

Abstract
Background
Expression of d-xylose isomerase having high catalytic activity in Saccharomyces cerevisiae (S. cerevisiae) is a prerequisite for efficient and economical production of bioethanol from cellulosic biomass. Although previous studies demonstrated functional expression of several xylose isomerases (XI) in S. cerevisiae, identification of XIs having higher catalytic activity is needed. Here, we report a new strategy to improve xylose fermentation in the S. cerevisiae strain IR-2 that involves an evolutionary engineering to select top-performing XIs from eight previously reported XIs derived from various species.
Results
Eight XI genes shown to have good expression in S. cerevisiae were introduced into the strain IR-2 having a deletion of GRE3 and XKS1 overexpression that allows use of d-xylose as a carbon source. Each transformant was evaluated under aerobic and micro-aerobic culture conditions. The strain expressing XI from Lachnoclostridium phytofermentans ISDg (LpXI) had the highest d-xylose consumption rate after 72 h of micro-aerobic fermentation on d-glucose and d-xylose mixed medium. To enhance LpXI catalytic activity, we performed random mutagenesis using error-prone polymerase chain reaction (PCR), which yielded two LpXI candidates, SS82 and SS92, that showed markedly improved fermentation performance. The LpXI genes in these clones carried either T63I or V162A/N303T point mutations. The SS120 strain expressing LpXI with the double mutation of T63I/V162A assimilated nearly 85 g/L d-glucose and 35 g/L d-xylose to produce 53.3 g/L ethanol in 72 h with an ethanol yield of approximately 0.44 (g/g-input sugars). An in vitro enzyme assay showed that, compared to wild-type, the LpXI double mutant in SS120 had a considerably higher Vmax (0.107 µmol/mg protein/min) and lower Km (37.1 mM).
Conclusions
This study demonstrated that LpXI has the highest d-xylose consumption rate among the XIs expressed in IR-2 under micro-aerobic co-fermentation conditions. A combination of novel mutations (T63I and V162A) significantly improved the enzymatic activity of LpXI, indicating that LpXI-T63I/V162A would be a potential construct for highly efficient production of cellulosic ethanol.
Electronic supplementary material
The online version of this article (10.1186/s13068-019-1474-z) contains supplementary material, which is available to authorized users.
Keywords: Saccharomyces cerevisiae, Xylose isomerase, Lachnoclostridium phytofermentans, Mutagenesis, Bioethanol, Error-prone PCR, Metabolic engineering
Background
Industrial production of second-generation bioethanol (also called cellulosic ethanol) produced from lignocellulosic biomass such as corn stover, rice straw or sugarcane bagasse is critical to avoid competition with food crops that are predominantly used for production of first-generation bioethanol [1]. Despite various trials, commercial production of cellulosic ethanol still faces technological difficulties in sustainable supply of raw materials, delignification to liberate cellulose and hemicellulose from these complexes, effective carbohydrate depolymerization to produce free sugars and fermentation of mixed sugars to produce ethanol as well as high production costs [2]. In particular, the insufficient ability of microorganisms to metabolize pentose sugars, particularly d-xylose, which comprises a substantial portion of herbaceous hemicelluloses, is associated with low yields of bioethanol from lignocellulosic biomass [2, 3]. The budding yeast Saccharomyces cerevisiae (S. cerevisiae) is a potential host candidate for industrial ethanol production due to its high fermentation activity, resistance to ethanol and robustness in the presence of compounds that inhibit fermentation. Significant efforts have been made over the past two decades to develop engineered S. cerevisiae strains having modified pathways that enhance d-xylose metabolism, but the critical genes needed to optimize d-xylose metabolism in yeast remain unclear.
Two different metabolic pathways have been proposed for the initial conversion step of d-xylose by S. cerevisiae [4]. The first, a redox pathway catalyzed by NADPH-dependent xylose reductase (XR) followed by NAD+-dependent xylitol dehydrogenase (XDH), involves different coenzyme specificities of XR and XDH that cause a co-factor imbalance and subsequent accumulation of byproduct xylitol. Although attempts to address this problem including adaptive evolution, alteration of co-factor dependency and fine-tuning of enzyme expression levels have been partially successful in reducing xylitol production [5–9], the accumulation of xylitol remains problematic. The second pathway is the direct isomerization of d-xylose by d-xylose isomerase (XI), which would be superior to the redox pathway, since co-factor imbalance and xylitol accumulation do not occur. However, XI-based pathways predominate in bacteria and these enzymes are difficult to express functionally in yeast. The first attempts to obtain bacterial XIs encoded by xylA genes that can function in S. cerevisiae were unsuccessful, likely due to improper folding and cytoplasmic insolubility of the expressed protein [10–12]. In 1996, Walfridsson et al. [13] first reported that XI from the extreme thermophiles Thermus thermophilus could be expressed in an active form in S. cerevisiae, although the optimal temperature of the XI for function, 85 °C, was unsuitable for practical applications and only 4% of maximum activity was retained at 30 °C. Later, the first XI from the anaerobic fungus Piromyces sp. E2 was expressed in yeast, but the recombinant strain consumed d-xylose slowly [14]. Successful expression of XIs in S. cerevisiae was subsequently reported by several research groups in succession: Orpinomyces sp. ukk1 [15–17], Lachnoclostridium (previously known as Clostridium) phytofermentans ISDg [18, 19], Ruminococcus flavefaciens 17 [20], Prevotella ruminicola TC2-24 [21], Burkholderia cenocepacia J2315 [22, 23], Ruminiclostridium (previously known as Clostridium) cellulolyticum H10 [24] and Streptomyces rubiginosus [25]. Although the recombinant S. cerevisiae strains expressing the different XIs functioned to some extent, which XIs would be best suited for industrial ethanol production was still unclear.
In 2012, Lee and colleagues [26] subjected XI from Piromyces sp. E2 to three rounds of directed evolution and generated XI mutants containing six mutations (E15D, E114G, E129D, T142S, A177T and V433I) that had increased d-xylose consumption rates and in turn improved aerobic growth rates and ethanol production. The mutated XI exhibited a 77% increase in the Vmax value and a twofold lower Km compared to the wild-type. Back-substitution analyses showed that the E15D and T142S mutations in particular contributed to the improved catalytic activity of this XI [26]. Site-directed mutagenesis was also used to improve the XI enzyme activity of R. flavefaciens [20]. A G179A mutation, at a position close to the d-xylose binding site, showed a 15% increase in activity over the corresponding wild-type, and the 5′-P10 modification, in which the first 10 amino acids are replaced by the corresponding 12 amino acids from Piromyces sp. E2 XI, produced a 26.8% increase in activity over the wild-type while maintaining a Km value of 66.7 mM [20]. In addition, Waltman and colleagues [25] carried out rational enzyme engineering of S. rubiginosus XI to produce several variants (e.g., D215N) that show significantly lower affinity for d-xylose at < pH 6. Although these mutated XIs have improved performance in anaerobic fermentation, they should be reexamined in a common industrial strain under identical fermentation conditions. In this study, we evaluated the catalytic activities of previously reported XIs under identical fermentation conditions using a common parental strain SS29, a haploid strain derived from the diploid strain IR-2 that has a deletion of the endogenous xylose reductase GRE3, and directed evolution of genes encoding LpXI to improve the catalytic activities of XI for efficient and cost-effective bioethanol production from cellulosic biomass.
Results
Growth kinetics of transformants expressing XIs in d-xylose medium under aerobic conditions
A list of strains used in this study is shown in Additional file 1: Table S1. Eight currently known XI genes (xylA) derived from microorganisms were expressed in yeast and the ability of the transformants to metabolize d-xylose was assessed. Codon-optimization of XI gene nucleotide sequences was performed to maximize translational efficiency in S. cerevisiae and the genes were cloned into the low copy number expression vector pUG35. The XI genes under the control of the stress-inducible HSP12 promoter and an additional xylulokinase gene (XKS1) under the control of the strong promoter PGK1 were expressed in the S. cerevisiae strain SS29 with disrupted endogenous xylose reductase gene (GRE3). Although the function of GRE3 in consumption of d-xylose by S. cerevisiae is unclear, we nonetheless disrupted this gene to ensure that it would not compete with the exogenous XI during d-xylose metabolism. In addition, to maintain the enhanced d-xylose metabolic flow by the introduced XIs, we increased the expression level of XKS1 using a strong PGK1 promoter. These plasmids carrying the eight different XIs and a control vector lacking XI genes were used to transform the host strain SS29 derived from the diploid IR-2 to generate the strains termed SS36 to SS44 (see “Methods” section).
We first examined the growth kinetics of each strain in high-concentration d-xylose media YPX50 under aerobic conditions (Fig. 1). Among the XI-expressing strains, SS40 (OspXI) had the highest specific growth rate of 0.71 h−1 (Table 1). SS42 (LpXI) exhibited similar, but slower, growth kinetics to SS40 with a specific growth rate of 0.62 h−1 (Fig. 1 and Table 1). SS38 (PrXI), SS39 (RfXI) and SS41 (PspXI) showed aerobic growth on xylose media but had lower growth rates than did SS40 and SS42 (Table 1). SS37 (BcXI), SS43 (RcXI), SS44 (SrXI) and the control strain SS36 grew poorly or at undetectable levels under these culture conditions (Table 1).
Fig. 1.
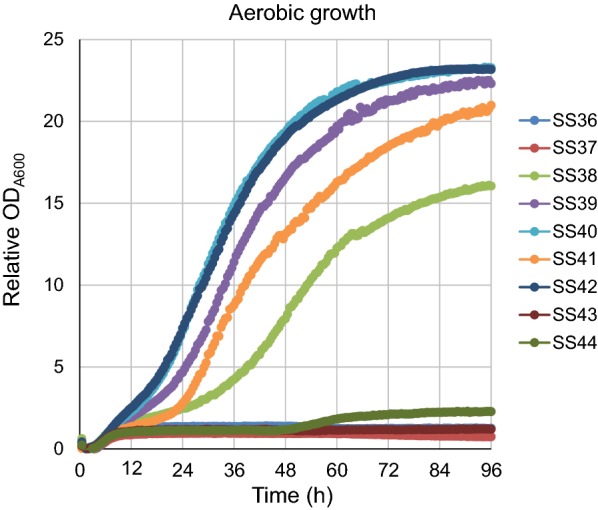
Growth kinetics of a control strain without XI and strains containing plasmid vector expressing various XIs in xylose media as the sole carbon source. Strains were cultured under aerobic conditions using YPX50 medium containing G418 (200 µg/mL). Cells for strain SS36 (blue), SS37 (red), SS38 (yellow green), SS39 (purple), SS40 (light blue), SS41 (orange), SS42 (dark blue), SS43 (brown) and SS44 (green) were initially inoculated at 0.1 U ODA600 and grown at 30 °C with shaking at 1000 rpm. Growth was measured using an Infinite 200 PRO microplate reader. Cell density was measured hourly. The dots indicate mean value of biological triplicates
Table 1.
Comparison of specific aerobic growth rates in YPX50 medium for strains harboring XIs
| Strain | Name | Origin | Specific growth rates (µ) (h−1) |
|---|---|---|---|
| SS36 | a | a | NA |
| SS37 | BcXI | Burkholderia cenocepacia J2315 | NA |
| SS38 | PrXI | Prevotella aff. ruminicola TC2-24 | 0.39 ± 0.05 |
| SS39 | RfXI | Ruminococcus flavefaciens 17 | 0.58 ± 0.03 |
| SS40 | OspXI | Orpinomyces sp. ukk1 | 0.71 ± 0.07 |
| SS41 | PspXI | Piromyces sp. E2 | 0.51 ± 0.10 |
| SS42 | LpXI | Lachnoclostridium phytofermentans ISDg | 0.62 ± 0.09 |
| SS43 | RcXI | Ruminiclostridium cellulolyticum H10 | NA |
| SS44 | SrXI | Streptomyces rubiginosus | 0.03 ± 0.01 |
The mean specific growth rate with standard deviations of biological triplicates are shown
NA: aerobic growth in YPX50 medium was very poor or showed no growth
aSS36 was a control strain carrying a plasmid vector lacking any XI genes
Comparison of xylose consumption and ethanol production of wild-type XI-expressing strains in glucose and xylose co-fermentation under micro-aerobic conditions
The fermentation activity of each XI-expressing strain was evaluated in the d-glucose and d-xylose mixed medium YPD85X35 at high cell density (initial ODA600 = 20 for each) and 30 °C under micro-aerobic conditions. During the fermentation, changes in residual sugar concentrations (d-glucose and d-xylose) and metabolic products (xylitol, glycerol, acetate and ethanol) were measured by high-performance liquid chromatography (HPLC). Although all tested strains consumed d-glucose within 12 h, differences were observed in d-xylose consumption and ethanol production after glucose depletion. Among the XI-expressing strains, SS42 (LpXI) showed the most efficient d-xylose consumption and ethanol production with 47.6 g/L ethanol produced from 85 g/L d-glucose and 35 g/L d-xylose in 72 h. The amount of residual d-xylose was dramatically decreased to 10 g/L in 72 h relative to the control strain SS36 (Fig. 2 and Additional file 2: Table S2). SS39 (RfXI) produced 43.9 g/L ethanol with consumption of approximately 17 g/L d-xylose (Fig. 2 and Additional file 2: Table S2). Unexpectedly, SS40 (OspXI), which showed the fastest growth under aerobic conditions in YPX50 medium, had poor fermentation performance under these conditions. SS40 consumed only 12.6 g/L d-xylose in 72 h, approximately half that consumed by SS42 (Fig. 2 and Additional file 2: Table S2). SS38 (PrXI) behaved similarly to SS40 (OspXI) (Fig. 2 and Additional file 2: Table S2). The other four strains SS37 (BcXI), SS41 (PspXI), SS43 (RcXI) and SS44 (SrXI) consumed essentially the same amount of d-xylose as the control strain SS36 and no improvement in xylose consumption and ethanol production was observed with XI expression (Fig. 2 and Additional file 2: Table S2).
Fig. 2.

Fermentation performance of a control strain without XI and strains containing plasmid vectors expressing various XIs in glucose/xylose co-fermentation under micro-aerobic conditions. Batch fermentation examinations were performed using cultures inoculated with cells at high density (ODA600 = 20) under micro-aerobic conditions. A control strain lacking XI (SS36) and eight strains expressing XIs (SS37 to SS44) were grown in 70 mL YPD85X35 medium containing G418 (200 µg/mL) at 30 °C. Samples of culture medium were collected at 0, 1, 3, 6, 12, 24, 36, 48, 60 and 72 h (10 times) for HPLC analysis of metabolites. The XIs were introduced episomally via low copy number plasmids into the same parental strain, SS29. The dots and error bars in the panels represent the mean concentrations and standard deviations, respectively, of the following metabolites measured in biological triplicates: glucose (blue), xylose (red), xylitol (yellow green), glycerol (purple), acetate (light blue) and ethanol (orange). The details for the metabolite concentrations are shown in Additional file 2: Table S2
Comparison of xylose consumption and ethanol productivity by strains expressing previously characterized XI mutations in glucose and xylose co-fermentation under micro-aerobic conditions
Several amino acids substitutions identified in directed evolution studies of RfXI and PspXI were recently found to improve xylose metabolism [20, 25]. We thus compared the fermentation performance of strains expressing RfXI and PspXI carrying these known mutations under the same conditions. The resulting strains, SS45 (RfXI-G179A), SS46 (PspXI-E15D) and SS49 (PspXI-N370S), exhibited better fermentation performance than the parental strains SS39 (RfXI) and SS41 (PspXI), respectively, in co-fermentation with d-glucose and d-xylose (Fig. 2 and Additional file 2: Tables S2, Additional file 3: Figure S1 and Additional file 4: Table S3). However, these mutations had little effect on d-xylose consumption. Moreover, no improvement in d-xylose consumption by strains SS47 (PspXI-C54R), SS48 (PspXI-T142S), SS50 (PspXI-C54R/N370S) and SS51 (PspXI-E51D/T142S) was observed (Additional file 3: Figure S1 and Additional file 4: Table S3). The ethanol production and d-xylose consumption of SS42 (LpXI) were substantially better than those of SS45 (RfXI-G179A), SS46 (PspXI-E15D) and SS49 (PspXI-N370S).
Identification of beneficial LpXI mutations and characterization of growth and fermentation properties of resulting clones
Based on the above results for the eight XI genes, LpXI was chosen for expression in S. cerevisiae and used for protein engineering to improve its catalytic function. We carried out random mutagenesis of the LpXI gene using error-prone PCR. In brief, plasmids containing mutated LpXI were introduced into the parental strain SS29 and the growth activity on plates containing higher concentrations of d-xylose (80 g/L d-xylose, YPX80) was examined. Approximately, 2.4 × 105 yeast transformants were obtained, and in a growth assay on YPX80 plates, 24 colonies grew well. Repeated examination of growth assay on YPX80 plates to eliminate false-positive clones from the LpXI mutant library yielded 11 transformants as evolved strains containing mutated LpXI. These 11 LpXI expression plasmids were prepared from the transformants and the nucleotide sequences of the LpXI coding regions were determined by Sanger sequencing (Table 2). The aerobic growth properties and fermentation activity in micro-aerobic co-fermentation of d-glucose and d-xylose for the transformants were evaluated under the same experimental condition as described above. In the growth assay, all candidates showed improved growth phenotypes even on high-concentration xylose plates and liquid media containing 80 g/L xylose as the sole carbon source compared to that for the control strain SS42 (Table 2). In particular, the mutant clone M6-13 (LpXI-N233I, episomally) had approximately 4.0-fold higher specific growth rates relative to the parental strain SS42 (Table 2). A fermentation assay indicated that all 11 clones had superior fermentation ability, as evidenced by exhaustion of the input d-xylose within 72 h and production of at least 52 g/L ethanol relative to the control strain SS42 (wild-type LpXI, episomally) (Fig. 2 and Additional file 2: Table S2, Additional file 5: Figure S2 and Additional file 6: Table S4).
Table 2.
Mutations identified in LpXI-expressing plasmids and relative specific growth rate (µ/µ0) of isolated clones in YPX80 medium under aerobic cultivations
| Clone | Substitutions in LpXI | Specific growth rate (µ) (h−1)a | Relative growth rate (µ/µ0) |
|---|---|---|---|
| M6-13 | N223I | 1.07 ± 0.06 | 3.97 |
| M6-11 | D207G | 0.98 ± 0.12 | 3.63 |
| M6-21 | R191K/E192K | 0.95 ± 0.11 | 3.54 |
| M6-22 | L304S | 0.93 ± 0.09 | 3.44 |
| M6-15 | L78S | 0.92 ± 0.18 | 3.43 |
| M6-7 | Y13H/D228 V/P233P | 0.86 ± 0.16 | 3.20 |
| M6-10 | T273A | 0.82 ± 0.07 | 3.06 |
| M6-6 | T133T/K136T/A176T | 0.82 ± 0.31 | 3.04 |
| M6-19 | E114G | 0.75 ± 0.08 | 2.80 |
| M6-20 | V162A/T273T/N303T | 0.49 ± 0.03 | 1.81 |
| M6-2 | T63I/A121A | 0.30 ± 0.03 | 1.13 |
| SS42 | Wild-type | 0.27 ± 0.00 | 1.00 |
aMean ± standard deviations
Micro-aerobic co-fermentation of d-glucose and d-xylose by strains carrying wild-type and mutated LpXI at the AUR1 locus
As shown above, strains that grow well under aerobic culture conditions and have effective performance in micro-aerobic co-fermentation of d-glucose and d-xylose were successfully obtained, and the amino acid substitutions in the mutated XI were identified. To verify critical mutations for improved fermentation activity in micro-aerobic co-fermentation of d-glucose and d-xylose, wild-type and mutated LpXIs with XKS1 were integrated at the AUR1 chromosome locus in SS29. The fermentation performance of the control strain SS81 (wild-type LpXI) was roughly the same as that of SS42 (wild-type LpXI, episomally) with 12.2 g/L and 47.7 g/L of d-xylose and ethanol, respectively, produced in 72 h (Figs. 2 and 3 and Additional file 2: Table S2 and Additional file 7: Table S5). Of the 11 transformants, SS82 (LpXI-T63I/A121A, corresponding to the M6-2 clone) and SS92 (LpXI-V162A/T273T/N303T, corresponding to the M6-20 clone) showed remarkable improvement in xylose consumption and ethanol productivity in micro-aerobic co-fermentation of d-glucose and d-xylose compared to the control strain SS81 (wild-type LpXI). Within 72 h, SS82 and SS92 both assimilated almost all available d-xylose, with residual d-xylose dramatically reduced to 2.51 g/L and 1.45 g/L, concurrent with the production of 52.2 g/L and 52.4 g/L ethanol, respectively (Fig. 3 and Additional file 7: Table S5). The other strains SS89 (LpXI-L78S, corresponding to the M6-15 clone) and SS93 (LpXI-R191K/E192K, corresponding to the M6-21 clone) also showed a slight improvement in d-xylose consumption and ethanol production (Fig. 3 and Additional file 7: Table S5). The other 7 strains, SS84 (LpXI-T133T/K136T/A176T, corresponding to the M6-6 clone), SS85 (LpXI-Y13H/D228V/P233P, corresponding to the M6-7 clone), SS86 (LpXI-T273A, corresponding to the M6-10 clone), SS87 (LpXI-D207G, corresponding to the M6-11 clone), SS88 (LpXI-N223I, corresponding to the M6-13 clone), SS91 (LpXI-E114G, corresponding to the M6-19 clone) and SS94 (LpXI-L304S, corresponding to the M6-22 clone), showed no significant improvement in d-xylose consumption even though the growth of the corresponding clones carrying the mutated LpXI plasmids was much faster than that of the control strain in high-concentration xylose media under aerobic conditions and the performance of glucose/xylose co-fermentation was high (Additional file 8: Figure S3 and Additional file 9: Table S6).
Fig. 3.

Fermentation performance of wild-type strains and strains with mutated LpXIs integrated into the chromosome in glucose/xylose co-fermentation under micro-aerobic conditions. Batch fermentation examinations were performed under the same conditions as in Fig. 2. The fermentation properties of control strain SS81 (wild-type LpXI) and four representative strains having improved d-xylose consumption rates are shown: SS82 (LpXI-T63I/A121A), SS89 (LpXI-L78S), SS92 (LpXI-V162A/T273T/N303T) and SS93 (LpXI-R191K/E192K). The mutated LpXI expression cassettes were introduced into the AUR1 locus of the chromosome of the parental strain SS29 via homologous recombination. The dots and error bars in the panels represent the mean concentrations and standard deviations, respectively, of the following metabolites in biological triplicates: glucose (blue), xylose (red), xylitol (yellow green), glycerol (purple), acetate (light blue) and ethanol (orange). The details for the metabolite concentrations are shown in Additional file 7: Table S5
Characterization of LpXI mutation(s) in SS82 (LpXI-T63I/A121A) and SS92 (LpXI-V162A/T273T/N303T)
Both SS82 (LpXI-T63I/A121A) and SS92 (LpXI-V162A/T273T/N303T) showed higher fermentation activity in micro-aerobic co-fermentation of d-glucose and d-xylose, but it was unclear which mutation(s) was responsible for the improvement in d-xylose catalytic activity. The nucleotide variations at residues 121 and 273 in SS82 and SS92 produced no amino acid substitution. As such, we focused on three amino acid substitutions T63I, V162A and N303T, and determined whether one, two or all three mutations would be required to provide beneficial effects for d-xylose consumption and ethanol production in micro-aerobic co-fermentation of d-glucose and d-xylose. The fermentation performance of SS104 (LpXI-V162A) was obviously higher than that of the control strain SS81 (wild-type LpXI) but did not exceed that of SS92 (LpXI-V162A/T273T/N303T) (Figs. 3 and 4 and Additional file 7: Table S5 and Additional file 10: Table S7). On the other hand, strain SS105 (LpXI-N303T) showed no improvement in fermentation performance, and the amount of d-xylose consumption was much less than that of the control strain SS81 (wild-type LpXI) (Figs. 3 and 4 and Additional file 7: Table S5 and Additional file 10: Table S7). Furthermore, SS120 (LpXI-T63I/V162A) showed superb fermentation ability as evidenced by production of 53.3 g/L ethanol and consumption of nearly all available d-xylose within 72 h. SS92 (LpXI-V162A/T273T/N303T) and SS120 (LpXI-T63I/V162A) had the fastest rates of d-xylose consumption among all the strains tested (Fig. 4). A comparison of fermentation indices showed a d-xylose consumption rate in 36 h (Cxyl36) for SS82, SS92 and SS120 of 0.68 g/L/h, 0.76 g/L/h and 0.75 g/L/h and 0.47 g/L/h, 0.48 g/L/h and 0.49 g/L/h, respectively, at 72 h (Cxyl72) (Table 3), which were calculated as increases of 57%, 58% and 64%, respectively, over that of control strain SS81 (Cxyl72, 0.30 g/L/h). The ethanol yield of these three strains reached 0.44 g/g-input sugars in 72 h (Table 3). The strain carrying the single mutation, SS105 (LpXI-N303T), had no improvement in any of the indices. The responsible mutation in SS92 was thus V162A, as N303T alone did not improve fermentation efficiency. Taken together, we concluded that LpXI carrying the double mutation T63I/V162A is the most suitable mutant for functional expression in yeast and for optimal glucose/xylose co-fermentation.
Fig. 4.

Fermentation performance of strains with single and double mutations in LpXI integrated into the chromosome in glucose/xylose co-fermentation under micro-aerobic conditions. Batch fermentation examinations were performed under the same conditions as for Figs. 2 and 3. The fermentation properties of strains are shown for SS104 (LpXI-V162A, single mutation, from SS92), SS105 (LpXI-N303T, single mutation, from SS92) and SS120 (LpXI-T63I/V162A, combination of mutations from SS82 and SS92, respectively). The mutated LpXI expression cassettes were introduced into the AUR1 locus of the chromosome of the parental strain SS29 via homologous recombination. The dots and error bars in the panels represent the mean concentrations and standard deviations, respectively, of the following metabolites in biological triplicates: glucose (blue), xylose (red), xylitol (yellow green), glycerol (purple), acetate (light blue) and ethanol (orange). The details for the metabolite concentrations are shown in Additional file 10: Table S7
Table 3.
Comparison of fermentation performance of improved strains with mutated LpXI
| Strain | LpXI | Cxyl36 (g/L/h)a | Cxyl72 (g/L/h)b | Yeth72 (g/g-input sugars)c |
|---|---|---|---|---|
| SS81 | Wild-type | 0.36 ± 0.08 | 0.30 ± 0.08 | 0.39 ± 0.03 |
| SS82 | T63I/A121A | 0.68 ± 0.06 | 0.47 ± 0.01 | 0.44 ± 0.01 |
| SS92 | V162A/T273T/N303T | 0.76 ± 0.04 | 0.48 ± 0.02 | 0.44 ± 0.01 |
| SS104 | V162A | 0.59 ± 0.05 | 0.46 ± 0.01 | 0.43 ± 0.01 |
| SS105 | N303T | 0.32 ± 0.06 | 0.25 ± 0.02 | 0.40 ± 0.01 |
| SS120 | T63I/V162A | 0.75 ± 0.05 | 0.49 ± 0.00 | 0.44 ± 0.01 |
aCxyl36: d-xylose consumption rate in 36 h
bCxyl72: d-xylose consumption rate in 72 h
cYeth72: ethanol yield in 72 h per input sugars
Determination of catalytic activities of mutated LpXIs
To confirm the fermentation assay results and determine the underlying mechanisms associated with improved LpXI performance, the catalytic activities of wild-type and mutated LpXIs were assessed using an in vitro enzymatic assay. In brief, total proteins were extracted from each strain expressing wild-type and mutated LpXIs after 24 h of micro-aerobic co-fermentation and the total protein concentration was adjusted to 1 mg/mL before XI activity was measured in terms of decreases in the amount of NADH (see “Methods”). LpXI-T63I/A121A (SS82) had a 44% increase in the catalytic activity (Vmax) over wild-type and a Km value of 29.3 mM. On the other hand, the Vmax for LpXI-V162A/T273T/N303T (SS92) showed no increase in catalytic activity over wild-type, but the Km of 28.4 mM was significantly lower than the 41.8 mM for wild-type (Table 4, Additional file 11: Table S8). Notably, LpXI-T63I/V162A (SS120) showed the highest Vmax among the mutated LpXIs (Table 4).
Table 4.
Kinetic properties of wild-type and mutated LpXIs
| Strain | LpXI | V amax | Km (mM) |
|---|---|---|---|
| SS81 | Wild-type | 0.064 ± 0.002 | 41.8 ± 0.8 |
| SS82 | T63I/A121A | 0.092 ± 0.001 | 29.3 ± 2.6 |
| SS92 | V162A/T273T/N303T | 0.066 ± 0.001 | 28.4 ± 2.3 |
| SS104 | V162A | 0.063 ± 0.002 | 29.5 ± 2.2 |
| SS105 | N303T | 0.030 ± 0.001 | 19.1 ± 0.6 |
| SS120 | T63I/V162A | 0.107 ± 0.002 | 37.1 ± 2.3 |
The mean and standard deviations of biological triplicates are indicated
aμmol mg protein−1 min−1
Discussion
Cellulosic ethanol, which is produced from non-edible biomass, has advantages over first-generation biomass ethanol made from food starch [1–3]. Despite its potential practical uses and the construction of several demonstrational or commercial plants, large-scale production of ethanol from cellulose and hemicellulose has not yet been realized. In addition to high cellulase production costs, the cost of cellulosic biomass as an inedible agricultural waste is rising, such that the total production costs for cellulosic ethanol production will likely further increase. Although multiple studies have shown the potential for practical use of XI enzyme expressed by S. cerevisiae and engineered strains harboring XI have been developed for production of cellulosic ethanol, the fermentation ability of these strains remains too low for practical applications. Therefore, development of more productive and practical ethanologenic microorganisms that can metabolize not only d-glucose from cellulose but also other sugars, especially d-xylose, from hemicellulose, is needed.
As mentioned above, pioneering studies showed the difficulties in functional expression of bacterial XI in yeast [15–26]. In addition, there are few direct comparisons of XI expression efficiency, catalytic activity and fermentation performance using the same platform and under the same conditions [18]. For commercial production of cellulosic ethanol, one of the most critical issues is the need to identify the most effective XI for glucose/xylose co-fermentation. In this study, we first employed an identical platform and growth conditions to comprehensively evaluate the d-xylose consumption and fermentation performance of strains expressing one of eight previously reported XIs. The control strain SS36 that carried no XI did not grow on YPX50 media, but five of eight XI-expressing strains, SS38 (PrXI), SS39 (RfXI), SS40 (OspXI), SS41 (PspXI) and SS42 (LpXI), grew well aerobically, clearly indicating that these five XIs, in particular, LpXI and OspXI, can be functionally expressed in yeast. However, LpXI and OspXI showed quite different traits in terms of co-fermentation. Xylose consumption was dramatically improved in the strain expressing LpXI, but not in the strain with OspXI. These inconsistent results could be attributed to differences in functional expression of XI or its protein folding under aerobic growth and micro-aerobic fermentation conditions, although further investigation is needed to explore these possibilities. Interestingly, the habitat environment of the microorganisms from which XI originated could be responsible for these different behaviors. OspXI and PspXI are genes derived from anaerobic fungi that are part of the ruminal microflora of herbivorous animals. Moreover, previous studies demonstrated that recombinant yeast strains expressing these XIs could consume d-xylose [14, 18, 25, 27, 28]. In this study, we used the same promoter, HSP12, for XI expression so that the expression level of each XI should be similar. However, even with similar XI expression levels, the protein lifetime or folding related to XI enzymatic activity could differ between aerobic and micro-aerobic conditions. To the best of our knowledge, the sustainability of XI proteins in yeast under aerobic and micro-aerobic conditions has not yet been examined. However, recent studies focusing on XI protein folding demonstrated that co-expression with Escherichia coli (E. coli) GroEL could facilitate the functional expression of xylose and arabinose isomerase genes from E. coli as well as d-psicose isomerase from Agrobacterium tumefaciens in S. cerevisiae [29]. Moreover, Propionibacterium acidipropionici XI is functional in yeast when co-expressed with the molecular chaperones GroEL and GroES [30]. This approach may facilitate functional expression of XI enzymes in yeast.
To better understand requirements for producing ethanol from cellulosic biomass, the relationship between cell growth and fermentation performance under given fermentation conditions must be considered. Of course, cell growth is a crucial issue for fermentation because the number of cells during fermentation directly affects the rate of sugar consumption and ethanol production as well as d-xylose metabolism. In this study, we used the fermentation medium YPD85X35 that includes sugar concentrations that reflect those of typical cellulosic biomass hydrolysates [31, 32]. The model medium contains high concentrations of sugars, which are needed for commercial production of cellulosic ethanol, particularly to produce higher concentrations of ethanol (e.g., 50 g/L or more) that allow efficient distillation. The sugar consumption rates integrate the viable cell number and the consumption rate per cell as a function of time. The true catalytic activities of XIs in examination of fermentation are thought to be proportional to the consumption rate of d-xylose per cell. However, because the viable cell number changes over time, this value cannot be determined in real time. Therefore, to avoid differences in growth rates between XI-containing strains, high numbers of cells (ODA600 = 20) that prevent further cell proliferation were inoculated for examinations of XI activity in micro-aerobic fermentation. Brat and colleagues [18] previously reported that heterogenous expression of LpXI conferred the ability to metabolize d-xylose on both industrial and laboratory yeast strains. Consistent with our results, they showed that strains containing LpXI had superior d-xylose consumption and growth on media having d-xylose as a sole carbon source, but their study did not determine whether LpXI-expressing strains had the best fermentation performance in micro-aerobic glucose/xylose co-fermentation [18]. In our results, both SS42 (wild-type LpXI, episomally) and SS81 (wild-type LpXI, integrated into the chromosome) consumed 85 g/L d-glucose in 12 h and subsequently consumed 25 g/L d-xylose in 72 h. Such incomplete consumption and remaining d-xylose in the fermentation process could be directly associated with decreased ethanol yield. Our results clearly showed that LpXI has the best potential among the eight XIs tested for ethanol production in micro-aerobic glucose/xylose co-fermentation, but the amount of expressed XI protein and its catalytic activity was still low and requires further improvement for use on a commercial scale.
To improve the catalytic activities of LpXI, we carried out evolutionary molecular engineering using error-prone PCR to introduce one or two amino acid mutations in the coding region. At least 90% of plasmids had several mutations per open reading frame (ORF) of LpXI (2.0 mutations on average), indicating that the in vitro mutagenesis was successful (data not shown). After large-scale selection, we determined the amino acid substitutions for 24 candidates that could grow well on high-concentration d-xylose plates (YPX80) and that had mutated LpXI. After eliminating false-positives 11 candidates remained, and of these, two strains, SS82 (LpXI-T63I/A121A) and SS92 (LpXI-V162A/T273T/N303T), consumed nearly all d-glucose and d-xylose within 72 h. Co-fermentation examination using either SS104 (LpXI-V162A) or SS105 (LpXI-N303T) clearly indicated that V162A was the mutation responsible for the improved performance of SS92 (LpXI-V162A/T273T/N303T). Meanwhile, SS120 carrying the LpXI double mutation T63I and V162A derived from the different two-mutant clones showed that these mutations produced no synergistic improvement in fermentation performance relative to the single mutants. Biochemical assays showed that the Vmax of LpXI-T63I/V162A (SS120, 0.107 µmol/mg protein/min) was 1.7-fold higher than that of wild-type LpXI (SS81, 0.064 µmol/mg protein/min), while the Km value increased to 37.1 mM, which was higher than that for the singly mutated LpXIs. This result could explain why SS120 (LpXI-T63I/V162A) did not show improved catalytic activities relative to the singly mutated LpXIs. Furthermore, the yeast strains used in this study had no exogenous d-xylose transporters, such that incorporation of d-xylose was the rate-limiting process and may have contributed to low intracellular concentrations of d-xylose. As such, attempts to lower the XI Km could be effective in promoting d-xylose metabolism by S. cerevisiae cells. Under our experimental conditions, PspXI had the lowest Km among the eight XIs at 19.7 mM, which was similar to the 20 mM reported by Kuyper et al. [14], but lower than other previously reported Km values for PspXI that ranged between 49.9 and 87.0 mM [18, 21, 25]. The second-best Km measured for this study was for OspXI (27.6 mM), but as mentioned above, both PspXI and OspXI were unsuitable for glucose/xylose co-fermentation. Although the wild-type LpXI Km of 45.7 mM was comparatively moderate, it was substantially higher than that of hexokinases for d-glucose (1.0 mM) [33].
To compare the performance of engineered C5C6 yeast harboring XIs in glucose/xylose co-fermentation, we adopted four indices: (i) the time to complete consumption of input sugars (T); the consumption rate of d-xylose at (ii) the end of co-fermentation (Cxyl(T)) and (iii) the halfway point of the fermentation period (Cxyl(T/2)); and (iv) the ethanol yield at the end of co-fermentation (Yeth(T)). In typical glucose/xylose co-fermentation, microbes first consume d-glucose via carbon catabolite repression, such that T is the practical time to complete consumption of d-xylose. In this study, the T value was defined as about 72 h based on the results for SS92 and SS120. These indices depend on cells in the initial inoculum. The Cxyl72 value represents the amount of d-xylose consumption in 72 h and in particular, the consumption of d-xylose is represented by the difference between the input and residual sugars, so we measured the precise concentrations of input sugars in the medium for each experiment. Cxyl36 is an index for the d-xylose consumption rate during the early phase of xylose fermentation and also depends on the time needed for d-glucose consumption. For example, if glucose consumption is delayed, d-xylose consumption should also be delayed as active cells do not increase as well. The theoretical ethanol yields of d-glucose and d-xylose are 0.51 (g/g-input sugars). The best ethanol yield seen for SS82, SS92 and SS120 in this study was 0.44 (g/g-input sugars). In particular, the LpXI-V162A/T273T/N303T and LpXI-T63I/V162A mutations were effective for improving the xylose utilization rate. The T63I and V162A substitutions in LpXI generated by error-prone PCR contributed to an increase in the d-xylose consumption rate by 1.64- and 1.11-fold, respectively, over that of wild-type LpXI in micro-aerobic glucose/xylose co-fermentation. These novel mutations provide insight for further improvement of XI enzyme activity to develop C5C6 yeast and facilitate commercial production of cellulosic ethanol.
Currently, the best-performing reported C5C6 yeast strain GS1.11-26, which contains wild-type LpXI for d-xylose metabolism, can consume 36 g/L d-glucose and 35 g/L d-xylose within 12 h and has an ethanol yield of 0.46 [19]. This strain could quickly consume almost all d-xylose in just 13 h in a semi-aerobic glucose/xylose co-fermentation with an initial cell density of 1.3 g-dry cell weight/L [19]. Interestingly, the heterogenous gene LpXI in the GS1.11-26 strain was inserted close to an ARS1529 sequence and was amplified by about ninefold for both chromosome alleles [34]. Thus, the amount or activities of XI enzymes would still be rate-limiting for d-xylose metabolism. The fermentation performance for SS120 is not yet comparable to the GS1.11-26 strain. To develop strains with superior fermentation performance in terms of d-xylose consumption and ethanol production rates, expression of d-xylose metabolism genes must first be optimized.
Selection of a promoter to drive expression of a foreign gene is also critical. In this study, we chose the S. cerevisiae HSP12 promoter to drive XI expression in all strains. This promoter was selected based on the assumption that co-fermentation environments represent a stressful condition for yeast because micro-aerobic culture conditions restrict energy supplies. An approach to determine a promoter that includes an artificial promoter to drive gene expression during micro-aerobic fermentation may be needed. Wild-type LpXI used in GS1.11-26 was driven by the PYK2 promoter, a paralog of the CDC19 pyruvate kinase gene in yeast [19]. Because carbon catabolite repression reduces PYK2 expression in the presence of glucose [35], LpXI should best be expressed during the d-xylose assimilating phase. Factors affecting promoter activity under fermentation conditions, such as temperature, pH, sugar composition and the presence of fermentation inhibitors as well as the timing of gene expression should be considered.
Regulation of coordinated metabolic flux in the d-xylose metabolic pathway is another important consideration for promoter selection. We recently reported that balanced expression of pentose phosphate pathway (PPP) genes would maximize the d-xylose consumption rates during high-temperature glucose/xylose co-fermentation and that the necessary PPP genes for optimization of d-xylose consumption and ethanol production differ between strains with XI and XR-XDH [36, 37]. Furthermore, as mentioned above, we have not yet optimized d-xylose uptake, which can be facilitated by controlled expression of hexose transporters including HXT7, HXT5, GAL2, HXT1 and HXT2 [38]. Several recent studies engineered these hexose/pentose transporters to promote specific uptake of d-xylose in yeast (reviewed in [4]). Using d-xylose media to screen mutant clones made by site-directed mutagenesis, the glucose-insensitive xylose transporters HXT7-N370S and GAL2-N376F were identified [39]. This glucose insensitivity is an important property for utilization of d-xylose during glucose/xylose co-fermentation because these transporters cannot incorporate d-xylose in the presence of d-glucose due to the higher affinity for d-glucose compared to d-xylose.
Despite the numerous pioneering studies, whether the XR-XDH pathway or XI pathway is superior for yeast-mediated biomass fermentation remains controversial. We believe that metabolic pathways using XI will have advantages over those using XR-XDH for several reasons. For the XR-XDH pathway, the production of byproducts such as xylitol cannot be avoided and the dependence of XR on NADPH should produce a coenzyme imbalance. On the other hand, XI could surmount these issues as the XI protein can directly convert d-xylose to d-xylulose. Indeed, based on our results and those of previous studies, the amount of xylitol byproduct in glucose/xylose co-fermentation was negligible, resulting in an ethanol yield that was even higher than that for the high-performance XR-XDH strain NAPX37 [40]. In any case, the d-xylose consumption rate for SS120 was still tenfold lower than that for d-glucose. The XR-XDH pathway and the XI pathway both have advantages and disadvantages, and which is superior remains a topic of debate. Nonetheless, to develop strains that can be used for the commercial production of cellulosic ethanol, it will be necessary to increase the consumption rate of d-xylose and ethanol productivity by several fold as well as the ethanol yield.
Conclusions
Here, we compared the d-xylose metabolism of eight previously reported XIs expressed in the yeast strain IR-2 grown on d-glucose and d-xylose that mimics a typical biomass hydrolysate to determine the most suitable XI for industrial ethanol production. Comparative analyses of transformants harboring these XIs in micro-aerobic fermentation showed that LpXI has the highest d-xylose consumption rate in 72 h. Random mutagenesis of LpXI using error-prone PCR yielded two novel and beneficial mutations (T63I and V162A) that were shown to be responsible for improved catalytic activities relative to wild-type. The strain SS120 expressing LpXI-T63I/V162A with deletion of the GRE3 gene and overexpression of the XKS1 gene produced a maximum rate of ethanol production of 53.3 g/L ethanol from about 85 g/L d-glucose and 35 g/L d-xylose for 72 h under micro-aerobic fermentations. This strain could assimilate nearly all available d-xylose, in contrast to the control strain SS82 expressing wild-type LpXI that left 10 g/L d-xylose. To our knowledge, the novel LpXI-T63I/V162A mutant is a superior d-xylose isomerase that could be used to improve the capacity of industrial S. cerevisiae strains to efficiently metabolize d-xylose and produce maximum amounts of bioethanol.
Methods
Strains, media and culture conditions
Yeast strains used in this study are listed in Additional file 1: Table S1. S. cerevisiae SS29 (MATα ho::bleMX6 gre3::hphMX6), a haploid strain derived from diploid IR-2, was used as a common host strain [41, 42]. Yeast cells were grown at 30 °C in YPD medium [10 g/L yeast extract (BD Biosciences, Sparks, MD), 20 g/L Bacto™ peptone (BD Biosciences), 20 g/L d-glucose (Sigma-Aldrich, St. Louis, MO)], YPX50 medium [10 g/L yeast extract, 20 g/L Bacto™ peptone, 50 g/L d-xylose (Sigma-Aldrich)], YPX80 medium (10 g/L yeast extract, 20 g/L Bacto™ peptone, 80 g/L d-xylose), or YPD85X35 medium (10 g/L yeast extract, 20 g/L Bacto™ peptone, 85 g/L d-glucose, 35 g/L d-xylose). For solid plates, 20 g/L Bacto™ agar (BD Biosciences) was added to the respective liquid media. For selection of transformants, several antibiotics were added to culture media: G418 (200 µg/mL; Thermo Fisher Scientific/Life Technologies, Carlsbad, CA), Zeocin (400 µg/mL; Thermo Fisher Scientific/Life Technologies), hygromycin B (200 µg/mL; Thermo Fisher Scientific/Life Technologies), and/or Aureobasidin A (0.5 µg/mL; Takara-Bio Inc., Shiga, Japan). Escherichia coli strain DH5α (Nippon Gene, Co. Ltd., Tokyo, Japan, or BioDynamics Laboratory Inc., Tokyo, Japan) was used for cloning, plasmid propagation, and construction of a mutated XI library. DH5α cells were grown at 37 °C in Luria–Bertani (LB) broth (BD Biosciences) supplemented with 100 µg/mL ampicillin (Wako Pure Chemical Industries, Ltd., Osaka, Japan). Yeast and bacterial strains were stored at − 80 °C in 15% glycerol.
Design, synthesis and cloning of XI genes
DNA fragments were amplified by polymerase chain reaction (PCR) with KOD-Plus-Neo DNA polymerase (TOYOBO, Osaka, Japan) using the primers listed in Additional file 12: Table S9. An expression vector pUG35-kan was constructed based on a low copy number expression vector pUG35 (GenBank Acc. No. AF298787) as follows: first, for construction of pUG35-MET25, pUG35 was digested with SacI and XbaI, treated with a DNA Blunting Kit (Takara-bio), and self-ligated with T4 DNA Ligase (Takara-Bio). A DNA fragment including multiple cloning sites and the CYC1 terminator of pLTex321sV5H was digested with SmaI and KpnI and introduced into the SmaI and KpnI sites, resulting in pUG35-MET25-EGFP3 + MCS. The kanr (G418 resistant) gene expression cassette (kanMX) was amplified by PCR using a primer set (Kan exu F NdeI and Kan exu R KpnI) containing a NdeI or KpnI site and using the pUG6 plasmid as a template [43]. The amplified DNA fragment was digested with NdeI and KpnI and introduced into NdeI and KpnI sites of pUG35-MET25-EGFP3 + MCS, resulting in pUG35-kan. Eight XIs that were previously demonstrated to be functional upon expression in S. cerevisiae were selected: Piromyces sp. E2 [14], Orpinomyces sp. ukk1 [15–17], Lachnoclostridium phytofermentans ISDg (LpXI) [18, 19], Ruminococcus flavefaciens 17 [20], Prevotella ruminicola TC2-24 [21], Burkholderia cenocepacia J2315 [22, 23], Ruminiclostridium cellulolyticum H10 [24] and Streptomyces rubiginosus [25, 26]. The original nucleotide and amino acid sequences of these XIs were obtained from the National Center for Biotechnology Information (NCBI). The synthetic nucleotide sequences of these XI genes were optimized based on codon usage tables derived from highly expressed genes in S. cerevisiae [44] and were synthesized by the GeneArt® gene synthesis service (Thermo Fisher Scientific/Life Technologies). The sequences included SpeI and KpnI sites for subcloning. The accession numbers of the codon-optimized nucleotide sequences are PspXI (LC424543), OspXI (LC424542), LpXI (LC424544), RfXI (LC424541), PrXI (LC424540), BcXI (LC424539), RcXI (LC424545) and SrXI (LC424546). These codon-optimized XI genes were amplified by PCR with the specific primer sets *XIopt_F1 and *XIopt_R1 (where the asterisks are specific for the species of XI listed above). The XhoI sites were added in the reverse primers *XIopt_R1 for directional cloning. The amplified and purified DNA fragments were digested with XhoI and then subcloned into SmaI and XhoI sites of pLTex321sV5H [45] to add a V5-6xHis tag at the C-terminus of each XI gene for purification of XI proteins. The SpeI and KpnI fragments containing the XI gene expression cassette, including the HSP12 promoter, XI gene, and CYC1 terminator, were transferred into the SpeI and KpnI sites of the low copy vector pUG35-kan. In addition, the d-xylulokinase (XKS1) expression cassette including the PGK1 promoter and XKS1 derived from S. cerevisiae was amplified by PCR with a primer set (TCYC1-PPGK1 F and pUG35-TCYC1 R) and pAUR-XKXDHXR plasmid as a template [46]. The fragment was tandemly subcloned into the KpnI site of pUG35-Kan harboring each XI gene cassette using an In-Fusion® HD Cloning Kit (Takara-Bio) according to the manufacturer’s instructions to generate XI expression plasmids pUG35-kan-pHSP12-*XI-tCYC1-pPGK1-XKS1-tCYC1. XI expression was driven by the stress-responsive HSP12 promoter, which can induce expression of downstream genes during the d-xylose consumption phase that occurs after glucose depletion, and the XKS1 gene was overexpressed under the control of the 3-phosphoglycerate kinase gene (PGK1) promoter. All DNA sequences of the constructed plasmids were verified by Sanger sequencing (Eurofins Genomics Inc., Tokyo Japan).
Growth assays under aerobic cultivation
Yeast cells were pre-cultured in YPD medium supplemented with an appropriate antibiotic until the stationary phase was reached. The cells were then washed once with sterilized water before inoculation into growth assay media. YPX50 medium was used for cultivation to determine d-xylose assimilation activity of strains expressing XIs under aerobic conditions. Yeast cells at a final ODA600 of 0.1 were inoculated into 100 µL YPX50 medium in 96-well microplates. Yeast strain growth was then monitored using an Infinite® 200 PRO microplate reader (Tecan, Männedorf, Switzerland). During 4 days of cultivation with continuous shaking at 30 °C, the absorbance (600 nm) of each well containing individual yeast strain cultures was measured every hour. Growth assays for each strain were performed in triplicate. The specific growth rate μ (h−1) and its mean ± SD were calculated from the slope of a line fitted to the exponential growth curve.
Fermentation assays under micro-aerobic cultivation
Fermentation assays were carried out as described previously with slight modifications [36, 37]. In brief, cells were pre-cultured to the stationary phase at 30 °C in 50 mL YPD medium supplemented with an appropriate antibiotic and washed once with YPD85X35 medium. Cells (initial cell density of 20 ODA600 unit) were then inoculated into 70 mL fresh YPD85X35 media in 100-mL flasks capped with rubber stoppers with inserted syringe needles to release CO2. The flasks were also fitted with T-shape stopcocks for ventilation and sampling. During fermentation, 500 µL from each culture medium was collected via the sampling port at specified intervals (0, 1, 3, 6, 12, 24, 36, 48, 60 and 72 h). Samples were stored at − 80 °C until assessment by HPLC. All fermentation assays were performed in triplicate unless otherwise stated. To determine the concentration of sugars and fermentation products such as glucose, xylose, xylitol, glycerol, acetate and ethanol in the culture media, specimens were diluted fivefold with deionized water and analyzed by an HPLC instrument coupled to a refractive index detector (Jasco, Tokyo, Japan) and fitted with an Aminex HPX-87H column and Cation H Refill Guard column (Bio-Rad, Hercules, CA). HPLC assays were performed at 65 °C with 5 mM H2SO4 buffer as the mobile phase, a flow rate of 0.6 mL/min, and an injection volume of 25 µL.
Construction of an LpXI mutant library
A library of randomly mutated LpXI genes was generated using the Diversify PCR Random Mutagenesis Kit (Clontech Laboratories, Inc., Mountain View, CA) according to the manufacturer’s instructions to perform error-prone PCR with the primer set oSS62 XI-F_HSP12s and oSS74 XI-Rc from pUG35-kan-pHSP12-CpXI-tCYC1-pPGK1-XKS1-tCYC1. To produce a mutation rate of 2.7 mutations per 1 kb by error-prone PCR, the following reaction conditions were used: 38 µL PCR-grade water, 5 μL TITANIUM Taq Buffer (10×), 2 µL MnSO4 (8 mM), 1 μL dGTP (2 mM), 1 μL Diversify dNTP Mix (50×), 1 μL primer mix (10 μM each), 1 μL template DNA (1 ng/μL), and 1 μL TITANIUM Taq Polymerase. The purified PCR products were cloned into plasmids to generate a library of more than 104 clones. A linearized vector was prepared using inverse PCR with the primer set oSS63 XI-R_HSP12as and oSS83 06_LpXI-Fc from pUG35-kan-pHSP12-LpXI-tCYC1-pPGK1-XKS1-tCYC1 and the KOD-Plus-NEO DNA polymerase. The randomly mutagenized LpXI genes were fused to the linearized vector using an In-Fusion® HD Cloning Kit (Clontech). An aliquot of the reaction solution was used to transform Giga Competent Cells (DH5α, BioDynamics Laboratory, Inc.) that were then used to amplify a plasmid library harboring mutated LpXI genes. To estimate the mutation rate of the LpXI gene, randomly selected plasmids were purified from E. coli transformants in the library using a QIAprep Spin Miniprep kit (QIAGEN) and the plasmid nucleotide sequences were determined by Sanger sequencing.
Identification of beneficial mutations in LpXI
The plasmid library harboring mutated LpXI was purified from E. coli transformants and the purified plasmids were introduced into the host strain SS29. To determine the suitable experimental conditions for selection of yeast transformants harboring mutated LpXIs that had improved enzymatic characteristics, the growth rate of SS42 strains expressing wild-type LpXI on YPX plates having varying d-xylose concentrations (20–100 g/L) was first tested. These assays indicated that 80 g/L d-xylose inhibited growth of the control strain SS42 and thus YPX80 plates were used to assess the mutant LpXI library. The total number of transformants was 2.4 × 105, indicating that each variant was examined in 24 yeast transformants on average. After the first selection, plasmids containing mutated LpXI genes were prepared from mature colonies that were then used to re-transform SS29 to exclude naturally occurring mutations that were unique to chromosomal rather than plasmid DNA. To determine the LpXI amino acid substitutions, coding regions in the selected clones were verified by DNA sequencing using four primers, oSS62 (XI-F_HSP12s), oSS74 (XI-Rc) and LpXIopt_Seq_F1, and LpXIopt_Seq_R1. For the final verification, each mutated LpXI and XKS1 expression cassette was introduced into the chromosome in the SS29 strain by homologous recombination to prevent effects from variation in LpXI gene copy number. The homologous recombination was carried out by first subcloning an SpeI/SphI fragment containing the mutated LpXI genes from the plasmid pUG35-kan-pHSP12-LpXI-tCYC1-pPGK1-XKS1-tCYC1 into pAUR101 (Takara Bio). The LpXI gene then integrated into the SS29 strain chromosome at the AUR1 locus. The ability of the resulting strains harboring mutated LpXI to metabolize d-xylose in co-fermentation in YPD85X35 medium under micro-aerobic conditions at 30 °C was then examined.
Construction of artificially mutated LpXI genes by separation and combination of mutations found in the mutant library
Site-directed mutagenesis of LpXI was performed to construct expression plasmids harboring mutated LpXI genes (LpXI-V162A, LpXI-N303T and LpXI-T63I/V162A). To introduce the mutations into LpXI, the pAUR-kan-pHSP12-LpXI-tCYC1-pPGK1-XKS1-tCYC1 plasmid was fragmentated by inverse PCR using KOD-Plus-Neo DNA polymerase with oSS106 06_LpXI_V162A and oSS107 06_LpXI_V162Aas as primers. An amplified DNA fragment was self-ligated with T4 DNA Ligase, resulting in the plasmid harboring LpXI-V162A. The other mutated LpXIs (LpXI-N303T and LpXI-T63I/V162A) were constructed in the same manner using oSS108 (06_LpXI_N303T) and oSS109 (06_LpXI_N303Tas) or oSS106 (06_LpXI_V162A) and oSS107 (06_LpXI_V162Aas) as primers and the plasmid (pAUR-kan-pHSP12-LpXI-tCYC1-pPGK1-XKS1-tCYC1). The nucleotide sequences of expression plasmids harboring mutated LpXI genes were validated by Sanger sequencing.
Measurement of d-xylose isomerase catalytic activity
The catalytic activities of d-xylose isomerase in yeast strains harboring LpXI were examined according to a previously described method [14]. In brief, cell lysates from yeast cells of three independent cultivations after 24 h of fermentation in YPD85X35 medium were prepared using CelLytic™ Y Cell Lysis Reagent for Yeast Cells (Sigma-Aldrich, St. Louis, MO) and Protease Inhibitor Cocktail for use with fungal and yeast extracts (Sigma-Aldrich) with 0.5-mm zirconium beads. Cell extract protein concentrations were determined using Pierce™ 660 nm Protein Assay Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. The catalytic activities of cell lysates were assayed at 30 °C by measuring changes in absorbance at 340 nm using an Infinite® 200 PRO spectrophotometer (Tecan), which reflected decreases in the amount of NADH in 100 µL of reaction mixture containing 100 mM Tris–HCl buffer (pH 8.0), 10 mM MgCl2, 0.3 mM β-NADH (Oriental Yeast Co. Ltd., Tokyo, Japan), 2 U sorbitol dehydrogenase (Sigma-Aldrich), and 1 mg/mL-cell lysate. The reaction was started by addition of d-xylose at varying concentrations (10, 50, 100 and 500 mM) as a substrate. Each Km and Vmax were determined by the Lineweaver–Burk equation (1/ν = (Km/Vmax) × 1/[S] + 1/Vmax) and a plot of 1/[S] for the x-axis and 1/ν for the y-axis with a linear approximation. The molar extinction coefficient of 6.25 (mM−1 cm−1) at 340 nm for NADH was used to calculate of Vmax (µmol mg protein−1 min−1). The assay was performed in biological triplicates.
Additional files
Additional file 1: Table S1. Saccharomyces cerevisiae strains used in this study.
Additional file 2: Table S2. Metabolic profiles of recombinant S. cerevisiae strains expressing various XIs in glucose/xylose co-fermentation.
Additional file 3: Figure S1. Fermentation performance of strains containing plasmid vector expressing previously reported mutated XIs in glucose/xylose co-fermentation under micro-aerobic conditions. Batch fermentation assays were performed under the same conditions as for Figs. 2, 3, 4. The fermentation properties of seven strains harboring previously reported mutations for RfXI and PspXI are: SS45 (RfXI-G178A), SS46 (PspXI-E15D), SS47 (PspXI-C54R), SS48 (PspXI-T142S), SS29 (PspXI-N370S), SS50 (PspXI-C54R, N370S) and SS51 (PspXI-E51D, T142S). These mutated XIs were episomally introduced on low copy number plasmids into the same parental strain, SS29. The dots in the panels represent the concentrations of the following metabolites in a single experiment performed with biological triplicates: glucose (blue), xylose (red), xylitol (yellow green), glycerol (purple), acetate (light blue) and ethanol (orange). The details for metabolite concentrations are shown in Additional file 4: Table S3.
Additional file 4: Table S3. Metabolic profiles of recombinant S. cerevisiae strains expressing previously characterized RfXI and PspXI mutants in glucose/xylose co-fermentation.
Additional file 5: Figure S2. Fermentation performance of eleven clones selected in the second growth assay that contained plasmid vectors with mutated LpXI in glucose/xylose co-fermentation under micro-aerobic conditions. Batch fermentation assays were performed under the same conditions as Figs. 2, 3, 4. The fermentation properties were measured for eleven clones containing plasmid vectors carrying mutated LpXIs generated using error-prone PCR: M6-2, M6-6, M6-7, M6-10, M6-11, M6-13, M6-15, M6-19, M6-20, M6-21 and M6-22. The amino acid substitutions in each clone are shown in Table 2. The dots and error bars in the panels represent the mean concentrations and standard deviations, respectively, of the following metabolites in biological triplicates; glucose (blue), xylose (red), xylitol (yellow green), glycerol (purple), acetate (light blue) and ethanol (orange). The details for metabolite concentrations are shown in Additional file 6: Table S4.
Additional file 6: Table S4. Metabolic profiles of recombinant S. cerevisiae strains expressing evolved LpXIs from plasmid vectors in glucose/xylose co-fermentation.
Additional file 7: Table S5. Metabolic profiles of recombinant S. cerevisiae strains expressing mutated LpXIs in glucose/xylose co-fermentation.
Additional file 8: Figure S3. Micro-aerobic fermentation of strains expressing mutated XIs on medium containing d-glucose and d-xylose. Batch fermentation assays were performed under the same conditions as for Fig. 2. The fermentation properties were measured for seven strains without improved d-xylose consumption rates: SS84 (LpXI-K136T/A176T), SS85 (LpXI-Y13H/D228V), SS86 (LpXI-T273A), SS87 (LpXI-D207G), SS88 (LpXI-N223I), SS91 (LpXI-E114G) and SS94 (LpXI-L304S). These mutated LpXI expression cassettes were introduced into the AUR1 locus of the SS29 parental strain chromosome. The dots in the panels represent the concentrations of the following metabolites in a single experiment performed in biological triplicate: glucose (blue), xylose (red), xylitol (yellow green), glycerol (purple), acetate (light blue) and ethanol (orange). The details of metabolite concentrations are shown in Additional file 9: Table S6.
Additional file 9: Table S6. Metabolic profiles of recombinant S. cerevisiae strains expressed mutated LpXIs in a glucose/xylose co-fermentation.
Additional file 10: Table S7. Metabolic profiles of recombinant S. cerevisiae strains expressed mutated LpXIs in a glucose/xylose co-fermentation.
Additional file 11: Table S8. Kinetic analysis of mutated LpXIs
Additional file 12: Table S9. Oligonucleotides used in this study.
Acknowledgements
This study was supported by the New Energy and Industrial Technology Development Organization (NEDO; Project No. P13011).
Abbreviations
- YPD
yeast extract/peptone/dextrose
- YPX
yeast extract/peptone/xylose
- LB
Luria–Bertani
- OD
optical density
- XI
xylose isomerase
- XR
xylose reductase
- XDH
xylitol dehydrogenase
- HPLC
high-performance liquid chromatography
- PCR
polymerase chain reaction
- HMF
5-hydroxymethyl-2-furfural
- OspXI
xylose isomerase derived from Orpinomyces sp. Ukk1
- LpXI
xylose isomerase derived from Lachnoclostridium (previously known as Clostridium) phytofermentans ISDg
- RfXI
xylose isomerase derived from Ruminococcus flavefaciens 17
- PrXI
xylose isomerase derived from Prevotella ruminicola TC2-24
- BcXI
xylose isomerase derived from Burkholderia cenocepacia J2315
- RcXI
xylose isomerase derived from Ruminiclostridium (previously known as Clostridium) cellulolyticum H10
- SrXI
xylose isomerase derived from Streptomyces rubiginosus
- XKS1
xylulokinase derived from S. cerevisiae
- pPGK1
promoter of 3-phosphoglycerate kinase derived from S. cerevisiae
- pHSP12
promoter of heat shock protein 12 derived from S. cerevisiae
- tCYC1
terminator of cytochrome c, isoform 1 derived from S. cerevisiae
- PPP
pentose phosphate pathway
Authors’ contributions
TSe and TSa designed and performed all the experiments. TSe and KEF analyzed and summarized the data obtained. YKo, SO and YKa managed and supported the overall project, which included this study. TSe drafted the manuscript and KEF supervised and wrote the manuscript. All authors read and approved the final manuscript.
Funding
Funding sources are listed in the “Acknowledgements”.
Availability of supporting data
All data supporting the conclusions of this article are included within the manuscript and additional files.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Taisuke Seike, Email: taisuke.seike@riken.jp.
Yosuke Kobayashi, Email: kobayashi.y@biomt.onmicrosoft.com.
Takehiko Sahara, Email: t-sahara@aist.go.jp.
Satoru Ohgiya, Email: s.ohgiya@aist.go.jp.
Yoichi Kamagata, Email: y-kamagata@aist.go.jp.
Kazuhiro E. Fujimori, Phone: +81-29-849-1228, Email: k-fujimori@aist.go.jp
References
- 1.Robak K, Balcerek M. Review of second generation bioethanol production from residual biomass. Food Technol Biotechnol. 2018;56:174–187. doi: 10.17113/ftb.56.02.18.5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee J. Biological conversion of lignocellulosic biomass to ethanol. J Biotechnol. 1997;56:1–24. doi: 10.1016/S0168-1656(97)00073-4. [DOI] [PubMed] [Google Scholar]
- 3.Hendriks ATWM, Zeeman G. Pretreatments to enhance the digestibility of lignocellulosic biomass. Bioresour Technol. 2009;100:10–18. doi: 10.1016/j.biortech.2008.05.027. [DOI] [PubMed] [Google Scholar]
- 4.Hou J, Qiu C, Shen Y, Li H, Bao X. Engineering of Saccharomyces cerevisiae for the efficient co-utilization of glucose and xylose. FEMS Yeast Res. 2017;17:fox034. doi: 10.1093/femsyr/fox034. [DOI] [PubMed] [Google Scholar]
- 5.Jeppsson M, Bengtsson O, Franke K, Lee H, Hahn-Hägerdal B, Gorwa-Grauslund MF. The expression of a Pichia stipitis xylose reductase mutant with higher Km for NADPH increases ethanol production from xylose in recombinant Saccharomyces cerevisiae. Biotechnol Bioeng. 2006;93:665–673. doi: 10.1002/bit.20737. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe S, Saleh AA, Pack SP, Annaluru N, Kodaki T, Makino K. Ethanol production from xylose by recombinant Saccharomyces cerevisiae expressing protein engineered NADP+-dependent xylitol dehydrogenase. J Biotechnol. 2007;130:316–319. doi: 10.1016/j.jbiotec.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 7.Karhumaa K, Fromanger R, Hahn-Hägerdal B, Gorwa-Grauslund MF. High activity of xylose reductase and xylitol dehydrogenase improves xylose fermentation by recombinant Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2007;73:1039–1046. doi: 10.1007/s00253-006-0575-3. [DOI] [PubMed] [Google Scholar]
- 8.Matsushika A, Sawayama S. Efficient bioethanol production from xylose by recombinant Saccharomyces cerevisiae requires high activity of xylose reductase and moderate xylulokinase activity. J Biosci Bioeng. 2008;106:306–309. doi: 10.1263/jbb.106.306. [DOI] [PubMed] [Google Scholar]
- 9.Zeng WY, Tang YQ, Gou M, Sun ZY, Xia ZY, Kida K. Comparative transcriptomes reveal novel evolutionary strategies adopted by Saccharomyces cerevisiae with improved xylose utilization capability. Appl Microbiol Biotechnol. 2017;101:1753–1767. doi: 10.1007/s00253-016-8046-y. [DOI] [PubMed] [Google Scholar]
- 10.Sarthy AV, McConaughy BL, Lobo Z, Sundstrom JA, Furlong CE, Hall BD. Expression of the Escherichia coli xylose isomerase gene in Saccharomyces cerevisiae. Appl Environ Microbiol. 1987;53:1996–2000. doi: 10.1128/aem.53.9.1996-2000.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amore R, Wilhelm M, Hollenberg CP. The fermentation of xylose—an analysis of the expression of Bacillus and Actinoplanes xylose isomerase genes in yeast. Appl Microbiol Biotechnol. 1989;30:351–357. doi: 10.1007/BF00296623. [DOI] [Google Scholar]
- 12.Moes CJ, Pretorius IS, van Zyl WH. Cloning and expression of the Clostridium thermosulfurogenesd-xylose isomerase gene (xylA) in Saccharomyces cerevisiae. Biotechnol Lett. 1996;18:269–274. doi: 10.1007/BF00142943. [DOI] [Google Scholar]
- 13.Walfridsson M, Bao X, Anderlund M, Lilius G, Bülow L, Hahn-Hägerdal B. Ethanolic fermentation of xylose with Saccharomyces cerevisiae harboring the Thermus thermophilus xylA gene, which expresses an active xylose (glucose) isomerase. Appl Environ Microbiol. 1996;62:4648–4651. doi: 10.1128/aem.62.12.4648-4651.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuyper M, Harhangi HR, Stave AK, Winkler AA, Jetten MS, de Laat WT, den Ridder JJ, Op den Camp HJ, van Dijken JP, Pronk JT. High-level functional expression of a fungal xylose isomerase: the key to efficient ethanolic fermentation of xylose by Saccharomyces cerevisiae? FEMS Yeast Res. 2003;4:69–78. doi: 10.1016/S1567-1356(03)00141-7. [DOI] [PubMed] [Google Scholar]
- 15.Madhavan A, Tamalampudi S, Srivastava A, Fukuda H, Bisaria VS, Kondo A. Alcoholic fermentation of xylose and mixed sugars using recombinant Saccharomyces cerevisiae engineered for xylose utilization. Appl Microbiol Biotechnol. 2009;82:1037–1047. doi: 10.1007/s00253-008-1818-2. [DOI] [PubMed] [Google Scholar]
- 16.Madhavan A, Tamalampudi S, Ushida K, Kanai D, Katahira S, Srivastava A, Fukuda H, Bisaria VS, Kondo A. Xylose isomerase from polycentric fungus Orpinomyces: gene sequencing, cloning, and expression in Saccharomyces cerevisiae for bioconversion of xylose to ethanol. Appl Microbiol Biotechnol. 2009;82:1067–1078. doi: 10.1007/s00253-008-1794-6. [DOI] [PubMed] [Google Scholar]
- 17.Tanino T, Hotta A, Ito T, Ishii J, Yamada R, Hasunuma T, Ogino C, Ohmura N, Ohshima T, Kondo A. Construction of a xylose-metabolizing yeast by genome integration of xylose isomerase gene and investigation of the effect of xylitol on fermentation. Appl Microbiol Biotechnol. 2010;88:1215–1221. doi: 10.1007/s00253-010-2870-2. [DOI] [PubMed] [Google Scholar]
- 18.Brat D, Boles E, Wiedemann B. Functional expression of a bacterial xylose isomerase in Saccharomyces cerevisiae. Appl Environ Microbiol. 2009;75:2304–2311. doi: 10.1128/AEM.02522-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demeke MM, Dietz H, Li Y, Foulquié-Moreno MR, Mutturi S, Deprez S, Den Abt T, Bonini BM, Liden G, Dumortier F, Verplaetse A, Boles E, Thevelein JM. Development of a d-xylose fermenting and inhibitor tolerant industrial Saccharomyces cerevisiae strain with high performance in lignocellulose hydrolysates using metabolic and evolutionary engineering. Biotechnol Biofuels. 2013;6:89. doi: 10.1186/1754-6834-6-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aeling KA, Salmon KA, Laplaza JM, Li L, Headman JR, Hutagalung AH, Picataggio S. Co-fermentation of xylose and cellobiose by an engineered Saccharomyces cerevisiae. J Ind Microbiol Biotechnol. 2012;39:1597–1604. doi: 10.1007/s10295-012-1169-y. [DOI] [PubMed] [Google Scholar]
- 21.Hector RE, Dien BS, Cotta MA, Mertens JA. Growth and fermentation of d-xylose by Saccharomyces cerevisiae expressing a novel d-xylose isomerase originating from the bacterium Prevotella ruminicola TC2-24. Biotechnol Biofuels. 2013;6:84. doi: 10.1186/1754-6834-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Figueiredo Vilela L, de Mello VM, Reis VC, Bon EP, Gonçalves Torres FA, Neves BC, Eleutherio EC. Functional expression of Burkholderia cenocepacia xylose isomerase in yeast increases ethanol production from a glucose–xylose blend. Bioresour Technol. 2013;128:792–796. doi: 10.1016/j.biortech.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 23.de Figueiredo Vilela L, de Araujo VPG, de Sousa Paredes R, da Silva Bon EP, Torres FAG, Neves BC, Eleutherio ECA. Enhanced xylose fermentation and ethanol production by engineered Saccharomyces cerevisiae strain. AMB Express. 2015;5:1–7. doi: 10.1186/s13568-014-0092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harcus D, Dignard D, Lépine G, Askew C, Raymond M, Whiteway M, Wu C. Comparative xylose metabolism among the ascomycetes C. albicans, S. stipitis and S. cerevisiae. PLoS ONE. 2013;8:e80733. doi: 10.1371/journal.pone.0080733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waltman MJ, Yang ZK, Langan P, Graham DE, Kovalevsky A. Engineering acidic Streptomyces rubiginosusd-xylose isomerase by rational enzyme design. Protein Eng Des Sel. 2014;27:59–64. doi: 10.1093/protein/gzt062. [DOI] [PubMed] [Google Scholar]
- 26.Lee SM, Jellison T, Alper HS. Directed evolution of xylose isomerase for improved xylose catabolism and fermentation in the yeast Saccharomyces cerevisiae. Appl Environ Microbiol. 2012;78:5708–5716. doi: 10.1128/AEM.01419-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harhangi HR, Akhmanova AS, Emmens R, van der Drift C, de Laat WT, van Dijken JP, Jetten MS, Pronk JT, Op den Camp HJ. Xylose metabolism in the anaerobic fungus Piromyces sp. strain E2 follows the bacterial pathway. Arch Microbiol. 2003;180:134–141. doi: 10.1007/s00203-003-0565-0. [DOI] [PubMed] [Google Scholar]
- 28.Van Maris AJ, Winkler AA, Kuyper M, De Laat WT, Van Dijken JP, Pronk JT. Development of efficient xylose fermentation in Saccharomyces cerevisiae: xylose isomerase as a key component. Adv Biochem Eng Biotechnol. 2007;108:179–204. doi: 10.1007/10_2007_057. [DOI] [PubMed] [Google Scholar]
- 29.Xia PF, Zhang GC, Liu JJ, Kwak S, Tsai CS, Kong II, Sung BH, Sohn JH, Wang SG, Jin YS. GroE chaperonins assisted functional expression of bacterial enzymes in Saccharomyces cerevisiae. Biotechnol Bioeng. 2016;113:2149–2155. doi: 10.1002/bit.25980. [DOI] [PubMed] [Google Scholar]
- 30.Temer B, Dos Santos LV, Negri VA, Galhardo JP, Magalhães PHM, José J, Marschalk C, Corrêa TLR, Carazzolle MF, Pereira GAG. Conversion of an inactive xylose isomerase into a functional enzyme by co-expression of GroEL-GroES chaperonins in Saccharomyces cerevisiae. BMC Biotechnol. 2017;17:71. doi: 10.1186/s12896-017-0389-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang GC, Liu JJ, Kong II, Kwak S, Jin YS. Combining C6 and C5 sugar metabolism for enhancing microbial bioconversion. Curr Opin Chem Biol. 2015;29:49–57. doi: 10.1016/j.cbpa.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Moysés DN, Reis VC, de Almeida JR, de Moraes LM, Torres FA. Xylose Fermentation by Saccharomyces cerevisiae: challenges and prospects. Int J Mol Sci. 2016;17:207. doi: 10.3390/ijms17030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rose M. Molecular and biochemical characterization of the hexokinase from the starch-utilizing yeast Schwanniomyces occidentalis. Curr Genet. 1995;27:330–338. doi: 10.1007/BF00352102. [DOI] [PubMed] [Google Scholar]
- 34.Demeke MM, Foulquié-Moreno MR, Dumortier F, Thevelein JM. Rapid evolution of recombinant Saccharomyces cerevisiae for xylose fermentation through formation of extra-chromosomal circular DNA. PLoS Genet. 2015;11:e1005010. doi: 10.1371/journal.pgen.1005010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boles E, Schulte F, Miosga T, Freidel K, Schlüter E, Zimmermann FK, Hollenberg CP, Heinisch JJ. Characterization of a glucose-repressed pyruvate kinase (Pyk2p) in Saccharomyces cerevisiae that is catalytically insensitive to fructose-1,6-bisphosphate. J Bacteriol. 1997;179:2987–2993. doi: 10.1128/jb.179.9.2987-2993.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi Y, Sahara T, Suzuki T, Kamachi S, Matsushika A, Hoshino T, Ohgiya S, Kamagata Y, Fujimori KE. Genetic improvement of xylose metabolism by enhancing the expression of pentose phosphate pathway genes in Saccharomyces cerevisiae IR-2 for high-temperature ethanol production. J Ind Microbiol Biotechnol. 2017;44:879–891. doi: 10.1007/s10295-017-1912-5. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi Y, Sahara T, Ohgiya S, Kamagata Y, Fujimori KE. Systematic optimization of gene expression of pentose phosphate pathway enhances ethanol production from a glucose/xylose mixed medium in a recombinant Saccharomyces cerevisiae. AMB Express. 2018;8:139. doi: 10.1186/s13568-018-0670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sedlak M, Ho NW. Characterization of the effectiveness of hexose transporters for transporting xylose during glucose and xylose co-fermentation by a recombinant Saccharomyces yeast. Yeast. 2004;21:671–684. doi: 10.1002/yea.1060. [DOI] [PubMed] [Google Scholar]
- 39.Farwick A, Bruder S, Schadeweg V, Oreb M, Boles E. Engineering of yeast hexose transporters to transport d-xylose without inhibition by d-glucose. Proc Natl Acad Sci USA. 2014;111:5159–5164. doi: 10.1073/pnas.1323464111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li YC, Mitsumasu K, Gou ZX, Gou M, Tang YQ, Li GY, Wu XL, Akamatsu T, Taguchi H, Kida K. Xylose fermentation efficiency and inhibitor tolerance of the recombinant industrial Saccharomyces cerevisiae strain NAPX37. Appl Microbiol Biotechnol. 2016;100:1531–1542. doi: 10.1007/s00253-015-7167-z. [DOI] [PubMed] [Google Scholar]
- 41.Kuriyama H, Seiko Y, Murakami T, Kobayashi H. Continuous ethanol fermentation with cell recycling using flocculating yeast. J Ferment Technol. 1985;63:159–165. [Google Scholar]
- 42.Sahara T, Fujimori KE, Nezuo M, Tsukahara M, Tochigi Y, Ohgiya S, Kamagata Y. Draft genome sequence of Saccharomyces cerevisiae IR-2, a useful industrial strain for highly efficient production of bioethanol. Genome Announc. 2014;2:e01160-13. doi: 10.1128/genomeA.01160-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Güldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 1996;24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akashi H. Translational selection and yeast proteome evolution. Genetics. 2003;164:1291–1303. doi: 10.1093/genetics/164.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohgiya S, Sahara T. Protein production at low temperature by yeast Saccharomyces cerevisiae. Biosci Ind. 2007;65:130–131. [Google Scholar]
- 46.Matsushika A, Watanabe S, Kodaki T, Makino K, Inoue H, Murakami K, Takimura O, Sawayama S. Expression of protein engineered NADP+-dependent xylitol dehydrogenase increase ethanol production from xylose in recombinant Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2008;81:243–255. doi: 10.1007/s00253-008-1649-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Saccharomyces cerevisiae strains used in this study.
Additional file 2: Table S2. Metabolic profiles of recombinant S. cerevisiae strains expressing various XIs in glucose/xylose co-fermentation.
Additional file 3: Figure S1. Fermentation performance of strains containing plasmid vector expressing previously reported mutated XIs in glucose/xylose co-fermentation under micro-aerobic conditions. Batch fermentation assays were performed under the same conditions as for Figs. 2, 3, 4. The fermentation properties of seven strains harboring previously reported mutations for RfXI and PspXI are: SS45 (RfXI-G178A), SS46 (PspXI-E15D), SS47 (PspXI-C54R), SS48 (PspXI-T142S), SS29 (PspXI-N370S), SS50 (PspXI-C54R, N370S) and SS51 (PspXI-E51D, T142S). These mutated XIs were episomally introduced on low copy number plasmids into the same parental strain, SS29. The dots in the panels represent the concentrations of the following metabolites in a single experiment performed with biological triplicates: glucose (blue), xylose (red), xylitol (yellow green), glycerol (purple), acetate (light blue) and ethanol (orange). The details for metabolite concentrations are shown in Additional file 4: Table S3.
Additional file 4: Table S3. Metabolic profiles of recombinant S. cerevisiae strains expressing previously characterized RfXI and PspXI mutants in glucose/xylose co-fermentation.
Additional file 5: Figure S2. Fermentation performance of eleven clones selected in the second growth assay that contained plasmid vectors with mutated LpXI in glucose/xylose co-fermentation under micro-aerobic conditions. Batch fermentation assays were performed under the same conditions as Figs. 2, 3, 4. The fermentation properties were measured for eleven clones containing plasmid vectors carrying mutated LpXIs generated using error-prone PCR: M6-2, M6-6, M6-7, M6-10, M6-11, M6-13, M6-15, M6-19, M6-20, M6-21 and M6-22. The amino acid substitutions in each clone are shown in Table 2. The dots and error bars in the panels represent the mean concentrations and standard deviations, respectively, of the following metabolites in biological triplicates; glucose (blue), xylose (red), xylitol (yellow green), glycerol (purple), acetate (light blue) and ethanol (orange). The details for metabolite concentrations are shown in Additional file 6: Table S4.
Additional file 6: Table S4. Metabolic profiles of recombinant S. cerevisiae strains expressing evolved LpXIs from plasmid vectors in glucose/xylose co-fermentation.
Additional file 7: Table S5. Metabolic profiles of recombinant S. cerevisiae strains expressing mutated LpXIs in glucose/xylose co-fermentation.
Additional file 8: Figure S3. Micro-aerobic fermentation of strains expressing mutated XIs on medium containing d-glucose and d-xylose. Batch fermentation assays were performed under the same conditions as for Fig. 2. The fermentation properties were measured for seven strains without improved d-xylose consumption rates: SS84 (LpXI-K136T/A176T), SS85 (LpXI-Y13H/D228V), SS86 (LpXI-T273A), SS87 (LpXI-D207G), SS88 (LpXI-N223I), SS91 (LpXI-E114G) and SS94 (LpXI-L304S). These mutated LpXI expression cassettes were introduced into the AUR1 locus of the SS29 parental strain chromosome. The dots in the panels represent the concentrations of the following metabolites in a single experiment performed in biological triplicate: glucose (blue), xylose (red), xylitol (yellow green), glycerol (purple), acetate (light blue) and ethanol (orange). The details of metabolite concentrations are shown in Additional file 9: Table S6.
Additional file 9: Table S6. Metabolic profiles of recombinant S. cerevisiae strains expressed mutated LpXIs in a glucose/xylose co-fermentation.
Additional file 10: Table S7. Metabolic profiles of recombinant S. cerevisiae strains expressed mutated LpXIs in a glucose/xylose co-fermentation.
Additional file 11: Table S8. Kinetic analysis of mutated LpXIs
Additional file 12: Table S9. Oligonucleotides used in this study.
Data Availability Statement
All data supporting the conclusions of this article are included within the manuscript and additional files.