

ABSTRACT
Here we present miR-CATCHv2.0, an implemented experimental method that allows the identification of the microRNA species directly bound to an RNA of interest. After cross-linking of microRNA::RNA::Ago2 complexes using formaldehyde, the RNA is fragmented using sonication and then subjected to affinity purification using two sets of biotinylated tiling probes (ODD and EVEN). Finally, enriched microRNA species are retrieved by means of small RNA sequencing coupled with an ad hoc analytical workflow.
In BRAFV600E mutant A375 melanoma cells, miR-CATCHv2.0 allowed us to identify 20 microRNAs that target X1, the most abundant isoform of BRAF mRNA. These microRNAs fall into different functional classes, according to the effect that they exert (decrease/increase in BRAFV600E mRNA and protein levels) and to the mechanism they use to achieve it (destabilization/stabilization of X1 mRNA or decrease/increase in its translation). microRNA-induced variations in BRAFV600E protein levels are most of the times coupled to consistent variations in pMEK levels, in melanoma cell proliferation in vitro and in sensitivity to the BRAF inhibitor vemurafenib in a xenograft model in zebrafish. However, microRNAs exist that uncouple the degree of activation of the ERK pathway from the levels of BRAFV600E protein.
Our study proposes miR-CATCHv2.0 as an effective tool for the identification of direct microRNA-target interactions and, by using such a tool, unveils the complexity of the post-transcriptional regulation to which BRAFV600E and the ERK pathway are subjected in melanoma cells.
KEYWORDS: Direct microRNA-target binding, affinity purification, BRAF-X1 mRNA, transcript stability, transcript translation, ERK signalling, melanoma
Introduction
MicroRNAs (miRNAs) are small single stranded non-coding RNAs (21-25nt) that affect the expression of target mRNAs by binding mainly to the 3ʹUTR of target genes. This usually results in the inhibition of protein production due to mRNA degradation or translational repression [1] . However, examples exist of microRNAs that sustain the expression of target mRNAs by increasing RNA stability or translation [2].
Although many aspects of microRNA involvement in cancer have been elucidated [3,4], the identification of their direct targets, which is the pre-requisite for a full understanding of the role that they play in the cell, remains a challenging issue. In order to overcome the many limitations of in silico target prediction algorithms [5,6], quite a few experimental methods have been developed that allow the identification of the microRNA species physically bound to an RNA of interest. In such methods, the 3ʹUTR under study is exogenously administered and used as bait, either as a 3ʹ-end biotinylated molecule [7] or as a chimerical transcript containing MS2 RNA hairpin motifs [8,9]. Alternatively, in the miR-CATCH method we previously developed, a cross-linking step using formaldehyde coupled with an affinity-purification step using a biotinylated DNA antisense probe are used to pull down the endogenous target mRNA of interest, together with all the microRNA species bound to it [10–14].
BRAF kinase belongs to the RAS/RAF/MEK/ERK signalling pathway and plays a crucial role in human cancer. Approximately 7% of all cancer cases carry a BRAF mutation, including 50–60% of melanomas. The most frequent mutation consists of a nucleotide substitution transforming Val at position 600 into Glu (V600E). This mutation renders BRAF independent of RAS activation and constitutively active as a monomer [15,16]. BRAFV600E is causally linked to cancer, as it has been shown in animal models [17]. Furthermore, selective inhibitors of BRAFV600E kinase activity [BRAFi, such as vemurafenib (vem)] have recently contributed to increase the life expectancy of metastatic melanoma patients, becoming a valid example of molecularly driven precision medicine [18].
In spite of the heavy exploitation of BRAFV600E for cancer modelling and targeted therapy, a thorough study of the regulation of BRAF expression has never been undertaken. With the aim to fill this gap and possibly uncover new strategies that can lead to a more efficient and specific targeting of the mutant kinase [19], we undertook a systematic analysis of BRAF transcripts in melanoma. We found that BRAF mRNA exists as a pool of three variants, two of which (BRAF-ref and BRAF-X1) encode for proteins with unique amino acid sequences at the C-terminus. Furthermore, the X1 isoform has a unique 3ʹ untranslated region (3ʹUTR) that is substantially larger than the 3ʹUTR of the reference BRAF transcript (~7 kb vs ~80 b) and is expressed at the highest level in cell lines and patient samples. Therefore, X1 is likely to give the most prominent contribution to microRNA-mediated post-transcriptional regulation of BRAF expression [20,21].
Here, we present miR-CATCHv2.0, an upgraded version of the miR-CATCH method in which, after cross-linking of microRNA::RNA::Ago2 complexes using formaldehyde, the RNA is fragmented using sonication and then subjected to affinity purification using not only one, but two sets of biotinylated tiling probes (ODD and EVEN). Enriched microRNA species are then retrieved by small RNA sequencing coupled with an ad hoc analytical workflow. miR-CATCHv2.0 allowed us to identify 20 microRNAs that directly bind to BRAF-X1 3ʹUTR in melanoma cells. We also show that some among these microRNAs are negative regulators of gene expression, while others are positive regulators. Furthermore, we demonstrate that they act by different mechanisms (decrease/increase of mRNA stability or translation). Finally, we use in vitro and in vivo assays to describe the variable consequences that the microRNA-mediated regulation of the X1 isoform has on ERK signalling and on melanoma cell biology.
Results
Optimization of the miR-CATCHv2.0 experimental method
The experimental steps of miR-CATCHv2.0, which we used to identify the microRNAs bound to the X1 3ʹUTR, are summarized in Figure 1(a), upper: A375 melanoma cells were cross-linked and then lysed using a sonicator. The lysate was incubated with biotinylated DNA probes complementary to the target. In order to maximize the recovery of the intended 3ʹUTR, we used multiple 20-mer antisense and non-overlapping DNA probes. Specifically, we used 12 probes that are located approximately 100nt apart and that tile the entire length of the X1 3ʹUTR in its 1.35kb long version, which is the one that we initially identified [20] (Figure 1(a), lower; Supplementary Table S1). Furthermore, in order to minimize artefacts, we doubled each capture by splitting the probes into two pools (ODD & EVEN), as reported in the ChiRP approach [22]. After probe hybridization, the affinity-purified RNA-protein complexes were isolated using magnetic streptavidin beads, then they were eluted and finally subjected to small RNA sequencing (RNA-seq) for the identification and quantification of bound microRNA species. The detailed description of each experimental step is provided as supplementary information.
Figure 1.
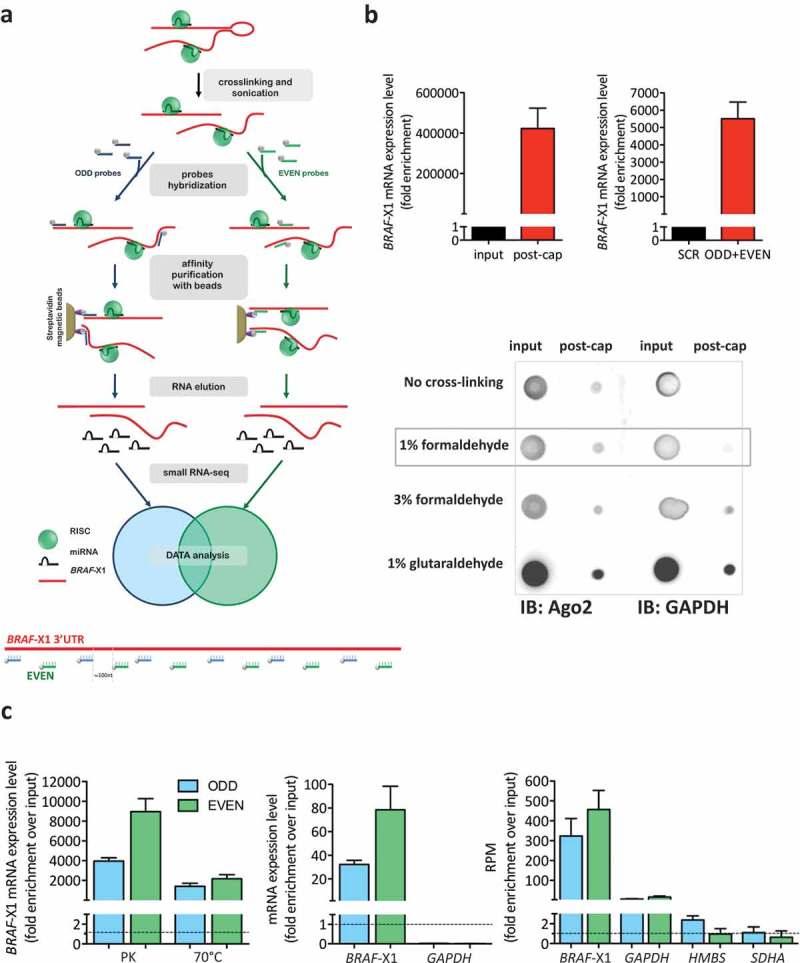
Optimization of miR-CATCHv2.0 experimental method.
(a). (upper) Workflow of miR-CATCHv2.0. Cells are cross-linked and sonicated. Two independent sets of biotinylated tiling probes (ODD probes and EVEN probes) are hybridized to the target mRNA (BRAF-X1 3ʹUTR). microRNA::mRNA::Ago2 complexes are then purified using magnetic streptavidin beads. After stringent washes, RNA is eluted using proteinase K and subjected to small RNA-seq. (lower) Design of antisense biotinylated tiling probes. The probes are located approximately 100nt apart along BRAF-X1 3ʹUTR and are grouped into ‘ODD’ and ‘EVEN’ sets based on their position.
(b). (upper) The capture performed with tiling probes allows to obtain a strong enrichment in BRAF-X1 compared to un-captured samples (input, left) or a scrambled probe (SCR, right). (lower) Effects of different cross-linking reagents on the purification of Ago2. After cross-linking with the indicated reagents, cells were subjected to sonication (12 rounds on cells cross-linked with 1% formaldehyde, 18 rounds on cells cross-linked with 3% formaldehyde, 18 rounds on cells cross-linked with 1% glutaraldehyde). Immunoblot assay shows that cross-linking performed with 1% formaldehyde allows effective Ago2 purification with the lowest background (measured as GAPDH purification). (c). (left) The release of BRAF-X1 mRNA from cross-linked proteins is more effective when treatment with proteinase K (PK) is used rather than heating at 70°C. (middle, right). Both the ODD and the EVEN pools of probes can capture BRAF-X1 mRNA, as measured by qRT-PCR (middle) and by counting the reads belonging to BRAF-X1 in the small RNA-seq (right). The levels of the unrelated GAPDH, HMBS and SDHA mRNAs are measured to show that the capture is selective for the intended mRNA.The graphs in this figure represent the mean ± SEM of three independent experiments.
The cross-linking step is fundamental for the subsequent co-precipitation of microRNA::mRNA::Ago2 complexes, but cells subjected to cross-linking are resistant to chemical lysis by detergents [23]. Therefore, in order to obtain the solubilization of microRNA::mRNA::Ago2 complexes, we opted for mechanical lysis by sonication. This method was chosen because it comes with the additional advantage of causing the shearing of RNA molecules, which, in turn, lowers steric hindrance, hence favours probe binding. Due to the probe distribution described above, we reasoned that sheared fragments of RNA need to be at least 200nt long, in order to be recognized by at least one ODD and one EVEN probe. Keeping this in mind, we compared different experimental protocols and found that 3 rounds of sonication performed on unfixed cells, 12 rounds performed on cells cross-linked with 1% formaldehyde, 18 rounds performed on cells cross-linked with 3% formaldehyde or 18 rounds performed on cells cross-linked with 1% glutaraldehyde are optimal to shear the vast majority of RNA molecules into the desired 200-500nt long fragments (Supplementary Figure S1).
Then, we tested whether the tiling probes can efficiently capture fragments of the intended target. qRT-PCR analysis confirmed that a capture performed using all tiling probes together on unfixed RNA subjected to three rounds of sonication causes a strong enrichment in the X1 3ʹUTR compared to the cell lysate prior the procedure (input, Figure 1(b), upper left). Furthermore, we ensured that this enrichment is robust by comparing it with the one obtained by performing a capture with a scrambled probe that does not recognize any sequence in the human transcriptome (SCR, Figure 1(b), upper right). Interestingly, in both cases the enrichment of target mRNA is several orders of magnitude higher than that obtained using only one probe and no sonication [10,12–14], confirming the effectiveness of the variations that have been introduced in the experimental protocol.
Next, we evaluated the cross-linking ability of different fixing reagents. As a read out, we measured the amount of Ago2 recovered together with the sheared RNA fragments. All cross-linkers tested were effective at precipitating Ago2. However, 1% formaldehyde was selected because it yielded the highest signal-to-noise ratio (Figure 1(b), lower).
As far as the elution step is concerned (it is required to release the RNA from cross-linked proteins), we evaluated two different strategies: digestion with proteinase K (PK), as performed in the ChIRP protocol [22], and reversion of formaldehyde cross-linking by heating. After elution, the levels of X1 3ʹUTR were measured by qRT-PCR and treatment with PK was chosen because it was more efficient compared to the heating procedure (Figure 1(c), left).
Once the cross-linking (1% formaldehyde), sonication (12 rounds) and elution (proteinase K) steps were optimized, three independent captures were performed and a very robust enrichment in the X1 3ʹUTR was obtained with both the ODD and the EVEN probe sets, as measured by qRT-PCR (Figure 1(c), middle) and by counting the number of reads belonging to X1 3ʹUTR via small RNA-seq (Figure 1(c), right). Conversely, the enrichment of housekeeping genes such as GAPDH, HMBS and SDHA was very mild, if any, confirming the selectivity of the ODD and EVEN probes for the X1 3ʹUTR (Figure 1(c), middle and right).
Identification of X1-targeting microRNAs from small RNA-seq data
The analysis of small RNA-seq data was performed using a bespoke approach and led to the identification of 20 microRNA species directly bound to the X1 3ʹUTR as those showing an enrichment in the pool of microRNAs obtained by affinity purification (miR-CATCHv2.0) compared to the full microRNA profile (microRNAome) of A375 cells. Specifically, the following steps were undertaken.
Identification of the microRNA species obtained by small RNA-seq of captured RNAs
The microRNA species present in each of the three independent ODD and EVEN captures (blue and green circles in Figure 2(a), respectively) were identified by comparing reads with sequences present in the miRBase repository (miRBase release 21), while their abundances were measured in reads per million (RPM). In each of the three experiments, ODD and EVEN captures showed a high degree of positive correlation (see Supplementary Table S2, second column). However, we reasoned that the relative abundance of microRNAs is proportional to the relative abundance of the captured target. Therefore, taking advantage of the fact that we introduced a sonication step, hence RNA molecules and in particular the X1 mRNA are cut into small pieces and captured during library construction, we quantified the X1 3ʹUTR by counting the number of reads that map to its sequence and were retrieved in each capture. Then, we used the X1-normalized RPM value as a scaling factor. By adding this step, we managed to increase correlation levels (see Supplementary Table S2, third column).
Figure 2.
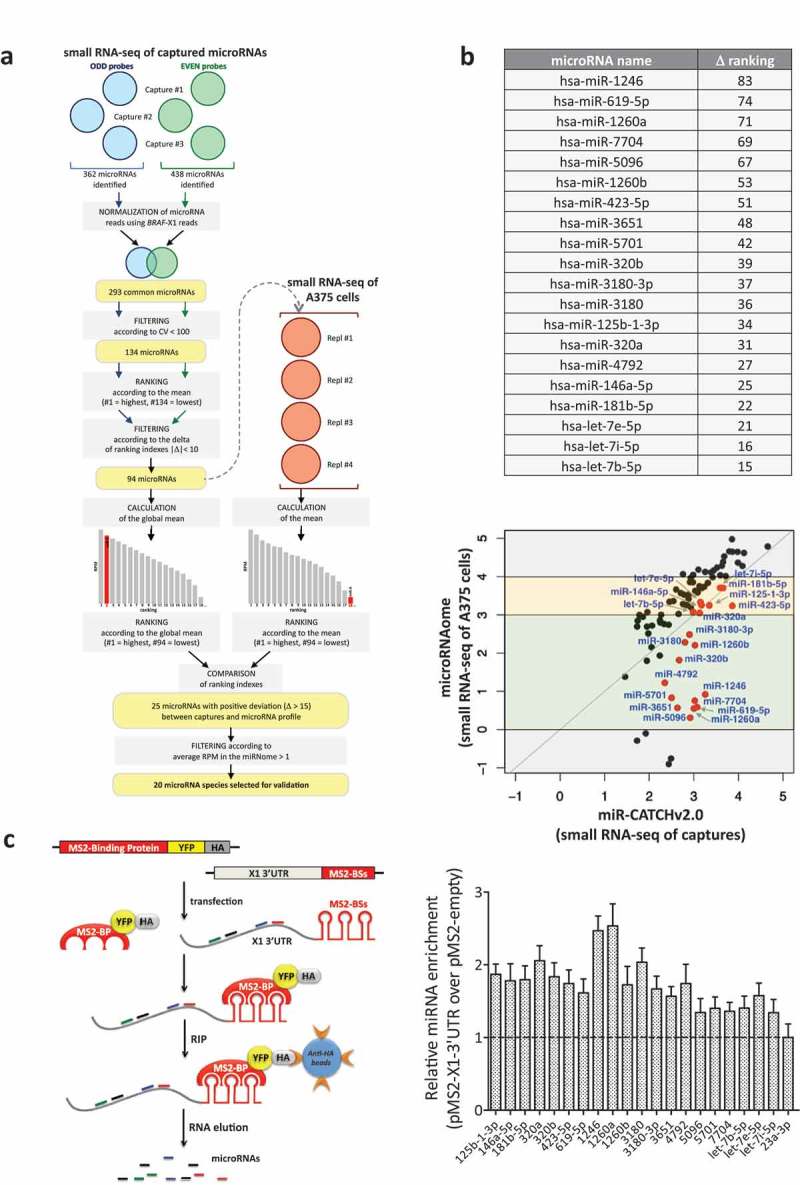
Identification of X1-binding microRNAs from small RNA-seq data.
(a). Description of the analytical steps followed in order to select the microRNAs to be validated (see text for details).
(b). (upper) List of the 20 selected microRNAs, ordered by difference in ranking index between the small RNA-seq of A375 cells and the small RNA-seq of captured microRNAs (see column N in Supplementary Table S3). (lower) Expression levels (log10(RPM mean)) of the 94 microRNAs in the small RNA-seq of the captures (X axis) and of A375 cells (Y axis). As indicated by the red dots, which represent the 20 selected microRNAs, overall our analytical approach prioritizes for experimental analysis the microRNA species that have intermediate expression levels in A375 cells (yellow and green middle boxes), while it discards those that are expressed at very high or very low levels (top and bottom grey boxes, respectively).
(c). (left) Cartoon describing the MS2-tagged RNA affinity purification (MS2-TRAP) assay (modified from [24]). This assay is based on the use of two plasmids. The pMS2-X1-3ʹUTR plasmid (right) expresses 12 copies of the MS2 binding site (BS), which is characterized by a defined secondary structure; furthermore, the presence of a multiple cloning site upstream of the MS2-BSs allows the insertion and co-expression of BRAF-X1 3ʹUTR. The other plasmid is called pMS2-BP (left) and expresses the MS2-Binding Protein (MS2-BP) fused with YFP and the HA-Tag. The MS2-BP, which is able to bind to MS2-BS, can be efficiently immunoprecipitated using the anti-HA antibody. Upon the co-transfection of the two plasmids inside the cells, a ribonucleoproteic complex is formed between the MS2-BP-YFP-HA chimerical protein and the BRAF-X1 3ʹUTR/MS2-BSs, to which endogenous microRNAs are physically bound. Such complex can be immunoprecipitated using anti-HA sepharose beads, so that, upon RNA extraction, the bound microRNAs can be quantified by qRT-PCR. (right) The qRT-PCR detection of the microRNAs identified by miR-CATCHv2.0 was performed on BRAF-X1 3ʹUTR immunoprecipitated using the MS2-TRAP assay. miR-23a-3p, which is depleted in the captures compared to the microRNA profile of A375 cells, is taken as negative control. The graphs in this figure represent the mean ± SEM of three independent experiments.
A total of 362 and 438 microRNAs were consistently identified in the three captures performed with ODD and EVEN probes (blue and green circles, respectively). Among them, 69 were identified using only the ODD probes, 145 using only the EVEN probes and 293 using both probe sets. For further analysis, we decided to focus on the 293 common microRNAs.
Selection of the microRNAs enriched in the captures compared to the microRNA profile of A375 cells
Among the 293 microRNAs, we decided to discard those that showed very different measurements in the three captures performed with the same pool of probes. Therefore, we selected the 134 microRNAs with coefficient of variation (CV) < 100.
These 134 microRNAs were ranked according to their mean abundance, separately, in the three ODD and three EVEN captures (#1 is the most abundant and #134 is the least abundant). The two lists were subsequently compared to identify concordant ranking: the 94 microRNAs selected were those whose ranking differs less than 10 positions between ODD and EVEN captures (see Supplementary Table S3 for a list).
The global mean of these 94 microRNAs (i.e. the mean of their abundance between captures performed with ODD and EVEN probes) was calculated and they were ranked according to mean values (#1 is the most abundant and #94 is the least abundant). In parallel, the mean abundance of the same 94 microRNAs was calculated using 4 small RNA sequencing datasets of A375 available to us (red circles) and they were ranked according to these mean values (again, #1 is the most abundant and #94 is the least abundant). Then, the ranking positions that each of the 94 microRNAs has in the small RNA-seq of captured microRNAs was compared with the ranking position that it has in the small RNA-seq of A375 cells. The 25 microRNAs that show a higher ranking position in the former versus the latter (positive deviation (Δ) > 15) were selected, as we reasoned that these are enriched. miR-A is shown as an example: ranking position in the small RNA-seq of A375 cells = 18; ranking position in the small RNA-seq of captured RNAs = 2; Δ = 16.
Among the 25 microRNAs, we excluded those that are expressed at very low levels in the small RNA-seq of A375 cells (less that 1 RPM on average). In so doing, 20 microRNA species were retained for further study (Figure 2(b), upper and Supplementary Figure S2).
Evaluation of the terms of enrichment of the 20 top scoring microRNAs
In Figure 2(b), lower we report the expression levels of the larger set of 94 microRNAs in the small RNA-seq of captured RNAs (miR-CATCHv2.0, X axis) versus the small RNA-seq of A375 cells (microRNAome, Y axis). According to the latter, these 94 microRNAs fall into four groups: those with very low or very high expression levels (grey boxes, log10(RPM mean) <0 or >4); those with intermediate-low expression levels (green box, log10(RPM mean) between 0 and 3; those with intermediate-high expression levels (yellow box, log10(RPM mean) between 3 and 4). As indicated by the red dots, which represent the 20 selected microRNAs, overall our analytical approach prioritizes for experimental analysis the microRNA species that have intermediate expression levels in A375 cells (green and yellow middle boxes), while it discards those that are expressed at very high or very low levels (top and bottom grey boxes, respectively).
The microRNAs expressed at very high levels are discarded because they tend to fall above the bisector line, which in turn means that their levels are higher in the microRNA profile compared to the captures. In other words, although captured, they are not enriched. This behaviour might be due to the fact that miR-CATCHv2.0 experiences some sort of saturation at high endogenous microRNA concentrations.
Conversely, the microRNAs that are expressed at very low levels are not prioritized for a ‘biological’ reason. As an example, miR-4508 is retrieved with 379 RPM on average using ODD probes and 229 RPM on average using EVEN probes, while it is undetectable in A375 cells (0 reads in the small RNA-seq of A375 cells). Therefore, it is highly enriched in the captures compared to the microRNA profile (it falls well below the bisector line in the bottom grey box) and in theory it is worth selecting. In spite of the robust enrichment, we still decided to discard it because we assume that the very low number of molecules present inside the cells makes its binding to BRAF-X1 inconsequential under a biological point of view [25].
As mentioned above, the 20 lead microRNAs have all intermediate expression levels. However, some are expressed at intermediate-low levels in the microRNA profile and show an absolute enrichment in the captures (green box, below the bisector line), while others are expressed at intermediate-high levels in the microRNA profile and were selected because in the captures they show a higher ranking position, in spite of the fact that their expression is not higher (yellow box, along the bisector line). Therefore, their enrichment has to be considered a relative one.
Technical validation of the direct binding of the 20 top scoring microRNAs to X1 mRNA
To confirm the binding of the 20 top-scoring microRNAs to the X1 3ʹUTR, we used the MS2-tagged RNA affinity purification (MS2-TRAP) assay (see Figure 2(c), left for a description [8]). Indeed, all 20 microRNAs show increased expression levels in presence of the exogenously transfected X1 3ʹUTR compared to the empty plasmid, which is indicative of direct binding (Figure 2(c), right). These findings attest the miR-CATCHv2.0 protocol and the subsequent bioinformatic analysis as reliable tools for the identification of microRNA species physically bound to an RNA of interest.
The microRNA Recognition Elements (MREs) of the 20 microRNA along the X1 3ʹUTR were mapped using RNAhybrid prediction algorithm (https://omictools.com/rnahybrid-tool) and the strongest MRE candidate for each microRNA is shown in Supplementary Figure S3.
Functional validation of the 20 BRAFV600E-X1-targeting microRNAs identified through miR-CATCHv2.0
We have previously demonstrated that X1 is the most abundant isoform of BRAF mRNA, irrespective of the mutational status of its coding region [20]. Therefore, we consider the 20 microRNAs identified through miR-CATCHv2.0 as both wtBRAF- and BRAFV600E-targeting. However, since they were identified in A375 cells, which carry a homozygous BRAFV600E mutation, from now on we will refer to them as BRAFV600E-X1-targeting.
In order to assess whether these 20 microRNAs are negative or positive regulators of BRAFV600E expression, we evaluated the effect of their overexpression on the levels of endogenous BRAFV600E-X1 mRNA. Each microRNA was transiently transfected in A375 cells as a double-stranded si-miRNA and 24 h later BRAFV600E-X1 was measured by qRT-PCR. As shown in Figure 3(a), the microRNAs cluster into three classes: those that induce a ≥ 20% decrease in BRAFV600E-X1 mRNA levels (miR-146a-5p, miR-423-5p, miR-1246 and miR-5701, Class I, red); those that induce a ≥ 20% increase in BRAFV600E-X1 mRNA levels (miR-125b-1-3p, miR-181b-5p, miR-3180, miR-3180-3p and miR-3651, Class II, blue); and those that do not alter BRAFV600E-X1 mRNA levels, that is cause a decrease or an increase < 20% (miR-320a, miR-320b, miR-619-5p, miR-1260a, miR-1260b, miR-4792, miR-5096, miR-7704, let-7b-5p, let-7e-5p and let-7i-5p, Class III, black). These three classes of microRNAs were studied separately.
Figure 3.
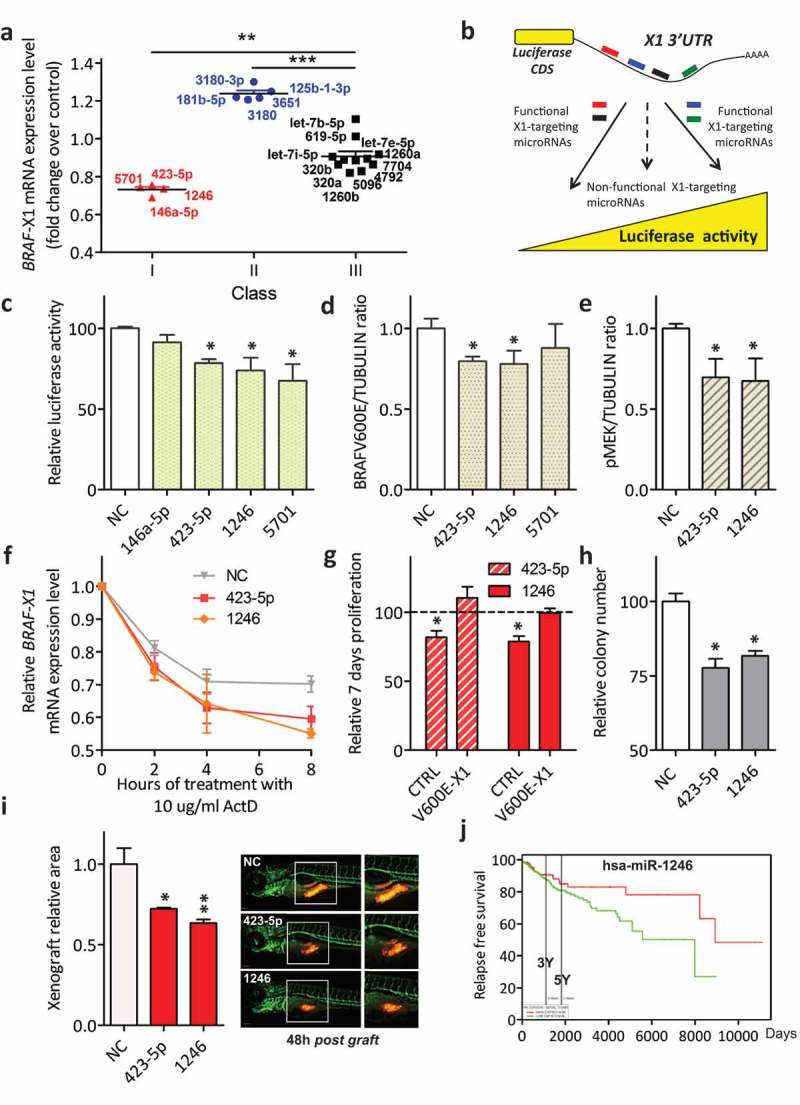
Classification of BRAFV600E-X1-binding microRNAs and functional validation of those that cause a decrease in BRAFV600E-X1 mRNA levels (Class I).
(a). The 20 microRNAs identified using miR-CATCHv2.0 were transiently transfected as si-miRNAs into A375 cells and were stratified in three different classes, according to the effect that they elicit on BRAFV600E-X1 mRNA levels 24 h later. Class I microRNAs (red) cause a ≥ 20% decrease in BRAFV600E-X1 mRNA levels compared to si-NC; Class II microRNAs (blue) cause a ≥ 20% increase in BRAFV600E-X1 mRNA levels; Class III microRNAs (black) do not alter BRAFV600E-X1 mRNA levels.
(b). Schematic representation of the Luciferase reporter assay. The pMIR-X1-3ʹUTR plasmid, in which the X1 3ʹUTR is cloned downstream of a Luciferase CDS, is transfected in HCT116 Dicer-/- cells together with the appropriate si-miRNAs. If a si-miRNA is functional, a decrease or increase in Luciferase activity is observed.
(c). miR-423-5p, miR-1246 and miR-5701 Class I microRNAs are functional because they cause a decrease in Luciferase activity.
(d-e). Quantification of BRAFV600E (d) and pMEK (e) protein levels, as detected by immunoblot 48 h after the transfection of si-miR-423-5p, si-miR-1246 and si-miR-5701 in A375 melanoma cells.
(f). Real-time PCR quantification of BRAF-X1 mRNA in A375 cells that were first transfected with the indicated siRNAs and 24 h later treated with 10 ug/ml Actinomycin D (ActD) for the indicated number of hours.
(g). Effect of the overexpression of miR-423-5p and miR-1246 on the proliferation of A375 melanoma cells and dependency on BRAFV600E-X1. Compared to si-NC, the transfection of si-miR-423-5p and si-miR-1246 causes a decrease in cell proliferation. Such a decrease is abrogated by the concomitant overexpression of the microRNA-insensitive BRAFV600E-X1 CDS, which indicates that it depends on the targeting of this protein. CTRL: A375 cells stably infected with the empty pCW lentiviral vector, transfected with si-miR-619-5p or si-let-7b-5p and induced with 2 ug/ml doxycycline for 48 h. V600E-X1: A375 cells stably infected with pCW-BRAFV600E-X1-CDS lentiviral vector, transfected with si-miR-619-5p or si-let-7b-5p and induced with 2 ug/ml doxycycline for 48 h. The proliferation of A375 cells stably infected with pCW-BRAFV600E-X1-CDS lentiviral vector, transfected with si-NC and induced with 2 ug/ml doxycycline for 48 h did not show a statistically significant difference compared to the proliferation of A375 cells stably infected with the empty pCW lentiviral vector, transfected with si-NC and induced with 2 ug/ml doxycycline for 48 h (which was taken as reference for all the other experimental conditions).
(h). Effect of the overexpression of miR-423-5p and miR-1246 on the colony forming ability of A375 melanoma cells.
(i). Effect of the overexpression of miR-423-5p and miR-1246 on the growth of A375-mCherry cells xenografted in zebrafish embryos. About 48 h after the transfection with si-miR-423-5p or si-miR-1246, cells were injected in 48 h post fertilization embryos and allowed to grow for an additional 48 h. At the end of this period, the size of the red cell masses was measured. The pictures are taken from 1 out of 3 independent experiments performed, all with comparable outcome. Scale bar: 100 um. The graphs in this figure represent the mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
(j). In the skin cutaneous melanoma dataset available at TCGA, patients with higher levels of miR-1246 (above the mean, red) show a better relapse free survival than patients with lower levels of miR-1246 (below the mean, green). n = 501. Samples above the mean: 160, 19 events. Samples below the mean: 341, 60 events. p = 0.0499.
Class I microRNAs are negative regulators of the stability of BRAFV600E-X1 mRNA
Given that Class I microRNAs (miR-146a-5p, miR-423-5p, miR-1246 and miR-5701) decrease BRAFV600E-X1 mRNA levels, we reasoned that they are ‘classical’ microRNAs that act as negative regulators of gene expression. Indeed, we show that Luciferase activity decreases when a reporter construct, in which the X1 3ʹUTR is cloned downstream of Luciferase CDS, is transiently transfected in HCT116 Dicer-/- cells together with si-miR-423-5p, si-miR-1246 or si-miR-5701 (Figure 3(b,c)). Furthermore, we used appropriate ΔMRE mutants of the reporter construct to validate the predicted MREs and confirm direct binding of the microRNAs under study to a specific portion of the X1 3ʹUTR (Supplementary Figures S4 and S5). We also show that, in the case of si-miR-423-5p and si-miR-1246, the decrease in endogenous BRAFV600E-X1 mRNA levels is coupled with a decrease in BRAFV600E protein levels and, consequently, of pMEK (Figure 3(d,e)). Finally, we demonstrate that these two microRNAs negatively affect BRAFV600E-X1 mRNA levels because they impair its stability (as reported in Figure 3(f), they accelerate mRNA decay in presence of Actinomycin D [26]).
Next, we studied whether miR-423-5p and miR-1246 alter the biological properties of A375 cells. Consistent with decreased BRAFV600E protein levels and attenuated ERK signalling, we found that they both cause a decrease in cell proliferation and in colony forming ability in vitro (Figure 3(g–h)). They also cause a decrease in the growth of tumour masses xenografted into zebrafish embryos (Figure 3(i)). Importantly, by performing a rescue experiment in presence of the microRNA-insensitive BRAFV600E-X1 CDS, we also provide experimental evidence that the microRNAs under testing exert their biological effects by targeting the X1 isoform of BRAFV600E (Figure 3(g) and Supplementary Figure S6). Finally, we found that, in melanoma patients, lower levels of miR-1246 are associated with worse relapse free survival, which further attests the oncosuppressive role of this microRNA (Figure 3(j)).
Class II microRNAs are positive regulators of the stability of BRAFV600E-X1 mRNA
The ≥ 20% increase in BRAFV600E-X1 levels suggested us that Class II microRNAs (miR-125b-1-3p, miR-181b-5p, miR-3180, miR-3180-3p and miR-3651) work as positive regulators of target gene expression. Indeed, we found that all five microRNAs increase X1 3ʹUTR reporter construct activity (Figure 4(a)) and two out of five (miR-3180 and miR-3651) increase endogenous BRAFV600E protein levels as well (Figure 4(b)). These two microRNAs were studied further.
Figure 4.
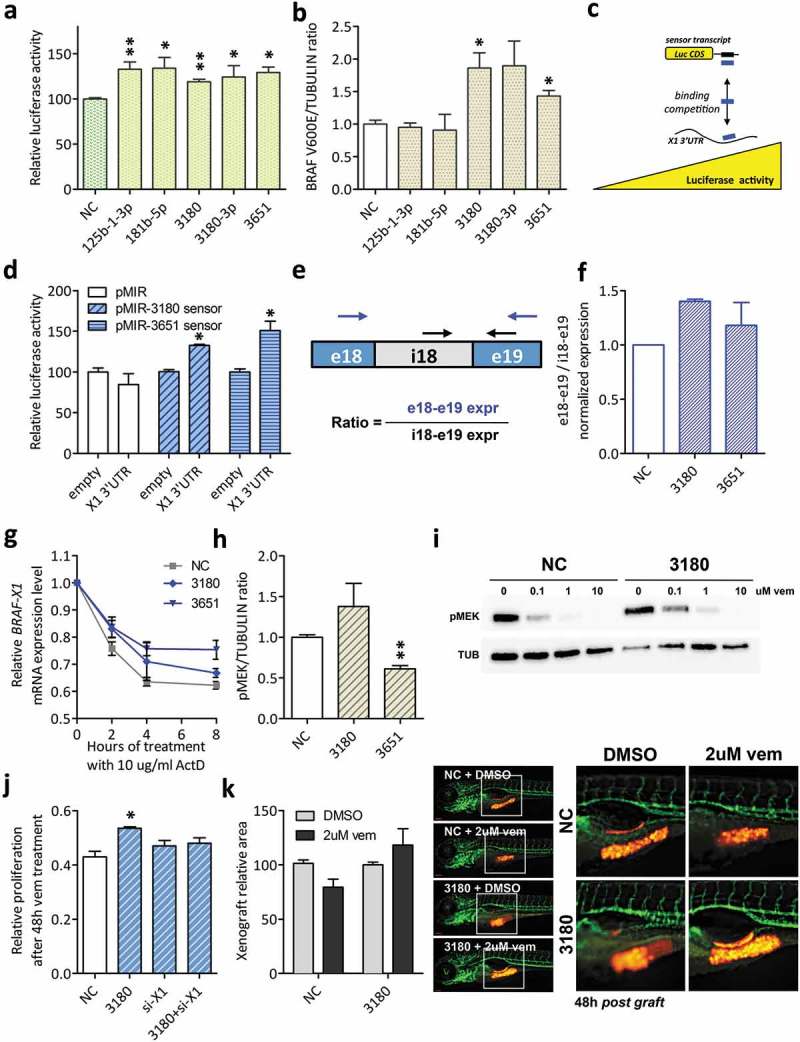
Functional validation of BRAFV600E-X1-binding microRNAs that cause an increase in BRAFV600E-X1 mRNA levels (Class II).
(a). Luciferase assay performed in HCT116 Dicer-/- cells indicates that miR-125b-1-3p, miR-181b-5p, miR-3180, miR-3180-3p and miR-3651 are functional, as they cause an increase in reporter gene activity.
(b). Quantification of BRAFV600E protein levels, as detected by immunoblot 48 h after the transfection of si-miR-125b-1-3p, si-miR-181b-5p, si-miR-3180, si-miR-3180-3p and si-miR-3651 in A375 melanoma cells.
(c–d). Sensor constructs for miR-3180 and miR-3651. (c). Schematic representation of the use of sensor constructs for X1-binding microRNAs. If the X1 3ʹUTR binds to a given microRNA species, the corresponding sensor transcript is released and an increase in Luciferase activity is observed. Blue rectangle: microRNA under study. Black rectangle: sequence complementary to that of the microRNA, expressed downstream of Luciferase CDS. (d). The sensor constructs for miR-3180 and miR-3651 show an increase in Luciferase activity in the presence of the X1 3ʹUTR, confirming its ability to bind to these microRNAs.
(e–f). The ratio between the expression level of exon 18 – exon 19 (taken as measure of mature BRAFV600E-X1 mRNA) and the expression level of intron 18 – exon 19 (taken as measure of BRAFV600E-X1 primary transcript) increases in the presence of si-miR-3180 and si-miR-3651. In (e), the position of the qRT-PCR primer pairs used is indicated by arrows (blue for e18-e19 and black for i18-e19). In (f), the results of real-time PCR analysis performed on A375 cells transfected with the indicated siRNAs are reported.
(g). Real-time PCR quantification of BRAF-X1 mRNA in A375 cells that were first transfected with the indicated siRNAs and 24 h later treated with 10 ug/ml Actinomycin D (ActD) for the indicated number of hours.
(h). Quantification of pMEK protein levels, as detected by immunoblot 48 h after the transfection of si-miR-3180 and si-miR-3651 in A375 melanoma cells.
(i). Immunoblot of pMEK performed on A375 cells that were transfected with si-NC or si-miR-3180 and 48 h later treated with the indicated doses of vemurafenib for 1 h.
(j). Effect of the overexpression of si-miR-3180 on the proliferation of A375 melanoma cells in presence of vem and dependency on BRAFV600E-X1. A375 cells were transfected with si-NC, si-miR-3180, si-X1 or si-miR-3180 + si-X1, treated with 2 uM vemurafenib for 48 h and then allowed to grow for 7 days. Compared to si-NC, the transfection of si-miR-3180 causes an increase in resistance of A375 cells to vem (measured as increase in proliferation). Such effect is abrogated by the concomitant knock-down of BRAFV600E-X1 by means of the co-transfection of si-X1 (an siRNA that targets the 3ʹUTR of BRAF-X1 mRNA [21]), which in turn indicates that the microRNA acts by targeting this protein.
(k). Effect of the overexpression of si-miR-3180 on the growth of A375-mCherry cells xenografted in zebrafish embryos. Cells were transfected with si-miR-3180 and treated with 2 uM vemurafenib for 48 h. They were then injected in 48 h post fertilization embryos and allowed to grow for an additional 48 h. At the end of this period, the size of red cell masses was measured (left). The pictures on the right are taken from one out of three independent experiments performed, all with comparable outcome. Scale bar: 100 um. The graphs in this figure represent the mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01.
First, their ability to bind to X1 mRNA was confirmed taking advantage of sensor constructs. In these constructs, the sequence complementary to that of the intended microRNA is cloned downstream of Luciferase CDS, so that the less microRNA is available for binding, the less the reporter mRNA is repressed, leading to higher Luciferase activity (Figure 4(c)). The sensors for miR-3180 or miR-3651 were transfected in HTC116 Dicer-/- cells together with the appropriate si-miRNAs, as well as the empty pMS2 or the pMS2-X1-3ʹUTR plasmid (see Figure 2(c)). Luciferase activity resulted higher in presence of pMS2-X1-3ʹUTR compared to pMS2 and this is a read out of the fact that both microRNAs bind to the X1 3ʹUTR, releasing the sensor (Figure 4(d)). Then, the binding of miR-3180 and miR-3651 to the MREs predicted by RNAhybrid algorithm was established using ΔMRE mutant constructs (Supplementary Figure S7). In addition, we show that miR-3180 and miR-3651 cause an increase BRAFV600E-X1 mRNA expression because they potentiate its stability at the post-transcriptional level: in Figure 4(e–f) we report the increase in the expression of BRAFV600E-X1 exons over introns that occurs in presence of si-miR-3180 and si-miR-3651, a result that rules out a transcriptional mode of action [27]. Furthermore, in Figure 4(g) we demonstrate that si-miR-3180 and si-miR-3651 slow down the decay of BRAFV600E-X1 mRNA in presence of Actinomycin D [26].
At the biological level, however, the overexpression of miR-3180 or miR-3651 does not cause the expected increase in the colony forming ability of A375 cells nor in their proliferation rate in vitro or in zebrafish (Supplementary Figure S8(a–c)). For miR-3651, we speculate that this is due to the fact that the increase in BRAFV600E levels not only is not coupled with the expected increase in pMEK levels, but the opposite rather occurs (pMEK levels decrease, Figure 4(h)), so that the net effect is null. Conversely, we hypothesized that the effects of the more sustained ERK signalling produced by miR-3180 overexpression become fully evident only when cells are exposed to a stress, such as the inhibition of BRAFV600E kinase activity by vemurafenib. Indeed, we observed that, when A375 are transfected with si-miR-3180, pMEK inhibition is achieved at higher doses of vemurafenib (Figure 4(i)) and, in turn, sensitivity to the drug decreases both in vitro (Figure 4(j)) and in vivo in zebrafish embryos (Figure 4(k)). Furthermore, by performing a rescue experiment in which the transfection of si-miR-3180 is coupled with that of si-X1 (an siRNA that targets the 3ʹUTR of BRAF-X1), we confirmed that miR-3180 exerts its protective activity against vem through the targeting of BRAFV600E-X1 (Figure 4(j)).
Class III microRNAs are negative or positive regulators of the translation of BRAFV600E-X1 mRNA
Since Class III microRNAs do not alter BRAFV600E-X1 mRNA levels, we used Luciferase assay to establish whether they are negative or positive regulators of target gene expression (i.e. whether they cause a decrease or an increase in BRAFV600E protein levels). Figure 5(a) shows that si-miR-619-5p, si-miR-7704, si-miR-let-7b-5p, si-let-7e-5p and si-let-7i-5p cause a decrease in the reporter activity, while si-miR-1260a and si-miR-1260b cause an increase. Consistently, we found that the transfection of si-miR-619-5p, si-miR-7704, si-let-7b-5p and si-let-7e-5p in A375 cells causes a decrease in endogenous BRAFV600E levels, while the transfection of si-miR-1260a causes an increase (Figure 5(b)). miR-619-5p, miR-7704, let-7b-5p and let-7e-5p (negative regulators of BRAFV600E expression, Class IIIa) and miR-1260a (positive regulator of BRAFV600E expression, Class IIIb) were studied further. We established that they bind directly to their predicted MREs (Supplementary Figure S9 and S10), then we focused on their molecular mechanism of action, as well as on their effects on ERK signalling and on the biological properties of A375 cells.
Figure 5.
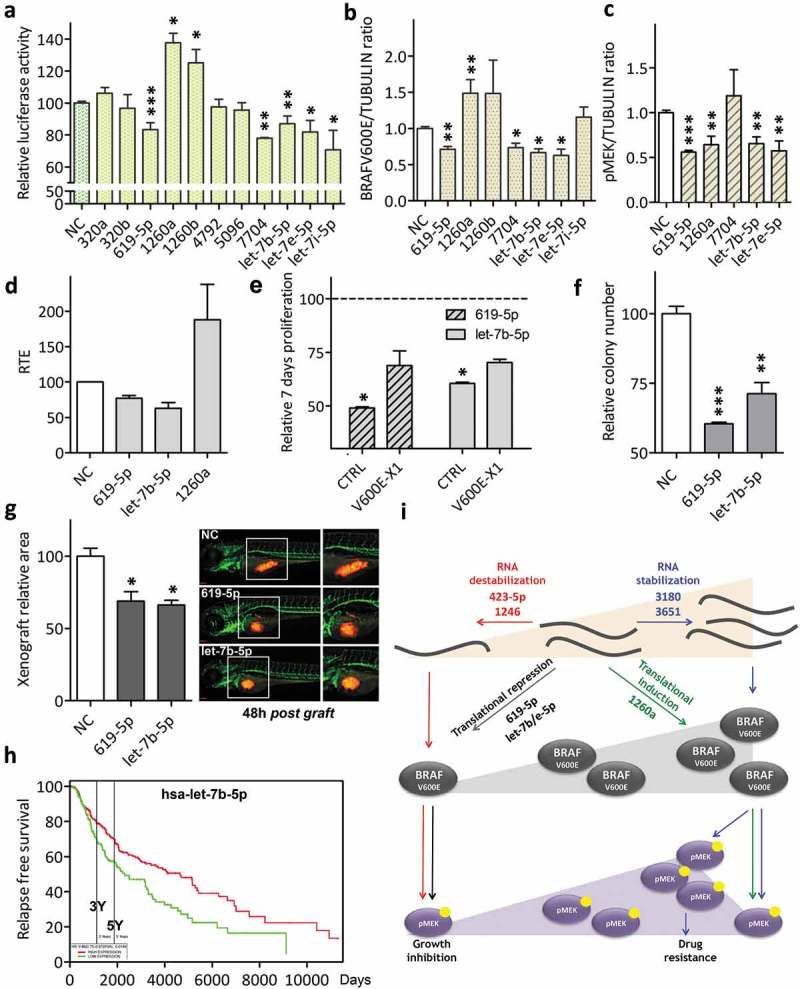
Functional validation of BRAFV600E-X1 binding microRNAs that do not alter BRAFV600E-X1 mRNA levels and rather affect its translation (Class III).
(a). Luciferase assay performed in HCT116 Dicer-/- cells indicates that miR-619-5p, miR-1260a, miR-1260b, miR-7704, let-7b-5p, let-7e-5p and let-7i-5p are functional, as they cause a decrease (miR-619-5p, miR-7704, let-7b-5p, let-7e-5p and let-7i-5p, Class IIIa) or an increase (miR-1260a and miR-1260b, Class IIIb) in reporter gene activity.
(b-c). Quantification of BRAFV600E (b) and pMEK (c) protein levels, as detected by immunoblot 48 h after the transfection of si-miR-619-5p, si-miR-1260a, si-miR-1260b, si-miR-7704, si-let-7b-5p, si-let-7e-5p and si-let-7i-5p into A375 melanoma cells.
(d). Compared to si-NC, the Relative Translation Efficiency (RTE) of Luciferase-X1-3ʹUTR chimerical construct, which is the ratio between Luciferase protein activity and Luciferase mRNA level [28], is lower in presence of si-miR-619-5p and si-let-7b-5p and higher in presence of si-miR-1260a.
(e). Effect of the overexpression of miR-619-5p and let-7b-5p on the proliferation of A375 melanoma cells and dependency on BRAFV600E-X1. Compared to si-NC, the transfection of si-miR-619-5p and si-let-7b-5p causes a net decrease in cell proliferation. Such effect is partially abrogated by the concomitant overexpression of the microRNA-insensitive BRAFV600E-X1 CDS, which indicates that the microRNA acts, at least in part, by targeting of this protein. CTRL: A375 cells stably infected with the empty pCW lentiviral vector, transfected with si-miR-619-5p or si-let-7b-5p and induced with 2 ug/ml doxycycline for 48 h. V600E-X1: A375 cells stably infected with pCW-BRAFV600E-X1-CDS lentiviral vector, transfected with si-miR-619-5p or si-let-7b-5p and induced with 2 ug/ml doxycycline for 48 h. The proliferation of A375 cells stably infected with pCW-BRAFV600E-X1-CDS lentiviral vector, transfected with si-NC and induced with 2 ug/ml doxycycline for 48 h did not show a statistically significant difference compared to the proliferation of A375 cells stably infected with the empty pCW lentiviral vector, transfected with si-NC and induced with 2 ug/ml doxycycline for 48 h (which was taken as reference for all the other experimental conditions).
(f). Effect of the overexpression of miR-619-5p and let-7b-5p on colony forming ability of A375 melanoma cells.
(g). Effect of the overexpression of miR-619-5p and let-7b-5p on the growth of A375-mCherry cells xenografted into zebrafish embryos. About 48 h after the transfection with si-miR-619-5p or si-let-7b-5p, the cells were injected in 48 h post fertilization embryos and allowed to grow for an additional 48 h. At the end of this period, the size of red cell masses was measured. The pictures are taken from one out of three independent experiments performed, all with comparable outcome. Scale bar: 100 um. The graphs in this figure represent the mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
(h). In the skin cutaneous melanoma dataset available at TCGA, patients with higher levels of let-7b-5p (above the mean, red) show a better overall survival than patients with lower levels of miR-1246 (below the mean, green). n = 501. Samples above the mean: 287, 116 events. Patients below the mean: 214, 96 events. p = 0.0149.
(i). Cartoon summarizing the mechanism of action of the X1-targeting microRNAs identified in this article and their effect on the biological properties of melanoma cells.
The decrease in BRAFV600E protein levels caused by miR-619-5p, let-7b-5p and let-7e-5p is accompanied by the expected decrease in pMEK levels (Figure 5(c)). As far as miR-619-5p and let-7b-5p are concerned (let-7b-5p was chosen as representative member of the let-7 family), we confirmed that the negative regulation of BRAFV600E expression that they produce is not due to accelerated decay of BRAFV600E-X1 mRNA (Supplementary Figure S11), but rather impaired translation. The Relative Translation Efficiency (RTE) [28] of the Luciferase-X1 3ʹUTR construct results in fact lower when miR-619-5p or let-7b-5p are overexpressed (Figure 5(d)).
At the biological level, we found that the decreased BRAFV600E protein levels and attenuated ERK signalling caused by miR-619-5p and let-7b-5p are accompanied by a decrease in cell proliferation (Figure 5(e)), in colony forming ability (Figure 5(f)) and in the growth of tumour masses xenografted into zebrafish embryos (Figure 5(g)). Analogously to what described above for Class I microRNAs, we also performed a rescue experiment in presence of the microRNA-insensitive BRAFV600E-X1 CDS and confirmed that both microRNAs exert these biological effects by targeting the X1 isoform of BRAFV600E (Figure 5(e) and Supplementary Figure S6). Finally, we found that, in melanoma patients, lower levels of let-7b-5p are associated with worse prognosis, confirming its role as an oncosuppressor (Figure 5(h)).
Neither miR-1260a affects the stability of BRAFV600E-X1 mRNA (Supplementary Figure S11) but it causes an increase rather than a decrease in its translation (Figure 5(d)). Yet, the consequent increase in BRAFV600E protein levels is not coupled with the expected increase in pMEK levels and the opposite rather occurs (Figure 5(c)). In turn, miR-1260a-overexpressing A375 cells do not show enhanced colony formation and proliferation ability in vitro, nor when xenografted in zebrafish embryos (Supplementary Figure S12).
Discussion
In this work, we use an improved version of our previously published miR-CATCH method [10,11] in order to identify the microRNA species that physically bind to the 3ʹUTR of the X1 isoform of BRAF. We also establish the molecular mechanism of action of these microRNAs and show the distinct effect that they exert on the expression of BRAFV600E itself, on the output of the ERK pathway, as well as on the biological properties of melanoma cells (Figure 5(i)).
Inspired by chromatin isolation by RNA purification (ChIRP)-based protocols [22,29] and by the adaptation of these protocols to the study of the binding of single microRNAs [30], in the upgraded miR-CATCHv2.0 we improved the affinity purification of the mRNA of interest by introducing a sonication-based RNA fragmentation step, as well as by using not only one, but several biotinylated probes that are tiled along the X1 3ʹUTR and are divided into two pools (ODD and EVEN). In addition, we used small RNA sequencing, rather than real-time PCR, in order to obtain the unbiased identification of all the microRNAs species physically bound to the X1 3ʹUTR [12]. Finally, the prioritization of the microRNAs to be validated was performed establishing their enrichment over a baseline, represented by the microRNA profile of A375 cells. The advantages offered by miR-CATCHv2.0 compared to the original miR-CATCH method are listed below.
First, by causing RNA fragmentation, the sonication step that was introduced simplifies probe design. A precise study of secondary and tertiary RNA structures in order to choose ‘open’ single-stranded regions is no longer required. Furthermore, RNA fragmentation enhances the stochastic accessibility of the target to the probes.
Second, since several antisense probes that target the same RNA molecule are used, miR-CATCHv2.0 allows to greatly increase specificity for the transcript of interest, while minimizing the noise due to aspecific probe binding. Furthermore, the probes can be designed to target the whole RNA molecule or just a portion of it. In the case of messenger RNAs, this in turn means that miR-CATCHv2.0 allows to identify microRNAs that bind to the 5ʹUTR, the ORF or the 3ʹUTR [31]. Finally, the splitting of the probes into two independent pools implies that each biological sample acts as a control for the other. Therefore, the robustness of the obtained results is increased. Interestingly, we obtained that the microRNAs identified using the pool of ODD probes and the pool of EVEN probes may not overlap in full, conceivably due to differences in binding efficiencies among probes, to the competition between probes and microRNA molecules for the binding to specific RNA regions or to non-random fragmentation of the target [32]. For example, in the case of the BRAF-X1 3ʹUTR, we retrieved 69 microRNA species using ODD probes only and 145 using EVEN probes only. In order to increase the total number of microRNA candidates and probably get a more comprehensive picture of microRNA-mediated post-transcriptional regulation of the RNA of interest, combining the two lists of microRNAs obtained with ODD and EVEN probes might be considered, besides intersecting them as we did.
Third, the small RNA-seq analysis included in the miR-CATCHv2.0 allows the de novo discovery of all the endogenous microRNA species bound to the target under study, irrespective of their identity (already deposited in miRBASE or novel), of the rules governing their binding (canonical or non-canonical [33]) and of the fact that they are predicted or not by target prediction algorithms. This is particularly useful when new or alternative transcript variants are under study, like in the case of BRAF-X1, since they are not included in the output of most target prediction algorithms [6]. In this respect, the comprehensiveness of our innovative methodology is comparable to that of techniques such as Ago-HITS-CLIP [34], Ago-PAR-CLIP [35], Ago-iPAR-CLIP [36] or CLASH [37] and yet the fact that it is tailored on a specific mRNA renders its execution and bioinformatic analysis within the reach of virtually any microRNA researcher.
Forth, the workflow we adopted in order to analyse the small RNA-seq output and select enriched microRNAs, which is focused on the comparison between the ranking that a given microRNA has in the pool of microRNAs obtained by affinity purification and the ranking that the same microRNA has in the microRNAome of the cells in which the affinity purification was performed, does not diminish the feasibility of our method: many microRNA profiles are publicly available in various repositories (e.g. the cancer genome atlas (TCGA, cancergenome.nih.gov), sequence read archive (SRA, www.ncbi.nlm.nih.gov/sra), gene expression omnibus (GEO, www.ncbi.nlm.nih.gov/geo)) and can be exploited for the purpose, without the need for annotating new ones. In addition, it comes with many advantages. In particular, it provides a reference point that takes into account the different expression levels displayed by microRNAs inside the cells and highlights the consequences that this might have on binding. In turn, this can be particularly useful when different experimental conditions are compared (e.g. different drug treatments, different developmental stages), because it can help understanding whether the gain/loss of the binding of a given microRNA is due to a change in its interaction with the target (the microRNA expression levels are unchanged, but its binding site becomes available or, on the contrary, is not available any longer) or to a change in its expression levels (the microRNA becomes able to bind to the target/cannot bind to the target anymore because it gets expressed/is no longer expressed). In light of all these considerations, we propose to use the appropriate microRNAome as the functional equivalent of the genome used in the original ChIRP protocol [22]: the microRNAome is the variable baseline necessary in order to identify enriched microRNAs, in the same way that the genome is the flat and universal baseline necessary to identify enriched DNA fragments.
Although in melanoma the most frequent alteration remains the single nucleotide substitution that renders BRAF protein constitutively active (e.g. BRAF 1799T>A, BRAFV600E) [38], the amplification of the mutant BRAFV600E allele has been recently reported as an event that quite frequently co-occurs [39–41]. Furthermore, it is renowned as one of the most common mechanisms of acquired resistance to BRAF and/or MEK inhibitors [42–46]. Finally, there are cancers types, such as breast cancer, in which the amplification of the BRAF gene is in fact more frequent than the mutation of the protein (http://www.cbioportal.org/). If we couple these data with our discovery that the most abundant isoform of BRAF mRNA is the X1, which is characterized by a 3ʹUTR that is extremely long (up to 7 kb [21]) and whose length is maintained in cancer compared to normal tissues [21,47], it is clear that the study of the microRNA-mediated post-transcriptional regulation of BRAF mRNA deserves to be undertaken systematically. Some microRNAs that target the less abundant reference BRAF isoform have already been reported and studied [48–50]. On the contrary, the microRNAs that were identified by us, using the miR-CATCHv2.0 method, are the first reported to specifically target the X1 isoform (Supplementary Figure S13). Interestingly, aside from let-7b-5p [51–53], these microRNAs were all of unknown function. Therefore, our work contributes to expand the list not only of mRNAs under microRNA regulation, but also of microRNAs with validated targets.
The mode of action of X1-targeting microRNAs highlights additional advantages offered by the miR-CATCHv2.0 method.
Not all the microRNA species that physically bind to the X1 3ʹUTR are functional: miR-146a-5p, miR-320a, miR-320b, miR-4792 and miR-5096 fail to alter the expression of a reporter gene, while miR-5701, miR-125b-1-3p, miR-181b-5p, miR-3180-3p, miR-1260b and let-7i-5p fail to alter the expression of the endogenous protein. This is not an uncommon event in the context of microRNA-target interactions [54,55] and might represent a mechanism by which the target RNA in turn regulates the availability hence the activity of microRNA molecules [56]. Conversely, the microRNAs that do alter X1 expression can be further classified into different classes, according to the net effect that they elicit: negative regulators act by decreasing the stability or translation of X1 mRNA (Class I and Class IIIa, respectively) and positive regulators act by increasing the stability or translation of X1 mRNA (Class II and Class IIIb, respectively). The identification of all the abovementioned groups of microRNAs would have not been possible without the employment of an experimental discovery tool that, like miR-CATCHv2.0, is totally unbiased. Neither would it have been possible without a ‘neutral’ attitude during the calling of the microRNA species bound to the target of interest. A bias towards microRNAs that act as negative regulators of gene expression and against microRNAs that act as positive regulators of gene expression would have likely led to an underestimation of the number and the impact of the latter group of microRNAs, in turn preventing a thorough assessment of the post-transcriptional regulatory circuits that revolve around our gene of interest.
It is also worth noticing that the microRNAs that affect BRAFV600E-X1 expression levels (miR-423-5p, miR-1246, miR-3180, miR-3651, miR-619-5p, miR-7704, let-7b-5p, let-7e-5p and miR-1260a) do not adhere to strict seed-seed match pairing rules. Using experimental methods like miR-CATCHv2.0 that are capable of recovering microRNAs physically associated with their targets, it is in fact common to identify non-canonical and yet functional microRNA-target interactions that are missed by classical prediction algorithms, such as TargetScan [20,33]. Specifically, many of the microRNAs predicted by TargetScan (see ref [20] for a list) are present in the extended group of 94 microRNAs. Yet, aside from miR-181b-5p, their enrichment is not so pronounced as that of the 20 top scoring microRNAs, which in turn suggests that overall the affinity for X1 3ʹUTR of ‘non-canonical’ microRNAs is higher than that of ‘canonical’ ones.
The mode of action of X1-targeting microRNAs also contributes to unveil the richness and complexity of the post-transcriptional regulation to which the ERK signalling pathway is subjected.
On one side, our data indicate that the pathway responds more promptly to a decrease rather than an increase in BRAFV600E protein levels. The microRNA-mediated downregulation of BRAFV600E levels is in fact accompanied by a decrease in pMEK levels in the case of miR-423-5p, 1246, 619-5p, let-7b and let-7e, while it appears inconsequential only in the case of miR-7704. Conversely, none of the microRNAs that increase BRAFV600E protein levels (miR-3180, miR-3651 and miR-1260a) also cause an increase in pMEK levels. In the case of miR-3651 and 1260a, an unexpected decrease in pMEK levels is evident. Conceivably, this can be ascribed to the fact that the microRNAs have other targets with an impact on the ERK pathway. Yet, it calls into question the ‘dogma’ that BRAF is the only kinase of MEK and MEK the only target of BRAF [57–60]. In the case of miR-3180, the signalling is indeed more sustained, but this becomes evident only when a stress condition is introduced (treatment with vemurafenib), as it has been observed when BRAFV600E overexpression occurs due to selective genomic amplification [42]. Overall, the failure of pMEK to increase in parallel with BRAFV600E might be attributable to the fact that too high levels of pMEK are toxic for melanoma cells and mechanisms are in place to avoid that [61,62].
On the other side, we found that the microRNAs that sustain BRAFV600E levels by increasing the stability or the translation of X1 mRNA prevail over those that produce the opposite effect (in Supplementary Figure S14(a) we show that Class II activators (miR-3180 and miR-3651) prevail over Class I repressors (miR-1246 and miR-5701, respectively); analogously, in Supplementary Figure S14(b) we show that Class IIIb activator miR-1260a prevails over Class IIIa repressors miR-619-5p, let-7b-5p and miR-7704).
Therefore, the microRNA-mediated post-transcriptional regulation of X1 seems to maintain BRAFV600E protein level within an optimal range, not too high so that the potential toxicity associated with the hyper-phosphorylation of MEK is avoided, and not too low so that the proliferative capacities of the cells are sustained in full. The subtle variations in protein levels caused by all four classes of microRNAs are indeed consistent with an overall buffering function designated to confer a high degree of robustness to the levels of BRAFV600E mRNA [63].
Finally, the formal demonstration that the X1 isoform is a microRNA target renders it a potential competing endogenous RNA [56] and prompts further investigation of its coding-independent functions. Interestingly, miR-3180 and 3651 increase the expression levels of BRAFV600E-X1 mRNA and are sequestered by the X1 3ʹUTR (Figure 4(d)). Furthermore, in the steady state, the increase in BRAFV600E-X1 mRNA is translated into an increase in BRAFV600E-X1 protein (Figure 4(b)), but not in an increase in ERK signalling (Figure 4(h)). Therefore, we speculate that, when stabilized by miR-3180 or miR-3651, BRAFV600E mRNA might exert a non-coding function and affect the expression of additional miR-3180 and 3651 targets [64,65].
In summary, miR-CATCHv2.0 is a method that can contribute to refining knowledge about the post-transcriptional regulation of any gene of interest. In particular, the use of miR-CATCHv2.0 for the identification of BRAF-X1 targeting microRNAs provides new insights into the complex post-transcriptional regulation to which the ERK pathway is subjected in melanoma, unveiling new fields of investigation and, possibly, expanding the portfolio of therapeutic opportunities.
Materials and Methods
Available as Supplementary Information.
Funding Statement
This work was supported by Istituto Toscano Tumori and Istituto per lo Studio, la Prevenzione e la Rete Oncologica [start up funding to LP]; Italian Ministry of Health [grant number GR-2011-02348535 to LP]; and Science Foundation Ireland [Technology Innovation Development Award number 12/TIDA/B2265 to CMG]. It was also partially supported by Associazione Italiana Ricerca sul Cancro [grant number MFAG 17095 to LP]. Funding for open access charge: Istituto per lo Studio, la Prevenzione e la Rete Oncologica.
Acknowledgements
The authors are grateful to: L. Tedeschi for her help with the purification of microRNA mimics; all members of the Poliseno lab and the Greene lab for insightful discussions.
Author contributions
AM, SV, CMG and LP conceived the project and designed the experiments; AM, SV, SM, EG, MR performed the experiments; RDA analysed the small RNA-seq data; GC analysed patients data; LeP, MP, CMG and LP supervised the project; all authors contributed to data analysis; LP wrote the manuscript with the help of all authors.
Availability
The microRNA reads obtained from miR-CATCHv2.0 and from A375 cells are accessible through GEO Super Series accession number GSE117642 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117642).
The prognostic values of microRNAs are available at the PROGmiR v2 prognostic miRNA database (http://xvm145.jefferson.edu/progmir).
The scripts of the analysis of small RNA-seq data are available at https://github.com/Romina/mirCTCH2analysis/tree/master.
Disclosure statement
The authors have no interest to disclose.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
References
- [1].Iwakawa HO, Tomari Y.. The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol. 2015;25:651–665. [DOI] [PubMed] [Google Scholar]
- [2].Valinezhad Orang A, Safaralizadeh R, Kazemzadeh-Bavili M. Mechanisms of miRNA-mediated gene regulation from common downregulation to mRNA-specific upregulation. Int J Genomics. 2014;2014:970607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Acunzo M, Romano G, Wernicke D, et al. MicroRNA and cancer–a brief overview. Adv Biol Regul. 2015;57:1–9. [DOI] [PubMed] [Google Scholar]
- [4].Fattore L, Costantini S, Malpicci D, et al. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget. 2017;8:22262–22278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tarang S, Weston MD. Macros in microRNA target identification: a comparative analysis of in silico, in vitro, and in vivo approaches to microRNA target identification. RNA Biol. 2014;11:324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Poliseno L, Pandolfi PP. PTEN ceRNA networks in human cancer. Methods. 2015;77-78:41–50. [DOI] [PubMed] [Google Scholar]
- [7].Shi M, Han W, Spivack SD. A quantitative method to identify microRNAs targeting a messenger RNA using a 3‘UTR RNA affinity technique. Anal Biochem. 2013;443:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yoon JH, Srikantan S, Gorospe M. MS2-TRAP (MS2-tagged RNA affinity purification): tagging RNA to identify associated miRNAs. Methods. 2012;58:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Braun J, Misiak D, Busch B, et al. Rapid identification of regulatory microRNAs by miTRAP (miRNA trapping by RNA in vitro affinity purification). Nucleic Acids Res. 2014;42:e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hassan T, Smith SG, Gaughan K, et al. Isolation and identification of cell-specific microRNAs targeting a messenger RNA using a biotinylated anti-sense oligonucleotide capture affinity technique. Nucleic Acids Res. 2013;41:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vencken S, Hassan T, McElvaney NG, et al. miR-CATCH: microRNA capture affinity technology. Methods Mol Biol. 2015;1218:365–373. [DOI] [PubMed] [Google Scholar]
- [12].De Santi C, Vencken S, Blake J, et al. Identification of MiR-21-5p as a functional regulator of mesothelin expression using MicroRNA capture affinity coupled with next generation sequencing. PLoS One. 2017;12:e0170999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Palfi A, Hokamp K, Hauck SM, et al. microRNA regulatory circuits in a mouse model of inherited retinal degeneration. Sci Rep. 2016;6:31431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Griffith A, Kelly PS, Vencken S, et al. miR-CATCH identifies biologically active miRNA regulators of the pro-survival gene XIAP, in Chinese Hamster ovary cells. Biotechnol J. 2018;13:e1700299. [DOI] [PubMed] [Google Scholar]
- [15].Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16:281–298. [DOI] [PubMed] [Google Scholar]
- [16].Dankner M, Rose AAN, Rajkumar S, et al. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. 2018;37:3183–3199. [DOI] [PubMed] [Google Scholar]
- [17].Dankort D, Curley DP, Cartlidge RA, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ascierto PA, Agarwala SS, Ciliberto G, et al. Future perspectives in melanoma research “melanoma bridge”, Napoli, November 30th–3rd December 2016. J Transl Med. 2017;15:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].McArthur GA. Combination therapies to inhibit the RAF/MEK/ERK pathway in melanoma: we are not done yet. Front Oncol. 2015;5:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Marranci A, Tuccoli A, Vitiello M, et al. Identification of BRAF 3‘UTR isoforms in melanoma. J Invest Dermatol. 2015;135:1694–1697. [DOI] [PubMed] [Google Scholar]
- [21].Marranci A, Jiang Z, Vitiello M, et al. The landscape of BRAF transcript and protein variants in human cancer. Mol Cancer. 2017;16:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chu C, Qu K, Zhong FL, et al. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Niranjanakumari S, Lasda E, Brazas R, et al. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods. 2002;26:182–190. [DOI] [PubMed] [Google Scholar]
- [24].Tuccoli A, Vitiello M, Marranci A, et al. Methods for the identification of PTEN-targeting microRNAs. Methods Mol Biol. 2016;1388:111–138. [DOI] [PubMed] [Google Scholar]
- [25].Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013;14:475–488. [DOI] [PubMed] [Google Scholar]
- [26].Ma F, Liu X, Li D, et al. MicroRNA-466l upregulates IL-10 expression in TLR-triggered macrophages by antagonizing RNA-binding protein tristetraprolin-mediated IL-10 mRNA degradation. J Immunol. 2010;184:6053–6059. [DOI] [PubMed] [Google Scholar]
- [27].Gaidatzis D, Burger L, Florescu M, et al. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat Biotechnol. 2015;33:722–729. [DOI] [PubMed] [Google Scholar]
- [28].Lin CC, Liu LZ, Addison JB, et al. A KLF4-miRNA-206 autoregulatory feedback loop can promote or inhibit protein translation depending upon cell context. Mol Cell Biol. 2011;31:2513–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chu C, Zhang QC, Da Rocha ST, et al. Systematic discovery of Xist RNA binding proteins. Cell. 2015;161:404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Roy-Chaudhuri B, Valdmanis PN, Zhang Y, et al. Regulation of microRNA-mediated gene silencing by microRNA precursors. Nat Struct Mol Biol. 2014;21:825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu C, Mallick B, Long D, et al. CLIP-based prediction of mammalian microRNA binding sites. Nucleic Acids Res. 2013;41:e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Poptsova MS, Il‘Icheva IA, Nechipurenko DY, et al. Non-random DNA fragmentation in next-generation sequencing. Sci Rep. 2014;4:4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Seok H, Ham J, Jang ES, et al. MicroRNA target recognition: insights from transcriptome-wide non-canonical interactions. Mol Cells. 2016;39:375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chi SW, Zang JB, Mele A, et al. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hafner M, Landthaler M, Burger L, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jungkamp AC, Stoeckius M, Mecenas D, et al. In vivo and transcriptome-wide identification of RNA binding protein target sites. Mol Cell. 2011;44:828–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Helwak A, Kudla G, Dudnakova T, et al. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fiskus W, Mitsiades N. B-Raf inhibition in the clinic: present and future. Annu Rev Med. 2016;67:29–43. [DOI] [PubMed] [Google Scholar]
- [39].Cancer Genome Atlas N Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hayward NK, Wilmott JS, Waddell N, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545:175–180. [DOI] [PubMed] [Google Scholar]
- [41].Stagni C, Zamuner C, Elefanti L, et al. BRAF gene copy number and mutant allele frequency correlate with time to progression in metastatic melanoma patients treated with MAPK inhibitors. Mol Cancer Ther. 2018. [DOI] [PubMed] [Google Scholar]
- [42].Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nathanson KL, Martin AM, Wubbenhorst B, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res. 2013;19:4868–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xue Y, Martelotto L, Baslan T, et al. An approach to suppress the evolution of resistance in BRAFV600E-mutant cancer. Nat Med. 2017;23:929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Villanueva J, Infante JR, Krepler C, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4:1090–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Moriceau G, Hugo W, Hong A, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell. 2015;27:240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Park HJ, Ji P, Kim S, et al. 3‘ UTR shortening represses tumor-suppressor genes in trans by disrupting ceRNA crosstalk. Nat Genet. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang Z, Ma B, Ji X, et al. MicroRNA-378-5p suppresses cell proliferation and induces apoptosis in colorectal cancer cells by targeting BRAF. Cancer Cell Int. 2015;15:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu SM, Lu J, Lee HC, et al. miR-524-5p suppresses the growth of oncogenic BRAF melanoma by targeting BRAF and ERK2. Oncotarget. 2014;5:9444–9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fattore L, Mancini R, Acunzo M, et al. miR-579-3p controls melanoma progression and resistance to target therapy. Proc Natl Acad Sci U S A. 2016;113:E5005–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Fu TY, Chang CC, Lin CT, et al. Let-7b-mediated suppression of basigin expression and metastasis in mouse melanoma cells. Exp Cell Res. 2011;317:445–451. [DOI] [PubMed] [Google Scholar]
- [52].Schultz J, Lorenz P, Gross G, et al. MicroRNA let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Res. 2008;18:549–557. [DOI] [PubMed] [Google Scholar]
- [53].Yamada S, Tsukamoto S, Huang Y, et al. Epigallocatechin-3-O-gallate up-regulates microRNA-let-7b expression by activating 67-kDa laminin receptor signaling in melanoma cells. Sci Rep. 2016;6:19225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Agarwal V, Bell GW, Nam JW, et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Cloonan N. Re-thinking miRNA-mRNA interactions: intertwining issues confound target discovery. Bioessays. 2015;37:379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Salmena L, Poliseno L, Tay Y, et al. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Matallanas D, Birtwistle M, Romano D, et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2:232–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Park ER, Eblen ST, Catling AD. MEK1 activation by PAK: a novel mechanism. Cell Signal. 2007;19:1488–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Stuart SA, Houel S, Lee T, et al. A phosphoproteomic comparison of B-RAFV600E and MKK1/2 inhibitors in melanoma cells. Mol Cell Proteomics. 2015;14:1599–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gopalbhai K, Jansen G, Beauregard G, et al. Negative regulation of MAPKK by phosphorylation of a conserved serine residue equivalent to Ser212 of MEK1. J Biol Chem. 2003;278:8118–8125. [DOI] [PubMed] [Google Scholar]
- [61].Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Leung GP, Feng T, Sigoillot FD, et al. Hyperactivation of MAPK signaling is deleterious to RAS/RAF mutant melanoma. Mol Cancer Res. 2018;78:965–966. [DOI] [PubMed] [Google Scholar]
- [63].Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processe s. Cell. 2012;149:515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Gilot D, Migault M, Bachelot L, et al. A non-coding function of TYRP1 mRNA promotes melanoma growth. Nat Cell Biol. 2017;19:1348–1357. [DOI] [PubMed] [Google Scholar]
- [65].Migault M, Donnou-Fournet E, Galibert MD, et al. Definition and identification of small RNA sponges: focus on miRNA sequestration. Methods. 2017;117:35–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.