

P-TEFb activation by cAMP-PKA signaling promotes cystogenic gene transcription elongation and disease progression in ADPKD.
Abstract
Positive transcription elongation factor b (P-TEFb) functions as a central regulator of transcription elongation. Activation of P-TEFb occurs through its dissociation from the transcriptionally inactive P-TEFb/HEXIM1/7SK snRNP complex. However, the mechanisms of signal-regulated P-TEFb activation and its roles in human diseases remain largely unknown. Here, we demonstrate that cAMP-PKA signaling disrupts the inactive P-TEFb/HEXIM1/7SK snRNP complex by PKA-mediated phosphorylation of HEXIM1 at serine-158. The cAMP pathway plays central roles in the development of autosomal dominant polycystic kidney disease (ADPKD), and we show that P-TEFb is hyperactivated in mouse and human ADPKD kidneys. Genetic activation of P-TEFb promotes cyst formation in a zebrafish ADPKD model, while pharmacological inhibition of P-TEFb attenuates cyst development by suppressing the pathological gene expression program in ADPKD mice. Our study therefore elucidates a mechanism by which P-TEFb activation by cAMP-PKA signaling promotes cystogenesis in ADPKD.
INTRODUCTION
Genome-wide studies have demonstrated that pausing of RNA polymerase II (Pol II) in promoter-proximal regions is a key regulatory step of transcriptional regulation in most metazoans (1). Pol II pausing is largely established by two pause-inducing complexes: negative elongation factor (NELF) and 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole sensitivity-inducible factor (DSIF). Release of paused Pol II into productive elongation is triggered by positive transcription elongation factor b (P-TEFb), an evolutionarily conserved kinase comprising cyclin-dependent kinase 9 (CDK9) and cyclin T1 or T2 (CycT1 or CycT2) (2). P-TEFb phosphorylates NELF and DSIF, as well as serine-2 and serine-5 residues in the C-terminal domain (CTD) of the largest subunit of Pol II. Phosphorylation of these targets by P-TEFb is essential for releasing paused Pol II and promoting productive RNA synthesis (3).
Because of the pivotal role of P-TEFb in regulating Pol II transcription, its kinase activity is subject to tight control in cells. The majority of P-TEFb is sequestered into a large inhibitory 7SK small nuclear ribonucleoprotein (7SK snRNP) complex, where P-TEFb kinase is catalytically inactive (4). 7SK snRNP complex consists of 7SK snRNA, hexamethylene bisacetamide–induced mRNA-encoded protein 1 or 2 (HEXIM1 or HEXIM2), La-related protein 7 (LARP7), and the methylphosphate capping enzyme (5). As a central component in this inhibitory complex, HEXIM1 is responsible for both the incorporation of P-TEFb into the P-TEFb/HEXIM1/7SK snRNP complex and the inhibition of P-TEFb kinase activity (6, 7). Various signals disrupt the inactive P-TEFb/HEXIM1/7SK snRNP complex by phosphorylation of HEXIM1 at distinct sites, including threonine-270 and serine-278, tyrosine-271 and tyrosine-274, and serine-158, which are phosphorylated by phosphatidylinositol 3-kinase/AKT, extracellular signal–regulated kinase, and protein kinase C θ, respectively (8–11). In addition, various transcriptional regulators can directly liberate P-TEFb from the P-TEFb/HEXIM1/7SK snRNP complex. These P-TEFb–releasing factors include HIV-1 transactivator of transcription, super elongation complex, bromodomain-containing protein 4 (Brd4), splicing and transcription regulator serine and arginine rich splicing factor 2 (SRSF2), histone demethylase jumonji domain containing 6 (JMJD6), transcription factor (TF) p53, signal transducers and activators of transcription 3 (STAT3), c-Myc, and nuclear factor κB (NF-κB). These P-TEFb–releasing factors recruit P-TEFb to target genes, allowing it to phosphorylate NELF, DSIF, and Pol II to enable pause release and transcription elongation.
Cyclic adenosine monophosphate (cAMP) acts as a key second messenger that couples extracellular signals to intracellular responses (12). Misregulation of the cAMP pathway leads to a variety of human diseases, including autosomal dominant polycystic kidney disease (ADPKD) (13, 14). ADPKD is a common, life-threatening monogenetic disorder and a leading genetic cause of end-stage renal disease, affecting more than 12.5 million individuals worldwide (15). ADPKD is mainly caused by mutations in the PKD1 or PKD2 genes encoding polycystin-1 (PC1) or PC2, respectively. The clinical hallmark of this disease is the progressive expansion of renal tubule–derived cysts. Excessive cell proliferation, a major pathological feature, drives the relentless growth of cysts, ultimately leading to destruction of the normal renal parenchyma and of its function (16). Widespread transcriptional misregulation has been observed in ADPKD, although the molecular mechanisms underlying cystogenic gene transcription remain largely unknown (17).
cAMP exerts many of its cellular functions by binding to and activating protein kinase A (PKA), which, in turn, phosphorylates a number of cytoplasmic and nuclear proteins (12). In the nucleus, the coupling of cAMP-PKA signaling to transcription initiation has been well established (18). However, the mechanisms of action of cAMP-regulated transcription elongation and its roles in human diseases are largely unknown. In the present study, we report that activation of cAMP-PKA signaling induces serine-158 phosphorylation of HEXIM1, disassembles the inactive P-TEFb/HEXIM1/7SK snRNP complex, and activates P-TEFb kinase activity. Aberrant activation of P-TEFb in ADPKD accelerates cyst formation by stimulating pathological gene transcription elongation.
RESULTS
Activation of cAMP-PKA signaling releases P-TEFb from the inactive P-TEFb/HEXIM1/7SK snRNP complex
cAMP-PKA mediates the transduction of extracellular signals to extensive nuclear transcriptional responses by phosphorylating cAMP-dependent TFs (12). Given that P-TEFb can be released from the inactive P-TEFb/HEXIM1/7SK snRNP complex in response to stress or specific cellular signals (4), we sought to investigate the potential roles of cAMP in regulation of P-TEFb activation. As shown in Fig. 1 (A and B), treatment with the cAMP agonist forskolin (FSK) for 1 hour in 293T cells led to rapid dissociation of P-TEFb from HEXIM1 and 7SK snRNA. To explore whether cAMP-promoted P-TEFb release is mediated by PKA, we pretreated the cells with H-89, a PKA inhibitor, for 30 min before FSK treatment. Pretreatment with H-89 completely blocked the cAMP-promoted dissociation of P-TEFb from P-TEFb/HEXIM1/7SK snRNP (Fig. 1, B and C). Furthermore, ectopic expression of the catalytic subunit of PKA (PKAc) disrupted the association between P-TEFb and 7SK snRNP (Fig. 1, D to F). Glycerol gradient fractionation analysis demonstrated that the majority of P-TEFb is found in the large P-TEFb/HEXIM1/7SK snRNP complex in control cells. In cells expressing PKAc, there was a marked shift of P-TEFb from the large inactive form of the complex to the small active form (Fig. 1G). Thus, we conclude that activation of cAMP-PKA pathway alters the P-TEFb equilibrium by releasing P-TEFb from the inactive P-TEFb/HEXIM1/7SK snRNP complex.
Fig. 1. cAMP-PKA signaling promotes P-TEFb release from P-TEFb/HEXIM1/7SK snRNP complex.
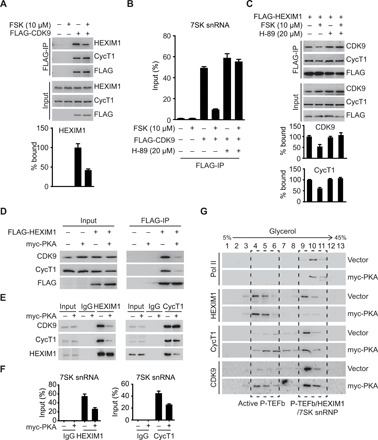
(A) Co-immunoprecipitation (Co-IP) assays examining the interactions between FLAG-CDK9 and endogenous HEXIM1 in 293T cells with or without FSK treatment. (B) RNA-IP assays examining the associations between FLAG-CDK9 and 7SK snRNA in 293T cells with or without FSK treatment. (C) Co-IP assays examining the interactions between FLAG-HEXIM1 and endogenous P-TEFb in 293T cells treated with the indicated agents. (D) Co-IP assays examining the interactions between FLAG-HEXIM1 and endogenous P-TEFb in 293T cells transfected with vector or myc-PKA. (E) Co-IP assays examining the interactions between endogenous HEXIM1 and P-TEFb in 293T cells. (F) RNA-IP assays examining the associations between endogenous HEXIM1 (left) or P-TEFb (right) and 7SK snRNA in 293T cells as in (E). (G) Protein extracts from 293T cells transfected with vector or myc-PKA were separated by centrifugation through a 5 to 45% continuous glycerol gradient. Collected fractions were analyzed by Western blot analysis. Data represent as means ± SEM of three replicates in (B) and (F). IgG, immunoglobulin G.
PKA disrupts the P-TEFb/HEXIM1/7SK snRNP complex by phosphorylating HEXIM1 at serine-158
Signal-dependent posttranslational modifications of P-TEFb/HEXIM1/7SK snRNP components are essential for affecting the release of transcriptionally active P-TEFb. The above results suggest that the catalytic activity of PKA is required for cAMP-PKA–promoted P-TEFb release. We therefore asked whether PKA promotes the shift of P-TEFb equilibrium via direct phosphorylation of one or more P-TEFb/HEXIM1/7SK snRNP components. For this purpose, we first examined whether endogenous CDK9, CycT1, and HEXIM1 were phosphorylated by PKA using an anti-PKA substrate antibody. As shown in Fig. 2A, CDK9 and HEXIM1, but not CycT1, were recognized by this anti-PKA substrate antibody in the presence of PKAc expression, suggesting that CDK9 and HEXIM1 are phosphorylated by PKA in vivo.
Fig. 2. PKA-mediated Ser158phosphorylation of HEXIM1 disrupts the P-TEFb/HEXIM1/7SK snRNP complex.
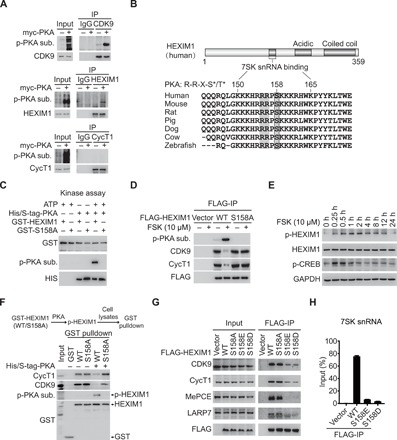
(A) PKA phosphorylates CDK9 and HEXIM1 but not CycT1. 293T cell lysates immunoprecipitated with the indicated antibodies were analyzed by Western blotting using a p-PKA substrate antibody. (B) Cross-species amino acid sequence alignment of HEXIM1. S* and T* denote phosphorylated serine and threonine, respectively. (C) In vitro PKA kinase assays containing GST-HEXIM1 (WT and S158A) as substrates. His/S-tag-PKA was detected by histidine (His) tag antibody. (D) Co-IP analysis of the interactions between the indicated FLAG-HEXIM1 and P-TEFb in 293T cells treated with or without FSK. (E) Western blot analysis of p-HEXIM1 and p-CREB in 293T cells treated with FSK. GAPDH, glyceraldehyde-phosphate dehydrogenase. (F) GST pulldown assays examining the interactions between CycT1 and GST-HEXIM1 (WT and S158A) with or without previous incubation with S-tag-PKA. (G) Co-IP assays examining the interactions between the indicated FLAG-HEXIM1 and other components of 7SK snRNP complex. (H) RNA-IP assays examining the associations between the indicated FLAG-HEXIM1 and 7SK snRNA. Data represent as means ± SEM of three replicates.
To identify potential PKA phosphorylation site(s) in HEXIM1, we searched the HEXIM1 sequence for PKA substrate consensus sites (R(R/K)X(S*/T*)). Sequence comparison showed that serine-158 (S158) of HEXIM1 lies within a canonical PKA phosphorylation site. The region between positions 150 and 165 of HEXIM1 is evolutionarily conserved from fish to human in a cross-species alignment (Fig. 2B). This motif contains clusters of positively charged residues and is responsible for binding to 7SK snRNA, and S158 is located at its center. S158 phosphorylation adds negative charge to the protein, which may decrease the ability of HEXIM1 to bind to 7SK snRNA and cause the disruption of 7SK snRNP. We first sought to examine whether PKA directly phosphorylates HEXIM1. We purified recombinant S-tagged PKA and glutathione S-transferase (GST)–tagged HEXIM1 [wild type (WT) and S158A] expressed from Escherichia coli. S-tag pulldown assays demonstrated that PKA directly interacted with both WT and S158A mutant HEXIM1 (fig. S1A). In vitro kinase assays showed that PKA directly phosphorylated HEXIM1 at S158 (Fig. 2C). As shown in Fig. 2D and fig. S1B, phosphorylation of HEXIM1 by PKA was remarkably elevated in the presence of FSK. A mutant version of HEXIM1, harboring the substitution S158A, failed to be phosphorylated, indicating that S158 is the major PKA phosphorylation site in vivo.
To further characterize HEXIM1 S158 phosphorylation by PKA in vivo, we generated a phospho-specific HEXIM1 (p-HEXIM1) antibody against the epitope containing the phospho-Ser158 residue of HEXIM1. As shown in fig. S1C, the anti–p-HEXIM1 antibody specifically recognized the phosphorylated peptide. The level of p-HEXIM1 was elevated in extracts from cells transfected with WT HEXIM1 in the presence of FSK. As expected, the S158A mutant of HEXIM1 had negligible immunoreactivity with the anti–p-HEXIM1 antibody (fig. S1D), further verifying the antibody’s specificity. Next, we tested whether this antibody could recognize endogenous p-HEXIM1 in vivo. As shown in Fig. 2E, FSK treatment led to a pronounced and transient increase in p-HEXIM1 level, with a maximum rise observed after 2 hours, followed by a gradual decline to basal level by 24 hours. By contrast, the level of cAMP response element–binding protein (CREB) phosphorylation (p-CREB) at serine-133 reached maximum within 30 min and rapidly reverted to basal level by 4 hours. These data indicate that the cAMP-induced phosphorylation kinetics of different downstream effector proteins varied in cells. The specificity of this antibody was further confirmed by immunostaining analysis. As shown in fig. S1E, FSK treatment specifically increased the nuclear staining of p-HEXIM1. Furthermore, overexpression of WT PKA, but not the kinase-inactive K72H mutant, strongly induced S158 phosphorylation of endogenous HEXIM1 (fig. S1F). The above results demonstrate that the p-HEXIM1 antibody could specifically recognize endogenous HEXIM1 phosphorylated at S158 in a PKA-dependent manner. Thus, we conclude that HEXIM1 is a bona fide nuclear substrate of PKA in vitro and in vivo.
To determine the role of PKA-mediated S158 phosphorylation of HEXIM1 in P-TEFb complex assembly, we phosphorylated recombinant GST-HEXIM1 (WT and S158A) with PKA in vitro and performed GST pulldown assays with 293T cell lysates. As shown in Fig. 2 (D and F), PKA-mediated phosphorylation of WT, but not S158A mutant, HEXIM1 substantially decreased the interaction between HEXIM1 and P-TEFb in vitro and in vivo. Next, we introduced phosphomimetic mutations of HEXIM1 to analyze the effects of HEXIM1 phosphorylation on P-TEFb/HEXIM1/7SK snRNP assembly in vivo. To this end, S158 of HEXIM1 was mutated to generate S158D or S158E. The amounts of CDK9, CycT1, and 7SK snRNA coprecipitated with these mutants decreased substantially compared to WT HEXIM1 (Fig. 2, G and H), indicating that S158 phosphorylation of HEXIM1 blocks P-TEFb/HEXIM1/7SK snRNP complex assembly.
Residue serine-347 of CDK9 lies within a PKA target consensus site (RRKGS347Q) and has previously been shown to be phosphorylated by PKA in vitro (19). We confirmed that PKA also phosphorylated S347 of CDK9 in vivo (fig. S2A). To determine the role of S347 phosphorylation of CDK9 in P-TEFb/HEXIM1/7SK snRNP assembly, we generated S347A and S347E mutants of CDK9. As shown in fig. S2 (B and C), association of these mutants with HEXIM1 and 7SK snRNA was maintained, indicating that phosphorylation of CDK9 at S347 does not affect P-TEFb/HEXIM1/7SK snRNP complex assembly.
P-TEFb kinase activity is increased in mouse and human ADPKD kidneys
Dysregulation of cAMP signaling contributes to cyst formation and enlargement by stimulating both cell proliferation and fluid secretion (13, 14). The tissue level of cAMP is elevated in cystic kidneys (20, 21). Both in vitro and in vivo studies have suggested that PKA is the main downstream effector of cAMP in cystogenesis (22, 23). Since PKA phosphorylates HEXIM1 at S158 in vitro and in vivo, we sought to determine the status of S158 phosphorylation of HEXIM1 and the P-TEFb/HEXMI1/7SK snRNP complex assembly in ADPKD kidneys. For this purpose, we first analyzed the cAMP level and PKA activity in immortalized Pkd1 null (PN) and Pkd1 heterozygous (PH) renal epithelial cells. As shown in fig. S3 (A and B), the basal and FSK-induced cAMP levels and PKA activities were higher in the PN cells than in the PH cells. We observed that the basal level of HEXIM1 phosphorylation is increased, and the association of P-TEFb with HEXIM1 is decreased in the PN cells compared to the PH cells (fig. S3, C and D), suggesting that the P-TEFb/HEXIM1/7SK snRNP complex is disrupted in cystic cells.
Next, we used an orthologous ADPKD mouse model. Pkd1fl/fl;Cre/Esr1+ (Pkd1−/−) and control Pkd1fl/fl;Cre/Esr1− mice were injected with tamoxifen at postnatal day 10 (P10) to inactivate the Pkd1 gene. We examined phosphorylation of HEXIM1 in kidneys of 29-day-old mice. As analyzed by immunoblotting and immunostaining with the p-HEXIM1 antibody, phosphorylation level of HEXIM1 was markedly increased in cyst-lining epithelial cells in the ADPKD mouse model (Fig. 3, A and B). Next, we tested S158 phosphorylation of HEXIM1 in human ADPKD patient samples. As shown in Fig. 3 (C and D), S158 phosphorylation was strongly elevated in human ADPKD kidneys. We double-stained mouse and human cystic kidneys with the p-HEXIM1 antibody and tubular markers Dolichos biflorus agglutin (DBA) or Lotus tetragonolobus lectin (LTL). We detected prominent p-HEXIM1 signals in both LTL-positive proximal tubules and DBA-positive collecting ducts (fig. S3, E to H).
Fig. 3. P-TEFb kinase activity is increased in ADPKD kidneys.
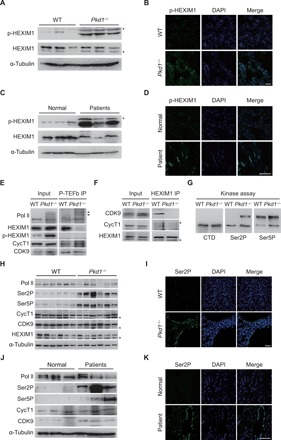
(A) Western blot analysis of p-HEXIM1 in kidneys from WT and Pkd1−/− mice. (B) Immunofluorescence analysis of p-HEXIM1 level in kidneys from WT and Pkd1−/− mice. (C) Western blot analysis of p-HEXIM1 in normal human kidneys and ADPKD kidneys. (D) Immunofluorescence analysis of p-HEXIM1 level in human kidneys. (E and F) Co-IP assays examining the interactions between P-TEFb and HEXIM1 in WT and Pkd1−/− mice kidneys. The tissue lysates were immunoprecipitated with CDK9 and CycT1 antibody in (E). Arrows indicated phosphorylated Pol II. (G) Kidney lysates immunoprecipitated by the anti–P-TEFb antibody were subjected to in vitro kinase assay, using GST-Pol II CTD as substrates. (H) Western blot analysis of kidney lysates from WT and Pkd1−/− mice. (I) Immunofluorescence analysis of Pol II CTD Ser2P in kidneys from WT and Pkd1−/− mice. (J) Western blot analysis of protein lysates from normal human kidneys and ADPKD kidneys. (K) Immunofluorescence analysis of Pol II Ser2P levels in human kidneys. Each gel line was obtained from an independent mouse (A and H) or human (C and J). “*” indicates nonspecific bands. Scale bars, 50 μm. DAPI, 4′,6-diamidino-2-phenylindole.
Since phosphorylation of HEXIM1 at S158 disrupts the P-TEFb/HEXIM1/7SK snRNP complex and HEXIM1 is highly phosphorylated in ADPKD kidneys, we hypothesized that the inactive complex may be disassembled to release active P-TEFb in ADPKD. As shown in Fig. 3 (E and F), the association between HEXIM1 and P-TEFb decreased significantly in kidneys from Pkd1−/− mice compared with WT controls, indicating that the inactive complex is disrupted and active P-TEFb is released in cystic kidney cells. Note that P-TEFb interacted with the hyper-phosphorylated form of Pol II in cystic, but not in WT, kidneys. P-TEFb did not associate with p-HEXIM1 in either WT or cystic kidneys. P-TEFb phosphorylates Pol II CTD at serine-2 (Ser2P) and serine-5 (Ser5P) in vitro. To assess the activation of P-TEFb in ADPKD, we immunoprecipitated endogenous P-TEFb kinase complex from kidney samples and performed in vitro kinase assays using GST-CTD as the substrate. As shown in Fig. 3G, the catalytic activity of P-TEFb was higher in Pkd1−/− kidneys than in control kidneys.
To further examine the kinase activity of P-TEFb in vivo, we analyzed Ser2P and Ser5P in ADPKD kidneys. As shown in Fig. 3H, whereas the total Pol II level remained unchanged, Ser2P and Ser5P were strongly elevated in Pkd1−/− mice compared with WT controls. Protein levels of CycT1 and CDK9 were similar between WT and Pkd1−/− mice. Expression of HEXIM1 was slightly decreased in Pkd1−/− mice compared with WT mice. Strong nuclear staining of Ser2P and Ser5P was observed in Pkd1−/− kidneys, most prominently in cyst-lining epithelial cells (Fig. 3I and fig. S3I). Compared with renal tubules in normal human kidney samples, the Ser2P and Ser5P levels were greatly increased in tubular cysts in kidney samples from ADPKD patients (Fig. 3, J and K, and fig. S3J). Together, these data demonstrate that P-TEFb kinase is aberrantly activated in cystic kidneys.
Disruption of the P-TEFb/HEXIM1/7SK snRNP complex promotes pronephric cyst formation in zebrafish pkd2 morphants
The P-TEFb/HEXIM1/7SK snRNP complex is functionally conserved among vertebrate classes from fish to human (5). To determine the roles of P-TEFb complex assembly and activation in cystogenesis in vivo, we first confirmed the association between P-TEFb and Hexim1 in zebrafish (fig. S4A). Next, we used a well-characterized zebrafish ADPKD model. Morpholino (MO)–induced knockdown of pkd2 results in cystic kidney, hydrocephalus, and dorsal axis curvature in zebrafish embryos (24). Co-injection of the hexim1 and pkd2 MOs caused a more severe cystic kidney phenotype than that observed in embryos co-injected with pkd2 and control MOs (Fig. 4, A and B). The efficiency of MO-mediated knockdown of Pkd2 and Hexim1 proteins was confirmed by Western blotting (Fig. 4C). These data demonstrate that knockdown of hexim1 aggravates cyst formation in vivo.
Fig. 4. Disruption of the P-TEFb/HEXIM1/7SK snRNP complex accelerates cystogenesis in zebrafish pkd2 morphants.
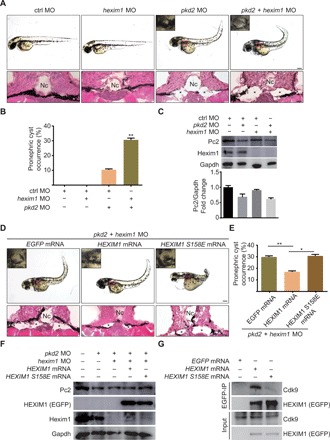
(A) Representative pictures of zebrafish embryos injected with the indicated MO. Notochord was indicated by “Nc.” Asterisks indicate dilated pronephric tubules. Pictures were taken at 2 days post fertilization (dpf). (B) Quantification of the incidences of pronephric cyst occurrence in zebrafish embryos from the indicated groups. (C) Western blot analysis of Pc2 and Hexim1 expression in zebrafish embryos from the indicated groups. The Pc2 density was normalized using the Gapdh level. (D) Representative pictures of zebrafish embryos injected with the indicated MOs and mRNAs. Pictures were taken at 2 dpf. (E) Quantification of the incidences of pronephric cyst occurrence in zebrafish embryos from the indicated groups. (F) Western blot analysis of Pc2, Hexim1, and exogenous EGFP-HEXIM1 levels in zebrafish embryos from the indicated groups. (G) Co-IP assays examining the interactions between exogenous EGFP-HEXIM1 (WT and S158E) and zebrafish CDK9. Scale bars, 200 μm (top) and 10 μm (bottom) in (A) and (D). Quantification data (n > 50) represent means ± SEM of three replicates in (B) and (E). *P < 0.05, **P < 0.01. Two-tailed unpaired Student’s t test was used for statistical analysis.
To determine whether modulation of cystogenesis by HEXIM1 is dependent on its P-TEFb sequestration function, we injected WT and S158E mutant human HEXIM1 mRNA into pkd2/hexim1 double morphants. Compared with EGFP mRNA injection, injection of WT human HEXIM1 mRNA significantly decreased cystogenesis, whereas injection of S158E mutant HEXIM1 mRNA failed to rescue the cystic kidney phenotype (Fig. 4, D and E). The expression of exogenous enhanced green fluorescent protein (EGFP)–HEXIM1 (WT and S158E) was examined by fluorescence imaging and Western blotting (fig. S4B and Fig. 4F). Human WT HEXIM1, but not the S158E mutant, interacted with fish Cdk9 (Fig. 4G), indicating that this regulatory mechanism is evolutionarily conserved. Together, these genetic studies demonstrate that disruption of P-TEFb/HEXIM1/7SK snRNP by Ser158 phosphorylation of HEXIM1 activates P-TEFb and accelerates cystogenesis in vivo.
Inhibition of P-TEFb suppresses cystogenesis in embryonic kidney and 3D-MDCK cyst models
Given that P-TEFb is hyperactivated in ADPKD, we next sought to determine whether inhibition of P-TEFb could delay cyst formation and growth. We first used an embryonic kidney cyst model. We dissected embryonic metanephric kidneys from WT CD1 mice at embryonic day 13.5 (E13.5). Under basal culture conditions, kidney explants grew in size over 6 days in culture. Treatment with 8-Br-cAMP (100 μM) resulted in tubule dilation and cyst formation (23). Dose-response experiments showed that cyst development was highly sensitive to flavopiridol (FP), a potent P-TEFb inhibitor (fig. S5A) (25, 26). FP reduced the fractional cyst area by 50% at 40 nM and by 75% at 80 nM (fig. S5B). Thus, these data suggest that P-TEFb inhibition attenuates cyst formation in ex vivo kidney organ cultures.
Next, we used an established three-dimensional (3D)–Madin-Darby canine kidney (MDCK) cyst model. MDCK cells form cysts when grown in collagen gels in the presence of FSK. The established cysts (day 4) were treated with low concentrations of FP (10, 50, and 100 nM) for 8 days in the presence of 10 μM FSK. FP dose dependently diminished cyst enlargement (fig. S6, A and B). Treatment with FP had little or no effect on the total number of established colonies in all groups, indicating that exposure to FP was marginally toxic to MDCK cells at concentrations up to 100 nM (fig. S6C). The effects of P-TEFb inhibition on cystogenesis were further evaluated by lentivirus-mediated delivery of a catalytically inactive, dominant-negative human CDK9 (D167N) mutant (dnCDK9) into MDCK cells. Overexpressing this inactive CDK9 mutant significantly reduced the diameter and number of cysts (fig. S6, D to F). The interaction between human dnCDK9 and canine CycT1 was confirmed by co-IP analysis (fig. S6G). Last, to further investigate the requirement of P-TEFb in cyst formation and growth, we knocked down CDK9 in MDCK cells using three different short hairpin RNAs. The diameter and number of cysts in CDK9-silenced MDCK cells were remarkably smaller than those of control cells (fig. S6, H to J). The knockdown efficiency was confirmed by Western blotting (fig. S6K). Collectively, these data demonstrate that the inhibition of P-TEFb kinase slows cyst formation and growth in vitro.
To investigate the contribution of PKA-mediated HEXIM1 phosphorylation to P-TEFb hyperactivation in the MDCK cyst model, we first examined the phosphorylation of HEXIM1 and the association between P-TEFb and HEXIM1 in the presence of PKA. As shown in fig. S7 (A and B), overexpression of PKA induced phosphorylation of endogenous HEXIM1 and disrupted the association between HEXIM1 and P-TEFb in MDCK cells. Overexpression of the phospho-resistant HEXIM1 (S158A) significantly reduced cyst growth (fig. S7, C and D). Furthermore, PKA-mediated phosphorylation of WT, but not S158A mutant, HEXIM1 markedly decreased the interaction between HEXIM1 and P-TEFb (fig. S7E). The amount of 7SK snRNA coprecipitating with WT HEXIM1 was considerably lower than that with the S158A mutant (fig. S7F) in MDCK cells. Together, these data indicate that the S158 phosphorylation of HEXIM1 blocks P-TEFb/HEXIM1/7SK snRNP complex assembly and retards cystogenesis in the MDCK cyst model.
FP delays cyst growth in the early- and late-onset ADPKD mouse model
Since P-TEFb is hyperactivated in cystic kidneys and P-TEFb inhibition suppresses cystogenesis in vitro, we next tested whether inhibiting P-TEFb activity would affect cyst growth and other renal pathologies in vivo. First, we examined the effects of FP treatment on cystogenesis in the early-onset ADPKD mouse model. A single injection of tamoxifen was given at P10 to induce Pkd1 gene deletion. FP (3.5 mg/kg) treatment was started from P11 and continued for 3 weeks. Treatment was applied once daily for 5 days, followed by two rest days (Fig. 5A). Administration of FP decreased the kidney size, kidney weight–to–body weight (KW/BW) ratio and cystic index (Fig. 5, B to E) compared with vehicle treatment. FP treatment significantly improved kidney function, as assessed by blood urea nitrogen (BUN) (Fig. 5F). Furthermore, we observed that FP decreased cyst epithelial proliferation, kidney injury, inflammation, and fibrosis (fig. S8, A to D). The elevated Ser2P level in Pkd1−/− kidneys was significantly reduced by FP treatment, indicating that FP inhibited P-TEFb kinase activity in vivo (fig. S8, E and F). These data suggest that inhibition of P-TEFb attenuates disease progression in the early-onset ADPKD mouse model.
Fig. 5. FP treatment inhibits renal cyst growth in the early- and late-onset ADPKD mouse model.
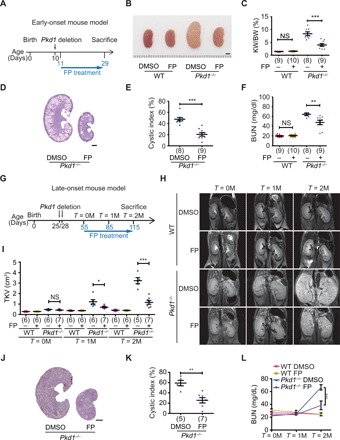
(A) Schematic view of experimental design in the early-onset ADPKD model. (B) Representative images of P29 kidneys from WT and Pkd1−/− mice treated with dimethyl sulfoxide (DMSO) or FP. (C) Ratios of kidney weight to body weight in the indicated groups of mice. (D) Hematoxylin and eosin (H&E) staining of kidney sections from Pkd1−/− mice treated with DMSO or FP. (E) Cystic index of H&E-stained kidney sections from Pkd1−/− mice treated with DMSO or FP. (F) Plasma BUN levels of P29 mice from the indicated groups. (G) Schematic view of the experimental design in the late-onset ADPKD model. (H) Representative magnetic resonance imaging (MRI) images of kidneys from the indicated groups. (I) Total kidney volumes (TKVs) were calculated from MRI images acquired at the indicated time points. (J) H&E staining of kidney sections from Pkd1−/− mice treated with DMSO or FP. (K) Cystic index of H&E-stained kidney sections as in (J). (L) Plasma BUN levels of P115 mice from the indicated groups. Scale bars, 2 mm (B, D, and J). Data represent as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. Two-tailed unpaired Student’s t test was used for statistical analysis. NS, not significant. (Photo credit: Zhiheng Liu, Tianjin Medical University).
To more closely resemble human ADPKD progression and to evaluate long-term effects of FP treatment, we used a late-onset ADPKD mouse model and addressed whether FP could reduce cyst growth in animals with established ADPKD (27). To this end, Pkd1 deletion was induced by injection of tamoxifen twice, on P25 and P28. FP (5 mg/kg) treatment was started 1 month later (P55, T = 0M). FP was administered once daily for 5 days, followed by two rest days (Fig. 5G). To follow disease progression longitudinally, we monitored these mice for 2 months using in vivo magnetic resonance imaging (MRI). We acquired MRI images at P55 (T = 0M), P85 (T = 1M), and P115 (T = 2M). Compared with vehicle-treated control groups, we found that FP treatment delayed cyst growth as indicated by decreased MRI-based total kidney volume (TKV), kidney size, cyst index, KW/BW ratio, BUN levels, and cyst epithelial proliferation (Fig. 5, H to L, and fig. S8, G to I). We observed no systemic toxicity such as body weight loss (fig. S8J) or behavioral changes. Together, these results demonstrate that pharmacologically targeting P-TEFb delays ADPKD progression in the late-onset mouse model.
Inhibition of P-TEFb attenuates a pathological gene expression program of ADPKD by regulating Pol II pause release
To determine the transcriptional effects of P-TEFb inhibition in vivo, we performed RNA sequencing (RNA-seq) in kidney tissues from WT and Pkd1−/− mice treated with vehicle or FP. Consistent with previous findings (28, 29), gene expression profiles of Pkd1−/− mice differed significantly from those of age-matched WT littermates: 1253 genes were down-regulated and 1384 genes were up-regulated in Pkd1−/− mice relative to WT mice (Fig. 6A). In agreement with our observation that low-dose FP did not alter kidney structure or function in WT mice (Fig. 5, B and F), only five differentially expressed genes were identified in WT mice treated with FP (two down-regulated and three up-regulated) (Fig. 6B). By contrast, we identified a total of 515 genes as differentially expressed in Pkd1−/− mice in the presence of FP treatment, of which 354 genes were down-regulated and 161 genes were up-regulated (Fig. 6C), suggesting that the gene expression program in ADPKD was extremely sensitive to P-TEFb inhibition. P-TEFb is a transcriptional activator and is hyperactivated in ADPKD. Therefore, our further analysis focused on the 1384 up-regulated genes in Pkd1−/− kidney. Notably, many of these up-regulated genes were either partially or completely rescued by FP treatment (Fig. 6D). We defined “FP-rescued genes” as genes whose up-regulation in ADPKD (ADPKD-induced genes) is reversed by FP treatment. Compared to non–FP-rescued genes, FP treatment significantly reduced the expression of FP-rescued genes (Fig. 6, E and F). Two representative FP-rescued genes are shown in Fig. 6G. Gene ontology analysis revealed that FP-rescued transcripts were significantly enriched in pathways related to cystogenesis, including cell proliferation, cell differentiation, inflammation, and fibrosis (Fig. 6H). Thus, these data indicate that P-TEFb inhibition suppresses the expression of cystogenic genes in ADPKD.
Fig. 6. Inhibition of P-TEFb attenuates a pathological gene expression program of ADPKD by regulating Pol II pause release.
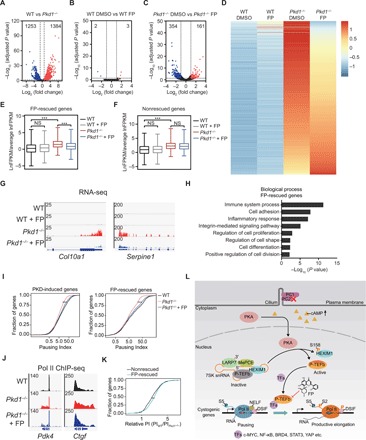
(A to C) Volcano plots showing differentially expressed genes in kidneys from WT versus Pkd1−/− (A), WT DMSO versus WT FP (B), and Pkd1−/− DMSO versus Pkd1−/− FP (C) mice. (D) Heatmap of gene expression values of kidneys from the indicated groups. Rows show Z scores calculated for each group. (E and F) Boxplots of relative gene fragments per kilobase million (FPKM) values of kidneys from WT, WT FP, Pkd1−/−, and Pkd1−/− FP mice. (G) Gene tracks showing representative RNA-seq profiles of two genes (Col10a1 and Serpine1) in kidneys from the indicated groups. (H) Gene ontology enrichment analysis of FP-rescued genes. (I) Cumulative curve of Pol II pausing index (PI) for PKD-induced genes (left) and FP-rescued genes (right). (J) Gene tracks showing representative ChIP-Seq profiles of two genes (Pdk4 and Ctgf) in kidneys from the indicated groups. (K) Cumulative curve of relative pausing index for FP-rescued genes and nonrescued genes. (L) Working model of activation and function of P-TEFb in ADPKD. **P < 0.01, ***P < 0.001. One-way ANOVA with Tukey’s post hoc test was used for statistical analysis in (E) and (F). Two-tailed unpaired Student’s t test was used in (I) and (K).
The main function of P-TEFb is to regulate transcription elongation by promoting Pol II pause release. Therefore, we hypothesize that the functional rescue of the pathological transcriptional program by FP relies on modulating P-TEFb–mediated Pol II pause release and transcription elongation. To test this hypothesis, we examined Pol II distribution by chromatin immunoprecipitation sequencing (ChIP-seq) analysis in kidney tissues. We used a pausing index (PI) to measure transcriptional pause release. The PI was calculated as the ratio of Pol II density within the promoter-proximal region to that within the gene body. As shown in Fig. 6 (I and J), the PI in Pkd1−/− kidney was lower than that in WT kidney, and FP treatment significantly increased the PI in Pkd1−/− kidney at all ADPKD-induced and FP-rescued genes.
Since the PI of both FP-rescued and nonrescued genes decreased in ADPKD kidneys, we further investigated why P-TEFb inhibition specifically down-regulates the FP-rescued genes, but not the nonrescued genes. We hypothesized that the extent to which pausing contributes to gene expression varies between the FP-rescued and nonrescued genes. We calculated the relative PI for all ADPKD-induced genes; the relative PI is the ratio of PI in WT to PI in ADPKD. As shown in Fig. 6K, the relative PI of FP-rescued genes was significantly higher than that of nonrescued genes. These data suggest that Pol II pause release is greater at the FP-rescued genes than at the nonrescued genes, which may confer higher sensitivity to P-TEFb inhibition on the FP-rescued genes. Together, these data suggest that P-TEFb inhibition suppresses gene expression programs that are activated in ADPKD by abrogating abnormal Pol II pause release.
DISCUSSION
The P-TEFb equilibrium between active and inactive states is dynamically regulated in response to intra- and extracellular signals. Given the vital roles of P-TEFb in regulating transcription elongation, misregulation of P-TEFb equilibrium and aberrant activation of P-TEFb are implicated in several human diseases. Activation of P-TEFb has been observed in pathological cardiac hypertrophy. Overexpression of CycT1 or deletion of HEXIM1 results in cardiac hypertrophy in vivo (30, 31). Ectopic activation of P-TEFb by depletion of LARP7 blocks mammary epithelial cell differentiation (32). Activation of P-TEFb after HEXIM1 dissociation is required for satellite cell proliferation and the repression of early myogenic differentiation (33). In ADPKD, cAMP-PKA signaling is central to cyst formation and enlargement. We found that cAMP-PKA signaling activates P-TEFb kinase by releasing P-TEFb from the inactive P-TEFb/HEXIM1/7SK snRNP complex in cystic kidney cells. Activation of P-TEFb promotes pathological gene expression and accelerates cystogenesis in ADPKD. The active P-TEFb has been reported to be recruited to target genes by direct interaction with TFs, including c-Myc, Brd4, NF-κB, Stat3, and yes-associated protein (YAP). Many of these TFs are downstream effectors of signaling pathways that are dysregulated in ADPKD (13). Among these TFs, Brd4 is a major P-TEFb recruitment factor to active chromatin templates once P-TEFb is released from 7SK snRNP (34, 35) and may synergize with P-TEFb in activating the pathological gene expression program and promoting cystogenesis. Thus, P-TEFb may function as a hub to integrate multiple regulatory signals and coactivate a broad transcriptional network involving various TFs. The target genes of these TFs in ADPKD have not yet been fully clarified, however, and further investigation is therefore needed to define their target genes and the extent to which P-TEFb–mediated transcription elongation contributes to the expression of these targets in ADPKD.
Abundant evidence points to the importance of cAMP-PKA signaling in modulating gene transcription through phosphorylation of nuclear TFs, among which CREB is the best studied (12). PKA phosphorylates CREB at S133, which induces CREB transcriptional activity. CREB is highly phosphorylated in cystic cells and inhibiting CREB activity suppresses cystogenesis (36), indicating that PKA is active in the nucleus of cystic kidney cells. We found that cAMP-PKA signaling induces phosphorylation of HEXIM1 at S158 and that the abnormal hyperphosphorylation of HEXIM1 in ADPKD disrupts P-TEFb/HEXIM1/7SK snRNP complex and activates P-TEFb. Earlier studies on cAMP-PKA–regulated gene transcription focused solely on the transcription initiation of cAMP-responsive genes (18). In the present study, we identified the P-TEFb/HEXIM1/7SK snRNP complex as a novel nuclear effector of cAMP-PKA signaling and elucidated a new mechanism by which P-TEFb–controlled transcription elongation is regulated.
Regulation of the P-TEFb equilibrium between inactive and active states has been shown to play vital roles in the control of cell differentiation and proliferation (37). Defective differentiation and excessive proliferation of renal tubular epithelial cells are key hallmarks of ADPKD (13, 38). The proliferation of fully differentiated tubule epithelial cell ceases before birth in normal kidney. PC1 and PC2 are essential for normal differentiation and tight control of tubular epithelial cell proliferation. Defects in these two proteins dysregulate multiple signaling pathways, initiate a pathological cascade of gene expression, lock the renal epithelial cells in an abnormal differentiated state, and stimulate uncontrolled cell proliferation. In this study, we found that, in normal differentiated renal tubular epithelial cells, P-TEFb is sequestered into the P-TEFb/HEXIM1/7SK snRNP complex and its activity is suppressed. However, in cystic cells, P-TEFb is released from the inactive complex and hyperactivated. Disruption of the P-TEFb/HEXIM1/7SK snRNP complex and activation of P-TEFb promotes cystic cell proliferation. Thus, regulation of the P-TEFb equilibrium plays critical roles in the renal tubular epithelial cell fate decision between differentiation and proliferative growth.
Accumulating evidence reveals marked similarities in pathological features and dysregulated signaling pathways between ADPKD and cancer (39). In cancer cells, aberrant activation of P-TEFb serves to support a high transcriptional demand for sustained cell proliferation (40). The present study demonstrates that hyperactivation of P-TEFb accelerates cystogenesis in ADPKD. Thus, P-TEFb–promoted cell proliferation represents a new commonality between ADPKD and cancer. The similarities between ADPKD and cancer cells suggest potential avenues for repurposing existing cancer drugs for ADPKD treatment. Inhibiting transcriptional regulators, including BET bromodomain proteins and histone deacetylases, has been shown to be effective for treating both ADPKD and cancers (24, 41). Targeting P-TEFb is currently emerging as a promising therapeutic strategy for cancer treatment (26, 40). FP is now actively investigated in the treatment of a wide variety of chronic diseases including hematological malignancies, neurological disorders, cardiovascular disease, and infectious diseases (25, 26). In ADPKD, the cystogenic cAMP-PKA signaling activates P-TEFb and its target genes in cyst-forming cells. Therefore, inhibition of P-TEFb may selectively target cystic epithelial cells while sparing normal parenchyma. In addition, functional studies in the early- and late-onset ADPKD animal models provide a further rationale for developing P-TEFb inhibitors as potential therapeutic agents for ADPKD.
MATERIALS AND METHODS
Cell culture
Human embryonic kidney 293T and MDCK I cell lines were purchased from the American Type Culture Collection. Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and maintained in a 37°C incubator with 5% CO2. PKD1 null (PN) and heterozygous (PH) cells were provided by Y. Cai (Yale University, New Haven, CT) and cultured as described previously (42).
Zebrafish studies
Zebrafish experiments were approved by the Institutional Animal Care and Use Committee of Tongji University. Embryos were raised at 28°C in E3 solution. MOs were obtained from Gene Tools. Sequences of MOs are listed as follows: hexim1, 5′-TAATAAGCTCCATAACTGCACACTC-3′ (43); pkd2, 5′-AGGACGAACGCGACTGGAGCTCATC-3′ (24); and a standard control, 5′-CCTCTTACCTCAGTTACAATTTATA-3′. For rescue experiments, 200 pg of human HEXIM1 mRNA or HEXIM1 S158E mRNA was co-injected with pkd2 MO and hexim1 MO into one-cell–stage embryos. For total lysates, embryos were deyolked and homogenized completely in 2× SDS-loading buffer and then boiled for 10 min. For IP, embryo lysates were prepared in NP-40 buffer. Lysates were then incubated at 4°C with GFP-Trap, followed by Western blotting analysis.
Mice and drug treatment
Pkd1fl/fl;Cre/Esr1+ mice were generated as described previously (44). To induce Cre recombinase activity, tamoxifen (Sigma, T5648) dissolved in corn oil (Sigma, C8267) was injected intraperitoneally. Tamoxifen was administrated at P10 (10 mg/kg) to generate the early-onset model and at P25 to P28 (250 mg/kg) to generate the late-onset model. For FP (Selleck, S1230; dissolved in 5% DMSO in saline) treatment, the early-onset mice were injected intraperitoneally at a dosage of 3.5 mg/kg each day for five consecutive days from P11 to P29. For the late-onset model, mice were treated at a dosage of 5 mg/kg each day for five consecutive days from P55 to P115. Both female and male mice were used for the early-onset model. Only male mice were used for the late-onset model. Total area and cyst area measurement were evaluated using the National Institutes of Health ImageJ software. The cyst index was calculated as the ratio of cyst area to total area. BUN was measured in plasma using the QuantiChrom Urea Assay Kit (DIUR, BioAssay Systems).
MRI imaging
MRI scanning was performed on a 7.0-T PharmaScan 70/16 ultra shielding (US) (Bruker, Switzerland) equipped with a 40-mm radio frequency (RF) coil. Mice were anesthetized with 1.5 to 2.5% isoflurane inhalation. Kidneys were placed to the center of the RF coil. Respiratory sensor was used to monitor mice breathing, body temperature was maintained around 37°C. MRI scanning was gated using the respiratory triggering system. TKV was evaluated from contiguous images by manually encircling kidney parenchyma and summing up the products of areas and slice thickness. Parameters of MRI studies included coronal T2-Turbo RARE sequences; repetition time/echo time, 2970/33 ms; image size, 256 × 256; field of view, 40 mm by 40 mm; slice thickness, 0.5 mm; and slice numbers, 12 to 25 (depending on the kidney size).
Immunoblotting
Cells and tissue samples were lysed in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitor cocktail, phosphatase inhibitors cocktail, and dithiothreitol (DTT). After incubating at 4°C for 30 min, cell or kidney lysates were cleared and denatured in SDS protein loading buffer. Protein samples were separated by SDS–polyacrylamide gel electrophoresis gels, transferred to nitrocellulose membranes, and immunoblotted with primary antibodies. The antibody information was listed in Supplementary Materials and Methods.
Immunofluorescence
The kidney specimens were embedded in 100% optimal cutting temperature (O.C.T.). Tissue sections or cells were blocked and incubated with primary antibody against Ser2P Pol II CTD (Abcam, ab-5095), Ser5P Pol II CTD (Abcam, ab-5131), fluorescein-DBA (Vector Lab, FL-1031), fluorescein-LTL (Vector Lab, FL-1321), or p-HEXIM1. After primary antibody incubation, sections were incubated with goat anti-rabbit Alexa Fluor 488 secondary antibody (Thermo Fisher Scientific). Sections were imaged using a fluorescence microscope (FV1000, Olympus).
IP and RNA-IP assays
Kidneys from WT and Pkd1−/− mice or cells were lysed with NP-40 buffer [150 mM NaCl, 1.0% NP-40, 50 mM tris-HCl (pH, 8.0) containing protease inhibitor cocktail, 20 mM DTT, and 2 mM phenylmethylsulfonyl fluoride (PMSF)]. Corresponding antibodies were added into tissue lysates and incubated overnight at 4°C. Proteins were immunoprecipitated by Dynabeads Protein G for 1 hour at 4°C. After three washes with IP buffer, the beads were boiled for 10 min for Western blotting analysis. RNA-IP was performed as described previously (45).
Protein purification and pulldown
S-tag-PKA, GST-HEXIM1, and mutant GST-HEXIM1 (S158A) fusion proteins were expressed in E. coli BL21 (DE3). After induction with isopropyl-β-d-thiogalactopyranoside (IPTG) overnight at 16°C, cells were sonicated in GST lysis buffer [50 mM tris-HCl (pH, 8.0), 100 mM NaCl and 1% Triton X-100, 0.2 mM PMSF, and protease inhibitor cocktail] and cleared by centrifugation. Lysates were purified on S-protein agarose or glutathione agarose. After washing three times with lysis buffer, GST-fusion proteins were eluted twice with elution buffer containing 10 mM reduced glutathione and 1 mM DTT. For pulldown assay, the S-tag-PKA immobilized on S-protein agarose was incubated with GST-HEXIME1 or mutant GST-HEXIM1 (S158A) overnight at 4°C. After washing three times, the proteins were boiled for 10 min and were detected by Coomassie blue staining or Western blotting.
Kinase assay
Immunoprecipitated P-TEFb and P-TEFb/HEXIM1 complexes on protein G magnetic beads from WT and Pkd1−/− kidneys were incubated with 500 ng of GST-CTD in kinase reaction buffer [20 mM Hepes (pH 7.6) and 5 mM MgCl2) supplemented with 2 mM DTT and 1 mM adenosine 5′-triphosphate (ATP) for 12 hours at 30°C. The reaction was stopped by addition of SDS-loading buffer. For phosphorylation of recombinant HEXIM1, purified S-tag-PKA fusion protein on S-protein agarose was incubated with recombinant HEXIM1 or mutant HEXIM1 (S158A) in kinase reaction buffer for 6 hours.
RNA isolation and RT-qPCR
Total RNA was isolated from whole kidneys or cells using TRIzol (Invitrogen). After RNA isolation, total RNA was reverse-transcribed by the cDNA Synthesis Kit (Roche). Gene-specific primers were provided in Supplementary Materials and Methods.
RNA-seq analysis
After reads filtering, clean reads were mapped to reference transcripts using Bowtie2, and then, gene expression level was calculated with RNA-seq by Expectation-Maximization (RSEM). On the basis of the gene expression level, differential expression genes were identified using DESeq2 algorithms. Fold change of ≤−2 or ≥2 and adjusted P < 0.05 were considered significantly differentially expression. Gene ontology analysis was performed using DAVID 6.8. FP-rescued genes were defined as either (i) genes whose expressions in FP-treated Pkd1−/− mice were returned to levels in WT mice (≤1.5-fold increase in FP-treated Pkd1−/− versus WT mice, adjusted P < 0.05) or (ii) genes whose expressions were restored ≥50% (adjusted P < 0.05) after FP treatment (Pkd1−/− versus FP-treated Pkd1−/− mice).
ChIP-seq analysis
ChIP assays were performed mainly as previously described (45). Briefly, kidney tissues were minced finely and fixed with 1% formaldehyde at room temperature (RT) for 10 min. Cross-linking was quenched with 150 mM glycine at RT for 5 min. Nuclei were harvested and chromatin were fragmented to a size range of 200 to 500 bp with 25U benzonase (Millipore, 70664) for 15 min at RT. IP was performed using 2 μg of the Pol II antibody (Cell Signaling Technology, 14958). Purified DNA was sequenced on the BGISEQ-500 platform. The quality control of ChIP-seq data was performed by FastQC software. Clean reads were mapped to reference genome (GRCm38/mm10) using Bowtie2. Promoter region was defined as ±300 bp around the transcription start site (TSS). Gene body region was defined as +300 bp from the TSS to +3000 bp past the transcription termination site (TTS). Reads were counted using BEDtools software. PI was calculated using R-3.4.3.
Study approval
Zebrafish studies were approved by the Institutional Animal Care and Use Committee of Tongji University. Mice studies were approved by the Ethical Committee of Tianjin Medical University (permit number: SYXK 2009-0001). ADPKD and normal kidney specimens were obtained from Changzheng Hospital, Shanghai. All patients provided informed consent. Human studies were approved by the Ethics Committee of Tianjin Medical University and the Ethics Committee of Changzheng Hospital.
Statistical analysis
GraphPad Prism 7.0 was used for statistical analysis. Results were presented as means ± SEM for at least three independent experiments. Unpaired two-tailed Student’s t test was used to compare two groups of independent samples, and one-way or two-way analysis of variance (ANOVA) with Tukey’s post hoc test was used to compare multiple groups. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by grants (81670612 and 81873595 to C.M., 31571336 to L.Z., and 81770658 and 31571337 to Y. Chen) from The National Natural Science Foundation of China, grants (15JCYBJC54100 to L.Z. and 15JCYBJC54000 to Y. Chen) from Tianjin Municipal Science and Technology Commission, grants (2016YFC0901502 to C.M. and 2017YFA0504102 to Y. Chen) from The National Key Research and Development Program of China, grant (2017ZZ02009 to C.M.) from Shanghai Top Priority Clinical Disciplines Construction Project, and a grant (to Y.C.) from The Excellent Talent Project of Tianjin Medical University. Author contributions: Y.S. performed animal and biochemistry studies. Z.L. performed drug screening, MDCK analysis, and genome-wide studies. X.C., Z.M., and J.L. performed biochemistry studies. Y. Lu performed mouse and zebrafish studies. C.H. performed drug screening. Z.Z. and M.J.L. performed bioinformatics analysis. T.L. provided expertise on MRI analysis. D.X. and Y. Li provided expertise on zebrafish studies. M.W. provided expertise on mouse studies. Y. Cao provided expertise on zebrafish studies and edited the manuscript. B.Y. provided expertise on MDCK and embryonic kidney studies. C.M. conceived the project, provided human specimens, and edited the manuscript. L.Z. and Y. Chen conceived, designed, and supervised the project, analyzed data, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. For the RNA-seq and ChIP-seq data, they can be found at the Gene Expression Omnibus database under accession number GSE128524. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw3593/DC1
Supplementary Materials and Methods
Fig. S1. PKA phosphorylates HEXIM1 at serine-158.
Fig. S2. Phosphorylation of CDK9 at serine-347 by PKA does not affect P-TEFb/HEXIM1/7SK snRNP complex assembly.
Fig. S3. The phosphorylation level of HEXIM1 is increased in cystic epithelial cells.
Fig. S4. The interaction between P-TEFb and Hexim1 is conserved in zebrafish.
Fig. S5. FP inhibits cystogenesis in the embryonic kidney model.
Fig. S6. Inhibition of CDK9 attenuates cyst development in the MDCK cyst model.
Fig. S7. HEXIM1 phosphorylation and P-TEFb/HEXIM1/7SK snRNP complex assembly in the MDCK cyst model.
Fig. S8. Effect of FP on cystogensis in the early- and late-onset ADPKD mouse models.
REFERENCES AND NOTES
- 1.Adelman K., Lis J. T., Promoter-proximal pausing of RNA polymerase II: Emerging roles in metazoans. Nat. Rev. Genet. 13, 720–731 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou Q., Li T., Price D. H., RNA polymerase II elongation control. Annu. Rev. Biochem. 81, 119–143 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen F. X., Smith E. R., Shilatifard A., Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 19, 464–478 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Quaresma A. J. C., Bugai A., Barboric M., Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res. 44, 7527–7539 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marz M., Donath A., Verstraete N., Nguyen V. T., Stadler P. F., Bensaude O., Evolution of 7SK RNA and its protein partners in metazoa. Mol. Biol. Evol. 26, 2821–2830 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Yik J. H., Chen R., Nishimura R., Jennings J. L., Link A. J., Zhou Q., Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 12, 971–982 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Michels A. A., Fraldi A., Li Q., Adamson T. E., Bonnet F., Nguyen V. T., Sedore S. C., Price J. P., Price D. H., Lania L., Bensaude O., Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 23, 2608–2619 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Contreras X., Barboric M., Lenasi T., Peterlin B. M., HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLOS Pathog. 3, 1459–1469 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mbonye U. R., Wang B., Gokulrangan G., Chance M. R., Karn J., Phosphorylation of HEXIM1 at Tyr271 and Tyr274 promotes release of P-TEFb from the 7SK snRNP complex and enhances proviral HIV gene expression. Proteomics 15, 2078–2086 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim Y. K., Mbonye U., Hokello J., Karn J., T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J. Mol. Biol. 410, 896–916 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujinaga K., Barboric M., Li Q., Luo Z., Price D. H., Peterlin B. M., PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucleic Acids Res. 40, 9160–9170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sassone-Corsi P., The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 4, a011148 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris P. C., Torres V. E., Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Invest. 124, 2315–2324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallace D. P., Cyclic AMP-mediated cyst expansion. Biochim. Biophys. Acta 1812, 1291–1300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grantham J. J., Autosomal dominant polycystic kidney disease. N. Engl. J. Med. 359, 1477–1485 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Grantham J. J., Mulamalla S., Swenson-Fields K. I., Why kidneys fail in autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 7, 556–566 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Chapin H. C., Caplan M. J., The cell biology of polycystic kidney disease. J. Cell Biol. 191, 701–710 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniel P. B., Walker W. H., Habener J. F., Cyclic AMP signaling and gene regulation. Annu. Rev. Nutr. 18, 353–383 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Garber M. E., Mayall T. P., Suess E. M., Meisenhelder J., Thompson N. E., Jones K. A., CDK9 autophosphorylation regulates high-affinity binding of the human immunodeficiency virus type 1 tat–P-TEFb complex to TAR RNA. Mol. Cell. Biol. 20, 6958–6969 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamaguchi T., Nagao S., Kasahara M., Takahashi H., Grantham J. J., Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am. J. Kidney Dis. 30, 703–709 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Putnam W. C., Swenson S. M., Reif G. A., Wallace D. P., Helmkamp G. M. Jr., Grantham J. J., Identification of a forskolin-like molecule in human renal cysts. J. Am. Soc. Nephrol. 18, 934–943 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Ye H., Wang X., Constans M. M., Sussman C. R., Chebib F. T., Irazabal M. V., Young W. F. Jr., Harris P. C., Kirschner L. S., Torres V. E., The regulatory 1α subunit of protein kinase A modulates renal cystogenesis. Am. J. Physiol. Renal Physiol. 313, F677–F686 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magenheimer B. S., St John P. L., Isom K. S., Abrahamson D. R., De Lisle R. C., Wallace D. P., Maser R. L., Grantham J. J., Calvet J. P., Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na+,K+,2Cl− co-transporter-dependent cystic dilation. J. Am. Soc. Nephrol. 17, 3424–3437 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Cao Y., Semanchik N., Lee S. H., Somlo S., Barbano P. E., Coifman R., Sun Z., Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc. Natl. Acad. Sci. U.S.A. 106, 21819–21824 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krystof V., Baumli S., Furst R., Perspective of cyclin-dependent kinase 9 (CDK9) as a drug target. Curr. Pharm. Des. 18, 2883–2890 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srikumar T., Padmanabhan J., Potential use of flavopiridol in treatment of chronic diseases. Adv. Exp. Med. Biol. 929, 209–228 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Chiaravalli M., Rowe I., Mannella V., Quilici G., Canu T., Bianchi V., Gurgone A., Antunes S., D’Adamo P., Esposito A., Musco G., Boletta A., 2-Deoxy-d -glucose ameliorates PKD progression. J. Am. Soc. Nephrol. 27, 1958–1969 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menezes L. F., Lin C. C., Zhou F., Germino G. G., Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine 5, 183–192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song X., Di Giovanni V., He N., Wang K., Ingram A., Rosenblum N. D., Pei Y., Systems biology of autosomal dominant polycystic kidney disease (ADPKD): Computational identification of gene expression pathways and integrated regulatory networks. Hum. Mol. Genet. 18, 2328–2343 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Sano M., Abdellatif M., Oh H., Xie M., Bagella L., Giordano A., Michael L. H., DeMayo F. J., Schneider M. D., Activation and function of cyclin T–CDK9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat. Med. 8, 1310–1317 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Mascareno E., Galatioto J., Rozenberg I., Salciccioli L., Kamran H., Lazar J. M., Liu F., Pedrazzini T., Siddiqui M. A. Q., Cardiac lineage protein-1 (CLP-1) regulates cardiac remodeling via transcriptional modulation of diverse hypertrophic and fibrotic responses and angiotensin II-transforming growth factor β (TGF-β1) signaling axis. J. Biol. Chem. 287, 13084–13093 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He N., Jahchan N. S., Hong E., Li Q., Bayfield M. A., Maraia R. J., Luo K., Zhou Q., A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol. Cell 29, 588–599 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong P., Chen K., Huang B., Liu M., Cui M., Rozenberg I., Chaqour B., Pan X., Barton E. R., Jiang X.-C., Siddiqui M. A. Q., HEXIM1 controls satellite cell expansion after injury to regulate skeletal muscle regeneration. J. Clin. Invest. 122, 3873–3887 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Z., Yik J. H., Chen R., He N., Jang M. K., Ozato K., Zhou Q., Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19, 535–545 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Jang M. K., Mochizuki K., Zhou M., Jeong H. S., Brady J. N., Ozato K., The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19, 523–534 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Kakade V. R., Tao S., Rajagopal M., Zhou X., Li X., Yu A. S. L., Calvet J. P., Pandey P., Rao R., A cAMP and CREB-mediated feed-forward mechanism regulates GSK3β in polycystic kidney disease. J. Mol. Cell Biol. 8, 464–476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Q., Yik J. H. N., The Yin and Yang of P-TEFb regulation: Implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol. Mol. Biol. Rev. 70, 646–659 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calvet J. P., Polycystic kidney disease: Primary extracellular matrix abnormality or defective cellular differentiation? Kidney Int. 43, 101–108 (1993). [DOI] [PubMed] [Google Scholar]
- 39.Seeger-Nukpezah T., Geynisman D. M., Nikonova A. S., Benzing T., Golemis E. A., The hallmarks of cancer: Relevance to the pathogenesis of polycystic kidney disease. Nat. Rev. Nephrol. 11, 515–534 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franco L. C., Morales F., Boffo S., Giordano A., CDK9: A key player in cancer and other diseases. J. Cell. Biochem. 119, 1273–1284 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Zhou X., Fan L. X., Peters D. J., Trudel M., Bradner J. E., Li X., Therapeutic targeting of BET bromodomain protein, Brd4, delays cyst growth in ADPKD. Hum. Mol. Genet. 24, 3982–3993 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu M., Chen M., Jing Y., Gu J., Mei S., Yao Q., Zhou J., Yang M., Sun L., Wang W., Hu H., Wuthrich R. P., Mei C., The C-terminal tail of polycystin-1 regulates complement factor B expression by signal transducer and activator of transcription 1. Am. J. Physiol. Renal Physiol. 310, F1284–1294 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Tan J. L., Fogley R. D., Flynn R. A., Ablain J., Yang S., Saint-Andre V., Fan Z. P., Do B. T., Laga A. C., Fujinaga K., Santoriello C., Greer C. B., Kim Y. J., Clohessy J. G., Bothmer A., Pandell N., Avagyan S., Brogie J. E., van Rooijen E., Hagedorn E. J., Shyh-Chang N., White R. M., Price D. H., Pandolfi P. P., Peterlin B. M., Zhou Y., Kim T. H., Asara J. M., Chang H. Y., Young R. A., Zon L. I., Stress from nucleotide depletion activates the transcriptional regulator HEXIM1 to suppress melanoma. Mol. Cell 62, 34–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Y., Chen M., Zhou J., Lv J., Song S., Fu L., Chen J., Yang M., Mei C., Interactions between macrophages and cyst-lining epithelial cells promote kidney cyst growth in Pkd1-Deficient Mice. J. Am. Soc. Nephrol. 29, 2310–2325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y., Zhang L., Jones K. A., SKIP counteracts p53-mediated apoptosis via selective regulation of p21Cip1 mRNA splicing. Genes Dev. 25, 701–716 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw3593/DC1
Supplementary Materials and Methods
Fig. S1. PKA phosphorylates HEXIM1 at serine-158.
Fig. S2. Phosphorylation of CDK9 at serine-347 by PKA does not affect P-TEFb/HEXIM1/7SK snRNP complex assembly.
Fig. S3. The phosphorylation level of HEXIM1 is increased in cystic epithelial cells.
Fig. S4. The interaction between P-TEFb and Hexim1 is conserved in zebrafish.
Fig. S5. FP inhibits cystogenesis in the embryonic kidney model.
Fig. S6. Inhibition of CDK9 attenuates cyst development in the MDCK cyst model.
Fig. S7. HEXIM1 phosphorylation and P-TEFb/HEXIM1/7SK snRNP complex assembly in the MDCK cyst model.
Fig. S8. Effect of FP on cystogensis in the early- and late-onset ADPKD mouse models.