

Abstract
Proteins exist as ensembles of interconverting states on a complex energy landscape. A complete, molecular-level understanding of their function requires knowledge of the populated states and thus the experimental tools to characterize them. Infrared (IR) spectroscopy has an inherently fast time scale that can capture all states and their dynamics with, in principle, bond-specific spatial resolution, and 2D IR methods that provide richer information are becoming more routine. Although application of IR spectroscopy for investigation of proteins is challenged by spectral congestion, the issue can be overcome by site-specific introduction of amino acid side chains that have IR probe groups with frequency-resolved absorptions, which furthermore enables selective characterization of different locations in proteins. Here, we briefly introduce the biophysical methods and summarize the current progress toward the study of proteins. We then describe our efforts to apply site-specific 1D and 2D IR spectroscopy toward elucidation of protein conformations and dynamics to investigate their involvement in protein molecular recognition, in particular mediated by dynamic complexes: plastocyanin and its binding partner cytochrome f, cytochrome P450s and substrates or redox partners, and Src homology 3 domains and proline-rich peptide motifs. We highlight the advantages of frequency-resolved probes to characterize specific, local sites in proteins and uncover variation among different locations, as well as the advantage of the fast time scale of IR spectroscopy to detect rapidly interconverting states. In addition, we illustrate the greater insight provided by 2D methods and discuss potential routes for further advancement of the field of biomolecular 2D IR spectroscopy.
Graphical Abstract

INTRODUCTION
A complete understanding of protein function must go beyond the static structure–function paradigm of biochemistry and include the range of structures adopted in solution and how they interconvert. Proteins are described by hierarchical energy landscapes with states that interconvert on a range of length and time scales, including slower, large-scale motions among conformations but also fast, smaller-scale motions among substates within a conformation, and even faster motions among substates within substates (Figure 1).1 Further contributing to complexity, proteins are heterogeneous spatially, such that different parts could show different dynamics that variably contribute to function. Thus, an ideal experimental method to study protein dynamics would provide both high spatial and temporal resolution. A range of spectroscopic techniques have been applied, including NMR visible, and EPR spectroscopy, each with their advantages and limitations. IR spectroscopy is emerging as a powerful alternative approach due to its potential for combined spatial and temporal resolution. IR chromophores can be as small as a bond, and the time scale of IR spectroscopy (ps) is very fast.
Figure 1.

Time scales of protein motion (left) and illustration of example protein energy landscape and relationship to IR spectral features (right). Multiple distinct bands reflect population of multiple conformations, while inhomogeneous broadening of a band reflects the populations of substates at lower tiers in the energy landscape.
Any spectroscopic approach fundamentally relies on the sensitivity of a chromophore to its environment to serve as a local reporter. The frequency of a vibration, for example, can vary due to electrostatic, hydrogen bonding, or other short-range interactions.2,3 The observation of multiple distinct absorption bands for a single vibration provides evidence that it experiences multiple distinct environments or that the protein adopts multiple conformational states (Figure 1). If the relative transition dipole strengths of the bands are known or at least similar, then the relative populations of the states can be determined from the integrated absorbance of the bands. However, a vast number of states make up a protein energy landscape, many of which are associated with subtle structural variations that do not lead to distinct bands but rather a distribution of frequencies, i.e., inhomogeneous broadening. A key advantage of IR spectroscopy is the ability to directly capture the majority of states, even those involving small scale changes that fluctuate on fast time scales. IR spectroscopy also can in principle characterize specific parts of proteins, similarly to NMR spectroscopy, but the difference in time scale makes them complementary techniques. Because NMR dephasing times are slow, NMR resonances have narrow homogeneous line widths, providing high spectral selectivity. IR dephasing times are very fast, leading to broad line widths that impair spectral resolution. However, because the energy scale of NMR spectroscopy is small, the associated time scale is slow, such that the signals from different states that rapidly interconvert coalesce to their average. In comparison, IR spectroscopy can capture states with faster dynamics. For example, NMR resonances separated in energy by 100 Hz would be averaged if the associated states interconverted faster than an ~1 ms time scale, whereas IR bands separated by 5 cm–1 would have to interconvert faster than an ~1 ps time scale. Thus, while the broad line widths of IR bands hinder the resolution of distinct species, the line widths also contain information about inhomogeneity, which enables detection of all populated states, including those that rapidly interconvert.
2D IR SPECTROSCOPY
While linear spectroscopy cannot provide a rigorous measure of the inhomogeneous broadening, due to its convolution with homogeneous line broadening, nor provide information about the underlying dynamics, the inhomogeneity and associated dynamics can be unmasked via multidimensional techniques, in particular 2D IR spectroscopy. Multidimensional spectroscopy at optical frequencies has recently advanced significantly, and a number of excellent texts covering the theory and methods are now available.4–6 In short, the techniques generate 2D spectra, where one axis reports the frequencies of vibrations initially labeled, or pumped, while the other axis probes the frequencies of vibrations that are correlated, because they either belong to the same species or are in some manner coupled. The time between the initial pumping and later probing is typically referred to as the waiting time (Tw). The bands along the diagonal of a 2D spectrum reflect those found in the linear spectrum, while bands off the diagonal associate the vibrational transitions. For example, coupling between two distinct vibrations leads to cross bands, such as those observed between amide backbone vibrations due to their strong transition dipole coupling.7–10 Alternatively, two absorption bands at distinct frequencies may correspond to a single probe in two distinct environments or states, for example, those associated with two protein conformations. In this case, cross bands between the absorptions report that the vibration was pumped at one frequency but later probed at the other, indicating that the interconversion of the states took place in the intervening time period Tw. The increase in cross band amplitude in 2D IR spectra taken as a function of Tw directly follows the exchange dynamics.11 For example, myoglobin conformational states associated with different orientations of a histidine side chain in the binding pocket lead to distinct absorption bands, and the conformational exchange has been directly followed through the appearance of distinct cross bands in Tw-dependent 2D IR spectra.12
In contrast to conformational states that differ by large-scale structural changes, the numerous substates that make up a conformation typically manifest as inhomogeneous broadening that results in the elongation of a band along the diagonal of the 2D spectrum (Figure 2). However, similarly as for distinct bands, the amplitude off the diagonal increases with longer Tw as the substates underlying the inhomogeneous distribution have increasingly more time to interconvert. The interconversion results in 2D spectra that appear less elongated, such that the dynamics among the inhomogeneous distribution (spectral diffusion) can be followed by analysis of the Tw -dependent 2D band shapes. The spectral line broadening and dynamics can be conveniently described by a frequency–frequency correlation function (C(t)), which can be determined from 2D IR data by established methods.13 The time scales of spectral diffusion reflect the barrier heights among the substates that underlie the inhomogeneous broadening, providing insight into the ruggedness of the local energy landscape. This information is useful for assessing the role of substate heterogeneity and dynamics in protein function but also can provide guidance for assigning distinct bands to the same or different conformational states. Such information from 2D experiments, for example, has been applied for our investigations of the conformational dynamics involved in cytochrome P450 activity (vide infra).14,15
Figure 2.

Inhomogeneous broadening results in elongation of 2D bands along the diagonal (right). Interconversion among the underlying states with increasing Tw reduces the elongation. Analysis of the 2D line shapes can yield a frequency–frequency correlation function that describes the spectral diffusion within the inhomogeneous frequency distribution (left).
IR SPECTROSCOPY OF PROTEINS
IR spectroscopy of amide backbone vibrations has been historically utilized as a routine approach to estimate the secondary structural content of proteins.16 However, because of the large number of amide vibrations and their delocalized nature, the information provided is global rather than site-specific. Indeed, the lack of spectral selectivity due to the spectral congestion has been a key hindrance in the widespread application of IR spectroscopy for the study of proteins. The issue of spectral congestion can be avoided by use of ligands with vibrations in a “transparent window” of a protein IR spectrum (1900–2500 cm–1; Figure 3) as probes of the ligands’ binding sites in the proteins.17–19 For example, a carbon monoxide (CO) ligand has long been used to characterize hemeprotein active sites.18,20 The application of such ligand probes, however, is limited in scope to the specific proteins that bind them and, moreover, to the one location in a protein where the ligand binds.
Figure 3.

Representative protein IR spectrum highlighting the transparent frequency window and the structures of frequency-resolved probes used in our studies.
A more generally applicable approach is the introduction of IR probes with frequencies in the transparent window by appending them to amino acid side chains. The side chains of proteins not only commonly mediate critical interactions in biochemical processes, but they are also the part of proteins directly subjected to manipulation by evolution. A wide variety of such transparent window side chain probes have been developed and applied toward the study of protein function, and several excellent reviews are available.19,21,22 When choosing a probe, one must balance the desire for strong signals, small size to minimize perturbation, sensitive solvatochromism, and practical feasibility of site-specific incorporation. The first demonstration of the use of frequency-resolved probes of protein side chains utilized carbon–deuterium (C–D) bonds specifically incorporated into cytochrome c.23 C–D bonds can in principle provide nonperturbative probes of any side chain.19,24–27 Another nonperturbative probe with a vibrational frequency around 2550 cm–1, at the edge of the transparent window, is the thiol group of native cysteine residues.28,29 While ideal as probes of the native protein, the transition dipole strengths of both are relatively small, which makes their absorptions challenging to discern, even in the absence of other allowed absorptions. Unnatural probes can provide more intense absorptions. For this reason, cyano- and azido-functionalized side chains have been utilized widely.30–38 Strong carbonyl probes can be generated by functionalization of side chains with metal complexes, ketones, or esters, although incorporation is more prone to be perturbative due to their relatively large size.39–44 Alkynes are also under development as probes with vibrations in the transparent window.45 Both alkyne and C–D labeling of lipids and proteins also have been utilized for vibrational imaging.46,47 Rather than their use as probes of specific locations of proteins, this application uses the spectroscopic handle for detecting the spatial locations of the biomolecules in a cell.
A consideration in addition to the spectroscopic properties of an IR probe is the practical need to selectively introduce it at a specific location in a protein. Solid phase peptide synthesis (SPPS) can be used to directly incorporate virtually any labeled amino acid anywhere in a sequence of up to ~50 residues, enabling access to specific labeling of peptides and some small folded protein domains.48,49 Larger labeled proteins can be constructed via native chemical ligation or other conformationally assisted ligation reactions from multiple synthesized peptides or protein fragments generated via recombinant expression or expressed protein ligation.19,49–53 Residue-selective labeling with deuterated amino acids or analogues similar to natural amino acids, such as the methionine analogue azidohomoalanine (Aha), can be achieved through expression in defined media in an auxotrophic host or one that cannot efficiently biosynthesize the amino acid.24,27,34,54 This approach is most useful for labeling rare amino acids, as they may be present at only one or a few positions, or in proteins that can tolerate the mutational removal of all but one instance of an amino acid. Unique residues also can be functionalized to generate site-specific IR probes through chemical post-translational modification.31,39,41,42,55 For example, chemical modification of unique cysteine residues to introduce thiocyanate groups is becoming a popular route for introducing IR probes.31,35–37,55 Alternatively, cell-free methods to isotopically label any residue in a protein are now available, but production of quantities needed for biophysical studies remains challenging.56,57 An approach for the facile introduction of different unnatural amino acids at virtually any specific site in any protein is nonsense (amber) suppression, in which an unnatural amino acid is incorporated during ribosomal protein synthesis in response to an amber stop codon introduced into the gene sequence at the desired site of labeling.58,59 The scope of this approach is limited by the availability of tRNA and aminoacyl tRNA synthetases that are laboratory-evolved for incorporation of the desired unnatural amino acid within the expression host. Nonetheless, the approach is established for selective introduction of many IR probes, including C–D bonds, nitriles, and azides.25,33,60
While IR spectroscopy is becoming a popular approach for the study of proteins, 2D IR spectroscopy of proteins remains limited. As with linear IR spectroscopy, 2D IR spectroscopy was first directed at the vibrations of diatomic ligands of hemeproteins and the amide backbone of peptides.7,61 A carbon monoxide ligand of a heme generates what is among the strongest signals possible for characterization of a protein and thus has been leveraged to study the active site of a number of hemeproteins by our group and other groups.14,15,61–63 A variety of ligands with frequency-resolved vibrations, ranging from polyatomic ions to small molecule inhibitors and modified peptides, have been exploited as IR probes for the characterization of their binding sites.64–69 Unlike such ligand probes, amide backbone vibrations can be characterized for all proteins. A particular advantage of 2D compared to linear spectroscopy of amide vibrations is the fourth power rather than square dependence on the transition dipole strength, which accentuates the signals of amide and other vibrations with transition dipole strengths greater than water.6 In addition, the spreading of the spectra along the diagonal in 2D plots facilitates spectral deconvolution. Nonetheless, spectral congestion of the amide frequency region typically prevents site-specific interpretation. Isotopic labeling of specific residues or protein segments shifts the amide frequencies; however, except for some impressive examples, its utility remains limited to peptides and small proteins.10,70–75 In a few cases, native side chains of proteins have been utilized as probes for 2D IR spectroscopy, such as the thiol of cysteine and carboxylate side chains.29,76 As native vibrations, these are attractive probes, although their weak transition dipole strengths or spectral congestion, respectively, could present challenges for their widespread use in larger proteins.
To develop an approach that is generally applicable for characterization of any location in any protein, our and other groups have been pursuing 2D IR spectroscopy of side chains with probe vibrations in the transparent frequency window.40,41,77–82 Hochstrasser and co-workers first demonstrated 2D IR spectroscopy of two cyanophenylalanine (CNF) probes incorporated by SPPS into the 35-residue peptide HP35 that folds into a small domain.77 The HP35 peptide was concurrently investigated by Fayer and co-workers, who used single CNF probes to characterize folding of the wild-type and a mutant peptide,78,83 and later, this system was further studied by Gai and co-workers.84 Fayer and co-workers first extended site-specific 2D IR spectroscopy to full sized proteins through use of azidophenylalanine specifically introduced into myoglobin via amber suppression.79 Thiocyanate groups introduced through cyanylation of native cysteine residues have been employed to characterize the dynamics of dihydrofolate reductase by Cheatum and co-workers and hemoglobin by Brendenbeck and co-workers.80,85 Aha specifically incorporated into both a PDZ domain, and its peptide ligand has been employed in several studies by the Hamm group directed at protein folding and allostery of binding.68,81,82,86 Aha is attractive as a 2D IR probe due to its strong absorbance and convenient incorporation as a methionine analogue via auxotrophic expression. Particularly intense 2D IR probes have been generated by the covalent attachment of metal carbonyl complexes to unique amino acid side chains.40–42 These probes might be too large and perturbative to place in a protein core but are useful for characterization of protein surface properties, such as surface hydration, as reported by Kubarych and co-workers.40
Although 2D IR spectroscopy with transparent window probes is actively under development, it remains in a relatively early stage. Thus far, the approach has almost exclusively been applied to demonstrate 2D IR spectroscopy of a single probe in a protein, and most studies have not yet taken advantage of its intrinsic power to investigate the spatial heterogeneity of proteins by characterization of different sites to elucidate how specific parts contribute to function. Toward expanding the approach in this direction, a recent study by the Hamm group employed multiple Aha probes to investigate allostery of a Pdz domain.81 They acquired difference 2D IR spectra induced by photoisomerization of a covalently attached azobenzene linker, which was used to model the conformational change associated with ligand binding. Unexpectedly, the 2D IR difference spectra could be detected for only one of six labeled sites. Nonetheless, the study demonstrated differential 2D IR spectral responses among sites within a protein domain. Recently, we applied Tw-dependent 2D IR spectroscopy with multiple CNF probes to obtain site-specific information about the heterogeneity and dynamics throughout a Src homology 3 (SH3) domain to investigate their involvement in molecular recognition,87 which is described in more detail below.
OUR CONTRIBUTIONS TO THE FIELD OF SITE-SPECIFIC IR SPECTROSCOPY OF PROTEINS
Our research program has aimed to take advantage of site-specific IR spectroscopy toward advancing our understanding of protein molecular recognition. In particular, we have focused on proteins that form dynamic complexes that are challenging to investigate with the more conventional biophysical techniques: plastocyanin (Pc) and its binding partner cytochrome f (cyt f), cytochrome P450s (P450s) and substrates or redox partners, and SH3 domains and proline-rich (PR) peptide motifs. These proteins play critical roles in diverse cellular processes, including photosynthesis, metabolism, and signaling; however, in each case, their biological function is mediated via the formation of relatively low affinity and dynamic complexes.88–90 The complexes are thought to involve a heterogeneity of states, including those that rapidly interconvert. Through the use of IR spectroscopy with site-specific labeling, we have aimed to characterize the proteins and their complexes with high spatial resolution and capture the heterogeneity of states and fluctuations with high temporal resolution, to gain new information for generating a better molecular-level understanding of dynamic protein recognition.
While investigating protein molecular recognition, we also have aimed to push forward the experimental method of site-specific IR spectroscopy as a general approach to investigate protein biophysics. With this consideration, we have explored probes with frequencies in the transparent frequency window that are anticipated to be minimally perturbative while still providing sufficiently intense absorptions to make feasible the characterization of samples at low mM concentrations (Figure 3). C–D bonds are not only the least perturbative probes, but they also provide intrinsic probes of the protein itself.19,23–27 While in many ways they are our ideal probes, their small transition dipole strengths make their absorptions difficult to discern, and their use as 2D IR probes is not yet feasible. Nonetheless, we believe that their appeal makes their development worthy of further exploration. For 2D IR studies of the hemeprotein P450, we have leveraged the intense signals from a CO ligated to the heme.14,15 For our efforts to develop site-specific 2D IR spectroscopy as a tool to investigate any part of any protein, we have focused on the cyano (CN) group of CNF as a 2D IR probe. The CN group is extrinsic to the protein but relatively small in comparison to other options and, importantly, is a sufficiently strong chromophore to make possible 2D IR spectroscopy of singly labeled proteins.87 Furthermore, the CN probe has other attractive spectroscopic properties. Fermi resonances do not typically complicate spectral interpretation, and the vibrational lifetime, which determines the temporal decay of the 2D bands, is sufficiently long to enable the characterization of spectral diffusion.78 Finally, CNF can easily be site-specifically incorporated via amber suppression.60
In the following, we highlight some of our efforts to use the transparent window probes in combination with linear and 2D IR spectroscopy to gain insight into the molecular recognition underlying the biological function of the three protein systems mentioned above. First, we describe our application of C–D probes with linear IR spectroscopy to detect subtle changes in ligand–metal interactions in the blue copper protein Pc induced by binding to its electron transfer partner cyt f providing new information to suggest a molecular mechanism for the associated decrease in Pc midpoint potential.27 We then describe 2D IR spectroscopy with a CO ligand as a probe of the active site of a P450 to uncover how conformational dynamics contribute to the regioselective activity with different substrates and to the effector role played by redox partner binding.14,15 These studies provide evidence for the importance of fast dynamics in protein function and illustrate the richer information provided by 2D over linear methods. Finally, we describe our studies of SH3-mediated molecular recognition of PR motifs from the perspective of both protein domain and ligand.26,87 We first describe how linear spectroscopy of C–D probes introduced in the PR ligand can capture states that are in rapid exchange on the NMR time scale. We then describe our use of 2D IR spectroscopy with CNF probes to characterize the heterogeneity and dynamics at different sites in the SH3 domain and uncover variation in how the sites are involved in the molecular recognition of the PR ligand.
BINDING-INDUCED CHANGE IN THE COPPER SITE OF Pc INVESTIGATED BY IR SPECTROSCOPY
Unlike electron transfer between small molecules, biological electron transfer occurs between redox centers embedded within environments that are structurally and electrostatically heterogeneous and undergo fluctuations over a wide range of time scales.91 Furthermore, interprotein electron transfer typically involves transient protein–protein interactions in highly dynamic complexes.89,92 An example of central importance in photosynthesis is the complex formed to mediate electron transfer between the blue copper protein Pc and cyt f (Figure 4A).89,93
Figure 4.

(A) Structural model of the Pc–cyt f complex (PDB ID: 1TU2). The inset provides an expanded view of the Cu site of Pc with the introduced C–D probes at Met97 highlighted in purple. (B) FT IR spectra of d3Met97 of Cu(I)Pc (red), Cu(II)Pc (blue), Zn(II)Pc (green), and Co(II)Pc (black). (C) FT IR spectra of d3Met97 of free Cu(I)Pc (red) and Cu(II)Pc (blue) and cyt f-bound Cu(I)Pc (black) and Cu(II)Pc (green). Reproduced with permission from ref 27. Copyright 2016 American Chemical Society.
The reactivity of the metal site in Pc and other proteins is well-known to be modulated by the protein environment.94 In Pc, the metal coordination geometry is thought to arise from a weak and elongated bond between Cu and an axial methionine (Met97) ligand, which is compensated by a strong and shortened bond with a cysteine ligand (Cys89) (Figure 4A).95 Comparison of the metal sites among blue copper proteins suggests that the strength of the bonding with the axial ligand can modulate reactivity, for instance, by affecting the midpoint potentials or reorganization energies.96 While Pc has been extensively studied, how the axial Met determines the geometry and bonding of the Cu site and, moreover, how formation of the complex with cyt f might impact the metal site reactivity is not well understood. For example, complexation with cyt f was shown long ago to decrease the midpoint potential of Pc,97 but the mechanism is unclear. One challenge is that the redox reaction of course involves both the reduced Cu(I) and oxidized Cu(II) states of Pc, but most spectroscopic methods are not able to characterize the reduced state due to its filled d10 orbital configuration. In contrast, IR spectroscopy is applicable to both redox states. To enable the use of IR spectroscopy to selectively investigate the metal site of Pc, we incorporated C–D probes at the methyl group of the axial Met97 ligand (d3Met97).27
The C–D vibrations of d3Met97 were highly sensitive to the interaction of the adjacent sulfur-based orbitals with the positively charged metal center, making them useful reporters of the Cu–Met97 bond (Figure 4B). Specifically, the symmetric stretch of d3Met97 showed a shift to higher frequency upon oxidation of Cu(I) Pc to Cu(II) Pc. Then, a series of Pc variants were compared to assign the source of the frequency change to either the ionic interaction of Met97 with the metal center or covalent mixing of the unoccupied d orbitals of Cu(II) with the protein ligand orbitals. Briefly, comparison of Cu(I) Pc to Zn(II)-substituted Pc and a state of Cu(I) Pc with a shorter Cu–Met bond formed at low pH was used to assess the effect on the C–D frequency of increasing the ionic interaction with a filled d10 metal center, where bonding is exclusively ionic. Then, Zn(II) Pc was compared to the Cu(II) or Co(II) Pc to assess the effect due to the Cys89 or Met97 ligand interaction with the unoccupied dx2-y2 or dz2 metal orbitals, respectively, within the context of a constant ionic interaction with metal centers of the same nominal charge. Altogether, this analysis revealed that the increase in the C–D frequency upon oxidation of Cu(I) to Cu(II) Pc resulted from a stronger ionic interaction with the formally doubled metal charge, but the effect was reduced by mixing of the Cys89 ligand orbitals with the unoccupied dx2-y2 orbital of Cu(II).
After calibrating the C–D probes at d3Met97, IR spectroscopy was applied to characterize the effects of binding to cyt f (Figure 4C). Association led to an increase in the C–D frequency, consistent with a stronger interaction between Met97 and the Cu ion in the complex. A stronger interaction between the Cu ion and the axial ligand is expected to decrease the midpoint potential of Pc,95 as is known to occur upon complexation with cyt f.97 The IR data suggest that the functional change in Pc results from an interaction with cyt f that induces a slight contraction of the Met97–Cu bond. Altogether, the study illustrates how IR spectroscopy of C–D probes can provide molecular-level information about how the metal sites of metalloproteins are controlled by protein–protein interactions and demonstrates the utility of the high structural resolution afforded by IR spectroscopy of C–D probes for the nonperturbative characterization of localized changes in proteins.
FUNCTIONAL DYNAMICS OF CYTOCHROME P450s
P450s are hemeprotein oxidases involved in a wide variety of metabolic and catabolic reactions in organisms throughout all kingdoms of life.98,99 The ability of these enzymes to insert oxygen atoms at inactivated carbon centers with high regio- and enantioselectivity makes them a subject of considerable interest to synthetic chemists and for biotechnology. In addition, P450s play central roles in drug metabolism, and their activities are key determinants of drug pharmacokinetics.100 Most P450s share a common multistep catalytic cycle, which includes substrate binding, two reduction steps, uptake of two protons, and formation of an oxo-ferryl species called compound I, a highly reactive intermediate that initiates substrate oxidation.101 Despite extensive study, many aspects of P450 activity remain poorly understood. A missing factor in our current picture is the role of dynamics. For instance, P450 homologues can vary greatly in their substrate specificity as well as in the regio- and enantioselectivity of their activity on different substrates.98,99 Because P450s have similar structures and catalytic mechanisms, protein flexibility is often evoked to explain the variation in activity;102–104 however, we lack a rigorous molecular-level understanding of how conformational dynamics are involved. In addition, conformational changes are known to be induced upon the binding of substrates,105,106 but whether dynamics participate in other steps in the catalytic cycle is still unknown. Toward addressing these outstanding questions, we have applied 2D IR spectroscopy to measure P450 conformations and dynamics and unravel their contributions to function.
FAST DYNAMICS PLAYS A CRITICAL ROLE IN THE REGIOSELECTIVITY
Our studies have focused on the archetypical P450, P450cam (CYP101A1), from Pseudomonas putida, which hydroxylates camphor in the metabolism of the compound as a carbon source.99 P450cam hydroxylation of camphor shows complete regioselectivity to produce 100% 5-hydroxylcamphor.107 P450cam is also active with the structurally related substrates norcamphor and thiocamphor but with low and intermediate regioselectivity (45 and 63% 5-hydroxy product), respectively.107,108 The product-determining step in the P450cam catalytic cycle is the reaction of the activated oxygen of the compound I intermediate with a particular carbon center of the substrate. Therefore, what determines the regioselectivity of P450cam on the substrates is either (1) the intrinsic reactivity of the substrate or (2) how the enzyme binds the substrate within the binding site to control its mobility during the limited lifetime of the reactive compound I.109 Previous calculations have indicated that the difference in the intrinsic reactivity of the carbon centers of camphor, thiocamphor, and norcamphor does not explain the observed product distributions. To investigate how the active site environment and dynamics might contribute to the regioselectivity, we applied 2D IR spectroscopy to characterize the complexes with each of the three substrates. As P450cam is a hemeprotein, CO could be ligated to the heme center for use as a vibrational probe of the active site.
The linear spectra of the CO probe for the camphor and norcamphor complexes showed symmetric, Gaussian absorption bands, suggestive of environments of a single conformational state (Figure 5A). However, the absorptions were shifted from each other, implying some difference in the states. In contrast, the linear spectra of the CO for the thiocamphor complex were more complex, and adequate fitting required a superposition of three Gaussian bands, indicative of three underlying absorptions due to three distinct states (Figure 5B). Interestingly, the two dominant bands showed the same frequency and line width as the two bands individually measured for the camphor and norcamphor complexes. These results indicate that the camphor and norcamphor complexes adopt distinct conformational states, associated with high and low regioselectivity, respectively, whereas the thiocamphor complex populates both states, with the intermediate regioselectivity likely resulting from a combination of the activity associated with each.
Figure 5.

(A) IR spectra of the P450cam–CO complex with camphor (blue) and norcamphor (red). (B) IR spectrum of the thiocamphor complex (black) and multicomponent fit (blue, red, and gray). (C) Frequency–frequency correlation functions determined for the P450cam–CO complex with camphor (blue), norcamphor (red), and thiocamphor (black). (D) Model for conformational dynamics underlying P450cam regioselectivity. See text for details. Structures of substrates are shown above the mechanism. Reproduced with permission from ref 14. Copyright 2015 American Chemical Society.
To further investigate the origins of the high or low regioselectivity in each conformation, Tw-dependent 2D IR spectra were acquired and analyzed to determine the dynamics within the inhomogeneous distribution of frequencies of each absorption band (Figure 5C), providing insight into the local energy landscape within each associated conformation. Interestingly, the dynamics reported by the CO at the active site of the substrate complexes were correlated with regioselectivity. Specifically, the complexes with camphor, thiocamphor, and norcamphor, which are hydroxylated with high, intermediate, and low regioselectivity, showed slow, intermediate, and fast dynamics, respectively. As found for the linear spectrum, the 2D data for the thiocamphor complex could be well modeled by a superposition of two states with the dynamics of the camphor and norcamphor complexes. Altogether, the spectral data indicate that substrate complexes of P450cam adopt two major conformational states and that a particular substrate can selectively stabilize one or the other. However, what appears to be the key factor that determines the regioselectivity on a substrate is the dynamics among the substates within the populated conformation. A model consistent with the data is shown in Figure 5D. When P450cam adopts the norcamphor-stabilized conformation (ECAMS), dynamics within the active site on time scales faster than the lifetime of compound I allow for multiple carbon centers to approach and react with the activated oxygen, leading to low regioselectivity. In contrast, in the camphor-stabilized conformation (ENORS), the dynamics within the active site are slower, and the substrate is held more rigidly such that only the 5-carbon can approach the reactive compound I species and be hydroxylated. In the thiocamphor complex, both of the conformations are populated but do not interconvert during the lifetime of compound I; thus, the reactions proceed among both populations in parallel, leading to the intermediate distribution of products. 2D IR spectroscopy thus provides a model for the mechanism of regioselectivity in which the fast dynamics within a conformation play a central role.
DETECTION OF A CONFORMATIONAL CHANGE IN P450cam INDUCED BY REDOX PARTNER BINDING
A second aspect of P450cam activity illuminated by 2D IR spectroscopy is the mechanism of the effector role of the redox partner, putidaredoxin (Pdx) (Figure 6A). Pdx provides both electrons in the two reduction steps in the catalytic cycle of P450cam. Whereas the first reduction can be mediated by any reductant with appropriate potential, the second reduction requires Pdx.111 The reason why is a long-standing question. The complex formed by P450cam and Pdx, like most electron transfer partners, is low affinity and highly dynamic,90 which challenges investigation by conventional biophysical methods. Although it is well-known that a conformational change is induced by substrate binding,105,106 whether a similar or different conformational change occurs upon binding Pdx is currently under intense investigation.112–114 We again took advantage of heme-ligated CO as a vibrational probe of the P450cam active site to characterize how binding of Pdx affects the conformations and dynamics.15
Figure 6.

(A) Structural model (PDB ID: 4JWU) of the complex of P450cam (blue) and Pdx (pink). The CO probe is circled in blue. (B) FT IR spectra (black lines) and fits (shaded bands) of heme-ligated CO for the camphor complex of P450cam in the absence of Pdx (top) and in complex with Pdx (bottom). The black dotted line is the diagonal slice of the 2D spectrum obtained for Tw = 0.25 ps. (C) Tw-dependent 2D IR spectra of the CO probe for the camphor complex of P450cam in the absence of Pdx (top) and in the complex with Pdx (bottom). The white dotted lines represent the center frequencies of each spectral component. Adapted with permission from ref 15. Copyright 2018 Ramos, Basom, Thielges.
The CO-ligated ferric state of the camphor complex of P450cam, a model for the ferric, oxygen-ligated state of the enzyme involved in the second reduction step, was characterized in the absence and presence of Pdx (Figure 6B). As observed previously, the absorption for the camphor complex was well fit by a single Gaussian band,14 suggesting that the CO experiences a single environment. In contrast, the absorption in the Pdx complex was shifted to lower frequency and slightly asymmetrical. Whereas this asymmetry hinted at the presence of two underlying bands, they were more obvious from comparison of the linear FT IR spectrum and the diagonal of the 2D spectrum. The band at higher frequency is accentuated in the 2D diagonal, indicating that the two CO vibrations have different transition dipole strengths. Analysis of the component bands from fitting the linear and 2D spectra indicated that the transition dipole strength of the vibration at higher frequency was about 1.3-fold greater, information that enabled accurate quantification of the relative populations of the states from the relative band areas.
Another observation from analysis of the Tw-dependent 2D spectra was that the two CO vibrations have different lifetimes. Because the lifetime of the vibration at lower frequency was shorter, its 2D band decayed more rapidly than the band at higher frequency. The result was that at sufficiently long Tw, when the dominant band at lower frequency was mostly decayed, the presence of the second minor band at higher frequency was unequivocally revealed (Figure 6C). Furthermore, analysis of the Tw-dependent 2D shapes found differences in the spectral diffusion within the bands, indicating distinct local energy landscapes within the associated conformations, providing evidence that the bands reflect two distinct conformational states. The data suggest that the Pdx complex populates two states, which we attribute to a loosely bound encounter state and more tightly associated state, according to the prevalent model for complexes of electron transfer partners.92 The minor band (25%) showed similar average frequency and spectral dynamics as the free enzyme, and thus was interpreted as the encounter state, whereas the major band (75%) found at lower frequency with distinct dynamics was attributed to a more tightly engaged state. In agreement with our interpretation, NMR studies of the P450cam–Pdx complex are consistent with an ensemble of states,90 but their rapid exchange on the NMR time scale complicates analysis of the populations. 2D IR spectroscopy provided clear evidence that Pdx binding induces the partial population of a new conformation of P450cam, which could likely underlie the effector role of Pdx in reduction. Moreover, 2D IR enabled quantification of the distinct populations observed.
Src HOMOLOGY 3 DOMAIN-MEDIATED MOLECULAR RECOGNITION OF COGNATE PROLINE-RICH MOTIFS
To mediate rapid responses via cellular signaling, molecular recognition within protein interaction networks often involves low affinity, highly dynamic protein–protein complexes. Such complexes, for example, are formed by SH3 domains and PR linear recognition motifs. SH3 domains are among the most abundant domains in human and other eukaryotic proteomes, and their recognition of PR motifs underlies a range of diverse signaling cascades.88 Moreover, they have become an archetypical system for investigating biological molecular recognition. SH3 domains recognize a PR motif that contains a core PxxP sequence, where x is typically proline or a hydrophobic amino acid. Crystal structures of the complexes of SH3 domains and peptides consisting of the PR recognition motif show that the PR ligand adopts a polyproline II secondary structure in which the proline residues pack within grooves on the SH3 surface formed by three conserved Tyr residues. As these models indicate that binding is mediated by hydrophobic interactions, the change in entropy is expected to be favorable due to the release of water molecules, yet most studies have found SH3–PR peptide complex formation is entropically unfavorable. The unfavorable entropy changes must arise from restriction in the conformational freedom of the SH3 domain, PR sequence, or water. While the system has been extensively studied via a wide range of experimental techniques, the role of conformational heterogeneity and dynamics in their molecular recognition is still not well understood.
SITE-SPECIFIC IR SPECTROSCOPY CAN RESOLVE RAPIDLY INTERCONVERTING STATES OF THE PR LIGAND
We have used IR spectroscopy with site-selectively introduced transparent window probes to gain molecular insight into how SH3 domains recognize cognate ligands.26 Our initial studies have focused on the yeast Sho1 Src homology 3 (SH3Sho1) domain-mediated recognition of a PR peptide from its physiological target, the protein Pbs2 (pPbs2). Interaction of SH3Sho1 and pPbs2 was shown to be specific within the yeast proteome,115 so the system provides a case in which the binding of the proteins, rather than spatial or temporal regulation of their expression alone, is known to be crucial for their function. We introduced C–D bonds as transparent window probes by incorporation of (Cαd1,Cδd2)proline during synthesis of pPbs2 and then used the probes to site-specifically characterize the changes along the peptide induced by binding to SH3Sho1. The symmetric and asymmetric stretching vibrations of the CδD2 group showed reasonably intense absorptions that facilitated analysis. As anticipated, the spectra of the peptides labeled at the proline residues of the recognition motif were the most sensitive to binding. A less expected observation was that the absorption bands of single vibrational modes showed asymmetry (Figure 7A,B). In addition, second derivative analysis and band fitting indicated that the spectra consisted of multiple overlapping bands. The observation of multiple bands for single vibrational modes provides evidence that the C–D probes experience multiple, distinct environments.
Figure 7.
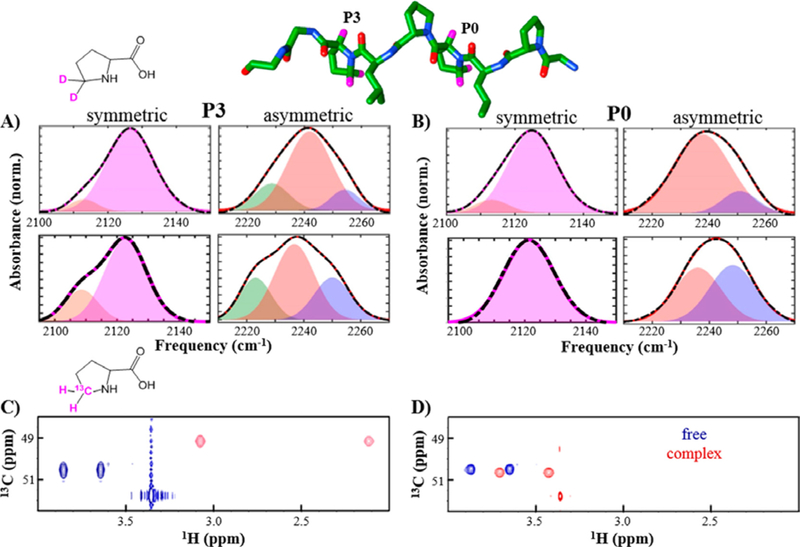
IR and NMR spectra of specifically labeled pPbs2. Shown is a structural model of pPbs2 with locations of introduced C–D probes at motif residues P0 and P3 highlighted (pink, top). The IR absorptions for the symmetric (left plots) and asymmetric stretches (right plots) of the CδD2 probes for the free pPbs2 (top plots) and complex with SH3Sho1 (bottom plots) are shown for (A) P3 and (B) P0. 13C–1H HSQC (800 MHz) spectral region with resonances for the equivalent nuclei are shown for (C) P3 and (D) P0 for free pPbs2 (blue) and SH3Sho1 complex (red). Adapted from ref 26 with permission. Copyright 2016 American Chemical Society.
For comparison, we prepared the pPbs2 peptide labeled at the same residues with the 13C-proline for characterization by NMR spectroscopy (Figure 7C,D). The 1H13C HSQC NMR spectra showed single resonances for the 1H/13C nuclei associated with C–D bonds that showed multiple IR absorptions. Considering that interactions sufficient to alter the IR frequencies of the bonds are likely to also affect the NMR resonances given the relative energy scales of the techniques, the presence of only single NMR resonances indicates that the states uncovered by IR spectroscopy are in rapid exchange on the NMR time scale. These results illustrate the value of the high inherent temporal resolution of IR spectroscopy to ensure the detection of all protein heterogeneity that potentially contributes to function. We have extended these studies by performing a more systematic investigation of a set of PR peptides, combined with support from molecular dynamics and density functional theory calculations, and have assigned the multiple absorptions found for the asymmetric stretch of the CδD2 group of proline to distinct backbone conformations.116 Currently, we are working to elucidate how the binding-induced changes in these backbone conformations along the PR ligand contribute to the unfavorable entropy changes characteristic of SH3-PR motif recognition.
SITE-SPECIFIC 2D IR SPECTROSCOPY UNCOVERS VARYING ENGAGEMENT OF THE SH3 DOMAIN WITH THE PR LIGAND
We have also investigated the molecular recognition mediated by SH3 domains from the perspective of the protein.87 So that we could use 2D IR spectroscopy to site-specifically measure the frequency inhomogeneity and dynamics, we utilized CNF as a probe. CNF was introduced via amber suppression at six different sites in the SH3Sho1 (Figure 8A). CNF substituted each of the three conserved Tyr residues, Tyr8, Tyr10, and Tyr54, that form the binding surface (CNF8, CNF10, and CNF54), Tyr16 within the so-called RT loop (CNF16), Phe20 in a more buried location (CNF20), or Tyr2 that is distant from the ligand binding surface (CNF2). We then applied linear and 2D IR spectroscopy to characterize their microenvironments and how their nature and heterogeneity change upon binding the pPbs2 ligand.
Figure 8.

(A) Structural model of the complex of SH3Sho1 and pPbs2 (PDB ID: 2VKN) showing locations of CNF incorporation. (B) FT IR spectra of the unligated state (colored lines) and the pPbs2 complex (black lines); difference spectra are displayed in black below each set of spectra. (C) Correlation functions (points) and fits (lines) for unligated SH3Sho1 (colored) and the pPbs2 complex (black). Reproduced from ref 87 with permission. Copyright PCCP Owner Societies.
The variation in the spectra of the CNF probes and how they are affected by pPbs2 binding reveal distinct environments throughout the SH3 domain and provide information with which to build a picture of the dynamic complex in solution (Figure 8). The absorptions of residue CNF2, the site distant from the recognition surface, as anticipated, showed no change either in its average frequency or spectral dynamics. Similarly, CNF16 was not highly sensitive to pPbs2 binding. In contrast, association with pPbs2 impacted all of the other probes, indicating that their local environments were changed upon complex formation.
The linear absorptions of the three probes placed along the binding surface, CNF8, CNF10, and CNF54, and the most buried probe, CNF20, shifted in frequency upon binding to pPbs2, indicative of perturbation to the average environment, but the changes were rather small (Figure 8B). In comparison, 2D IR spectroscopy uncovered relatively dramatic effects to the inhomogeneous broadening and the spectral diffusion, as reflected by the C(t) shown in Figure 8C. These results indicate that the heterogeneity of the environments of the probes and the rates of interconversion among the underlying states are impacted by complexation. With assistance from molecular dynamics (MD) simulations, the 2D data support a molecular picture for how the different probes’ environments are affected by complex formation. At CNF20, no change in inhomogeneity occurred upon ligand binding but the dynamics became faster. Correspondingly, MD simulations indicated that CNF20 does not undergo substantial changes in its interactions with its surrounding environment nor does it contact the ligand. Nonetheless, the perturbed dynamics suggest that the binding event is sensed at CNF20, although the residue is not part of the interaction surface. In comparison to CNF20 and all other sites, CNF10 showed the largest increase in inhomogeneity associated with states that slowly interconvert, suggestive of greater exposure to slower protein motions in the complex. In MD simulations, CNF10 experiences a heterogeneous protein environment where it interacts with numerous different residues, many of which are impacted by ligand binding. In contrast, CNF54 showed the opposite behavior, as the association with pPbs2 induced a decrease in inhomogeneity associated with slowly interconverting states, indicative of exposure to a more homogeneous environment with slowly fluctuating interactions. MD simulations suggested that this change results from the packing of CNF54 between the side chains of the two motif prolines of the ligand, where it experiences a relatively homogeneous environment. Surprisingly, CNF8 showed no change in inhomogeneity or dynamics upon ligand binding, despite that it replaced a conserved Tyr residue that packs against the ligand in the crystal structure. The insensitivity suggests that in solution the complex of SH3Sho1 and pPbs2 might not be as tightly engaged near the location of CNF8.
2D IR spectroscopy revealed site-specific heterogeneity and dynamics that depend significantly on ligand binding, including at the probes placed at the adjacent conserved Tyr residues that form the binding surface. As side chain dynamics play a key role in entropy changes of molecular recognition by other proteins,117,118 we speculate that the site-specific changes in the heterogeneity and dynamics uncovered in our study of pPbs2 recognition by SH3Sho1 may make important contributions to binding thermodynamics. For example, the results indicate that part of the recognition surface, specifically the location of CNF54, could contribute more substantially to the unfavorable binding entropy than other locations in the protein, such as those probed by CNF10, and particularly CNF8. Information about binding-induced changes to heterogeneity and dynamics should help address the long-standing question about the molecular origins of the large unfavorable entropy contributions typical of SH3-mediated molecular recognition.
Altogether, the data underscore how generating a complete molecular description of protein recognition and other aspects of function requires the ability to measure conformational heterogeneity and dynamics with residue-specific precision. This study illustrates site-specific 2D IR spectroscopy as an experimental approach with sufficient spatial and temporal resolution to tackle the complexity of proteins. In addition to the SH3 domain, we have applied the approach to uncover site-dependent involvement of residues in the molecular recognition of Pc and cyt f.119 Indeed, the intrinsic power of IR spectroscopy lies in its ability to measure rapidly interconverting states at multiple specific locations in a protein with high spatial detail to test if and how each contributes to function. The elucidation of differences in heterogeneity and dynamics throughout a protein, and how they change in a biologically relevant process, is an essential step toward the longer-term aspiration of using biomolecular 2D IR spectroscopy as a routine approach to study protein biophysics.
FUTURE DIRECTIONS
Site-specific incorporation of frequency-resolved probes has extended the applicability of IR spectroscopy from the characterization of global secondary structure to the detailed characterization of local side chain environments at any specific location. In addition to the desired properties of IR probes already discussed, another characteristic of importance for 2D IR spectroscopy is the probe’s vibrational lifetime. The lifetime determines the rate of decay of 2D bands with Tw, so it ultimately limits the time window over which 2D IR spectroscopy can directly follow vibrational dynamics. As probes with longer lifetimes would enable characterization of dynamics over longer time scales, their development is actively being pursued.120–124 A common strategy is to decouple the probe vibration from others within the molecule to disrupt intramolecular energy relaxation, for instance, by attachment of the probe group to a heavy atom. This rationale has motivated the characterization of a number of thio- and selenocyanate functionalized amino acids.120,121,124 Our group for example has synthesized and evaluated selenocyanophenylalanine (CNSeF) as a potential probe with a significantly extended lifetime.124 However, this and most reported extended lifetime probes have not yet been incorporated into a protein or utilized in practice to study function. For CNSeF, issues with its instability during incorporation still need to be addressed. An accompanying challenge is that probes with longer lifetimes tend to also show weaker absorptions. Thus far, the frequency-resolved IR probe with the longest lifetime (~30 ps) previously utilized for study of a protein is the thiocyanate group generated from derivatization of cysteine,80,85 but its small transition dipole strength challenges application for 2D studies of dynamics at single sites.
While the vibrational lifetime of a probe limits the measurement of equilibrium dynamics directly via analysis of Tw-dependent 2D IR cross bands, interrogation of kinetic processes on longer time scales is possible via nonequilibrium 2D IR spectroscopy.125,126 Nonequilibrium experiments involve triggering a process and the collection of 2D IR difference spectra at successive time intervals to characterize the subsequent dynamics. A key advantage of nonequilibrium experiments is the better detection limits possible via acquisition of difference compared to absolute spectra as a result of the more accurate background subtraction. For example, measurement of absolute 2D IR spectra for transparent window probes typically requires 1 mM sample concentrations,79,80,85,87 whereas a difference 2D IR spectrum of an Aha probe at 10-fold lower concentration of 0.1 mM has been obtained.86 However, nonequilibrium experiments require greater instrument complexity and the ability to repetitively trigger a biologically relevant process with high efficiency. In the first nonequilibrium 2D IR study reported by Hamm and co-workers, a UV pulse was used to trigger a reversible azobenzene photoswitch placed in the backbone of a cyclic octapeptide.127 Similar photoswitches have since been employed by the group to trigger nonequilibrium studies of peptide folding and protein–ligand binding.72,81,82,86,125 Tokmakoff and co-workers have developed a 2D IR spectrometer capable of reaction triggering via temperature jump and have applied the approach to study the kinetics of protein folding and protein–protein binding.128–130 However, not all samples are amenable to repetitive triggering to average the temporal response over many experiments. To overcome this issue for the study of peptide aggregation kinetics, Zanni and co-workers have developed a mid-infrared pulse shaper with “rapid-scan” capability to track peptide aggregation in real time.131,132 Although still relatively underutilized, nonequilibrium 2D IR spectroscopy has potential for investigation of a range of kinetic processes and to further improve sensitivity of 2D IR spectroscopy.
Another exciting avenue for improving sensitivity is surface-enhanced vibrational spectroscopy, an approach based on the large enhancement of signal strengths of molecules near the surface of noble metals due to excitation of local plasmon resonances.133–135 The technique has been extensively developed for linear spectroscopy and could provide even greater benefits for 2D methods due to expected squared greater signal enhancement. Surface enhancement of 2D IR signals was first demonstrated by Hamm and co-workers using capped colloidal gold nanoparticles and later at the surface of metal-coated ATR prisms.136,137 Even stronger signal enhancements can be achieved using optical nanoantennas with dimensions fabricated to tune plasmonic transitions to be resonant with the molecular frequencies.138–141 Signal enhancements as large as 7 orders of magnitude have been reported.141 The strong enhancements occur only for molecules near the metal surface, typically within a few nm, making possible the selective characterization of proteins or other biomolecules confined within a monolayer. We envision the approach will be particularly powerful for the study of monolayers of membrane proteins within lipid bilayers. Linear surface enhanced IR spectroscopy has previously been applied to the study of membrane proteins, such as the electron transfer and protein–protein interactions of cytochrome c oxidase and photosystem I.142,143 Because of the greater signal enhancement possible for nonlinear spectroscopic signals, 2D IR studies of membrane proteins and other biochemical systems that are hindered by low solubility could be achieved by surface enhancement.
Low sensitivity due to the weak signals and limited sample concentrations is a principle challenge of using 2D IR spectroscopy for the characterization of proteins. Spectral selectivity is another major issue but can be alleviated through use of frequency-resolved probes. The field is beginning to tackle these challenges,144 and a number of studies have demonstrated site-specific 2D IR spectroscopy of single side chain probes in proteins.40,41,79,81,82 An obvious extension of the approach is the measurement of the interaction of two probes to generate an IR experiment analogous to the well-established biophysical techniques of FRET, NOE, or DEER spectroscopy. As the strength of the coupling of two IR probes depends on the distance and orientation of the two probes, the amplitude and polarization dependence of cross bands can provide constraints for modeling the structure of proteins. For example, Hochstrasser and co-workers have shown the capability for structure determination of small peptides through measurement of couplings between amide vibrations. 145 Coupling between two side chain probes has been detected via the observation of 2D cross bands for a trimer peptide146 but not yet for larger peptides or proteins. Specific introduction, into a protein, of two probes with distinct frequencies increases the challenges of sample preparation but is nonetheless feasible. Multiple unnatural amino acids have been introduced into proteins via amber suppression, albeit typically with lower efficiency and yield.147 Alternately, two probes could be introduced by SPPS into peptides and larger labeled proteins then generated via native chemical ligation. Measurement of the coupling between two probes in different proteins is another possibility. Along these lines, a recent study showed the possibility to measure vibrational energy transfer between multiple unnatural azido-labeled amino acids selectively introduced in a PDZ domain and peptide ligand, demonstrating a potential method for determination of distances between sites within or between proteins.148
While unnatural amino acids with vibrations that provide strong, frequency-resolved signals are currently the most promising candidates as 2D IR probes of proteins, our long-term aspiration is to utilize C–D bonds introduced at native amino acid residues to directly interrogate the protein and eliminate concerns about perturbation. At this time, application of C–D bonds as probes even via linear spectroscopy is challenging and methodological improvement is needed to facilitate widespread adoption. Endeavors to apply 2D IR spectroscopy with C–D probes will benefit from selection of deuterated amino acids, such as (d10)leucine, that have vibrations with transition dipole strengths larger than water, such that the fourth power enhancement of the 2D will facilitate their detection in aqueous protein samples.149,150 Another possibility for enabling detection of C–D vibrations is to take advantage of coupling to stronger transitions, potentially those of the amide backbone, and detect the C–D vibrations via cross bands.150,151 Because the amplitude of cross bands depends on the transition dipole strengths of both vibrations, the cross bands arising from C–D bonds coupled to strong transitions could be more intense than the fundamental bands on the diagonal. Pursuit of these experiments, and moreover general progress in the development of biomolecular 2D IR spectroscopy, will require the combination of new biochemical methods for labeling proteins alongside recent instrumental developments, including dual frequency or broad band implementations of 2D IR experiments.
SUMMARY AND CONCLUSIONS
IR spectroscopy with frequency-resolved probes is emerging as a powerful approach to characterize specific parts of proteins while ensuring detection of all conformational heterogeneity, including states that undergo rapid interconversion. Notably, we detected multiple IR absorptions but single NMR resonances for the atoms associated with the same bonds in a peptide, thus uncovering multiple populated states by IR that are in fast exchange on the NMR time scale. In addition, we illustrated the potential rich insight provided by 2D IR spectroscopy. For example, 2D IR spectroscopy unequivocally revealed the presence of two underlying bands that provided evidence for a conformational change of P450cam. Comparison of linear and 2D diagonal spectra yielded the relative transition dipole strengths of the probe vibration for each state to enable rigorous quantification of the populations. Our studies also revealed how the measurement of spectral diffusion within different bands is useful for assigning them to the same or distinct conformational states, as well as providing insight into the dynamics among the underlying substates. Notably, the spectral diffusion measured for substrate complexes of P450cam suggested that the dynamics among substates within a populated conformation are central to the regioselectivity of catalysis, supporting the controversial idea that fast dynamics can play an important role in protein function.
The heterogeneity and dynamics at any specific location in a protein can be measured by 2D IR spectroscopy of frequency-resolved side chain probes, motivating our and others’ efforts to develop better 2D IR probes. While 2D IR spectroscopy of unique sites in proteins has been demonstrated for a number of probes, further extension of the approach to multiple, distinct sites is needed to take full advantage of the method’s potential for spatial resolution. Although the field of 2D IR spectroscopy has advanced in the past decades, the applications to proteins and other biomolecules remain relatively undeveloped. Further advances in methodology and application to additional systems and questions should generate a powerful, routine biophysical method that provides site-specific information with high temporal resolution. The new experimental tools of 2D IR spectroscopy open up the investigation of potentially unappreciated contributions to the functional biophysics of proteins.
ACKNOWLEDGMENTS
We thank Indiana University and the Department of Energy (DE-F0A-0000751), National Science Foundation (1552996), and National Institutes of Health (GM114500) for funding. S.R. thanks the Indiana University Chemical and Quantitative Biology Training Grant (T32 GM109825) for partial support.
Biographies
Sashary Ramos completed her B.S. in chemistry at San Diego State University in 2015. She is currently a 4th year analytical chemistry student at Indiana University in Megan Thielges’ group. Her doctoral research has focused on the development and application of multidimensional infrared spectroscopy for characterization of protein conformational dynamics in diverse biological systems.
Megan C. Thielges received her Ph.D. in biophysics at The Scripps Research Institute (La Jolla, CA) in 2009 and completed postdoctoral studies in multidimensional IR spectroscopy at Stanford University. In 2012, she joined the faculty at Indiana University and became an associate professor in 2018. Her primary research interests center around elucidating the conformations and dynamics of proteins and understanding how they are involved in function. In support of these goals, her research program aims to advance experimental methods of site-specific linear and multidimensional infrared spectroscopy for characterizing protein dynamics with high spatial and temporal detail.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Frauenfelder H; Sligar SG; Wolynes PG The Energy Landscapes and Motions of Proteins. Science 1991, 254, 1598–1603. [DOI] [PubMed] [Google Scholar]
- (2).Boxer SG Stark Realities. J. Phys. Chem. B 2009, 113, 2972–2983. [DOI] [PubMed] [Google Scholar]
- (3).Blasiak B; Londergan CH; Webb LJ; Cho M Vibrational Probes: From Small Molecule Solvatochromism Theory and Experiments to Applications in Complex Systems. Acc. Chem. Res. 2017, 50, 968–976. [DOI] [PubMed] [Google Scholar]
- (4).Mukamel S Principles of Nonlinear Optics and Spectroscopy; Oxford University Press: New York, 1995. [Google Scholar]
- (5).Cho M Two-Dimensional Optical Spectroscopy; CRC Press: Boca Raton, FL, 2009. [Google Scholar]
- (6).Hamm P; Zanni M Concepts and Methods of 2D Infrared Spectroscopy; Cambridge University Press: New York, 2011; p 286. [Google Scholar]
- (7).Hamm P; Lim M; Hochstrasser RM Structure of the Amide I Band of Peptides Measured by Femtosecond Nonlinear-Infrared Spectroscopy. J. Phys. Chem. B 1998, 102, 6123–6138. [Google Scholar]
- (8).Ganim Z; Chung HS; Smith AW; DeFlores LP; Jones KC; Tokmakoff A Amide I Two-Dimensional Infrared Spectroscopy of Proteins. Acc. Chem. Res. 2008, 41 , 432–441. [DOI] [PubMed] [Google Scholar]
- (9).Maekawa H; De Poli M; Moretto A; Toniolo C; Ge NH Toward Detecting the Formation of a Single Helical Turn by 2D IR Cross Peaks Between Amide-I and -II Modes. J. Phys. Chem. B 2009, 113, 11775–11786. [DOI] [PubMed] [Google Scholar]
- (10).Woys AM; Almeida AM; Wang L; Chiu CC; McGovern M; de Pablo JJ; Skinner JL; Gellman SH; Zanni MT Parallel β-Sheet Vibrational Couplings Revealed by 2D IR Spectroscopy of an Isotopically Labeled Macrocycle: Quantitative Benchmark for the Interpretation of Amyloid and Protein Infrared Spectra. J. Am. Chem. Soc. 2012, 134, 19118–19128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Fayer MD Dynamics of Liquids, Molecules, and Proteins Measured with Ultrafast 2D IR Vibrational Echo Chemical Exchange Spectroscopy. Annu. Rev. Phys. Chem. 2009, 60, 21–38. [DOI] [PubMed] [Google Scholar]
- (12).Ishikawa H; Kwak K; Chung JK; Kim S; Fayer MD Direct Observation of Fast Protein Conformational Switching. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8619–8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kwak K; Park S; Finkelstein IJ; Fayer MD Frequency-Frequency Correlation Functions and Apodization in Two-Dimensional Infrared Vibrational Echo Spectroscopy: A New Approach. J. Chem. Phys. 2007, 127, 124503. [DOI] [PubMed] [Google Scholar]
- (14).Basom EJ; Spearman JW; Thielges MC Conformational Landscape and the Selectivity of Cytochrome P450cam. J. Phys. Chem. B 2015, 119, 6620–6627. [DOI] [PubMed] [Google Scholar]
- (15).Ramos S; Basom EJ; Thielges MC Conformational Change Induced by Putidaredoxin Binding to Ferrous CO-ligated Cytochrome P450cam Characterized by 2D IR Spectroscopy. Front. Mol. Biosci. 2018, 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Barth A Infrared Spectroscopy of Proteins. Biochim. Biophys. Acta, Bioenerg. 2007, 1767, 1073–1101. [DOI] [PubMed] [Google Scholar]
- (17).Park ES; Boxer SG Origins of the Sensitivity of Molecular Vibrations to Electric Fields: Carbonyl and Nitrosyl Stretches in Model Compounds and Proteins. J. Phys. Chem. B 2002, 106, 5800–5806. [Google Scholar]
- (18).Spiro TG; Wasbotten IH CO as a Vibrational Probe of Heme Protein Active Sites. J. Inorg. Biochem. 2005, 99, 34–44. [DOI] [PubMed] [Google Scholar]
- (19).Adhikary R; Zimmermann J; Romesberg FE Transparent Window Vibrational Probes for the Characterization of Proteins With High Structural and Temporal Resolution. Chem. Rev. 2017, 117, 1927–1969. [DOI] [PubMed] [Google Scholar]
- (20).Anfinrud PA; Han C; Hochstrasser RM Direct Observations of Ligand Dynamics in Hemoglobin by Subpicosecond Infrared Spectroscopy. Proc. Natl. Acad. Sci U. S. A. 1989, 86, 8387–8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kim H; Cho M Infrared Probes for Studying the Structure and Dynamics of Biomolecules. Chem. Rev. 2013, 113, 5817–5847. [DOI] [PubMed] [Google Scholar]
- (22).Ma J; Pazos IM; Zhang W; Culik RM; Gai F Site-Specific Infrared Probes of Proteins. Annu. Rev. Phys. Chem. 2015, 66, 357–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chin JK; Jimenez R; Romesberg FE Direct Observation of Protein Vibrations by Selective Incorporation of Spectroscopically Observable Carbon-Deuterium Bonds in Cytochrome c. J. Am. Chem. Soc. 2001, 123, 2426–2427. [DOI] [PubMed] [Google Scholar]
- (24).Thielges MC; Case DA; Romesberg FE Carbon-Deuterium Bonds as Probes of Dihydrofolate Reductase. J. Am. Chem. Soc. 2008, 130, 6597–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Groff D; Thielges MC; Cellitti S; Schultz PG; Romesberg FE Efforts Toward the Direct Experimental Characterization of Enzyme Microenvironments: Tyrosine100 in Dihydrofolate Reductase. Angew. Chem. Int. Ed. 2009, 48, 3478–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Horness RE; Basom EJ; Mayer JP; Thielges MC Resolution of Site-Specific Conformational Heterogeneity in Proline- Rich Molecular Recognition by Src Homology 3 Domains. J. Am. Chem. Soc. 2016, 138, 1130–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Le Sueur AL; Schaugaard RN; Baik M-H; Thielges MC Methionine Ligand Interaction in a Blue Copper Protein Characterized by Site-Selective Infrared Spectroscopy. J. Am. Chem. Soc. 2016, 138, 7187–7193. [DOI] [PubMed] [Google Scholar]
- (28).Bare GH; Alben JO; Bromberg PA Sulfhydryl Groups in Hemoglobin. A New Molecular Probe at the Interface Studied by Fourier Transform Infrared Spectroscopy. Biochemistry 1975, 14, 1578–1583. [DOI] [PubMed] [Google Scholar]
- (29).Kozinski M; Garrett-Roe S; Hamm P 2D-IR Spectroscopy of the Sulhydryl Band of Cysteines in the Hydrophobic Core of Proteins. J. Phys. Chem. B 2008, 112, 7645–7650. [DOI] [PubMed] [Google Scholar]
- (30).Getahun Z; Huang C-Y; Wang T; De Leon B; DeGrado WF; Gai F Using Nitrile-Derivitized Amino Acids as Infrared Probes of Local Environments. J.Am. Chem. Soc. 2003, 125, 405–411. [DOI] [PubMed] [Google Scholar]
- (31).Fafarman AT; Webb LJ; Chuang JI; Boxer SG Site-Specific Conversion of Cysteine Thiols into Thiocyanate Creates an IR Probe for Electric Fields in Proteins. J. Am. Chem. Soc. 2006, 128, 13356–13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Oh K; Lee JC; Joo C; Han H; Cho M ß-Azidoalanine as an IR Probe: Application to Amyloid Aβ (16–22) Aggregation. J. Phys. Chem. B 2008, 112, 10352–10357. [DOI] [PubMed] [Google Scholar]
- (33).Ye S; Huber T; Vogel R; Sakmar TP FTIR Analysis of GPCR Activation using Azido Probes. Nat. Chem. Biol. 2009, 5, 397–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Taskent-Sezgin H; Chung J; Banerjee PS; Nagarajan S; Dyer RB; Carrico I; Raleigh DP Azidohomoalanine: A Conformationally Sensitive IR Probe of Protein Folding, Protein Structure, and Electrostatics. Angew. Chem. Int. Ed. 2010, 49, 7473–7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Stafford AJ; Ensign DL; Webb LJ Vibrational Stark Effect Spectroscopy at the Interface of Ras and Rap1A Bound to the Ras Binding Domain of Ra1GDS Reveals an Electrostatic Mechanism for Protein-Protein Interaction. J. Phys. Chem. B 2010, 114, 15331–15344. [DOI] [PubMed] [Google Scholar]
- (36).McMahon HA; Alfieri KN; Clark KA; Londergan CH Cyanylated Cysteine: A Covalently Attached Vibrational Probe of Protein-Lipid Contacts. J. Phys. Chem. Lett. 2010, 1, 850–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Liu CT; Layfield JP; Stewart RJ 3rd; French JB; Hanoian P; Asbury JB; Hammes-Schiffer S; Benkovic SJ Probing the Electrostatics of Active Site Microenvironments along the Catalytic Cycle for Escherichia coli Dihydrofolate Reductase. J. Am. Chem. Soc. 2014, 136, 10349–10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Basom EJ; Maj M; Cho M; Thielges MC Site-specific Characterization of Cytochrome P450cam Conformations by Infrared Spectroscopy. Anal. Chem. 2016, 88, 6598–6606. [DOI] [PubMed] [Google Scholar]
- (39).Varenne A; Salmain M; Brisson C; Jaouen G Transition Metal Carbonyl Labeling of Proteins. A Novel Approach to a Solid-Phase Two-Site Immunoassay Using Fourier Transform Infrared Spectroscopy. Bioconjugate Chem. 1992, 3, 471–476. [DOI] [PubMed] [Google Scholar]
- (40).King JT; Kubarych KJ Site-specific Coupling of Hydration Water and Protein Flexibility Studied in Solution with Ultrafast 2D-IR Spectroscopy. J. Am. Chem. Soc. 2012, 134, 18705–18712. [DOI] [PubMed] [Google Scholar]
- (41).Woys AM; Mukherjee SS; Skoff DR; Moran SD; Zanni MT A Strongly Absorbing Class of Non-Natural Labels for Probing Protein Electrostatics and Solvation with FTIR and 2D IR Spectroscopies. J. Phys. Chem. B 2013, 117, 5009–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Peran I; Oudenhoven T; Woys AM; Watson MD; Zhang TO; Carrico I; Zanni MT; Raleigh DP General Strategy for the Bioorthogonal Incorporation of Strongly Absorbing, Solvation-Sensitive Infrared Probes into Proteins. J. Phys. Chem. B 2014, 118, 7946–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Fried SD; Bagchi S; Boxer SG Measuring Electrostatic Fields in Both Hydrogen-Bonding and Non-Hydrogen-Bonding Environments Using Carbonyl Vibrational Probes. J. Am. Chem. Soc. 2013, 135, 11181–11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Pazos IM; Ghosh A; Tucker MJ; Gai F Ester Carbonyl Vibration as a Sensitive Probe of Protein Local Electric Field. Angew. Chem., Int. Ed. 2014, 53, 6080–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zheng ML; Zheng DC; Wang J Non-Native Side Chain IR Probe in Peptides: Ab Initio Computation and 1D and 2D IR Spectral Simulation. J. Phys. Chem. B 2010, 114, 2327–2336. [DOI] [PubMed] [Google Scholar]
- (46).van Manen HJ; Lenferink A; Otto C Noninvasive Imaging of Protein Metabolic Labeling in Single Human Cells Using Stable Isotopes and Raman Microscopy. Anal. Chem. 2008, 80, 9576–9582. [DOI] [PubMed] [Google Scholar]
- (47).Yamakoshi H; Dodo K; Okada M; Ando J; Palonpon A; Fujita K; Kawata S; Sodeoka M Imaging of Edu, an Alkyne-Tagged Cell Proliferation Probe, by Raman Microscopy. J. Am. Chem. Soc. 2011, 133, 6102–6105. [DOI] [PubMed] [Google Scholar]
- (48).Merrifield RB Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar]
- (49).Kent SB Total Chemical Synthesis of Proteins. Chem. Soc. Rev. 2009, 38, 338–351. [DOI] [PubMed] [Google Scholar]
- (50).Wallace CJA Understanding Cytochrome c Function: Engineering Protein Structure by Semisynthesis. FASEB J. 1993, 7, 505–515. [DOI] [PubMed] [Google Scholar]
- (51).Dawson PE; Kent SBH Synthesis of Native Proteins by Chemical Ligation. Annu. Rev. Biochem. 2000, 69, 923–960. [DOI] [PubMed] [Google Scholar]
- (52).Muir TW Semisynthesis of Proteins by Expressed Protein Ligation. Annu. Rev. Biochem. 2003, 72, 249–289. [DOI] [PubMed] [Google Scholar]
- (53).Ottesen JJ; Bar-Dagan M; Giovani B; Muir TW An Amalgamation of Solid Phase Peptide Synthesis and Ribosomal Peptide Synthesis. Biopolymers 2008, 90, 406–414. [DOI] [PubMed] [Google Scholar]
- (54).Kiick KL; Saxon E; Tirrell DA; Bertozzi C R Incorporation of Azides into Recombinant Proteins for Chemo-selective Modification by the Staudinger Ligation. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Degani Y; Neumann H; Patchornik A Selective Cyanylation of Sulfhydryl Groups. J. Am. Chem. Soc. 1970, 92, 6969–6971. [DOI] [PubMed] [Google Scholar]
- (56).Kigawa T; Yabuki T; Yoshida Y; Tsutsui M; Ito Y; Shibata T; Yokoyama S Cell-free Production and Stable-Isotope Labeling of Milligram Quantities of Proteins. FEBS Lett. 1999, 442, 15–19. [DOI] [PubMed] [Google Scholar]
- (57).Zemella A; Thoring L; Hoffmeister C; Kubick S Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems. ChemBio Chem 2015, 16, 2420–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wang L; Xie J; Schultz PG Expanding the Genetic Code. Annu. Rev. Biophys. Biomol Struct. 2006, 35, 225–249. [DOI] [PubMed] [Google Scholar]
- (59).Liu CC; Schultz PG Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem. 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- (60).Schultz KC; Supekova L; Ryu Y; Xie J; Perera R; Schultz PG A Genetically Encoded Infrared Probe. J. Am. Chem. Soc. 2006, 128, 13984–13985. [DOI] [PubMed] [Google Scholar]
- (61).Rella CW; Kwok A; Rector K; Hill JR; Schwettman HA; Dlott DD; Fayer MD Vibrational Echo Studies of Protein Dynamics. Phys. Rev. Lett. 1996, 77, 1648–1651. [DOI] [PubMed] [Google Scholar]
- (62).Merchant KA; Thompson DE; Xu Q-H; Williams RB; Loring RF; Fayer MD Myoglobin-CO Conformational Substate Dynamics: 2D Vibrational Echoes and MD Simulations. Biophys. J. 2002, 82, 3277–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Thielges MC; Chung JK; Fayer MD Protein Dynamics in Cytochrome P450 Molecular Recognition and Substrate Specificity Using 2D IR Vibrational Echo Spectroscopy. J. Am. Chem. Soc. 2011, 133, 3995–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Lim M; Hamm P; Hochstrasser RM Protein Fluctuations are Sensed by Stimulated Infrared Echoes of the Vibrations of Carbon Monoxide and Azide Probes. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 15315–15320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Fang C; Bauman JD; Das K; Remorino A; Arnold E; Hochstrasser RM Two-Dimensional Infrared Spectra Reveal Relaxation of the Nonnucleoside Inhibitor TMC278 Complexed with HIV-1 Reverse Transcriptase. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Hill SE; Bandaria JN; Fox M; Vanderah E; Kohen A; Cheatum CM Exploring the Molecular Origins of Protein Dynamics in the Active Site of Human Carbonic Anhydrase II. J. Phys. Chem. B 2009, 113, 11505–11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Bandaria JN; Dutta S; Nydegger MW; Rock W; Kohen A; Cheatum CM Characterizing the Dynamics of Functionally Relevant Complexes of Formate Dehydrogenase. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 17974–17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Bloem R; Koziol K; Waldauer SA; Buchli B; Walser R; Samatanga B; Jelesarov I; Hamm P Ligand Binding Studied by 2D IR Spectroscopy Using the Azidohomoalanine Label. J. Phys. Chem. B 2012, 116, 13705–13712. [DOI] [PubMed] [Google Scholar]
- (69).Simpson N; Hunt NT Ultrafast 2D-IR Spectroscopy of Haemoproteins. Int. Rev. Phys. Chem. 2015, 34, 361–383. [Google Scholar]
- (70).Torres J; Kukol A; Goodman JM; Arkin IT Site-Specific Examination of Secondary Structure and Orientation Determination in Membrane Proteins: The Peptidic 13C18O Group as a Novel Infrared Probe. Biopolymers 2001, 59, 396–401. [DOI] [PubMed] [Google Scholar]
- (71).Shim SH; Gupta R; Ling YL; Strasfeld DB; Raleigh DP; Zanni MT Two-Dimensional IR Spectroscopy and Isotope Labeling Defines the Pathway of Amyloid Formation with Residue-Specific Resolution. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6614–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Backus EHG; Bloem R; Donaldson PM; Ihalainen JA; Pfister R; Paoli B; Caflisch A; Hamm P 2D-IR Study of a Photoswitchable Isotope-Labeled a-Helix. J. Phys. Chem. B 2010, 114, 3735–3740. [DOI] [PubMed] [Google Scholar]
- (73).Jones KC; Peng CS; Tokmakoff A Folding of a Heterogeneous ß-Hairpin Peptide from Temperature-jump 2D IR Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 2828–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Buchanan LE; Dunkelberger EB; Tran HQ; Cheng PN; Chiu C-C; Cao P; Raleigh DP; de Pablo JJ; Nowick JS; Zanni MT Mechanism of IAPP Amyloid Fibril Formation Involves an Intermediate with a Transient β-sheet. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 19285–19290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Kratochvil HT; Carr JK; Matulef K; Annen AW; Li H; Maj M; Ostmeyer J; Serrano AL; Raghuraman H; Moran SD; et al. Instantaneous Ion Configurations in the K+ Ion Channel Selectivity Filter Revealed by 2D IR Spectroscopy. Science 2016, 353, 1040–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Edington SC; Gonzalez A; Middendorf TR; Halling DB; Aldrich RW; Baiz CR Coordination to Lanthanide Ions Distorts Binding Site Conformation in Calmodulin. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E3126–E3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Urbanek DC; Vorobyev DY; Serrano AL; Gai F; Hochstrasser RM The Two Dimensional Vibrational Echo of a Nitrile Probe of the Villin HP35 Protein. J. Phys. Chem. Lett. 2010, 1, 3311–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Chung JK; Thielges MC; Fayer MD Dynamics of the Folded and Unfolded Villin Headpiece (HP35) Measured with Ultrafast 2D IR Vibrational Echo Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 3578–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Thielges MC; Axup JY; Wong D; Lee HS; Chung JK; Schultz PG; Fayer MD Two-Dimensional IR Spectroscopy of Protein Dynamics Using Two Vibrational Labels: A Site-Specific Genetically Encoded Unnatural Amino Acid and an Active Site ligand. J. Phys. Chem. B 2011, 115, 11294–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Rock W; Li YL; Pagano P; Cheatum CM 2D IR Spectroscopy using Four-Wave Mixing, Pulse Shaping, and IR Upconversion: A Quantitative Comparison. J. Phys. Chem. A 2013, 117, 6073–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Stucki-Buchli B; Johnson PJM; Bozovic O; Zanobini C; Koziol KL; Hamm P; Gulzar A; Wolf S; Buchenberg S; Stock G 2D-IR Spectroscopy of an AHA Labeled Photoswitchable PDZ2 Domain. J. Phys. Chem. A 2017, 121, 9435–9445. [DOI] [PubMed] [Google Scholar]
- (82).Zanobini C; Bozovic O; Jankovic B; Koziol KL; Johnson PJM; Hamm P; Gulzar A; Wolf S; Stock G Azidohomoalanine: A Minimally Invasive, Versatile, and Sensitive Infrared Label in Proteins To Study Ligand Binding. J. Phys. Chem. B 2018, 122, 10118–10125. [DOI] [PubMed] [Google Scholar]
- (83).Chung JK; Thielges MC; Lynch SR; Fayer MD Fast Dynamics of HP35 for Folded and Urea-Unfolded Conditions. J. Phys. Chem. B 2012, 116, 11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Ma J; Pazos IM; Gai F Microscopic Insights into the Protein-Stabilizing Effect of Trimethylamine N-Oxide (TMAO). Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 8476–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).van Wilderen LJGW; Kern-Michler D; Muller-Werkmeister HM; Bredenbeck J Vibrational Dynamics and Solvatochromism of the Label SCN in Various Solvents and Hemoglobin by Time Dependent IR and 2D-IR Spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 19643–19653. [DOI] [PubMed] [Google Scholar]
- (86).Johnson PJM; Koziol KL; Hamm P Quantifying Biomolecular Recognition with Site-Specific 2D Infrared Probes. J. Phys. Chem. Lett. 2017, 8, 2280–2284. [DOI] [PubMed] [Google Scholar]
- (87).Ramos S; Horness RE; Collins JA; Haak D; Thielges MC Site-Specific 2D IR Spectroscopy: A General Approach for the Characteriztion of Protein Dynamics with High Spatial and Temporal Resolution. Phys. Chem. Chem. Phys. 2019, 21, 780–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Zarrinpar A; Bhattacharyya RP; Lim WA The Structure and Function of Proline Recognition Domains. Sci. Signaling 2003, 2003, Re8. [DOI] [PubMed] [Google Scholar]
- (89).Cruz-Gallardo I; Diaz-Moreno I; Diaz-Quintana A; De la Rosa MA The Cytochrome f-plastocyanin Complex as a Model to Study Transient Interactions Between Redox Proteins. FEBS Lett. 2012, 586, 646–652. [DOI] [PubMed] [Google Scholar]
- (90).Hiruma Y; Hass MA; Kikui Y; Liu WM; Olmez B; Skinner SP; Blok A; Kloosterman A; Koteishi H; Lohr F; et al. The Structure of the Cytochrome P450cam-putidaredoxin Complex Determined by Paramagnetic NMR Spectroscopy and Crystallography. J. Mol. Biol. 2013, 425, 4353–4365. [DOI] [PubMed] [Google Scholar]
- (91).Skourtis SS; Waldeck DH; Beratan DN Fluctuations in Biological and Bioinspired Electron-Transfer Reactions. Annu. Rev. Phys. Chem. 2010, 61, 461–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Ubbink M Dynamics in Transient Complexes of Redox Proteins. Biochem. Soc. Trans. 2012, 40, 415–418. [DOI] [PubMed] [Google Scholar]
- (93).Sykes AG Plastocyanin and the Blue Copper Proteins. Struct. Bonding (Berlin, Ger.) 1990, 75, 175–224. [Google Scholar]
- (94).Warren JJ; Lancaster KM; Richards JH; Gray HB Inner- and Outer-Sphere Metal Coordination in Blue Copper Proteins. J. Inorg. Biochem. 2012, 115, 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Solomon EI; Szilagyi RK; DeBeer George S; Basumallick L Electronic Structures of Metal Sites in Proteins and Models Contributions to Function in Blue Copper Proteins. Chem. Rev. 2004, 104, 419–458. [DOI] [PubMed] [Google Scholar]
- (96).Malmstrom BG; Wittung-Stafshede P Effects of Protein Folding on Metalloprotein Redox-Active Sites: Electron-Transfer Properties of Blue and Purple Copper Proteins. Coord. Chem. Rev. 1999, 185–186, 127–140. [Google Scholar]
- (97).Malkin R; Knaff DB; Bearden AJ The Oxidation-Reduction Potential of Membrane-Bound Chloroplast Plastocyanin and Cytochrome f. Biochim. Biophys. Ada, Bioenerg. 1973, 305, 675–678. [DOI] [PubMed] [Google Scholar]
- (98).Sigel A; Sigel H; Sigel RKO The Ubiquitous Roles of Cytochrome P450 Proteins; John Wiley & Sons, Ltd: West Sussex, England, 2007. [Google Scholar]
- (99).de Montellano Ortiz P. R Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th ed; Springer International Publishing: New York, 2015; p 912. [Google Scholar]
- (100).Guengerich FP Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [DOI] [PubMed] [Google Scholar]
- (101).Denisov IG; Makris TM; Sligar SG; Schlichting I Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]
- (102).Scott EE; He YA; Wester MR; White MA; Chin CC; Halpert JR; Jonhnson EF; Stout CD An Open Conformation of Mammalian Cytochrome P450 2B4 at 1.6-A Resolution. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 13196–13201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Ekroos M; Sjogren T Structural Basis for Ligand Promiscuity in Cytochrome P450 3A4. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13682–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Hendrychova T; Anzenbacherova E; Hudecek J; Skopalik J; Lange R; Hildebrandt P; Otyepka M; Anzenbacher P Flexibility of Human Cytochrome P450 Enzymes: Molecular Dynamics and Spectroscopy Reveal Important Function-Related Variations. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 58–68. [DOI] [PubMed] [Google Scholar]
- (105).Lee YT; Wilson RF. Rupniewski I.; Goodin DB. P450cam Visits an Open Conformation in the Absence of Substrate. Biochemistry 2010, 49, 3412–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Asciutto EK; Dang M; Pochapsky SS; Madura JD; Pochapsky TC Experimentally Restrained Molecular Dynamics Simulations for Characterizing the Open States of Cytochrome P450cam. Biochemistry 2011, 50, 1664–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Atkins WM; Sligar SG The Roles of Active Site Hydrogen Bonding in Cytochrome P-450cam as Revealed by Site-Directed Mutagenesis. J. Biol. Chem. 1988, 263, 18842–18849. [PubMed] [Google Scholar]
- (108).Loida P; Sligar SG; Paulsen MD; Arnold GE; Ornstein RL Stereoselective Hydroxylation of Norcamphor by Cytochrome P450cam. J. Biol. Chem. 1995, 270, 5326–5330. [DOI] [PubMed] [Google Scholar]
- (109).Ortiz de Montellano PR; Stearns R A. Timing of the Radical Recombination Step in Cytochrome P-450 Catalysis with Ring-Strained Probes. J. Am. Chem. Soc. 1987, 109, 3415–3420. [Google Scholar]
- (110).Collins JR; Loe GH Theoretical Study of the Product Specificity in the Hydroxylation of Camphor, Norcamphor, 5,5-Difluorocamphor, and Pericyclocamphanone by Cytochrome P-45Ocam. J. Biol. Chem. 1988, 263, 3164–3170. [PubMed] [Google Scholar]
- (111).Tyson CA; Lipscomb JD; Gunsalus IC The roles of putidaredoxin and P450cam in methylene hydroxylation. J. Biol. Chem. 1972, 247, 5777–5784. [PubMed] [Google Scholar]
- (112).Hollingsworth SA; Poulos TL Molecular Dynamics of the P450cam-Pdx Complex Reveals Complex Stability and Novel Interface Contacts. Protein Sci. 2015, 24, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Liou SH; Mahomed M; Lee YT; Goodin DB Effector Roles of Putidaredoxin on Cytochrome P450cam Conformational States. J. Am. Chem. Soc. 2016, 138, 10163–10172. [DOI] [PubMed] [Google Scholar]
- (114).Batabyal D; Richards LS; Poulos TL Effect of Redox Partner Binding on Cytochrome P450 Conformational Dynamics. J. Am. Chem. Soc. 2017, 139, 13193–13199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Zarrinpar A; Park S-H; Lim WA Optimization of Specificity in a Cellular Protein Interaction Network by Negative Selection. Nature 2003, 426, 676–680. [DOI] [PubMed] [Google Scholar]
- (116).Bukowski GS; Thielges MC Residue-Specific Conformational Heterogeneity of Proline-Rich Sequences Uncovered via Infrared Spectroscopy. Anal. Chem. 2018, 90, 14355–14362. [DOI] [PubMed] [Google Scholar]
- (117).Palmer AG III NMR Probes of Molecular Dynamics: Overview and Comparison with Other Techniques. Annu. Rev. Biophys. Biomol Struct. 2001, 30, 129–155. [DOI] [PubMed] [Google Scholar]
- (118).Wand AJ; Sharp KA Measuring Entropy in Molecular Recognition by Proteins. Annu. Rev. Biophys. 2018, 47, 41–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Ramos S; Le Sueur AL; Horness RE; Specker JT; Collins JA; Thibodeau KE; Thielges MC Heterogeneous and Highly Dynamic Interface in Plastocyanin–Cytochrome f Complex Revealed by Site-Specific 2D-IR Spectroscopy. J. Phys. Chem. B 2019, 123, 2114–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Park K-H; Jeon J; Park Y; Lee S; Kwon H-J; Joo C; Park S; Han H; Cho M Infrared Probes Based on Nitrile- Derivatized Prolines: Thermal Insulation Effect and Enhanced Dynamic Range. J. Phys. Chem. Lett. 2013, 4, 2105–2110. [Google Scholar]
- (121).Levin DE; Schmitz AJ; Hines SM; Hines KJ; Tucker MJ; Brewer SH; Fenlon EE Synthesis and Evaluation of the Sensitivity and Vibrational Lifetimes of Thiocyanate and Selenocyanate Infrared Reporters. RSC Adv. 2016, 6, 36231–36237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Rodgers JM; Zhang W; Bazewicz CG; Chen J; Brewer SH; Gai F Kinetic Isotope Effect Provides Insight into the Vibrational Relaxation Mechanism of Aromatic Molecules: Application to Cyano-phenylalanine. J. Phys. Chem. Lett. 2016, 7, 1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Le Sueur AL; Ramos S; Ellefsen JD; Cook SP; Thielges MC Evaluation of p-(13C,15N-cyano)phenylalanine as an Extended Timescale 2D IR Probe of Proteins. Anal. Chem. 2017, 89, 5254–5260. [DOI] [PubMed] [Google Scholar]
- (124).Ramos S; Scott KJ; Horness RE; Le Sueur AL.; Thielges MC. Extended timescale 2D IR Probes of Proteins: p-cyanoselenophenylalanine. Phys. Chem. Chem. Phys. 2017, 19, 10081–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Cervetto V; Hamm P; Helbing J Transient 2D-IR Spectroscopy of Thiopeptide Isomerization. J. Phys. Chem. B 2008, 112, 8398–8405. [DOI] [PubMed] [Google Scholar]
- (126).Ghosh A; Ostrander JS; Zanni MT Watching Proteins Wiggle: Mapping Structures with Two-Dimensional Infrared Spectroscopy. Chem. Rev. 2017, 117, 10726–10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Bredenbeck J; Helbing J; Behrendt R; Renner C; Moroder L; Wachtveitl J; Hamm P Transient 2D-IR Spectroscopy: Snapshots of the Nonequilibrium Ensemble during the Picosecond Conformational Transition of a Small Peptide. J. Phys. Chem. B 2003, 107, 8654–8660. [Google Scholar]
- (128).Chung HS; Ganim Z; Jones KC; Tokmakoff A Transient 2D IR Spectroscopy of Ubiquitin Unfolding Dynamics. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14237–14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Jones KC; Ganim Z; Peng CS; Tokmakoff A Transient Two-dimensional Spectroscopy with Linear Absorption Corrections Applied to Temperature-jump Two-Dimensional Infrared. J. Opt. Soc. Am. B 2012, 29, 118–129. [Google Scholar]
- (130).Zhang XX; Jones KC; Fitzpatrick A; Peng CS; Feng CJ; Baiz CR; Tokmakoff A Studying Protein-Protein Binding through T-Jump Induced Dissociation: Transient 2D IR Spectroscopy of Insulin Dimer. J. Phys. Chem. B 2016, 120, 5134–5145. [DOI] [PubMed] [Google Scholar]
- (131).Strasfeld DB; Ling YL; Shim SH; Zanni MT Tracking Fiber Formation in Human Islet Amyloid Polypeptide with Automated 2D-IR spectroscopy. J. Am. Chem. Soc. 2008, 130, 6698–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Moran SD; Woys AM; Buchanan LE; Bixby E; Decatur SM; Zanni MT Two-Dimensional IR Spectroscopy and Segmental 13C Labeling Reveals the Domain Structure of Human γD-Crystallin Amyloid Fibrils. Proc. Natl. Acad. Sci U. S. A. 2012, 109, 3329–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Hartstein A; Kirtley JR; Tsang JC Enhancement of the Infrared Absorption from Molecular Monolayers with Thin Metal Overlayers. Phys. Rev. Lett. 1980, 45, 201–204. [Google Scholar]
- (134).Osawa M Surface-Enhanced Infrared Absorption In Optics Near-Field and Surface Plasmon Polaritons; Kawata S, Ed.; Springer: Berlin, Heidelberg, 2001; pp 163–187. [Google Scholar]
- (135).Aroca R Surface-enhanced Vibrational Spectroscopy; John Wiley & Sons: West Sussex, England, 2006. [Google Scholar]
- (136).Donaldson PM; Hamm P Gold Nanoparticle Capping Layers: Structure, Dynamics, and Surface Enhancement Measured Using 2D-IR Spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 634–638. [DOI] [PubMed] [Google Scholar]
- (137).Kraack JP; Lotti D; Hamm P Ultrafast, Multidimensional Attenuated Total Reflectance Spectroscopy of Adsorbates at Metal Surfaces. J. Phys. Chem. Lett. 2014, 5, 2325–2329. [DOI] [PubMed] [Google Scholar]
- (138).Selig O; Siffels R; Rezus YL Ultrasensitive Ultrafast Vibrational Spectroscopy Employing the Near Field of Gold Nanoantennas. Phys. Rev. Lett. 2015, 114, 233004. [DOI] [PubMed] [Google Scholar]
- (139).Gandman A; Mackin RT; Cohn B; Rubtsov IV; Chuntonov L Radiative Enhancement of Linear and Third-Order Vibrational Excitations by an Array of Infrared Plasmonic Antennas. ACS Nano 2018, 12, 4521–4528. [DOI] [PubMed] [Google Scholar]
- (140).Mackin RT; Cohn B; Gandman A; Leger JD; Chuntonov L; Rubtsov IV Surface-Enhanced Dual-Frequency Two-Dimensional Vibrational Spectroscopy of Thin Layers at an Interface. J. Phys. Chem. C 2018, 122, 11015–11023. [Google Scholar]
- (141).Morichika I; Kusa F; Takegami A; Sakurai A; Ashihara S Antenna-Enhanced Nonlinear Infrared Spectroscopy in Reflection Geometry. J. Phys. Chem. C 2017, 121, 11643–11649. [Google Scholar]
- (142).Ataka K; Heberle J Biochemical Applications of Surface-Enhanced Infrared Absorption Spectroscopy. Anal. Bioanal. Chem. 2007, 388, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Ataka K; Stripp ST; Heberle J Surface-enhanced Infrared Absorption Spectroscopy (SEIRAS) to Probe Monolayers of Membrane Proteins. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 2283–2293. [DOI] [PubMed] [Google Scholar]
- (144).Koziol KL; Johnson PJ; Stucki-Buchli B; Waldauer SA; Hamm P Fast Infrared Spectroscopy of Protein Dynamics: Advancing Sensitivity and Selectivity. Curr. Opin. Struct. Biol. 2015, 34, 1–6. [DOI] [PubMed] [Google Scholar]
- (145).Remorino A; Hochstrasser RM Three-Dimensional Structures by Two-Dimensional Vibrational Spectroscopy. Acc. Chem. Res. 2012, 45, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Chalyavi F; Gilmartin PH; Schmitz AJ; Fennie MW; Tucker MJ Synthesis of 5-Cyano-Tryptophan as a Two-Dimensional Infrared Spectroscopic Reporter of Structure. Angew. Chem. Int. Ed. 2018, 57, 7528–7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Chatterjee A; Sun SB; Furman JL; Xiao H; Schultz PG A Versatile Platform for Single- and Multiple-Unnatural Amino Acid Mutagenesis in Escherichia coli. Biochemistry 2013, 52, 1828–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Baumann T; Hauf M; Schildhauer F; Eberl KB; Durkin PM; Deniz E; Löffler JG; Acevedo-Rocha CG; Jaric J; Martins BM; et al. Ortsaufgeloste Beobachtung von Schwingungse-nergietransfer durch ein genetisch codiertes ultraschnelles Heizelement. Angew. Chem. 2019, 131, 2925–2930. [Google Scholar]
- (149).Schade M; Moretto A; Crisma M; Toniolo C; Hamm P Vibrational Energy Transport in Peptide Helices after Excitation of C-D Modes in Leu-d^. J. Phys. Chem. B 2009, 113, 13393–13397. [DOI] [PubMed] [Google Scholar]
- (150).Naraharisetty SRG; Kasyanenko VM; Zimmermann J; Thielges MC; Romesberg FE; Rubtsov IV C-D Modes of Deuterated Side Chain of Leucine as Structural Reporters via Dual-frequency Two-dimensional Infrared Spectroscopy. J. Phys. Chem. B 2009, 113, 4940–4946. [DOI] [PubMed] [Google Scholar]
- (151).Naraharisetty SRG; Kurochkin DV; Rubtsov IV C-D Modes as structural reporters via dual-frequency 2DIR spectroscopy. Chem. Phys. Leit. 2007, 437, 262–266. [Google Scholar]