


Abstract
BRCA1-associated basal-like breast cancer originates from luminal progenitor cells. Breast epithelial cells from cancer-free BRCA1 mutation carriers are defective in luminal differentiation. However, how BRCA1 deficiency leads to lineage-specific differentiation defect is not clear. BRCA1 is implicated in resolving R-loops, DNA-RNA hybrid structures associated with genome instability and transcriptional regulation. We recently showed that R-loops are preferentially accumulated in breast luminal epithelial cells of BRCA1 mutation carriers. Here, we interrogate the impact of a BRCA1 mutation-associated R-loop located in a putative transcriptional enhancer upstream of the ERα-encoding ESR1 gene. Genetic ablation confirms the relevance of this R-loop-containing region to enhancer-promoter interactions and transcriptional activation of the corresponding neighboring genes, including ESR1, CCDC170 and RMND1. BRCA1 knockdown in ERα+ luminal breast cancer cells increases intensity of this R-loop and reduces transcription of its neighboring genes. The deleterious effect of BRCA1 depletion on transcription is mitigated by ectopic expression of R-loop-removing RNase H1. Furthermore, RNase H1 overexpression in primary breast cells from BRCA1 mutation carriers results in a shift from luminal progenitor cells to mature luminal cells. Our findings suggest that BRCA1-dependent R-loop mitigation contributes to luminal cell-specific transcription and differentiation, which could in turn suppress BRCA1-associated tumorigenesis.
INTRODUCTION
Women who harbor germline mutations in BRCA1 have increased lifetime risk of developing breast cancer (1,2). BRCA1-associated breast tumors are usually early onset, hormone receptor-negative and basal-like, yet originate from luminal progenitor cells (3–6). Luminal progenitor cells from precancerous breast tissue of BRCA1 mutation carriers are compromised in differentiation to ER-positive mature luminal cells (7–10). However, it remains unclear how BRCA1 cancer-predisposing mutations preferentially block luminal epithelial differentiation of these cells of origin for BRCA1-associated tumors. BRCA1 contributes to a variety of cellular processes, including DNA repair, DNA replication stress reduction, cell cycle checkpoint, transcriptional regulation, chromatin remodeling, and ubiquitination (11–13). Though BRCA1 functions in promoting DNA repair and resolving DNA replication stress have been extensively investigated, loss of these BRCA1 functions that are ubiquitously important to all proliferating cells may not be sufficient to explain tissue- and sex-specific cancer development in BRCA1 mutation carriers. Rather, regulation of tissue- and cell type-dependent gene transcription by BRCA1, in combination with its well-documented roles in maintenance of genomic stability, is more likely to fully account for BRCA1-dependent tumor suppression in selective tissues (14–16).
R-loops have a three-strand nucleic acid structure that comprises a nascent RNA strand hybridized with the DNA template strand, leaving the non-template DNA single-stranded. Initially thought to be mere byproducts of transcription with little biological consequences, R-loops have been shown to be important regulators of gene expression and a major threat to genome stability (17–25). Recent in vitro cell line studies have shown that BRCA1/BRCA2 knockdown resulted in accumulation of R-loops (26–30). Using sorted primary cells from precancerous breast tissue, we recently found that R-loops are preferentially accumulated in luminal epithelial cells from BRCA1 mutation-carrying breast tissue (29). Furthermore, these BRCA1 mutation-associated R-loops tend to be localized at transcription start sites (TSS). Of note, a number of these R-loop-associated genes are involved in luminal fate determination and differentiation, such as XBP1, GATA3, CEBPB and FOXC1 (29), raising the distinct possibility that BRCA1-dependent modulation of R-loop dynamics at these gene loci could contribute to BRCA1 functions in luminal cell differentiation and tissue-specific tumor suppression.
In the current study, we chose a BRCA1-associated R-loop for in-depth functional analysis. This R-loop is located in a noncoding region upstream of ESR1 locus, which encodes estrogen receptor α (ERα). We first used genetic editing to demonstrate the functional relevance of this R-loop-containing region in transcriptional activation of ESR1 and other neighboring genes. We further investigated antagonism between BRCA1 depletion and R-loop removal on gene transcription in this genomic region. Lastly, we found that R-loops removal by ectopic RNase H1 promoted a switch from primary luminal progenitor cells to mature luminal cells isolated from BRCA1 mutation carriers.
MATERIALS AND METHODS
Human tissue sample procurement
Cancer-free breast tissues were obtained with informed patient consent from women undergoing either cosmetic reduction mammoplasty or prophylactic mastectomy, following protocols approved by the Institutional Review Board at the University of Texas Health Science Center at San Antonio.
Cell culture, siRNA transfection and lentiviral infection
MCF7 was purchased from ATCC and cultured in high glucose DMEM (Thermo Fisher Scientific; 11965) supplemented with 10% fetal bovine serum (FBS), 100 unit ml−1 penicillin and 100 μg ml−1 streptomycin (Thermo Fisher Scientific; 15140122). Non-targeting control siRNA pools (D-001810-10) and human CCDC170 siRNA SMARTpool (L-014568-01-0005) were purchased from Dharmacon. Individual siRNAs were synthesized from Sigma-Aldrich (see Table 1 for target sequences). siRNA knockdown experiments were performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific; 13778150) following the manufacturer's instruction. Briefly, 20 nM of siRNA was transfected with 25 μl of RNAiMAX reagent. Experiments were carried out three days after siRNA transfection.
Table 1.
siRNA target sequences and PCR primer sequences
| siRNA oligoes | ||
| Name | Target sequences | |
| siBRCA1 | GAAGCCAGCTCAAGCAATA | |
| siRMND1 | GAAAATCGGTGAACTCTTT | |
| Primer sequences for RT-PCR | ||
| Name | Forward primer | Reverse primer |
| BRCA1 | ACCTTGGAACTGTGAGAACTCT | TCTTGATCTCCCACACTGCAATA |
| CCDC170 | GTCGTCATGAGCCTGGACTG | ACCGGGACTTCCGAAAGATG |
| RMND1 | AGGAGCTGCTGTGTTTTGGA | CCCAGTGTACCAGTGCGATT |
| ESR1 | ATCTCTCTGGCGCTTGTGTT | TGCTACGAAGTGGGAATGATGA |
| RNASEH1 | AGGAATCGGCGTTTACTGGG | CTCTTTGGTTTGTCTGCCGC |
| CITED2 | CCTAATGGGCGAGCACATACA | GGGGTAGGGGTGATGGTTGA |
| PPP1R14C | TGGAGCAGCTGGGTCAG | TCTCTTCATCACTGTCTGCATCA |
| MTHFD1L | CTGCCTTCAAGCCGGTTCTT | TTTCCTGCATCAAGTTGTCGT |
| PLEKHG1 | CTCCCCGGGTGAAGACTGAT | GCACTTCAAGACGCAACTGG |
| AKAP12 | GAGATGGCTACTAAGTCAGCGG | CAGTGGGTTGTGTTAGCTCTTC |
| ZBTB2 | ATCAGACCAGTGAGTGTGTCC | ACTTGATGCCCTGTTCTAATCG |
| SYNE1 | TGCCATTCGACCGGAATTGG | GTTCTGTTTCGGCGATAGTGAA |
| FBXO5 | CATGCGTGATAGACCCTCCACA | TCACACTTCATTTTGACAGAAAGGG |
| SOD2 | AAACCTCAGCCCTAACGGTG | CACGTTTGATGGCTTCCAGC |
| GATA3 | GCCCCTCATTAAGCCCAAG | TTGTGGTGGTCTGACAGTTCG |
| PGR | ATGGAAGGGCAGCACAACTA | CGACATGCTGGGCAGTTTTT |
| FOXA1 | CCAGGATGTTAGGAACTGTGA | GAGTAGGCCTCCTGCGTG |
| XBP1 | CTGAGTCCGCAGCAGGTG | GGAGATGTTCTGGAGGGGTG |
| CEBPB | TTTGTCCAAACCAACCGCAC | TAAATAACACCACGGGCGGGACTB |
| 18S rRNA | AACGGCTACCACATCCAAGG | GGGAGTGGGTAATTTGCGC |
| ACTB | AGGCACCAGGGCGTGAT | GCCCACATAGGAATCCTTCTGAC |
| Primer sequences for DRIP | ||
| Name | Forward primer | Reverse primer |
| ESR1-RE | TGGACGTTAACTGCAGCCCA | TCATCCCATAATCAGGACCATCAAT |
| Geneless | CCTGTGTCTGACATATGATTGGTTT | CCATTGCCTGGCTGTTATTTG |
| Primer sequences for 3C | ||
| Name | Primer sequence | |
| ESR1-RE_BamHI_F | CGAACCTAGCACAGTGAAAGTTGCCCATA | |
| ESR1-TSS_BamHI _F | CACCTCCTCCATGTGGCTTGTACCTGTTT | |
| CCDC170-TSS_BamHI _F | GTGAACCAATATCTCTGTGAGACCCTGCT | |
| RMND1-TSS_BamHI _F | GAGAAAGAAATAAGGGGACCTGGGGAACC | |
| RMND1-TTS_BamHI_F | CCCCAGGAGAAGGTGGGATTAGAAGTCA | |
| RMND1-GB_BamHI_F | CACGGGGCCTGGCCTAAGTATTGTTAAA | |
| CCDC170-GB_BamHI_F | TCCCCAGCTGCCTCCTGTTACTCTCTTT | |
| CCDC170-GB2_BamHI_F | CTCCATGGGCCTCAGAGAAATTTAGCATC | |
| ESR1-GB_BamHI_F | ACCATGCCCAGCTAATTGCAGGTGATTT | |
| GAPDH_Ctrl_F | TCCTCCTGTTTCATCCAAGC | |
| GAPDH_Ctrl_R | TAGTAGCCGGGCCCTACTTT | |
Human RNASEH1 gene that encodes the nuclear form of the protein (M27; Addgene 65782) was subcloned into pCDH-EF1-MCS-T2A-Puro lentivector (System Biosciences; CD520A-1) via EcoRI/BamHI restriction sites. Empty vector (EV) and RNase H1 (RH1) lentiviruses were produced in HEK293T cells by co-transfecting cells with the lentiviral vectors and the corresponding packaging plasmids using Lipofectamine 2000 (Thermo Fisher Scientific; 11668019). Viral supernatant was collected 48 h after transfection and was passed through a 0.45 μM filter (Foxx Life Sciences; 146-2313-RLS). Lentiviruses were titered using a Quantitative PCR-Based Lentivirus Titration Kit (Applied Biological Materials; LV900) following the manufacturer's instruction. MCF7 cells infected with the same titer of either empty vector (EV) or RNase H1 (RH1) lentivirus were selected using 2 μg ml−1 puromycin (Gibco; A11138-03). Single clones with highest RNase H1 overexpression were picked for the ensuing experiments.
CRISPR deletion clones
The target sequence flanking ESR1-RE R-Loop region was submitted to online CRISPR Design Tool (http://crispr.mit.edu/), and the sgRNAs with highest score were chosen. Corresponding oligonucleotides were synthesized by a commercial source (Sigma) and subcloned into the pSpCas9 (BB)-2A-Puro plasmid (pX459, Addgene #62988) following previously published protocol (31). The sgRNA sequences are ESR1-RE_L1: GAAGGAATTAGCGTGAGTC; ESR1-RE_L2: AGCGTGAGTCCAGAGTAGA; ESR1-RE_R1: TGTCAATAAAACCGAGTTTC; ESR1-RE_R2: GGATAGCTCAGGAATACCAG.
MCF7 cells were co-transfected with two pX459 plasmids containing the sgRNA flanking the targeted region, using Lipofectamine 2000 (Invitrogen). Transfected cells were first selected with puromycin (2 μg ml−1) for 72 h, followed by single-cell sorting into 96-well plates. After propagation of individual clones, genomic DNA was extracted and screened for deletion events. PCR primers were designed to cover sequences outside of the deleted region. The PCR primers used are, CRISPR_F1: GAAGTGGATCTACCATGGGTGT; CRISPR_R1: TGTGGAATGGAAAACAAAATGATGA. The PCR-screened deletion clones were further validated by Sanger sequencing.
Quantitative RT-PCR analysis
Total RNA was extracted using Trizol (Invitrogen) and used for random hexamer-based reverse-transcription (ImProm-II™ Reverse Transcription System, Promega) according to the manufacturer's protocol. RT-PCR was performed using an ABI-7300 sequence detection system using SYBR Green qPCR master mix (Thermo Fisher Scientific; FERK0221). Each measurement was performed in duplicate and expression levels of either ACTB or 18S rRNA were used for normalization. The primers used for quantitative RT-PCR are listed in Table 1.
Antibodies
Antibodies used for Western blotting are: mouse anti-BRCA1 1:200 (Calbiochem; OP92), rabbit anti-CCDC170 1:1,000 (Atlas Antibodies; HPA027185), rabbit anti-RMND1 1:500 (Sigma; HPA031399), rabbit anti-RNASEH1 1:5000 (Proteintech; 15606-1-AP), rabbit anti-β-ACTIN 1:7,000 (Cell Signaling Technology; 4967S), mouse anti-VINCULIN 1:2,000 (Proteintech; 66305-I), rabbit anti-GAPDH 1:10 000 (Cell Signaling Technology; 2118), mouse anti-α-Tubulin 1:10 000 (Calbiochem; CP06). Rabbit anti-RNASEH1 1:500 (Proteintech; 15606-1-AP) was used for immunofluorescence.
S9.6 antibody for detecting RNA-DNA hybrids was purified from the mouse S9.6 hybridoma cell line (ATCC; HB8730). Briefly, suspension cell cultures were grown in antibody-producing media (high glucose DMEM, 2% FBS, 2 mM l-glutamine, 1 mM sodium pyruvate, 100 unit ml−1 penicillin and 100 μg ml−1 streptomycin) for ∼9 days. Cultured medium was centrifuged at 1000 rpm for 5 min and sterilized through a 0.22 μM filter. Concentrated supernatant was first diluted with 1× volume of protein A IgG binding buffer (Pierce; 21001) and purified using the Protein A Plus Spin Kit following manufacture's protocol (Pierce; 89978). Fractions containing high concentrations of antibody were dialyzed with Spectra/Por molecular porous membrane tubing (MWCO 12–14k, Spectrum; 132678) in PBS at 4°C overnight. The antibody concentration was measured and aliquots were stored at −80°C.
Chromatin conformation capture (3C) assay
3C experiments were carried out essentially as described previously with minor modification (32,33). Briefly, ∼1 × 107 cells were crosslinked with 1% formaldehyde at room temperature for 10 min. Cross-linking reactions were quenched by addition of glycine at the final concentration of 125 mM. Nuclei of crosslinked cells were purified by incubating the cells with lysis buffer (10 mM Tris–HCl, pH 8.0, 10 mM NaCl, 0.2% NP-40, 1 μg ml−1 leupeptin, 1 μg ml−1 aprotinin, 1 μg ml−1 pepstatin and 1 mM PMSF) for 15 min on ice, then passed through a 21 G needle 15 times, incubated on ice for 10 min, and finally centrifuged at 2200 g at 4°C for 5 min. Resultant nuclei were re-suspended in restriction enzyme buffer. SDS was added to the nuclei at a final concentration of 0.1%. Samples were incubated on a 65°C shaker for 10 min and kept on ice afterwards. Triton-X-100 was added to quench SDS at a final concentration of 1%. Two criteria were used in choosing the appropriate restriction enzyme. First, sequences after deletion in knockout clones do not fortuitously generate a new site for the chosen restriction enzyme. Second, the primer sequences do not overlap with the deleted region. Samples were digested with 400 U of BamHI (New England Biolabs; R3136L) overnight at 37°C. To inactivate the restriction enzyme, digested samples were treated with SDS at a final concentration of 1.6% on a shaker for 30 min at 65°C. Samples were ligated in ligation buffer (1% Triton-X-100, 0.8 mg BSA, 50 mM Tris–HCl, pH 8.0, 10 mM MgCl2, 10 mM DTT and 2 mM ATP) with 300 U of T4 DNA ligase (Thermo Fisher Scientific; EL0011) for 4 h at 16°C under a diluted condition that favors ligation events between crosslinked fragments. Ligated samples were treated with 0.5 mg Proteinase K at 65°C for 4 h, followed by an additional 0.5 mg of Proteinase K treatment at 65°C overnight to reverse cross-linking. DNA fragments were extracted with phenol-chloroform and precipitated with 0.3 M sodium acetate (pH 5.6) and ethanol. Samples were treated with RNase A at 37°C for 2 h, followed by phenol-chloroform extraction and ethanol precipitation. DNA was dissolved in TE at 4°C overnight. For quantification, serially diluted 3C products were analyzed by PCR to determine the linear range. 3C samples within the linear range were analyzed by PCR and normalized by reference to a region within GAPDH. 3C primers were listed in Table 1.
DRIP
DRIP was performed as previously described (19). Briefly, MCF7 cells were first lysed with SDS/Proteinase K treatment at 37°C. After overnight incubation, total nucleic acid was extracted by phenol-chloroform extraction in Phase Lock Gel™ tubes (5 PRIME; 2302840) and ethanol/sodium acetate precipitation. DNA was fragmented using a cocktail of restriction enzymes (BsrG1, EcoR1, HindIII, SspI, XbaI) overnight at 37°C. DNA was purified by phenol-chloroform extraction and ethanol precipitation. For DRIP, 4.4 μg of digested DNA was incubated with 10 mg S9.6 antibody in binding buffer (10 mM sodium phosphate, 140 mM NaCl, 0.05% Triton X-100 in TE) overnight at 4 °C. Dynabeads were added the following day for 2 h. Bound Dynabeads were then washed with binding buffer three times at room temperature. DNA was eluted in elution buffer (50 mM Tris pH 8, 10 mM EDTA, 0.5% SDS, Proteinase K) for 45 min at 55°C. DRIP DNA was purified as described above. Locus-specific DRIP signals were assessed by RT-PCR using primers listed in Table 1.
Slot blot
Total nucleic acid was extracted from cells and digested with a cocktail of restriction enzymes as described in the DRIP assay. 1 μg of digested DNA from each sample was diluted in 500 μl TE and spotted on a Nylon membrane (Amersham), using a slot blot apparatus and vacuum suction (GE Healthcare; 80609558). For total DNA control, membrane was denatured for 10 min in 0.5 M NaOH, 1.5 M NaCl, and neutralized for another 10 min in 1 M NaCl, 0.5 M Tris–HCl pH 7.0. Membranes were then UV-crosslinked, blocked in 5% milk/TBST, and incubated overnight at 4°C with mouse S9.6 antibody or single-strand DNA antibody (Millipore; MAB3868). Blots were washed three times with TBST and incubated in secondary antibody for 2 h at room temperature before developing on a film using ECL Western Blotting Detection Reagent (Thermo Fisher Scientific; 34580).
Dissociation of breast tissue and flow cytometry
Fresh unfixed human breast tissue was processed as previously described (29). Briefly, tissue was minced with scalpels and then dissociated enzymatically for 18 h. An epithelium-rich organoid was obtained by centrifugation at 100 g for 3 min. Organoids were further dissociated by sequential digestion with gentle pipetting for 1–2 min in 0.05% trypsin, washed once with HBSS supplemented with 2% FBS and then 2 min in 5 mg ml–1 dispase plus 0.1 mg ml–1 deoxyribonuclease I (DNase I). Single-cell suspension was obtained by filtering through a 40-μm strainer (BD Biosciences) to remove remaining cell aggregates. Single cells were plated in ultralow attachment plates (Corning) in the presence of lentivirus (EV or RH1) and 10 μg ml−1 polybrene. Three days after infection, single cells were harvested from lentivirus-infected primary epithelial cells using trypsin and stained for flow cytometry analysis.
For flow cytometry, infected cells were pre-blocked with 10% rat serum for 10 min and then labeled with an allophycocyanin-conjugated rat antibody to human CD49f (clone GOH3, R&D Systems) and FITC-conjugated mouse antibody to human EpCAM (clone VU1-D9, StemCell Technologies). Cells were also labeled with hematopoietic and endothelial cells markers using biotin-conjugated mouse antibodies to human CD45 (clone HI30, eBiosciences), human CD235a (clone HIR2, eBiosciences) and human CD31 (clone WM59, eBiosciences), followed by pacific blue-conjugated streptavidin (Invitrogen). 7-ADD (BD Bioscience) was added to cells before analysis for live/dead cell discrimination. Flow cytometry analyses were performed using an LSR-II flow cytometer (Becton Dickinson).
Statistical analysis
Mean difference comparison from two groups using unpaired student t-test was used throughout the experiments. All statistics were conducted on GraphPad Prism software. Data in bar and dot graphs are means ± s.e.m. Whiskers in box and whiskers plots represent maximum and minimum value in the group. P < 0.05 was considered significant.
RESULTS
BRCA1-associated R-Loop accumulation at a non-coding region upstream of ESR1 locus
We previously conducted DNA/RNA immunoprecipitation-sequencing (DRIP-seq) to survey genome-wide R-loop dynamics in different cell types of precancerous breast tissues. We reported luminal lineage-specific R-loop accumulation in BRCA1 mutation carriers (BRCA1mut/+) versus non-carriers (BRCA1+/+) (29). Given that germline BRCA1 mutations predominantly predispose cancers in breast and ovary, two major estrogen responsive tissues (34–36), we scrutinized R-loop intensity surrounding the ESR1 locus and found a single R-loop peak in the noncoding region between ESR1 and its neighboring gene CCDC170 (Figure 1A). In contrast, no appreciable R-loop signals were found in the ESR1 TSS, gene body, or transcription termination site (Figure 1A, data not shown). Of note, this intergenic R-loop peak, which covers a region of approximately 250 base pairs (bp), is only present in luminal progenitor and mature luminal cells, not basal or stromal cells, of BRCA1 mutation carriers (Figure 1A). Pretreatment with RNase H, which degrades RNA in R-loops, significantly reduced the R-loop signal (Supplementary Figure S1), corroborating the anti-R-loop antibody specificity. We confirmed this BRCA1-associated R-loop by locus-specific DRIP using the clinical samples (Figure 1B). In addition, BRCA1 knockdown in MCF7, an ERα-positive luminal breast cancer cell line, significantly increased R-loop intensity at the same genomic location (Figure 1C and Supplementary Figure S1B), thus further implicating BRCA1 in regulation of this particular R-loop.
Figure 1.

BRCA1-associated R-Loop accumulation at a non-coding region upstream of ESR1 locus. (A) Alignment of DRIP-seq profiles in primary human breast cells with functional annotations of other published genomic data in MCF7 cells through ENCODE project. DRIP-seq was done in four sorted populations: luminal progenitor (LP), mature luminal (ML), stromal cells, and basal epithelial cells. Each track is an overlay of four individual non-carriers (BRCA1+/+) or four BRCA1 mutation carriers (BRCA1mut/+) indicated by different colors. ENCODE ChIP-seq profiles of various transcription factors are labeled for each track. The super-enhancer region defined by a published study in MCF7 cells is represented by a red bar (40). All sequence data are aligned to human reference genome, hg19. (B) Validation of DRIP-seq result using locus-specific DRIP. *P < 0.05 by Student's t-test. Error bar: s.e.m. (C) Locus-specific DRIP in control and BRCA1 knockdown MCF7 cells. Experiments were performed three times. *P < 0.05 by Student's t-test. Error bar: s.e.m.
By mining publicly available genomic databases, we found that the BRCA1-associated R-loop resides within previously identified cis-regulatory elements for ESR1 (37,38), including a super-enhancer in MCF7 cells (39,40) (Figure 1A). Moreover, ENCODE project database (41) clearly shows that this R-loop peak coincides precisely with multiple functional genomic annotations, including histone H3K27ac (mark for active transcription), chromatin binding of RNA polymerase II (Pol II), and binding of site-specific transcription factors GATA3, FOXM1 and CEBPB (Figure 1A). Of note, all three transcription factors have been previously implicated in breast luminal epithelial cell fate determination (42–44). Taken together, these intriguing correlations suggest a functional link between the BRCA1-associated R-loop accumulation and luminal cell-specific transcription.
Deletion of the R-loop-encompassing region impairs transcription of neighboring genes
To validate the functional relevance of the region encompassing the R loop of interest, we used CRISPR-Cas9 to delete an approximately 550 bp genomic sequence that includes the BRCA1-associated R-loop in parental MCF7 cells (Figure 2A). A total of eight knockout (KO) clones from two sets of independent sgRNA were identified by PCR and subsequently confirmed by DNA sequencing (Supplementary Figure S2A and B). As a control, BRCA1 expression was unchanged in the KO clones (Supplementary Figure S2C). The length of deletion ranges from 540 to 563 bp. We refer to this region ESR1 regulatory element (ESR1-RE). To minimize spurious clonal effects, multiple KO clones and control clones were used throughout our functional study.
Figure 2.

Deletion of ESR1-RE in MCF7 cells affects neighboring genes expression. (A) RT-PCR analysis of gene expression level in knockout clones (n = 8) compared to control clones (n = 8). Box and whiskers graphs show relative gene expression levels in knockout clones over control clones. The relative location of the knockout region is indicated by a red arrow. Statistically affected genes are shown in red and unaffected genes in blue. Experiments were performed three times. Error bar = s.e.m. *P < 0.05 by Student's t-test. (B–D) Western analysis showing protein level of ERα (B), CCDC170 (C) and RMND1 (D) in a control (Ctrl) and a knockout clone (KO). siRNA knockdown was done in parental MCF7 cells.
We found that ESR1-RE deletion significantly reduced mRNA levels of both upstream (CCDC170, RMND1, ZBTB2, AKAP12 and MTHFD1L; Figure 2A) and downstream genes (ESR1 and SYNE1). The ESR1-RE deletion did not have any appreciable effect on more distant genes including upstream PPP1R14C (1.5 Mb) and CITED2 (8 Mb), and downstream FBXO5 (1.3 Mb) and SOD2 (12 Mb; Figure 2A). It has been shown that enhancers tend to mediate transcription of genes within a topologically associating domain (TAD) (45–48). However, chromatin interaction can occur between TADs (49–51). In this regard, it is worth noting that the ESR1-RE deletion affects genes spanning more than one TAD (Supplementary Figure S2D).
To corroborate the mRNA findings, we assessed abundance of the protein products of the three genes most proximal to ESR1-RE (CCDC170, RMND1, and ESR1). Levels of all three proteins were substantially reduced in the KO clones versus their control counterparts (Figure 2B–D and Supplementary Figure S3). Identity of the corresponding protein bands in the western analysis was further confirmed by inclusion of either ERα-negative cells (for ERα) or siRNA knockdown in parental MCF7 cells (for CCDC170 and RMND1; Figure 2B–D). Together, these data support the notion that ESR1-RE serves as a transcriptional enhancer to activate transcription of both upstream and downstream neighboring genes.
ESR1-RE is important for chromosomal looping
According to the prevailing chromatin looping model, transcriptional enhancers regulate their target gene transcription by physically bringing enhancers and promoters to close proximity, looping out the intervening chromosomal sequences (52). To determine whether ESR1-RE is important for looping with the promoters of neighboring genes, we performed chromosome conformation capture (3C) assay in control and ESR1-RE KO clones (32,33,53,54) (Figure 3A). We chose BamH1 restriction enzyme and specific PCR primers that allowed us to detect possible looping events between a chromosomal region in the vicinity of ESR1-RE and the promoters of ESR1, CCDC170, and RMND1 in both WT and KO clones (Figure 3A). As negative controls, 3C experiments with no ligase or crosslinking reagent yielded no ligation product, corroborating the robustness of the assay (Supplementary Figure S4A). We used a set of primers spanning the transcription start site (TSS), gene body (GB), and transcription termination site (TTS) of the neighboring genes. Prominent ligation products were detected for each of the TSS-specific reactions (Supplementary Figure S4B), demonstrating specificity of the enhancer-promoter looping. Furthermore, sequencing of the promoter-specific 3C ligation products in WT control cells confirmed ligation of the ESR1-RE end to the corresponding TSS ends (Supplementary Figure S4C). Taken together, these results rigorously validate physical looping between the intergenic ESR1-RE region and the neighboring promoters in WT cells. In contrast, looping efficiencies in the ESR1-RE KO clones were significantly reduced compared to their WT counterparts (Figure 3B–C and Supplementary Figure S4D). Combined with the mRNA analysis, the 3C assay strongly suggests an enhancer-like transcriptional regulatory role of ESR1-RE.
Figure 3.
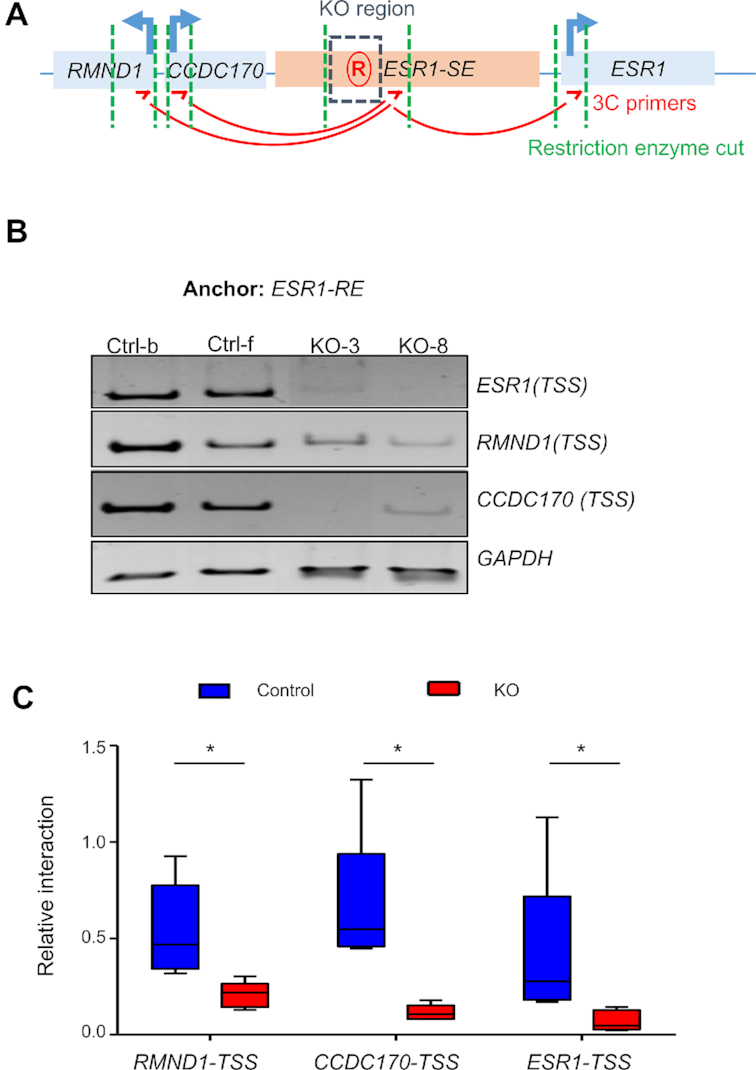
ESR1-RE supports chromosome looping. (A) Schematic representation of the region in chromosome conformation capture (3C) assay. Genes are illustrated as blue box, ESR1 super-enhancer region in pink, and the ESR1-RE region in blue dashed box. Restriction enzyme cutting sites are shown in green dashed lines. 3C primers (red arrow) are designed in same direction for looped ligation product. (B) 3C ligation products from two control and two KO clones were analyzed by polyacrylamide gel electrophoresis. BamHI was used for restriction enzyme cutting. (C) Quantification of the 3C intensity. Five control clones and five knockout clones were used for quantification. Representing graph is average of two independent experiments. Intensity is normalized to GAPDH. Whiskers represent maximum and minimum value in the group. *P < 0.05 by Student's t-test.
Functional antagonism between BRCA1 and R-loop accumulation at ESR1-RE
Because BRCA1 knockdown (KD) increases R-loop intensity at ESR1-RE (Figure 1C), we asked whether BRCA1 depletion also affects transcription of the neighboring genes and if so, whether the effect of BRCA1 KD is mediated through ESR1-RE. BRCA1 was knocked down to a similar efficiency in WT control and ESR1-RE KO clones (Supplementary Figure S5). In control clones with intact ESR1-RE, BRCA1 KD reduced mRNA levels of ESR1, CCDC170, and RMND1 (blue in Figure 4A–C). In comparison, the deleterious effects of BRCA1-KD on these transcripts in ESR1-RE KO clones were significantly ameliorated (red in Figure 4A–C). These results support the notion that BRCA1 facilitates transcription of ESR1, CCDC170 and RMND1 at least partially through ESR1-RE.
Figure 4.

BRCA1 facilitates transcription partially through suppressing R-loop accumulation. (A–C) Fold change (BRCA1 KD/siRNA control) of gene expression levels for ESR1 (A), CCDC170 (B), and RMND1 (C) in control clones (n = 8) and ESR1-RE KO clones (n = 8). Results were average of three independent experiments. Error bar: s.e.m. *P < 0.05 by Student's t-test. (D–F) Fold change (BRCA1 KD/siRNA control) of mRNA levels for ESR1 (D), CCDC170 (E), and RMND1 (F) expression level in empty vector (EV) clones (n = 3) and RH1 overexpression clones (n = 3) after BRCA1 knockdown. Results were average of three independent experiments. Error bar: s.e.m. *P < 0.05 by Student's t-test. (G) 3C ligation products from empty vector (EV) and RNase H1 overexpression (RH1) MCF7 cells analyzed on polyacrylamide gel. BamHI was used for restriction enzyme digestion. (H) Quantification of 3C intensity. Results were average of three independent experiments. Error bar: s.e.m. *P < 0.05 by Student's t-test. Western analysis of RNASEH1 protein level was also shown as an inlet.
Next, we asked whether BRCA1-dependent R-loop attenuation contributes to its role in transcriptional regulation. To this end, we overexpressed the nuclear form of RNase H1 (M27), which has been used to reduce R-loop intensity in cell culture systems (25,55–57). We confirmed elevated RNase H1 mRNA and protein levels in MCF7 cell clones with the stably integrated RNase H1 expression vector (Supplementary Figure S6A and B). Furthermore, we verified that RNase H1 overexpression markedly reduced R-loop levels both globally (Supplementary Figure S6C) and locally at ESR1-RE (Supplementary Figure S6D). We knocked down BRCA1 in empty vector (EV) control and RNase H1-overexpressing (RH1) clones (Supplementary Figure S6E), and subsequently compared the effects of BRCA1 KD on transcription in EV and RH1 cells. We found that BRCA1 KD-associated reduction in mRNA levels of ESR1 and CCDC170 was partially rescued by RNase H1 overexpression (Figure 4D and E). A similar trend, albeit not statistically significant, was observed for RMND1 (Figure 4F). This result suggests that BRCA1 regulates gene expression at least partially through R-loop down-regulation. We next perform 3C assay in control and RNase H1-overexpressing MCF7 cells. Efficiency of looping between ESR1-RE and the promoters of ESR1, CCDC170 and RMND1 was substantially increased in RNase H1-overexpressing cells (Figure 4G and H). Altogether, our study points to a functional consequence of BRCA1-modulated R-loop on the activity of a transcriptional enhancer at the ESR1 locus.
RNase H1 overexpression promotes shift from luminal progenitors to mature luminal cells of BRCA1 mutation carriers
The antagonistic relationship between BRCA1 and R-loops raises the distinct possibility that the latter could mediate differentiation blockage of BRCA1 mutation-carrying luminal epithelial cells (6,7,58). To test this notion, we employed an ex vivo system to culture and manipulate primary breast epithelial cells isolated from cancer-free BRCA1+/+ (n = 5) and BRCA1mut/+ tissue donors (n = 5) (59–61). Breast epithelial cells cultured in suspension were infected with lentiviruses that overexpress RNase H1 (RH1). Three days later, infected cells were stained for EpCAM and CD49f and analyzed by flow cytometry for the relative abundance of luminal progenitor (LP; EpCAM+ CD49f+) cells and mature luminal (ML; EpCAM+ CD49f−) cells (Figure 5A). We confirmed by RT-PCR that RNase H1 was overexpressed to a comparable degree in BRCA1+/+ and BRCA1mut/+ samples (Supplementary Figure S7A). Consistent with published finding of blockage of luminal differentiation in BRCA1 mutation carriers (6,62), we observed a trend of elevated luminal progenitors and reduced mature luminal cell population in BRCA1mut/+ cohort versus BRCA1+/+ cohort without ectopic RNase H1 expression, although it was not statistically significant (P = 0.062 for columns 1 and 5, P = 0.123 for 3 and 7 in Figure 5B). RNase H1 overexpression in BRCA1+/+ control cohort did not affect abundance of luminal progenitors or mature luminal cells (compare column 1 and 2, 3 and 4 in Figure 5B). In contrast, ectopic RNase H1 in BRCA1mut/+ cohort resulted in an appreciable shift (P < 0.05) from luminal progenitors to mature luminal cells (compare column 5 and 6, 7 and 8 in Figure 5B and Supplementary Figure S7B). Consistent with the flow cytometry data, RNase H1 overexpression also resulted in significantly elevated mRNA levels of luminal genes including PGR and GATA3, and a trend of increase for ESR1, FOXA1 and XBP1 in BRCA1mut/+ versus BRCA1+/+ cells (Figure 5C–H). Collectively, our results suggest that R-loop elimination overcomes BRCA1 mutation-associated blockage of luminal epithelial differentiation.
Figure 5.

RNase H1 overexpression in breast epithelial cells of BRCA1 mutation carriers promotes shift from luminal progenitors to mature luminal cells. (A) Schematic diagram showing ex vivo culturing system for lentivirus infection to overexpress RNase H1 in primary breast epithelial cells. (B) Bar graph showing the percentage of luminal progenitors and mature luminal cells in non-carriers (BRCA1+/+, n = 5) and BRCA1 mutation carriers (BRCA1mut/+,n = 5) with (red, RH1) and without (blue, EV) RNase H1 overexpression. Error bar: s.e.m. *P < 0.05 by Student's t-test. (C–H) Fold change (RH1/EV) of expression level of luminal fate genes in BRCA1+/+ and BRCA1mut/+ breast epithelial cells. Error bar: s.e.m.
DISCUSSION
In this study, we characterized a transcription regulatory element upstream of the ESR1 locus that is associated with R-loop accumulation in BRCA1 mutation-carrying breast epithelium. Our work establishes that this chromosomal region is important for transcriptional activation of adjacent genes and enhancer-promoter chromosome looping. We further show that BRCA1 facilitates transcription of ESR1 and its neighboring genes at least partly through this cis-acting regulatory element and by mitigating R-loop accumulation. Lastly, reduction of global R-loop levels in BRCA1 mutation-carrying breast epithelium results in lower luminal progenitor cell population, higher mature luminal cell population and augmented expression of key luminal fate genes. Collectively, our data strongly suggest that BRCA1-modulated R-loop accumulation has functional consequences in luminal gene transcription and epithelial differentiation.
In normal breast development, mammary stem cells undergo differentiation through lineage-restricted myoepithelial or luminal progenitors, which further differentiate into mature cells that constitute the glandular structure (63). The observed effects of R-loop removal in BRCA1-depleted luminal breast cancer cells and primary BRCA1mut/+ luminal epithelial cells suggest an impact of BRCA1-mediated R-loop resolution on luminal gene transcription and lineage-specific differentiation. Based on the current literature and our new findings, we propose that R-loop accumulation in BRCA1 mutation carriers at the enhancer and/or promoter regions of luminal-specific genes (this study and (29)) contributes to transcriptional downregulation of luminal fate-related gene expression (Figure 6). This in turn could result in luminal differentiation blockage and expansion of undifferentiated progenitor cell compartment. Because R-loop accumulation also contributes to genome instability (25), BRCA1 mutation-carrying luminal progenitor cells with accumulated R-loops and compromised ability of maintaining genome integrity (28,64–66) could be the prime target for further oncogenic events that ultimately lead to basal-like BRCA1-associated breast cancer.
Figure 6.

Model depicting the proposed roles of BRCA1 in R-loop resolution and luminal epithelial differentiation. In the process of normal breast differentiation and development, luminal fate genes regulate luminal progenitor cells differentiate into mature luminal cells. Mutated BRCA1 causes elevated R-loops at enhancer and/or promoter regions of luminal fate genes and downregulate their expression. Aberrant expansion of ER-negative luminal progenitor cells could be the target of other oncogenic hits, leading to basal-like BRCA1-associated tumor formation.
ESR1 and its two neighboring genes affected by BRCA1 deficiency, CCDC170 and RMND1, have been implicated in sporadic breast cancer (67–71). For example, CCDC170 protein is involved in Golgi-associated microtubules dynamics. Its truncation in breast cancer has been suggested to alter cell polarity/motility and drive tumor initiation/progression (70). In addition, co-expression and co-regulation of ESR1, CCDC170 and RMND1 have been reported in breast cancer specimens (69), and their allele-specific expression could account for the association between variants at the ESR1 locus and ERα negative breast cancer (68,71). Furthermore, tumor-specific ESR1-CCDC170 rearrangements have been reported in breast cancer (67,72,73), accentuating the clinical relevance of this genomic region to tumorigenesis. It remains to be determined whether R-loop accumulation in this genomic region also contributes to the previously reported genetic and transcriptional alterations in sporadic breast cancer.
ESR1-RE is part of a larger intergenic noncoding chromosomal region (6q25.1) that contains a number of breast cancer risk-associated single nucleotide polymorphisms (SNPs) (74–76). More recent analyses also demonstrated that SNPs in the same region are associated with breast cancer risk for BRCA1 mutation carriers (77). These risk loci are enriched in regulatory elements that function by altering gene expression (78). In fact, some of these SNPs have been found to affect gene promoter activity, chromosome looping efficiency and transcription factor binding (38,68,79). The striking alignment of the BRCA1-associated R-loop peak with the ChIP peaks at the same region for a number of transcription factors involved in luminal differentiation underscores an intimate relationship between R-loop dynamics and transcription factor binding. While SNPs can permanently alter binding affinity of the corresponding transcription factor, R-loops accumulated within a transcription factor binding site could interfere with chromatin binding of transcription factors and thus dampen transcription of their target genes in a transient and reversible manner. In support, disruption of chromatin structure by R-loops inhibits the binding and function of epigenetic regulators PRC2 and DNMT3B (19,55). Thus R-loops can alter the chromatin landscape by modulating the binding of chromatin regulatory factors. Alternatively, R-loop formation could create physical hindrance that weakens long-distance chromatin interactions.
We recognize obvious differences between primary non-cancerous breast epithelial cells and MCF7 breast cancer cell line used in the current study. For example, due to oncogene-mediated rewiring of the transcriptional program that governs ESR1 expression, MCF7 tumor cells could become less dependent on BRCA1-mediated R-loop removal at the ESR1-RE locus. This would explain the less robust effect of BRCA1 knockdown in MCF7 cells on the level of R-loop enhancement at ESR1-RE versus the difference between BRCA1+/+ and BRCA1mut/+ primary normal breast epithelial cells. How R-loop dynamics is influenced by tumorigenesis merits further investigation.
The fact that R-loop removal by RNase H1 overexpression only partially rescues BRCA1 KD effect on transcription suggests that BRCA1 likely uses R-loop-independent mechanisms to regulate transcription. In this regard, accumulating evidence supports a role of BRCA1 in epigenetic regulation through its intrinsic E3 ligase activity or interactions with histone acetyltransferases (80–82). BRCA1 is also known for its function in high-order chromatin reorganization (81,83). Furthermore, BRCA1 interacts with RNA polymerase II (84), DNA-binding transcription factors including GATA3 (85), and transcription elongation factor NELF-B/COBRA1 (83). Of note, there is a complex regulatory interplay between BRCA1 and ERα (36,86–88). BRCA1 was shown to interact with ERα and inhibit ERα-mediated transactivation upon estrogen stimulation (89–91). On the other hand, BRCA1 can be recruited to the ERα promoter and induce ERα expression in a BRD7-dependent manner (88,92). Collectively, BRCA1 could act through multiple mechanisms to regulate transcriptional program in breast luminal epithelium and suppress luminal-to-basal transition in BRCA1-associated tumorigenesis.
Overexpression of RNase H1 is a widely used and efficient way to reduce global cellular levels of R-loop (25,55,56). However, one limitation of this approach is the inability to confine R loop removal to a specific locus. While our current study does not allow us to pinpoint the specific R-loop location(s) the reduction of which mitigates the blockage of luminal differentiation, the effect of RNase H1 overexpression on luminal cell differentiation could be due to changes of R-loop dynamics in multiple enhancer and promoter regions. Regardless the extent of R-loop involved in blocking BRCA1-associated luminal differentiation, our current study using clinical samples provides compelling evidence for a functional link between these two biological events in breast epithelium of BRCA1 mutation carriers. This is in agreement with our previous finding of a significant overlap between BRCA1 mutation-associated R-loops and luminal signature genes (29). Future work is warranted to examine a direct contribution of luminal R-loop accumulation to BRCA1-associated breast cancer. A better understanding of transcription-related function of BRCA1 in R-loop resolution and regulation of the cell of origin of BRCA1-associated breast tumors could potentially inform development of novel approaches for cancer-risk assessment and reduction.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Chi Zhang, Sabrina Smith, Xiujie Sun, Bogang Wu, Lisa Sanchez and Michael Garcia for technical assistance.
Author contributions: R.L., Y.H., H.T-H.W., A.B. and I.J. conceived and supervised the project. R.L., Y.H. and H-C.C. designed the experiments. H-C.C., X.Z., J.L., X.Z. and J.C. performed the experiments. H-C.C., X.Z., Y.H. and R.L. analyzed the data. H-C.C. and R.L. wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [CA220578 to R.L., CA212674 to Y.H., T32CA148724 to H-C.C.]; Department of Defense [W81XWH-17-1-0007 to Y.H. and W81XWH-17-1-0008 R.L.]; Cancer Prevention and Research Institute of Texas (CPRIT) [RP170126]; Mays Cancer Center at UT Health San Antonio [NIH-NCI P30 CA054174]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Milne R.L., Osorio A., Cajal T.R., Vega A., Llort G., de la Hoya M., Diez O., Alonso M.C., Lazaro C., Blanco I. et al.. The average cumulative risks of breast and ovarian cancer for carriers of mutations in BRCA1 and BRCA2 attending genetic counseling units in Spain. Clin. Cancer Res. 2008; 14:2861–2869. [DOI] [PubMed] [Google Scholar]
- 2. Fackenthal J.D., Olopade O.I.. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat. Rev. Cancer. 2007; 7:937–948. [DOI] [PubMed] [Google Scholar]
- 3. Turner N., Tutt A., Ashworth A.. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer. 2004; 4:814–819. [DOI] [PubMed] [Google Scholar]
- 4. Molyneux G., Geyer F.C., Magnay F.A., McCarthy A., Kendrick H., Natrajan R., Mackay A., Grigoriadis A., Tutt A., Ashworth A. et al.. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010; 7:403–417. [DOI] [PubMed] [Google Scholar]
- 5. Proia T.A., Keller P.J., Gupta P.B., Klebba I., Jones A.D., Sedic M., Gilmore H., Tung N., Naber S.P., Schnitt S. et al.. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011; 8:149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lim E., Vaillant F., Wu D., Forrest N.C., Pal B., Hart A.H., Asselin-Labat M.L., Gyorki D.E., Ward T., Partanen A. et al.. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009; 15:907–913. [DOI] [PubMed] [Google Scholar]
- 7. Liu S., Ginestier C., Charafe-Jauffret E., Foco H., Kleer C.G., Merajver S.D., Dontu G., Wicha M.S.. BRCA1 regulates human mammary stem/progenitor cell fate. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:1680–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buckley N.E., tSaoir C.B.N.A., Blayney J.K., Oram L.C., Crawford N.T., D’Costa Z.C., Quinn J.E., Kennedy R.D., Harkin D.P., Mullan P.B.. BRCA1 is a key regulator of breast differentiation through activation of Notch signalling with implications for anti-endocrine treatment of breast cancers. Nucleic Acids Res. 2013; 41:8601–8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Furuta S., Jiang X., Gu B., Cheng E., Chen P.L., Lee W.H.. Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:9176–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burga L.N., Tung N.M., Troyan S.L., Bostina M., Konstantinopoulos P.A., Fountzilas H., Spentzos D., Miron A., Yassin Y.A., Lee B.T. et al.. Altered proliferation and differentiation properties of primary mammary epithelial cells from BRCA1 mutation carriers. Cancer Res. 2009; 69:1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Venkitaraman A.R. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science. 2014; 343:1470–1475. [DOI] [PubMed] [Google Scholar]
- 12. Christou C.M., Kyriacou K.. BRCA1 and its network of interacting partners. Biology (Basel). 2013; 2:40–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang X.W., Li R.. BRCA1-dependent transcriptional regulation: implication in tissue-specific tumor suppression. Cancers. 2018; 10:513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang S., Sharan S.K.. The role of epigenetic transcriptional regulation in BRCA1-mediated tumor suppression. Transcription. 2013; 4:24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Savage K.I., Harkin D.P.. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015; 282:630–646. [DOI] [PubMed] [Google Scholar]
- 16. Mullan P.B., Quinn J.E., Harkin D.P.. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene. 2006; 25:5854–5863. [DOI] [PubMed] [Google Scholar]
- 17. Stirling P.C., Chan Y.A., Minaker S.W., Aristizabal M.J., Barrett I., Sipahimalani P., Kobor M.S., Hieter P.. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 2012; 26:163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sollier J., Stork C.T., Garcia-Rubio M.L., Paulsen R.D., Aguilera A., Cimprich K.A.. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell. 2014; 56:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ginno P.A., Lott P.L., Christensen H.C., Korf I., Chedin F.. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 2012; 45:814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Castellano-Pozo M., Garcia-Muse T., Aguilera A.. R-loops cause replication impairment and genome instability during meiosis. EMBO Rep. 2012; 13:923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huertas P., Aguilera A.. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell. 2003; 12:711–721. [DOI] [PubMed] [Google Scholar]
- 22. Loomis E.W., Sanz L.A., Chedin F., Hagerman P.J.. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014; 10:e1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Svejstrup J.Q. The interface between transcription and mechanisms maintaining genome integrity. Trends Biochem Sci. 2010; 35:333–338. [DOI] [PubMed] [Google Scholar]
- 24. Aguilera A., Garcia-Muse T.. R loops: from transcription byproducts to threats to genome stability. Mol. Cell. 2012; 46:115–124. [DOI] [PubMed] [Google Scholar]
- 25. Skourti-Stathaki K., Proudfoot N.J.. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014; 28:1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bhatia V., Barroso S.I., Garcia-Rubio M.L., Tumini E., Herrera-Moyano E., Aguilera A.. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014; 511:362–365. [DOI] [PubMed] [Google Scholar]
- 27. Hill S.J., Rolland T., Adelmant G., Xia X., Owen M.S., Dricot A., Zack T.I., Sahni N., Jacob Y., Hao T. et al.. Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev. 2014; 28:1957–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hatchi E., Skourti-Stathaki K., Ventz S., Pinello L., Yen A., Kamieniarz-Gdula K., Dimitrov S., Pathania S., McKinney K.M., Eaton M.L. et al.. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell. 2015; 57:636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X., Chiang H.C., Wang Y., Zhang C., Smith S., Zhao X., Nair S.J., Michalek J., Jatoi I., Lautner M. et al.. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017; 8:15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shivji M.K.K., Renaudin X., Williams C.H., Venkitaraman A.R.. BRCA2 regulates transcription elongation by RNA polymerase II to prevent R-Loop accumulation. Cell Rep. 2018; 22:1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Naumova N., Smith E.M., Zhan Y., Dekker J.. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 2012; 58:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hagege H., Klous P., Braem C., Splinter E., Dekker J., Cathala G., de Laat W., Forne T.. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2007; 2:1722–1733. [DOI] [PubMed] [Google Scholar]
- 34. Lee E.Y., Abbondante S.. Tissue-specific tumor suppression by BRCA1. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:4353–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kauff N.D., Satagopan J.M., Robson M.E., Scheuer L., Hensley M., Hudis C.A., Ellis N.A., Boyd J., Borgen P.I., Barakat R.R. et al.. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N. Engl. J. Med. 2002; 346:1609–1615. [DOI] [PubMed] [Google Scholar]
- 36. Hu Y. BRCA1, hormone, and tissue-specific tumor suppression. Int. J. Biol. Sci. 2009; 5:20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eeckhoute J., Keeton E.K., Lupien M., Krum S.A., Carroll J.S., Brown M.. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 2007; 67:6477–6483. [DOI] [PubMed] [Google Scholar]
- 38. Bailey S.D., Desai K., Kron K.J., Mazrooei P., Sinnott-Armstrong N.A., Treloar A.E., Dowar M., Thu K.L., Cescon D.W., Silvester J. et al.. Noncoding somatic and inherited single-nucleotide variants converge to promote ESR1 expression in breast cancer. Nat. Genet. 2016; 48:1260–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hnisz D., Schuijers J., Lin C.Y., Weintraub A.S., Abraham B.J., Lee T.I., Bradner J.E., Young R.A.. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell. 2015; 58:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A., Hoke H.A., Young R.A.. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. The ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carr J.R., Kiefer M.M., Park H.J., Li J., Wang Z., Fontanarosa J., DeWaal D., Kopanja D., Benevolenskaya E.V., Guzman G. et al.. FoxM1 regulates mammary luminal cell fate. Cell Rep. 2012; 1:715–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. LaMarca H.L., Visbal A.P., Creighton C.J., Liu H., Zhang Y., Behbod F., Rosen J.M.. CCAAT/enhancer binding protein beta regulates stem cell activity and specifies luminal cell fate in the mammary gland. Stem Cells. 2010; 28:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kouros-Mehr H., Kim J.W., Bechis S.K., Werb Z.. GATA-3 and the regulation of the mammary luminal cell fate. Curr. Opin. Cell Biol. 2008; 20:164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A.. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ing-Simmons E., Seitan V.C., Faure A.J., Flicek P., Carroll T., Dekker J., Fisher A.G., Lenhard B., Merkenschlager M.. Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res. 2015; 25:504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pombo A., Dillon N.. Three-dimensional genome architecture: players and mechanisms. Nat. Rev. Mol. Cell Biol. 2015; 16:245–257. [DOI] [PubMed] [Google Scholar]
- 48. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith E.M., Lajoie B.R., Jain G., Dekker J.. Invariant TAD boundaries constrain Cell-Type-Specific looping interactions between promoters and distal elements around the CFTR locus. Am. J. Hum. Genet. 2016; 98:185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gibcus J.H., Dekker J.. The hierarchy of the 3D genome. Mol. Cell. 2013; 49:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gasperini M., Hill A.J., McFaline-Figueroa J.L., Martin B., Kim S., Zhang M.D., Jackson D., Leith A., Schreiber J., Noble W.S. et al.. A Genome-wide framework for mapping gene regulation via cellular genetic screens. Cell. 2019; 176:377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li W.B., Notani D., Rosenfeld M.G.. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat. Rev. Genet. 2016; 17:207–223. [DOI] [PubMed] [Google Scholar]
- 53. Dekker J. Two ways to fold the genome during the cell cycle: insights obtained with chromosome conformation capture. Epigenet. Chromatin. 2014; 7:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dekker J. The three ‘C’ s of chromosome conformation capture: controls, controls, controls. Nat. Methods. 2006; 3:17–21. [DOI] [PubMed] [Google Scholar]
- 55. Chen P.B., Chen H.V., Acharya D., Rando O.J., Fazzio T.G.. R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat. Struct. Mol. Biol. 2015; 22:999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Skourti-Stathaki K., Kamieniarz-Gdula K., Proudfoot N.J.. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 2014; 516:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Suzuki Y., Holmes J.B., Cerritelli S.M., Sakhuja K., Minczuk M., Holt I.J., Crouch R.J.. An upstream open reading frame and the context of the two AUG codons affect the abundance of mitochondrial and nuclear RNase H1. Mol. Cell Biol. 2010; 30:5123–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ginestier C., Liu S., Wicha M.S.. Getting to the root of BRCA1-deficient breast cancer. Cell Stem Cell. 2009; 5:229–230. [DOI] [PubMed] [Google Scholar]
- 59. Welm B.E., Dijkgraaf G.J., Bledau A.S., Welm A.L., Werb Z.. Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell. 2008; 2:90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iggo R., Richard E.. Lentiviral transduction of mammary epithelial cells. Methods Mol. Biol. 2015; 1293:137–160. [DOI] [PubMed] [Google Scholar]
- 61. Ginestier C., Hur M.H., Charafe-Jauffret E., Monville F., Dutcher J., Brown M., Jacquemier J., Viens P., Kleer C.G., Liu S. et al.. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007; 1:555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nolan E., Vaillant F., Branstetter D., Pal B., Giner G., Whitehead L., Lok S.W., Mann G.B. Kathleen Cuningham Foundation Consortium for Research into Familial Breast, C. . Kathleen Cuningham Foundation Consortium for Research into Familial Breast, C. Rohrbach K. et al.. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat. Med. 2016; 22:933–939. [DOI] [PubMed] [Google Scholar]
- 63. Visvader J.E., Stingl J.. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 2014; 28:1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sedic M., Skibinski A., Brown N., Gallardo M., Mulligan P., Martinez P., Keller P.J., Glover E., Richardson A.L., Cowan J. et al.. Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nat. Commun. 2015; 6:7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Konishi H., Mohseni M., Tamaki A., Garay J.P., Croessmann S., Karnan S., Ota A., Wong H.Y., Konishi Y., Karakas B. et al.. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:17773–17778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Savage K.I., Matchett K.B., Barros E.M., Cooper K.M., Irwin G.W., Gorski J.J., Orr K.S., Vohhodina J., Kavanagh J.N., Madden A.F. et al.. BRCA1 deficiency exacerbates estrogen-induced DNA damage and genomic instability. Cancer Res. 2014; 74:2773–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Veeraraghavan J., Tan Y., Cao X.X., Kim J.A., Wang X., Chamness G.C., Maiti S.N., Cooper L.J., Edwards D.P., Contreras A. et al.. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat. Commun. 2014; 5:4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dunning A.M., Michailidou K., Kuchenbaecker K.B., Thompson D., French J.D., Beesley J., Healey C.S., Kar S., Pooley K.A., Lopez-Knowles E. et al.. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet. 2016; 48:374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dunbier A.K., Anderson H., Ghazoui Z., Lopez-Knowles E., Pancholi S., Ribas R., Drury S., Sidhu K., Leary A., Martin L.A. et al.. ESR1 is co-expressed with closely adjacent uncharacterised genes spanning a breast cancer susceptibility locus at 6q25.1. PLoS Genet. 2011; 7:e1001382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jiang P., Li Y., Poleshko A., Medvedeva V., Baulina N., Zhang Y., Zhou Y., Slater C.M., Pellegrin T., Wasserman J. et al.. The protein encoded by the CCDC170 breast cancer gene functions to organize the Golgi-Microtubule network. EBioMedicine. 2017; 22:28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yamamoto-Ibusuki M., Yamamoto Y., Fujiwara S., Sueta A., Yamamoto S., Hayashi M., Tomiguchi M., Takeshita T., Iwase H.. C6ORF97-ESR1 breast cancer susceptibility locus: influence on progression and survival in breast cancer patients. Eur. J. Hum. Genet. 2015; 23:949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sakarya O., Breu H., Radovich M., Chen Y., Wang Y.N., Barbacioru C., Utiramerur S., Whitley P.P., Brockman J.P., Vatta P. et al.. RNA-Seq mapping and detection of gene fusions with a suffix array algorithm. PLoS Comput. Biol. 2012; 8:e1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Robinson D.R., Kalyana-Sundaram S., Wu Y.M., Shankar S., Cao X., Ateeq B., Asangani I.A., Iyer M., Maher C.A., Grasso C.S. et al.. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011; 17:1646–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Turnbull C., Ahmed S., Morrison J., Pernet D., Renwick A., Maranian M., Seal S., Ghoussaini M., Hines S., Healey C.S. et al.. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 2010; 42:504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zheng W., Long J., Gao Y.T., Li C., Zheng Y., Xiang Y.B., Wen W., Levy S., Deming S.L., Haines J.L. et al.. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat. Genet. 2009; 41:324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sueta A., Ito H., Kawase T., Hirose K., Hosono S., Yatabe Y., Tajima K., Tanaka H., Iwata H., Iwase H. et al.. A genetic risk predictor for breast cancer using a combination of low-penetrance polymorphisms in a Japanese population. Breast Cancer Res. Treat. 2012; 132:711–721. [DOI] [PubMed] [Google Scholar]
- 77. Antoniou A.C., Kartsonaki C., Sinilnikova O.M., Soucy P., McGuffog L., Healey S., Lee A., Peterlongo P., Manoukian S., Peissel B. et al.. Common alleles at 6q25.1 and 1p11.2 are associated with breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum. Mol. Genet. 2011; 20:3304–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. et al.. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012; 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cowper-Sal lari R., Zhang X., Wright J.B., Bailey S.D., Cole M.D., Eeckhoute J., Moore J.H., Lupien M.. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet. 2012; 44:1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pao G.M., Janknecht R., Ruffner H., Hunter T., Verma I.M.. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. PNAS. 2000; 97:1020–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhu Q., Pao G.M., Huynh A.M., Suh H., Tonnu N., Nederlof P.M., Gage F.H., Verma I.M.. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011; 477:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bochar D.A., Wang L., Beniya H., Kinev A., Xue Y., Lane W.S., Wang W., Kashanchi F., Shiekhattar R.. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000; 102:257–265. [DOI] [PubMed] [Google Scholar]
- 83. Ye Q., Hu Y.F., Zhong H., Nye A.C., Belmont A.S., Li R.. BRCA1-induced large-scale chromatin unfolding and allele-specific effects of cancer-predisposing mutations. J. Cell Biol. 2001; 155:911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Scully R., Anderson S.F., Chao D.M., Wei W., Ye L., Young R.A., Livingston D.M., Parvin J.D.. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:5605–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tkocz D., Crawford N.T., Buckley N.E., Berry F.B., Kennedy R.D., Gorski J.J., Harkin D.P., Mullan P.B.. BRCA1 and GATA3 corepress FOXC1 to inhibit the pathogenesis of basal-like breast cancers. Oncogene. 2012; 31:3667–3678. [DOI] [PubMed] [Google Scholar]
- 86. Rosen E.M., Fan S., Ma Y.. BRCA1 regulation of transcription. Cancer Lett. 2006; 236:175–185. [DOI] [PubMed] [Google Scholar]
- 87. Gorski J.J., Kennedy R.D., Hosey A.M., Harkin D.P.. The complex relationship between BRCA1 and ERalpha in hereditary breast cancer. Clin. Cancer Res. 2009; 15:1514–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Harte M.T., O’Brien G.J., Ryan N.M., Gorski J.J., Savage K.I., Crawford N.T., Mullan P.B., Harkin D.P.. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res. 2010; 70:2538–2547. [DOI] [PubMed] [Google Scholar]
- 89. Fan S., Ma Y.X., Wang C., Yuan R.Q., Meng Q., Wang J.A., Erdos M., Goldberg I.D., Webb P., Kushner P.J. et al.. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001; 20:77–87. [DOI] [PubMed] [Google Scholar]
- 90. Fan S., Ma Y.X., Wang C., Yuan R.Q., Meng Q., Wang J.A., Erdos M., Goldberg I.D., Webb P., Kushner P.J. et al.. p300 Modulates the BRCA1 inhibition of estrogen receptor activity. Cancer Res. 2002; 62:141–151. [PubMed] [Google Scholar]
- 91. Fan S., Wang J., Yuan R., Ma Y., Meng Q., Erdos M.R., Pestell R.G., Yuan F., Auborn K.J., Goldberg I.D. et al.. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science. 1999; 284:1354–1356. [DOI] [PubMed] [Google Scholar]
- 92. Hosey A.M., Gorski J.J., Murray M.M., Quinn J.E., Chung W.Y., Stewart G.E., James C.R., Farragher S.M., Mulligan J.M., Scott A.N. et al.. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. J. Natl. Cancer Inst. 2007; 99:1683–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.