

Abstract
Human CST (CTC1-STN1-TEN1) is an RPA-like complex that associates with G-rich single-strand DNA and helps resolve replication problems both at telomeres and genome-wide. We previously showed that CST binds and disrupts G-quadruplex (G4) DNA in vitro, suggesting that CST may prevent in vivo blocks to replication by resolving G4 structures. Here, we demonstrate that CST binds and unfolds G4 with similar efficiency to RPA. In cells, CST is recruited to telomeric and non-telomeric chromatin upon G4 stabilization, even when ATR/ATM pathways were inhibited. STN1 depletion increases G4 accumulation and slows bulk genomic DNA replication. At telomeres, combined STN1 depletion and G4 stabilization causes multi-telomere FISH signals and telomere loss, hallmarks of deficient telomere duplex replication. Strand-specific telomere FISH indicates preferential loss of C-strand DNA while analysis of BrdU uptake during leading and lagging-strand telomere replication shows preferential under-replication of lagging telomeres. Together these results indicate a block to Okazaki fragment synthesis. Overall, our findings indicate a novel role for CST in maintaining genome integrity through resolution of G4 structures both ahead of the replication fork and on the lagging strand template.
INTRODUCTION
Telomeres are specialized nucleoprotein structures that maintain genome stability by preventing chromosome ends from being detected as DNA damage and triggering end-to-end fusion through unwanted DNA repair reactions (1–3). In mammalian cells, the telomeric DNA consists of thousands of TTAGGG•AATCC repeats which terminate with a short region of ssDNA on the G-rich strand. This repeated sequence DNA is packaged by shelterin, a six protein complex that sequesters the DNA terminus to prevent inappropriate DNA damage signaling (4,5). Mammalian telomeres also associate with a ssDNA-binding complex called CST (CTC1–STN1–TEN1) that is essential for multiple aspects of telomere replication (6–8). Telomeres are particularly challenging to replicate due their G-rich sequence and terminal structure (3,9). The repetitive G-rich DNA tends to stall the replication fork, while the inability of DNA polymerase to replicate the DNA 5′ end leads to progressive telomere shortening unless additional sequence is added to the DNA terminus. This telomere elongation is achieved by telomerase which extends the G-strand and DNA polymerase which then synthesizes the complementary C-strand.
The CTC1 and STN1 subunits of CST were originally identified as DNA polymerase α-primase (Pol α) cofactors that increase the affinity of Pol α for ssDNA templates (10). More recent studies have shown that one role of CST is in regulation of Pol α switching from RNA and DNA synthesis (11), a process that is essential to generate Okazaki fragments during lagging strand replication. In vivo studies have shown that CST participates in multiple stages of telomere replication. First, it aids in replication of the G-rich DNA duplex. Depletion of CST subunits slows replication through this region and leads to telomere fragility and sudden telomere loss, suggesting that CST either prevents replication fork stalling or facilitates fork restart (12–14). CST then regulates telomerase to prevent G-strand overextension (15,16). Finally, CST is required to engage Pol α for C-strand synthesis so that the ssDNA generated by telomerase is converted to new telomere duplex (12,17–19). In the absence of CST, the lack of C-strand synthesis leads to progressive telomere shortening with each round of replication, similar to a telomerase knockout. CST also has less well understood non-telomeric roles related to recovery from replication stress (20,21). When hydroxylurea is used to stall replication, CST promotes replication restart by increasing the firing of dormant replication origins (14,21).And CST was shown to localize to GC-rich loci in response to replication stress where it functions to prevent fragile site expression (22). A more recent study has shown that CST recruits Pol α to DNA damage sites and plays an essential role in filling in the resected DSBs through interaction with the shieldin complex (23).
CST is structurally similar to Replication Protein A (RPA), the ssDNA binding protein that directs DNA replication, repair, and recombination in eukaryotic cells (24–26). The structural conservation encompasses the OB-folds, winged helix domains and the dimerization interface of the two smaller subunits (STN1-TEN1 and RPA2-RPA3). Like RPA, CST appears to contact DNA via multiple OB folds leading to a dynamic mode of DNA binding. Binding and release of individual OB folds is thought to underlie the capacity of both proteins to melt DNA structure and regulate DNA association and activity of interaction partners such as DNA polymerases. Although CST and RPA share many similarities in terms of structure and DNA binding activity, they are architecturally different and they play quite distinct roles during DNA replication and repair (27). Moreover, CST binds preferentially to G-rich ssDNA and, unlike ssDNA-bound RPA, it does not trigger ATR activation (18,26,28). Thus, CST appears to have evolved a unique role by serving to resolve a variety of replication problems without activation of DNA damage signaling.
Currently the mechanism by which CST facilitates replication through G-rich dsDNA is unclear. However, the recent discovery that CST can unwind G-quadruplex (G4) DNA (27), suggests that the role of CST is to prevent or resolve replication fork stalling at G4 structures. G4s form in G-rich DNA due to the ability of guanine to form thermodynamically stable Hoogsteen base-paired quartets (29). The human genome contains thousands of regions with G4-forming potential (30). These regions are becoming appreciated as an important genomic feature because of their capacity to form stable G4 structures that interrupt the DNA duplex (31,32). The G4 DNA is thought to play roles in various cellular processes including transcriptional activation, stimulation of meiotic recombination, chromatin assembly and rudimentary telomere capping (33,34). Despite serving these beneficial regulatory functions, G4 structures can also have adverse effects on genome stability because they impose a structural barrier to DNA replication. Any G4 structures formed as a result of transcription may later block passage of the DNA replication machinery (35,36). Replication may also be blocked by G4 that forms when ssDNA is exposed during lagging strand synthesis.
The recent discovery that CST can resolve G4 DNA in vitro, suggested that CST may also remove G4 structures in vivo and this property could explain the ability of CST to facilitate replication through telomeres and other G-rich regions. However, no prior studies have addressed whether CST has in vivo roles in G4 resolution. We now provide evidence that that CST is indeed needed to prevent G4 accumulation in cells and that STN1/CST loss slows bulk DNA replication after G4 stabilization. At telomeres, combined STN1 depletion and G4 stabilization causes replication-associated defects on both sister chromatids but lagging-strand telomere replication is most heavily affected. Our results indicate that the G4 resolving activity of CST is necessary to prevent a range of replication defects.
MATERIALS AND METHODS
Cell culture
HeLa, HeLa 1.2.11 cell clones and U2OS cells were cultured in RPMI 1640, HCT116 cells in McCoy's 5A media containing 10% FBS, antibiotics and glutamine. As previously reported, the STN1sh HeLa 1.2.11 cells were made by transducing cells with lentivirus viral encoding STN1 shRNA sh6 (5′-CAAGGCAATTCATAGTATA) and sh7 (5′-GCAATTCATAGTATATTTA) (14). The rescue cell line was made by retroviruses transfection of an shRNA-resistant FLAG-STN1 allele with 5-point mutations. The CST oe (over expression) cell line was made by transient transfection using HA-CTC1, Flag-STN1 and TEN1 plasmid. The level of STN1 was determined by western blot using the OBFC1 antibody (abcam, ab89250).
Cells were treated for 24 h with 50 μM TmPyP4 or 10 μM PDS to induce G4 formation, which caused only modest cell cycle arrest. While TMPyP4 is often used to stabilize G4 structure, it has also been found to bind dsDNA at high in vivo concentration. PDS binds more specifically to telomeric G4s but we found it difficult to detect telomere signal by FISH, due to the high background. Thus, TmPyP4 was used for most telomere FISH experiments, and PDS was used for other studies. For ATM and ATR inhibition: 10 μM KU55933 (Selleck Chemical) and 2 μM ETP-46464 (Sigma) were added at the same time as the G4 stabilizer was used.
CST and RPA expression
Sf9 cells were co-infected with baculovirus encoding Flag-CTC1, His-STN1 and TEN1 or Flag-RPA1, His-RPA2 and RPA3. Two-step protein purification was performed essentially as described (18). Briefly, following cell lysis the protein complex was bound to nickel-Sepharose beads (GE 17526801), eluted with imidazole, then bound to FLAG beads (Sigma A2220) and eluted with 3xFLAG-peptide (Sigma F4799). Protein was stored in 1 mg/ml BSA and 15% glycerol at –80°C after flash freezing in liquid nitrogen.
EMSA assays
For Kd analysis, CST (0.5–10 nM) was incubated with 32P-labeled Tel21 (0.05 nM) in binding buffer (10 mM Tris pH 7.4, 1 mM DTT) containing either 150 mM KCl or 150 mM LiCl for 2 h at 4°C to reach reaction equilibrium. DNA binding was then monitored by EMSA. Samples were separated in 0.7% agarose gels with 1× TAE for 1 h at 90 V and 4°C and then quantified by PhosphorImaging. To determine Kd,app, the amount of bound versus free DNA was quantified using ImageQuantTL software. Data were fit to a one site specific saturation binding equation using GraphPad prism software.
CD spectra
Tel21 DNA (0.5 μM) was dissolved in 10 mM Tris (pH 7.4) with 150 mM KCl or 150 mM LiCl, heated at 95°C for 5 min, and then cooled down to room temperature over night. CD spectrum was collected from 320 to 220 nm at 1-nm bandwidth on a Chirascan Plus CD spectropolarimeter (Applied Photophysics) with a 10-mm pathlength at 25°C. Buffer blank correction was made for both samples.
Fluorescence resonance energy transfer
Tel21 DNA labeled with fluorescein (FAM) at the 5′-end and tetramethylrhodamine (TAMRA) at the 3′-end, were purchased from Takara Biotech (Dalian, China).) The FRET was performed as previously described (37,38), the DNA (1 μM) was incubated with 25 or 100 nM protein in 10 mM Tris (pH 7.4) buffer containing 150 mM NaCl for 2 h at 4°C. Fluorescence measurements were carried out on a Spex Fluorolog-3 spectrofluorometer (HORIBA Jobin Yvon, France) at 25°C. The excitation and emission slits were both 5 nm. Excitation was set at 480 nm, and emission was collected from 490 to 650 nm. Lamp fluctuations were corrected by using the reference channel. A buffer blank was subtracted for all spectra.
Immunofluorescence (IF)
Cells were grown in chamber slides and fixed in 4% paraformaldehyde for 15 min, washed three times with PBS, permeabilized with 0.15% Triton X-100 for 15 min, washed three times for 5 min with PBS, blocked with 10% BSA at 37°C for 1 h in humidified chamber, incubated with primary antibodies for overnight at 4°C, washed with PBS three times, incubated with secondary antibody at room temperature for 1 h, and finally washed three times in PBS. Slides were treated with a cold ethanol series, dried in the dark, and DAPI-containing mounting medium (Vector Labs) was applied for visualization. Images were taken under a Nikon ECLIPSE Ti fluorescence microscope with a 100× objective. Antibodies used were as follows: Flag-tag, Sigma-Aldrich, F3165; TRF2, Millipore, 05-521; 53BP1, NOVUSBIO, NB100-304; HA-tag, Immunoway, YM3003; Dylight 488-anti-mouse IgG (ThermoFisher) and Dylight 549-anti-rabbit IgG (ThermoFisher).
Chromatin immunoprecipitation
As previously described (18), cells were fixed with 1% formaldehyde for 20 min then treated with 200 mM glycine for 10 min to quench the reaction. Cell were pelleted by centrifugation, suspended in swelling buffer (25 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.25% Triton X-100 and protease inhibitors) on ice for 10 min, pelleted and incubated in sonication buffer (50 mM HEPES, pH 7.9, 150 mM NaCl, 1 mM EDTA, 0.1% Sodium deoxycholate, 0.1% SDS, 1% Triton X-100 and protease inhibitors) and sonicated for 20 min in a sonication system (Dajin). Samples were centrifuged at 14 000 rpm for 10 min and the supernatant used for ChIP. Samples containing supernatant (0.3 mg protein), antibody (10 μg TPP1, abcam, ab54685; 5 μg Flag, Sigma-Aldrich, F3165; 15 μg Pol α, Santa Cruz, sc-5921; 15 μg Pol δ, Santa Cruz, sc-10784; 15 μg Pol ϵ, Santa Cruz, sc-398582) and 1 μg yeast RNA were incubated overnight at 4°C. Protein A/G PLUS agarose beads (Santa Cruz) were then added and samples incubated for 1 h at 4°C. Beads were washed sequentially with wash buffer A (20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 150 mM NaCl), buffer B (20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 500 mM NaCl), buffer C (20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.5% NP40, 0.5% sodium deoxycholate, 250 mM LiCl) and TE buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA). The immunoprecipitate was eluted in 450 μl elution buffer (1% SDS, 0.1 M NaHCO3), and cross-linking was reversed by incubation at 65°C overnight. The eluate was brought to 10 mM EDTA, 40 mM Tris–HCl, pH 6.8 and treated with RNase A at 37°C for 1 h and protease K at 55°C for another hour and the DNA purified by phenol–chloroform extracttel/ion. The input and precipitated DNAs were analyzed by slot blot hybridization with Tel/Alu probes or real-time PCR with tubulin primers at intron 2 (sense: 5′-TATTTGGAAACCGTCACCC antisense: 5′-CTTAGGCTCCGTCCCTGT). The background from the no antibody control was subtracted and the amount of precipitated DNA was calculated as a percentage of the corresponding input.
Telomere FISH
FISH was performed on metaphase spreads from methanol/acetic acid fixed cells as previously described (18), with FITC G-strand probe (5′ CCCTAACCCTAACCCTAA, Biosynthesis) or TelG-Cy3 PNA C-strand probe (5′ GGGTTAGGGTTAGGGTTA, Biosynthesis). Images were taken at a constant exposure time. Multiple Telomere Signals (MTS) and Signal free ends (SFE) are quantified by eye.
Telomere Southern blot
Purified genomic DNAs were restriction digested then run on 1% agarose gels. The gels were denatured, dried and hybridized with 32P-labeled (TAAGGG)3TTA probe to the telomere. Signal was quantified by PhosphorImager, and mean telomere length was determined by dividing each lane into 100 boxes using ImageQuant and applying the formula ΣSig/Σ(SigI/LI), where ΣSig is the sum of the signal from all 100 boxes, SigI is the signal in an individual box, and LI corresponds to the average length of the DNA in that box as determined using DNA markers and a standard curve. To determine the relative amount of telomeric DNA in each sample, the total telomere signal from individual lanes was divided by the signal from the β-actin as loading control.
Analysis of genomic DNA and telomere replication rates
Whole genome replication rates were determined based on EdU uptake (14). Cells were cultured with or without PDS for 24 h, then pulsed with 50 μM EdU for 45 min. Cells were stained for EdU uptake using Click-It AlexaFluor488 according to the manufacturer's instructions. To quantify EdU uptake, the average fluorescence (AFU) of individual nuclei was determined with ImageJ using particle analysis with watershed. The defined regions of interest (ROI) were overlaid onto the image with the EdU signal. The mean AFU was then acquired for each ROI. These numbers were used to determine the average AFU of all nuclei and to show the distribution of nuclei with different ranges of AFU/EdU uptake. At least 700 or 400 nuclei were scored for each independent experiment.
For telomere replication analysis, HeLa cells were released into S phase after a double-thymidine block and cultured in the media containing 50 μM PDS for 4 h, then cells were washed and pulse labeled with BrdU (100 mM) for 2 h. Genomic DNA was isolated by high-salt precipitation. Leading and lagging strand daughter telomeres were separated in CsCl density gradients as described (17). Gradients were fractionated and the amount of telomeric DNA in each fraction was determined by slot blot using 32P-labeled C-strand probe. The percentage of newly synthesized telomere was calculated by dividing the replicated leading or lagging strand peak by the sum of all peaks.
RESULTS
CST and RPA unfold telomeric G-quadruplex DNA with similar efficiency
Although both CST and RPA can unfold G4 DNA (27,38–41) their relative efficiency in removal of this structure has not been examined. Thus, as a first step towards understanding the biological importance of the CST G4 resolving activity, we directly compared the ability of CST and RPA to bind and unfold G4 DNA formed by a telomeric oligonucleotide Tel21 (GGG(TTAGGG)3. In initial experiments we used circular dichroism to confirm Tel21 folding into a G4 structure (Supplementary Figure S1). Consistent with previous reports (42), when Tel21 was incubated in buffer containing 150 mM KCl, the CD revealed two positive peaks at 265 and 295 nm indicating G4 formation. As expected, incubation in 150 mM LiCl destabilized the G4 structure. We next used electrophoretic mobility shift assays (EMSA) to compare CST and RPA binding to Tel21 in the presence of either 150 mM LiCl or KCl (Figure 1A–E). 32P-labeled Tel21 was incubated in LiCl or KCl containing buffer for 30 min to allow DNA folding/unfolding, purified CST or RPA was added for 30 min, then samples were separated in native agarose gels. The EMSA analysis indicated that in conditions favoring ssDNA over G4 formation (150 M LiCl), CST and RPA bound Tel21 with similar efficiency (Kd(app) 1.3 nM for CST versus 1.9 nM for RPA). However, in 150 nM KCl, where Tel21 formed a G4, CST showed slightly lower affinity than RPA (Kd(app) 8.5 nM for CST versus 5.5 nM for RPA). (Figure 1A–E)).
Figure 1.

CST and RPA bind telomeric ssDNA and disrupt G4 structure. (A–E) Comparison of CST and RPA binding to Tel21 in 150 mM LiCl or KCl. (A, C) EMSAs showing CST (A) or RPA (C) binding. Reactions contained the indicated amounts of protein and 0.05 nM DNA. (B, D) Quantification of CST (B) or RPA (D) binding to Tel21 in LiCl versus KCl. Data were fit to a one site specific model to obtain binding isotherms and Kd(app). Mean ± SEM, n = 3 independent experiments each with a different protein preparation. (E) Comparison of Kd(app) for CST and RPA binding to Tel21 in LiCl or KCl. (F) Emission spectra for 5′(FAM)-Tel21-(TAMRA)3′ following addition of indicated amount of CST or RPA and excitation with 494 nM light. Reactions contained 150 mM KCl. (G) FRET showing the disruption of G4 by CST (Red) and RPA (Blue). Donor fluorescence monitored in real-time upon addition of 100 nM CST or RPA. The base line was set to 0 and the maximum value for scan was set to 100. The red line was 50, where 50% of substrates formed G4 structure. CST or RPA was added at the 50 seconds time point. Related to Supplementary Figure S1.
Since CST bound G4 DNA, we next used Fluorescence Resonance Energy Transfer (FRET) to compare the capacity of CST and RPA to unfold G4 structure. The FRET substrate was double-labeled Tel21 that had fluorescein (FAM) at the 5′ end as the FRET donor and tetramethylrhodamine (TAMRA) at the 3′ end as the FRET receptor (Figure 1F, top). As we previous demonstrated, when this oligomer forms a G4, the two fluorophores become closely juxtaposed, leading to FRET and suppression of the donor fluorescence (37,43). To assess G4 unfolding, we added either CST or RPA to the folded Tel21, excited the donor fluorophore with 480 nm light and examined the emission spectrum. As anticipated, addition of RPA caused a concentration-dependent reduction in FRET, as seen by an increase in donor emission (518 nm) and a concomitant decrease in acceptor emission (580 nm). This loss of FRET indicated increased distance between the two fluorophores due to G4 unfolding (43). Addition of CST also resulted in a concentration-dependent decrease in donor emission and an increase in acceptor emission indicating that, like RPA, CST can unfold G4 structure.
To compare the kinetics of G4 unfolding by CST and RPA, we performed a time course study where donor fluorescence was measured before and after CST or RPA addition. Double-labeled Tel21 was incubated in 150 mM KCl to promote G4 formation, then 100 nM proteins was added to maximize unfolding. To our surprise, we found that it took 5 s for CST to resolve 50% of the G4 structure, but 20 s for RPA, suggesting CST was more efficient for G4 resolving compare to RPA (Figure 1G). Thus, although CST has a slightly lower affinity for Tel21 G4 DNA than RPA, CST appears to initiate G4 opening more rapidly at saturating protein concentrations. Despite the subtle differences in Kd(app) and rate of G4 unfolding, our data indicate that CST and RPA generally recognize and unfold G4 DNA with similar efficiency. This finding suggests that CST and RPA are equally well equipped for in vivo G4 removal. Although, CST is less abundant than RPA, local concentrations may be quite high due to interaction with chromatin-associated proteins such as the telomere protein TPP1 (15,44).
CST is recruited to telomeric and non-telomeric DNA in response to G4 formation
To address the in vivo significance of the CST G4-resolving activity, we next examined whether STN1 subcellular distribution is altered after G4 formation. STN1 localization was examined by indirect immunofluorescence using FLAG antibody and a previously established HeLa cell line expressing FLAG-tagged STN1(STN1sh6-Res) (21). The level of STN1 was determined by western blot (Supplementary Figure S2A). G4 was induced by treating the cells with the G4 stabilizing ligands TmPyP4 (50 μM for 24 h) or PDS (10 μM for 24 h). The analysis revealed only a few STN1 foci in DMSO treated control cells whereas TmPyP4 or PDS treatment resulted in a large number of foci. With either drug, the number of cells with 10–20 foci increased ∼1.5-fold and the number with >20 foci increased ∼2-fold (Figure 2A and B). To determine whether the STN1 foci were present at telomeres, we performed telomere FISH to monitor the extent of FLAG-STN1 and telomere co-localization. The FISH revealed that ∼20% of the STN1 foci were present at telomeres (∼17.6% for TmPyP4 and ∼23.1% for PDS) (Figure 2C). These results demonstrate that G4 formation triggers STN1 redistribution and accumulation at specific sites in the nucleus. They further suggested that CST may be recruited to chromatin to aid in the resolution of both telomeric and non-telomeric G4 structures.
Figure 2.

CST is recruited to telomeres after G4 stabilization. (A) Co-localization of FLAG-STN1 and telomeres in HeLa cells after treatment with G4 stabilizers TmPyP4 or PDS. Red, FLAG-STN1 immunostaining; green, telomere FISH; blue, DAPI. (B) Quantification showing increased numbers of FLAG-STN1 foci following treatment with TmPyP4 or PDS. (C) Quantification of FLAG-STN1 foci at telomeres after treatment with G4 ligands. (D, E) ChIP analysis to examine changes in localization of the indicated proteins to telomeres or Alu repeats after PDS treatment with or without 10 μM ATM inhibitor (KU55933) and 2μM ATR inhibitor (ETP-46464). (D) Slot-blot analysis of DNA precipitated with antibody to TPP1, FLAG (STN1), pol α, pol δ or pol ϵ. Blot was hybridized with a telomere (Telo) or Alu probe. (E) Quantification of ChIP. Telo/Alu ChIP signals and Tubulin ChIP-PCR value were normalized against input DNA. Values are mean ± SEM, n = 3 experiments, * P < 0.05. Related to Supplementary Figure S3.
A recent study reported that CST localizes to DNA damage sites through interaction with 53BP1-shiedin, and the localization of CST to DSBs is diminished by inhibition of ATM and ATR signaling pathways. It is also established that G4 formation can induce double and single strand DNA breaks. Thus, to examine whether the CST chromatin association was through direct interaction with G4 or dependent on the DDR pathway activation, we used Chromatin Immunoprecipitation (ChIP) assays to quantify changes in CST abundance on telomeric DNA and Alu repeats following 24 h PDS or TmPyP4 treatment with or without ATM/ATR inhibitors. The ChIP revealed that both PDS and TmPyP4 cause an increase in the amount of STN1 and CTC1 association with telomeric DNA (Figure 2D, E and Supplementary Figure S3A–C). The increase in telomere localization after drug treatment provides strong support for CST recruitment to chromatin following G4 formation. The increase in CST at Alu repeats is also likely to reflect G4 stabilization as Alu sequences contain runs of Gs and additional G-repeats can be present in the adjacent genomic DNA (45). Thus, to examine the effect of G4 ligands on CST association with a non G-rich sequence, we used ChIP-PCR to evaluate the amount of STN1 on intron 2 of the tubulin gene locus (Figure 2E). However, there was no significant change in STN1 association after PDS exposure, implying that the increase in STN1 and CTC1 on telomeric and Alu DNA reflected localized G4 formation rather than a genome-wide effect on DNA replication. Interestingly, we consistently observed that the level STN1 binding to G-rich chromatin was significantly increased upon PDS treatment, even when ATM and ATR signaling pathway was suppressed (Figure 2D–E), suggesting the interaction of CST with the G4 DNA is independent of DDR activation. Since CST interacts with the telomere protein TPP1 (15), we also asked whether PDS affects chromatin association of TPP1. However, ChIP revealed no change in TPP1 abundance, indicating that an increase in chromatin-bound TPP1 is unlikely to underlie the G4-induced recruitment of CST. We did observe some localization of TPP1 to Alu sites, it is possible that this reflects a genome-wide role for shelterin (46,47).
Given that DNA polymerases are required to re-initiate replication after fork stalling, and that CST interacts directly with DNA Pol α (10,48), we next asked whether G4 stabilization also affects DNA polymerase association with telomeres or Alu repeats. Cells were again treated with PDS for 24 h to allow G4 accumulation and ChIP or ChIP-PCR was used to quantify DNA Pol α, Pol δ and Pol ϵ association. At the tubulin gene, PDS treatment had no significant effect on polymerase association. In contrast, at telomeres, PDS treatment caused a clear increase in DNA Pol α and Pol δ but only a modest, non-statistically significant, increase in Pol ϵ. Since Pol α and Pol δ are required for lagging strand replication while Pol ϵ is needed for leading strand replication, this result implies that G4 stabilization predominantly affects lagging strand replication at telomeres. At Alu repeats, PDS treatment only caused a significant increase in Pol α accumulation, raising the possibility of a different outcome with increased primer synthesis by Pol α but no subsequent primer extension by Pol δ or ϵ. Despite this difference, it is notable that G4 stabilizers cause a simultaneous increase in CST and Pol α at telomeres and Alu repeats because this fits with the known role of CST in enhancing priming by Pol α (10,11). Overall, our data suggested that CST might prevent G4s from having adverse effects on DNA replication either by resolving the DNA structure and/or stimulating re-priming after fork stalling.
G4 accumulation is inversely proportional to CST level
Given the ability of CST to bind and unfold G4 DNA in vitro (27), we predicted that the level of G4 accumulation in vivo would be determined by CST abundance, such that cells would have reduced G4 foci after CST overexpression and increased foci after CST depletion. To test this prediction, we first monitored G4 abundance in cells transiently overexpressing CTC1, STN1 and TEN1 (CST oe, Supplementary Figure S2B–D) (21). G4 formation was detected by immunofluorescence using the antibody BG4, which was raised against human telomeric DNA (49). Prior to antibody staining, fixed cells were subjected to cytoplasmic extraction and RNase treatment to remove any RNA G4. Additionally, the specificity of the antibody for DNA versus RNA G4 was confirmed by showing that DNase1-treatment of cells removed BG4 staining whereas RNase treatment did not (Supplementary Figure S4). When we examined G4 accumulation in control cells, as reported previously (49), we observed G4 foci throughout the cell with some foci co-localizing with a subset of telomeres (Figure 3A–C). Strikingly, overexpression of CST reduced the number of G4 foci both at telomeres and elsewhere in the nucleus. When cells were treated with TmPyP4, we saw the expected large increase in G4 foci, however again CST overexpression greatly reduced the number of foci throughout the cell, including at telomeres (Figure 3A–C).
Figure 3.

CST prevents G4 accumulation. (A) Localization of G4 and telomere DNA in control (HeLa) or CST overexpression (HeLa oeCST) HeLa cells. Red, imunolocalization of G4 DNA; green, telomere FISH; blue, DAPI. Arrows indicate telomere and G4 co-localization. Arrows indicate telomere and G4 co-localization. Quantification of total (B) and telomeric (C) G4 foci. Values are mean ± SEM, n = 3 experiments, *P < 0.05, **P < 0.01. (D) Localization of G4 and telomere DNA in control (NT sh) or STN1-depleted (STN sh) HeLa cells with two different G4 antibody BG4 (top) and D1 (bottom) in the present or absent of TmPyP4. NT, non-target control shRNA. Red, imunolocalization of G4 DNA; Green, telomere FISH; Blue, DAPI. Arrows indicate telomere and G4 co-localization. Arrows indicate telomere and G4 co-localization. Quantification of total (E) and telomeric (F) G4 foci. Values are mean ± SEM, n = 3 experiments, *P < 0.05, **P < 0.01. Related to Supplementary Figure S4 and S5.
We next examined the effect of reduced CST on G4 accumulation by monitoring G4 foci in cells expressing an shRNA to STN1 (49) (Supplementary Figure S2A). As an additional control to confirm specific staining, we used two different G4 antibodies (BG4 and D1) to detect G4 foci (50) (Figure 3D–F). Unlike the BG4 antibody, D1 detects parallel G4 structures within telomeres. Regardless of which antibody was used, STN1 depletion caused a significant increase in G4 foci throughout the nucleus and at telomeres. Interestingly, we observed that treatment with G4 ligands in conjunction with STN1/CST lost has a synergistic effect on G4 formation at both telomeric and non-telomeric sequence. Taken together, our experiments with CST overexpressing and STN1 depleted cells indicate that the number of G4 foci in a cell is inversely proportional to CST abundance. This finding points to an in vivo role for CST in regulating G4 accumulation.
Since CST can bind G4 DNA, we next asked whether STN1 localizes to G4s in vivo. Interestingly, immunostaining revealed that STN1 and G4 foci rarely co-localized (Supplementary Figure S5A). One interpretation of this result is that once CST binds a G4 in vivo, it is rapidly unwound. This interpretation fits well with the robust G4 unwinding activity exhibited by CST in vitro (Figure 1, (27)). However, it is also possible that CST binding to the G4 blocks antibody recognition of the G4. Either way, our finding that CST prevents G4 accumulation, together with the previously described affinity of CST for G-rich ssDNA, lead us to suggest two mechanisms by which CST may prevent G4 accumulation. First CST may bind newly formed ssDNA (e.g. the G-rich lagging strand template during telomere replication) to prevent G4 formation. Second, CST may bind and disrupt previously formed G4 structures (e.g. those formed as a result of transcription (51)). Both aspect of CST function would help prevent blocks to DNA replication.
CST limits G4 accumulation in G1
To address whether CST functions to prevent G4 accumulation outside of DNA replication, we examined the effect of STN1 depletion on G4 foci formation in G1 cells. Control and STN1 sh cells were synchronized in G1 using a double thymidine block and then harvested for G4 immunlocalization using BG4 antibody (Figure 4A and B). In some cases, TmPyP4 was added during the second thymidine block. Staining with G4 antibody revealed that STN1 depletion caused a significant increase in G4 at telomeres and elsewhere in the genome of G1 cells. This increase was magnified by TmPyP4 treatment. However, when the effect of STN1 depletion was compared for the G1-synchronized versus unsynchronized cells, it became apparent that loss of CST has a larger effect on G4 accumulation in non G1 cells (Figure 4B versus Figure 3E and F). This was particularly apparent at telomeres. The increase in G4 during G1-phase may reflect ongoing transcription throughout the genome. The larger increase in G4 foci outside of G1 suggests that substantial G4 accumulation occurs as a result of DNA replication.
Figure 4.

STN1 depletion induces G4 formation in G1 phase. (A) Localization of G4 and telomere DNA in control (NT sh) or STN1-depleted (STN sh) HeLa cells. NT, non-target control shRNA. Red, imunolocalization of G4 DNA; green, telomere FISH; blue, DAPI. Arrows indicate telomere and G4 co-localization. Arrows indicate telomere and G4 co-localization. FACS data showing G1 synchronization is shown to the right. (B) Time line of double thymidine block and TMPyP4 treatment. (C) Quantification of total (left) and telomeric (right) G4 foci. Values are mean ± SEM, n = 3 experiments, *P < 0.05, **P < 0.01.
We were interested in why STN1 depletion should cause an increase in telomeric G4 foci in G1 cells. Since CST depletion can cause an increase in G-overhang length (12,14,16,17), one possible explanation was that the longer overhangs might now fold into a G4 structure. To investigate whether the increased G4 correlates with overhang elongation, we examined G4 foci and G-overhang length following transient STN1 knock down using siRNA (Supplementary Figure S6A–E). Western blot analysis revealed that STN1 levels were reduced by day 2 after transfection, reached a minimum at day 4 and were partially restored by day 6. The timing of G4 focus accumulation correlated quite well with STN1 protein levels as most foci were seen at days 2 and 4 but the number of foci dropped substantially by day 6. In contrast, the kinetics of G-overhang elongation was quite different as G-overhang length kept increasing and reached a maximum at day 6 (Supplementary Figure S6A and B). Since, maximum overhang length was observed at a time when the number of G4 foci had declined, we conclude that accumulation of telomeric G4 foci in response to STN1 depletion is largely unrelated to G-overhang elongation. An alternative reason is that telomeres are transcribed and the displaced strand of the resulting R-loops may fold into a G4 structure. Thus, we suspected that the depletion of TERRA would decrease G4 formation during G1 phase. To test for this, we knocked down TERRA using a LNA probe (Supplementary Figure S7A) in cells synchronized in G1 by double thymidine block. The numbers of G4 foci were then quantified by immunostaining (Supplementary Figure S7B–D). Interestingly, we observed a significant decrease in telomeric G4 but not non-telomeric G4 after TERRA depletion, indicating telomeric G4 accumulation was caused by telomere transcription.
CST prevents G4-induced inhibition of DNA replication
If CST facilitates DNA replication by removing or preventing G4, loss of CST should result in a slowing of replication and decreased nucleotide uptake. We therefore asked whether STN1 depletion affects EdU uptake during treatment with G4 stabilizers. STN1 sh and control cells were treated with PDS for 24 h with EdU added to the culture medium for the last 45 min of treatment (Figure 5A). The cells were fixed and the EdU reacted with fluorophore (Figure 5B). The cells were then imaged and the fluorescence intensity of individual nuclei was quantified. The resulting data are presented both as the mean fluorescence intensity of all nuclei (Supplementary Figure S8A) and the percentage of individual nuclei that exhibit a specific level of fluorescence (Arbitrary Fluorescence Units, AFU) (Figure 5C, Supplementary Figure S8B). As anticipated, the control cells showed a decrease in the average fluorescence intensity of all nuclei following 24 h PDS treatment, indicating that EdU uptake was suppressed by G4 formation (Figure 5A–C, Supplementary Figure S8A–B). The decrease in EdU incorporation likely reflects replication fork stalling at G4 DNA (31,52). STN1 depletion caused a further decline in EdU uptake in the PDS-treated cells. This decline was observed both as a decrease in the average fluorescence of all nuclei (Supplementary Figure S8A) and a decrease in the percent of nuclei with the highest levels of EdU incorporation (AFU > 30) (Figure 5C). The decline in EdU uptake was largely rescued by overexpression of an sh-resistant FLAG-STN1 allele in the STN1-sh cells (14) (Figure 5A–C, Supplementary Figure S8A and B). Importantly, without PDS treatment, the levels of EdU uptake were similar in the STN1 sh and control cells (Supplementary Figure S8A and B) indicating that STN1 depletion in the absence of PDS does not cause a significant difference in the number of cells in S-phase or the rate of replication. We therefore conclude that STN1/CST helps promote passage of the replication fork through genomic regions that are prone to G4 formation. This role in replication most likely stems from the capacity of CST to prevent G4 formation during replication or to unwind previously formed G4. It may also reflect the ability of CST to re-prime replication in situations where fork stalling leads to polymerase dissociation (27).
Figure 5.
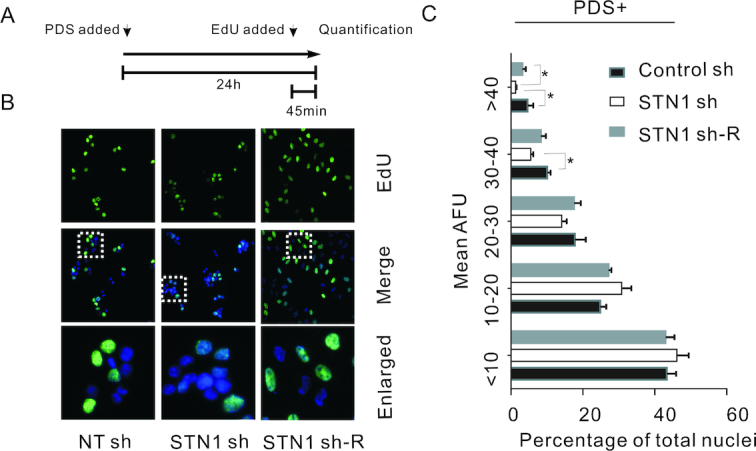
Depletion of CST slows bulk DNA replication upon G4 stabilization. (A) Experimental timeline. HeLa cells were treated with PDSs for 24 h and Edu was pulsed in the last 45 min. (B) EdU incorporation by the indicated PDS-treated cell lines. Blue, DAPI; green, EdU. White boxes indicate regions enlarged in bottom panel. White boxes indicate regions enlarged in bottom panel. (C) Quantification of EdU uptake in the presence of PDS, as measured by mean fluorescence intensity (mean ± SEM, n = 3 experiments, *P < 0.05). Each bar indicates the percent of nuclei within the indicated AFU range. AFU, arbitrary fluorescence units. Related to Supplementary Figure S3 to Supplementary Figure S7.
Combined STN1 depletion and G4 stabilization induces telomere loss.
Since CST functions in multiple aspects of telomere replication, we next examined how G4 stabilization affects telomere integrity in cells that lack STN1. In initial experiments, we examined how the combination of STN1 depletion and PDS treatment affects telomere length. DNA was isolated from control and STN1 sh Hela or U2OS cells after 48 h growth with or without PDS, and Telomere Restriction Fragments (TRF) were examined by Southern blot (Figure 6A and Supplementary Figure S9A). The TRF analysis indicated that mean telomere length was not significantly altered by STN1 depletion consistent with previous reports (6,7). Telomere length also remained essentially unchanged by PDS treatment and by PDS treatment combined with STN1 depletion (Figure 6A and B, Supplementary Figure S9A and B). However, the combination of G4 stabilization and STN1 depletion lead to a striking decline in the overall intensity of the telomere hybridization signal. To determine the magnitude of the decline, blots were hybridized with probe to the actin gene to allow normalization for DNA loading. Quantification revealed that the telomere signal decreased by about one third in both HeLa and U2OS cells (Figure 6C). Given the lack of apparent telomere shortening, this result suggested that a subset of chromosomes had lost essentially all of their telomeric DNA. This sudden telomere loss after combined STN1 depletion and PDS treatment is reminiscent of the what is seen in fission yeast where deletion or mutation of Stn1 lead to dramatic loss of telomere signal due to blocks to telomeric/subtelomeric DNA synthesis (53,54).
Figure 6.
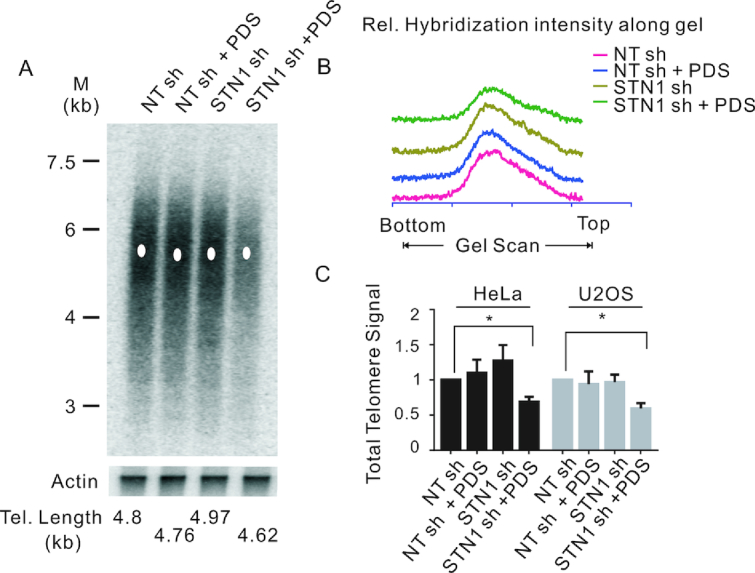
STN1 depletion and G4 stabilization cooperate to cause sudden telomere loss. (A) Southern blot showing telomeric restriction fragments from NT sh and STN1 sh HeLa cells grown with or without 10 μM PDS for 48 h. Probe was (T2AG3)3TTA (top) or β-actin (bottom). M, molecular weight markers. The white dot indicates the mean telomere length. (B) Scans showing relative intensity of hybridization signal. Scans for each sample were from top to bottom of blot. (C) Quantification of total telomere hybridization signal from the indicated HeLa and U2OS cell lines grown with or without PDS. Related to Supplementary Figure S8.
G4-induced telomere dysfunction is exacerbated by STN1 depletion
CST has previously been shown to facilitate replication through the telomere duplex, with loss of CST leading to slowing of BrdU uptake (17). CST depletion also leads to two another hallmarks of disturbed telomere replication: the fragile telomere phenotype where individual chromatids exhibit multiple telomere FISH signals (MTS) and sporadic signal free ends (SFE) where telomeres lack detectable FISH signals (14,16). Our finding that CST can resolve G4 structure suggested that that CST may facilitate telomere duplex replication by decreasing G4 formation during lagging strand synthesis and/or by preventing G4 accumulation as a result of telomere transcription during TERRA production (Supplementary Figure S7B–D) (55). Moreover, if CST functions in this manner, the detrimental effects of CST loss on telomere structure should be increased by treatment with G4 stabilizers. To test this prediction, we examined the combined effect of STN1 depletion and TmPyP4 exposure on telomere integrity.
In initial experiments, we prepared metaphase spreads from STN1 sh and NT sh cells grown with and without TmPyP4 for 48 h and used telomere FISH to monitor the levels of MTS and SFE. The FISH was performed with probes to both the G-and C-strands so we could determine whether any SFE reflected loss of one or both DNA strands (Figure 7A). As previously observed (14), quantification of MTS revealed an ∼2-fold increase after STN1 knockdown regardless of whether we used the G- or C-strand probe (Figure 7B–E). When the cells were treated with TmPyP4, the combined STN1 depletion and G4 stabilization lead to a synergistic increase in MTS being detected with either probe. Since G4 stabilization exacerbated the effect of STN1 loss, this result implies that CST averts replication problems by preventing or removing G4 structure. Moreover, the similar frequency of MTS observed with the G- and C-strand probes implies that CST removes G4 structure ahead of the replication fork as fork stalling at a G4 block should affect both daughter telomeres equally.
Figure 7.

Combined STN1 depletion and G4 stabilization results in preferential loss of telomeric C-strand DNA. (A) Model showing how G4 formation may block DNA synthesis during telomere replication. Red indicates C-strands (identified in telomere FISH using Cy3-TTAGGG)3TTA (red) probe), green indicates G-strands (identified using FITC-(CCCAAT)3CCC (green) probe). (B–E) Telomere FISH with control NT sh or STN1 sh HeLa cells grown with or without 50 μM TmPyP4 for 48 h. (B–C) FISH with probe to G-strand. (D–E) FISH with probe to C-strand. (B, D) Representative images of metaphase spreads showing multiple telomere signals (MTS, yellow arrows) and signal free ends (SFE, white arrows). (C, E) Quantification of MTSs and SFEs (mean ± SEM, n = 3 experiments, *P < 0.05, **P < 0.01, ***P <0.001).
A remarkably different result was observed when we quantified SFE. As previously reported, STN1 depletion did not increase the frequency of detected using either the G- or the C-strand probe (3). Treatment of the control NT sh cells with TmPyP4 also had no effect on SFE detected using the G-strand probe but caused a small increase in SFE detected using the C-strand probe. However, when we combined TmPyP4 treatment with STN1 knockdown we saw an ∼4-fold increase in SFE with the G-strand probe and an ∼16-fold increase with the C-strand probe. Thus, G4 stabilization caused preferential loss of one strand (C-strand) of the telomeric DNA. Since leading and lagging strand synthesis are normally tightly coupled, this specific loss of C-strand DNA is unlikely to result from replication fork collapse leading to a DSB as this would not favor loss of one particular strand. However, TmPyP4 treatment is expected to cause G4 accumulation on the parental G-rich strand as a result of ssDNA exposure during Okazaki fragment synthesis. The G4s could then block lagging strand (C-strand) synthesis thus causing loss of C-strand DNA (Figure 7A) and the increase in SFE detected with the C-strand probe (Figure 7D and E). In contrast, TmPyP4 should not bind to leading strand replication intermediates and hence the drug is unlikely to cause loss of the parental C-rich strand. We therefore infer that combined STN1 depletion and G4 stabilization causes preferential inhibition of lagging strand replication.
A block in lagging strand synthesis should lead to regions of ssDNA on the parental G-rich strand. This ssDNA would then be expected to trigger DNA damage signals. To test whether combined STN1 depletion and G4 stabilization leads to increased damaging signaling, we used immunostaining with antibody to 53BP1 and the telomere protein TRF2 to monitor the appearance of telomere dysfunction induced DNA damage foci (TIFs). Consistent with previous studies (12), a modest increase in TIFs was observed after STN1 knockdown. Likewise, the TmPyP4 caused a slight increase in TIFs in the control (NT sh) cells. However, combined STN1 depletion and TmPyP4 treatment elicited robust TIFs formation (Supplementary Figure S10A and B). This increase in damage signals at chromosome ends that remain bound by TRF2 fits with partial inhibition of lagging strand synthesis as many telomeres would be expected to retain sufficient dsDNA to bind TRF2 while still eliciting a DNA damage response CST depletion slows lagging strand telomere replication after G4 stabilization
CST depletion slows lagging strand telomere replication after G4 stabilization
Since our FISH data suggested that combined STN1 depletion and G4 stabilization preferentially inhibited lagging strand telomere replication, we set out to directly test for this by quantifying the fraction of telomeres replicated by leading versus lagging strand synthesis within a set time period. To achieve this, we labeled S-phase cells with BrdU and then used CsCl gradients to separate telomeres newly replicated by leading or lagging strand synthesis (termed leading or lagging telomeres) and quantified the amount of newly replicated telomeric DNA.
HeLa NT sh and STN1 sh clones were synchronized at G1/S with a double-thymidine block, then released into media with or without PDSs and allowed to progress through S-phase for 4 hrs. They were then pulsed labelled with BrdU for 2 h and harvested (Figure 8A and Supplementary Figure S11A). DNA was isolated, restriction digested and subjected to CsCl density gradient centrifugation to separate the replicated from unreplicated telomeres (Figure 8B) (56). The gradients were fractionated and the relative amount of telomeric DNA in each fraction was determined by slot-blot hybridization using a telomere C-strand probe (Figure 8C). Telomeres replicated by leading strand synthesis incorporate two BrdU molecules per telomeric repeat (UUAGGG) and hence sediment at a higher density than telomeres replicated by lagging strand synthesis (CCCUAA) and both are separated from any unreplicated telomeric DNA, which has no BrdU incorporation (Figure 8B).
Figure 8.

STN1 depletion preferentially decreases lagging strand telomere replication after G4 induction. (A) Experimental timeline. (B) Strategy to separate replicated leading and lagging strand telomeres. Diagram shows BrdU labeling and sedimentation profile for leading, lagging and unreplicated telomeres. Top of the CsCl gradient is to the left. (C) Detection of leading and lagging telomeres by slot blot hybridization with (TTAGGG)3TTA probe. (D) Quantification of telomeric DNA hybridization from each gradient fraction. (E) Percent of newly replicated leading and lagging strand telomere signals relative to the total telomere signal. Data are representative of three independent experiments (mean ± SEM, n = 3, *P < 0.05, **P < 0.01). Related to Supplementary Figure S9.
Quantification of the hybridization signal from replicated leading and lagging telomeres indicated that PDS treatment decreased replication of lagging but not leading telomeres as might be expected given the G-rich nature of the lagging-strand template (Figure 8D and E). STN1 depletion decreased the replication of both leading and lagging telomeres suggesting a general slowing of replication through the telomere dsDNA. The combination of STN1 depletion and PDS treatment had no further effect on leading telomere replication. In contrast, lagging telomeres exhibited a significant decrease in newly replicated DNA relative to either single treatment. The preferential effect of the combined treatment on lagging telomeres indicates that CST (STN1) plays an important role in facilitating lagging strand telomere replication in response to G4-induced replication stress.
DISCUSSION
Although timely resolution of G4 DNA is essential to prevent stalling of DNA replication and ensuing genomic instability, the mechanisms for G4 removal are still not fully understood (31,57). Here we identify CST as a novel player in this process. We show that mammalian CST unwinds G4 structure in vitro, prevents G4 accumulation in vivo and facilitates DNA replication through G4-containing sequence. Although CST is particularly important for telomere replication, its roles in G4 resolution are not limited to telomeres. Induction of G4 structure increases CST localization to both telomeric and non-telomeric chromatin and loss of CST slows bulk genomic DNA replication after G4 stabilization. At telomeres, combined CST depletion and G4 stabilization causes dramatic telomere loss and decreased lagging-strand telomere replication. Thus, the capacity of CST to prevent or remove G4 is essential to maintain telomere integrity. Our findings lead to a model where CST facilitates Okazaki fragment synthesis by resolving or preventing G4 structure on the single-stranded G-rich template DNA. Our data also support a role for CST in preventing replication blocks by removing G4 structures that accumulate ahead of the replication fork, most likely as a result of transcription earlier in the cell cycle.
Failure of lagging-strand synthesis occurs when the exposed template DNA forms secondary structures that block passage of the replicative polymerases (58). Here, we demonstrate the sensitivity of lagging-strand replication to G4 blocks by showing that G4 stabilizers specifically cause the lagging-strand polymerases (Pol α and Pol δ) to accumulate at telomeres. We also show that loss of CST greatly exacerbates problems associated with G4 stabilization to cause G4-induced failure of lagging strand synthesis. The result is a reduction in replicated lagging strand telomeres and loss of telomeric C-strand DNA. These findings provide the first direct evidence that CST facilitates DNA replication by resolving blocks to lagging strand synthesis. Although multiple factors affect telomere replication (59–63), it is most unusual to observe the preferential loss of one strand of telomeric DNA. This is because problems associated with telomere dsDNA replication generally cause replication fork stalling. Subsequent fork collapse leads to a DSB and loss of both DNA strands from one or other sister chromatid (64,65). Our finding that CST depletion causes a specific reduction in C-strand DNA implies a lack of replication fork collapse despite the perturbation of lagging strand synthesis. Given that leading and lagging strand synthesis are normally tightly coupled (66), this phenomenon could be explained if initiation of Okazaki fragment synthesis by Pol α remains unaffected. We therefore suggest that G4 formation on the lagging strand template blocks Okazaki fragment extension by DNA Pol α and/or Pol δ rather than the primer initiation step (Figure 9). The outcome would be incomplete C-strand synthesis (Figure 7) and accumulation of ssDNA without stalling of the replication fork, a situation that is consistent with recent studies indicating that coordination of leading- and lagging-strand synthesis may not be required for DNA duplication (67).
Figure 9.

Model for how CST prevents preferential loss of telomeric C-strand DNA following G4 stabilization. Top: In the absence of STN1/CST, replication of the leading strand telomere is unaffected, however formation of G4 structure on the lagging strand template blocks Okazaki fragment synthesis. Bottom: CST/ STN1 resolves or prevents G4 formation. CST may also recruit DNA polα primase to reinitiate replication after G4 resolution.
While the lagging strand template is a prime site for G4 accumulation at telomeres and other G-rich sequences, G4 structures also form during transcription (29,68). They tend to occur on the displaced G-rich strands of R-loops and once formed they stabilize the R-loop (69). The resulting G4/R-loop combination can then block passage of the replication fork. Since, TERRA transcription makes telomeres especially prone to R-loop formation, efficient G4 removal ahead of the replication fork is essential to prevent fork stalling and collapse (70). Evidence that CST participates in removal of G4 from telomeric dsDNA comes from our finding that combined CST depletion and G4 stabilization (i) increases G4 accumulation in G1 cells and (ii) causes MTS levels to increase on both leading and lagging telomeres. Since MTS are a hallmark of replication fork stalling (62), the latter result supports a role for CST in removing G4 structure ahead of the replication fork. Replication fork stalling and collapse into a DSB is also likely to explain the ∼1.5% of telomeres that exhibit SFE with loss of G-strand DNA after CST depletion and G4 stabilization (Figure 7C).
Although the current study is focused more heavily on the role of CST in telomere replication, we show that CST depletion also decreases bulk genomic DNA replication after G4 stabilization. Thus it seems likely that the ability of CST to prevent or resolve G4 will be important for efficient DNA replication at G-rich regions throughout the genome. Interestingly, although STN1(CST) depletion leads to obvious G4 accumulation (Figures 3D and E, 4A and B), we did not observe significant changes in the rate of bulk genomic DNA replication. One explanation for this observation might be that G4 formation is relatively rare in bulk genomic DNA relative to within the telomeric tract. Thus, other protein factors (e.g. ssDNA binding proteins, helicases and nucleases) may be sufficient to resolve the structure and maintain replication.
The detrimental effects of G4 on DNA replication and genome stability likely explains why cells have evolved a wide range of factors to remove these structures (e.g. ssDNA binding proteins, helicases and nucleases) (38,71–73). Depending on the context of the G4, specific factors may be better suited for G4 prevention or resolution. For example, RPA and CST have similar ssDNA binding and G4 unfolding activities (38,74). However, use of CST to resolve G4s during DNA replication may be advantageous because CST can unfold G4 structures more rapidly than RPA and CST-coated ssDNA does not cause ATR activation (Figure 1 and (18)). Helicases are another major group of proteins known to promote efficient resolution of G4 structure (75). However, engagement of a helicase on G4-containing DNA requires either an adjacent region of ssDNA for loading or interaction with another protein. For example, TRF1 is needed to recruit the BLM helicase to telomeres for G4 resolution (76). In contrast to helicases, CST can bind to and unwind G4 in the absence of adjacent ssDNA (27).
Going forward it will be important to explore how CST interfaces with other G4 resolving activities in a cell. It could be that CST serves as a first line of defense against G4-induced replication issues by coating the exposed ssDNA to prevent G4 formation without activating DNA damage signaling. It will be interesting to determine whether CST also promotes helicase engagement on the ssDNA (e.g. WRN, BLM or RTEL). CST-mediated G4 regulation may be also important for processes beyond DNA replication. G4 structures are expected to form at thousands of sites in the genome where they can affect a wide range of cellular processes including transcription and translation. Interestingly, mutations in CST cause Coats plus, a lethal disease with very pleiotrophic symptoms (77–79). While some symptoms mirror those of telomere maintenance syndromes such as dyskeratosis congenita, others are unique. It is possible that the diverse symptoms of Coats plus patients reflect both altered gene expression and genome-wide replication issues due to loss of normal CST G4-resolving activity.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Shankar Balasubramanian (University of Cambridge, UK) for providing BG4 antibody (plasmid). Dr Zhishu Huang (Sun Yat-sen University) for kindly providing us the D1 antibody. We thank Dr Xuyang Feng's comment and help on the TERRA knockdown.
Author contributions: Y.L. and R.L. designed and performed the ChIP assay, G4 staining and contributed to other experiments. Y.N.X. and T.F.L. designed and executed the MTS and TIF analysis. They also contributed to other experiments. M.M.Z. performed the EdU incorporation analysis and contributed to other experiments. B.W. performed the CsCl gradient experiment. B.W. and R.L. designed and performed the TERRA depletion experiment. Q.L. performed the southern blot. F.W. designed and directed the project and prepared the manuscript. C.M.P. contributed to project design and manuscript preparation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2017YFC1001904]; National Natural Science Foundation of China [91649102, 31771520, 31471293, 21177091, 81772243, 21647008, 81501386, 81671054, 81771135]; Natural Science Foundation of Tianjin [17JCYBJC42700]; PUMC Youth Fund and PUMC Fundamental Research Funds for the Central Universities [2018RC310020;2017310026]; National Institutes of Health [RO1041803 to C.M.P.]. Funding for open access charge: Ministry of Science and Technology [2017YFC1001904].
Conflict of interest statement. None declared.
REFERENCES
- 1. O'Sullivan R.J., Karlseder J.. Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010; 11:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arnoult N., Karlseder J.. Complex interactions between the DNA-damage response and mammalian telomeres. Nat. Struct. Mol. Biol. 2015; 22:859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stewart J.A., Chaiken M.F., Wang F., Price C.M.. Maintaining the end: roles of telomere proteins in end-protection, telomere replication and length regulation. Mut. Res. 2012; 730:12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sfeir A., de Lange T.. Removal of shelterin reveals the telomere end-protection problem. Science. 2012; 336:593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim J.K., Liu J., Hu X., Yu C., Roskamp K., Sankaran B., Huang L., Komives E.A., Qiao F.. Structural basis for shelterin bridge assembly. Mol.Cell. 2017; 68:698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miyake Y., Nakamura M., Nabetani A., Shimamura S., Tamura M., Yonehara S., Saito M., Ishikawa F.. RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol. Cell. 2009; 36:193–206. [DOI] [PubMed] [Google Scholar]
- 7. Surovtseva Y.V., Churikov D., Boltz K.A., Song X., Lamb J.C., Warrington R., Leehy K., Heacock M., Price C.M., Shippen D.E.. Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol. Cell. 2009; 36:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wellinger R.J. The CST complex and telomere maintenance: the exception becomes the rule. Mol. Cell. 2009; 36:168–169. [DOI] [PubMed] [Google Scholar]
- 9. Ruggiero E., Richter S.N.. G-quadruplexes and G-quadruplex ligands: targets and tools in antiviral therapy. Nucleic Acids Res. 2018; 46:3270–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goulian M., Heard C.J.. The mechanism of action of an accessory protein for DNA polymerase alpha/primase. J. Biol. Chem. 1990; 265:13231–13239. [PubMed] [Google Scholar]
- 11. Ganduri S., Lue N.F.. STN1-POLA2 interaction provides a basis for primase-pol alpha stimulation by human STN1. Nucleic Acids Res. 2017; 45:9455–9466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang C., Dai X., Chai W.. Human Stn1 protects telomere integrity by promoting efficient lagging-strand synthesis at telomeres and mediating C-strand fill-in. Cell Res. 2012; 22:1681–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gu P., Min J.N., Wang Y., Huang C., Peng T., Chai W., Chang S.. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J. 2012; 31:2309–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stewart J.A., Wang F., Chaiken M.F., Kasbek C., Chastain P.D. 2nd, Wright W.E., Price C.M.. Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J. 2012; 31:3537–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen L.Y., Redon S., Lingner J.. The human CST complex is a terminator of telomerase activity. Nature. 2012; 488:540–544. [DOI] [PubMed] [Google Scholar]
- 16. Kasbek C., Wang F., Price C.M.. Human TEN1 maintains telomere integrity and functions in genome-wide replication restart. J. Biol. Chem. 2013; 288:30139–30150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang F., Stewart J.A., Kasbek C., Zhao Y., Wright W.E., Price C.M.. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep. 2012; 2:1096–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feng X., Hsu S.J., Kasbek C., Chaiken M., Price C.M.. CTC1-mediated C-strand fill-in is an essential step in telomere length maintenance. Nucleic Acids Res. 2017; 45:4281–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng X., Hsu S.J., Bhattacharjee A., Wang Y., Diao J., Price C.M.. CTC1-STN1 terminates telomerase while STN1-TEN1 enables C-strand synthesis during telomere replication in colon cancer cells. Nat. Commun. 2018; 9:2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boccardi V., Razdan N., Kaplunov J., Mundra J.J., Kimura M., Aviv A., Herbig U.. Stn1 is critical for telomere maintenance and long-term viability of somatic human cells. Aging Cell. 2015; 14:372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang F., Stewart J., Price C.M.. Human CST abundance determines recovery from diverse forms of DNA damage and replication stress. Cell Cycle. 2014; 13:3488–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chastain M., Zhou Q., Shiva O., Fadri-Moskwik M., Whitmore L., Jia P., Dai X., Huang C., Ye P., Chai W.. Human CST facilitates Genome-wide RAD51 recruitment to GC-Rich repetitive sequences in response to replication stress. Cell Rep. 2016; 16:1300–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mirman Z., Lottersberger F., Takai H., Kibe T., Gong Y., Takai K., Bianchi A., Zimmermann M., Durocher D., de Lange T.. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature. 2018; 560:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun J., Yu E.Y., Yang Y., Confer L.A., Sun S.H., Wan K., Lue N.F., Lei M.. Stn1-Ten1 is an Rpa2-Rpa3-like complex at telomeres. Genes Dev. 2009; 23:2900–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bhattacharjee A., Stewart J., Chaiken M., Price C.M.. STN1 OB fold mutation alters DNA binding and affects selective aspects of CST function. PLoS Genet. 2016; 12:e1006342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen R., Wold M.S.. Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. BioEssays. 2014; 36:1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bhattacharjee A., Wang Y., Diao J., Price C.M.. Dynamic DNA binding, junction recognition and G4 melting activity underlie the telomeric and genome-wide roles of human CST. Nucleic Acids Res. 2017; 45:12311–12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hom R.A., Wuttke D.S.. Human CST prefers G-Rich but not necessarily telomeric sequences. Biochemistry. 2017; 56:4210–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rhodes D., Lipps H.J.. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015; 43:8627–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huppert J.L., Balasubramanian S.. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005; 33:2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vannier J.B., Pavicic-Kaltenbrunner V., Petalcorin M.I., Ding H., Boulton S.J.. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012; 149:795–806. [DOI] [PubMed] [Google Scholar]
- 32. Li X.M., Zheng K.W., Zhang J.Y., Liu H.H., He Y.D., Yuan B.F., Hao Y.H., Tan Z.. Guanine-vacancy-bearing G-quadruplexes responsive to guanine derivatives. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:14581–14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guilbaud G., Murat P., Recolin B., Campbell B.C., Maiter A., Sale J.E., Balasubramanian S.. Local epigenetic reprogramming induced by G-quadruplex ligands. Nat. Chem. 2017; 9:1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zimmer J., Tacconi E.M., Folio C., Badie S., Porru M., Klare K., Tumiati M., Markkanen E., Halder S., Ryan A. et al.. Targeting BRCA1 and BRCA2 deficiencies with G-Quadruplex-Interacting compounds. Mol. Cell. 2016; 61:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paeschke K., Bochman M.L., Garcia P.D., Cejka P., Friedman K.L., Kowalczykowski S.C., Zakian V.A.. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature. 2013; 497:458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sarkies P., Reams C., Simpson L.J., Sale J.E.. Epigenetic instability due to defective replication of structured DNA. Mol. Cell. 2010; 40:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mergny J.L., Maurizot J.C.. Fluorescence resonance energy transfer as a probe for G-quartet formation by a telomeric repeat. Chembiochem. 2001; 2:124–132. [DOI] [PubMed] [Google Scholar]
- 38. Salas T.R., Petruseva I., Lavrik O., Bourdoncle A., Mergny J.L., Favre A., Saintome C.. Human replication protein A unfolds telomeric G-quadruplexes. Nucleic Acids Res. 2006; 34:4857–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Audry J., Maestroni L., Delagoutte E., Gauthier T., Nakamura T.M., Gachet Y., Saintome C., Geli V., Coulon S.. RPA prevents G-rich structure formation at lagging-strand telomeres to allow maintenance of chromosome ends. EMBO J. 2015; 34:1942–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Safa L., Delagoutte E., Petruseva I., Alberti P., Lavrik O., Riou J.F., Saintome C.. Binding polarity of RPA to telomeric sequences and influence of G-quadruplex stability. Biochimie. 2014; 103:80–88. [DOI] [PubMed] [Google Scholar]
- 41. Ray S., Qureshi M.H., Malcolm D.W., Budhathoki J.B., Celik U., Balci H.. RPA-mediated unfolding of systematically varying G-quadruplex structures. Biophys. J. 2013; 104:2235–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mergny J.L., Lacroix L.. UV melting of G-Quadruplexes. Curr.Protoc. Nucleic Acid Chem. 2009; doi:10.1002/0471142700.nc1701s37. [DOI] [PubMed] [Google Scholar]
- 43. Wang F., Tang M.L., Zeng Z.X., Wu R.Y., Xue Y., Hao Y.H., Pang D.W., Zhao Y., Tan Z.. Telomere- and telomerase-interacting protein that unfolds telomere G-quadruplex and promotes telomere extension in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:20413–20418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wan M., Qin J., Songyang Z., Liu D.. OB fold-containing protein 1 (OBFC1), a human homolog of yeast Stn1, associates with TPP1 and is implicated in telomere length regulation. J. Biol. Chem. 2009; 284:26725–26731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Batzer M.A., Deininger P.L.. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002; 3:370–379. [DOI] [PubMed] [Google Scholar]
- 46. Martinez P., Thanasoula M., Carlos A.R., Gomez-Lopez G., Tejera A.M., Schoeftner S., Dominguez O., Pisano D.G., Tarsounas M., Blasco M.A.. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat. Cell Biol. 2010; 12:768–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang D., Xiong Y., Kim H., He Q., Li Y., Chen R., Songyang Z.. Human telomeric proteins occupy selective interstitial sites. Cell Res. 2011; 21:1013–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Casteel D.E., Zhuang S., Zeng Y., Perrino F.W., Boss G.R., Goulian M., Pilz R.B.. A DNA polymerase-{alpha}{middle dot}primase cofactor with homology to replication protein A-32 regulates DNA replication in mammalian cells. J. Biol. Chem. 2009; 284:5807–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Biffi G., Tannahill D., McCafferty J., Balasubramanian S.. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013; 5:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zheng X.H., Nie X., Fang Y., Zhang Z., Xiao Y., Mao Z., Liu H., Ren J., Wang F., Xia L. et al.. A cisplatin derivative Tetra-Pt(bpy) as an oncotherapeutic agent for targeting ALT cancer. J. Natl.Cancer Inst. 2017; 109:doi:10.1093/jnci/djx061. [DOI] [PubMed] [Google Scholar]
- 51. Fleming A.M., Zhu J., Ding Y., Burrows C.J.. 8-Oxo-7,8-dihydroguanine in the context of a gene promoter G-Quadruplex is an On-Off switch for transcription. ACS Chem. Biol. 2017; 12:2417–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Crabbe L., Verdun R.E., Haggblom C.I., Karlseder J.. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004; 306:1951–1953. [DOI] [PubMed] [Google Scholar]
- 53. Matmati S., Vaurs M., Escandell J.M., Maestroni L., Nakamura T.M., Ferreira M.G., Geli V., Coulon S.. The fission yeast Stn1-Ten1 complex limits telomerase activity via its SUMO-interacting motif and promotes telomeres replication. Sci. Adv. 2018; 4:eaar2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Takikawa M., Tarumoto Y., Ishikawa F.. Fission yeast Stn1 is crucial for semi-conservative replication at telomeres and subtelomeres. Nucleic Acids Res. 2017; 45:1255–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maestroni L., Matmati S., Coulon S.. Solving the telomere replication problem. Genes. 2017; 8:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao Y., Abreu E., Kim J., Stadler G., Eskiocak U., Terns M.P., Terns R.M., Shay J.W., Wright W.E.. Processive and distributive extension of human telomeres by telomerase under homeostatic and nonequilibrium conditions. Mol. Cell. 2011; 42:297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gilson E., Geli V.. How telomeres are replicated. Nat. Rev. Mol. Cell Biol. 2007; 8:825–838. [DOI] [PubMed] [Google Scholar]
- 58. Schiavone D., Guilbaud G., Murat P., Papadopoulou C., Sarkies P., Prioleau M.N., Balasubramanian S., Sale J.E.. Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J. 2014; 33:2507–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li F., Kim H., Ji Z., Zhang T., Chen B., Ge Y., Hu Y., Feng X., Han X., Xu H. et al.. The BUB3-BUB1 complex promotes telomere DNA replication. Mol. Cell. 2018; 70:395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nera B., Huang H.S., Lai T., Xu L.. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat. Commun. 2015; 6:10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sarek G., Vannier J.B., Panier S., Petrini J.H.J., Boulton S.J.. TRF2 recruits RTEL1 to telomeres in S phase to promote t-loop unwinding. Mol. Cell. 2015; 57:622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sfeir A., Kosiyatrakul S.T., Hockemeyer D., MacRae S.L., Karlseder J., Schildkraut C.L., de Lange T.. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009; 138:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. d’Alcontres M.S., Palacios J.A., Mejias D., Blasco M.A.. TopoIIalpha prevents telomere fragility and formation of ultra thin DNA bridges during mitosis through TRF1-dependent binding to telomeres. Cell Cycle. 2014; 13:1463–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sirbu B.M., McDonald W.H., Dungrawala H., Badu-Nkansah A., Kavanaugh G.M., Chen Y., Tabb D.L., Cortez D.. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 2013; 288:31458–31467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Trenz K., Smith E., Smith S., Costanzo V.. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006; 25:1764–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Georgescu R.E., Yao N., Indiani C., Yurieva O., O’Donnell M.E.. Replisome mechanics: lagging strand events that influence speed and processivity. Nucleic Acids Res. 2014; 42:6497–6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Graham J.E., Marians K.J., Kowalczykowski S.C.. Independent and stochastic action of DNA polymerases in the replisome. Cell. 2017; 169:1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maizels N., Gray L.T.. The G4 genome. PLoS Genet. 2013; 9:e1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Abd El-Moneam N.M., Shreadah M.A., El-Assar S.A., Nabil-Adam A.. Protective role of antioxidants capacity of Hyrtios aff. Erectus sponge extract against mixture of persistent organic pollutants (POPs)-induced hepatic toxicity in mice liver: biomarkers and ultrastructural study. Environ. Sci. Pollut. Res. Int. 2017; 24:22061–22072. [DOI] [PubMed] [Google Scholar]
- 70. Joo H.N., Van Thi Nguyen T., Chae H.K., Seo Y.J.. pH-Dependant fluorescence switching of an i-motif structure incorporating an isomeric azobenzene/pyrene fluorophore. Bioorg. Med. Chem. Lett. 2017; 27:2415–2419. [DOI] [PubMed] [Google Scholar]
- 71. Leon-Ortiz A.M., Svendsen J., Boulton S.J.. Metabolism of DNA secondary structures at the eukaryotic replication fork. DNA Repair. 2014; 19:152–162. [DOI] [PubMed] [Google Scholar]
- 72. Lin W., Sampathi S., Dai H., Liu C., Zhou M., Hu J., Huang Q., Campbell J., Shin-Ya K., Zheng L. et al.. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013; 32:1425–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zaug A.J., Podell E.R., Cech T.R.. Human POT1 disrupts telomeric G-quadruplexes allowing telomerase extension in vitro. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:10864–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Marechal A., Zou L.. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015; 25:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sauer M., Paeschke K.. G-quadruplex unwinding helicases and their function in vivo. Biochem. Soc. Trans. 2017; 45:1173–1182. [DOI] [PubMed] [Google Scholar]
- 76. Zimmermann M., Kibe T., Kabir S., de Lange T.. TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev. 2014; 28:2477–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Anderson B.H., Kasher P.R., Mayer J., Szynkiewicz M., Jenkinson E.M., Bhaskar S.S., Urquhart J.E., Daly S.B., Dickerson J.E., O'Sullivan J. et al.. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat. Genet. 2012; 44:338–342. [DOI] [PubMed] [Google Scholar]
- 78. Simon A.J., Lev A., Zhang Y., Weiss B., Rylova A., Eyal E., Kol N., Barel O., Cesarkas K., Soudack M. et al.. Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J. Exp. Med. 2016; 213:1429–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Walne A.J., Bhagat T., Kirwan M., Gitiaux C., Desguerre I., Leonard N., Nogales E., Vulliamy T., Dokal I.S.. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica. 2013; 98:334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.