

Abstract
Autism spectrum disorder (ASD) is a markedly heterogeneous condition with a varied phenotypic presentation. Its high concordance among siblings, as well as its clear association with specific genetic disorders, both point to a strong genetic etiology. However, the molecular basis of ASD is still poorly understood, although recent studies point to the existence of sex-specific ASD pathophysiologies and biomarkers. Despite this, little is known about how exactly sex influences the gene expression signatures of ASD probands. In an effort to identify sex-dependent biomarkers and characterise their function, we present an analysis of a single paired- end post-mortem brain RNA-Seq data set and a meta-analysis of six blood-based microarray data sets. Here, we identify several genes with sex-dependent dysregulation, and many more with sex-independent dysregulation. Moreover, through pathway analysis, we find that these sex-independent biomarkers have substantially different biological roles than the sex-dependent biomarkers, and that some of these pathways are ubiquitously dysregulated in both post- mortem brain and blood. We conclude by synthesizing the discovered biomarker profiles with the extant literature, by highlighting the advantage of studying sex-specific dysregulation directly, and by making a call for new transcriptomic data that comprise large female cohorts.
Keywords: transcriptome, sex-specific, biomarkers, ASD, autism
1. Introduction
Autism Spectrum Disorder (ASD) is a markedly heterogeneous condition with a varied phenotypic presentation and a spectrum of disability for those affected. As a neurodevelopmental disorder, the ASD syndrome is characterised by social abnormalities, language abnormalities, and stereotyped behavioural patterns [Bailey et al. (1996)]. The presence of a genetic link in ASD etiology is well- established [Miles (2011); Miyauchi and Voineagu (2013)], as first evidenced by ASD concordance among siblings and by a clear association between ASD and specific genetic disorders (e.g., Fragile X mental retardation) [Bailey et al. (1996)]. This link has prompted a number of transcriptomic studies (e.g., [Hertz-Picciotto et al. (2006); Glatt et al. (2012); Gupta et al. (2014a)]) to identify gene expression signatures as a biomarker that might help elucidate the etiology of ASD and aid in its diagnosis (an important objective since early diagnosis and therapy is shown to improve outcomes in ASD [Elder et al. (2017)]). However, despite the number of transcriptomic studies performed, the pathophysiology and biomarker profile of ASD are still not known. Rather, these studies have tended to produce inconsistent results, suggesting wide heterogeneity among both the individual patients and the study populations, although several studies have converged to find associations among neuron-specific genes [Gupta et al. (2014b); Parikshak et al. (2016); Voineagu et al. (2011)]. Indeed, ASD may not have one signature at all, but instead multiple diverging signatures [Tylee et al. (2017a)].
Transcriptomic studies of ASD probands typically use cells collected from either post-mortem brains or blood in order to estimate the mRNA abundance for thousands of gene transcripts, by way of microarray technology or massively parallel high-throughput sequencing (RNA-Seq). Since many expressed transcripts are a precursor to structural or functional proteins, these studies can provide an insight into the functional state of a cell, capturing the common pathway for hereditary predisposition and environmental exposure. Although post-mortem brain studies have an advantage in that they look directly at the tissue of interest, blood-based studies can identify clinically useful biomarkers while also serving as a reliable proxy for gene expression in the brain [Tylee et al. (2013)], though a complete understanding of ASD pathophysiology and its biomarker profile will likely require careful consideration of both lines of evidence. To date, more than a dozen studies have measured the transcriptomic profiles of ASD probands and controls, the results of which have been summarised by two separate meta-analyses [Ch’ng et al. (2015); Ning et al. (2015)] and one mega-analysis [Tylee et al. (2017a)].
Sex is often called a risk factor for ASD, and it is stated that the risk for a male to have ASD is four to five times higher than that for females [Werling et al. (2016); Christensen et al. (2016)] (although the magnitude of this difference may be partly due to diagnostic biases [Lai et al. (2015)]). A similar observation, that the increased male risk is even higher among high-functioning ASD probands [Fombonne (1999)], likewise suggests that sex-specific mechanisms could influence ASD pathophysiology and its biomarker profile. Further evidence for sex-specific mechanisms is found in recent transcriptomic and functional-imaging studies. For example, Tylee et al., using transformed lymphoblastoid cell lines, found evidence for sex-specific differential regulation of genes and pathways among ASD probands [Tylee et al. (2017)]. Similarly, Trabzuni et al. found sex-specific differences in alternative splicing in adult human brains, including for a well-known ASD risk gene NRXN3 [Trabzuni et al. (2013)]. Functional brain connectivity studies using fRMI imaging have also identified sexual heterogeneity among ASD probands, showing dysregulation in sexually dimorphic brain regions across two large studies [Floris et al. (2018); Lai et al. (2013)]. Moreover, recent work by Mitra et al. found evidence for pleiotropy between common single nucleotide polymorphisms (SNPs) for secondary sex characteristics and ASD risk, as well as sex heterogeneity on the X-chromosome, through a comprehensive SNP mega-analysis combining 12 individual data sets from diverse genetic backgrounds [Mitra et al. (2016)]. Taken together, it seems plausible that sex could interact with other genetic and environmental factors to create sex-specific ASD pathophysiologies and biomarker profiles.
As ASD is more common in males, it suggests that females may have some underlying protection whereby a higher risk load is required for them to become afflicted [Robinson et al. (2013)]. One hypothesis posits that ASD itself reflects a shift towards “extreme maleness” such that males are necessarily predisposed [Baron-Cohen (2002)]. In support of this, females with ASD do harbour more and larger copy number variants than males with ASDs [Levy et al. (2011)], and moreover exhibit differential penetrance given the same genetic etiology [Lionel et al. (2014)], although Mitra et al. found no evidence for an increased SNP load in females [Mitra et al. (2016)]. Unfortunately, however, the increased prevalence of ASD in males has led to the exclusion of females from many transcriptomic studies (e.g., [Hu et al. (2009); Sarachana et al. (2010); Alter et al. (2011)]), making it difficult to understand the male skew in ASD prevalence. Indeed, individual studies are often underpowered to detect subtle sex-specific differences, if they contain female subjects at all. When female subjects are included, sex is typically modelled as a simple covariate rather than an ASD-sex interaction, meaning that only sex-independent, and not sex-dependent, biomarkers are discovered. When male ASD is contrasted with female ASD, it typically involves loosely comparing simple sex- specific differences in a statistically anticonservative manner (e.g., noting differential expression in males but not females, or vice versa). To our knowledge, no study has looked at whether gene expression signatures show a sex-autism interaction across multiple studies and human tissues.
Using a meta-analysis of six blood-based microarray data sets and an exploratory analysis of a single paired-end post-mortem brain RNA-Seq data set, we present an analysis of transcriptomic data that focuses on comparing sex-dependent and sex-independent ASD biomarkers across multiple tissues. By modelling the interaction of sex and ASD directly, we identify biomarkers and functional pathways that show sex-differences in ASD probands that are different than those in control subjects. Then, for those biomarkers that show no interaction, we pool male and female probands to look for sex-independent biomarkers. Our results suggest that, despite low power, some genes have FDR-adjusted significant sex-dependent interactions, while even more have significant sex- independent main effects. Subsequent pathway analysis further shows that these sex-independent biomarkers have substantially different biological roles than the sex-dependent biomarkers, and that some of these pathways are dysregulated in both post-mortem brain and blood.
2. Methods
2.1. Data acquisition
2.1.1. Microarray data
We collected multiple microarray data sets to perform a meta-analysis of sex-autism interactions and main effects of ASD (i.e., sex-independent effects, where males and females are pooled). We referenced two prior meta-analyses [Ch’ng et al. (2015); Ning et al. (2015)], and one mega-analysis [Tylee et al. (2017a)], to prepare a list of data sets to study. Of these data sets, we excluded any study that (a) measured transcript expression from brain tissue, (b) had no female cases, (c) used cell lines (i.e., GSE37772 and GSE43076), or (d) treated cells (i.e., GSE32136, treated with PPA). Six data sets remained after exclusion, as described in Table 1.
Table 1:
This table details all studies included in the meta-analysis, and the number of probes available after establishing a final probe set. We filtered each data set so that all subjects in the ASD group had a diagnosis of autism spectrum disorder. Some typically developing subjects from the Glatt et al. data sets were considered “Type-1 errors”.
| Study ID | Probes (Intersect) | Females (TD) | Males (TD) | Females (ASD) | Males (ASD) |
|---|---|---|---|---|---|
| GSE6575 | 39561 | 3 | 9 | 8 | 36 |
| GSE18123 | 19532 | 34 | 48 | 24 | 80 |
| Glatt et al. Wave I | 28424 | 28 | 40 | 11 | 49 |
| Glatt et al. Wave II | 28424 | 35 | 56 | 28 | 85 |
| CHARGE | 39561 | 15 | 75 | 15 | 103 |
| Kong et al. 2013 | 19532 | 7 | 10 | 7 | 46 |
Data acquired from the Gene Expression Omnibus (GEO) [Barrett and Edgar (2006)] (i.e., GSE6575 [Gregg et al. (2008)] and GSE18123 [Kong et al. (2012)]) were acquired already normalised and were not modified further. The other data sets (i.e., the Glatt et al. Wave I and Wave II data [Glatt et al. (2012)], the CHARGE study data [Hertz-Picciotto et al. (2006)], and the Kong et al. 2013 data [Kong et al. (2013)]) each underwent RMA normalization, quantile normalization, and base-2 logarithm transformation. We filtered each data set so that all subjects in the ASD group had a diagnosis of autism spectrum disorder. Some typically developing subjects from the Glatt et al. data sets were considered “Type-1 errors”.
2.1.2. RNA-Seq data
We searched for relevant publicly available RNA-Seq data using the Gene Expression Omnibus (GEO) [Barrett and Edgar (2006)] with the term (“expression profiling by high throughput sequencing”[DataSet Type] AND (“autism spectrum disorder”[MeSH Terms] OR “autistic disorder”[MeSH Terms])) AND “homo sapiens”[Organism] (query made January 2018). We restricted eligible data sets to those sequenced with paired-end and non-poly-A-selected libraries. After excluding any data sets that used cell lines or did not have female cases, only one experiment, GSE107241 [Wright et al. (2017)], remained. These data comprise a RiboZero Gold paired-end RNA-Seq data set from 52 postmortem dorsolateral prefrontal cortex tissue samples (10 ASD males, 3 ASD females, 30 control males, and 9 control females). No other data met this search criteria.
Prior to alignment and quantification, raw RNA-Seq reads were trimmed using Trimmomatic (docker image quay.io/biocontainers/trimmomatic:0.36–4) [Bolger et al. (2014)] and quality control metrics were recorded (before and after trimming) using FastQC (docker image biocontainers/fastqc:0.11.5) [Andrews (2010)]. We aligned trimmed reads and quantified expression using Salmon (docker image combinelab/salmon:0.9.0) [Patro et al. (2017)] as run in pseudo-quantification mode with a k-mer index of length 31. For the reference, we concatenated a human coding reference (i.e., GRCh38.90.cds) with the corresponding non-coding reference (i.e., GRCh38.90.ncrna).
2.2. Meta-analysis of microarray data
Before proceeding with the meta-analysis, we established a set of probes for each microarray platform that represent genes also represented by probes in the other platforms. In other words, we established a final probe set based on the intersection of unique gene symbols present in all microarray platforms under study.
For each microarray data set, and for each probe (i.e., of those representing genes found in all data sets), we performed differential expression analysis using limma (Version 3.34) [Smyth (2004)], applying the following steps: (1) fit a single model with the formula ~ASD + Sex + ASD:Sex + Age where ASD and Sex are each two-level factors (except GSE6575, where the Age covariate is unknown), (2) define contrasts for the sex-autism interaction and for the sex-independent main effects, and (3) measure the differential expression for each contrast using the eBayes procedure. In other words, we fit a single model but pulled out the appropriate contrasts in two steps.
Next, we transformed platform-specific probe p-values to HGNC symbol p-values using AnnotationDbi (available from Bioconductor [Huber et al. (2015)]). We resolved many-to-one mapping ambiguities by FDR-adjusting the minimum p-value of all probes for a given gene symbol (i.e., calculating a within-gene FDR correction). We then used Fisher’s method to perform a meta-analysis of the p-values obtained from the differential expression analysis. For K studies, Fisher’s method scores each gene based on negative two times the sum of the logarithm of the p-values:
This score follows a χ2 distribution with 2K degrees of freedom [Mosteller and Fisher (1948)]. Thus, for each gene, we computed a p-value directly from this score. We corrected for multiple testing using the Benjamini-Hochberg procedure [Benjamini and Hochberg (1995)].
2.3. Differential expression analysis of RNA-Seq data
We used DESeq2 (Version 3.6) [Love et al. (2014)] to test for differential transcript expression within the Salmon-generated counts. We applied a conservative expression filter (i.e., at least 10 estimated counts per-gene in every sample) to the raw count matrix to ensure that the high variability of lowly expressed transcripts did not bias results due to the small group sizes. For each transcript that passed the expression filter, a single model was fit using the formula ~ASD + Sex + ASD:Sex + Age (where Age is the age of death). Interaction and sex-independent main effects were then extracted from the model by specifying the relevant contrasts to the DESeq2::results function. Again, we fit a single model but pulled out the appropriate contrasts in two steps. We corrected for multiple testing using the Benjamini-Hochberg procedure [Benjamini and Hochberg (1995)].
2.4. Adjustment of latent batch effects
To ensure that latent batch effects did not inflate the discovery of false positives, we performed all analyses above with adjustment for batch effects using sva (Version 3.26) [Leek et al. (2012); Leek (2014)], applying the following steps: (1) estimate the number of surrogate variables while specifying the ASD * Sex interaction as the variable of interest (via a 4-level factor that contains ‘MaleASD’, ‘FemaleASD’, ‘MaleControl’, and ‘FemaleControl’) and Age as an adjustment variable, (2) use the sva function (or, in the case of Salmon-generated counts, the svaseq function) to estimate the surrogate variables, and (3) include the surrogate variables in the differential expression model(s) described above. Generally speaking, using sva yielded more conservative results than not using sva. All tables and figures show results generated with sva except where otherwise noted.
2.5. Pathway analysis and knowledge integration
We performed pathway analysis using GSEA (Version 3.0) [Subramanian et al. (2005)] in PreRanked mode with classic enrichment and 1,000 permutations. Enrichment scores were calculated for specific MSigDB (Version 6.1) [Subramanian et al. (2005); Liberzon et al. (2011)] gene sets, including the curated KEGG (c2.cp.kegg [Kanehisa et al. (2017)]), Gene Ontology Biological Process (c5.bp) [The Gene Ontology Consortium (2017)], Reactome (c2.cp.reactome) [Fabregat et al. (2018)], and MSigDB Hallmark (h.all) [Liberzon et al. (2015)] sets.
Based on the nature of the analyses, input rank lists were prepared differently for the RNA-Seq and microarray results. For the RNA-Seq analysis, we ranked transcripts based on the p-value, p, and the direction of the fold-change, FC:
where positive FC refers to up-regulation in ASD for the sex-independent main effects. Then, these transcript-level ranks were converted into gene-level ranks based on the top transcript-level rank. For the microarray meta-analysis, we ranked genes using the χ2 test statistic (as calculated from Fisher’s method). Note that since this latter metric is agnostic to the direction of expression changes (i.e., only large χ2 test statistics suggest dysregulation), we focused here on pathways enriched with a positive score (effectively making this pathway enrichment test one-tailed). In both cases, we set α = 0.30.
3. Results
3.1. Evidence for sex-dependent autism biomarkers
By modelling the sex-autism interaction directly, we can detect gene expression signatures that have differential dysregulation in male ASD probands when compared with female ASD probands. In other words, we can find sexually dimorphic ASD biomarkers (e.g., a gene up-regulated in male ASD but not in female ASD, or vice versa). Despite small study sizes, and disproportionately fewer females, we find some evidence for a sex-autism interaction among biomarkers, especially throughout the microarray meta-analysis data.
From the analysis of the RNA-Seq data derived from post-mortem brain tissue, we find no transcripts with significant (FDR-adjusted p-value < 0.05) sex-dependent dysregulation, although one of these transcripts showed a significant interaction prior to batch correction with sva. To illustrate what a sex-autism interaction might look like, Figure 1 shows the per-group expression profiles for the two transcripts with the largest interaction effect (i.e., those with the smallest corrected p-value). Table 2 characterises those transcripts with the most sex-dependent dysregulation.
Figure 1:
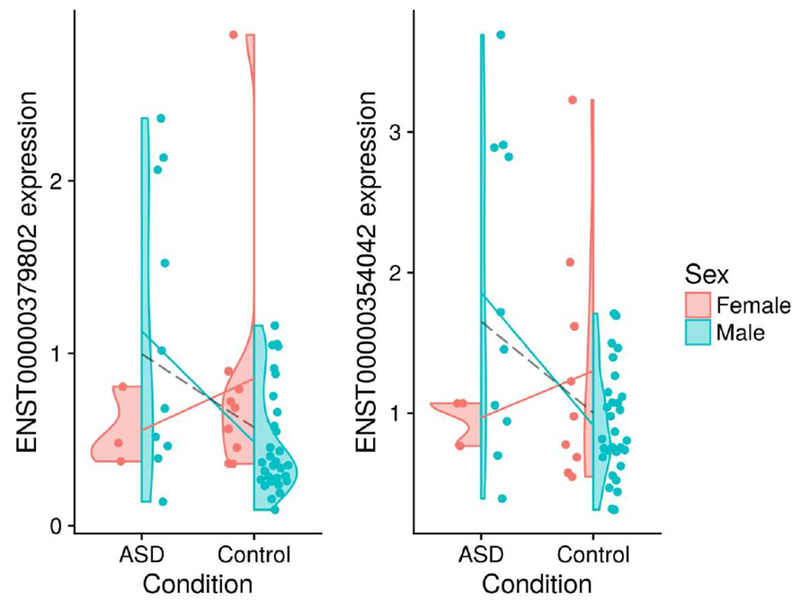
These violin plots show the base-2 logarithm-transformed expression for the two transcripts with the largest interaction effect from the RNA-Seq data (i.e., those with the smallest corrected p-value). The solid lines show sex-specific mean expression differences. The dashed line shows the sex-independent (i.e., pooled) mean expression difference. The left and right transcripts are associated with the DSP and SLC13A4 genes, respectively.
Table 2:
This table shows SVA-adjusted results for the sex-autism interaction for the RNA-Seq data (sorted by FDR-adjusted p-value). Note that FDR-adjusted p-values are also shown for an analysis performed without the adjustment of latent batch effects.
| Transcript ID | Gene symbol | Transcript biotype | Log 2 FC | P-adj SVA | P-adj (no SVA) |
|---|---|---|---|---|---|
| ENST00000354042 | SLC13A4 | protein coding | 3.27 | 0.293 | 0.1136846 |
| ENST00000379802 | DSP | protein coding | 3.19 | 0.293 | 0.6534814 |
| ENST00000262551 | OGN | protein coding | 2.97 | 0.299 | 0.8169099 |
| ENST00000371625 | PTGDS | protein coding | 1.74 | 0.299 | 0.0329544 |
| ENST00000223357 | AEBP1 | protein coding | 1.85 | 0.529 | 0.8713166 |
From the meta-analysis of the blood-based microarray data, we find four genes with significant (FDR-adjusted) sex-dependent dysregulation: TTF2, UTY, KCNJ8, and NCS1. Table 3 characterises those genes with the most sex-dependent dysregulation. For Fisher’s method, a very small p-value in only one study could cause the meta-analysis to post a significant result [Tseng et al. (2012)]. Therefore, it is useful to inspect visually how each study contributed to the results of the meta-analysis. Figure 2 shows how each study contributed to the meta-analysis findings by plotting the aggregate Fisher score for each gene with large sex-dependent dysregulation, along with the study-wise nominal significance (unadjusted p-value < 0.05). Notably, several of the most significantly dysregulated genes are at least nominally significant in more than one study.
Table 3:
This table shows genes with the most sex-dependent dysregulation (and their chromosomal position) for the meta-analysis results, sorted by Fisher score and adjusted p-value. In addition, this table shows the Fisher score and adjusted p-value calculated for an analysis repeated without the adjustment of latent batch effects.
| Location | Fisher | Fisher p-adj | Fisher (no SVA) | Fisher p-adj (noSVA) | |
|---|---|---|---|---|---|
| TTF2 | 1p13.1 | 52.80272 | 0.0080909 | 30.30688 | 1.0000000 |
| UTY | Yq11.221 | 49.17813 | 0.0352693 | 45.32043 | 0.1644908 |
| KCNJ8 | 12p12.1 | 48.80700 | 0.0409512 | 41.60015 | 0.7040022 |
| NCS1 | 9q34.11 | 48.69443 | 0.0428454 | 33.16947 | 1.0000000 |
| RAP2C | Xq26.2 | 47.97863 | 0.0571022 | 25.06737 | 1.0000000 |
| CHST11 | 12q23.3 | 47.29425 | 0.0750785 | 21.86910 | 1.0000000 |
| MAP1B | 5q13.2 | 47.18032 | 0.0785676 | 44.44789 | 0.2320088 |
| CRHR1 | 17q21.31 | 47.15296 | 0.0794259 | 43.43498 | 0.3450932 |
| PAFAH1B1 | 17p13.3 | 46.36425 | 0.1087281 | 17.26173 | 1.0000000 |
| SH3BGR | 21q22.2 | 46.35975 | 0.1089168 | 32.82178 | 1.0000000 |
| PAK3 | Xq23 | 45.67211 | 0.1430652 | 46.45645 | 0.1048620 |
| FMO1 | 1q24.3 | 44.77510 | 0.2038808 | 32.85009 | 1.0000000 |
| TROVE2 | 1q31.2 | 44.42306 | 0.2341680 | 21.84958 | 1.0000000 |
| BNC2 | 9p22.3-p22.2 | 44.40285 | 0.2360233 | 39.13101 | 1.0000000 |
| DCUN1D1 | 3q26.33 | 44.08602 | 0.2672865 | 40.02468 | 1.0000000 |
| HECA | 6q24.1 | 44.06996 | 0.2689620 | 33.92752 | 1.0000000 |
| RORA | 15q22.2 | 43.52061 | 0.3335278 | 34.11106 | 1.0000000 |
| ARHGAP35 | 19q13.32 | 43.40139 | 0.3494222 | 41.54671 | 0.7186470 |
| CAMK1D | 10p13 | 43.34605 | 0.3570412 | 23.46336 | 1.0000000 |
| SLC27A6 | 5q23.3 | 43.26356 | 0.3687130 | 42.46235 | 0.5041354 |
| SNX13 | 7p21.1 | 43.07811 | 0.3963678 | 27.35225 | 1.0000000 |
| ACKR2 | 3p22.1 | 43.06703 | 0.3980622 | 36.05068 | 1.0000000 |
| KCNN2 | 5q22.3 | 42.76385 | 0.4479560 | 44.03212 | 0.2731511 |
| TEX35 | 1q25.2 | 42.72490 | 0.4547740 | 40.79263 | 0.9608426 |
| TMEM56 | 1p21.3 | 42.56794 | 0.4833811 | 34.90678 | 1.0000000 |
| SYTL4 | Xq22.1 | 42.49851 | 0.4965789 | 40.68910 | 0.9997724 |
| AZGP1 | 7q22.1 | 42.48820 | 0.4985441 | 27.40362 | 1.0000000 |
Figure 2:
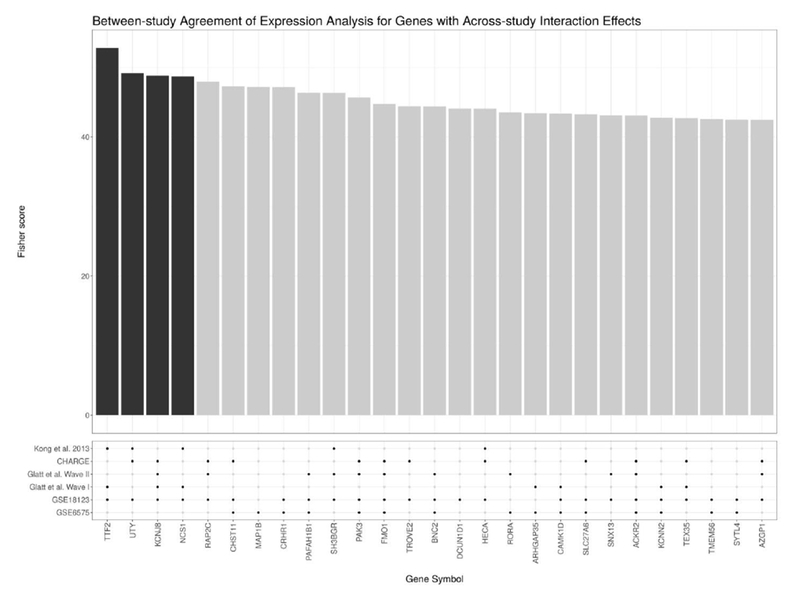
This figure shows the genes with the most significant sex-dependent dysregulation (i.e., a sex-autism interaction) according to the meta-analysis of the microarray data. Above, the bar plot shows the χ2 score for each gene as calculated using Fisher’s method (where the dark bars indicate that the gene has an FDR-adjusted p-value < 0.05). Below, the dot plot shows whether a gene showed a nominally significant sex-dependent dysregulation at an unadjusted p-value < 0.05 for a given study. Note that most genes selected for by the meta-analysis show at least nominal significance across multiple studies.
3.2. Evidence for sex-independent autism biomarkers
In situations where a sex-autism interaction is not detectable, we can proceed to measure sex- independent main condition effects by pooling male ASD probands with female ASD probands (and male controls with female controls), without having to model sex as a covariate. Genes with significant sex-independent main effects have large unidirectional effect sizes in male ASD probands, female ASD probands, or both. Yet, because the interaction is tested first, we can interpret the main condition effects as sex-independent.
From the analysis of the RNA-Seq data derived from post-mortem brain tissue, we find seven transcripts with significant (FDR-adjusted p-value < 0.05) sex-independent differential expression. Of these, only one transcript showed significant up-regulation in ASD (with all others showing down-regulation). Figure 3 shows the expression profile for the two transcripts with the most significant sex-independent main effects. Table 4 characterises those transcripts with significant sex-independent dysregulation. Interestingly, several of the transcripts called differentially expressed by the analysis are annotated as non-coding RNA species.
Figure 3:
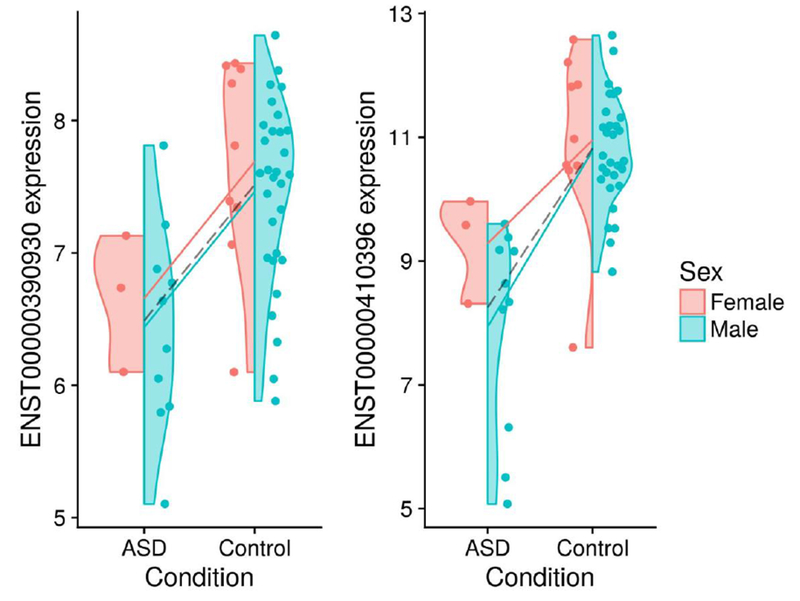
These violin plots show base-2 logarithm-transformed expression for the two most significant main effects (i.e., of the ASD condition) from the RNA-Seq data. The solid lines show sex-specific mean expression differences. The dashed line shows the sex-independent (i.e., pooled) mean expression difference. The left and right transcripts are associated with the SNORD17 and RN U 2 − 2P genes, respectively.
Table 4:
This table shows SVA-adjusted results for the main effects (i.e., of the ASD condition) for the RNA-Seq data (sorted by FDR-adjusted p-value). Note that FDR-adjusted p-values are also shown for an analysis performed without the adjustment of latent batch effects.
| Transcript ID | Gene symbol | Transcript biotype | Log 2 FC | P-adj (SVA) | P-adj (no SVA) |
|---|---|---|---|---|---|
| ENST00000390930 | SNORD17 | snoRNA | −2.98 | 1.54e-05 | 0.0000102 |
| ENST00000410396 | RNU2-2P | snRNA | −4.76 | 4.04e-05 | 0.0000000 |
| ENST00000613119 | snRNA | −3.23 | 9.18e-05 | 0.0000000 | |
| ENST00000258526 | PLXNC1 | protein coding | 0.48 | 0.00468 | 0.4273372 |
| ENST00000393775 | IGSF11 | protein coding | −1.18 | 0.00468 | 1.0000000 |
| ENST00000459255 | SCARNA10 | snoRNA | −1.71 | 0.00468 | 0.0014803 |
| ENST00000618786 | RN7SL1 | misc RNA | −1.35 | 0.0124 | 0.0026454 |
From the meta-analysis of blood-based microarray data, we find over 20 genes with significant (FDR-adjusted) sex-independent dysregulation. Table 5 characterises those genes with the most sex-independent dysregulation. As in Figure 2, Figure 4 shows how each study contributed to the meta-analysis findings by plotting the aggregate Fisher score for each gene with large sex- independent dysregulation, along with the study-wise nominal significance (unadjusted p-value < 0.05). Again, most genes selected as statistically significant by the meta-analysis are at least nominally significant in more than one study.
Table 5:
This table shows genes with the most sex-independent dysregulation (and their chromosomal position) for the meta-analysis results, sorted by Fisher score and adjusted p-value. In addition, this table shows the Fisher score and adjusted p-value calculated for an analysis repeated without the adjustment of latent batch effects.
| Location | Fisher | Fisher p-adj | Fisher (no SVA) | Fisher p-adj (noSVA) | |
|---|---|---|---|---|---|
| ARHGAP35 | 19q13.32 | 69.93173 | 0.0000060 | 59.58783 | 0.0004856 |
| GIMAP8 | 7q36.1 | 59.63083 | 0.0004774 | 52.83287 | 0.0079551 |
| UCHL3 | 13q22.2 | 55.73392 | 0.0024208 | 26.61452 | 1.0000000 |
| SPART | 13q13.3 | 55.39389 | 0.0027862 | 42.18348 | 0.5444711 |
| MAGED2 | Xp11.21 | 54.65897 | 0.0037735 | 31.43801 | 1.0000000 |
| ZNF721 | 4p16.3 | 54.38812 | 0.0042188 | 46.99662 | 0.0833787 |
| TCEAL8 | Xq22.1 | 54.30254 | 0.0043699 | 31.39363 | 1.0000000 |
| KIF13B | 8p12 | 53.86902 | 0.0052226 | 44.83078 | 0.1953472 |
| COX19 | 7p22.3 | 51.12904 | 0.0160066 | 51.00574 | 0.0167250 |
| POLR1A | 2p11.2 | 50.82535 | 0.0181073 | 32.52993 | 1.0000000 |
| NUCB2 | 11p15.1 | 50.20003 | 0.0233305 | 49.91295 | 0.0260057 |
| EIF3A | 10q26.11 | 49.84351 | 0.0269484 | 47.30791 | 0.0736972 |
| GNG5 | 1p22.3 | 49.73328 | 0.0281743 | 23.65365 | 1.0000000 |
| HNRNPA3P1 | 10q11.21 | 49.54482 | 0.0304006 | 51.23881 | 0.0152201 |
| KLF1 | 19p13.13 | 49.43419 | 0.0317868 | 35.96265 | 1.0000000 |
| GNPDA1 | 5q31.3 | 48.90133 | 0.0393978 | 41.09008 | 0.8257563 |
| SART3 | 12q23.3 | 48.88131 | 0.0397143 | 52.43134 | 0.0093729 |
| CCNC | 6q16.2 | 48.87932 | 0.0397440 | 16.14519 | 1.0000000 |
| UBE2A | Xq24 | 48.87429 | 0.0398222 | 23.54974 | 1.0000000 |
| ESF1 | 20p12.1 | 48.70005 | 0.0427109 | 39.78234 | 1.0000000 |
| ZNF740 | 12q13.13 | 48.63177 | 0.0438971 | 46.72013 | 0.0930420 |
| MTERF4 | 2q37.3 | 48.49211 | 0.0464282 | 37.44203 | 1.0000000 |
| CCP110 | 16p12.3 | 47.96099 | 0.0574500 | 28.55662 | 1.0000000 |
| JUND | 19p13.11 | 47.85553 | 0.0599253 | 37.46297 | 1.0000000 |
| MTERF1 | 7q21.2 | 47.82145 | 0.0607453 | 29.44710 | 1.0000000 |
| ZNF569 | 19q13.12 | 47.57334 | 0.0670814 | 36.67860 | 1.0000000 |
| PGM1 | 1p31.3 | 46.90219 | 0.0876872 | 37.26441 | 1.0000000 |
| ECI2 | 6p25.2 | 46.79028 | 0.0916801 | 51.52112 | 0.0135729 |
| ARHGAP8 | 22q13.31 | 46.67208 | 0.0960926 | 46.17090 | 0.1155403 |
Figure 4:
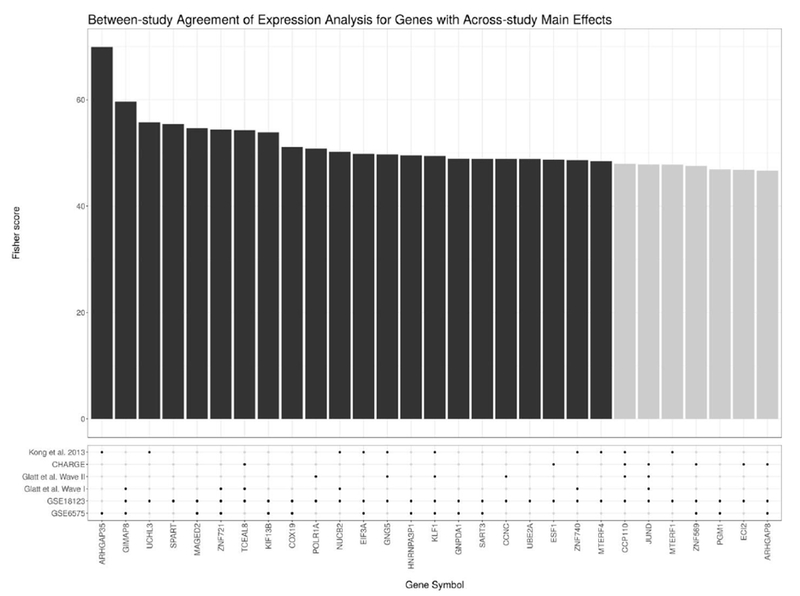
This figure shows the genes with the most significant sex-independent main effects (i.e., of the ASD condition) according to the meta-analysis of the microarray data. Above, the bar plot shows the χ2 score for each gene as calculated using Fisher’s method (where the dark bars indicate that the gene has an FDR-adjusted p-value < 0.05). Below, the dot plot shows whether a gene showed a nominally significant sex-independent main effect at an unadjusted p-value < 0.05 for a given study. Note that most genes selected for by the meta-analysis show at least nominal significance across multiple studies.
3.3. Pathway enrichment of ASD biomarkers
In an effort to summarise the biological relevance of the biomarker profiles generated above, we used the complete ranked lists of the differentially expressed transcripts and genes in four separate gene set enrichment analyses to identify common differentially regulated pathways. Four enrichment profiles were generated using the sex-dependent RNA-Seq brain biomakers, sex-independent RNA-Seq brain biomarkers, sex-dependent microarray blood biomarkers, and sex-independent microarray blood biomarkers.
Figure 5 shows the KEGG pathways enriched by the biomarkers as ranked by the analysis of the RNA-Seq data. Interestingly, all significant enrichment occurred in the same direction. Figure 6 shows the KEGG pathways enriched by the biomarkers as ranked by the analysis of the microarray data. Unlike the RNA-Seq enrichment analysis, the meta-analysis enrichment analysis is agnostic to direction. Figure 7 compares the overlap between these significant pathways. For the sex-dependent analyses, no pathways are enriched in both the RNA-Seq and microarray data. However, for the sex-independent analyses, two pathways are enriched in both data (i.e., Ribosome and Proteasome), though this observation might have occurred by chance. Note that we also tested for enrichment among the Gene Ontology Biological Process, Reactome, and MSigDB Hallmarks gene sets, all of which show more examples of overlap between the separate sex-independent analyses (see the Supplementary Information for more details). We make the complete pathway enrichment results for the interaction and main effects found in the RNA-Seq and microarray data sets all available at at https://doi.org/10.5281/zenodo.1421429.
Figure 5:
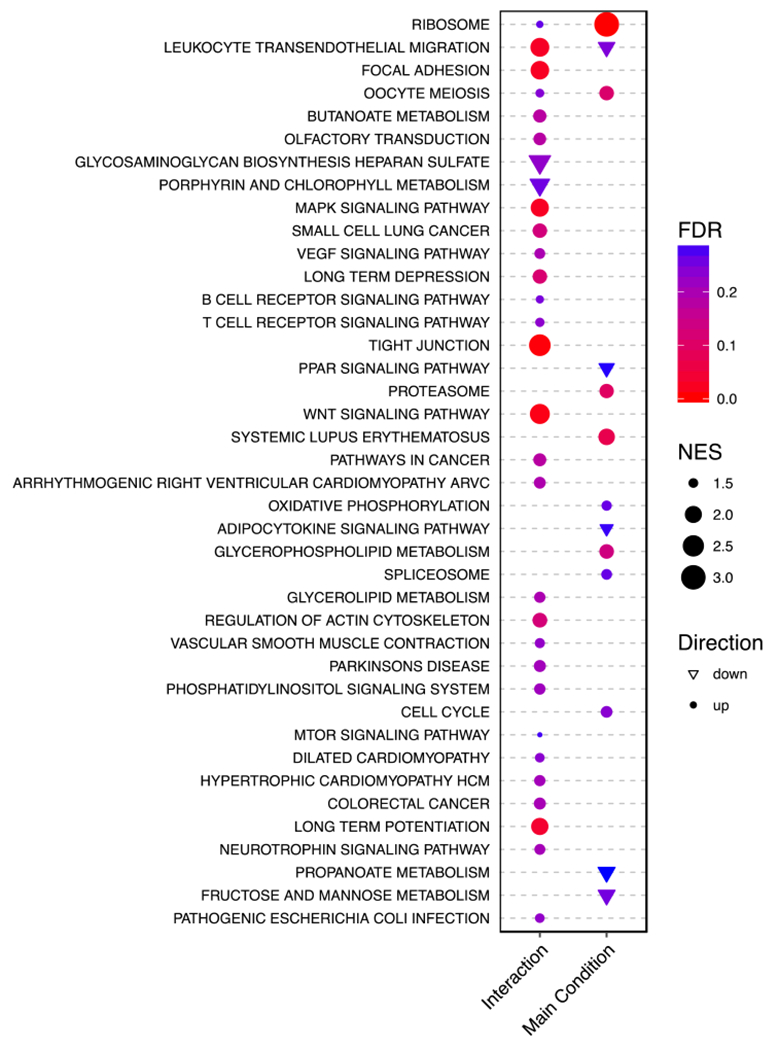
This dot plot shows results from a GSEA of the RNA-Seq data against the MSigDB KEGG pathways. For the sex-autism interaction and the main effect results, a KEGG pathway (y-axis) has a circle (or triangle) if it is enriched (or depleted). The size of the points indicates the absolute normalised enrichment score. The colour indicates the FDR. Note that only points with an FDR < 0.3 are plotted (see Methods). The enrichment score is defined as the degree to which a gene set is over-represented at the top or bottom of a ranked gene list as calculated by GSEA. The ratio of this to the expected enrichment score of all permutations is the normalised enrichment score (NES).
Figure 6:
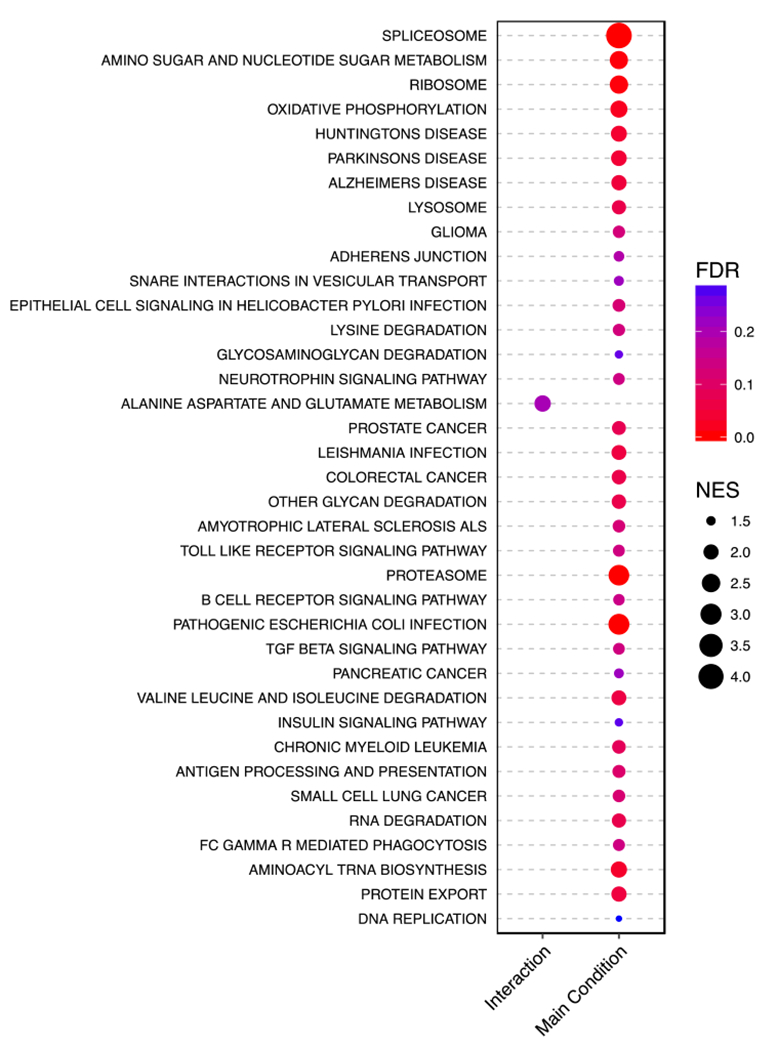
This dot plot shows results from a GSEA of the meta-analysis data against the MSigDB KEGG pathways. For the sex-autism interaction and the main effect results, a KEGG pathway (y-axis) has a circle if it is enriched. The size of the points indicates the absolute normalised enrichment score. The colour indicates the FDR. Note that only points with an FDR < 0.3 are plotted (see Methods). The enrichment score is defined as the degree to which a gene set is over- represented at the top or bottom of a ranked gene list as calculated by GSEA. The ratio of this to the expected enrichment score of all permutations is the normalised enrichment score (NES).
Figure 7:
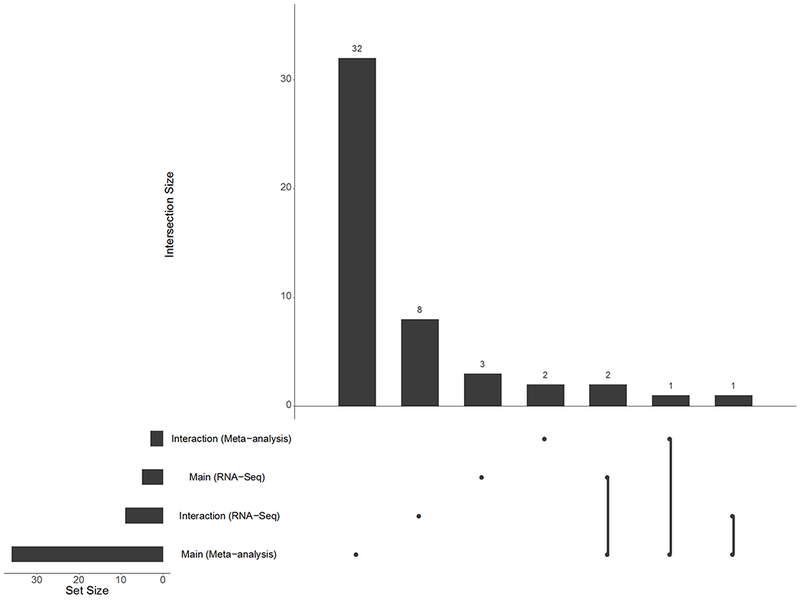
This UpSet plot [Lex et al. (2014)] shows set intersections (and their sizes) from a GSEA of four results against the MSigDB KEGG pathways. Set identity is indicated by the joined lines. Set size is indicated by the top bar chart. The bar chart on the left shows the total set size for each individual GSEA run. Results are filtered using a liberal FDR threshold of FDR < 0.15 for the RNA-Seq data and FDR < 0.3 for the meta-analysis data (see Methods).
4. Discussion
In this report, we present an analysis of several ASD transcriptomic studies, including a meta- analysis of six blood-based microarray data sets and an exploratory analysis of RNA-Seq data derived from post-mortem brain. In both analyses, we focus on identifying sex-dependent and sex-independent biomarker profiles for ASD by modelling the sex-autism interaction directly. In addition to identifying transcript and gene biomarkers, we use gene set enrichment analysis to summarise the observed dysregulation at the pathway level, contrasting sex-dependent pathway enrichment with sex-independent pathway enrichment. In doing so, we find that some pathways are across both tissues (i.e., Ribosome and Proteasome), though this observation might have occurred by chance.
Despite small sample sizes in all studies, we found evidence for the existence of some sex- dependent biomarkers in human tissue. The meta-analysis identified four genes with significant (FDR-adjusted) sex-dependent dysregulation in the blood: TTF2, UTY, KCNJ8, and NCS1. One of these, TTF2, plays an important role in normal thyroid development [De Felice and Di Lauro (2004)]. Interestingly, a loss of thyroid hormone homoeostasis has been linked to ASD [Berbel et al. (2014); Khan et al. (2014)]. Since it is well-known that thyroid diseases have a sex-specific presentation [Bauer et al. (2014)], it seems plausible that thyroid abnormalities could contribute to a sexually dimorphic ASD signature. Some thyroid-disrupting environmental chemicals have also been linked to an altered risk for autism [Lyall et al. (2017); Braun et al. (2014)], including one study showing sexually dimorphic associations [Lyall et al. (2017)]. The other, U T Y , is a Y-chromosome gene (with considerable homology to an X-chromosome homolog), making any interpretation of its differential dysregulation difficult. Two other genes, KCNJ8 and NCS1, are involved in potassium and calcium channel activity, respectively, the latter of which has been implicated in autism [Handley et al. (2010)]. Although the RNA-Seq analysis did not yield any significant interactions, it is not surprising considering this data set contained only three female ASD probands. Nevertheless, the large (albeit non-significant) effect sizes warrant repeat studies with bigger cohorts and more female ASD probands.
By modelling the sex-autism interaction directly, we are able to follow-up the sex-dependent analysis with a secondary sex-independent analysis for any transcript or gene whose expression did not significantly interact with sex. Using the same regression model, we contrast the pooled male ASD probands and female ASD probands against the pooled male controls and female controls to calculate the main effects (which we can interpret as sex-independent biomarkers). Here, over twenty transcripts and genes exceeded the threshold for FDR-adjusted significance. Interestingly, for the RNA-Seq data, several of the significant biomarkers are not protein-coding genes, highlighting the value of using non-poly-A-selected libraries to quantify both coding and non-coding transcripts. For the microarray meta-analysis, several of the sex-independent biomarkers are associated with key neurodevelopmental processes, including some X-chromosome genes. For example, MAGED2, differentially expressed in ASD probands, is located on an X-linked intellectual disability hotspot (i.e., Xp11.2) [Langnaese et al. (2001); Moey et al. (2016)] which, if causally relevant, could contribute to the male risk bias.
For both the microarray meta-analysis and the RNA-Seq analysis, we tested the ranked sex- dependent and sex-independent biomarker profiles separately for pathway-level enrichment. We found some pathway enrichment for the sex-dependent profiles, and even more for the sex-independent profiles. Importantly, very few of the enriched pathways were the same for both the interaction and main effects. This suggests that males and females exhibit unique pathway-level signatures that, if causally relevant, might further suggest the existence of both sex-specific and common ASD pathophysiologies. Although few KEGG pathways are enriched among the sex-dependent results, there are dozens of significantly enriched sex-dependent pathways across other tested gene sets (see Supplementary Information for more details). Among the pathways enriched in the sex- independent meta-analysis results, there are a number of pathways for known neurodevelopmental and neurodegenerative diseases, including Huntingtons, Parkinsons, Alzheimers, and amyotrophic lateral sclerosis (ALS), suggesting that at least some of these ASD biomarkers may have functions important to general brain health. Considering that the sex-dependent and sex-independent biomarkers and pathways differ, it seems plausible that molecular diagnostics could benefit from modelling sex-specific processes directly.
In addition to finding that pathway enrichment differs considerably between the sex-dependent and sex-independent biomarker profiles, we found that some sex-independent pathways (e.g., Ribosome and Proteasome) were enriched across both the RNA-Seq and microarray data. Interestingly, this overlap exists despite the fact that analyses were performed on different human tissues, and with different transcript quantification assays. In fact, more than fifty Gene Ontology pathways were enriched among both sets of ranked sex-independent biomarkers, even though no gene products showed significant differential expression in both data. This overlap is consistent with a broad literature supporting common pathway-level signatures across the widely heterogeneous population of ASD probands. If true, it may be advantageous to model pathway-level dysregulation directly, for example in machine learning applications Quinn et al. (2018).
When we compare our pathway enrichments to the previous ASD mega-analysis pathway enrichments [Tylee et al. (2017b)], we observe several complementary results. First, we found positive enrichment of the MAPK pathway in our sex-dependent RNA-Seq results, agreeing with the male- specific enrichment of Mek targets found in the Tylee et al. study [Tylee et al. (2017b)]. Second, we found an enrichment of the ribosome-related pathway in both of our sex-independent analyses, agreeing with the ribosome-related pathway enrichment identified by the sex-independent mega-analysis [Tylee et al. (2017b)]. Third, we found an enrichment of the Toll-like receptor (TLR) signalling pathway in our sex-independent meta-analysis results, agreeing with the TLR 3 and 4 signalling pathway enrichment identified by the sex-independent mega-analysis [Tylee et al. (2017b)]. Importantly, these complementary results exist despite considerable differences in statistical methodology and data set inclusion. The Wright et al. study which generated the RNA-Seq study did not test for KEGG enrichment. However, we both found SNORD17 to be differentially expressed independent of sex [Wright et al. (2017)].
Our analysis is not without limitations. First, although we used sva to adjust for latent batch effects, it is still possible that some residual batch effects remain because they coincide with the diagnostic label (e.g., undocumented co-morbidities or medication use). This would confound the discovered biomarker profile, causing spurious results. Second, as with any observational study, it is impossible to conclude whether the gene expression signatures, and their associated pathways, are causally related to ASD. Third, this analysis is likely under-powered to detect all sex-autism interactions, owing to the small sample sizes and disproportionately smaller female cohorts. Yet, based on the extant literature, which clearly highlights sex as an ASD risk factor, and the results published here, we believe that modelling the sex-autism interaction should become a mainstay of ASD transcriptomic research. Advantageously, interaction modelling is compatible with the most commonly used softwares for batch-effect correction [Leek et al. (2012)], RNA-Seq analysis [Love et al. (2014)], and microarray analysis [Smyth (2004)]. Yet, this analytical technique cannot offer any benefit if transcriptomic studies continue to systematically exclude female subjects (e.g., [Hu et al. (2009); Sarachana et al. (2010); Alter et al. (2011)]). Although there seems to exist a strong skew in the prevalence of male ASD, this very fact underlies the importance of studying female ASD: a complete understanding of the molecular basis of ASD will require the intentional study of both sex-dependent and sex-independent mechanisms, as well as their differences and commonalities.
Supplementary Material
5. Acknowledgements
SCL and TPQ designed the experiments, performed the analyses, and drafted the manuscript. JL provided statistical expertise and helped revise the manuscript. SWK, IHP, and SJG contributed data and helped revise the manuscript. TMC, SV, and TN supervised the project and helped revise the manuscript. This research was partially supported by the Australian Government through the Australian Research Council’s Linkage Projects funding scheme (LP140100240). SWK is supported by a grant from the National Institute of Health (R01MH107205). IHP is supported by grants from the National Institute of Environmental Health Sciences (1R01ES015359; 2R01ES015359; 1P30ES023513; 3P01ES011269), the National Institute of Health (UG3OD023365), and the United States Environmental Protection Agency (RD83543201).
References
- Alter MD, Kharkar R, Ramsey KE, Craig DW, Melmed RD, Grebe TA, Bay RC, Ober-Reynolds S, Kirwan J, Jones JJ, Turner JB, Hen R, and Stephan DA (2011, February). Autism and increased paternal age related changes in global levels of gene expression regulation. PloS One 6(2), e16715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC a Quality Control Tool for High Throughput Sequence Data [Online].
- Bailey A, Phillips W, and Rutter M (1996, January). Autism: towards an integration of clinical, genetic, neuropsychological, and neurobiological perspectives. Journal of Child Psychology and Psychiatry, and Allied Disciplines 37(1), 89–126. [DOI] [PubMed] [Google Scholar]
- Baron-Cohen S (2002, June). The extreme male brain theory of autism. Trends in cognitive sciences 6(6), 248–254. [DOI] [PubMed] [Google Scholar]
- Barrett T and Edgar R (2006). Gene Expression Omnibus (GEO): Microarray data storage, submission, retrieval, and analysis. Methods in enzymology 411, 352–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Glenn T, Pilhatsch M, Pfennig A, and Whybrow PC (2014, February). Gender differences in thyroid system function: relevance to bipolar disorder and its treatment. Bipolar Disorders 16(1), 58–71. [DOI] [PubMed] [Google Scholar]
- Benjamini Y and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 57(1), 289–300. [Google Scholar]
- Berbel P, Navarro D, and Román GC (2014). An evo-devo approach to thyroid hormones in cerebral and cerebellar cortical development: etiological implications for autism. Frontiers in endocrinology 5, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JM, Kalkbrenner AE, Just AC, Yolton K, Calafat AM, Sjödin A, Hauser R, Webster GM, Chen A, and Lanphear BP (2014, March). Gestational Exposure to Endocrine-Disrupting Chemicals and Reciprocal Social, Repetitive, and Stereotypic Behaviors in 4- and 5-Year-Old Children: The HOME Study. Environmental Health Perspectives. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch’ng C, Kwok W, Rogic S, and Pavlidis P (2015, October). Meta-Analysis of Gene Expression in Autism Spectrum Disorder. Autism Research 8(5), 593–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen DL, Baio J, Braun KVN, Bilder D, Charles J, Constantino JN, Daniels J, Durkin MS, Fitzgerald RT, Kurzius-Spencer M, Lee L-C, Pettygrove S, Robinson C, Schulz E, Wells C, Wingate MS, Zahorodny W, and Yeargin-Allsopp M (2016, April). Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years — Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR. Surveillance Summaries 65(3), 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice M and Di Lauro R (2004, October). Thyroid Development and Its Disorders: Genetics and Molecular Mechanisms. Endocrine Reviews 25(5), 722–746. [DOI] [PubMed] [Google Scholar]
- Elder JH, Kreider CM, Brasher SN, and Ansell M (2017). Clinical impact of early diagnosis of autism on the prognosis and parent-child relationships. Psychology research and behavior management 10, 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, Milacic M, Roca CD, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Viteri G, Weiser J, Wu G, Stein L, Hermjakob H, and D’Eustachio P (2018, January). The Reactome Pathway Knowledgebase. Nucleic Acids Research 46(D1), D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floris DL, Lai M-C, Nath T, Milham MP, and Di Martino A (2018, December). Network-specific sex differentiation of intrinsic brain function in males with autism. Molecular Autism 9(1), 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fombonne E (1999, July). The epidemiology of autism: a review. Psychological medicine 29(4), 769–86. [DOI] [PubMed] [Google Scholar]
- Glatt SJ, Tsuang MT, Winn M, Chandler SD, Collins M, Lopez L, Weinfeld M, Carter C, Schork N, Pierce K, and Courchesne E (2012, September). Blood-based gene expression signatures of infants and toddlers with autism. Journal of the American Academy of Child and Adolescent Psychiatry 51(9), 934–944.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg JP, Lit L, Baron CA, Hertz-Picciotto I, Walker W, Davis RA, Croen LA, Ozonoff S, Hansen R, Pessah IN, and Sharp FR (2008, January). Gene expression changes in children with autism. Genomics 91(1), 22–29. [DOI] [PubMed] [Google Scholar]
- Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, West AB, and Arking DE (2014a, December). Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nature Communications 5, 5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, West AB, and Arking DE (2014b, December). Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nature Communications 5, 5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley MTW, Lian L-Y, Haynes LP, and Burgoyne RD (2010, May). Structural and functional deficits in a neuronal calcium sensor-1 mutant identified in a case of autistic spectrum disorder. PloS One 5(5), e10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Picciotto I, Croen LA, Hansen R, Jones CR, van de Water J, and Pessah IN (2006, July). The CHARGE study: an epidemiologic investigation of genetic and environmental factors contributing to autism. Environmental Health Perspectives 114(7), 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu VW, Sarachana T, Kim KS, Nguyen A, Kulkarni S, Steinberg ME, Luu T, Lai Y, and Lee NH (2009, April). Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: evidence for circadian rhythm dysfunction in severe autism. Autism Research: Official Journal of the International Society for Autism Research 2(2), 78–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, Gottardo R, Hahne F, Hansen KD, Irizarry RA, Lawrence M, Love MI, MacDonald J, Obenchain V, Oleś AK, Pagès H, Reyes A, Shannon P, Smyth GK, Tenenbaum D, Waldron L, and Morgan M (2015, February). Orchestrating high-throughput genomic analysis with Bioconductor. Nature Methods 12(2), 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, and Morishima K (2017). KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research 45(D1), D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Harney JW, Zavacki AM, and Sajdel-Sulkowska EM (2014, April). Disrupted brain thyroid hormone homeostasis and altered thyroid hormone-dependent brain gene expression in autism spectrum disorders. Journal of physiology and pharmacology : an official journal of the Polish Physiological Society 65(2), 257–72. [PubMed] [Google Scholar]
- Kong SW, Collins CD, Shimizu-Motohashi Y, Holm IA, Campbell MG, Lee I-H, Brewster SJ, Hanson E, Harris HK, Lowe KR, Saada A, Mora A, Madison K, Hundley R, Egan J, McCarthy J, Eran A, Galdzicki M, Rappaport L, Kunkel LM, and Kohane IS (2012). Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PloS One 7(12), e49475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong SW, Shimizu-Motohashi Y, Campbell MG, Lee IH, Collins CD, Brewster SJ, Holm IA, Rappaport L, Kohane IS, and Kunkel LM (2013, May). Peripheral blood gene expression signature differentiates children with autism from unaffected siblings. Neurogenetics 14 (2), 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M-C, Lombardo MV, Auyeung B, Chakrabarti B, and Baron-Cohen S (2015, January). Sex/gender differences and autism: setting the scene for future research. Journal of the American Academy of Child and Adolescent Psychiatry 54(1), 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M-C, Lombardo MV, Suckling J, Ruigrok ANV, Chakrabarti B, Ecker C, Deoni SCL, Craig MC, Murphy DGM, Bullmore ET, and Baron-Cohen S (2013, September). Biological sex affects the neurobiology of autism. Brain 136(9), 2799–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langnaese K, Kloos D, Wehnert M, Seidel B, and Wieacker P (2001). Expression pattern and further characterization of human MAGED2 and identification of rodent orthologues. Cytogenetic and Genome Research 94(3–4), 233–240. [DOI] [PubMed] [Google Scholar]
- Leek JT (2014, December). svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Research 42(21), e161–e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, and Storey JD (2012, March). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics (Oxford, England) 28(6), 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ronemus M, Yamrom B, Lee Y.-h., Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, Buja A, Krieger A, Yoon S, Troge J, Rodgers L, Iossifov I, and Wigler M (2011, June). Rare De Novo and Transmitted Copy-Number Variation in Autistic Spectrum Disorders. Neuron 70(5), 886–897. [DOI] [PubMed] [Google Scholar]
- Lex A, Gehlenborg N, Strobelt H, Vuillemot R, and Pfister H (2014, December). UpSet: Visualization of Intersecting Sets. IEEE Transactions on Visualization and Computer Graphics 20 (12), 1983–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov J, and Tamayo P (2015, December). The Molecular Signatures Database Hallmark Gene Set Collection. Cell Systems 1(6), 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011, June). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27(12), 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionel AC, Tammimies K, Vaags AK, Rosenfeld JA, Ahn JW, Merico D, Noor A, Runke CK, Pillalamarri VK, Carter MT, Gazzellone MJ, Thiruvahindrapuram B, Fagerberg C, Laulund LW, Pellecchia G, Lamoureux S, Deshpande C, Clayton-Smith J, White AC, Leather S, Trounce J, Bedford H. Melanie, Hatchwell E, Eis PS, Yuen RKC, Walker S, Uddin M, Geraghty MT, Nikkel SM, Tomiak EM, Fernandez BA, Soreni N, Crosbie J, Arnold PD, Schachar RJ, Roberts W, Paterson AD, So J, Szatmari P, Chrysler C, Woodbury-Smith M, Lowry R. Brian, Zwaigenbaum L, Mandyam D, Wei J, Macdonald JR, Howe JL, Nalpathamkalam T, Wang Z, Tolson D, Cobb DS, Wilks TM, Sorensen MJ, Bader PI, An Y, Wu B-L, Musumeci SA, Romano C, Postorivo D, Nardone AM, Monica MD, Scarano G, Zoccante L, Novara F, Zuffardi O, Ciccone R, Antona V, Carella M, Zelante L, Cavalli P, Poggiani C, Cavallari U, Argiropoulos B, Chernos J, Brasch-Andersen C, Speevak M, Fichera M, Ogilvie CM, Shen Y, Hodge JC, Talkowski ME, Stavropoulos DJ, Marshall CR, and Scherer SW (2014, May). Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Human Molecular Genetics 23(10), 2752–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15(12), 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall K, Croen LA, Sjödin A, Yoshida CK, Zerbo O, Kharrazi M, and Windham GC (2017). Polychlorinated Biphenyl and Organochlorine Pesticide Concentrations in Maternal Mid-Pregnancy Serum Samples: Association with Autism Spectrum Disorder and Intellectual Disability. Environmental health perspectives 125(3), 474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall K, Croen LA, Weiss LA, Kharrazi M, Traglia M, Delorenze GN, and Windham GC (2017, August). Prenatal Serum Concentrations of Brominated Flame Retardants and Autism Spectrum Disorder and Intellectual Disability in the Early Markers of Autism Study: A Population- Based Case–Control Study in California. Environmental Health Perspectives 125(8), 087023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles JH (2011, April). Autism spectrum disorders–a genetics review. Genetics in medicine : official journal of the American College of Medical Genetics 13(4), 278–294. [DOI] [PubMed] [Google Scholar]
- Mitra I, Tsang K, Ladd-Acosta C, Croen LA, Aldinger KA, Hendren RL, Traglia M, Lavillaureix A, Zaitlen N, Oldham MC, Levitt P, Nelson S, Amaral DG, Hetrz-Picciotto I, Fallin MD, and Weiss LA (2016, November). Pleiotropic Mechanisms Indicated for Sex Differences in Autism. PLOS Genetics 12(11), e1006425.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi S and Voineagu I (2013). Autism susceptibility genes and the transcriptional landscape of the human brain. International review of neurobiology 113, 303–318. [DOI] [PubMed] [Google Scholar]
- Moey C, Hinze SJ, Brueton L, Morton J, McMullan DJ, Kamien B, Barnett CP, Brunetti-Pierri N, Nicholl J, Gecz J, and Shoubridge C (2016, March). Xp11.2 microduplications including IQSEC2, TSPYL2 and KDM5C genes in patients with neurodevelopmental disorders. European Journal of Human Genetics 24(3), 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosteller F and Fisher RA (1948). Questions and Answers. The American Statistician 2(5), 30–31. [Google Scholar]
- Ning LF, Yu YQ, GuoJi ET, Kou CG, Wu YH, Shi JP, Ai LZ, and Yu Q (2015, March). Meta-analysis of differentially expressed genes in autism based on gene expression data. Genetics and molecular research: GMR 14(1), 2146–2155. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard T. Grant, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, De La L, Ubieta T, Huang J, Lowe JK, Blencowe BJ, Horvath S, and Geschwind DH (2016). Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature Publishing Group 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods 14(4), 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn TP, Lee SC, Venkatesh S, and Nguyen T (2018, August). Improving the classification of neuropsychiatric conditions using gene ontology terms as features. bioRxiv, 393082. [DOI] [PubMed] [Google Scholar]
- Robinson EB, Lichtenstein P, Anckarsäter H, Happé F, and Ronald A (2013, March). Examining and interpreting the female protective effect against autistic behavior. Proceedings of the National Academy of Sciences of the United States of America 110(13), 5258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarachana T, Zhou R, Chen G, Manji HK, and Hu VW (2010, April). Investigation of post-transcriptional gene regulatory networks associated with autism spectrum disorders by microRNA expression profiling of lymphoblastoid cell lines. Genome Medicine 2(4), 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology 3, Article3. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005, October). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 102(43), 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium (2017, January). Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Research 45(D1), D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabzuni D, Ramasamy A, Imran S, Walker R, Smith C, Weale ME, Hardy J, Ryten M, and Consortium NABE (2013, November). Widespread sex differences in gene expression and splicing in the adult human brain. Nature Communications 4, 2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GC, Ghosh D, and Feingold E (2012, May). Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Research 40(9), 3785–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tylee DS, Espinoza AJ, Hess JL, Tahir MA, McCoy SY, Rim JK, Dhimal T, Cohen OS, and Glatt SJ (2017, March). RNA sequencing of transformed lymphoblastoid cells from siblings discordant for autism spectrum disorders reveals transcriptomic and functional alterations: Evidence for sex-specific effects. Autism Research 10(3), 439–455. [DOI] [PubMed] [Google Scholar]
- Tylee DS, Hess JL, Quinn TP, Barve R, Huang H, Zhang-James Y, Chang J, Stamova BS, Sharp FR, Hertz-Picciotto I, Faraone SV, Kong SW, and Glatt SJ (2017a, April). Blood transcriptomic comparison of individuals with and without autism spectrum disorder: A combined-samples mega-analysis. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics: The Official Publication of the International Society of Psychiatric Genetics 174(3), 181–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tylee DS, Hess JL, Quinn TP, Barve R, Huang H, Zhang-James Y, Chang J, Stamova BS, Sharp FR, Hertz-Picciotto I, Faraone SV, Kong SW, and Glatt SJ (2017b, April). Blood transcriptomic comparison of individuals with and without autism spectrum disorder: A combined-samples mega-analysis. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 174(3), 181–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tylee DS, Kawaguchi DM, and Glatt SJ (2013, October). On the outside, looking in: a review and evaluation of the comparability of blood and brain “-omes”. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics: The Official Publication of the International Society of Psychiatric Genetics 162B(7), 595–603. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, and Geschwind DH (2011, June). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474(7351), 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling DM, Parikshak NN, and Geschwind DH (2016, February). Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nature Communications 7, 10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright C, Shin JH, Rajpurohit A, Deep-Soboslay A, Collado-Torres L, Brandon NJ, Hyde TM, Kleinman JE, Jaffe AE, Cross AJ, and Weinberger DR (2017, May). Altered expression of histamine signaling genes in autism spectrum disorder. Translational psychiatry 7(5), e1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.