystems) capillary column (inner diameter, 200 μm; outer diameter, 360 μm) was rinsed with 100 mM EDTA at pH 8.9 at 10 μL/min for 10 min, ultrapure water at 10 μL/min for 10 min, and charged with 100 mM FeCl3 for 40 min. Excess iron was removed by washing with 0.1% HOAc at 10 μL/min for 10 min before loading the IP elution for 1 hour at room temperature. Nonspecific peptides were removed by washing with 25% MeCN, 1% HOAc, 100 mM NaCl for 10 min at 10 μL/min. The IMAC column was equilibrated with 0.1% HOAc for 10 min at 10 μl/min and bound phosphopeptides eluted onto a 10-cm self-packed C18 capillary precolumn [100 μm ID × 10 cm packed with 10 μm C18 beads (YMC gel, ODS-A, 12 nm, S-10 μm, AA12S11)] with 40 μL of 250 mM Na2HPO4, pH 8.0. The loaded precolumn was rinsed with 0.2 M acetic acid for 10 min before LC-MS analysis.
LC-MS/MS
The washed pre-column was connected in series with a self-packed analytical capillary column [50 μm ID × 12 cm packed with 5 μm C18 beads (YMC gel, ODS-AQ, 12 nm, S-5 μm, AQ12S05)] with an integrated electrospray tip (~1 μm orifice). Peptides were eluted using a 140-min gradient from aqueous (0.2 M acetic acid) to organic [0.2 M acetic acid, 70% (v/v) MeCN] at a flow rate of 0.2 μL/min with a flow split >99%. Peptides were ionized with a spray voltage of 2 kV and injected directly into a Thermo Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer. The 15 most abundant precursor peaks were selected in information-dependent acquisition mode with an isolation width of 2 m/z, an AGC target of 3e6, a maximum injection time of 350 ms, and previously selected peaks excluded for 30 s.
MS data analysis
Raw mass spectral data files were searched against the SwissProt database containing Homo sapiens protein sequences (20,199 sequences), using Mascot version 2.4 (Matrix Science). TMT reporter quantification was extracted and isotope-corrected using Proteome Discoverer software (Thermo) and was further normalized based on median relative protein quantification ratios obtained from total protein expression analysis. For each peptide, relative quantification was represented as a ratio between ion intensities for experimental conditions and normalization channel.
Peptide chip array
Biological quadruplicates of BT474 cells were treated for 6 or 48 hours with 100 nM trastuzumab, 10 μM ARRY-380, the combination of 100 nM trastuzumab and 10 μM ARRY-380 or 100 nM DARPin 6L1G. Cells were washed with ice-cold PBS and scraped in PBS containing Halt phosphatase and Halt protease inhibitor cocktails (Pierce) on ice. Cells were centrifuged at 300 rpm for 2 min, and afterwards pellets were lysed by addition of M-PER mammalian extraction buffer (Pierce) containing both inhibitor cocktails for 30 min at 4°C on a rocker. Cell extracts were cleared by centrifugation for 10 min at 12,000 rpm at 4°C and protein concentrations were determined by BCA assays (Pierce). Aliquots of 1 mg/ml were prepared and snap-frozen in liquid nitrogen. 5 μg of protein extract was used for the PTK array protocol (V1.9) and 1.5 μg for the STK array protocol (V4.1). Measurements were performed on a Pamstation12 from PamGene (Wolvenhoek 10, ‘s-Hertogenbosch, Netherlands). Briefly, the PTK array was processed in a single-step reaction. Cell extracts, ATP and FITC-labelled pY20 antibody were incubated on the chip and the phosphorylation of the individual Tyr-peptides was followed by fluorescence detection in real time. The STK array was processed in a two- step reaction. First, the cell extracts, ATP and the primary antibody mixture was incubated with the chip for 110 min. Second, the reaction mix was removed and the secondary FITC-labelled antibody was added. Development of the fluorescence signal was detected again by Alexa488 fluorescence. Signal intensities were analyzed in the Bionavigator software (PamGen) as a function of time and expressed as log2 fold-ratio versus DMSO treatment after 6 or 48 hours, respectively.
Software and statistical analysis
One-sided Student’s f-test was performed in SigmaPlot. Heatmaps and Pearson cluster correlation was performed in Perseus from the Max Planck Institute of Biochemistry (39). Venn diagram analyses were performed in Venny2.0 (http://bioinfogp.cnb.csic.es/tools/venny/index.html). Direct interaction networks were generated in MetaCore V6.31 (https://portal.genego.com/). Functional protein association networks were generated in String database V10.5 (https://string-db.org/). Cognate kinase prediction was performed in an offline version of a group-based prediction system (GPS3.0) (http://gps.biocuckoo.org/userguide.php). High-throughput image analysis was performed in Gen5 Image+ from Biotek. Western blot analysis was performed in the Odyssey application software. Network plots were generated in Cytoscape 3.5.1 (http://cytoscape.org/) and analyzed by Network Analyzer 2.7 and ClusterMaker2.0 V1.1.0 (http://www.cgl.ucsf.edu/cytoscape/cluster/clusterMaker.html).
Supplementary Material
Acknowledgements:
We thank M. Wymann and his group from the Department of Biomedicine in Basel for their assistance with experiments performed on Pamstation12; S. Rangarajan and F. Naji from PamGene International (www.pamgene.com) for scientific support and analysis of PamChip imaging data; J. Grossmann from the Functional Genomics Center Zurich for scientific discussions; and P. Eusebi from the University of Perugia and G. Kratzer from the University of Zurich for their statistical review of the manuscript.
Funding: This work was supported by the Swiss Cancer Research foundation (SKL; grant 4147–02-2017 to A.P.), Schweizerischer Nationalfonds (SNF; grant 310030B_166676 to A.P.), and the National Institutes of Health (NIH; grants U54 CA210180 and R01 CA096504 to F.M.W.).
Footnotes
Author contributions: M.S. and A.P. designed the study, with contributions from F.K., A.S.G., R.T., and F.M.W. on the experimental design. A.S.G. performed mass spectrometry; F.K. performed protein expressions and purifications; M.S. performed all other experiments and all analyses. M.S and A.P. wrote the manuscript, with contributions from all authors.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (40) partner repository with the dataset identifier PXD011999. All other data needed to evaluate the conclusions in the paper are present in the in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Atlas N. Cancer Genome, Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR et al. , The landscape of cancer genes and mutational processes in breast cancer. Nature 486, 400–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M et al. , A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 12, 395–402 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Baselga J, Swain SM, Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer 9, 463–475 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX, Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 15, 429–440 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, Smith DJ, Landolfi S, Ramon y Cajal S., Arribas J et al. , Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 28, 803–814 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Tamaskovic R, Schwill M, Nagy-Davidescu G, Jost C, Schaefer DC, Verdurmen WP, Schaefer JV, Honegger A, Plückthun A, Intermolecular biparatopic trapping of ErbB2 prevents compensatory activation of PI3K/AKT via RAS-p110 crosstalk. Nat Commun 7, 11672 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, Moasser MM, Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 445, 437–441 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moulder SL, Borges VF, Baetz T, McSpadden T, Fernetich G, Murthy RK, Chavira R, Guthrie K, Barrett E, Chia SK, Phase I Study of ONT-380, a HER2 Inhibitor, in Patients with HER2(+)-Advanced Solid Tumors, with an Expansion Cohort in HER2(+) Metastatic Breast Cancer (MBC). Clin Cancer Res 23, 3529–3536 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Amin DN, Sergina N, Ahuja D, McMahon M, Blair JA, Wang D, Hann B, Koch KM, Shokat KM, Moasser MM, Resiliency and vulnerability in the HER2-HER3 tumorigenic driver. Sci Transl Med 2, 16ra–17 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sanchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E et al. , Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci U S A 108, 5021–5026 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N, AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19, 58–71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS, Angus SP, Collins KA, Granger DA, Reuther RA et al. , Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep 11, 390–404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rexer BN, Arteaga CL, Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog 17, 1–16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bose R, Molina H, Patterson AS, Bitok JK, Periaswamy B, Bader JS, Pandey A, Cole PA, Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc Natl Acad Sci U S A 103, 9773–9778 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandarlapaty S, Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov 2, 311–319 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Citri A, Yarden Y, EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol 7, 505–516 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Jost C, Schilling J, Tamaskovic R, Schwill M, Honegger A, Plückthun A, Structural basis for eliciting a cytotoxic effect in HER2-overexpressing cancer cells via binding to the extracellular domain of HER2. Structure 21, 1979–1991 (2013). [DOI] [PubMed] [Google Scholar]
- 19.McDonald ER 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K et al. , Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 170, 577–592 e510 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Campbell J, Ryan CJ, Brough R, Bajrami I, Pemberton HN, Chong IY, Costa-Cabral S, Frankum J, Gulati A, Holme H et al. , Large-Scale Profiling of Kinase Dependencies in Cancer Cell Lines. Cell Rep 14, 2490–2501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JLY et al. , Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov 2, 172–189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE, Bader GD, Mills GB et al. , Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 164, 293–309 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le XF, Claret FX, Lammayot A, Tian L, Deshpande D, LaPushin R, Tari AM, Bast RC Jr., The role of cyclin-dependent kinase inhibitor p27Kip1 in anti-HER2 antibody-induced G1 cell cycle arrest and tumor growth inhibition. J Biol Chem 278, 23441–23450 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Reddy RJ, Gajadhar AS, Swenson EJ, Rothenberg DA, Curran TG, White FM, Early signaling dynamics of the epidermal growth factor receptor. Proc Natl Acad Sci U S A 113, 3114–3119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ree AH, Flatmark K, Saelen MG, Folkvord S, Dueland S, Geisler J, Redalen KR, Tumor phosphatidylinositol 3-kinase signaling in therapy resistance and metastatic dissemination of rectal cancer: opportunities for signaling-adapted therapies. Crit Rev Oncol Hematol 95, 114–124 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Azevedo H, Moreira-Filho CA, Topological robustness analysis of protein interaction networks reveals key targets for overcoming chemotherapy resistance in glioma. Sci Rep 5, 16830 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dubovenko A, Nikolsky Y, Rakhmatulin E, Nikolskaya T, Functional Analysis of OMICs Data and Small Molecule Compounds in an Integrated “Knowledge-Based” Platform. Methods Mol Biol 1613, 101–124 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Su G, Kuchinsky A, Morris JH, States DJ, Meng F, GLay: community structure analysis of biological networks. Bioinformatics 26, 3135–3137 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen-Petersen BL, Carter CJ, Ohm AM, Reyland ME, Protein kinase Cdelta is required for ErbB2-driven mammary gland tumorigenesis and negatively correlates with prognosis in human breast cancer. Oncogene 33, 1306–1315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sikkema AH, Diks SH, den Dunnen WF, ter Elst A, Scherpen FJ, Hoving EW, Ruijtenbeek R, Boender PJ, de Wijn R, Kamps WA et al. , Kinome profiling in pediatric brain tumors as a new approach for target discovery. Cancer Res 69, 5987–5995 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Franklin RA, McCubrey JA, Kinases: positive and negative regulators of apoptosis. Leukemia 14, 2019–2034 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Xue Y, Liu Z, Cao J, Ma Q, Gao X, Wang Q, Jin C, Zhou Y, Wen L, Ren J, GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng Des Sel 24, 255–260 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P et al. , The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45, D362–D368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sulzmaier FJ, Jean C, Schlaepfer DD, FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 14, 598–610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS et al. , MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9, 1956–1967 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Wilkes EH, Terfve C, Gribben JG, Saez-Rodriguez J, Cutillas PR, Empirical inference of circuitry and plasticity in a kinase signaling network. Proc Natl Acad Sci U S A 112, 7719–7724 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.You D, Xin J, Volk A, Wei W, Schmidt R, Scurti G, Nand S, Breuer EK, Kuo PC, Breslin P et al. , FAK mediates a compensatory survival signal parallel to PI3K-AKT in PTEN-null T-ALL cells. Cell Rep 10, 2055–2068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamaskovic R, Simon M, Stefan N, Schwill M, Plückthun A, Designed ankyrin repeat proteins (DARPins) from research to therapy. Methods Enzymol 503, 101–134 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J, The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez- Riverol Y, Reisinger F, Ternent T et al. , 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, 11033 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.