

Abstract
Background
Shotgun metagenomes contain a sample of all the genomic material in an environment, allowing for the characterization of a microbial community. In order to understand these communities, bioinformatics methods are crucial. A common first step in processing metagenomes is to compute abundance estimates of different taxonomic or functional groups from the raw sequencing data.
Given the breadth of the field, computational solutions need to be flexible and extensible, enabling the combination of different tools into a larger pipeline.
Results
We present NGLess and NG-meta-profiler. NGLess is a domain specific language for describing next-generation sequence processing pipelines. It was developed with the goal of enabling user-friendly computational reproducibility. It provides built-in support for many common operations on sequencing data and is extensible with external tools with configuration files.
Using this framework, we developed NG-meta-profiler, a fast profiler for metagenomes which performs sequence preprocessing, mapping to bundled databases, filtering of the mapping results, and profiling (taxonomic and functional). It is significantly faster than either MOCAT2 or htseq-count and (as it builds on NGLess) its results are perfectly reproducible.
Conclusions
NG-meta-profiler is a high-performance solution for metagenomics processing built on NGLess. It can be used as-is to execute standard analyses or serve as the starting point for customization in a perfectly reproducible fashion.
NGLess and NG-meta-profiler are open source software (under the liberal MIT license) and can be downloaded from https://ngless.embl.de or installed through bioconda.
Electronic supplementary material
The online version of this article (10.1186/s40168-019-0684-8) contains supplementary material, which is available to authorized users.
Keywords: Metagenomics, Next-generation sequencing, Domain-specific language
Background
Over the last decade, metagenomics has increasingly been applied to the study of microbial communities. Most work has focused on human-associated habitats [1] with a particular emphasis on the human gut microbiome [2, 3]. However, the same methodologies have been used for studying other host-associated microbiota [4–6] or the marine microbiome [7]. Due to its size and complexity, several computational approaches have been proposed to handle these data, including bioinformatic pipelines combining different tools and approaches [8–11].
A typical metagenomics processing workflow can be divided into two distinct phases: in the first phase, raw data is processed (often using prebuilt reference databases) to generate a table of feature abundances (a profile). These features can be either taxonomic or functional annotations. Secondly, these profiles are analyzed (often in regards to relevant metadata) using statistical methods and packages such as phyloseq [12], vegan [13], or LEfSe [14]. In this work, we are focused on the first phase: namely obtaining functional and taxonomic profiles from raw metagenomic reads.
To this end, we present NG-meta-profiler, a collection of pre-configured pipelines based on the domain-specific language NGLess (Next Generation Language for less effortful analysis). Although NG-meta-profiler can be used as a standalone tool, the syntax and semantics of NGLess have been designed to be simple and human readable, allowing users to read or create their own pipelines, even without deep bioinformatics and programming knowledge. In other scientific contexts, domain-specific languages have been empirically found to increase productivity and user satisfaction [15, 16]. At the same time, NGLess is designed to enable perfect reproducibility of the computational process, an increasingly important concern [17–19].
Implementation
NGLess, a domain-specific language for NGS processing
NGLess is a domain-specific language which was designed specifically for next-generation sequence (NGS) processing. NGLess was initially designed by analyzing the intended use cases with potential users. NGLess is an imperative programming language, given that most bioinformaticians are familiar with languages using this paradigm [20]. The result contains types for concepts that were found to be important in the problem domain, such as ShortRead (representing a single short read) or ShortReadSet (representing a set of, possibly paired-end, short reads). Unlike most programming languages, values of these types are often represented by files on disk and not values in memory due to their large size.
We also conducted a user study (with a sample of convenience, with participants recruited locally and through social media) on an earlier version of the language, which revealed some issues in those earlier versions. Experience from tutorials on using the language was also used to simplify some aspects of the language. For example, in an earlier version of NGLess, there existed a construct to preprocess short-reads using pass-by-reference, where the argument to the preprocess function was modified. We found that changing this to pass-by-value led to fewer misunderstandings. Thus, the language now uses exclusively pass-by-value semantics. Another area where progress benefited from user input on beta versions is the quality of the error messages. Even when usage reports were due to user errors or malformed input files, we took this opportunity to improve the error reporting in NGLess to help the users diagnose and debug the underlying issue.
The built-in knowledge of the sequence processing domain allows for best-practices to be automatic. For example, our tool always collects quality control statistics without the user having to specify it as an additional computational step.
Domain knowledge enables the interpreter to perform computations more efficiently. For example, even though users write their pipeline script in a purely linear fashion, the interpreter can automatically detect when parallelization opportunities are available and take advantage of multiple processors in shared memory machines. In particular, loops are automatically parallelized, and format conversions (e.g., compression/decompression) use separate threads. When relying on external tools, NGLess can automatically detect possibilities to avoid intermediate files, while handling all format conversions internally.
Finally, error detection and reporting are significantly improved by having the tool be semantically aware of its goals. Given that debugging consumes a significant fraction of the time invested in developing computational pipelines, fast error detection can speed up the overall project. For example, it is possible to check whether inputs are readable and outputs are writable prior to starting interpretation. This benefits the user whenever they have made a mistake as errors are detected and reported immediately.
While introducing a novel language implies that the user needs to learn a new tool, the language is designed to be easily understandable to scientists familiar with the field. Alternatively, a Python interface to NGLess allows users familiar with that programming language to access NGLess functionality. Similarly, NG-meta-profiler is a command line tool that can be used directly without knowledge of NGLess.
In version 1.0, NGLess implements the following tasks: (1) preprocessing, (2) assembly, (3) open-reading frame (ORF) finding, (4) mapping to sequence databases, (5) filtering of mapping results, (6) profiling (up-to-date taxonomic and functional profiling databases are provided), and (7) summary plots. See Additional file 4: Text S1 and the online manual for a full description of the language, including the complete grammar in extended Back-Naur notation as well as documentation for all the built-in types, functions, and modules.
NG-meta-profiler: a fast metagenomics profiler
Using the NGLess framework, we have developed NG-meta-profiler, a collection of pipelines for taxonomic and functional profiling of metagenomes. These standard analyses can be run with a single command. However, they can also serve as a starting point for customization by the user, including extending them with novel tools.
These pipelines are defined in the NGLess language, and currently, there are workflows available for human, mouse, pig, and dog gut as well as marine metagenomes. In the future, we plan to continually update the resources (annotated gene catalogs and other databases) for these environments, as well as providing resources for other environments.
At a high level, NG-meta-profiler, performs the following operations: (1) preprocess the reads (performing quality-based trimming and filtering of short reads and, if appropriate, discarding reads that align to the host), (2) map the reads against a predefined gene catalog selected for that biome, and (3) use the predefined gene annotations to build a profile (see Fig. 1).
Fig. 1.

Schematic illustration of NG-meta-profiler
For preprocessing the short-reads, we use built-in NGLess constructs to perform quality based trimming and filtering of short reads. In NG-meta-profiler, we use the substrim function to trim reads. This function is built into NGLess and trims the sequence to the longest substring of its input such that all bases have at least the requested quality (the same approach was initially introduced by SolexaQA [21]). We keep only reads above quality value 25, which is the central point of the recommended range (20 to 30) in a previous study of the effects of read trimming [22]. Finally, we discard reads shorter than 45 base pairs as shorter reads are significantly more likely to align in multiple locations. These defaults can easily be changed by the user.
After preprocessing, it is necessary to remove reads that match the human genome. For this operation, we will first align the reads against the hg19 built-in reference.
As the default mapper is a local aligner (bwa-mem), it aligns reads even when only a short substring maps. To remove these spurious alignments, we remove very short alignments, namely those below 45 bp (see Additional file 1: Figure S1).
To obtain functional profiles from metagenomes, we use pre-classified gene catalogs as references, an approach that was previously shown to outperform per-read classification [8, 9, 23, 24].
Bundled databases and modules
As part of the first release of NG-meta-profiler, we bundle several genomes (including human and mouse) as well as gene catalogs for the human gut microbiome [25], the marine microbiome [7], and three non-human mammal microbiomes (pig [5], dog [6], and mouse [4]). These gene catalogs have been functionally annotated with eggnog-mapper [23] so that functional profiles can be generated by NGLess (see Table 1). In the current version, NG-meta-profiler can produce eggNOG orthologous group [26], KEGG orthologous groups (KO) [27], SEED [28], and BiGG [29] abundance profiles. Additionally, genomes of several organisms (see Additional file 4: Text S1) are also provided. These are used for filtering out host reads in host-associated metagenomes in NG-meta-profiler.
Table 1.
Gene catalogs bundled with NG-meta-profiler
All of these resources are automatically downloaded by NGLess the first time they are used so that the initial download is small (currently 15 MiB) and each user only downloads those resources they effectively use. In addition to these, the user can provide external references by specifying the file paths to the resources on disk or writing an external module (which can be defined using a text file).
Several operations in NGLess are performed with bundled software. De novo assembly is performed using MEGAHIT [30], which has been found to perform well for metagenomics [31, 32]. Open reading frame (ORF) finding is performed with Prodigal [33]. By default, mapping is performed using bwa [34], but minimap2 [35] is also provided and SOAPaligner [36] can be used. Additional built-in modules provide extra functionality as shown in Table 2.
Table 2.
NGLess built-in modules that add extra functionality
The bundled databases and modules were chosen because of their wide use at the time that NGLess 1.0 was released. However, they will be updated as appropriate (e.g., when new versions of the underlying databases are available).
Results and discussion
Benchmarking
We compared the performance of NG-meta-profiler with both MOCAT2 [9] and a pipeline based on calling bwa without preprocessing data and htseq-count [37] for profiling human gut [38] and ocean metagenomes [7]. For this benchmark, eight threads were used (except for the htseq-count software which only supports a single thread).
Results are presented in Fig. 2 (see also Additional file 2: Table S1). NG-meta-profiler clearly outperforms the other solutions in this task. When compared to MOCAT2, the NG-meta-profiler runs 11× faster in the marine benchmark, and 2.6× in the human gut metagenome benchmark. Compared to htseq-count, NGLess is 20–25× faster at building profiles from mapping results (for the marine and the human gut metagenomes, respectively). Even when limited to a single core (for a more direct comparison, as htseq-count does not support multiple cores), NGLess is still 11–16× faster (for the human gut and marine metagenomes, respectively).
Fig. 2.

Timing comparison of NGLess and other tools. Three replicates are shown for each tool. The bwa/htseq-count pipeline does not include preprocess and filtering steps
The larger ratio in the larger ocean microbial gene catalog (40 million genes) compared to the integrated gene catalog used for profiling the human gut (10 million genes) is evidence that NGLess scales better to very large catalogs.
Note that running the full NG-meta-profiler pipeline takes less time than the sum of the individual steps as the pipeline is optimized as a whole (e.g., it avoids generating unnecessary intermediate files).
The three pipelines can perform the same computation, with some minor differences. MOCAT2 defaults to using SOAPAligner [36], while NGLess defaults to using bwa [34], although both tools support both aligners. To test the quality of the results, metagenomes with known taxonomic composition were simulated using abundance and quality distributions derived from real samples. In particular, species distributions were taken from real samples, and reads were simulated from the corresponding genomes. The eggNOG-mapper [23] annotation of the genomes, weighted by the abundance of the corresponding species, was then considered the ground truth. All the three pipelines considered produce results in high agreement with the ground truth (see Table 3 and Additional file 3: Table S2).
Table 3.
Quality of results based on simulated data
| Environment | Tool | Mean | Std. dev. |
|---|---|---|---|
| Human gut | NGLess | 88.44 | 2.07 |
| MOCAT2 | 87.32 | 2.04 | |
| bwa/htseq-count | 85.96 | 1.84 | |
| Marine | NGLess | 82.26 | 6.29 |
| MOCAT2 | 83.07 | 6.42 | |
| bwa/htseq-count | 82.46 | 6.32 |
Shown are the average and std. dev. (over 8 simulations for each environment) of the Spearman rho between the output of each tool and the ground truth
Pipeline design with NGLess
Using NGLess allows the user to work at a higher level of abstraction than is the case when working directly with traditional command-line tools or generic workflow engines, by using a domain-specific language (DSL). Several tools had already used a DSL to define a computational pipeline. The classical Make tool, which was originally designed for compiling software, but has been used as the basis of a scientific pipeline system [39], uses its own internal language, as do the more modern pipelining tools Snakemake [40] and nextflow [41]. These tools operate by organizing the computation around calls to command-line software. As such, they are fully generic and can be used in a wide range of problems. NGLess trades off this generality to achieve higher usability and performance within its problem domain.
In this section, we illustrate the implementation in NGLess of elements of the human gut profiler (see Figs. 1 and 3) to demonstrate the language and the advantages of using NGLess.
Fig. 3.

Abridged version of the human profiler, written in the NGLess language
The first element in an NGLess script is the version declaration:![]()
This documents which version of the language the user wants to use. This allows the language to be updated while keeping backward compatibility for older scripts. Already, the current version of the interpreter can run more than one version of the language.
The user can then import helper modules. Specifying versions is required for reproducibility. The first module that needs to be imported is the mocat module. This module provides functions to load data that is organized such that each biological sample corresponds to a directory on disk, possibly containing multiple input files. This structure was used in MOCAT/MOCAT2 [8, 9], hence the name of the module. Secondly, we import the igc module to be able to use the integrated gene catalog (IGC) of the human gut [25].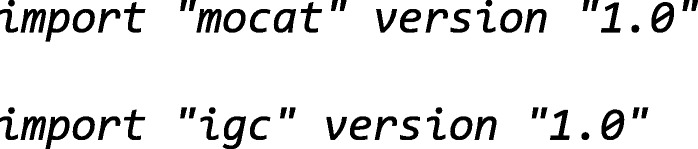
The first step in the pipeline is to load the data. The input directory is given as the first of the command line arguments (which are stored in an array named ARGV, following the convention from other programming languages), using the load_mocat_sample from the mocat module imported above:![]()
This function can read the FastQ data format in different configurations (single-end, paired-end, multi-lane), which allows transparently handling heterogeneity in read depth and different sequencing strategies.
NGLess is a statically typed language, with automatically inferred types. As such, input has type ShortReadSet, representing a variable number of paired-end and/or single-end files on disk. To perform quality based trimming and filtering of short reads, the built-in preprocess function is used to loop over all the input data:
As described above, substrim is a built-in NGLess function to trim short-reads based on quality. Alternatively, the user can opt for two additional trimming algorithms, endtrim and smoothtrim, depending on the sequencing platform and technology used to obtain the raw data (see Additional file 4: Text S1 and the online documentation for details on these functions). While performing the preprocess operation, the interpreter will also estimate qualities and generate quality-control plots for both the input and the preprocessed data. These are computed on the data as reads are loaded from disk for preprocessing and before writing it back to disk, so there is no extra input/output (I/O) generated. This behavior would be hard to achieve in standard pipelines where different tasks are often performed by independent software, requiring the same data to be loaded several times.
After preprocessing the short reads, it is necessary to remove reads that match the human genomes as they are not relevant for microbiome studies. For this operation, we will first align the reads against one of the human built-in references, namely hg19:![]()
As no aligner is specified explicitly, the default, bwa-mem [34], is used. Independently of which aligner is used, NGLess will ensure that the reference is correctly downloaded and indexed the first time it is used. NGLess will also uncompress and merge multiple input read files before passing the reads to the aligner, while streaming the data so that no additional intermediate files are generated. This is the case not just for the map function, but for all tools in NGLess. By using NGLess-based pipelines, the user does not need to be aware of whether these tools support compressed inputs or about subformat issues (e.g., interleaved FastQ files or SAM/BAM distinctions) as these transformations are implicit.
To remove spurious alignments (see Additional file 1: Figure S1), we filter the these aligned reads:
The nhuman object is of type MappedShortReadSet (the NGLess representation of the information in a SAM/BAM file [42]). In order to extract only the sequences (effectively, converting it back to a set of FastQ files), the as_reads function is used:![]()
Now, these sequences are mapped to the igc reference (the integrated gene catalog, imported earlier). To obtain an abundance table, the count function summarizes the mappings:
In this case, we quantify eggNOG orthologs groups [43], relying on gene annotations that are part of the igc reference. As the annotations are bundled together with the sequence databases, the user does not need to be concerned with the possibility of mismatched identifiers, or even specify the exact paths on disk to the corresponding file.
Finally, the igc_counts object can now be saved to disk.![]()
In this case, the file is saved to a subdirectory, namely outputs/. If the directory does not exist or if the user does not have permission to write to it, an error will be produced, as would be the case in any other pipeline design. With NGLess, however, the interpreter checks whether this directory exists and can be written to, as early as possible, namely before starting the execution of the pipeline. Thus, in case of error, the user receives a readable error immediately instead of needing to wait for that step of the process to be reached.
Pipelines developed with NGLess are reproducible
Unlike tools which are based on traditional programming languages, NGLess is designed from the ground up with reproducibility as a goal.
Every pipeline defined with NGLess includes a version declaration and every imported module specifies the particular version which is being imported. Documenting the version of all the tools and dependencies used for a given analysis is considered a best practice [44, 45], but is not always followed. By making it a requirement within the script, NGLess ensures that this best practice is adopted. At the same time, NGLess lowers the necessary effort when compared to having to record the version of all tools and dependencies manually. For example, the versions of samtools [42] and bwa [34] used internally (and shipped with NGLess) are implicitly fixed by specifying the NGLess version. To encourage that credit be given to the original authors, NGLess will print out references for any tools that are used, asking the user to cite them in publications.
Furthermore, although external configuration and command line options may change how results are computed (e.g., how many threads to use, where to store temporary files), the results do not depend on any information outside the script. This separation of implementation details from the data processing specification has the added potential of making the resulting code easier to port between systems [46].
Extensibility and integration into the wider ecosystem of bioinformatics tools
Pipelines defined with NGLess are easily extensible. We encourage users of NG-meta-profiler to customize these pipelines to their specific problems and to extend them as desired. Functions can be added to the language based on external software by specifying the interface in a text format and importing it from the main script.
In May 2018, we opened up to the community a repository of external modules (https://github.com/ngless-toolkit/ngless-contrib) where contributions of new integrations are accepted. At the moment, integration of MetaPhlAn2 (a tool for profiling microbial species based on taxa-specific marker genes [47]), mOTUs2 (an updated metagenomics profiler based on univeral marker genes [48]), and salmon (a tool which generates abundance profiles based on k-mers [49]) are available.
Alternatively, NGLess-based analyses can be integrated into larger pipelines. Most existing bioinformatics pipelines are performed with command-line based software. To facilitate integration with existing tools, we provide several tools based on NGLess with a command line interface. In addition, we provide Common Workflow Language (CWL) descriptions of these tools which enable their use as part of CWL workflows [50].
NGLess scripts can (with some limitations) be automatically exported as CWL tools in order to be embedded in larger projects. When passed the --export-cwl option, NGLess will output a CWL description of a given script which can then be embedded in a larger CWL-based pipeline.
Finally, NGLess can also be used as an embedded language within Python, a programming language that is widely used for scientific computing [15]. With this interface, NGLess-based pipelines can be defined by Python-based scripts.
Future developments
In addition to maintenance releases consisting of updates to the databases and their annotations, we plan to keep developing NG-meta-profiler and the underlying NGLess framework with a focus on readability and reproducibility. For example, one planned feature is to add support for automatic downloading data from public reference repositories such as the European Nucleotide Archive. Users will then be able to share a small readable script that would access publicly deposited data and process it in a perfectly reproducible fashion. Additionally, several improvements to performance are in progress.
Conclusions
NGLess puts forward a different approach for defining data analysis pipelines: the use of a domain-specific language for sequence analysis with reproducibility as a design concern.
Using this framework, we developed NG-meta-profiler which generates taxonomic and functional profiles from metagenomes based on prebuilt gene catalogs that are provided with the tool and downloaded on first use. When compared with other alternatives, NG-meta-profiler performs reference-based functional and taxonomic profiling much faster. Furthermore, this collection of scripts can be easily adapted and extended by the users within the NGLess framework to perform novel functions.
Availability and requirements
Project name: NG-meta-profiler and NGLess
Project home page: https://ngless.embl.de
Operating system(s): Linux, Mac OS X, and Windows
Programming language: Haskell/NGLess/Python/Bash
Other requirements: None
License: MIT License
Any restrictions to use by non-academics: None
Additional files
Figure S1. Fraction of spurious hits to the human genome that is incorrectly kept as a function of the minimum size of the alignment used to consider it a valid alignment. The benchmark simulated dataset was aligned to the human genome. As in this simulated dataset there should be no human reads, any alignment was considered spurious. (PDF 52 kb)
Table S1. Complete timing information for benchmark on real data for NGLess, MOCAT2, and bwa/htseq-count. (XLSX 11 kb)
Table S2. Full results on the synthetic benchmark, shown are the Spearman correlation values between the estimated OG abundances and the distribution being simulated. (XLSX 6 kb)
Text S1. The NGLess Language and Standard Library (PDF 129 kb)
Acknowledgements
The authors thank João Carriço (Instituto de Medicina Molecular, University of Lisbon) as well as the members of the Bork lab for helpful comments. We thank Anna Głazek (EMBL) for several patches to the code. Beta users of NGLess are thanked for their feedback and bug reports.
Funding
Funding was provided by the European Union’s Horizon 2020 Research and Innovation Programme (grant #686070; DD-DeCaF), the European Research Council (ERC) MicrobioS (ERC-AdG-669830), the Fundação para a Ciência e a Tecnologia (grant EXCL/EEI-ESS/0257/2012; DataStorm: Large-Scale Data management in cloud environments), and the European Molecular Biology Laboratory (EMBL).
Availability of data and materials
NGLess is available as open source software at https://github.com/ngless-toolkit/ngless and releases are archived under DOI 10.5281/zenodo.1040241. Additionally, NGLess is available as a bioconda package [51] and in container form (through biocontainers [52]). Documentation and tutorials can be found at https://ngless.embl.de. NG-meta-profiler is available at https://github.com/ngless-toolkit/ng-meta-profiler and released versions are archived under DOI 10.5281/zenodo.1310745. All the databases mentioned can be downloaded automatically with the NGLess tool or accessed using the DOI 10.5281/zenodo.1299267.
Jug [53] based scripts to download the data and run the benchmarks are available at https://github.com/ngless-toolkit/NGLess2018benchmark. This repository also contains the results of running the benchmarks on our servers and all downstream processing. The benchmarks were run on an Intel Xeon (CPU model “E7- 4830 @ 2.13GHz”) with 32 cores (64 virtual cores).
The sequence datasets used for the benchmark are available from the European Nucleotide Archive (ENA) (accession numbers: SAMEA2621229, SAMEA2621155, SAMEA2621033, SAMEA2467039, SAMEA2466896, and SAMEA2466965). The simulated datasets are available on Zenodo (DOI: 10.5281/zenodo.2539431). That resource contains the metagenomes, the underlying ground truth values, and a more complete description of the generation process.
Authors’ contributions
LPC conceived of the project and wrote the first draft of the manuscript. LPC, ATF, and PB coordinated and oversaw the study. LPC, RA, JHC, and PM developed the software and associated databases. LPC, RA, PM, JHC, and PB drafted the manuscript and Additional file 4: Text S1. LPC and RA designed and performed the benchmark studies. All authors contributed to the review of the manuscript before submission for publication. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Luis Pedro Coelho, Email: coelho@embl.de.
Renato Alves, Email: renato.alves@embl.de.
Paulo Monteiro, Email: paulomonteiro7.pm@gmail.com.
Jaime Huerta-Cepas, Email: huerta@embl.de.
Ana Teresa Freitas, Email: ana.freitas@tecnico.ulisboa.pt.
Peer Bork, Email: bork@embl.de.
References
- 1.Grice EA, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt TSB, Raes J, Bork P. The human gut microbiome: from association to modulation. Cell. 2018;172:1198–1215. doi: 10.1016/j.cell.2018.02.044. [DOI] [PubMed] [Google Scholar]
- 3.Gilbert JA, et al. Current understanding of the human microbiome. Nat. Med. 2018;24:392–400. doi: 10.1038/nm.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao L, et al. A catalog of the mouse gut metagenome. Nature Biotechnology. 2015;33:1103–1108. doi: 10.1038/nbt.3353. [DOI] [PubMed] [Google Scholar]
- 5.Xiao L, et al. A reference gene catalogue of the pig gut microbiome. Nature Microbiology. 2016;1:16161. doi: 10.1038/nmicrobiol.2016.161. [DOI] [PubMed] [Google Scholar]
- 6.Coelho LP, et al. Similarity of the dog and human gut microbiomes in gene content and response to diet. Microbiome. 2018;6:72. doi: 10.1186/s40168-018-0450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sunagawa S, et al. Structure and function of the global ocean microbiome. Science. 2015;348:1261359. doi: 10.1126/science.1261359. [DOI] [PubMed] [Google Scholar]
- 8.Kultima JR, et al. MOCAT: A metagenomics assembly and gene prediction toolkit. PLoS ONE. 2012;7:e47656. doi: 10.1371/journal.pone.0047656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kultima JR, et al. MOCAT2: A metagenomic assembly, annotation and profiling framework. Bioinformatics (Oxford, England) 2016;32:2520–2523. doi: 10.1093/bioinformatics/btw183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Treangen TJ, et al. MetAMOS: A modular and open source metagenomic assembly and analysis pipeline. Genome biology. 2013;14:R2. doi: 10.1186/gb-2013-14-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Narayanasamy S, et al. IMP: A pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biol. 2016;17:260. doi: 10.1186/s13059-016-1116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oksanen J, et al. The vegan package. Community ecology package. 2007;10:631–637. [Google Scholar]
- 14.Segata N, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prabhu, P. et al. A survey of the practice of computational science. in SC ’11: Proceedings of 2011 international conference for high performance computing, networking, storage and analysis 1–12 (2011). 10.1145/2063348.2063374
- 16.Johanson AN, Hasselbring W. Effectiveness and efficiency of a domain-specific language for high-performance marine ecosystem simulation: a controlled experiment. Empirical Software Engineering. 2017;22:2206–2236. doi: 10.1007/s10664-016-9483-z. [DOI] [Google Scholar]
- 17.Donoho DL, Maleki A, Rahman IU, Shahram M, Stodden V. Reproducible research in computational harmonic analysis. Computing in Science & Engineering. 2009;11:8–18. doi: 10.1109/MCSE.2009.15. [DOI] [Google Scholar]
- 18.Vandewalle P, Kovacevic J, Vetterli M. Reproducible research in signal processing. Signal Process Mag IEEE. 2009;26:37–47. doi: 10.1109/MSP.2009.932122. [DOI] [Google Scholar]
- 19.Fomel S. Reproducible research as a community effort: Lessons from the madagascar project. Computing in Science & Engineering. 2015;17:20–26. doi: 10.1109/MCSE.2014.94. [DOI] [Google Scholar]
- 20.Carey MA, Papin JA. Ten simple rules for biologists learning to program. PLoS Comput. Biol. 2018;14:e1005871. doi: 10.1371/journal.pcbi.1005871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cox MP, Peterson DA, Biggs PJ, SolexaQA At-a-glance quality assessment of illumina second-generation sequencing data. BMC Bioinformatics. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Del Fabbro C, Scalabrin S, Morgante M, Giorgi FM. An extensive evaluation of read trimming effects on illumina NGS data analysis. PLoS One. 2013;8:e85024. doi: 10.1371/journal.pone.0085024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huerta-Cepas J, et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Molecular Biology and Evolution. 2017;34:2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ugarte A, Vicedomini R, Bernardes J, Carbone A. A multi-source domain annotation pipeline for quantitative metagenomic and metatranscriptomic functional profiling. Microbiome. 2018;6:149. doi: 10.1186/s40168-018-0532-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, et al. An integrated catalog of reference genes in the human gut microbiome. Nature Biotechnology. 2014;32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 26.Huerta-Cepas J, et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanehisa M, et al. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Overbeek R, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King ZA, et al. BiGG models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016;44:D515–D522. doi: 10.1093/nar/gkv1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics. 2015;31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 31.Darling AE, et al. Critical assessment of metagenome interpretation—a benchmark of metagenomics software. Nature Methods. 2017;14:1063. doi: 10.1038/nmeth.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Awad S, Irber L, Titus Brown C. Evaluating metagenome assembly on a simple defined community with many strain variants. bioRxiv. 2017;155358.
- 33.Hyatt D, et al. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:1–11. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv. 2013;1303.3997.
- 35.Li H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li R, et al. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 37.Anders S, Pyl PT, Huber W. HTSeq–a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeller G., Tap J., Voigt A. Y., Sunagawa S., Kultima J. R., Costea P. I., Amiot A., Bohm J., Brunetti F., Habermann N., Hercog R., Koch M., Luciani A., Mende D. R., Schneider M. A., Schrotz-King P., Tournigand C., Tran Van Nhieu J., Yamada T., Zimmermann J., Benes V., Kloor M., Ulrich C. M., von Knebel Doeberitz M., Sobhani I., Bork P. Potential of fecal microbiota for early-stage detection of colorectal cancer. Molecular Systems Biology. 2014;10(11):766–766. doi: 10.15252/msb.20145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwab M, Karrenbach M, Claerbout J. Making scientific computations reproducible. Comput Sci Eng. 2000;2:61–67. doi: 10.1109/5992.881708. [DOI] [Google Scholar]
- 40.Köster J, Rahmann S. Snakemake–a scalable bioinformatics workflow engine. Bioinformatics (Oxford, England) 2012;28:2520–2522. doi: 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 41.Di Tommaso P, et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017;35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 42.Li H, et al. The sequence alignment/map format and samtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huerta-Cepas J, et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson G, et al. Best practices for scientific computing. PLoS Biology. 2014;12:e1001745. doi: 10.1371/journal.pbio.1001745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith AM, Katz DS, Niemeyer KE. Software citation principles. Peer J Comput Sci. 2016;2:e86. doi: 10.7717/peerj-cs.86. [DOI] [Google Scholar]
- 46.Johanson A, Hasselbring W. Software engineering for computational science: Past, present, future. Computing in Science Engineering. 2018:1–1.
- 47.Truong DT, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods. 2015;12:902–903. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- 48.Milanese A, et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 2019;10:1014. doi: 10.1038/s41467-019-08844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amstutz, P. et al. Common workflow language, v1.0. (2016). 10.6084/m9.figshare.3115156.v2
- 51.Grüning Björn, Dale Ryan, Sjödin Andreas, Chapman Brad A., Rowe Jillian, Tomkins-Tinch Christopher H., Valieris Renan, Köster Johannes. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nature Methods. 2018;15(7):475–476. doi: 10.1038/s41592-018-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.da Veiga Leprevost F, et al. BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics. 2017;33:2580–2582. doi: 10.1093/bioinformatics/btx192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coelho LP. Jug: Software for parallel reproducible computation in python. Journal of Open Research Software. 2017;5.
- 54.Mende DR, Sunagawa S, Zeller G, Bork P. Accurate and universal delineation of prokaryotic species. Nature Methods. 2013;10:881–884. doi: 10.1038/nmeth.2575. [DOI] [PubMed] [Google Scholar]
- 55.Sunagawa S, et al. Metagenomic species profiling using universal phylogenetic marker genes. Nature Methods. 2013;10:1196–1199. doi: 10.1038/nmeth.2693. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Fraction of spurious hits to the human genome that is incorrectly kept as a function of the minimum size of the alignment used to consider it a valid alignment. The benchmark simulated dataset was aligned to the human genome. As in this simulated dataset there should be no human reads, any alignment was considered spurious. (PDF 52 kb)
Table S1. Complete timing information for benchmark on real data for NGLess, MOCAT2, and bwa/htseq-count. (XLSX 11 kb)
Table S2. Full results on the synthetic benchmark, shown are the Spearman correlation values between the estimated OG abundances and the distribution being simulated. (XLSX 6 kb)
Text S1. The NGLess Language and Standard Library (PDF 129 kb)
Data Availability Statement
NGLess is available as open source software at https://github.com/ngless-toolkit/ngless and releases are archived under DOI 10.5281/zenodo.1040241. Additionally, NGLess is available as a bioconda package [51] and in container form (through biocontainers [52]). Documentation and tutorials can be found at https://ngless.embl.de. NG-meta-profiler is available at https://github.com/ngless-toolkit/ng-meta-profiler and released versions are archived under DOI 10.5281/zenodo.1310745. All the databases mentioned can be downloaded automatically with the NGLess tool or accessed using the DOI 10.5281/zenodo.1299267.
Jug [53] based scripts to download the data and run the benchmarks are available at https://github.com/ngless-toolkit/NGLess2018benchmark. This repository also contains the results of running the benchmarks on our servers and all downstream processing. The benchmarks were run on an Intel Xeon (CPU model “E7- 4830 @ 2.13GHz”) with 32 cores (64 virtual cores).
The sequence datasets used for the benchmark are available from the European Nucleotide Archive (ENA) (accession numbers: SAMEA2621229, SAMEA2621155, SAMEA2621033, SAMEA2467039, SAMEA2466896, and SAMEA2466965). The simulated datasets are available on Zenodo (DOI: 10.5281/zenodo.2539431). That resource contains the metagenomes, the underlying ground truth values, and a more complete description of the generation process.