

Abstract
Histone deacetylase (HDAC) activity is modulated in vivo by post-translational modifications and formation of multiprotein complexes. Novel chemical tools to study how these factors affect engagement of HDAC isoforms by HDAC inhibitors (HDACi) in cells and tissues are needed. In this study, a synthetic strategy to access chemically diverse photoreactive probes (PRPs) was developed and used to prepare seven novel HDAC PRPs 9–15. The class I HDAC isoform engagement by PRPs was determined in the biochemical assays and photolabeling experiments in live SET-2, HepG2, HuH7, and HEK293T cell lines and in mouse liver tissue. Unlike the HDAC protein abundance and biochemical activity against recombinant HDACs, the chemotype of the PRPs and the type of cells were key in defining engagement of HDAC isoforms in live cells. Our findings suggest that engagement of HDAC isoforms by HDACi in vivo may be substantially modulated in a cell and tissue type dependent manner.
Keywords: Histone deacetylase, Inhibitors, Photoaffinity labelling, Target engagement, Tissue labeling
Graphical Abstract
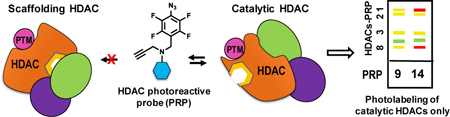
Engaged by inhibitors? The identity of the histone deacetylase isoforms engaged by inhibitors in vivo is one of the unresolved mysteries in epigenetic drug discovery. Using newly designed photoreactive probes, we show that their relative histone deacetylase isoform engagement in live cells in culture and mouse liver tissue is chemotype- and cell-type dependent and is substantially different from the isoform selectivity determined in the biochemical assays.
Introduction
Histone deacetylases (HDACs) are promising therapeutic targets.[1] Four HDAC inhibitors (HDACi) of multiple class I and II HDACs are FDA approved for the treatment of T-cell lymphoma and multiple myeloma. Since HDAC isoforms deacetylate both histone and non-histone proteins and generally play unique non-redundant functions,[2] it is hypothesized that selective HDACi will offer a desired therapeutic effect while minimizing toxicity. To investigate this hypothesis, many isoform-selective HDACi have been synthesized and tested (Figure 1).[3]
Figure 1.
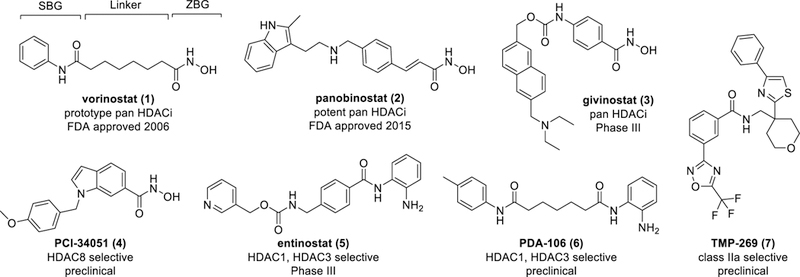
Representative HDACi 1–7 of diverse chemotypes in different stages of clinical development. The pharmacophore of HDACi is annotated on the structure of vorinostat (1). SBG: Surface Binding Group, ZBG: Zinc Binding Group.
HDACi potency and isoform selectivity are typically evaluated in biochemical assays under pseudo-equilibrium utilizing individual purified recombinant HDAC isoforms and a synthetic substrate. However, substrate specificity and HDAC catalytic activity, and hence biological function, of HDACs are often modulated in vivo via formation of protein-protein complexes and post-translation modifications,[4] which these assays do not recapitulate. It was also demonstrated that the catalytic activity of at least some of the HDACs is dispensable and HDACs can play a non-enzymatic structural (scaffolding) role.[2b] These modulatory mechanisms have been shown to be cell-state dependent and can be dysregulated in diseases and conditions.[5] A typical alternative approach in this case would be to use cellular phenotypic readouts. With few exceptions, HDAC isoform substrate specificity in live cells and in vivo is poorly understood, and these biochemical assays almost always remain the only avenue for measuring potency and selectivity of HDACi. HDACi are non-hydrolysable HDAC substrate analogues, and, therefore, the same mechanisms that control substrate specificity of HDACs should also affect binding of HDACi. Since cellular context is lost in the biochemical inhibitory assays, HDACi potency and selectivity may not accurately reflect the in situ target engagement of HDACi. Several experimental approaches, including HDAC photoreactive probes (PRPs), have emerged to assess HDACi target engagement within complex systems.[6] The importance of cellular context to study engagement of HDACs was further highlighted by a recent study by Bantscheff et al [6a] who showed that HDACi tethered to polymer beads exhibited chemotype- and deacetylase complex-dependent binding to HDAC isoforms. Cell permeable HDAC PRPs were reported by Cravatt et al,[6c, 7] Storer et al,[8] and our laboratory.[6f, 9] Despite the overall progress in applying PRPs to study engagement of HDACs in situ, synthetic accessibility to drug-like cell permeable HDAC PRPs with photoreactive groups able to generate short lived reactive intermediates to accurately report on target engagement is limited.
We have recently demonstrated that incorporation of a photoreactive tetrafluorophenyl azide (TFPA) group and an alkyne reporter into the surface binding group (SBG) resulted in a potent, relatively non-selective, and cell permeable photoreactive probe (8, Figure 2) that structurally resembles an FDA approved HDACi (1, Figure 1).[6f] Photolabeling with this probe showed that engagement of HDAC isoforms is dependent on the posttranslational modification state of HDACs, which is cell-type dependent and generally does not correlate with the selectivity profile measured in biochemical assays. The goal of the current study is to design and synthesize PRPs based on 1 and other common HDACi 2–7 (Figure 1) and validate them in target engagement studies in live cells in culture and in tissues in situ. To accomplish this goal, we developed a general synthetic strategy that incorporates a TFPA and an alkyne reporter groups as an integral part of the SBGs of HDACi, designed and synthesized seven novel PRPs 9–15 (Figure 2), evaluated their biochemical potency and selectivity for individual recombinant HDAC isoforms, and conducted photolabeling studies in four cell lines in culture and in mouse liver tissue.
Figure 2.
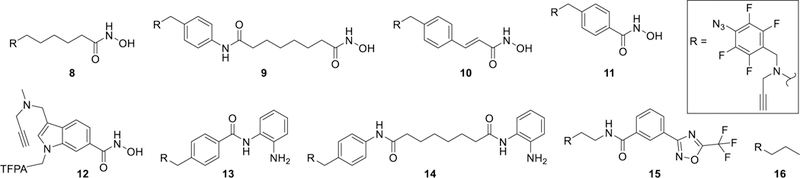
Tetrafluorophenylazide (TFPA) photoreactive probes 8–15 designed based on diverse chemotype HDACi and TFPA control compound 16.
Results
PRP Design and Chemical Synthesis
The PRPs synthesized in this work are based on diverse parent HDACi with varying ZBG, linker and SBG. Parent HDACi containing three ZBGs were chosen to synthesize PRPs 8–15. These include either hydroxamic acid (8–12), o-aminoanilide (13 and 14), or trifluoromethyloxadiazole (15) (Figure 2). Linker regions of the PRPs contained both saturated and unsaturated aliphatic and aromatic moieties similar to those in the reported HDACi (Figure 1). Although several photoreactive groups are accessible synthetically, the TFPA was chosen based on its similarity to aryl-based moieties present in the SBG of most HDACi, short half-life of the corresponding nitrene to capture interactions with HDACs (and possibly other proteins) while PRPs are still in the binding site of HDACs,[10] and fewer side-reactions that could decrease the yield of the PRP-protein adducts.[11] The alkyne handle was chosen because it is small, electronically inert, and stable in biological systems, yet reactive under copper(I)-catalyzed “click reaction” conditions. Given the extensive structure-activity relationships (SAR) available for most of the parent compounds, the TFPA and the alkyne moieties were placed at positions deemed to tolerate the same size substituents when possible. The ZBG and the linker regions were unaltered to maintain similarity to the parent HDACi.[12] All the parent HDACi and the corresponding PRPs were docked to the binding site of HDAC isoforms as described previously[13] to ensure that the TFPA and the reporter alkyne moieties do not interfere with the binding of the PRPs. The in silico physicochemical properties; octanol/water partition coefficient (SlogP), solubility (logS), and topological polar surface area (TPSA) of PRPs and the parent HDACi were calculated and compared to those of the parent HDACi (Figure S1 and Table S1, Supporting information). Generally, the differences in SlogP, logS, and TPSA between the parent HDACi and PRPs were found to be comparable or smaller than those for the previously published PRPs.
PRPs 8 and 9 were designed based on the chemotype of 1 (Figure 1). PRP 8 isosterically replaces the phenyl ring of 1 with the TFPA and alkyne moieties so that only a single phenyl ring is present in the SBG. PRP 9 includes the TFPA and alkyne handle extended off the phenyl ring of 1. PRP 10 was designed based on the chemotype of the potent pan HDACi panobinostat, 2 (Figure 1) and its published SAR.[14] PRP 11 represents aromatic hydroxamic acid class of HDACi similar to givinostat (3). The chemotype of HDAC8 selective compound PCI-34051 (4) was used to design PRP 12. Replacement of the p-methoxy phenyl substituent at the indole nitrogen with the TFPA group and attachment of the alkyne handle at the indole C-3 were chosen as both positions could tolerate substituents of similar electronic and steric properties while retaining HDAC8 selectivity.[15] PRPs 13 and 14 were based on the o-aminoanilide-based inhibitors selective for HDAC1 and HDAC3,[16] entinostat (5) and PDA-106 (6), respectively. PRP 15 was designed by replacing the SBG of TMP-269 (7), a class IIa selective inhibitor,[17] with TFPA and alkyne moieties. Compounds 16 (Figure 2 and Scheme S1, Supporting information) and S20 (Scheme S2, Supporting information) were designed to serve as a control for non-specific photolabeling and as a competitor lacking the alkyne tag, respectively.
To incorporate the TFPA and alkyne moieties into a structurally diverse set of HDACi chemotypes, the synthetic strategy needed to be amenable to the reaction conditions used in the synthesis of the parent compounds. For example, most parent compounds utilize reductive amination for amine synthesis, hydroxylamine hydrochloride reaction with esters to form hydroxamic acid ZBG, or acid catalyzed Boc-deprotection to form benzamide ZBG. We found that the TFPA was not stable in most of the standard reaction conditions used for the synthesis of the parent compounds (Scheme S3, Supporting information). To circumvent this issue, our optimized general strategy installs the TFPA in the final steps of the synthesis from a perfluorobenzyl (PFB) group, in the presence of a protected ZBG. Further deprotection of the ZBG in mild conditions allows to retain all the other moieties intact. Our optimized general strategy for HDAC PRPs synthesis is outlined in Scheme 1 and detailed for PRPs 9–15 in Schemes 2–6. The synthetic schemes for the non-HDACi control 16 and the competitor S20 are provided in Schemes S1 and S2, respectively.
Scheme 1.
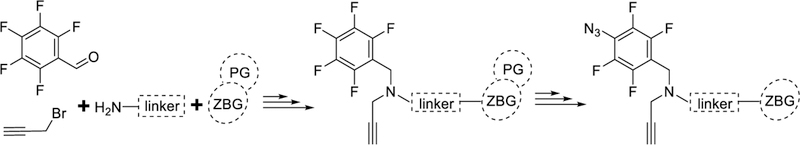
General synthetic strategy for PRPs 8–15. PG: protecting group.
The synthesis of PRPs 9 and 14 is shown in Scheme 2. A reductive amination reaction of pentafluorobenzaldehyde with p-(aminomethyl)aniline 17 gave secondary amine 18. Coupling of suberic acid monomethyl ester with either O-tritylhydroxylamine 19 or N-boc-1,2-phenylenediamine 20 followed by ester hydrolysis gave acids 21 or 22, respectively. Coupling of 18 with either 21 or 22 gave the secondary amines 23 or 24 that were alkylated with propargyl bromide resulting in 25 or 26, respectively. The intermediates 25 and 26 were subject to regioselective azidation to yield the TFPA derivatives 27 or 28, respectively. Detritylation of 27 was carried out under mild Lewis acid catalyzed conditions to give PRP 9. Deprotection of 28 under acidic conditions gave the o-aminoanilide PRP 14.
Scheme 2.
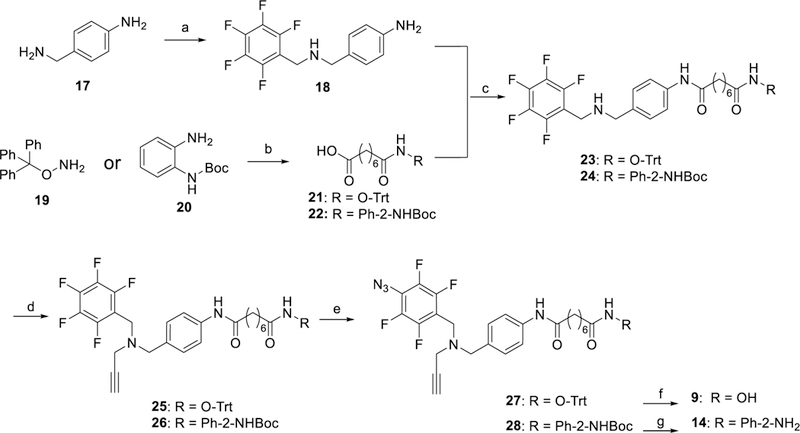
Synthesis of PRPs 9 and 14. Reagents and Conditions: a) 1. perfluorobenzaldehyde, DCM,18 h, 2. NaBH4, CH3OH, 0 °C→rt, 4 h; b) 1. octanedioic acid monomethyl ester, EDC•HCl, HOBt, DMAP, Et3N, CHCl3, 18 h, 2. NaOH, 2:1 MeOH/H2O, 8 h; c) EDC•HCl, HOBt, Et3N, CHCl3, 12 h; d) propargyl bromide, K2CO3, CH3CN, 16 h; e) NaN3, Bu4NN3, DMF, 75–80 °C, 18 h; f) MgBr2, DCM, 30 min; g) HCl, 1,4-dioxane, 0 °C, 3 h.
In Scheme 3, the synthesis of PRP 10 started with the procedure reported for its parent compound 2.[14] The resulting aldehyde 31 was reacted with propargylamine under reductive conditions to give 32 that was alkylated with PFB bromide to give the amino ester 33. A sequence of hydrolysis, tritylation, azidation, and deprotection yielded the final PRP 10. The synthesis of PRPs 11 and 13 is shown in Scheme 4. A reductive amination reaction of methyl 4-formylbenzoate 37 with propargyl amine gave the secondary amine 38 that was alkylated with PFB bromide and hydrolyzed to the corresponding acid 40. Coupling of acid 40 with either 19 or 20 followed by azidation and deprotection gave PRPs 11 or 13, respectively. PRP 12 was synthesized as shown in Scheme 5. Alkylation of N-1 nitrogen in commercially available indole 45 followed by reductive amination with N-methyl propargyl amine gave ester 47. Hydrolysis of 47 followed by reaction with 19 resulted in ester 49 that was subjected to azidation and deprotection to give PRP 12. The synthesis of PRP 15 is shown in Scheme 6. Trifluoromethyloxadiazole 53 was synthesized following previously reported procedure.[17] The perfluorophenyl group and the alkyne moieties were reacted with ethylene diamine 54 via reductive amination followed by alkylation to give 58. Next, coupling of 53 and 58, followed by azidation gave the final product 15.
Scheme 3.
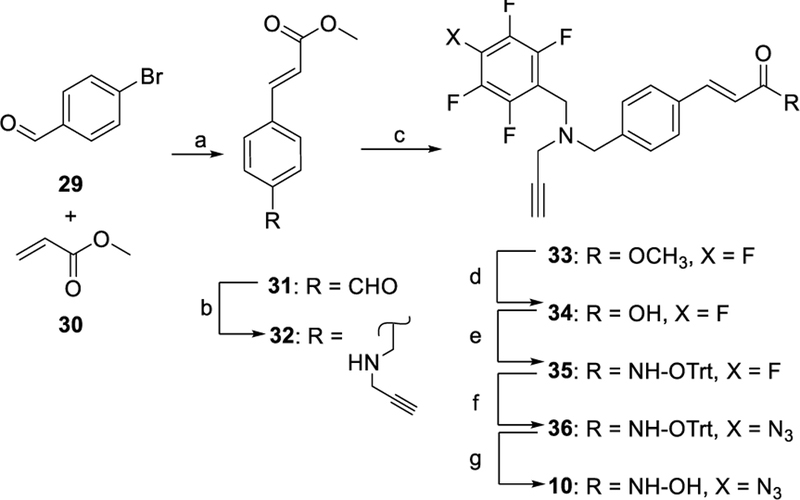
Synthesis of PRP 10. Reagents and conditions: a) Pd(OAc)2, NaOAc, NMP, 120 °C, 1 h; b) propargylamine, STAB-H, DCE, 16 h; c) perfluorobenzylbromide, K2CO3, CH3CN, 14 h; d) LiOH, 2:1 THF/H2O, 20 h; e) 19, EDC•HCl, HOBt, DMAP, Et3N, CHCl3, 6 h; f) NaN3, Bu4NN3, DMF,75–80 °C, 18 h; g) MgBr2, DCM, 3 h.
Scheme 4.
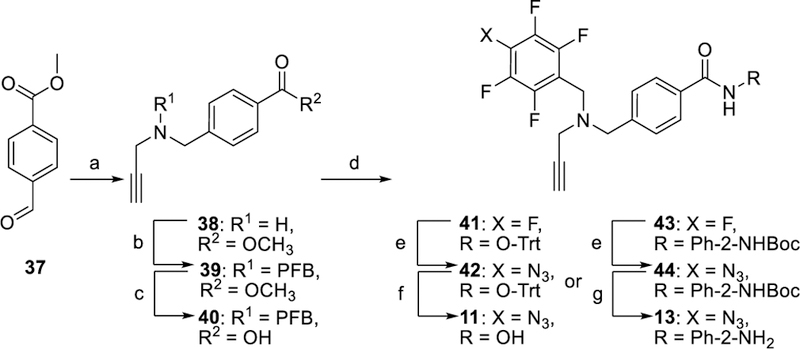
Synthesis of PRPs 11 and 13. Reagents and Conditions: a) 1. propargylamine, DCM, 16 h, 2. NaBH4, CH3OH, 0 °C→rt, 4 h; b) pentafluorobenzyl bromide, K2CO3, CH3CN, 16 h; c) LiOH, 6:3:1 THF/H2O/dioxane, 24 h; d) 19 for 41 or 20 for 43, EDC•HCl, HOBt, DMAP, Et3N, CHCl3, 12 h; e) NaN3, Bu4NN3, DMF, 75–80 °C, 18 h; f) MgBr2, DCM, 3 h; g) HCl, 1,4-dioxane, 0 °C, 6 h. PFB = perfluorobenzyl.
Scheme 5.
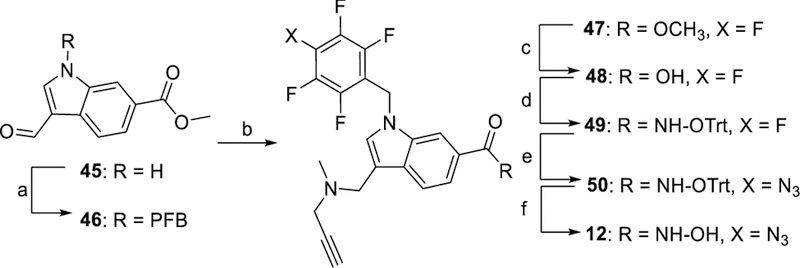
Synthesis of PRP 12. Reagents and conditions: a) perfluorobenzylbromide, NaH, DMF, 14 h; b) N-methylpropargylamine, STAB-H, DCE, 5 h; c) LiOH, 6:3:1 THF/H2O/dioxane, 12 h; d) 19, EDC•HCl, HOBt, DMAP, Et3N, CHCl3, 6 h; e) NaN3, Bu4NN3, DMF, 50 °C, 22 h; f) MgBr2, DCM, 3 h. PFB: perfluorobenzyl.
Scheme 6.
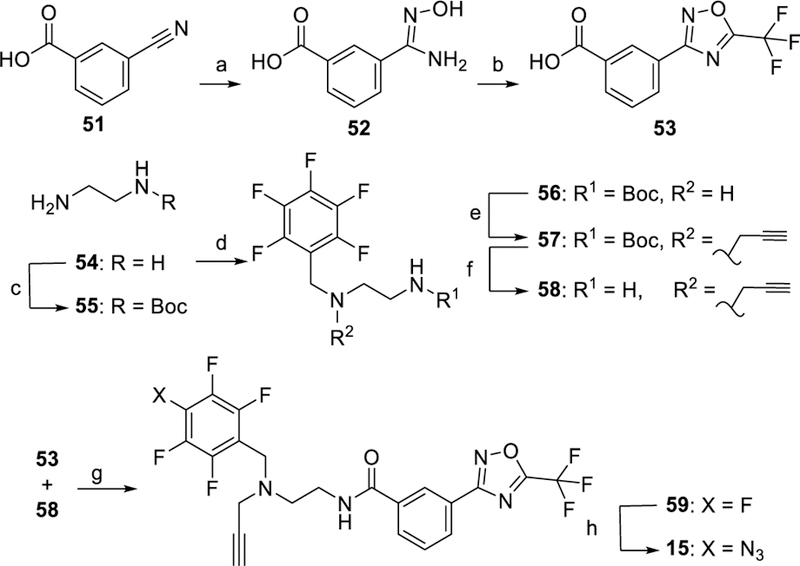
Synthesis of PRP 15. Reagents and conditions: a) NH2OH, Na2CO3, 1:1 MeOH/H2O, Reflux, 4 h; b) trifluoroacetic anhydride, 0→50 °C, pyridine, 5 h; c) (Boc)2O, CHCl3, 0 °C→rt; d) 1. perfluorobenzaldehyde, DCM, 8 h, 2. NaBH4, CH3OH, 0 °C–rt, 3 h; e) propargyl bromide, K2CO3, CH3CN, 48 h; f) HCl, 1,4-dioxane, 1 h; g) EDC•HCl, HOBt, DMAP, Et3N, CHCl3, 12 h; h) NaN3, Bu4NN3, DMF, 80 °C, 18 h.
As mentioned above, azidation of the PFB group was performed near the end of each synthesis as the resulting arylazido TFPA group was found to be unstable under the reaction conditions typically used to prepare most of the parent HDACi. Specifically, we found that the TFPA group could not withstand conditions necessary for reductive amination, palladium-catalyzed cross-coupling, and reactions of hydroxylamine hydrochloride under basic conditions including amidoxime formation from nitriles and hydroxamic acid formation from esters. Several rounds of experimentation with different conditions and synthetic routes showed that coupling of the corresponding carboxylic acid with O-trityl hydroxylamine permitted introduction and maintenance of the masked hydroxamic acid ZBG during all the steps of the synthesis (Scheme S3, A). Azidation reaction of the PFB group with trimethylsilyl azide resulted in low yields of the desired TFPA product irrespective of the temperature, solvent, and the duration of the reaction. It was found that the reaction between PFB-based intermediates and stochiometric amount of sodium azide and catalytic amount of tetrabutylammonium azide, which was adopted from previously reported procedure,[18] gave 25–95%, yields. The temperature and reaction time needed optimization for each chemotype to ensure complete conversion of PFB to TFPA. It was also important to monitor completion of the reaction using liquid chromatography mass-spectrometry analysis (LC-MS) and 19F NMR as the starting PFB derivatives often exhibited identical retention times and were inseparable by chromatography. Lowering reaction temperature required longer reaction times resulting in formation of additional side products and a reduced yield. The optimal temperature and duration of this reaction were ca. 80 °C and ca. 18 h, respectively, except for intermediate 50, for which this reaction had to be conducted at 50 °C for 22 h to obtain acceptable (≥ 25%) reaction yields (Scheme S3, C).
For hydroxamate PRPs, detritylation of intermediates 27, 36, 42 and 50 (Schemes 2–5) was carried out using a Lewis acid mediated deprotection.[19] Protic acids commonly used for deprotection, such as trifluoroacetic acid and hydrochloric acid, were found to be incompatible in deprotection of intermediates containing TFPA moiety, resulting in a complex mixture of products (Scheme S3, D). Deprotection of Boc intermediates 28 and 44 (Schemes 2 and 4) required an ice-cooled reaction for 3–6 h. Longer reaction times or reactions at room temperature gave the previously reported benzimidazole cyclization side product as detected by LC-MS (Scheme S3, E).[20]
It is also important to mention that mild hydrolysis conditions, LiOH in THF/water at room temperature, were used for hydrolysis of PFB esters 33, 39, and 47, whereas methyl esters of 21 and 22 were hydrolyzed with NaOH in methanol/water. The latter conditions in the case of 33, 39 and 47 or heating above 40 °C resulted in para-hydroxylated PFB side products (as detected by LC-MS and NMR) that were inseparable from the desired products in the subsequent reaction steps (Scheme S3, B).
Biochemical Selectivity
The PRPs and their parent compounds were tested for HDAC inhibitory activity against recombinant class I HDACs as well as HDAC4, as a representative member of class II isoforms, using a standard experimental procedure with a competitive fluorescence-based assay commonly used in the field.[21] Briefly, inhibition of HDAC1, 2, and 3 was measured using the fluorescent HDAC substrate Boc-L-Lys(Ac)-AMC and commercially available recombinant human HDAC1, 2, and 3, whereas inhibition of HDAC8 was measured using the BML-KI178 HDAC8 substrate and a recombinant human HDAC8.[22] The pIC50 values for PRPs 8–15 compared to their parent HDACi 1–7, are listed in Table 1 and their HDAC isoform selectivity profile is given in Table 2. The dose-response curves for all tested compounds are shown in Figure S2, Supporting information. The potency of PRPs was generally 10–100-fold lower than that of their parent compounds (Table 1), whereas the isoform selectivity profile of PRPs was comparable and in one case, PRP 13, HDAC3 selectivity was superior to that of its parent HDACi 5 (Table 2).
Table 1.
Biochemical pIC50 values of PRPs and their parent HDACi for HDAC1, 2, 3, 8 and 4.
| Compound | pIC50 ± SE[a] |
||||
|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC4 | |
| vorinostat (1) | 7.67 ± 0.01 | 6.92 ± 0.02 | 7.74 ± 0.02 | 6.61 ± 0.06 | NA[b] |
| PRP 8 | 6.26 ± 0.04 | 6.77 ± 0.04 | 6.96 ± 0.06 | 5.64 ± 0.11 | 4.42 ± 0.07 |
| PRP 9 | 6.13 ± 0.07 | 5.95 ± 0.08 | 6.46 ± 0.10 | 5.30 ± 0.10 | NA[b] |
| panobinostat (2) | 9.52 ± 0.10 | 9.70 ± 0.46 | 9.20 ± 0.20 | 6.41 ± 0.20 | NA[b] |
| PRP 10 | 5.56 ± 0.07 | 5.32 ± 0.05 | 5.69 ± 0.06 | 5.12 ± 0.09 | NA[b] |
| givinostat (3)[23] | 6.70[c] | 6.49[c] | 6.80[c] | 6.07[c] | NA[b] |
| PRP 11 | 5.43 ± 0.09 | 4.77 ± 0.01 | 4.96 ± 0.02 | 5.10 ± 0.06 | NA[b] |
| PCI-34051 (4) | 5.50 ± 0.08 | 4.60 ± 0.03 | 4.94 ± 0.04 | 8.48 ± 0.06 | NA[b] |
| PRP 12 | 3.33 ± 0.14 | 4.19 ± 0.06 | 5.30 ± 0.03 | 5.67 ± 0.08 | NA[b] |
| entinostat (5)[24] | 6.61[c] | 6.34[c] | 6.61[c] | <5[c] | NA[b] |
| PRP 13 | 5.09 ± 0.09 | 5.10 ± 0.12 | 5.82 ± 0.08 | < 3 | NA[b] |
| PDA-106 (6) | 6.33 ± 0.07 | 6.54 ± 0.05 | 7.66 ± 0.02 | 5.24 ± 0.09 | NA[b] |
| PRP 14 | 3.91 ± 0.05 | 3.37 ± 0.11 | 5.43 ± 0.08 | < 3 | NA[b] |
| TMP-269 (7) | 4.33 ± 0.10 | 4.44 ± 0.11 | 4.24 ± 0.07 | < 3 | 4.54 ± 0.11 |
| PRP 15 | 3.95 ± 0.14 | 4.21 ± 0.04 | < 3 | 2.81 ± 0.43 | 3.40 ± 0.78 |
pIC50 is equal to (- log IC50) and SE is standard error, both were calculated by non-linear regression analysis (enzyme-inhibitor model) using GraphPad prism 7.02. Values reported are the mean ± SE at least 2 replicate experiments; the numbers are rounded to three significant figures.
NA: less than 50% inhibition at 100 µM. The percent of inhibition at 10 and 100 µM is given in Table S2, Supporting information.
pIC50 of both 5 and 6 were calculated based on reported biochemical inhibition data for both using similar assay in the annotated references.
Table 2.
Biochemical selectivity profiles for PRPs 12–15 and their parent HDACi (4–7).
| Compound | Selectivity[a] |
|||||
|---|---|---|---|---|---|---|
| vorinostat (1) | 5.6 | 0.85 | 11 | 0.15 | 2.0 | 13 |
| PRP 8 | 0.31 | 0.20 | 4.2 | 0.65 | 13 | 21 |
| PRP 9 | 1.5 | 0.47 | 6.8 | 0.31 | 4.5 | 14 |
| Panobinostat (2) | 0.66 | 2.1 | 1300 | 3.2 | 1900 | 620 |
| PRP 10 | 1.7 | 0.74 | 2.8 | 0.43 | 1.6 | 3.7 |
| givinostat (3) | 1.6 | 0.79 | 4.3 | 0.49 | 2.6 | 5.4 |
| PRP 11 | 4.6 | 3.0 | 2.1 | 0.65 | 0.47 | 0.72 |
| PCI-34051 (4) | 7.9 | 3.6 | 0.0010 | 0.46 | 0.00013 | 0.00029 |
| PRP 12 | 0.14 | 0.011 | 0.0046 | 0.078 | 0.033 | 0.43 |
| entinostat (5) | 1.9 | 1.0 | > 40[b] | 0.54 | > 22[b] | > 41[b] |
| PRP 13 | 0.98 | 0.19 | > 120[b] | 0.19 | > 130[b] | > 660[b] |
| PDA-106 (6) | 0.62 | 0.047 | 12 | 0.076 | 20.0 | 260 |
| PRP 14 | 3.5 | 0.030 | > 8.1[b] | 0.0087 | > 2.3[b] | > 270[b] |
| TMP-269 (7) | 0.78 | 1.2 | > 21[b] | 1.6 | > 28[b] | > 17[b] |
| PRP 15 | 0.55 | > 8.9[b] | 14 | > 16[b] | 25 | 1.6 |
Selectivity ratios based on IC50 measurements; the numbers are rounded to two significant figures.
An estimate based on the highest concentration tested.
PRP 8, with high similarity to the structure of its parent HDACi 1, showed the highest potency among all the PRPs with pIC50 of 6.3, 6.8, 7, and 5.6 against HDAC1, HDAC2, HDAC3, and HDAC8, respectively (Table 1). PRP 9 was ca. 3-fold less potent than PRP 8 against all tested isoforms with pIC50 of 6.1, 6.0, 6.5 and 5.3 against HDAC1, HDAC2, HDAC3, and HDAC8, respectively (Table 1). PRP 10, with pIC50 of ca. 5.5 for HDACs 1–3, was 10-fold less potent than PRPs 8 and 9 and 10,000-fold less potent than the parent HDACi 2, which exhibited pIC50 of ca. 9.5 for HDACs 1–3. Potency of 10 against HDAC8, pIC50 of 5.1, was comparable to those of 8 and 9 and was 19-fold lower than that of 2, pIC50 of 6.4 (Table 1). PRP 11, with an average pIC50 of ca. 5.0, was 30-fold less potent than the parent HDACi 3, whose average pIC50 was ca. 6.5 against class I HDAC isoforms (Table 1). PRPs 8–11 showed little to no selectivity to any of class I HDAC isoforms (Table 2).
PRP 12 was 200-, 30-, and 2-fold more selective for HDAC8 (Table 2) with a pIC50 of 5.7 as compared to pIC50 of 3.3, 4.2, and 5.3 for HDAC 1, 2, and 3, respectively (Table 1). PRPs 13 and 14 did not inhibit HDAC8 at the maximum tested concentration, 1 mM (Table 1). PRP 13 exhibited pIC50 of 5.1, 5.1, and 5.8 for inhibition of HDAC1, 2, and 3, respectively (Table 1). PRP 14 had a pIC50 of 3.9, 3.4 and 5.4 against HDAC1, HDAC2 and HDAC3, respectively (Table 1). PRP 13 was 15-, 50-, and 3-fold more potent than 14 against HDACs 1, 2, 3, respectively. Both PRPs 13 and 14 showed selectivity for HDAC3 over HDAC1 and HDAC2 higher than that of their respective parent HDACi 5 and 6 (Table 2). We found that both PRP 15 and its parent HDACi 7, which was previously reported as being selective for class II HDACs,[17] were not selective for the class II HDAC4 and were generally weak inhibitors of all the tested isoforms, with pIC50 ranging between 3 and 4 for both compounds (Table 1). The latter finding highlights the variability in the results of the biochemical assays usually encountered due to differences in experimental conditions.
In-cell Photoaffinity Labeling
Following our previously published procedure[6f] outlined in Figure 3, photolabeling experiments were performed in live SET-2 (Figure 4), HepG2, HuH7 and HEK293T cells (Figure 5). Corresponding densitometric analysis of selected class I HDAC labeling is given in Figures S3 and S4 (Supporting information). Briefly, cells in culture were incubated with either DMSO or compound 16 as a control, PRP or a competitor HDACi and PRP and incubated for 40 min with PRPs 8–12 and 15 or for 3 h with PRPs 13 and 14, irradiated with UV light at 366 nm, washed, and lysed. The labeled lysate was subjected to “click reaction” conditions with an azide-conjugated 800CW IRDye and electrophoretically separated. Antibodies for individual HDAC isoforms were added followed by incubation with 680RD-IRDye conjugated secondary antibodies. Visualization in both 800 and 700 nm channels show PRP labeled proteins as well as HDAC antibody bound proteins, respectively. The PRP labeled bands (800 nm), that were absent from the controls and decreased by at least half upon addition of a competitor, were considered specific. These bands (800 nm) were assigned to a particular HDAC isoform if they counter-stained with an HDAC antibody (700 nm), Figure 3. The concentration of the PRPs was optimized to achieve the highest signal-to-noise ratio (Figures S5 and S6, Supporting information) and was kept within the HDACi concentration range commonly used in cell-based studies[15, 25] and typically observed in cancer patients undergoing HDACi based therapy.[26]
Figure 3.
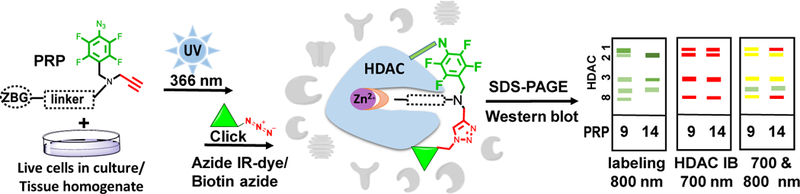
Experimental workflow of photoaffinity labeling in live cells and gel-based visualization of labeled proteins.
Figure 4.
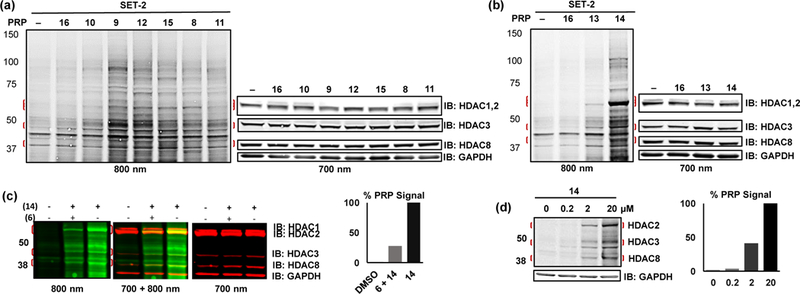
Photolabeling with PRPs 8–15 in live SET-2 cells. Labeling with (a) hydroxamate PRPs 8–12 and TFMO PRP 15, (b) o-aminoanilide PRPs 13 and 14. In each of (a) and (b), vehicle DMSO (–) and 16 were included as controls for nonspecific bands. (c) competition of 14 HDAC-labeled bands with the HDACi 6. (d) concentration dependent labeling of HDACs by 14. In (c) and (d), densitometry of HDAC2 labeling bands normalized to HDAC2/GAPDH signal is shown to the right of each gel.
Figure 5.
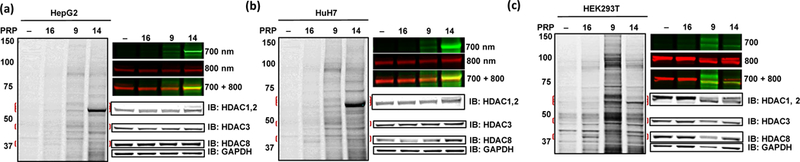
Photolabeling with PRPs 9 and 14 in (a) HepG2, (b) HuH7 and (c) HEK293T live cells. DMSO (–) and TFPA 16 were included as controls. IB with class I HDAC antibodies and GAPDH is shown to the right at the level of molecular weight for each protein. Top right part of each panel shows detection of HDAC2 labeling by IR dye (800 nm) and HDAC2 antibody (700 nm) and overlap of both labeling band and antibody (700 + 800 nm).
In SET-2 cells, the bands observed with TFPA control compound 16 were the same as those observed with the DMSO control (Figures 4a and 4b), indicating that the TFPA moiety itself would have little effect on the labeling with PRPs. Labeling of HDAC8 was difficult to interpret due to a non-specific band found in the control lanes at the same level as HDAC8 in SET-2 cells (Figure 4a). PRP 9, which is a relatively potent inhibitor of all class I HDACs (pIC50 ca. 6), showed labeling of HDACs 1–3 higher than that observed with 8 (Figure 4a and Figure S3a, Supporting information). PRPs 10 and 11 showed weak labeling of HDAC 1 and 2 isoforms only marginally different from the control (Figure 4a and Figure S3a, Supporting information). The HDAC8 selective PRP 12, showed weak labeling of HDAC1 and 2 and distinct labeling of HDAC3. Despite being a weak HDAC inhibitor in biochemical assay, PRP 15 labeled HDAC2 and 3, but not HDAC4 in live cells (Figure 4a and Figure S3a, Supporting information). The absence of HDAC4 labeling is likely due to low abundance of HDAC4 as we could not detect it by HDAC4 specific antibody in this type of cells. Unexpectedly, both HDAC3 selective PRPs 13 and 14 labeled mostly HDAC2, to a lesser extent HDAC3, and (Figure 4b and Figure S3b, Supporting information). Labeling of HDAC1 was indistinguishable from background. The labeling bands of HDAC2 and HDAC3 by PRP 14 were further confirmed by competition with the parent HDACi 6 (Figure 4c). PRP 14, although being a relatively weak HDAC2 inhibitor in the biochemical assay (pIC50 3.4), labeled HDAC2 even at concentrations as low as 200 nM (Figure 4d). In preliminary labeling experiments using recombinant HDAC1 (Figure S7, Supporting information), co-incubation with the covalent competitor S20 showed a ca. 75% decrease in labeling with PRP 8 compared to 25% decrease when co-incubated with 1. In live SET-2 cells, however, cellular levels of HDACs 1–3 and 8 were decreased upon co-treatment with S20 and both S20 and 1 had a comparable 50% decrease in labeling of HDAC2 with PRP 8 (Figure S7, Supporting information).
Photolabeling of 9 and 14 was further characterized in HepG2, HuH7, and HEK293T cells (Figure 5). In HepG2 (Figure 5a and Figure S4a, Supporting information) and HuH7 (Figure 5b and Figure S4b, Supporting information), labeling patterns were similar to those observed in SET-2 cells for both 9 and 14, with higher labeling of HDAC2 for PRP 14. In HEK293T (Figure 5c and Figure S4c, Supporting information), 14 maintained its superior labeling of HDAC2, however, to a lesser extent than in HepG2 and HuH7 cells. Labeling of HDAC1 in HEK293T was observed with both 9 and 14. Immunoblotting with anti-HDAC 1 and 2 antibodies showed only minor differences in the protein abundance in SET-2, HepG2, HuH7 and HEK293T cells, which does not explain the decreased labeling of HDAC2 by 14 in HEK293T cells (Figure S8, Supporting information).
In-tissue Photoaffinity Labeling
To investigate the feasibility of labeling HDACs with TFPA PRPs in tissues, PRP 8 was incubated with mouse liver homogenate and the PRP-labeled lysates were subject to photolabeling in a workflow similar to that optimized for live cells in culture (Figure 3). Briefly, minced fresh mouse tissue was incubated with either DMSO or PRP 8 or pretreated with 1 and then incubated with 8 and irradiated with 366 nm UV light. The amount of tissue was optimized to enable detection of class I HDAC isoforms. The labeled tissue was lysed, and the resulting lysate was reacted with the azide-conjugated biotin tag. The PRP labeled bands were detected by 800CW IRDye-conjugated streptavidin. HDAC-specific labeling bands were absent from DMSO control, decreased by competition with 1 and counterstained with HDAC-specific antibodies (Figure 6 and Figure S9, Supporting information). The band corresponding to HDAC8 and recognized with an anti-HDAC8 was relatively low in intensity compared to that in experiments with live cells. We attribute this to the use of anti-HDAC8 antibody raised against human HDAC8. PRP 8 labeled HDAC1–3 and 8 with the most pronounced bands for HDAC8 (Figure 6 and Figure S9, Supporting information).
Figure 6.
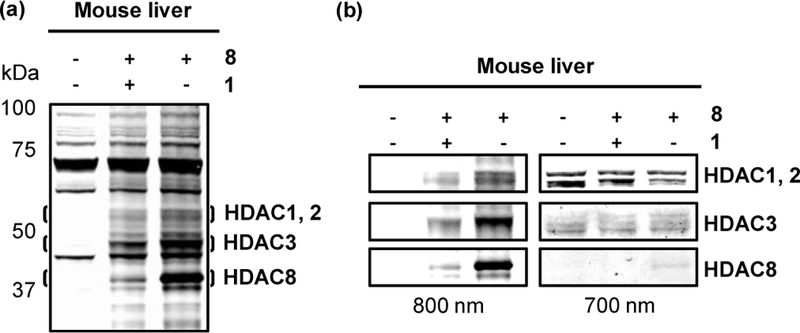
(a) Photolabeling with PRP 8 in mouse liver tissue, reacted with azide-conjugated biotin and incubated with 800CW IRDye-conjugated streptavidin (b) left: HDAC labeling bands as visualized in (a), right: immunoblotting with 680RD-IRDye conjugated HDAC1, 2, 3, and 8 antibodies.
Discussion
HDAC isoform engagement with HDACi is an important issue in development of HDAC-based therapeutics. It still needs a robust and readily implemented solution in typical biological settings and models. In this paper, we have designed and synthesized a series of novel HDAC PRPs (Figure 2) based on known HDACi (Figure 1) with diverse chemotypes and biochemical selectivity profiles. To ensure structural and chemical uniformity and direct systematic comparison of different chemotypes, the same photoreactive group and reporter handle were used for all PRPs in this study. We also developed a synthetic strategy that is compatible with the TFPA photoreactive moiety and other groups in the pharmacophore of HDACi (Scheme 1) that, we anticipate, will enable others to access even broader set of HDAC PRPs for target engagement studies.
We found that the convergent minimalistic synthetic approach described here resulted in the expected PRPs in overall acceptable yields with an individual yield for each reaction throughout the synthesis of at least 25%, which is important for scaling-up the synthesis of the PRPs. In the optimized synthetic scheme, unmasking of the photoreactive moiety was postponed to the last step, to avoid formation of side-products or losing the target compounds completely. Although PRPs containing TFPA and other azide-based photoreactive groups are typically stable in dark below 0 °C from several weeks to few months, they degrade upon prolonged storage. Ability to store a stock of non-photoreactive intermediates that can be readily converted to PRPs in a few straightforward experimental steps is expected to prevent the loss of PRP during storage and to yield consistent results in photolabeling experiments. The overall applicability of this synthetic strategy was demonstrated with three different ZBGs, two of which can be found in the majority of currently used HDACi. Although further SAR studies may be needed, the convergent approach developed here can be used to conduct the necessary optimization as well as to extend this strategy to other not yet explored HDACi chemotypes.
Incorporation of the photoreactive and reporter handle groups resulted in a variable moderate decrease in potency of the resulting PRPs compared to their parent HDACi. The biochemical activity of the PRPs against HDACs 1–3 and 8 was between 125 nM and 1 mM (Table 1) and the selectivity was similar to that of the parent HDACi (Table 2). In general, PRPs 8–15 displayed robust labeling of class I HDAC isoforms in cells in culture. Among all the PRPs tested, PRP 14 showed the best signal-to-noise ratio and was able to detect HDAC2 in SET-2 cells even at 200 nM (Figure 4d). We also demonstrated that PRP 8 can label class I HDAC isoforms in mouse liver tissue. To the best of our knowledge, this is the first published example of labeling HDACs in tissue with a small-molecule-based PRP. Unlike other commonly used in biology approaches to study HDAC engagement, application of PRPs is comparably faster, inexpensive, and, most importantly, can be done in the commonly used biological models without modifications. Our studies were conducted in live cells in culture without overexpression of HDACs, suggesting that the sensitivity of the newly designed PRPs and photolabeling approach in general are sufficient to detect endogenous levels of HDACs in cells with unaltered machinery. This would be particularly useful in situations where molecular biological manipulations with the cellular machinery are experimentally or methodologically undesirable or impossible.
The differences between the biochemical and cell-based photolabeling selectivity are observed for all the PRPs in this study. Our analysis shows that there are four types of differences. In the first type, less potent in biochemical assays PRPs 9 and 14 display higher labeling of HDACs compared to more potent congeners, 8 and 13, respectively (Figures 4a and 4b and Figure S3, Supporting information). In the second type, the isoform selectivity patterns of PRPs 12–15 determined in the biochemical assays do not match those observed by photolabeling in live cells. For instance, PRP 14, despite selectivity for HDAC3 in the biochemical assay (Table 2), showed pronounced labeling of HDAC2 in SET-2 (Figure 4b and Figure S3b, Supporting information), HepG2 (Figure 5a and Figure S4a, Supporting information), and HuH7 (Figure 5b and Figure S4b, Supporting information). In the third type, same PRPs targeted different HDAC isoforms in cell-type dependent manner. For example, PRR 14 showed higher HDAC2 labeling in HepG2 and HuH7 cells (Figures 5a and 5b) compared to that in HEK293T cells (Figure 5c), which could not be explained by the difference in isoform abundance (Figure S8, Supporting information). Whether this is due to higher activity of HDAC2 in HepG2 and HuH7 cell lines[27] and higher or equal to HDAC2 activity of HDAC1 in HEK293T cell line[2a, 28] remains to be investigated. In the fourth type, chemotype-dependent labeling profile was observed for PRPs of different ZBGs, o-aminoanilides 13 and 14 (Figure 4b, and Figure S3b, Supporting information) versus hydroxamates 8–12 (Figures 4a and Figure S3a, Supporting information). Regardless of variability in the labeling pattern within each class, the overall labeling trend is characteristic of each ZBG chemotype. The hydroxamate based-PRPs 8–12 display variable but consistent ability to label HDACs 1–3 and 8, whereas o-aminoanilide-based PRPs 13 and 14 labeled mostly HDAC2, to lesser extent HDAC3, and did not label HDAC1, which binding site is highly homologous to those of HDAC2 and HDAC3. This chemotype dependence of target engagement is consistent with the observations by Bantscheff et al[6a] and may be linked to the slow koff binding kinetics of the o-aminoanilide-based HDACi.[29] Although our efforts in this paper were focused on validation of photolabeling using class I HDACs, differences between the photolabeling and the biochemical profiles are expected for class II and IV HDACs as well. The overall discordance between the biochemical and cell-based relative selectivity is consistent with that recently demonstrated by us for PRP 8 in photolabeling of all zinc-dependent HDAC isoforms in different types of breast cancer cells.[6f] These findings further underscore the importance of the cellular context for target engagement.
This study shows that the observed differences between the biochemical and photolabeling profiles is a common phenomenon that does not depend on the abundance of the HDAC proteins and the biochemical potency of PRPs and is primarily driven by the chemotype of the PRP and the type of cells. By comparing the labeling of HDAC isoforms in different cell lines by the same PRP, we can exclude the differences in binding conformations, yields in click-reactions, and differences in photoactivation as factors for the discordance between the biochemical and the HDAC photolabeling profiles. It can, therefore, be argued that the cellular context affects photolabeling by PRPs, and more importantly HDAC isoform target engagement by HDACis, in several ways: 1) by altering the binding affinity and kinetics of the PRPs to HDACs via PTMs and/or conformational/accessibility changes due to multiprotein complex formation, and/or 2) by varying the relative abundance of the catalytically competent and catalytically inactive, e.g. scaffolding, HDACs (Figure 7). Both these scenarios individually or in combination would present themselves in the same way in the photolabeling experiments in live cells. Although we cannot exclude either of these scenarios, this report clearly demonstrates that the answer to the question “What HDAC isoform(s) does this HDAC ligand inhibit in cells/in vivo?” is not straightforward and that HDAC PRPs would be a useful tool to address it. We anticipate that the differences between the biochemical profiles and the HDAC isoform engagement in live cells, as determined by the PRPs, should also be observed for the non-photoreactive parent HDACi, and should be taken into consideration when analyzing the outcomes of ongoing clinical trials of isoform-selective HDACi.[30] Further studies of the phenomenon observed here may lead to exciting new discoveries in biology of HDAC and novel inhibitors with potency and selectivity engineered to be cell type selective.
Figure 7.
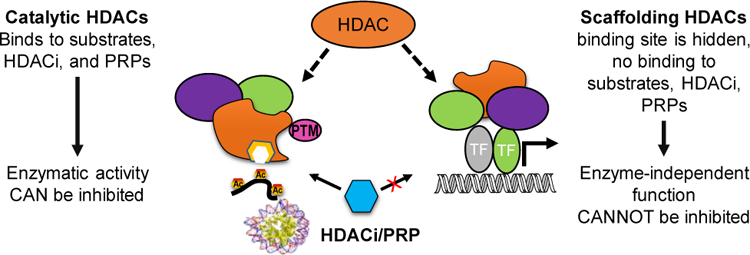
Hypothesis: Unlike scaffolding HDACs, catalytically active HDACs are able to bind substrates, small molecule HDACi, and HDAC PRPs. PTMs and HDAC protein complex components define whether the role of HDAC isoform is catalytic or scaffolding in cell-type, cell-cycle, and subcellular location manner.
Conclusions
In summary, a synthetic strategy to access diverse HDAC PRPs was developed. Novel HDAC PRPs 9–15 based on seven HDACi with diverse chemotypes 1–7 and biochemical selectivity profiles, were synthesized, tested in the HDAC 1–3, 4, and 8 biochemical assays, and validated in photolabeling experiments in live SET-2 cells. In addition, photolabeling with PRPs 9 and 14 was also validated in HepG2, HuH7, HEK293T cell lines, whereas PRP 8 was also tested for photolabeling capacity in mouse liver tissue. The synthetic strategy should be readily amenable for synthesis of PRPs with chemotypes of other HDACi. In general, the HDAC biochemical selectivity profiles of PRPs were similar to those of their parent HDACi. The cell-based photolabeling and the biochemical profiles of PRPs 9–15 were found to be substantially different from each other and did not correlate. The labeling by PRPs also did not show correlation with abundance of HDAC proteins in cells. Both the PRP chemotype and the cell type were key in defining what HDAC isoforms were labeled. This is the first report of cell-permeable uniform set of HDAC PRPs based on different HDACi chemotypes to probe engagement of multiple HDAC isoforms in live cells and tissues. Further extension of these studies to other types of cells and animal tissues as well as identification of the mechanisms responsible for modulation of HDACi selectivity and target engagement are in progress.
Experimental Section
1. Biochemical and Biological Procedure
1.1. Fluorogenic Enzymatic Assays for Class I HDAC Isoforms
Human recombinant HDAC1, HDAC2, HDAC3 (BPS Bioscience) and HDAC8 (In-house purified from E-coli) were diluted with assay buffer 1 (25 mM Tris–HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2, 1 mg/mL BSA) to give 4, 5, 1, and 8.5 ng/µL stocks of each isoform, respectively. Serial dilutions of the compounds/PRPs were made in assay buffer 2 (25 mM Tris–HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2) starting with the highest concentration at 1 mM. 10 µL of the enzyme stock and 30 µL of each of the serial dilutions were mixed in a black half-area, low protein binding 96 well plate (Corning) and pre-incubated for 5 minutes (3 hours for the o-aminoanilide derivatives, 6, 13 and 14) at room temperature (rt). Next, 10 µL of 125 µM Boc-L-Lys(Ac)-AMC (BLA) fluorescent substrate (Chem-Implex) for HDACs 1–3 or 10 µL of 25 µM Fluor de Lys®, BML-KI178 (Enzo Life Sciences) for HDAC8 was added to each well and incubated for 30 minutes at rt. The reaction was quenched by the addition of 50 µL of 1 mg/mL trypsin and 5 µM TSA in assay buffer 2 and incubated for an additional 30 minutes. The fluorescence signal was read at excitation wavelength 360 nm and emission wavelength 460 nm using a Synergy 4 hybrid microplate reader from BioTek. The statistical data analysis and IC50 values were determined using GraphPad Prism 7.02.
1.2. Fluorogenic Enzymatic Assay for HDAC4
HDAC4 inhibitory activity was determined using the fluorogenic HDAC4 assay kit (BPS Bioscience, catalogue # 50064) according to the manufacturer procedure. The statistical data analysis and IC50 values were determined using GraphPad Prism 7.02.
1.3. Cell Culture
The human megakaryoblastic cell line SET-2 was cultured in RPMI 1640 medium (Corning, VA, USA) supplemented with 10% heat-inactivated fetal bovine serum, FBS (Sigma Aldrich, St. Louis, MO, USA). Cells were seeded at the density of 1 × 105/ml of medium and were grown at 37°C with 5% CO2. Cells were harvested after 72 hours, collected by centrifugation, washed and viability was tested by trypan blue dye exclusion. Collected cells were then re-suspended in RPMI 1640 supplemented with 10% heat inactivated fetal calf serum. A total of 30 × 106 cells was spun down at 500 g for 10 min at 4 °C, re-suspended in 12 mL PBS and aliquoted into 0.5 mL on a 12-well plate for photolabeling experiments.
The human hepatocellular carcinoma HepG2 or HuH7, or the human embryonic kidney HEK293T cells were plated in 6-well plates (0.5 × 106 cells) and grown to 80% confluence in 2 mL culture medium (RPMI 1640 supplemented with 10% FBS, penicillin-streptomycin, and non-essential amino acids) in a humidified atmosphere containing 5% CO2 at 37 ºC. The medium was replaced with 1 mL PBS right before photolabeling experiments.
1.4. Cell-based Photolabeling
Cells were pretreated with 200 µM competitor for 15 minutes (or 3 h for 5 and 6) where applicable and then treated with 20 µM PRP or DMSO control. After incubation with the PRP at 37 °C for 30 min (or 3 h for PRPs 13 and 14), the cells were cooled on ice and irradiated with 366 nm light for 30 min. The medium was removed, and the cells gently washed twice with 2 mL PBS and then covered with 1 mL PBS. Cells were scraped/transferred from the plate into Eppendorf tubes, spun down at 1,000 g for 5 minutes at 4 °C, the supernatant removed, and the cells resuspended in whole cell lysis buffer (50 mM HEPES (pH 7.5), 150 mM NaCl, 1% Igepal CA-630, 5% glycerol and 1x protease inhibitor cocktail (Roche)) and 1x phosphatase inhibitor cocktail (ThermoFisher)). Samples were homogenized by vortexing, incubated on a rotating stand at 4 °C for 1 h, then spun down at 19,000g for 10 minutes at 4 °C and the protein concentrations were determined by bicinchoninic acid (BCA) assay. Next, 35 µg total lysates were incubated with IRDye® 800CW Azide Infrared dye (LI-COR) at concentration equal to that of the PRP, TCEP (1 mM), TBTA (0.1 mM), and CuSO4 (1 mM) for 90 minutes at room temperature. The samples were then frozen at −20 °C until analyzed by Western blot.
1.5. Mouse Tissue Photolabeling
129/SV mouse was exsanguinated by retro-orbital bleeding, sacrificed and fresh liver tissue (ca. 20 mg) was harvested and immediately homogenized in an Eppendorf tube (using a Teflon homogenizer) containing 500 µL of PBS. The tissue homogenate was centrifuged at 13,000g for 5 min at 4 °C. The supernatant was discarded, and the cells were resuspended with 1 mL of PBS and then centrifuged again at 13,000g for 5 min at 4 °C. The cells were resuspended in 1 mL PBS and plated in a 6-well plate at a 1:10 dilution. Cells were treated with either DMSO or10 µM PRP, or 200 µM SAHA then 10 µM PRP in PBS in a humidified atmosphere containing 5% CO2 at 37 ˚C (all treatments were done on cells from the same tissue preparation) for 40 minutes. Cells were then irradiated with UV light (366 nm) for 30 min on ice, harvested by centrifugation at 13,000g for 5 min at 4 °C, washed with 1 mL PBS and repelleted. The cells were lysed by addition of 100 µL cold lysis buffer (150 mM NaCl, 50 mM HEPES, 1% Igepal, 5% glycerol, 1X PIC, 1X Phosphatase inhibitor cocktail), homogenized by vortexing for 10 seconds at room temperature and placed on a rotating stand overnight at 4 °C. The lysate was clarified by centrifugation at 19,000g for 10 min at 4 °C. Protein concentration was determined using BCA assay and diluted to 2 mg/mL. 35 µg of each lysate was reacted a 0.1 mM azide-PEG3-biotin conjugate, Sigma Aldrich 762024 (1.7 mM stock in 1:4 DMSO:t-butanol) in the presence of 1 mM CuSO4, 0.1 mM TBTA, 1 mM TCEP for 90 minutes at room temperature. The reactions were then stored at −20 °C overnight. Precipitated proteins were pelleted at 6000g for 5 min at 4 °C. Proteins were washed with 250 µL methanol by brief sonication and repelleted, then resuspended in PBS containing 0.2% SDS by brief sonication. Proteins were then separated by SDS-PAGE and transferred to nitrocellulose membrane (as per 1.7. below). Membrane was blocked and incubated with IRDye® 800CW streptavidin (LI-COR) and labeled protein signal was recorded with Odyssey Sa scanner at 800 nm. Immunoblotting with HDAC specific antibodies was done as per 1.7.
1.6. Photolabeling of Recombinant Proteins
Recombinant HDAC1 (200 ng) in 10 µL whole cell lysis buffer (as per 1.4.) was pretreated with 100 mM SAHA (1) or S20 or DMSO control for 15 minutes and then treated with PRP 8 (10 mM) or DMSO control for 40-min. Samples were cooled on ice and irradiated with 366 nm light for 30 min then incubated with 0.1 mM azide-PEG3- biotin conjugate, Sigma Aldrich 762024 (2.5 mM stock in DMSO) in the presence of 1mM CuSO4, 0.1 mM TBTA, 1 mM TCEP for 90 minutes at room temperature. Samples were then diluted with loading buffer, separated by SDS-PAGE and transferred to nitrocellulose membrane (as per 1.7. below). Membrane was blocked and incubated with IRDye® 800CW streptavidin and labeled protein signal was recorded with Odyssey Sa scanner at 800 nm. Immunoblotting with HDAC specific antibodies was done as per 1.7.
1.7. Western Blot
Samples containing 30 µg protein were diluted with 4x LDS sample buffer containing DTT (Invitrogen), boiled for 5 minutes and separated by gel electrophoresis at 100 volts. Gels were transferred to nitrocellulose membranes with iBlot2 transfer system (P3 for 7 minutes) and visualized with Odyssey Sa imager at 800 nm. Membranes were then blocked with Odyssey blocking buffer for 2 hours at rt or overnight at 4 °C. The blots were incubated with desired antibodies (anti-HDAC1 (Abcam ab7028), anti-HDAC2 (Abcam, ab12169), anti-HDAC3 (Abcam, ab7030), anti-HDAC8 (Abcam, ab187139) and anti-HDAC4,5,9 (Abcam, ab131524)) at the recommended dilutions for each in Odyssey blocking buffer (LI-COR) overnight at 4 °C. The blots were then washed 3 × 5 minutes with PBST, incubated with relevant species of IRDye® 680RD secondary antibody (LI-COR) for 1 hour and visualized with Odyssey Sa imager at both 700 nm and 800 nm. If additional antibody probing was necessary, membranes were stripped with 0.2 N NaOH for 10 minutes and re-blockedwith Odyssey blocking buffer.
1.8. Ethics statement
All animal experiments were Institutional Animal Care and Use Committee (IACUC) approved and performed according to institutional guidelines with University of Illinois at Chicago Institutional Biosafety and Animal Care Committee approval (ACC Number: 17–012). All animal procedures were performed in the College of Medicine Research Building at the UIC and adhere to the policies of the NIH Office of Laboratory Animal Welfare (OLAW), the standards of the Animal Welfare Act, the Public Health Service Policy, and the Guide for the Care and Use of Laboratory Animals.
2. General Chemistry Methods
All the reagents and solvents were obtained from commercial sources and used without further purification. Reactions were performed under an inert atmosphere (nitrogen or argon) whenever anhydrous solvents were used. Reactions were monitored by thin-layer chromatography (TLC) using Merck 60F254 silica gel plates or by Liquid Chromatography-Mass Spectrometry on Shimadzu LCMS-2020 with either Waters, XSelect HSS CYANO 3.6 µm column or Agilent XBD-C18 3.5 μm column; dimensions 2.1 × 20 mm and UV detector at 254 nM. Chromatography purification was performed on Biotage Isolera Four instrument using pre-filled KP-Sil (normal phase) and KP-C18-HS (reverse phase) SNAP cartridges with UV detection at 254 and 280 nm. 1H, 13C and 19F NMR spectra were recorded on Bruker spectrometers at 400, 100 and 376 MHz, respectively. Chemical shifts were reported on δ scale in ppm with the deuterated solvent indicated as the internal reference. Coupling constants were reported in Hz and the standard abbreviations indicating multiplicity were used as follows: s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. High resolution mass spectrometry experiments (HRMS) were performed at either the Mass spectrometry, Metabolomics and Proteomics Facility at University of Illinois at Chicago or Mass spectrometry laboratory at University of Illinois at Urbana Champaign on either Thermo Finnigan LTQ FT ICR Hybrid Mass Spectrometer or Shimadzu LCMS IT-TOF Mass Spectrometer or Synapt G2-Si mass spectrometer.
3. Docking, Synthetic Procedures, and Compound Characterization
Detailed description of the docking, synthetic procedures, and compounds characterization is given in the supporting information.
Supplementary Material
Acknowledgements
This study was funded by the NCI/NIH grants R01 CA131970 (PAP) and R21 CA183627 (PAP), NHLBI/NIH R01 HL130760 (NM), NIAID/NIH R01 AI125401 (AMcL), the Alzheimer’s Drug Discovery Foundation grant ADDF #20101103 (PAP), the PhRMA Foundation Fellowship for Pharmacology and Toxicology (TYT), and Egyptian Government scholarship JS-2971 (SA).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Falkenberg KJ, Johnstone RW, Nat. Rev. Drug Discov 2014, 13, 673–691. [DOI] [PubMed] [Google Scholar]
- [2].a) Dovey OM, Foster CT, Cowley SM, PNAS 2010, 107, 8242–8247 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Haberland M, Montgomery RL, Olson EN, Nat. Rev. Genet 2009, 10, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K, Nature 2012, 489, 313–317 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, Yenamandra A, Locke K, Yuan J.-l., Bonine-Summers AR, Wells CE, Kaiser JF, Washington MK, Zhao Z, Wagner FF, Sun Z-W, Xia F, Holson EB, Khabele D, Hiebert SW, Cancer Cell 2010, 18, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Thaler F, Mercurio C, ChemMedChem 2014, 9, 523–526 [DOI] [PubMed] [Google Scholar]; b) Bieliauskas AV, Pflum MK, Chem. Soc. Rev 2008, 37, 1402–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D, Genes Dev 1999, 13, 1924–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) You A, Tong JK, Grozinger CM, Schreiber SL, PNAS 2001, 98, 1454–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Alland L, Muhle R, Hou H Jr., Potes J, Chin L, Schreiber-Agus N, DePinho RA, Nature 1997, 387, 49–55 [DOI] [PubMed] [Google Scholar]; d) Segre CV, Chiocca S, J. Biomed. Biotechnol 2011, 2011, 690848. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Eom GH, Kook H, Pharmacol. Ther 2014, 143, 168–180 [DOI] [PubMed] [Google Scholar]; f) Sun JM, Chen HY, Davie JR, J. Biol. Chem 2007, 282, 33227–33236 [DOI] [PubMed] [Google Scholar]; g) Pflum MK, Tong JK, Lane WS, Schreiber SL, J. Biol. Chem 2001, 276, 47733–47741 [DOI] [PubMed] [Google Scholar]; h) Qiu Y, Zhao Y, Becker M, John S, Parekh BS, Huang S, Hendarwanto A, Martinez ED, Chen Y, Lu H, Adkins NL, Stavreva DA, Wiench M, Georgel PT, Schiltz RL, Hager GL, Mol. Cell 2006, 22, 669–679 [DOI] [PubMed] [Google Scholar]; i) Zhang X, Ozawa Y, Lee H, Wen YD, Tan TH, Wadzinski BE, Seto E, Genes Dev 2005, 19, 827–839 [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Guenther MG, Barak O, Lazar MA, Mol. Cell. Biol 2001, 21, 6091–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Adikesavan AK, Karmakar S, Pardo P, Wang L, Liu S, Li W, Smith CL, Mol. Cell. Biol 2014, 34, 1246–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Frasor J, Danes JM, Funk CC, Katzenellenbogen BS, PNAS 2005, 102, 13153–13157 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Green AR, Burney C, Granger CJ, Paish EC, El-Sheikh S, Rakha EA, Powe DG, Macmillan RD, Ellis IO, Stylianou E, Breast Cancer Res. Treat 2008, 110, 427–437 [DOI] [PubMed] [Google Scholar]; d) Smith CL, Migliaccio I, Chaubal V, Wu MF, Pace MC, Hartmaier R, Jiang S, Edwards DP, Gutierrez MC, Hilsenbeck SG, Oesterreich S, Breast Cancer Res. Treat 2012, 136, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dumpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G, Nat. Biotechnol 2011, 29, 255–265 [DOI] [PubMed] [Google Scholar]; b) Robers MB, Dart ML, Woodroofe CC, Zimprich CA, Kirkland TA, Machleidt T, Kupcho KR, Levin S, Hartnett JR, Zimmerman K, Niles AL, Ohana RF, Daniels DL, Slater M, Wood MG, Cong M, Cheng YQ, Wood KV, Nat. Commun 2015, 6, 10091. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Salisbury CM, Cravatt BF, PNAS 2007, 104, 1171–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kuo HY, DeLuca TA, Miller WM, Mrksich M, Anal. Chem 2013, 85, 10635–10642 [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Padige G, Negmeldin AT, Pflum MK, J. Biomol. Screen 2015, 20, 1277–1285 [DOI] [PubMed] [Google Scholar]; f) Hanigan TW, Aboukhatwa SM, Taha TY, Frasor J, Petukhov PA, Cell Chem. Bio 2017, 24, 1356–1367 e1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Salisbury CM, Cravatt BF, J. Am. Chem. Soc 2008, 130, 2184–2194. [DOI] [PubMed] [Google Scholar]
- [8].Albrow VE, Grimley RL, Clulow J, Rose CR, Sun J, Warmus JS, Tate EW, Jones LH, Storer RI, Mol. Biosyst 2016, 1781–1789. [DOI] [PubMed]
- [9].He B, Velaparthi S, Pieffet G, Pennington C, Mahesh A, Holzle DL, Brunsteiner M, van Breemen R, Blond SY, Petukhov PA, J. Med. Chem 2009, 52, 7003–7013 [DOI] [PMC free article] [PubMed] [Google Scholar]; Abdelkarim H, Brunsteiner M, Neelarapu R, Bai H, Madriaga A, van Breemen RB, Blond SY, Gaponenko V, Petukhov PA, ACS Chem. Biol 2013, 8, 2538–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fleming SA, Tetrahedron 1995, 51, 12479–12520. [Google Scholar]
- [11].Schnapp K, Poe R, Leyva E, Soundararajan N, Platz MS, Bioconjug. Chem 1993, 4, 172–177. [DOI] [PubMed] [Google Scholar]
- [12].Marks PA, Breslow R, Nat. Biotechnol 2007, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- [13].Brunsteiner M, Petukhov PA, J. Mol. Model 2012, 18, 3927–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Remiszewski SW, Sambucetti LC, Bair KW, Bontempo J, Cesarz D, Chandramouli N, Chen R, Cheung M, Cornell-Kennon S, Dean K, Diamantidis G, France D, Green MA, Howell KL, Kashi R, Kwon P, Lassota P, Martin MS, Mou Y, Perez LB, Sharma S, Smith T, Sorensen E, Taplin F, Trogani N, Versace R, Walker H, Weltchek-Engler S, Wood A, Wu A, Atadja P, J. Med. Chem 2003, 46. [DOI] [PubMed]
- [15].Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ, Leukemia 2008, 22, 1026–1034. [DOI] [PubMed] [Google Scholar]
- [16].Chou CJ, Herman D, Gottesfeld JM, J. Biol. Chem 2008, 283, 35402–35409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, Baloglu E, Trump RP, Head MS, Hofmann GA, Murray-Thompson M, Schwartz B, Chakravorty S, Wu Z, Mander PK, Kruidenier L, Reid RA, Burkhart W, Turunen BJ, Rong JX, Wagner C, Moyer MB, Wells C, Hong X, Moore JT, Williams JD, Soler D, Ghosh S, Nolan MA, Nat. Chem. Biol 2013, 9, 319–325. [DOI] [PubMed] [Google Scholar]
- [18].Qin L, Sheridan C, Gao J, Org. Lett 2012, 14, 528–531. [DOI] [PubMed] [Google Scholar]
- [19].Yang SM, Lagu B, Wilson LJ, J. Org. Chem 2007, 72, 8123–8126. [DOI] [PubMed] [Google Scholar]
- [20].Beconi M, Aziz O, Matthews K, Moumne L, O’Connell C, Yates D, Clifton S, Pett H, Vann J, Crowley L, Haughan AF, Smith DL, Woodman B, Bates GP, Brookfield F, Burli RW, McAllister G, Dominguez C, Munoz-Sanjuan I, Beaumont V, PLoS One 2012, 7, e44498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Neelarapu R, Holzle DL, Velaparthi S, Bai H, Brunsteiner M, Blond SY, Petukhov PA, J. Med. Chem 2011, 54, 4350–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dowling DP, Gattis SG, Fierke CA, Christianson DW, Biochemistry 2010, 49, 5048–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li S, Fossati G, Marchetti C, Modena D, Pozzi P, Reznikov LL, Moras ML, Azam T, Abbate A, Mascagni P, Dinarello CA, J. Biol. Chem 2015, 290, 2368–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, Ortwine DF, Gunzner J, Modrusan Z, Neumann L, Koth CM, Lupardus PJ, Kaminker JS, Heise CE, Steiner P, J. Biol. Chem 2013, 288, 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Rosato RR, Almenara JA, Grant S, Cancer Res 2003, 63, 3637–3645 [PubMed] [Google Scholar]; b) Fortunati N, Marano F, Bandino A, Frairia R, Catalano MG, Boccuzzi G, Int. J. Oncol 2014, 44, 700–708 [DOI] [PubMed] [Google Scholar]; c) Tavakoli-Yaraki M, Karami-Tehrani F, Salimi V, Sirati-Sabet M, Tumour Biol 2013, 34, 241–249. [DOI] [PubMed] [Google Scholar]
- [26].Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM, J. Clin. Oncol 2005, 23, 3923–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Noh JH, Bae HJ, Eun JW, Shen Q, Park SJ, Kim HS, Nam B, Shin WC, Lee EK, Lee K, Jang JJ, Park WS, Lee JY, Nam SW, Cancer Res 2014, 74, 1728–1738. [DOI] [PubMed] [Google Scholar]
- [28].a) Brunmeir R, Lagger S, Seiser C, Int. J. Dev. Biol 2009, 53, 275–289 [DOI] [PubMed] [Google Scholar]; b) Chen S, Yao X, Li Y, Saifudeen Z, Bachvarov D, El-Dahr SS, Development 2015, 142, 1180–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Somanath P, Herndon Klein R, Knoepfler PS, PLoS One 2017, 12, e0185627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Vaidya AS, Karumudi B, Mendonca E, Madriaga A, Abdelkarim H, van Breemen RB, Petukhov PA, Bioorg. Med. Chem. Lett 2012, 22, 5025–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kral AM, Ozerova N, Close J, Jung J, Chenard M, Fleming J, Haines BB, Harrington P, Maclean J, Miller TA, Secrist P, Wang H, Heidebrecht RW Jr., Biochemistry 2014, 53, 725–734. [DOI] [PubMed] [Google Scholar]
- [30].Eckschlager T, Plch J, Stiborova M, Hrabeta J, Int. J. Mol. Sci 2017, 18. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.