

Resistance to first-line triazole antifungal agents among Aspergillus species has prompted the use of second-line therapy with echinocandins. As the number of Aspergillus-infected patients treated with echinocandins is rising, clinical observations of drug resistance are also increasing, indicating an emerging global health threat. Our knowledge regarding the development of clinical echinocandin resistance is largely derived from Candida spp., while little is known about resistance in Aspergillus. Therefore, it is important to understand the specific cellular responses raised by A. fumigatus against echinocandins. We discovered a new mechanism of resistance in A. fumigatus that is independent of the well-characterized FKS mutation mechanism observed in Candida. This study identified an off-target effect of CAS, i.e., ROS production, and integrated oxidative stress and sphingolipid alterations into a novel mechanism of resistance. This stress-induced response has implications for drug resistance and/or tolerance mechanisms in other fungal pathogens.
KEYWORDS: Aspergillus fumigatus, ROS, antifungal resistance, caspofungin, echinocandins, glucan synthase, glucan synthase inhibitors, lipids
ABSTRACT
Aspergillus fumigatus is a leading cause of invasive fungal infections. Resistance to first-line triazole antifungals has led to therapy with echinocandin drugs. Recently, we identified several high-minimum-effective-concentration (MEC) A. fumigatus clinical isolates from patients failing echinocandin therapy. Echinocandin resistance is known to arise from amino acid substitutions in β-(1,3)-d-glucan synthase encoded by the fks1 gene. Yet these clinical isolates did not contain mutations in fks1, indicating an undefined resistance mechanism. To explore this new mechanism, we used a laboratory-derived strain, RG101, with a nearly identical caspofungin (CAS) susceptibility phenotype that also does not contain fks1 mutations. Glucan synthase isolated from RG101 was fully sensitive to echinocandins. Yet exposure of RG101 to CAS during growth yielded a modified enzyme that was drug insensitive (4 log orders) in kinetic inhibition assays, and this insensitivity was also observed for enzymes isolated from clinical isolates. To understand this alteration, we analyzed whole-enzyme posttranslational modifications (PTMs) but found none linked to resistance. However, analysis of the lipid microenvironment of the enzyme with resistance induced by CAS revealed a prominent increase in the abundances of dihydrosphingosine (DhSph) and phytosphingosine (PhSph). Exogenous addition of DhSph and PhSph to the sensitive enzyme recapitulated the drug insensitivity of the CAS-derived enzyme. Further analysis demonstrated that CAS induces mitochondrion-derived reactive oxygen species (ROS) and that dampening ROS formation by antimycin A or thiourea eliminated drug-induced resistance. We conclude that CAS induces cellular stress, promoting formation of ROS and triggering an alteration in the composition of plasma membrane lipids surrounding glucan synthase, rendering it insensitive to echinocandins.
INTRODUCTION
Invasive fungal infections are a serious threat to human health, causing at least 1.5 million deaths worldwide each year (1). Such infections are common in immunocompromised patients and are associated with high rates of mortality: 30% to 40% for invasive candidiasis and 30% to 90% for invasive aspergillosis (2, 3). Patients with invasive or chronic pulmonary aspergillosis are typically treated with azoles as a first-line therapy. However, resistance to azoles is on the rise and second-line therapy with echinocandins is therefore becoming prominent (4).
Mechanisms of echinocandin resistance have been extensively studied in yeasts of the Candida genus. In these organisms, clinical resistance to echinocandins arises via mutations in the hot spot regions of FKS genes which encode the cell wall biosynthetic enzyme β-(1,3)-d-glucan synthase (5). While fks1 mutations have also been linked to resistance to echinocandins in Aspergillus fumigatus (6, 7), high-minimum-effective-concentration (MEC) echinocandin-resistant clinical strains of A. fumigatus containing a wild-type (WT) copy of fks1 have also been identified (8). In addition, it was reported that upregulation of glucan synthase may also result in reduced clinical drug response (9). These observations point to the clinical relevance of fks1 mutation-independent mechanisms for echinocandin resistance in A. fumigatus.
β-(1,3)-d-Glucan synthase catalyzes the addition of glucose monomers to the growing chain of β-(1,3)-d-glucan (10, 11). It is predicted to be an integral membrane protein with 16 transmembrane domains, a putative cytoplasmic N terminus, and a putative UDP-glucose binding sequence at the C terminus (12–16). Yet echinocandin binding and inhibition are not well defined. It has been speculated that the lipid-like tail of echinocandins may intercalate into the hot spot regions of glucan synthase, resulting in inhibition (17). In Candida glabrata, mutants in certain lipid biogenesis pathways showed differential susceptibilities to different echinocandins, a finding that was hypothesized to be due to changes in lipid composition near the enzyme (18). However, how the modulation of glucan synthase results in altered echinocandin sensitivity has not been examined.
RG101 is a spontaneous high-MEC A. fumigatus mutant derived from ATCC 13073 generated in Perlin laboratory. This strain is resistant to caspofungin (CAS) but contains no mutations in the fks1 gene (19). Therefore, to begin to examine fks1-independent mechanisms of echinocandin resistance in Aspergillus, we investigated the mechanism of RG101 resistance to CAS and then extended our findings to the echinocandin-resistant clinical isolates of A. fumigatus. Our study identified a novel stress-induced mechanism of echinocandin resistance in Aspergillus that is mediated by mitochondrion-derived reactive oxygen species (ROS). This clinically important mechanism induces drug insensitivity of glucan synthase by modulating its immediate lipid environment. It reflects an important adaptation response in fungal species.
RESULTS
A. fumigatus echinocandin resistance independent of fks1 mutations.
Clinical isolates of A. fumigatus obtained from patients with chronic pulmonary aspergillosis who failed echinocandin therapy were shown to have elevated MECs for both CAS and micafungin (MFG) (Table 1). DNA sequence analysis revealed no mutations in the fks1 gene open reading frame or promoter (data not shown), suggesting that the mechanism of echinocandin resistance in these strains was independent of the established FKS mechanism of well-characterized Candida species (4) and known to exist in Aspergillus fumigatus (6, 7). The level of fks1 expression was not increased upon CAS induction (see Fig. S1 in the supplemental material), indicating that overexpression of the drug target was not the mechanism of resistance in RG101.
TABLE 1.
Minimum effective concentrations of clinical isolates of A. fumigatus from patients with chronic pulmonary aspergillosis who failed echinocandin therapy
|
Aspergillus fumigatus
clinical isolate |
Isolate source | MECa
|
Nature of fks1 gene |
|
|---|---|---|---|---|
| CAS (μg/ml) |
MFG (μg/ml) |
|||
| 32458 | Sputum | 16 | 2 | WT |
| 32315 | Sputum | 16 | 2 | WT |
| 117 | Sputum | 16 | 4 | WT |
| 2770 | Sputum | 16 | 2 | WT |
| 68284B | Sputum | 4 | 4 | WT |
| ATCC 13073 | Human pulmonary lesion | 0.12 | 0.03 | WT |
| S679P | Laboratory-generated echinocandin-resistant mutant | 16 | 16 | Ser-679-Pro |
MEC, minimum effective concentration.
fks1 expression levels in RG101 under uninduced and CAS-induced conditions. RG101 conidia were grown for 16 h in YPD in the absence and presence of CAS (1 and 4 μg/ml), and expression levels of fks1 were compared using reverse transcription-PCR (RT-PCR). No significant differences in fks1 expression levels were seen under uninduced and CAS-induced conditions (P > 0.05). Download FIG S1, DOCX file, 0.02 MB (26.7KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To better understand this potential novel resistance mechanism, we turned our attention to a previously reported laboratory-generated strain of A. fumigatus called RG101, which exhibited a drug susceptibility phenotype comparable to those seen with the echinocandin-resistant and fks1 wild-type (WT) clinical isolates. The RG101 strain was spontaneously generated following CAS exposure of echinocandin-susceptible parental strain ATCC 13073 (19). The resulting mutant strain displayed an unusual paradoxical high-resistance phenotype but was devoid of any mutation in the fks1 gene. At 24 h, RG101 was sensitive to CAS with an MEC of 0.25 μg/ml, with the formation of characteristic rosette structures indicating growth inhibition. However, breakthrough growth began to manifest at 0.5 μg/ml, and at 1 and 8 μg/ml of CAS, this strain showed complete resistance. At 16 μg/ml, rosettes began to form again, indicative of drug sensitivity (Fig. 1A). By 30 h, full breakthrough was seen at all concentrations of CAS tested (0.25 to 8 μg/ml) (Fig. 1A). This phenotype, showing partial inhibition at low drug levels followed by full breakthrough growth at higher levels, suggested that there was drug-mediated induction of caspofungin resistance. As reported previously (19), RG101 was resistant to CAS and sensitive to all other antifungals, indicative of CAS-specific, inducible resistance phenotype (Fig. 1B).
FIG 1.

RG101 shows breakthrough growth in CAS. (A) Time-dependent changes in growth phenotypes of RG101 and ATCC 13073 in RPMI 1640 medium. At 24 h, the MEC of CAS for RG101 was 0.25 μg/ml, with the formation of characteristic rosettes indicating inhibition (red). However, breakthrough growth began to manifest at 0.5 μg/ml (green), and at between 1 and 8 μg/ml of CAS, this strain showed complete resistance. At 16 μg/ml, rosettes began to form again, indicative of growth inhibition. At 30 h, resistance of RG101 to CAS was seen at all concentrations of CAS tested. (B) Results of drug susceptibility testing of RG101 and ATCC 13073 at 24 h in the presence of different antifungals. RG101 was resistant only to CAS but was sensitive to other echinocandins, azoles, and polyenes. ATCC 13073 was sensitive to all antifungals.
To determine if RG101 was resistant to CAS in vivo, we tested the strain in a murine model of pulmonary invasive aspergillosis, in which neutropenic DBA/2 mice were infected with RG101 conidia. The lung burdens of mice treated with CAS, MFG, or vehicle are presented in Fig. 2. Animals treated with vehicle, CAS, and MFG showed 25.8, 22.6, and 5.2 ng fungal DNA per lung, respectively. No significant differences in lung burden were observed between the vehicle-treated and CAS-treated groups, whereas the MFG-treated animals showed an approximate 5-fold decrease in burden, suggesting that RG101 was resistant to CAS but sensitive to MFG in vivo.
FIG 2.
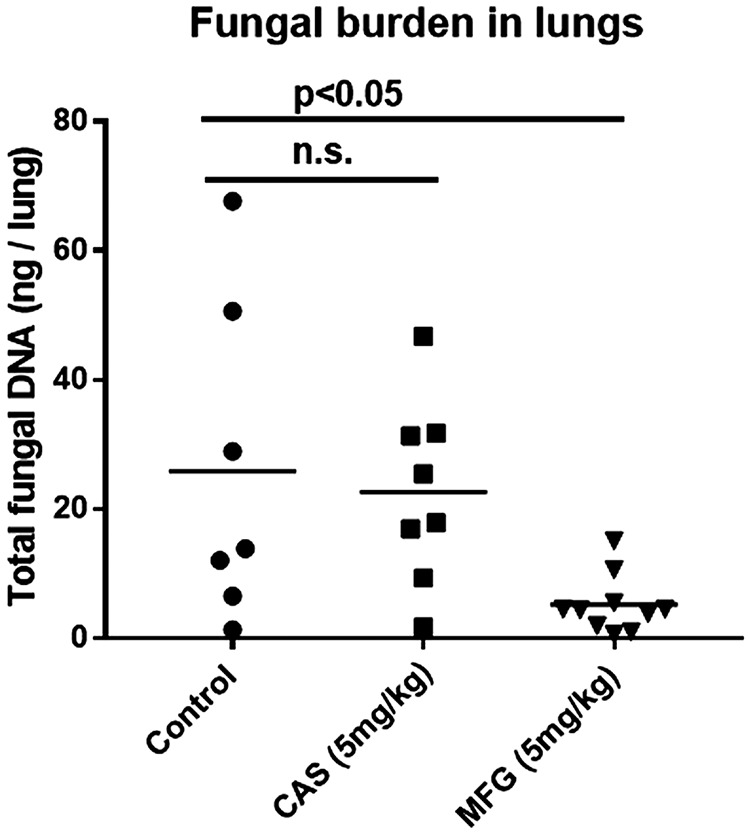
RG101 is resistant to caspofungin and sensitive to micafungin in vivo. Resistance of RG101 to CAS in vivo was tested in an acute pulmonary aspergillosis model. Inbred DBA/2 mice were rendered neutropenic and infected with 1 × 106 RG101 conidia intratracheally. The mice were then treated with CAS, micafungin, or vehicle daily for 5 days, and lung burdens were determined using qPCR. The average fungal lung burdens in animals treated with vehicle, CAS, and MFG were 25.8, 22.6, and 5.2 ng fungal DNA per lung, respectively. Statistical analysis showed no significant differences between the vehicle-treated and CAS-treated groups, whereas MFG-treated animals showed approximately 5-fold decrease in burden, suggesting that RG101 is resistant to CAS and sensitive to MFG in vivo.
Probing the fks1-independent resistance mechanism of RG101.
Echinocandin activity is known to induce compensatory changes in cell wall composition, resulting in cell wall alterations that can partially protect the fungus from the drug (20). We therefore examined potential ultrastructural changes in the cell wall upon CAS exposure using electron microscopy (EM) to obtain insights into the mechanism of resistance. In both scanning electron microscopy (SEM) and transmission electron microscopy (TEM) studies, RG101 grown in the presence of CAS did not show any ultrastructural cell wall damage. In contrast, the echinocandin-sensitive parent, ATCC 13073, showed extensive damage, irregular cell wall shape, and leaking of cellular contents (Fig. S2). We then examined the possibility that compensatory upregulation of other cell wall components, such as chitin, was driving the resistance in RG101. Biochemical analysis of cell wall composition by mass spectrometry (MS) and chitin content evaluation by calcofluor white staining showed no significant increase of chitin levels in RG101 upon CAS exposure (Fig. S3). We concluded that a compensatory increase in the level of chitin in the cell wall was not a likely mechanism of echinocandin resistance in RG101.
Cell wall ultrastructure of RG101 in the presence of CAS. Conidia of ATCC 13073 and RG101 strains were grown in liquid YPD in the presence of CAS (4 μg/ml) for 16 h and processed for SEM and TEM imaging. Electron microscopy images showed no morphological defects in RG101 grown in the presence of CAS, whereas the CAS-sensitive parental strain ATCC 13073 showed irregular mycelia and leaking cellular contents indicating damaged cell wall. Download FIG S2, DOCX file, 0.9 MB (964.4KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Evaluation of glucan and chitin contents of RG101 cell wall (A) Biochemical analysis of cell wall glucan levels using mass spectroscopy showing no reduction in glucan content in RG101 treated with CAS. (B and C) Fluorescence study using calcofluor white showing no increase in chitin levels in RG101 treated with CAS (4 μg/ml). In summary, no cell wall-associated changes were detected in RG101 treated with CAS, indicating complete resistance to the drug. Download FIG S3, DOCX file, 0.4 MB (369.2KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The fks1-independent mechanism of resistance is mediated by CAS-induced modification of glucan synthase.
To further examine whether glucan synthase of RG101 was intrinsically resistant to inhibition by CAS, RG101 and corresponding parental strain ATCC 13073 were grown in the absence and presence of resistance-inducing levels of CAS. The active enzyme was partially purified, and the sensitivity of the enzyme to echinocandins was analyzed in kinetic inhibition assays (21). We observed that glucan synthase isolated from RG101 cultured in the absence of CAS was fully sensitive to inhibition by CAS or MFG (Fig. 3A and B). However, when glucan synthase was isolated from RG101 grown in the presence of CAS, it was 4 to 5 log orders less sensitive (as determined by the enzyme’s higher 50% inhibitory concentration [IC50] value) than that of RG101 grown in the absence of the drug (Fig. 3A). This biochemical insensitivity was not observed with the WT parental strain ATCC 13073. The drug-mediated reduced sensitivity correlated with increasing effector concentrations of drug in the growth media; i.e., RG101 cells grown in the presence of higher concentrations of CAS had higher IC50 values and thus were more resistant to CAS inhibition (Fig. 3A). Strikingly, glucan synthase isolated from RG101 cultured in the presence of CAS was also insensitive to MFG (Fig. 3B). These results strongly suggested that culturing RG101 in the presence of a CAS-induced modification(s) in glucan synthase had rendered it largely insensitive to all echinocandin class drugs.
FIG 3.

CAS induces modification of β-glucan synthase in RG101 and several clinical isolates. Sensitivity of partially purified glucan synthase to different echinocandins was examined in vitro after isolating the enzyme from RG101 grown in different drug conditions. (A) Dose-dependent insensitivity of RG101 glucan synthase to CAS induced by CAS during mycelial growth of RG101. The IC50 of glucan synthase for RG101 grown in the absence of CAS [IC50 (no CAS)] was ∼0.01 ng/ml and for RG101 grown at 4 μg/ml CAS [IC50 (4 μg/ml CAS)] was high at >10,000 ng/ml. Previous exposure to CAS in RG101 culture induced insensitivity to CAS at the enzyme level in a dose-dependent fashion. (B) Previous exposure to CAS in culture induced insensitivity to micafungin at the enzyme level [IC50 (no CAS), <0.01 ng/ml; IC50 (4 μg/ml CAS), >10,000 ng/ml], indicating potential CAS-induced cross-resistance in cells. (C to E) Enzyme profiles similar to those seen with CAS were observed in clinical isolates 117 (C), 2770 (D), and 32458 (E). IC50 values for the three clinical isolates are as follows: for isolate 117, IC50 (no CAS) = ∼0.01 ng/ml and IC50 (4 μg/ml CAS) = >10,000 ng/ml; for isolate 2770, IC50 (no CAS) = ∼0.01 ng/ml and IC50 (4 μg/ml CAS) = >10,000 ng/ml; for isolate 32458, IC50 (no CAS) = ∼10 ng/ml and IC50 (4 μg/ml CAS) = ∼5,000 ng/ml.
To assess whether clinical isolates (Table 1) would show similar enzymatic behavior upon CAS induction, cells were cultured in the absence and presence of CAS (4 μg/ml), and the glucan synthase enzymes were isolated and evaluated in the kinetic inhibition assay. Indeed, three clinical isolates—117, 2770, and 32458—yielded enzymes with CAS-induced drug insensitivity, similar to RG101 (Fig. 3C and D and E), indicating that modification of glucan synthase induced by CAS is a conserved mechanism of echinocandin resistance in A. fumigatus.
CAS induces cross-resistance to other glucan synthase inhibitors in RG101.
Since glucan synthase of RG101 grown in the presence of CAS showed enzyme insensitivity not only to CAS but also to MFG, we examined whether cross-resistance was also observed in liquid growth culture. To this end, we performed drug susceptibility testing of ATCC 13073 and RG101 with different glucan synthase inhibitors individually and in combination with an inducing level (1 μg/ml) of CAS. RG101 was sensitive to micafungin (MFG), anidulafungin (ANF), rezafungin (RZF), and ibrexafungerp (IFG) when grown in the presence of each drug alone. However, when these drugs were present in combination with 1 μg/ml of CAS, cross-resistance was observed (Fig. 4). This suggested that CAS induces cross-resistance to other glucan synthase inhibitors in RG101, paralleling the kinetic inhibition studies.
FIG 4.

CAS induces cross-resistance to other glucan synthase inhibitors in RG101. RG101 was grown for 24 h in RPMI 1640 media containing 1 μg/ml of different glucan synthase inhibitors, including micafungin (MFG), anidulafungin (ANF), rezafungin (RZF), and ibrexafungerp (IFG), in both the absence and presence of CAS (1 μg/ml). While RG101 was susceptible to these drugs in the absence of CAS, as seen by characteristic formation of rosettes, it showed complete resistance in the presence of CAS, as seen by the presence of extended mycelia.
Nature of glucan synthase modification leading to resistance in RG101.
We hypothesized that CAS induces resistance in RG101 by direct modification of glucan synthase through posttranslational modifications (PTMs). To test this hypothesis, glucan synthase was isolated from RG101 grown in the absence and presence of CAS (1 μg/ml) and subjected to enzymatic digestion and analysis by a nano-LC-MS/MS (liquid chromatography-tandem mass spectrometry) method to identify any potential PTMs. The peptides covered a significant portion (up to 71.5%) of the enzyme, including the major hot spot regions known to confer resistance. This analysis resulted in three important findings (Fig. S4). First, the A. fumigatus glucan synthase enzyme was decorated with numerous PTMs—methylation, acetylation, and phosphorylation—throughout the length of the protein. Second, no PTMs were detected in the hot spot regions of the enzyme. Third, a subset of peptides had PTMs in the induced enzyme sample that were absent in the uninduced enzyme. However, only a very small fraction (<5%) of the enzyme population was modified, making it unlikely to account for the observed phenotype. On the basis of these data, we believed it improbable that any PTM present at such low abundance would be responsible for the resistance phenotype (Fig. S4).
PTMs identified in glucan synthase. Nano-LC/MS analysis of glucan synthase of RG101 after enzyme digestion covered 71.5% of the protein and detected several PTMs. (A) A list of different PTMs detected with specific amino acid locations. (B) A diagrammatic representation of glucan synthase 16 transmembrane domains and the overall distribution of PTMs detected. Most of the PTMs were studded in the cytosolic segments of the protein. No PTMs were detected in the two hot spot regions of the enzyme. (C) List of 8 peptides showing differences in PTMs between CAS-induced and uninduced enzymes. However, a very low fraction of the population of the enzyme was modified, which did not account for the observed phenotype in RG101. Download FIG S4, DOCX file, 0.2 MB (243.6KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
It was previously reported for Candida glabrata that a change in sphingolipid content could alter whole-cell susceptibility to CAS and MFG (18). Thus, a comprehensive lipidomics analysis was performed to compare lipid profiles of glucan synthase containing fractions derived from RG101 grown in the absence and presence of CAS (see Table S1 in the supplemental material). In samples derived from CAS-treated cells, two lipid molecules—dihydrosphingosine (DhSph) and phytosphingosine (PhSph)—were found in increased abundance (2-fold and 10-fold, respectively) compared to untreated cells (Fig. 5A and B). These lipid species also showed significantly increased abundance in enzyme preparations of clinical isolates (117, 2770, and 32458) treated with CAS (Fig. S5).
FIG 5.

Certain lipids affect the sensitivity of β-glucan synthase to CAS. (A and B) Lipidomics analysis of enriched glucan synthase from RG101 grown in the absence and presence (1 μg/ml) of CAS showed (A) >2-fold-increased abundance of dihydrosphingosine (DhSph) and (B) 10-fold-increased abundance of phytosphingosine (PhytoSph) in CAS-exposed cells. (C) Addition of DhSph and PhytoSph at a concentration of 150 μg/ml for 1 h to enriched glucan synthase extract in the enzyme assay made the enzyme insensitive to CAS [IC50 (no lipid) = ∼0.01 ng/ml; IC50 (150 μg/ml DhSph) = 0.1 ng/ml; IC50 (150 μg/ml PhytoSph) = ∼100 ng/ml], whereas addition of other lipids such as phytoceramide did not alter the enzyme property [IC50 (150 μg/ml C2 PhytoCer) = ∼0.01 ng/ml].
Abundance levels of DhSph and PhSph lipid species in clinical isolates: Glucan synthase of three clinical isolates listed in Table 1—2770, 117, and 32458—was tested for abundance levels of DhSph and PhSph under uninduced and induced conditions. Consistent with data from RG101, these clinical isolates also showed higher levels of DhSph (A) and PhSph (B) in CAS-induced enzymes than in preparations of uninduced enzymes. This indicated that CAS induces lipid modifications in clinical isolates, as observed in RG101. Download FIG S5, DOCX file, 0.04 MB (37.8KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Relative abundances of different lipid species in the microenvironment of glucan synthase. Three lipid subtypes (in red)—dihydrospingosine (DhSph), phytosphingosine (PhSph), and phytoceramide (C24)—were present in CAS-induced glucan synthase of RG101 at levels 3-, 10-, and 3-fold higher, respectively, than those seen with the uninduced preparation. DhSph and PhSph were used for further analysis. Download Table S1, DOCX file, 0.02 MB (17.5KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To further test whether DhSph and PhSph contribute to the insensitivity of the enzyme to CAS, these lipids were added exogenously to the kinetic inhibition assay reaction mixture containing the partially purified glucan synthase in detergent. Interestingly, each lipid dramatically altered the sensitivity profile of glucan synthase derived from RG101 grown in the absence of CAS. DhSph and PhSph increased the CAS IC50 value of the sensitive enzyme by at least 1 and 4 log orders, respectively (Fig. 5C). These effects were lipid dose dependent (data not shown). The observed effects were specific to DhSph and PhSph, as other lipid intermediates such as phytoceramide (C20) did not change the enzymatic sensitivity (Fig. 5C). This indicated that the composition of the lipid microenvironment surrounding the glucan synthase most likely altered its conformation in a way that is important for CAS enzyme inhibition.
CAS induces production of reactive oxygen species in A. fumigatus.
As described previously, the induction of resistance in RG101 was specific to CAS (Fig. 1), and the cells showed cross-resistance to other echinocandins in the presence of CAS. Thus, we investigated several potential factors that might differentiate CAS from other echinocandins and observed that only CAS strongly induced the production of reactive oxygen species (ROS) in cells (Fig. 6). A 2′,7′-dichlorofluorescin diacetate (DCFDA)-based fluorescence assay was used to measure ROS levels in A. fumigatus cells. After 1 h of drug exposure, CAS induced the highest levels of ROS in cells compared to other glucan synthase inhibitors (Fig. 6A). The levels of ROS induced by the drugs were very similar between the ATCC 13073 and RG101 strains. There was a dose-dependent increase in ROS production induced by CAS (Fig. 6B). Consistent with the induction of ROS, RNA-seq analysis of RG101 harvested in the presence and absence of CAS induction revealed that the oxidation-reduction pathway was one of the most highly differentially expressed pathways, consistent with the production of ROS induced by CAS (Fig. S6). Next, the effect of thiourea, a ROS scavenger, on ROS levels and CAS resistance was tested. As expected, treatment with 15 mM thiourea reduced the levels of ROS induced by CAS by 4-fold (Fig. 6B). Addition of the mitochondrial respiration inhibitor antimycin A (0.5 μg/ml) to the assay decreased ROS production induced by CAS by over 2-fold, indicating that CAS-induced ROS is generated in mitochondria (Fig. 6C). Importantly, thiourea reversed the CAS-induced breakthrough resistance phenotype of RG101 but had no effect on CAS resistance in the fks1-S679P strain (Fig. 6D). These results suggest that CAS indirectly induces the production of mitochondrial ROS, which in turn leads to downstream modification of glucan synthase and echinocandin resistance via changes in the lipid microenvironment.
FIG 6.

CAS induces ROS production. (A) Levels of ROS induced by different echinocandins as measured by a fluorescence assay using DCFDA dye. After 1 h of exposure of cells to echinocandins, CAS induced the highest levels of ROS production in the A. fumigatus RG101 and ATCC 13073 strains in comparison to other echinocandins. (B) Dose-dependent increase in ROS production induced by CAS. (C) Addition of thiourea, a ROS scavenger, at 15 mM reduced CAS-induced ROS production in RG101. (D) Addition of antimycin A, a mitochondrial respiration inhibitor, at 0.5 μg/ml reduced CAS-induced ROS production in RG101, indicating mitochondrion-associated ROS production induced by CAS. (E) Testing of susceptibility of ATCC 13073, RG101, and S679P strains with CAS (1 μg/ml) and thiourea (15 mM) revealed reversion of the resistance phenotype of RG101 but no change of the phenotype of ATCC 13073 and S679P.
Differentially expressed genes and gene ontology data from comparisons performed under CAS-induced and uninduced conditions in RG101. RG101 was grown in the absence and presence of CAS (1 μg/ml) for 16 h, and cells were isolated and processed for RNA-seq. Genes involved in the oxidation-reduction pathway were highly differentially expressed, consistent with the induction of ROS by CAS. Download FIG S6, DOCX file, 0.6 MB (661.5KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
In this study, a novel mechanism of resistance to echinocandins in Aspergillus fumigatus was investigated. Although azoles represent the primary treatment option for Aspergillus infections, therapeutic use of echinocandins is rising rapidly due to increasing emergence of azole resistance (22). The echinocandin caspofungin was first approved by the FDA in 2002 for use against Aspergillus infections refractory to conventional antifungal therapy (23). As the number of patients with Aspergillus infections being treated with echinocandins has expanded, reports of clinical failure of echinocandin therapy are emerging (6, 9). In Candida species, echinocandin resistance is tightly associated with FKS gene mutations that result in amino acid substitutions in hot spot regions of glucan synthase enzyme (21, 24–26). These mutations decrease the sensitivity of the enzyme to echinocandins by up to 3,000-fold (21, 25, 26). Additionally, certain cell wall stress adaptation response pathways become activated upon echinocandin exposure, resulting in elevated MIC values in vitro (27). However, these alterations are typically associated with drug tolerance and not with clinical failure. Drug tolerance represents an intermediate stage in the evolution of clinical resistance involving acquisition of characteristic FKS gene mutations. Tolerance mechanisms depend on fungal stress tolerance pathways, including cell wall integrity, protein kinase C, Ca2+- calcineurin/Crz1, and high-osmolarity glycerol (HOG) pathways (5). In one study, a systems biology approach was employed to infer regulatory networks and interactions during adaptation to CAS-induced stress in A. fumigatus (28). They confirmed the presence of cross talk of mitogen-activated protein (MAP) kinases corresponding to cell wall integrity and HOG signaling pathways upon CAS exposure. Data also suggested that intracellular transport was affected upon CAS exposure causing additional osmotic stress but that high concentrations of CAS reduced this osmotic stress, thus decreasing its toxicity. In this report, we describe a novel, stress-induced mechanism of echinocandin resistance in Aspergillus that is associated with clinical failure but independent of genetic changes leading to amino acid substitutions in the hot spot region of glucan synthase. This novel mechanism is mediated by an increase in ROS production induced prominently by CAS, resulting in a downstream modification of the lipid microenvironment, which likely alters the interaction of echinocandin drugs with glucan synthase.
Alternative mechanism of echinocandin resistance.
Echinocandin resistance in Aspergillus is a relatively new phenomenon that involves modulation of the drug-target interaction mediated via two separate mechanisms of action (MOA). The first MOA is highly conserved and occurs through mutations in the hot spot regions of glucan synthase. It has been shown that an engineered S679P amino acid substitution in glucan synthase in a laboratory-generated A. fumigatus strain rendered it highly resistant to all echinocandins (7). More recently, our laboratory reported an echinocandin-resistant clinical isolate of A. fumigatus carrying the point mutation F675S in the hot spot region of the fks1 gene (6). For the second MOA, echinocandin resistance in A. fumigatus has also been reported for high-MEC isolates from patients failing therapy in which there was no apparent genetic modification of fks1 (9). In this case, upregulation of fks1 was presumed to be a causal mechanism of resistance, comparable to what has been observed for gain-of-function mutations resulting in azole resistance in Candida albicans (29). In that report, clinical isolates from patients with chronic pulmonary aspergillosis who failed echinocandin therapy were analyzed and found to have elevated MECs with no mutations in the coding or noncoding regions of fks1. Furthermore, the strains did not show significant upregulation of fks1, pointing to a novel resistance mechanism. Previously, it was reported that a highly drug-resistant laboratory-generated strain, RG101, showed high-MEC CAS values with no apparent modification of the fks1 gene (19). That strain displayed paradoxical drug sensitivity in which growth was inhibited at low levels of CAS but showed frank resistance at drug levels above 0.5 μg/ml after 24 h. That phenotype was recapitulated in the present study, and RG101 was shown to be fully resistant at all drug levels after 30 h of growth. The strain also appeared to be resistant in vivo. However, it retained complete sensitivity to MFG at 24 and 48 h and in vivo. Transcriptional analysis of RG101 grown in the presence of CAS at breakthrough levels of drug revealed that the cells upregulated a variety of stress and tolerance pathways associated with cell wall biosynthesis and remodeling, as had been observed previously in Candida albicans (19). However, these compensatory responses were insufficient to account for prominent drug resistance. This conclusion is consistent with a study by Loiko and Wagener, who analyzed the paradoxical behavior of Aspergillus under conditions of CAS treatment and suggested that the paradoxical drug recovery reflected a restoration of enzyme function (30).
To further explore a novel mechanism directly impacting glucan synthase, we examined the enzyme isolated from untreated and CAS-treated RG101 cells to explore a potential modification(s) of the drug target. Indeed, we observed that the glucan synthase from RG101 grown in the presence of CAS was altered, rendering the enzyme insensitive to the drug (4-log order shift in sensitivity). Furthermore, the modified enzyme was subsequently found to be insensitive to MFG and other glucan synthase inhibitors. Consistently, CAS induced cross-resistance to all glucan synthase inhibitors in whole-cell growth inhibition assays. This indicated that CAS was inducing some form of enzyme modification that rendered cells resistant. The possibility of PTMs in the enzyme causing insensitivity to CAS was tested using LC-MS/MS. Several PTMs were detected in glucan synthase of RG101, but no modification was judged to be stable and strong enough to drive such a steady resistance phenotype. The role of the observed PTMs in enzyme function and/or regulation is being independently evaluated.
Further, potential lipid-associated modifications in the microenvironment of the enzyme were examined, and we found two types of lipids—dihydrosphingosine and phytosphingosine—that were highly abundant in product-entrapped glucan synthase after CAS induction compared to the results seen with an uninduced-enzyme preparation. Exogenous addition of these lipids to the sensitive glucan synthase of RG101 was found to have made the enzyme insensitive to CAS. It is noteworthy that the clinical isolates with resistance phenotypes showed a similar lipid profile. This suggests that changes in the lipid bilayer can alter the interaction of echinocandins with glucan synthase. The role of the lipid microenvironment in the susceptibility of fungi to echinocandins has not been studied extensively. However, a study showed that for some laboratory and clinical isolates of C. glabrata, defects in sphingolipid biosynthesis led to a mixed phenotype in which strains were resistant to CAS and hypersensitive to MFG (18, 31). The authors of that study hypothesized that changes in sphingolipid composition in the cell membrane weakened the interaction between glucan synthase and CAS and strengthened the interaction between the enzyme and MFG. A recent study of C. albicans showed that deletion mutants of genes encoding enzymes important for biosynthesis of very-long-chain fatty acids such as sphingolipids resulted in resistance to fluconazole, an azole antifungal agent targeting the ergosterol biosynthesis pathway (32). Our findings indicate that CAS-induced changes in sphingolipid composition in the microenvironment of glucan synthase alter the drug-binding affinity of echinocandins. Therefore, this apparent nexus of two drug classes with very different MOAs—azoles and echinocandins—suggests that sphingolipid biosynthesis may play an important role in fungal cell stress tolerance responses.
Oxidative stress response as a mediator of echinocandin resistance.
Studies have shown that the polyene drug amphotericin B (AmB) kills fungal cells by forming membrane pores but also acts as an oxidizing agent (33, 34), leading to the formation of ROS (35). The efficacy of the drug has been partially attributed to oxidative damage in cells through lipid peroxidation (36). Recently, a study in A. fumigatus showed that fungal cell membrane-targeting drugs such as itraconazole, terbinafine, and AmB induce mitochondrial ROS production, leading to lipid peroxidation and inhibition of fungal growth (37). Although all echinocandins share the same mechanism of action involving inhibition of glucan synthase, it is well documented that CAS induces in vitro phenotypic behavior (leading to high MICs) that is independent of genetic changes in FKS. For example, certain Candida strains grown in the presence of CAS show various degrees of paradoxical growth in which isolates are able to grow better at higher drug concentrations (38, 39). This effect is partially attributed to compensatory chitin deposition (40, 41), although this model is largely insufficient for Aspergillus (30). Overall, the biological mechanism behind these observations is not well understood. In this study, we investigated why CAS but not other echinocandins was able to induce modification of glucan synthase from RG101. We observed that CAS prominently induced ROS production, while the other echinocandins induced ROS weakly or not at all. The CAS-induced ROS was of mitochondrial origin, as it was blocked by antimycin A. Importantly, we showed a link between ROS production and development of resistance, as addition of ROS scavengers such as thiourea reduced ROS levels in cells and reversed the resistance phenotype of RG101. A few studies have linked increased ROS content to protein and lipid oxidation in cells (42). In one study in Saccharomyces cerevisiae, mitochondrial-respiration-deficient mutants were hypersensitive to CAS (42, 43). The most sensitive mutants were those with impaired mitochondrial biosynthesis of the phospholipids phosphatidylethanolamine and cardiolipin (43–46). Another study in S. cerevisiae showed that the TORC2/Ypk1 pathway functions as a ROS-sensing module and plays a central role in sphingolipid biosynthesis and homeostasis (47). How ROS production, induced by CAS, leads to altered lipid composition with respect to glucan synthase resulting in resistance is yet to be determined.
A new model for stress-induced drug resistance.
Our model for non-fks1 mutation-mediated echinocandin resistance in A. fumigatus is shown in Fig. 7. We propose that CAS induces production of ROS in mitochondria as an off-target effect. The other echinocandins showed poor inductive effect, most likely due to poor cell penetration. We believe that one of the downstream effects of increased ROS production in cells is altered regulation of lipid biosynthesis genes. This leads in turn to modified plasma membrane lipid composition in the microenvironment of glucan synthase, likely causing a conformational change in the enzyme, perhaps in a manner that prevents echinocandins from binding to the target and leads to resistance. Although our data demonstrate the CAS-specific insensitivity of the enzyme upon addition of dihydrosphingosine and phytosphingosine, we hypothesize that insensitivity to all echinocandins may occur with much higher concentrations of these modified lipids. Clinical isolates have phenotypes that highly similar but not identical to those seen with RG101, since they are also MFG resistant. However, these isolates were retrieved from patients with chronic pulmonary aspergillosis who had undergone prolonged treatment with MFG and other antifungal therapy. Such a chronic exposure to drugs could have triggered similar pathways such as ROS production leading to altered lipid composition. Alternatively, prolonged exposure to micafungin may have lowered the threshold for activation of sphingolipid biosynthesis pathway genes in these strains, leading to altered lipid composition in the microenvironment of the enzyme.
FIG 7.

Working model showing the non-fks1 mutation-mediated mechanism of resistance in RG101. (A) Addition of CAS during growth of RG101 altered the properties of glucan synthase, rendering it resistant to CAS, at both the cellular and enzyme levels. (B) Working model for CAS-induced resistance in RG101. CAS induces ROS production in cells. We hypothesize that high ROS levels alter the lipid composition in the microenvironment of glucan synthase, causing a conformational change in glucan synthase and leading to CAS resistance.
In conclusion, this study identified a novel, fks1-independent mechanism of echinocandin resistance in A. fumigatus that uses CAS-induced ROS-mediated changes in the lipid composition of the microenvironment of glucan synthase to alter the drug-target interaction. Other echinocandins may also induce off-target mitochondrial stress (although less robustly than CAS), resulting in lower ROS production. This is yet to be assessed with large collections of clinical isolates. Use of echinocandins to treat patients with Aspergillus infection is expanding, and we predict that this new mechanism of resistance will be an important clinical factor.
MATERIALS AND METHODS
Strains and isolates.
RG101 was derived from parental strain ATCC 13073 (U.S. clinical isolate collected from a lung in 2005) by regenerating spheroplasts in minimal medium containing a high (10 μg/ml) concentration of CAS (19). A. fumigatus clinical isolates 117, 2770, 32458, 32315, and 68284B were obtained from sputum samples of patients with chronic pulmonary aspergillosis at Wythenshawe Hospital, University of Manchester (the samples were provided by David W. Denning). Strain S679P (previously reported as strain S678P) (7) was engineered in the Perlin laboratory to harbor an amino acid substitution in the hot spot region (amino acid 679) making it resistant to all echinocandins.
Antifungal susceptibility testing.
Minimum effective concentrations (MECs) for A. fumigatus strains were determined according to CLSI document M38-A2 (48) and defined as the lowest drug concentration that led to the growth of small, rounded, compact hyphal forms compared to the hyphal growth seen in the drug-free growth control wells. The following drugs (supplier) were tested: caspofungin (Merck & Co., Inc.), micafungin (Astellas Pharma US, Inc.), anidulafungin (Pfizer Inc.), itraconazole (Sigma-Aldrich), voriconazole (Pfizer Inc.), and amphotericin B (Sigma-Aldrich). In addition, rezafungin and ibrexafungerp were obtained from Cidara Therapeutics and Scynexis Inc., respectively.
In vivo fungal burden analysis.
Resistance of RG101 to CAS in vivo was tested in an acute pulmonary aspergillosis model. Thirty inbred DBA/2 mice were rendered neutropenic and infected with 1 × 106 RG101 conidia intratracheally. The mice were then treated with CAS (5 mg/kg of body weight), MFG (5 mg/kg), or vehicle daily for 5 days, and lung burdens were determined using quantitative PCR (qPCR). Details of the experiment are provided in Text S1 in the supplemental material.
Supplemental Materials and Methods: in vivo fungal burden analysis, protein analysis by LC-MS/MS, and electron microscopy. Download Text S1, DOCX file, 0.03 MB (24.8KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Electron microscopy.
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) methods were followed as described previously (49) and in Text S1.
Cell wall carbohydrate analysis.
Strains ATCC 13073 and RG101 were grown in the absence and presence of CAS (4 μg/ml) in 6-well microtiter plates containing yeast extract-peptone-dextrose (YPD) broth for 22 h at 37°C. The cells were then lysed using a French press. The broken cell wall sample was processed for chemical analysis using mass spectrometry at the Complex Carbohydrate Research Center (CCRC) at the University of Georgia, Athens, GA.
Glucan synthase isolation and assay.
ATCC 13073 or RG101 was grown in liquid YPD medium at 37°C for 16 h. Partially purified glucan synthase was isolated by the product entrapment method, as previously described (21). Sensitivity to echinocandin drugs was measured by glucan synthase assay, using a 96-well multiscreen high-throughput screen filtration system (Millipore Corporation, Bedford, MA) in a final volume of 100 μl. Inhibition of the enzyme by the drug was analyzed after incubation of the enzyme with a radioactive substrate over a broad range of concentrations (0 to 10,000 ng/ml) of echinocandins for 1 h at room temperature. Radioactivity from the enzyme product, β-(1,3)-d-glucan, was measured. Inhibition profiles were determined using a sigmoidal response curve fitting algorithm with GraphPad Prism software (v 7.04).
To study the effect of lipids on the enzyme, dihydrosphingosine, phytosphingosine, or phytoceramide C2 (Sigma-Aldrich D6783, P2795, and C8105, respectively) was added directly to the assay mixture and coincubated with an echinocandin for 1 h at room temperature.
Protein analysis by LC-MS/MS.
To detect possible posttranslational modifications in glucan synthase, we assayed the enzyme from strain RG101 grown in the absence or presence (1 μg/ml) of CAS. To remove detergent from partially purified glucan synthase, the enzyme sample was spun at 39,000 rpm for 1 h in an ultracentrifuge. The pellets were resuspended in enzyme buffer. Protein samples were separated on SDS-PAGE, and bands of >180 kDa were collected and prepared for mass spectrometry (Text S1).
Lipid analysis.
Approximately 10 μg/ml of partially purified glucan synthase of RG101 and clinical isolates was analyzed for lipid content by LC-MS/MS (50) at the Lipidomics Shared Resource of the Medical University of South Carolina (http://www.hollingscancercenter.org/research/shared-resources/lipidomics/index.html).
Measurement of ROS production.
ROS production in mycelia was measured using a protocol previously described by Blatzer et al. (51) with a few changes. Suspensions of 104 spores/ml of ATCC 13073 and RG101 were grown in static culture in complete supplement mixture (CSM) medium for 16 h in a 96-well microplate at 37°C. After incubation, the mycelia were washed once with CSM, 10 μM 2′,7′–dichlorofluorescin diacetate (DCFDA) dye was added, and cells were incubated in the dark at 37°C for 30 min. Cells were then washed three times with phosphate-buffered saline (PBS), and a final concentration of 1 μg/ml of each echinocandin in CSM medium was added to different wells. Fluorescence intensity (excitation at 485 nm and emission at 530 nm) was measured for 1 h in a microplate reader (Tecan Infinite M200 Pro). Mycelia stained with DCFDA without any drug were used as a control.
RNA-seq.
RG101 conidia were grown for 16 h in YPD broth in the absence and presence of 1 μg/ml CAS at 37°C. Mycelia were washed twice with PBS. RNA was isolated using a Qiagen RNeasy plant minikit (catalog no. 74904). RNA-seq analysis was performed by LC Sciences, Houston, TX. StringTie was used to estimate expression levels for mRNAs by calculating fragments per kilobase per million (FPKM) as follows: FPKM = total number of exon fragments/number of mapped reads (millions) × exon length (in kilobases) (52). The differentially expressed mRNAs were selected with log2 (fold change) values of more than 1 or less than −1 and with a parametric F-test comparing nested linear models (P value, <0.05) by the use of R package Ballgown (53).
ACKNOWLEDGMENTS
We thank Milena Kordalewska and Elena Shopova for their expert assistance in experimental design and analysis of the results and Silke Steinbach for her technical expertise. We thank Erika Shor for her help with manuscript editing. We also thank Annie-Hee Lee, Nathaly Cabrera, Irina Kolesnikova, Yevgeniy Senin, Deborah Evans, and George Rasic for their great help with the animal experiments.
This work was supported by grants from the U.S. National Institutes of Health (AI109025) and Astellas Pharma (Reference Center for Molecular Evaluation of Drug Resistance to Echinocandin and Triazole Antifungal Drugs) to D.S.P. and by the Deutsche Forschungsgemeinschaft Collaborative Research Center TR124 FungiNet (project A1 and Z2) to A.A.B. and O.K.
D.S.P. receives funding from the U.S. National Institutes of Health and from contracts from Astellas, Scynexis, Cidara, and Amplyx. He serves on advisory boards for Astellas, Cidara, Amplyx, Scynexis, Matinas, and N8 Pharmaceuticals. In addition, D.S.P. has an issued U.S. patent concerning echinocandin resistance. D.W.D. and family hold founder shares in F2G Ltd., a University of Manchester spin-out antifungal discovery company. He acts or has recently acted as a consultant to Scynexis, Cidara, Quintiles, Pulmatrix, Pulmocide, Zambon, iCo Therapeutics, Roivant, and Fujifilm. In the last 3 years, he has been paid for talks on behalf of Astellas, Dynamiker, Gilead, Merck, Mylan, and Pfizer. He is a longstanding member of the Infectious Disease Society of America Aspergillosis Guidelines group, the European Society for Clinical Microbiology and Infectious Diseases Aspergillosis Guidelines group, and the British Society for Medical Mycology Standards of Care committee. The rest of us have no potential conflicts of interest.
We alone are responsible for the content and writing of the paper.
Footnotes
Citation Satish S, Jiménez-Ortigosa C, Zhao Y, Lee MH, Dolgov E, Krüger T, Park S, Denning DW, Kniemeyer O, Brakhage AA, Perlin DS. 2019. Stress-induced changes in the lipid microenvironment of β-(1,3)-d-glucan synthase cause clinically important echinocandin resistance in Aspergillus fumigatus. mBio 10:e00779-19. https://doi.org/10.1128/mBio.00779-19.
REFERENCES
- 1.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 2.Diekema D, Arbefeville S, Boyken L, Kroeger J, Pfaller M. 2012. The changing epidemiology of healthcare-associated candidemia over three decades. Diagn Microbiol Infect Dis 73:45–48. doi: 10.1016/j.diagmicrobio.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Jenks JD, Hoenigl M. 2018. Treatment of aspergillosis. J Fungi (Basel) 4:98. doi: 10.3390/jof4030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perlin DS. 2011. Current perspectives on echinocandin class drugs. Future Microbiol 6:441–457. doi: 10.2217/fmb.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlin DS. 2015. Echinocandin resistance in Candida. Clin Infect Dis 61(Suppl 6):S612–S617. doi: 10.1093/cid/civ791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jimenez-Ortigosa C, Moore C, Denning DW, Perlin DS. 2017. Emergence of echinocandin resistance due to a point mutation in the fks1 gene of Aspergillus fumigatus in a patient with chronic pulmonary aspergillosis. Antimicrob Agents Chemother 61:e01277-17. doi: 10.1128/AAC.01277-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rocha EM, Garcia-Effron G, Park S, Perlin DS. 2007. A Ser678Pro substitution in Fks1p confers resistance to echinocandin drugs in Aspergillus fumigatus. Antimicrob Agents Chemother 51:4174–4176. doi: 10.1128/AAC.00917-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Satish S, Jimenez-Ortigosa C, Perlin DS. 2018. Understanding the mechanistic basis of echinocandin resistance in Aspergillus fumigatus. Abstr Adv Aspergillosis, Lisbon, Portugal, 1–3 February 2018. [Google Scholar]
- 9.Arendrup MC, Garcia-Effron G, Buzina W, Mortensen KL, Reiter N, Lundin C, Jensen HE, Lass-Flörl C, Perlin DS, Bruun B. 2009. Breakthrough Aspergillus fumigatus and Candida albicans double infection during caspofungin treatment: laboratory characteristics and implication for susceptibility testing. Antimicrob Agents Chemother 53:1185–1193. doi: 10.1128/AAC.01292-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andaluz E, Guillen A, Larriba G. 1986. Preliminary evidence for a glucan acceptor in the yeast Candida albicans. Biochem J 240:495–502. doi: 10.1042/bj2400495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orlean PA. 1982. (1,3)-Beta-d-glucan synthase from budding and filamentous cultures of the dimorphic fungus Candida albicans. Eur J Biochem 127:397–403. doi: 10.1111/j.1432-1033.1982.tb06885.x. [DOI] [PubMed] [Google Scholar]
- 12.Ishiguro J, Saitou A, Duran A, Ribas JC. 1997. cps1+, a Schizosaccharomyces pombe gene homolog of Saccharomyces cerevisiae FKS genes whose mutation confers hypersensitivity to cyclosporin A and papulacandin B. J Bacteriol 179:7653–7662. doi: 10.1128/jb.179.24.7653-7662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly R, Register E, Hsu MJ, Kurtz M, Nielsen J. 1996. Isolation of a gene involved in 1,3-beta-glucan synthesis in Aspergillus nidulans and purification of the corresponding protein. J Bacteriol 178:4381–4391. doi: 10.1128/jb.178.15.4381-4391.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazur P, Morin N, Baginsky W, el-Sherbeini M, Clemas JA, Nielsen JB, Foor F. 1995. Differential expression and function of two homologous subunits of yeast 1,3-beta-d-glucan synthase. Mol Cell Biol 15:5671–5681. doi: 10.1128/MCB.15.10.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mio T, Adachi-Shimizu M, Tachibana Y, Tabuchi H, Inoue SB, Yabe T, Yamada-Okabe T, Arisawa M, Watanabe T, Yamada-Okabe H. 1997. Cloning of the Candida albicans homolog of Saccharomyces cerevisiae GSC1/FKS1 and its involvement in beta-1,3-glucan synthesis. J Bacteriol 179:4096–4105. doi: 10.1128/jb.179.13.4096-4105.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson JR, Douglas CM, Li W, Jue CK, Pramanik B, Yuan X, Rude TH, Toffaletti DL, Perfect JR, Kurtz M. 1999. A glucan synthase FKS1 homolog in cryptococcus neoformans is single copy and encodes an essential function. J Bacteriol 181:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson ME, Edlind TD. 2012. Topological and mutational analysis of Saccharomyces cerevisiae Fks1. Eukaryot Cell 11:952–960. doi: 10.1128/EC.00082-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Healey KR, Katiyar SK, Raj S, Edlind TD. 2012. CRS-MIS in Candida glabrata: sphingolipids modulate echinocandin-Fks interaction. Mol Microbiol 86:303–313. doi: 10.1111/j.1365-2958.2012.08194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardiner RE, Souteropoulos P, Park S, Perlin DS. 2005. Characterization of Aspergillus fumigatus mutants with reduced susceptibility to caspofungin. Med Mycol 43(Suppl 1):S299–S305. [DOI] [PubMed] [Google Scholar]
- 20.Walker LA, Gow NA, Munro CA. 2013. Elevated chitin content reduces the susceptibility of Candida species to caspofungin. Antimicrob Agents Chemother 57:146–154. doi: 10.1128/AAC.01486-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park S, Kelly R, Kahn JN, Robles J, Hsu MJ, Register E, Li W, Vyas V, Fan H, Abruzzo G, Flattery A, Gill C, Chrebet G, Parent SA, Kurtz M, Teppler H, Douglas CM, Perlin DS. 2005. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother 49:3264–3273. doi: 10.1128/AAC.49.8.3264-3273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meis JF, Chowdhary A, Rhodes JL, Fisher MC, Verweij PE. 5 December 2016, posting date. Clinical implications of globally emerging azole resistance in Aspergillus fumigatus. Philos Trans R Soc Lond B Biol Sci doi: 10.1098/rstb.2015.0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson MD, Perfect JR. 2003. Caspofungin: first approved agent in a new class of antifungals. Expert Opin Pharmacother 4:807–823. doi: 10.1517/14656566.4.5.807. [DOI] [PubMed] [Google Scholar]
- 24.Douglas CM, Marrinan JA, Li W, Kurtz MB. 1994. A Saccharomyces cerevisiae mutant with echinocandin-resistant 1,3-beta-d-glucan synthase. J Bacteriol 176:5686–5696. doi: 10.1128/jb.176.18.5686-5696.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Effron G, Lee S, Park S, Cleary JD, Perlin DS. 2009. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-beta-d-glucan synthase: implication for the existing susceptibility breakpoint. Antimicrob Agents Chemother 53:3690–3699. doi: 10.1128/AAC.00443-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Effron G, Park S, Perlin DS. 2009. Correlating echinocandin MIC and kinetic inhibition of fks1 mutant glucan synthases for Candida albicans: implications for interpretive breakpoints. Antimicrob Agents Chemother 53:112–122. doi: 10.1128/AAC.01162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfaller MA, Boyken L, Hollis RJ, Kroeger J, Messer SA, Tendolkar S, Diekema DJ. 2008. In vitro susceptibility of invasive isolates of Candida spp. to anidulafungin, caspofungin, and micafungin: six years of global surveillance. J Clin Microbiol 46:150–156. doi: 10.1128/JCM.01901-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altwasser R, Baldin C, Weber J, Guthke R, Kniemeyer O, Brakhage AA, Linde J, Valiante V. 2015. Network modeling reveals cross talk of MAP kinases during adaptation to caspofungin stress in Aspergillus fumigatus. PLoS One 10:e0136932. doi: 10.1371/journal.pone.0136932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhauser J, Rogers PD. 2008. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot Cell 7:1180–1190. doi: 10.1128/EC.00103-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loiko V, Wagener J. 24 January 2017, posting date. The paradoxical effect of echinocandins in Aspergillus fumigatus relies on recovery of the beta-1,3-glucan synthase Fks1. Antimicrob Agents Chemother doi: 10.1128/AAC.01690-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Healey KR, Katiyar SK, Castanheira M, Pfaller MA, Edlind TD. 2011. Candida glabrata mutants demonstrating paradoxical reduced caspofungin susceptibility but increased micafungin susceptibility. Antimicrob Agents Chemother 55:3947–3949. doi: 10.1128/AAC.00044-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao J, Wang H, Li Z, Wong AH-H, Wang Y-Z, Guo Y, Lin X, Zeng G, Liu H, Wang Y, Wang J. 2018. Candida albicans gains azole resistance by altering sphingolipid composition. Nat Commun 9:4495. doi: 10.1038/s41467-018-06944-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanden Bossche H, Warnock DW, Dupont B, Kerridge D, Sen Gupta S, Improvisi L, Marichal P, Odds FC, Provost F, Ronin O. 1994. Mechanisms and clinical impact of antifungal drug resistance. J Med Vet Mycol 32(Suppl 1):189–202. doi: 10.1080/02681219480000821. [DOI] [PubMed] [Google Scholar]
- 34.Brajtburg J, Powderly WG, Kobayashi GS, Medoff G. 1990. Amphotericin B: current understanding of mechanisms of action. Antimicrob Agents Chemother 34:183–188. doi: 10.1128/AAC.34.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamy-Freund MT, Ferreira VF, Schreier S. 1985. Mechanism of inactivation of the polyene antibiotic amphotericin B. Evidence for radical formation in the process of autooxidation. J Antibiot (Tokyo) 38:753–757. doi: 10.7164/antibiotics.38.753. [DOI] [PubMed] [Google Scholar]
- 36.Moore CB, Sayers N, Mosquera J, Slaven J, Denning DW. 2000. Antifungal drug resistance in Aspergillus. J Infect 41:203–220. doi: 10.1053/jinf.2000.0747. [DOI] [PubMed] [Google Scholar]
- 37.Shekhova E, Kniemeyer O, Brakhage AA. 2017. Induction of mitochondrial reactive oxygen species production by itraconazole, terbinafine, and amphotericin B as a mode of action against Aspergillus fumigatus. Antimicrob Agents Chemother 61:e00978-17. doi: 10.1128/AAC.00978-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stevens DA, Espiritu M, Parmar R. 2004. Paradoxical effect of caspofungin: reduced activity against Candida albicans at high drug concentrations. Antimicrob Agents Chemother 48:3407–3411. doi: 10.1128/AAC.48.9.3407-3411.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagener J, Loiko V. 28 December 2017, posting date. Recent insights into the paradoxical effect of echinocandins. J Fungi (Basel) doi: 10.3390/jof4010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fortwendel JR, Juvvadi PR, Perfect BZ, Rogg LE, Perfect JR, Steinbach WJ. 2010. Transcriptional regulation of chitin synthases by calcineurin controls paradoxical growth of Aspergillus fumigatus in response to caspofungin. Antimicrob Agents Chemother 54:1555–1563. doi: 10.1128/AAC.00854-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stevens DA, Ichinomiya M, Koshi Y, Horiuchi H. 2006. Escape of Candida from caspofungin inhibition at concentrations above the MIC (paradoxical effect) accomplished by increased cell wall chitin; evidence for beta-1,6-glucan synthesis inhibition by caspofungin. Antimicrob Agents Chemother 50:3160–3161. doi: 10.1128/AAC.00563-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shingu-Vazquez M, Traven A. 2011. Mitochondria and fungal pathogenesis: drug tolerance, virulence, and potential for antifungal therapy. Eukaryot Cell 10:1376–1383. doi: 10.1128/EC.05184-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dagley MJ, Gentle IE, Beilharz TH, Pettolino FA, Djordjevic JT, Lo TL, Uwamahoro N, Rupasinghe T, Tull DL, McConville M, Beaurepaire C, Nantel A, Lithgow T, Mitchell AP, Traven A. 2011. Cell wall integrity is linked to mitochondria and phospholipid homeostasis in Candida albicans through the activity of the post-transcriptional regulator Ccr4-Pop2. Mol Microbiol 79:968–989. doi: 10.1111/j.1365-2958.2010.07503.x. [DOI] [PubMed] [Google Scholar]
- 44.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. 2009. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, Brugger B, Westermann B, Langer T. 2009. The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J Cell Biol 184:583–596. doi: 10.1083/jcb.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tamura Y, Endo T, Iijima M, Sesaki H. 2009. Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J Cell Biol 185:1029–1045. doi: 10.1083/jcb.200812018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niles BJ, Joslin AC, Fresques T, Powers T. 2014. TOR complex 2-Ypk1 signaling maintains sphingolipid homeostasis by sensing and regulating ROS accumulation. Cell Rep 6:541–552. doi: 10.1016/j.celrep.2013.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.CLSI. 2008. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard. Document M38-A2, vol 28, no. 16 National Committee for Clinical Laboratory Standards, Wayne, Pa. [Google Scholar]
- 49.Nishiyama Y, Hasumi Y, Ueda K, Uchida K, Yamaguchi H. 2005. Effects of micafungin on the morphology of Aspergillus fumigatus. J Electron Microsc (Tokyo) 54:67–77. doi: 10.1093/jmicro/dfh100. [DOI] [PubMed] [Google Scholar]
- 50.Bielawski J, Pierce JS, Snider J, Rembiesa B, Szulc ZM, Bielawska A. 2010. Sphingolipid analysis by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). Adv Exp Med Biol 688:46–59. doi: 10.1007/978-1-4419-6741-1_3. [DOI] [PubMed] [Google Scholar]
- 51.Blatzer M, Jukic E, Posch W, Schöpf B, Binder U, Steger M, Blum G, Hackl H, Gnaiger E, Lass-Flörl C, Wilflingseder D. 2015. Amphotericin B resistance in Aspergillus terreus is overpowered by coapplication of pro-oxidants. Antioxid Redox Signal 23:1424–1438. doi: 10.1089/ars.2014.6220. [DOI] [PubMed] [Google Scholar]
- 52.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT. 2015. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol 33:243–246. doi: 10.1038/nbt.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
fks1 expression levels in RG101 under uninduced and CAS-induced conditions. RG101 conidia were grown for 16 h in YPD in the absence and presence of CAS (1 and 4 μg/ml), and expression levels of fks1 were compared using reverse transcription-PCR (RT-PCR). No significant differences in fks1 expression levels were seen under uninduced and CAS-induced conditions (P > 0.05). Download FIG S1, DOCX file, 0.02 MB (26.7KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Cell wall ultrastructure of RG101 in the presence of CAS. Conidia of ATCC 13073 and RG101 strains were grown in liquid YPD in the presence of CAS (4 μg/ml) for 16 h and processed for SEM and TEM imaging. Electron microscopy images showed no morphological defects in RG101 grown in the presence of CAS, whereas the CAS-sensitive parental strain ATCC 13073 showed irregular mycelia and leaking cellular contents indicating damaged cell wall. Download FIG S2, DOCX file, 0.9 MB (964.4KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Evaluation of glucan and chitin contents of RG101 cell wall (A) Biochemical analysis of cell wall glucan levels using mass spectroscopy showing no reduction in glucan content in RG101 treated with CAS. (B and C) Fluorescence study using calcofluor white showing no increase in chitin levels in RG101 treated with CAS (4 μg/ml). In summary, no cell wall-associated changes were detected in RG101 treated with CAS, indicating complete resistance to the drug. Download FIG S3, DOCX file, 0.4 MB (369.2KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PTMs identified in glucan synthase. Nano-LC/MS analysis of glucan synthase of RG101 after enzyme digestion covered 71.5% of the protein and detected several PTMs. (A) A list of different PTMs detected with specific amino acid locations. (B) A diagrammatic representation of glucan synthase 16 transmembrane domains and the overall distribution of PTMs detected. Most of the PTMs were studded in the cytosolic segments of the protein. No PTMs were detected in the two hot spot regions of the enzyme. (C) List of 8 peptides showing differences in PTMs between CAS-induced and uninduced enzymes. However, a very low fraction of the population of the enzyme was modified, which did not account for the observed phenotype in RG101. Download FIG S4, DOCX file, 0.2 MB (243.6KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Abundance levels of DhSph and PhSph lipid species in clinical isolates: Glucan synthase of three clinical isolates listed in Table 1—2770, 117, and 32458—was tested for abundance levels of DhSph and PhSph under uninduced and induced conditions. Consistent with data from RG101, these clinical isolates also showed higher levels of DhSph (A) and PhSph (B) in CAS-induced enzymes than in preparations of uninduced enzymes. This indicated that CAS induces lipid modifications in clinical isolates, as observed in RG101. Download FIG S5, DOCX file, 0.04 MB (37.8KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Relative abundances of different lipid species in the microenvironment of glucan synthase. Three lipid subtypes (in red)—dihydrospingosine (DhSph), phytosphingosine (PhSph), and phytoceramide (C24)—were present in CAS-induced glucan synthase of RG101 at levels 3-, 10-, and 3-fold higher, respectively, than those seen with the uninduced preparation. DhSph and PhSph were used for further analysis. Download Table S1, DOCX file, 0.02 MB (17.5KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differentially expressed genes and gene ontology data from comparisons performed under CAS-induced and uninduced conditions in RG101. RG101 was grown in the absence and presence of CAS (1 μg/ml) for 16 h, and cells were isolated and processed for RNA-seq. Genes involved in the oxidation-reduction pathway were highly differentially expressed, consistent with the induction of ROS by CAS. Download FIG S6, DOCX file, 0.6 MB (661.5KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental Materials and Methods: in vivo fungal burden analysis, protein analysis by LC-MS/MS, and electron microscopy. Download Text S1, DOCX file, 0.03 MB (24.8KB, docx) .
Copyright © 2019 Satish et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.