

Abstract
Costameres are signaling hubs at the sarcolemma and important contact points between the extracellular matrix and cell interior, sensing and transducing biomechanical signals into a cellular response. The transmembrane proteoglycan syndecan-4 localizes to these attachment points and has been shown to be important in the initial stages of cardiac remodeling, but its mechanistic function in the heart remains insufficiently understood. Here, we sought to map the cardiac interactome of syndecan-4 to better understand its function and downstream signaling mechanisms. By combining two different affinity purification methods with MS analysis, we found that the cardiac syndecan-4 interactome consists of 21 novel and 29 previously described interaction partners. Nine of the novel partners were further validated to bind syndecan-4 in HEK293 cells (i.e. CAVIN1/PTRF, CCT5, CDK9, EIF2S1, EIF4B, MPP7, PARVB, PFKM, and RASIP). We also found that 19 of the 50 interactome partners bind differently to syndecan-4 in the left ventricle lysate from aortic-banded heart failure (ABHF) rats compared with SHAM-operated animals. One of these partners was the well-known mechanotransducer muscle LIM protein (MLP), which showed direct and increased binding to syndecan-4 in ABHF. Nuclear translocation is important in MLP-mediated signaling, and we found less MLP in the nuclear-enriched fractions from syndecan-4−/− mouse left ventricles but increased nuclear MLP when syndecan-4 was overexpressed in a cardiomyocyte cell line. In the presence of a cell-permeable syndecan-4–MLP disruptor peptide, the nuclear MLP level was reduced. These findings suggest that syndecan-4 mediates nuclear translocation of MLP in the heart.
Keywords: proteoglycan, heart, cardiomyopathy, protein–protein interaction, nuclear translocation, protein complex, biomechanics, cysteine and glycine rich protein 3 (CSRP3), muscle LIM protein (MLP), syndecan-4
Introduction
In response to biomechanical stress and extracellular matrix cues, the heart is able to remodel its form and function to cope with increased demands. Major sites of biomechanical stress-sensing and transduction are the costamere and z-discs, which are the merging points between the sarcomere and sarcolemma in the cardiomyocytes, and focal adhesions, which are the linkage sites between the extracellular matrix (ECM)4 and cytoskeleton in fibroblasts (1–3). Molecules able to merge and coordinate signaling inward at these sites are likely key players in the biomechanical stress-sensing apparatus in the heart.
Syndecan-4 is a ubiquitously expressed transmembrane proteoglycan with a large ectodomain decorated with ECM-binding glycosaminoglycan chains, a transmembrane domain, and a short cytoplasmic tail that connects to intracellular-binding partners and the actin cytoskeleton (4). Because syndecan-4 holds no intrinsic enzymatic activity, direct signal transduction must occur through interactions with its cytoplasmic tail or indirectly through co-receptors like the integrins (4). Because of its membrane-spanning quality and ideal position at z-discs (5) and focal adhesions (6), syndecan-4 has attracted much attention as a potential mechanosensing and transducing molecule in the heart. When syndecan-4−/− mice are subjected to mechanical stress by inducing pressure overload of the left ventricle (LV), a reduction of the expected hypertrophy and ECM stiffness is observed, placing syndecan-4 as a player in the biomechanical stress-sensing apparatus (7, 8). Several direct or indirect syndecan-4 partners are implicated in cardiac remodeling and pathology, including PKCα (9), calcineurin (7), and the calcineurin-dependent transcription factor nuclear factor of activated T-cells (NFAT) (1, 2).
To better understand how syndecan-4 exerts its cellular function in the heart, we set out to map the cardiac syndecan-4 interactome. By combining two different large-scale affinity purification methods with MS (AP-MS), we identified 21 novel syndecan-4 interaction partners together with 29 partners previously described in the literature, which together make up the cardiac interactome. 38% of these 50 interactions were altered during LV pressure overload, including MLP, a known mechanotransducer directly involved in human cardiomyopathy (10, 11). We found a direct interaction between MLP and syndecan-4. Nuclear translocation of MLP, which is important for its ability to initiate hypertrophic gene expression (12), was reduced in syndecan-4−/− mice and in H9c2 rat cardiomyoblast cells treated with a cell-permeable syndecan-4–MLP disruptor peptide. In contrast, the nuclear MLP level increased when syndecan-4 was overexpressed in H9c2. Altogether, our data suggest syndecan-4 mediates nuclear translocation of MLP.
Results
Use of two different AP-MS approaches to capture syndecan-4 interaction partners
To increase confidence in our results, two different affinity purification (AP) approaches were used to identify syndecan-4 protein partners. In the first approach, we used biotinylated peptides covering the cytoplasmic part of syndecan-4 as bait, with or without phosphorylation at serine 179 (SDC4cyt and pS179–SDC4cyt, respectively) (Fig. 1A, panel i). A biotinylated scrambled peptide was used as a negative control (peptide sequences are given in Fig. 1B). The integrity of the peptides was first confirmed by immunoblotting. Both biotin–SDC4cyt and biotin–pS179–SDC4cyt were recognized by anti-syndecan-4cyt (custom-made with an internal epitope, Genscript) (Fig. S1A, upper panel), and biotin–pS179–SDC4 was only recognized by anti-pS179–syndecan-4 (custom-made, Genscript) (Fig. S1A, middle panel), whereas all three peptides were recognized by anti-biotin–HRP (Fig. S1A, lower panel). To confirm the binding capacity of the bait, biotin–SDC4cyt was shown to bind to the syndecan-4 C1-domain partner ezrin (13) in an ELISA-based interaction assay, whereas the scrambled control peptide did not (Fig. 1C). For the mass spectrometry (MS) analyses, equivalent amounts of the three peptides were separately incubated in rat LV lysate, and putative syndecan-4–interacting proteins were pulled out with anti-biotin–agarose beads. Nonspecific binders (light blue proteins in Fig. 1A) were removed by several wash steps, and the remaining bound proteins (light brown proteins) were eluted with 25 mm biotin before precipitation with ice-cold acetone, trypsin digestion, and identification by MS.
Figure 1.

Two different AP-MS strategies were used to identify novel syndecan-4 protein partners. A, schematic illustration of the experimental design. Precipitation of syndecan-4–interacting proteins was performed in panel i by fishing with a biotinylated peptide covering the syndecan-4 cytoplasmic part (biotin–SDC4cyt), or the syndecan-4 cytoplasmic part phosphorylated at serine 179 (biotin–pS179–SDC4cyt), or by precipitation of endogenous syndecan-4 using syndecan-4–specific antibodies in panel ii WT LV lysate or in panel iii ABHF LV lysate. A scrambled syndecan-4 control peptide, nonrelevant IgG, beads, and lysate from SHAM LVs were used as respective negative controls. Illustrations of the full-length transmembrane syndecan-4 and aortic banding are given at left. B, sequences of the three biotinylated peptides used in the pulldown approach in A. C, binding of biotin–SDC4cyt to ezrin was confirmed by ELISA (unpaired t test, ****, p < 0.0001). D, anti-SDC4 (KY/8.2) was overlaid arrays of immobilized overlapping 20-mer peptides covering the full-length syndecan-4 rat protein sequence. The core epitope is underlined. Immunoblotting without any primary antibody was used as negative control (lower panel) (n = 2, two independent peptide arrays syntheses). E, antibody core epitope is underlined in the alignment of rat syndecan 1–4 (DNA Star, Madison, WI). Black boxes indicate identical amino acids. F, anti-SDC4 (KY/8.2) (with an external epitope) precipitated endogenous syndecan-4 in lysates from both rat neonatal cardiomyocytes (CM) and cardiac fibroblasts (CFB). Nonrelevant rat IgG was used as negative control.
In the second approach, we used anti-syndecan-4 (KY/8.2) to precipitate endogenous syndecan-4 (Fig. 1A, panel ii). The specificity of the syndecan-4 (KY/8.2) antibody was first validated by epitope mapping. Anti-syndecan-4 (KY/8.2) was overlaid onto 20-mer peptides covering the full-length syndecan-4 rat protein, and ALPDDEDAGGL was recognized as the core epitope (Fig. 1D). Importantly, the ALPDDEDAGGL sequence was unique to syndecan-4 and not present in the three other syndecans (Fig. 1E). Consistently, anti-syndecan-4 (KY/8.2) did not recognize syndecan-1, -2, or -3 (Fig. S1B). Immunoprecipitation experiments confirmed that anti-syndecan-4 (KY/8.2) specifically precipitated syndecan-4 from both isolated rat neonatal cardiomyocytes (CM) and cardiac fibroblasts (CFB) (Fig. 1F).
Identification of syndecan-4–interacting proteins from AP-MS
Both AP approaches were performed in large scale and in three biological parallels, and each parallel was run in triplicate by MS. To eliminate potential false-positives, we excluded common contaminants such as ribosomal proteins, uncharacterized proteins, and pure nuclear and mitochondrial proteins. In the first approach using biotin–SDC4cyt or biotin–pS179–SDC4cyt versus biotin-scram (negative control) as bait, we identified 40 proteins that were significant in at least two experiments (n = 2, p < 0.05). No significant changes were observed when comparing the results of pulldown experiments with biotin–SDC4cyt and biotin–pS179–SDC4cyt. In the second approach using anti-syndecan-4 against endogenous syndecan-4, we identified 368 putative syndecan-4–interacting proteins (n = 3, p < 0.01). 21 novel partners were identified in LV lysate by both AP-MS approaches (Fig. 2, Venn diagram, pink circle). Those used were as follows: AP-3 complex subunit δ-1 (AP3D1); caveolae-associated protein 1 (CAVIN1/PTRF); t-complex protein 1 subunit ϵ (CCT5); cyclin-dependent kinase 9 (CDK9); cysteine and glycine-rich protein 3 (MLP/CSRP3); eukaryotic translation initiation factor 2 subunit 1 (EIF2S1); eukaryotic translation initiation factor 4B (EIF4B); protein 4.1 (EPB41); BRISC complex subunit Abraxas 2 (FAM175B/ABRAXAS2); GSH peroxidase 1 (GPX1); growth hormone-regulated TBC protein 1 (GRTP1); MAGUK p55 subfamily member 7 (MPP7); protein LYRIC (MTDH); β-parvin (PARVB); ATP-dependent 6-phosphofructokinase, muscle-type (PFKM); Ras-interacting protein 1 (RASIP1); Ras-related GTP-binding protein C (RRAGC); Na+/H+-exchange regulatory cofactor NHE–RF2 (SLC9A3R2); tight-junction protein ZO-2 (TJP2); tripartite motif-containing protein 72 (TRIM21); and uveal autoantigen with coiled-coil domains and ankyrin repeats (UACA). RRAGC, TRIM72, and UACA interacted with syndecan-4 only in the LV from rats with aortic-banded heart failure (ABHF) (Fig. 2, denoted with **) and is discussed further below. The 21 novel partners are summarized in Table 1 and in more detail in Table S1. A complete list of all proteins identified in either AP-MS approaches is given in Table S2.
Figure 2.
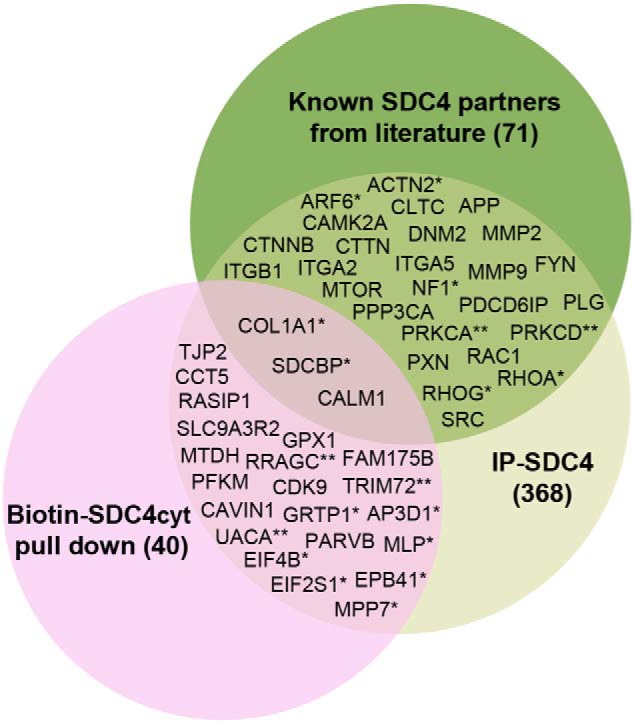
Venn diagram depicting the distribution of the putative syndecan-4 partners together with literature partners identified by the two different AP-MS strategies (p < 0. 01–0.05). *, interaction with syndecan-4 changed in ABHF (p < 0.05, n = 3); **, interaction with syndecan-4 only detected in ABHF (p < 0.05, n = 3). The syndecan-4 literature partners were detected at n = 1–3 (p < 0.01–0.05, see Table 2). SDCBP, CALM1, and COL1A1 were also detected in the pulldown approach, but only COL1A1 was significant.
Table 1.
21 novel syndecan-4–interacting proteins identified by two AP-MS approaches
| Gene | Protein (UniProt) | Evidence for interaction | Molecular function (HPRD) |
|---|---|---|---|
| AP3D1a | AP-3 complex subunit δ-1 | MS | Transporter activity |
| CAVIN1/PTRF | Caveolae-associated protein 1 | MS, co-IPb | Transcription regulator activity |
| CCT5 | T-complex protein 1 subunit ϵ | MS, co-IP | Chaperone activity |
| CDK9 | Cyclin-dependent kinase 9 | MS, co-IP | Protein serine/threonine kinase activity |
| EIF2S1a | Eukaryotic translation initiation factor 2 subunit 1 | MS, co-IP | Translation regulator activity |
| EIF4Ba | Eukaryotic translation initiation factor 4B | MS, co-IP | Translation regulator activity |
| EPB41a | Protein 4.1 | MS | Structural molecule activity |
| FAM175B/ABRAXAS2 | BRISC complex subunit Abraxas 2 | MS | Unknown |
| GPX1 | Glutathione peroxidase 1 | MS | Peroxidase activity |
| GRTP1a | Growth hormone-regulated TBC protein 1 | MS | GTPase activator activity |
| MLP/CSRP3a | Muscle LIM protein/cysteine and glycine-rich protein 3 | MS | Cytoskeletal protein binding |
| MPP7a | MAGUK p55 subfamily member 7 | MS, co-IP | Receptor signaling complex scaffold activity |
| MTDH | Protein LYRIC | MS | Unknown |
| PARVB | β-Parvin | MS, co-IP | Cytoskeletal protein binding |
| PFKM | ATP-dependent 6-phosphofructokinase, muscle type | MS, co-IP | Catalytic activity |
| RASIP1 | Ras-interacting protein 1 | MS, co-IP | Unknown |
| RRAGCc | Ras-related GTP binding protein C | MS | GTPase activity |
| SLC9A3R2 | Na+/H+ exchange regulatory cofactor NHE-RF2 | MS | Receptor signaling complex scaffold activity |
| TJP2 | Tight junction protein ZO-2 | MS | Cell adhesion molecule activity |
| TRIM72c | Tripartite motif-containing protein 72 | MS | Unknown |
| UACAc | Uveal autoantigen with coiled-coil domains and ankyrin repeats | MS | Unknown |
a Interaction with syndecan-4 was significantly changed in ABHF (p < 0.05, n = 3).
b co-IP, co-immunoprecipitation in HEK293.
c Interaction with syndecan-4 was only detected in ABHF (p < 0.05, n = 3).
We also performed an extensive literature search and a summary of the 71 direct or indirect literature syndecan-4 partners reported in different species, tissues, and cells is given in Table S3. Interestingly, 29 of the literature partners were also significant in our IP–syndecan-4 approach (Fig. 2, Venn diagram, green circle, and Table S3, p < 0.05). The remaining literature partners were either not significant or showed no interaction with syndecan-4 in LV lysates.
Confirmation of syndecan-4 interaction partners in HEK293
Novel syndecan-4 protein partners were validated for syndecan-4 binding in HEK293 cells. MPP7, PARVB, CDK9, EIF2S1, PFKM, CCT5, RASIP1, EIF4B, and CAVIN1 were cloned with either a 3×FLAG tag at the N terminus or a single FLAG tag at the C terminus and co-expressed with HA–syndecan-4 in HEK293 cells. The HEK293 lysates were further subjected to immunoprecipitation using anti-FLAG, thereby using the putative partner as bait and reversing the setup used for the MS analysis. Immunoblotting with anti-HA or anti-syndecan-4 revealed that syndecan-4 co-precipitated strongly with MPP7 (Fig. 3A), PARVB (Fig. 3B), CDK9 (Fig. 3C), EIF2S1 (Fig. 3D), PFKM (Fig. 3E), CCT5 (Fig. 3F), RASIP1 (Fig. 3G), EIF4B (Fig. 3H), and CAVIN1 (Fig. 3I) (n = 3). We detected binding between CAVIN1 and the 25-kDa syndecan-4 when probing with anti-syndecan-4 and not anti-HA, indicating that this band could represent the endogenous syndecan-4 core protein. Only nonspecific binding was observed in the negative controls 3×FLAG–ADD3 (γ-adducin) and 3×FLAG–STK39 (STE20/SPS1-related proline–alanine-rich protein kinase) (Fig. 3, J and K, respectively) (n = 3). Thus, we conclude that the syndecan-4 binding in Fig. 3, A–I, was not due to simple overexpression. Previous studies have shown unsuccessful translocation of MLP–FLAG, indicating that the FLAG tag disturbs MLP function (14). The syndecan-4–MLP interaction was therefore confirmed directly in rat LV lysate (Fig. 6A).
Figure 3.

Confirmation of syndecan-4 binding of selected putative partners in HEK293. HEK293 cells were co-transfected with HA-tagged syndecan-4 and 3×FLAG–MPP7 (A); 3×FLAG–PARVB (B); 3×FLAG–CDK9 (C); 3×FLAG–EIF2S1 (D); PFKM–FLAG (E); CCT5–FLAG (F); RASIP1–FLAG (G); EIF4B–FLAG (H); PTRF–FLAG (I); 3×FLAG–ADD3 (negative control) (J); or 3×FLAG–STK39 (negative control) (K). HEK293 lysates were subjected to immunoprecipitation using anti-FLAG. Immunoprecipitates (two upper most panels) and lysates (two lower most panels) were analyzed by immunoblotting with anti-HA or anti-FLAG. Expression of RASIP1 was too low to be detected without IP. The 25-kDa core syndecan-4 protein (arrow) interacted with PTRF-FLAG in I.
Figure 6.

Syndecan-4 binds directly to MLP and mediates its nuclear translocation. LV lysates from WT rat (A) and ABHF or SHAM rats (six individual rat hearts) (B) were subjected to immunoprecipitation using anti-syndecan-4 (KY/8.2, which has an extracellular epitope). Levels of immunoprecipitated MLP and syndecan-4 were analyzed by immunoblotting with anti-MLP and anti-syndecan-4cyt (which has an intracellular epitope). Nonrelevant rat IgG was used as a negative control. C, total MLP protein level in SHAM and ABHF lysates was analyzed through semiquantified densitometry analysis of immunoblots shown in Fig. S5A. D, syndecan-4 expression in SHAM and ABHF was analyzed by qPCR. E, recombinant MLP bound directly to syndecan-4 in an ELISA-based assay. A scrambled peptide was used as a negative control. Immunoblot analysis of MLP levels in nuclear-enriched fractions (F) and RIPA extracts of LV are from WT and syndecan-4−/− mice (G). Immunoblot analysis of MLP levels in H9c2 cells transduced with a syndecan-4 adenovirus or null virus (H), and H9c2 cells treated with a disruptor peptide (TAT–SDC4cyt) or a scrambled control peptide (TAT-scram) in presence of ISO (I) are shown. The ∼10–15-kDa syndecan-4 band represents the intracellular fragment after shedding of the extracellular domain and is visible when overexpressing syndecan-4 (52). Histone H3 was used as a loading marker for the nuclear-enriched fractions in F, H, and I. B and F–I, scatter plots show MLP levels semiquantified by densitometry analysis. J, protein content in cytoplasmic fractions of H9c2 cells treated with TAT–SDC4cyt and TAT-scram control peptide (n = 8). K, schematic illustration of the disruptor peptide experiment. Differences were tested using unpaired t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001, ns indicates not significant) (n = 3–8). Error bars, S.D.
Cardiac syndecan-4 interactome is enriched in cell communication/signal transduction, cardiovascular disease, stress-response proteins, and PDZ domains
To get an overview of the cardiac syndecan-4 interactome, the 21 novel partners together with the 29 literature partners (Venn diagram in Fig. 2) were clustered according to the biological process using the GO annotation data from the Human Protein Reference Database (HPRD) (Fig. 4). Most of the syndecan-4 partners were involved in cell communication/signal transduction, protein metabolism, and cell growth/maintenance. Within “cell communication/signal transduction” (right in Fig. 4), “enzymatic activity,” which includes serine/threonine/tyrosine kinases and phosphatases, formed the largest cluster, whereas “receptor activity” and “GTPase activity” were the next biggest clusters. An in silico interaction analysis using STRING 10.5 (http://string-db.org5 (15)) showed that the cardiac syndecan-4 interactome contained more interactions than expected from a random set of proteins, indicating that these proteins indeed form complexes (nodes, 50; edges, 166 (expected, 69), average node degree, 6.64; average local clustering coefficient; 0.621, PPI enrichment p value, <1.0e-16) (Fig. S2). According to STRING, 42% of the proteins in the cardiac syndecan-4 interactome were coupled to stress response (Fig. S2, labeled in yellow).
Figure 4.

Cardiac syndecan-4 interactome. Proteins in the syndecan-4 interactome are clustered according to biological processes using the HPRD (GO annotations). Proteins in “Cell communication/Signal transduction” are subdivided into molecular functions. The 21 novel syndecan-4 partners identified by the two AP-MS approaches are in pink. *, interaction changed in ABHF (p < 0.05, n = 3); **, interaction only detected in ABHF (p < 0.05, n = 3).
The cardiac syndecan-4 interactome was also analyzed by the DAVID functional annotation tool (15). “Metabolic” (count 25, 50%, 5.5 × 10−2) and “cardiovascular” (count 22, 44%, 4.3 × 10−2) were among the most enriched disease classes (Table 2). The cardiovascular disease class included the novel partners AP3D1, EPB41, GPX1, MLP, MPP7, PARVB, and RASIP1. Moreover, to get an insight into which signaling pathways the syndecan-4 interactome is involved, we used the Kyoto Encyclopedia of Genes and Genome (KEGG) pathways. The most enriched pathways were “proteoglycans in cancer” (30%, p value 9.7 × 10−13) and “focal adhesion” (26%, p value 4.9 × 10−10) (Table 3). The syndecan-4 interactome was also enriched in pathways involved in different cardiomyopathies like arrhythmogenic right ventricular cardiomyopathy (10%), hypertrophic cardiomyopathy (6%), and dilated cardiomyopathy (6%). To detect enriched protein domains in the syndecan-4 interactome, we used PFAM (16). PDZ was the most abundant PFAM domain and was found in the novel partners MPP7, SLC9A3R2, TJP2, and the literature partner SDCBP (Table 4).
Table 2.
Disease class enrichment for the syndecan-4 protein partners (DAVID)
| Disease class | Gene name | Count | p value |
|---|---|---|---|
| Metabolic | AP3D1, APP, CALM1, CAMK2A, COL1A1, MLP/CSRP3, CTNNB1, DNM2, FYN, GPX1, ITGA2, ITGB1, MMP2, MMP9, MPP7, MTOR, PDCD6IP, PFKM, PLG, PPP3CA, PRKCA, PRKCD, RHOA, SRC, TJP2 | 25 | 5.5 × 10−2 |
| Cardiovascular | ACTN2, AP3D1, APP, CALM1, COL1A1, MLP/CSRP3, DNM2, EPB41, FYN, GPX1, ITGA2, ITGB1, MMP2, MMP9, MPP7, NF1, PARVB, PLG, PDCD6IP, RASIP1, RHOA, RAC1 | 22 | 4.3 × 10−2 |
Table 3.
Enriched KEGG pathways in the syndecan-4 cardiac interactome
| Term | Gene name | Count | p value |
|---|---|---|---|
| Proteoglycans in cancer | CAMK2A, CTNNB1, CTTN, EIF4B, ITGA2, ITGA5, ITGB1, MMP2, MMP9, MTOR, PRKCA, PXN, RAC1, RHOA, SRC | 15 | 9.7 × 10−13 |
| Focal adhesion | ACTN2, COL1A1, CTNNB1, FYN, ITGA2, ITGA5, ITGB1, PARVB, PRKCA, PXN, RAC1, RHOA, SRC | 13 | 4.9 × 10−10 |
| Arrhythmogenic right ventricular cardiomyopathy | ACTN2, CTNNB1, ITGA2, ITGA5, ITGB1 | 5 | 5.8 × 10−4 |
| Hypertrophic cardiomyopathy | ITGA2, ITGA5, ITGB1 | 3 | 6.8 × 10−2 |
| Dilated cardiomyopathy | ITGA2, ITGA5, ITGB1 | 3 | 7.7 × 10−2 |
Table 4.
Enriched PFAM domains in the syndecan-4 cardiac interactome
| Term | Gene name | Count | p value |
|---|---|---|---|
| PDZ domain (also known as DHR or GLGF) | SLC9A3R2, MPP7, SDCBP, TJP2 | 4 | 5.4 × 10−3 |
| Src homology 3 domain | CTTN, FYN, SRC | 3 | 3.2 × 10−2 |
| Fibronectin type II domain | MMP2, MMP9 | 2 | 3.7 × 10−2 |
| Integrin α | ITGA2, ITGA5 | 2 | 4.7 × 10−2 |
Changes in the syndecan-4 interactome during development of heart failure
To identify changes within the syndecan-4 interactome during pressure overload, large scale IP–syndecan-4 experiments were performed in LV lysate isolated from ABHF and SHAM rats (Fig. 1A, panel iii). In total, 19 syndecan-4 interactions, involving novel and literature partners, were found to be either up- or down-regulated in ABHF compared with SHAM (Fig. S4 and Tables S1 and S3, right columns, n = 3, p < 0.05). These 19 proteins clustered into 11 different syndecan-4 networks (Fig. 5, novel partners in pink) according to STRING 10.5 and literature. The largest cluster contained the novel partners MLP and UACA and the literature partners PRKCD, PRKCA, ARF6, RHOA, RHOG, and ACTN2. The second largest cluster contained collagen α-1 (COL1A1) and other extracellular literature partners and is consistent with a role for syndecan-4 in matrix remodeling and fibrosis in heart (recently reviewed in Ref. 56).
Figure 5.

Changes in the cardiac syndecan-4 interactome during pressure overload. 11 syndecan-4 networks were changed during pressure overload. Arrows denote whether the syndecan-4 interaction was increased (↑) or decreased (↓). Novel partners identified are in pink. *, interaction significantly changed in ABHF (p < 0.05, n = 3); **, interaction only detected in ABHF (p < 0.05, n = 3). The STRING database 10.5 (https://string-db.org/)5 was used to generate small string maps.
Syndecan-4 mediates nuclear translocation of MLP
As MLP is a well-known mechanotransducer, we wanted to analyze its interaction with syndecan-4 in more detail. Consistent with the MS dataset (Figs. S3K and S4E), we showed that MLP co-precipitated stronger with anti-syndecan-4 (KY/8.2) than nonrelevant rabbit antibodies (rIgG) (Fig. 6A) and that the interaction was increased in ABHF compared with SHAM (Fig. 6B). Consistently, the total MLP protein level was increased in ABHF compared with SHAM (Fig. 6C), whereas the syndecan-4 expression was unchanged (Fig. 6D). An ELISA-based assay using biotin–SDC4cyt against recombinant MLP identified the syndecan-4–MLP interaction to be direct (Fig. 6E). To assess whether syndecan-4 influenced nuclear translocation of MLP, MLP was analyzed in nuclear-enriched fractions isolated from wildtype (WT) and syndecan-4 knockout (SDC4−/−) mouse hearts. The nuclear MLP level was reduced when syndecan-4 was absent (Fig. 6F, right panel), even though the total amount of MLP was equal in RIPA extracts of WT and SDC4−/− mouse hearts (Fig. 6G). On the other side, adenovirus-mediated overexpression of full-length syndecan-4 in an H9c2 rat cardiomyoblast cell line resulted in an increased nuclear MLP level compared with null virus (control) (Fig. 6H). The amount of syndecan-4 overexpressed and the distribution in cytoplasmic, nuclear, and membrane fractions is given in Fig. S5B. Because we found the syndecan-4–MLP interaction to be direct, breaking this bond should disrupt the biological function of this interaction. We generated a cell-permeable syndecan-4–MLP disruptor peptide by adding a TAT tag to the N terminus of the cytoplasmic tail of syndecan-4 (TAT–SDC4cyt) (upper panel in Fig. 6I), and we tested its effect on isoprenaline (ISO)-stimulated H9c2 cells. H9c2 cells show a similar hypertrophic response to ISO in primary neonatal rat cardiomyocytes in vitro (17). The time point of increased MLP nuclear entry in response to ISO was determined to be 6–12 h post-stimulation (Fig. S5C). In the presence of the disruptor peptide (TAT–SDC4cyt), the nuclear fraction of MLP was reduced compared with the TAT-tagged scrambled control peptide (Fig. 6I, lower panel). As a physiological readout, we also measured the protein content. Consistent with a role of MLP in gene transcription and hypertrophic growth (12), we observed less protein in the cytoplasmic fractions of cells treated with TAT–SDC4cyt compared with cells treated with TAT-scram (Fig. 6J). The principle of this disruptor peptide experiment is illustrated in Fig. 6K. Altogether, our data suggest syndecan-4 to be a mediator for nuclear translocation of MLP.
Discussion
Cardiac syndecan-4 interactome
An important step in understanding how syndecan-4 mediates its cardiac function is to determine its molecular interaction partners. In this study, we have identified the cardiac syndecan-4 protein interaction network of 21 novel partners and 29 described previously in the literature. This is the first experimental syndecan-4 interactome, and it is enriched with proteins involved in cell communication/signal transduction, stress response, metabolic, and cardiovascular disease.
In our analysis, proteins had to be detected by two different AP-MS approaches to be regarded as a novel syndecan-4 partner. This strategy might have favored more stable syndecan-4 interactions, and transient and weak interactions might have been lost. As input material, we used LVs lysed in a buffer containing 1% Triton and thus included all cellular compartments, except the nucleus, from all cardiac cell types. The complexity of the input material might have introduced false protein–protein interactions not relevant in vivo. However, false-positives seemed not to be a problem because all protein partners we tested also revealed syndecan-4 binding in HEK293, whereas nonrelevant proteins did not. It has also been noted by others that the cytoplasmic domain of syndecan-4 has a high number of interaction partners compared with size (18). The large number of partners can be explained by different types of interactions (direct/indirect) in different cardiac cell types and subcellular locations. Syndecan-4 locates to both costamere and z-disc (5), but it is also reported to localize to Golgi structures, intracellular vesicle membranes, and perinucleus during muscle differentiation (19).
Changes in the syndecan-4 interactome during development of heart failure
Interestingly, 19 proteins in the interactome (38%) showed an altered interaction with syndecan-4 in ABHF versus SHAM and clustered into 11 smaller syndecan-4 networks (Fig. 5). Altered interactions could be due to several factors, e.g. changes in availability, post-translational modifications, syndecan-4 oligomerization level, and/or competition between the protein partners for syndecan-4 binding.
One network contained SDCBP, which binds directly through its PDZ2 domain to the C-terminal FYA motif in syndecan-4 (20). SDCBP is involved in recycling of the syndecan to the plasma membrane (21) and exosome formation through an SDCBP-dependent recruitment of PDCD6IP (programmed cell death 6-interacting protein), another partner in the syndecan-4 interactome (Fig. 4), and the ESCRT-III complex (endosomal-sorting complex required for transport III) (22). Our findings indicate that these syndecan-4–dependent trafficking processes might change during pressure overload. SDCBP also negatively regulates syndecan-4 function in focal adhesion formation by reducing the syndecan-4–PKCα interaction and thus PKCα activation (20).
A second syndecan-4 network consisted of the two novel partners CAVIN1 and TRIM72, where the latter interaction was only identified in ABHF (Fig. 5). CAVIN1 plays an important role in caveolae formation and organization (23) and recruits TRIM72 in an emergency membrane repair response to protect against stress-induced loss of cardiomyocytes (24, 25). Our findings of a novel syndecan-4–CAVIN1–TRIM72 axis might suggest a compensatory role for syndecan-4 in cell membrane repair during pressure overload. Mutations in CAVIN1 are reported to cause skeletal muscle hypertrophy, lipodystrophy, and cardiac dysfunctions, including arrhythmias (25, 26).
A third syndecan-4 network contained the novel partner AP3D1 and the literature partner CLTC (27), a major protein of the polyhedral coat of coated pits. AP3D1, which showed an increased interaction with syndecan-4 (Fig. 5), is a component in the ubiquitous heterotetrameric AP-3 (adaptor protein complex 3) involved in intracellular trafficking and generation of vesicles with different cargo or protein compositions. The literature partner CLTC is suggested to play a cargo-selective role in this process (28). We speculate whether syndecan-4 is subjected to increased trafficking through an AP-3–dependent pathway during pressure overload and perhaps also plays a cargo-selective role. AP3D1 is reported to associate with myocardial infarction and atherosclerosis in a Japanese population (29).
A fourth syndecan-4 network contained the novel partner RRAGC and the literature partner serine/threonine-protein kinase mTOR (mammalian target of rapamycin) (Fig. 5). mTOR activity is regulated by assembly into two functionally distinct complexes, mTORC1 and mTORC2. mTORC1 is a major driver of cell growth and protein synthesis, triggered by growth factors, stress signals, energy, and nutrients (30). In response to amino acids, RRGACGDP targets mTORC1 to lysosomes where it is activated by Rheb (31, 32). Our findings of a novel syndecan-4–RRGAC interaction only in ABHF hearts (Fig. 5) could suggest that syndecan-4 is more involved in mTORC1 signaling in heart failure development. In contrast, activity of the mTORC2–Akt–eNOS axis depends on a syndecan-4–PKCα-dependent recruitment of mTORC2 to rafts in endothelial cells (33).
A fifth syndecan-4 network contained the two novel partners TJP2 and EPB41 (Fig. 5), where the latter partner links cell-surface membrane proteins to the actin cytoskeleton. EPB41 and TJP2 are reported to form a complex at cell–cell junctions, which are structures known to be important in maintaining mechanical integrity of cardiomyocytes and resulting in arrhythmias when absent (34, 35). Our findings of a decreased EPB41–syndecan-4 interaction (Fig. 5) indicate a reduction in the linkage between syndecan-4 and the actin cytoskeleton during pressure overload. Whether this interaction is lost at cell–cell junctions will have to be investigated in future studies. However, syndecan-4 showed an increased binding to the novel partner tight junction adaptor protein MMP7 (36), supporting a role for syndecan-4 at cell–cell junctions during pressure overload.
A sixth syndecan-4 network contained the two novel partners EIF4B and EIF2S1 (Fig. 5). EIF4B is involved in the initiation of translation and recruitment of the 40S ribosomal subunit to the mRNA, a process that is regulated by phosphorylation of EIF4B according to the cell's need, through the mTOR pathway, for example (37). EIF2S1 is also involved in translation initiation and can be regulated through phosphorylation. In response to cell stress, phosphorylation of EIF2S1 has been shown to inhibit protein synthesis except for selected stress-response mRNAs (38). More EIF2S1 and less EIF4B precipitated with syndecan-4 in ABHF, which could indicate competition between the two proteins and perhaps a role for syndecan-4 in directing translation in heart failure development.
The largest syndecan-4 network contained partners involved in actin remodeling, signal- and mechanotransduction (Fig. 5). PKCδ (PRKCD), PKCα (PRKCA), and UACA interacted only with syndecan-4 during pressure overload. PKCα is a well-known syndecan-4 partner and is involved in adhesion formation and contractility (9, 39). Upon dephosphorylation of pSer-179, syndecan-4 oligomerizes and activates PKCα and can thereby regulate the time and place for PKCα activity (40). PKC activation is recently found to be up-regulated at the costameres in heart disease (41), and we speculate whether this is due to an increased interaction with syndecan-4. In line with this, we have previously found reduced pSer-179–syndecan-4 levels in aortic stenosis patients and in mice after aortic banding (7). In contrast, the literature partner PKCδ phosphorylates syndecan-4 at serine 179, thus reducing PKCα activity and impairing endothelial cell function (42). The literature partners RHOA, RHOG, and ARF6 are involved in actin cytoskeletal remodeling (4). Syndecan-4 controls both suppression and activation of these GTPases, for example, it activates RHOG by first activating PKCα thereby triggering RHOG/caveolin-dependent endocytosis of integrins (4, 18). ARF6, which is also implicated in syndecan and integrin recycling (18, 21), interacts with the novel syndecan-4 partner UACA, another cytoskeletal remodeler (43). The network also revealed a novel syndecan-4–MLP–ACTN2 axis. The novel partner MLP is a well-known stress- and mechanotransducer in muscle cells and associates directly with human myopathies (11). The literature partner ACTN2 is an F-actin cross-linking scaffolding protein at z-disc and cell–cell junctions, where it couples receptors, like syndecan-4, to the actin cytoskeleton (35). A human hypertrophic cardiomyopathy mutation in MLP has been shown to destabilize the interaction between MLP and ACTN2 and a third component N-rap leading to myocyte disarray (44). The increased interaction between ACTN2 and MLP with syndecan-4 during pressure overload supports a role for syndecan-4 in mechanosignaling complexes.
Altogether, our data indicate that syndecan-4 associates with important cellular structures and functions, including focal adhesions, tight junctions, endocytosis, trafficking, mechanotransduction, and cytoskeleton remodeling during development of heart failure induced by pressure overload. These wide-ranging functions are probably explained by its many intracellular binding partners.
Syndecan-4 mediates nuclear translocation of MLP
The large multiprotein structures of z-discs have been suggested as major sites for sensing and transducing mechanical stimuli (1), and of particular interest are LIM domain proteins, which are able to shuttle to the nucleus, thus connecting mechanical stimuli with gene expression (45, 46). MLP, which we identified as a novel and direct syndecan-4 partner, contains two independent LIM domains that facilitate formation of multiprotein complexes (47). MLP interacts with structural and signaling proteins at the z-discs and translocates to the nucleus to initiate hypertrophic gene expression in response to biomechanical stress (11, 12, 14). Nuclear translocation of MLP is crucial for its mechanotransducing probabilities, and its nuclear accumulation associates with pathology (14). Interestingly, we found the syndecan-4–MLP interaction to be increased during pressure overload, suggesting a function for this complex in cardiac pathology. Our findings of a direct syndecan-4–MLP binding led us to develop a specific cell-permeable disruptor peptide to assess the biological function of this interaction. The syndecan-4–MLP disruptor peptide inhibited nuclear translocation of MLP and attenuated the increase in protein content following ISO stimulation of H9c2 cells, suggesting the nuclear translocation of MLP and the hypertrophic gene expression to be syndecan-4–dependent. The syndecan-4–MLP interaction also appears to be relevant in vivo, because less nuclear MLP was found in the syndecan-4−/− mouse LVs, an observation consistent with loss of concentric myocardial hypertrophy and rather LV dilatation and dysfunction during pressure overload (7, 12, 48).
The increased syndecan-4–MLP interaction can be due to changes in availability, structures, or post-translational modifications during pressure overload. We have previously shown reduced pSer-179–syndecan-4 levels in human aortic stenosis patients and in mice after pressure overload, enabling calcineurin binding and activation of the hypertrophic NFAT pathway (7). Perhaps the pSer-179 also works as a molecular switch for the MLP interaction and its nuclear translocation. Syndecan-4 has previously been found in the nuclear membrane and perinuclear compartment (19), and syndecan-1 has been suggested to translocate into the nucleus (49). Future studies are needed to investigate how syndecan-4 directs the translocation of MLP and whether syndecan-4 also goes into the nucleus. It will also be interesting to investigate whether the syndecan-4 binding overlaps with any of the MLP mutations associated with human hypertrophic cardiomyopathy (10, 50) or dilated cardiomyopathy (11). Collectively, these data indicate that syndecan-4 act upstream of MLP and mediates its nuclear translocation upon stress.
Experimental procedures
Animal handling
All animal handling was preapproved by the Norwegian National Animal Research Committee (FOTS ID 3820 for rats, and FOTS ID 6989 for SDC4−/− mice) and conformed to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication 85-23, revised 2011). Animals were stored with 12:12-h light/dark cycles and were given access to food and water ad libitum. For neonatal cell isolation, the permit of approval number IV1-17U was obtained.
Aorta banding in rats
Approximately 170-g male Wistar-Hannover rats (Taconic, Denmark) were anesthetized and ventilated with a mixture of 68% N2O, 29% O2 and 2–3% isoflurane through endotracheal intubation. The chest was opened, and aorta banding was induced by 3.0 silk suture tightened around the ascending aorta proximal to the brachiocephalic trunk. SHAM animals were used as controls and went through the same surgical procedure except tightening of the suture around the ascending aorta and buprenorphine was used as analgesic. Echocardiography was performed with a Vevo 2100 (VisualSonics, Canada) 6 weeks after surgery, before myocardial tissue samples were harvested in deep surgical anesthesia. The left and right ventricles were rapidly dissected and subsequently put into liquid nitrogen and stored at −70 °C for later molecular analysis. The criteria for congestive heart failure (ABHF) were increased lung weight (>2.0 g), increased posterior wall diameter (>1.9 mm), and left atrial diameter (>5.0 mm). Characteristics of the animals used in the large-scale AP-MS experiments are given in Table S4. For the qPCR and Western blot analysis, 23 additional animals were included using the same criteria as in Table S4.
Left ventricular lysates
Frozen rat or mouse LVs were pulverized in liquid nitrogen before homogenization with a Polytron 1200 homogenizer in three series of 1 min in lysis buffer (150 mm NaCl, 20 mm HEPES, pH 7.5, 1 mm EDTA, 0.5% Triton X-100 (Sigma)) supplemented with cOmplete protease inhibitor mixture (05050489001, Roche Applied Science, Switzerland) or in RIPA buffer (catalog no. R0278, Sigma) on ice. Following centrifugation at 100,000 × g for 60 min at 4 °C, supernatants were stored at −80 °C until analysis.
Antibodies
Anti-syndecan-4cyt (epitope: C-DLGKKPIYKKAPTN) and anti-pS179–syndecan-4 (epitope: EGpSYDLGKKPIYKK-C) were custom-made by Genscript Corp. Anti-syndecan-4 (KY/8.2, catalog no. 550350) was obtained from PharmingenTM. Anti-biotin–HRP (A-0185) and anti-FLAG (F1804) were obtained from Sigma. Anti-MLP was obtained from Santa Cruz Biotechnology (sc-166930) or Thermo Fisher Scientific (catalog no. PA5-19062). Anti-HA (catalog no. 3724) and anti-histone H3 (catalog no. 4499) were from Cell Signaling Technology, Inc. (The Netherlands). Nonrelevant rat IgG (catalog no. sc-2026) and anti-glyceraldehyde-3-phosphate dehydrogenase (catalog no. sc-20357) were from Santa Cruz Biotechnology. Anti-mouse IgG HRP (NA931V) and anti-rabbit IgG HRP (NA934V) affinity-purified polyclonal antibody (both from GE Healthcare) were used as secondary antibodies.
Peptides and recombinant proteins
The following peptides were synthesized to > 80% purity by Genscript: biotin–syndecan-4cyt, RMKKKDEGSYDLGKKPIYKKAPTNEFYA; biotin–pS179–syndecan-4cyt, RMKKKDEGpSYDLGKKPIYKKAPTNEFYA; biotin–scram, GTKYPKMDRGKLFKYKAKPEDNESAYIK; TAT–syndecan-4cyt, RKKRRQRRRRMKKKDEDSYDLGKKPIYKKAPTNEFYA; and TAT–scram, RKKRRQRRRGTKYPKMDRGKLFKYKAKPEDNESAYIK.
Recombinant 12×His-Ezrin (1–333) (rat, UniProt P31977) was made by Genscript. Recombinant MLP was obtained from Abnova (Taiwan) (H00008048-P01).
ELISA
The ELISA plate was first coated with 0.46 μg/well recombinant MLP or 10 μg/ml recombinant ezrin in 1× PBS and rotated ON at 4 °C. The next day, the wells were washed once in 1× PBS-T (0.1%) before blocking in PBS with 1% gelatin (G-1890, Sigma) for 1 h while rotating at room temperature. After blocking, the coated wells were incubated with either biotin–syndecan-4cyt or biotin–scram (control peptide) in 1× PBS-T (0.1%) for 2 h at 37 °C while rotating. Thereafter, the wells were washed five times in 1× PBS-T (0.1%) before incubating with anti-biotin–HRP (1:5000) for 30 min while rotating at room temperature. Then wells were washed five times in 1× PBS-T (0.1%) before adding Ultra TMB/ELISA solution (34028, Thermo Fisher Scientific) and incubated for 15–30 min while rotating at room temperature. Reaction was stopped by adding 2 n HCl, and the signal was measured by a plate reader at 450 nm (Hidex Sense multimodal microplate reader, Finland).
Epitope mapping
Rat syndecan-1–4 was synthesized as 20-mer with three amino acid offsets onto cellulose membranes by a MultiPep automated peptide synthesizer (INTAVIS Bioanalytical instruments AG, Germany) as described previously (51). After blocking, the membranes were incubated with anti-syndecan-4 ON at 4 °C. After washing three times in TBS-T, membranes were incubated with secondary antibodies for 1 h at room temperature, before washing another three times in TBS-T and developed as an immunoblot.
Primary rat neonatal cardiac myocytes and fibroblasts
Primary cardiomyocytes and fibroblasts were isolated from 1- to 3-day-old Wistar rats (Møllergaard Breeding and Research Center, Denmark). Hearts were excised, trimmed for atrial tissue, minced, and digested with a collagenase solution (Worthington Biochemical). The cell suspension was transferred to uncoated culture flasks with serum-containing medium in two rounds for 20 min at 37 °C for fibroblasts to attach. After attachment, fibroblasts were kept in fibroblast medium (Dulbecco's modified Eagle's medium (DMEM) (41965-039, Gibco, Life Technologies, Inc.)) supplemented with penicillin/streptomycin (P0781, Sigma) and fetal bovine serum (FBS) (FB-1001/500, Biosera, France). Myocytes were isolated as nonadherent cells and seeded onto 6-well culture plates precoated with gelatin/fibronectin (Sigma) and kept in plating medium consisting of DMEM (D1152, Sigma), M-199 (M2520, Sigma), penicillin/streptomycin, horse serum (14-403E, BioWhittaker), and fetal bovine serum (14-701F, BioWhittaker). The cells were cultured for up to 1 week in a 37 °C, 5% CO2-humidified incubator before lysis in RH buffer (1× PBS, 1% Triton (Sigma); 0.1% Tween (161-0781, Bio-Rad); supplemented with cOmplete protease (Roche Applied Science); and phosSTOP phosphatase inhibitor mixture (04906837001, Roche Applied Science)).
Cell lines, constructs, and transfection
HEK293 (ATCC® CRL-1573TM) and H9c2 (ATCC® CRL-1446TM) cells were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin (P0781, Sigma) at 37 °C in 5% CO2 in a humidified incubator. After 24 h in penicillin/streptomycin-free medium, HEK293 cells were transfected via the CaCl2 method. For each transfection, 8 μg of DNA in a CaCl2 solution (248 mm) was mixed with 500 μl of 2× HEPES (50 mm HEPES, 280 mm NaCl, 1.5 mm Na2HPO4, pH 7.0) in a dropwise manner and incubated at room temperature for 20–30 min before being transferred onto the cells. 24 h after transfection the cells were harvested in IP buffer (150 mm NaCl, 20 mm HEPES, pH 7.5, 1 mm EDTA, 1% Triton X-100 supplemented with complete protease inhibitor mixture (Roche Applied Science)). Cloning was done by Genscript, and all genes were tagged with either HA or FLAG tags in the pCEP4 or pcDNA3.1 vectors (Genscript). Constructs used were as follows: HA–SDC4 mouse (O35988); 3×FLAG–MPP7 rat (NM_001100575); 3×FLAG–PARVB rat (NM_001134780); 3×FLAG–CDK9 rat (NM_001007743); 3×FLAG–EIF2S1 rat (NM_019356); PFKM–FLAG human (NM_000289.5); CCT5–FLAG human (NM_012073.4); RASIP1–FLAG human (NM_017805.2); EIF4B–FLAG human (NM_001417.6); PTFR–FLAG human (NM 012232.5); 3×FLAG–ADD3 rat (NM XM_006231599.3) (negative control); and 3×FLAG–STK39 rat (NM_019362) (negative control).
Small-scale immunoprecipitation and immunoblotting
For all IPs, lysates were incubated with 2 μg of antibody and protein A/G–agarose beads (sc-2003, Santa Cruz Biotechnology) ON at 4 °C and unless otherwise stated washed three times in IP buffer (150 mm NaCl, 20 mm HEPES, pH 7.5, 1 mm EDTA, 1% Triton X-100 with cOmplete protease inhibitor mixture (Roche Applied Science)) before elution by boiling in 2× SDS loading buffer. In Fig. 6A, 200 μl (6.1 μg/μl) of rat LV lysate was used per IP, and 5 μl of lysate was used as input (2.5% of IP lysate). After precipitation, this IP was washed three times in PBS buffer. In Fig. 6B, 200 μl (2.0 μg/μl) of rat LV lysate was used per IP.
Lysates, immunoprecipitations, and peptides were analyzed on a 4–15 or 15% CriterionTM Tris-HCl precast gel (3450028 and 3450021, Bio-Rad) and blotted onto a polyvinylidene difluoride membrane (catalog no. 1704157, Bio-Rad). The membranes were blocked in 5% nonfat dried milk or 1% casein in TBS-T (TBS with 1% Tween 20 (161-0781, Bio-Rad)) for 1 h at room temperature. Membranes were thereafter incubated with primary antibodies for 1 h at room temperature or ON at 4 °C, followed by three 5-min wash in TBS-T before incubation with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Blots were developed using ECL Prime (RPN 2232, GE Healthcare). When the signal was too high, blots incubated with anti-MLP were washed in TBS-T before repeating development. For reprobing, the membranes were stripped in a RestoreTM Western blotting Stripping Buffer (21059, Thermo Fisher Scientific). Densitometric analysis was achieved with ImageJ (National Institutes of Health). Total protein concentration of the various samples was measured using the Micro BCA protein assay kit (23235, Thermo Fisher Scientific). Equal protein amounts were loaded per lane: 20 μg of nuclear fraction isolated from mouse hearts; 35.5 μg of RIPA extracts isolated from mouse hearts; 25 μg of nuclear fractions isolated from virus-transduced cells, and 11 μg of nuclear fractions isolated from ISO-stimulated cells.
Large-scale pulldown experiments and immunoprecipitations
For the large-scale pulldown experiments, LV lysates were pooled and mixed with 0.01 mm biotinylated peptides before rotation at 4 °C ON. Streptavidin-coated Dynabeads (DynabeadsTM M-270 streptavidin, catalog no. 65305, Life Technologies, Inc.) were washed three times in PBS before adding the peptide containing LV lysate and rotated for 40 min at room temperature. The beads were washed five times in 1 ml of PBS, and protein complexes were eluted in 250 μl of 25 mm biotin for 3 h at 60 °C. Proteins were precipitated in 1 ml of 4× ice-cold acetone and added glycoblue ON at −20 °C. After centrifugation, the pellets were air-dried before analysis by MS.
For the large-scale immunoprecipitation experiments, 10 μg/mg beads of anti-syndecan-4 or anti-rat IgG were coupled to magnetic dynabeads (DynabeadsTM antibody coupling kit, catalog no. 14311D, Thermo Fisher Scientific) according to the manufacturer's protocol. The antibody-coupled beads were incubated with LV lysates from WT, ABHF, or SHAM rat hearts and rotated ON at 4 °C. After washing three times in ice-cold PBS and two times in ice-cold MQ water to remove salts (52), interacting proteins were eluted in 0.1% TFA in 50% acetonitrile for 30 min while rotating at room temperature. The elution step was repeated once with fresh TFA before precipitation and centrifugation, and finally, the pellets were air-dried.
WT and syndecan-4−/− mice
Hearts were harvested from 9-week-old female WT littermates or syndecan-4−/− mice (53), snap-frozen in liquid nitrogen, and stored at −70 °C until fractionation with Compartment Protein Extraction kit (2145, Merck) according to the manufacturer's protocol.
Viral transduction
H9c2 cells were cultured to a confluency of about 80% before being transduced with adenovirus serotype 5 encoding mouse syndecan-4 (catalog no. ADV-271493, Vector Laboratories) or empty vector (null virus) (catalog no. 1300, Vector Laboratories). Virus titer used was 2 × 107 plaque-forming units/ml. After 24 h of incubation, the cells were washed in PBS and freshly added media. 48 h after induction, the cells were fractionated as described above.
Isoprenaline stimulation and treatment with cell-permeable peptides
H9c2 cells were serum-starved for 24 h before stimulation with 10 μm isoprenaline sulfate (ISO) (catalog no. 24-37-82, NAF, Norway) with or without cell-permeable peptides for 12 h (Fig. 6I), 6–12 h (Fig. 6J), and 1–24 h (Fig. S5C) and subsequently fractionated as described above.
RNA isolation and quantitative real-time PCR (qRT-PCR)
RNA was isolated from LV tissue using RNeasy mini (catalog no. 74104, Qiagen Norge, Oslo, Norway). RNA concentration was determined using the Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). For cDNA synthesis, iScript cDNA synthesis kit (catalog no. 170-8891, Bio-Rad), was used according to the manufacturer's protocol. Gene expression levels were determined using pre-designed TaqMan assays detected on a QuantStudio3 (Applied Biosystems by Thermo Fisher Scientific) and analyzed via the QuantStudio Design and Analysis Software. TaqMan assays used were Rn00561900_m1 (Sdc4) and Rn00820748_g1 (Rpl32) (LifeTech by Thermo Fisher Scientific). SDC4 expression was normalized to RPL32.
Mass spectrometry
The proteins were precipitated using 2D Clean Up kit (80-6484-51, GE Healthcare) according to the manufacturer's instructions. The precipitated proteins were dissolved in 40 μl of 0.2% ProteaseMAXTM surfactant, trypsin enhancer (Promega), in 50 mm NH4HCO3 followed by protein reduction, alkylation, and in-solution digestion with trypsin (Promega) ON at 37 °C. After digestion, the samples were centrifuged at 14,000 × g for 10 min, and trypsin was inactivated by adding 100 μl of 1% TFA, and the samples were again centrifuged at 14,000 × g for 10 min. Peptides were desalted and concentrated before MS by the STAGE-TIP method using a C18 resin disk (3M Empore). The peptides were eluted with 80 μl of 80% acetonitrile, 0.1% FA, dried, and solubilized in 7 μl of 0.1% FA for MS analysis.
Each peptide mixture was analyzed on an Easy nLC1000 nano-LC system connected to a quadrupole-Orbitrap (QExactive) mass spectrometer (ThermoElectron, Bremen, Germany) equipped with a nanoelectrospray ion source (EasySpray, ThermoElectron). For LC separation, we used an EasySpray column (C18, 2-μm beads, 100 Å, 75-μm inner diameter) (ThermoElectron) capillary with 25-cm bed length. The flow rate used was 0.3 μl/min, and the solvent gradient was 5% B to 30% B in 120 min and then 90% B wash in 20 min. Solvent A was aqueous 0.1% formic acid, and solvent B was 100% acetonitrile in 0.1% formic acid. Column temperature was kept at 60 °C.
The mass spectrometer was operated in the data-dependent mode to automatically switch between MS and MS/MS acquisition. Survey full-scan MS spectra (from m/z 400 to 1200) were acquired in the Orbitrap with resolution r = 70,000 at m/z 200 (after accumulation to a target of 3,000,000 ions in the quadruple). The method used allowed sequential isolation of the most intense multiply-charged ions, up to 10, depending on signal intensity, for fragmentation on the HCD cell using high-energy collision dissociation at a target value of 100,000 charges or maximum acquisition time of 100 ms. MS/MS scans were collected at 17,500 resolution at the Orbitrap cell. Target ions already selected for MS/MS were dynamically excluded for 30 s. General MS conditions were as follows: electrospray voltage, 2.1 kV; no sheath and auxiliary gas flow, heated capillary temperature of 250 °C, normalized HCD collision energy 25%. Ion selection threshold was set to 5e4 counts. Isolation width of 3.0 Da was used.
The resulting MS raw files were submitted to the MaxQuant software (version 1.5.2.8) for protein identification using the Andromeda search engine. Carbamidomethyl (C) was set as a fixed modification, and protein N-acetylation and methionine oxidation were set as variable modifications. First search peptide tolerance of 20 ppm and main search error of 6 ppm were used. Trypsin without proline restriction enzyme option was selected, with two allowed miscleavages. The minimal unique + razor peptide number was set to 1, and the allowed false discovery rate was 0.01 (1%) for peptide and protein identification. Label-free quantitation (LFQ) was employed with default settings. The UniProt database with “Rattus” entries was used for the database searches. Perseus software was used for the statistical analysis of the label-free quantitation results. Known contaminants as provided by MaxQuant and identified in the samples were excluded from further analysis. Proteins had to be identified with at least two peptides to be considered for further analysis.
Bioinformatics
For enrichment analysis, the Database for Annotation, Visualization and Integrated Discovery (DAVID), version 6.8, was used. Official gene names were imported into the search tool, and Homo sapiens was chosen for both species and background with the following setting: count (2) and EASE score (0.1) (Modified Fisher Exact p value) (54). For categorizing the interactome into Biological Process, the Human Protein Reference Database (HPRD) was used (55).
Statistics
For quantitative comparisons and determination of significance between the MS samples, LFQ intensities were loaded in Perseus (version 1.4.0.20). Those values were log2-transformed, and a t test was done. For the three large-scale biotin–pS179/syndecan-4 pulldown analyses (each run in triplicates), p values of 0.05 or below were accepted as statistically significant. For the three large-scale IP-SDCD4 analyses (each run in triplicates), p values of 0.01 or below were accepted as statistically significant. To get more inclusive data for the six AP-MS analyses, statistics without any correction were performed (Table S1). However, to be regarded as a true syndecan-4 partner, the protein had to be identified in at least five of these AP-MS analyses. The probability of the same random false-positives to occur in all five AP-MS analyses was regarded as low. For the large-scale IP-SDCD4 analysis in ABHF versus SHAM (run in triplicates), p values of 0.05 or below were accepted as statistically significant. ELISA-based and cell experiment data are presented with group means ± S.D. Statistical analysis was done in GraphPad Prism 7.01 using unpaired t test, and p < 0.05 was accepted as statistically significant.
Proteins used are cited in UniProt as follows: AP3D1, O14617; CAVIN1/PTRF, Q6NZI2; CCT5, P48643, CDK9, P50750; EIF2S1, P05198; EIF4B, P23588; EPB41, P11171; FAM175B/ABRAXAS2, Q15018; GPX1, P07203; GRTP1, Q5TC63; MLP/CSRP3, P50461; MPP7, Q5T2T1; MTDH, Q86UE4; PARVB, Q9HBI1; PFKM, P08237; RASIP1, Q5U651; RRAGC, Q9HB90; SDCBP, O00560; SLC9A3R2, Q15599; TJP2, Q9UDY2; TRIM72, Q6ZMU5; UACA, Q9BZF9.
Author contributions
S. B. M. and C. R. C. conceptualization; S. B. M., M. L., J. M. A., A. K., M. M., G. A. d. S., T. A. N., and C. R. C. data curation; S. B. M., M. L., G. A. d. S., and C. R. C. formal analysis; S. B. M. and C. R. C. validation; S. B. M. and C. R. C. investigation; S. B. M. and C. R. C. visualization; S. B. M., M. L., J. M. A., A. R., G. A. d. S., T. A. N., I. S., A.K., M.M., and C. R. C. methodology; S. B. M. and C. R. C. writing-original draft; S. B. M. and C. R. C. project administration; S. B. M., M. L., J. M. A., A. R., G. A. d. S., T. A. N., I. S., G. C., A.K., M.M., and C. R. C. writing-review and editing; G. A. d. S. and T. A. N. software; G. C. and C. R. C. supervision; C. R. C. funding acquisition.
Supplementary Material
Acknowledgments
We thank Solveig Sirnes, Hilde Jarstadmarken, Tone Lian, and Dina Behmen for help with transfections, cell isolation, and general technical assistance. We thank Astrid Tutturen and Maria Stensland for technical assistance regarding MS. We also thank Professor John R. Couchman for helpful advice on antibody selection. The Proteomics Core Facility is supported by the Core Facilities Program of the South-Eastern Norway Regional Health Authority.
This work was supported by Nasjonal Foreningen for Folkehelsen, Stiftelsen Kristian Gerhard Jebsen, The Research Council of Norway, Olav Raagholt og Gerd Meidel Raagholts stiftelse for forskning, Anders Jahre's Fund for the Promotion of Science and the South-Eastern Regional Health Authority, and Fondsstiftelsen from Oslo University Hospital, Norway. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5 and Tables S1–S4.
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier accession no. PXD011249.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- ECM
- extracellular matrix
- ABHF
- aortic-banded heart failure
- AP
- affinity purification
- AP-MS
- affinity purification coupled to mass spectrometry
- CFB
- cardiac fibroblast
- CM
- cardiomyocyte
- HPRD
- human protein reference database
- ISO
- isoprenaline
- KY/8.2
- commercially available anti-syndecan-4
- LV
- left ventricle
- ON
- overnight
- rIgG
- nonrelevant rabbit antibodies
- SHAM
- control operated
- SDC4cyt
- cytoplasmic domain of syndecan-4
- pS179-SDC4cyt
- cytoplasmic domain of syndecan-4 with phosphorylation at serine 179
- TAT–SDC4cyt
- cell-permeable disruptor peptide
- MLP
- muscle LIM protein
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- HRP
- horseradish peroxidase
- qPCR
- quantitative PCR
- mTOR
- mammalian target of rapamycin
- IP
- immunoprecipitation
- FA
- formic acid
- NFAT
- nuclear factor of activated T-cell
- LFQ
- label-free quantitation.
References
- 1. Frank D., and Frey N. (2011) Cardiac Z-disc signaling network. J. Biol. Chem. 286, 9897–9904 10.1074/jbc.R110.174268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haque Z. K., and Wang D. Z. (2017) How cardiomyocytes sense pathophysiological stresses for cardiac remodeling. Cell. Mol. Life Sci. 74, 983–1000 10.1007/s00018-016-2373-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Putten S., Shafieyan Y., and Hinz B. (2016) Mechanical control of cardiac myofibroblasts. J. Mol. Cell. Cardiol. 93, 133–142 10.1016/j.yjmcc.2015.11.025 [DOI] [PubMed] [Google Scholar]
- 4. Elfenbein A., and Simons M. (2013) Syndecan-4 signaling at a glance. J. Cell Sci. 126, 3799–3804 10.1242/jcs.124636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. VanWinkle W. B., Snuggs M. B., De Hostos E. L., Buja L. M., Woods A., and Couchman J. R. (2002) Localization of the transmembrane proteoglycan syndecan-4 and its regulatory kinases in costameres of rat cardiomyocytes: a deconvolution microscopic study. Anat. Rec. 268, 38–46 10.1002/ar.10130 [DOI] [PubMed] [Google Scholar]
- 6. Woods A., and Couchman J. R. (1994) Syndecan 4 heparan sulfate proteoglycan is a selectively enriched and widespread focal adhesion component. Mol. Biol. Cell 5, 183–192 10.1091/mbc.5.2.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Finsen A. V., Lunde I. G., Sjaastad I., Østli E. K., Lyngra M., Jarstadmarken H. O., Hasic A., Nygård S., Wilcox-Adelman S. A., Goetinck P. F., Lyberg T., Skrbic B., Florholmen G., Tønnessen T., Louch W. E., et al. (2011) Syndecan-4 is essential for development of concentric myocardial hypertrophy via stretch-induced activation of the calcineurin–NFAT pathway. PLoS One 6, e28302 10.1371/journal.pone.0028302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herum K. M., Lunde I. G., Skrbic B., Louch W. E., Hasic A., Boye S., Unger A., Brorson S. H., Sjaastad I., Tønnessen T., Linke W. A., Gomez M. F., and Christensen G. (2015) Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc. Res. 106, 217–226 10.1093/cvr/cvv002 [DOI] [PubMed] [Google Scholar]
- 9. Braz J. C., Gregory K., Pathak A., Zhao W., Sahin B., Klevitsky R., Kimball T. F., Lorenz J. N., Nairn A. C., Liggett S. B., Bodi I., Wang S., Schwartz A., Lakatta E. G., DePaoli-Roach A. A., et al. (2004) PKC-α regulates cardiac contractility and propensity toward heart failure. Nat. Med. 10, 248–254 10.1038/nm1000 [DOI] [PubMed] [Google Scholar]
- 10. Geier C., Perrot A., Ozcelik C., Binner P., Counsell D., Hoffmann K., Pilz B., Martiniak Y., Gehmlich K., van der Ven P. F., Fürst D. O., Vornwald A., von Hodenberg E., Nürnberg P., Scheffold T., et al. (2003) Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation 107, 1390–1395 10.1161/01.CIR.0000056522.82563.5F [DOI] [PubMed] [Google Scholar]
- 11. Knöll R., Hoshijima M., Hoffman H. M., Person V., Lorenzen-Schmidt I., Bang M. L., Hayashi T., Shiga N., Yasukawa H., Schaper W., McKenna W., Yokoyama M., Schork N. J., Omens J. H., McCulloch A. D., et al. (2002) The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111, 943–955 10.1016/S0092-8674(02)01226-6 [DOI] [PubMed] [Google Scholar]
- 12. Boateng S. Y., Senyo S. E., Qi L., Goldspink P. H., and Russell B. (2009) Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. J. Mol. Cell. Cardiol. 47, 426–435 10.1016/j.yjmcc.2009.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Granés F., Berndt C., Roy C., Mangeat P., Reina M., and Vilaró S. (2003) Identification of a novel Ezrin-binding site in syndecan-2 cytoplasmic domain. FEBS Lett. 547, 212–216 10.1016/S0014-5793(03)00712-9 [DOI] [PubMed] [Google Scholar]
- 14. Boateng S. Y., Belin R. J., Geenen D. L., Margulies K. B., Martin J. L., Hoshijima M., de Tombe P. P., and Russell B. (2007) Cardiac dysfunction and heart failure are associated with abnormalities in the subcellular distribution and amounts of oligomeric muscle LIM protein. Am. J. Physiol. Heart Circ. Physiol. 292, H259–H269 10.1152/ajpheart.00766.2006 [DOI] [PubMed] [Google Scholar]
- 15. Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N. T., Roth A., Bork P., Jensen L. J., and von Mering C. (2017) The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 10.1093/nar/gkw937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Finn R. D., Coggill P., Eberhardt R. Y., Eddy S. R., Mistry J., Mitchell A. L., Potter S. C., Punta M., Qureshi M., Sangrador-Vegas A., Salazar G. A., Tate J., and Bateman A. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 10.1093/nar/gkv1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watkins S. J., Borthwick G. M., and Arthur H. M. (2011) The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. In Vitro Cell. Dev. Biol. Anim. 47, 125–131 10.1007/s11626-010-9368-1 [DOI] [PubMed] [Google Scholar]
- 18. Roper J. A., Williamson R. C., and Bass M. D. (2012) Syndecan and integrin interactomes: large complexes in small spaces. Curr. Opin. Struct. Biol. 22, 583–590 10.1016/j.sbi.2012.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rønning S. B., Carlson C. R., Stang E., Kolset S. O., Hollung K., and Pedersen M. E. (2015) Syndecan-4 regulates muscle differentiation and is internalized from the plasma membrane during myogenesis. PLoS One 10, e0129288 10.1371/journal.pone.0129288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choi Y., Yun J. H., Yoo J., Lee I., Kim H., Son H. N., Kim I. S., Yoon H. S., Zimmermann P., Couchman J. R., Cho H. S., Oh E. S., and Lee W. (2016) New structural insight of C-terminal region of Syntenin-1, enhancing the molecular dimerization and inhibitory function related on Syndecan-4 signaling. Sci. Rep. 6, 36818 10.1038/srep36818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zimmermann P., Zhang Z., Degeest G., Mortier E., Leenaerts I., Coomans C., Schulz J., N'Kuli F., Courtoy P. J., and David G. (2005) Syndecan recycling (corrected) is controlled by syntenin–PIP2 interaction and Arf6. Dev. Cell 9, 377–388 10.1016/j.devcel.2005.07.011 [DOI] [PubMed] [Google Scholar]
- 22. Baietti M. F., Zhang Z., Mortier E., Melchior A., Degeest G., Geeraerts A., Ivarsson Y., Depoortere F., Coomans C., Vermeiren E., Zimmermann P., and David G. (2012) Syndecan–syntenin–ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 14, 677–685 10.1038/ncb2502 [DOI] [PubMed] [Google Scholar]
- 23. Hill M. M., Bastiani M., Luetterforst R., Kirkham M., Kirkham A., Nixon S. J., Walser P., Abankwa D., Oorschot V. M., Martin S., Hancock J. F., and Parton R. G. (2008) PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 132, 113–124 10.1016/j.cell.2007.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X., Xie W., Zhang Y., Lin P., Han L., Han P., Wang Y., Chen Z., Ji G., Zheng M., Weisleder N., Xiao R. P., Takeshima H., Ma J., and Cheng H. (2010) Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ. Res. 107, 76–83 10.1161/CIRCRESAHA.109.215822 [DOI] [PubMed] [Google Scholar]
- 25. Zhu H., Lin P., De G., Choi K. H., Takeshima H., Weisleder N., and Ma J. (2011) Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J. Biol. Chem. 286, 12820–12824 10.1074/jbc.C111.221440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rajab A., Straub V., McCann L. J., Seelow D., Varon R., Barresi R., Schulze A., Lucke B., Lützkendorf S., Karbasiyan M., Bachmann S., Spuler S., and Schuelke M. (2010) Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF–CAVIN mutations. PLoS Genet. 6, e1000874 10.1371/journal.pgen.1000874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ohkawara B., Glinka A., and Niehrs C. (2011) Rspo3 binds syndecan 4 and induces Wnt/PCP signaling via clathrin-mediated endocytosis to promote morphogenesis. Dev. Cell 20, 303–314 10.1016/j.devcel.2011.01.006 [DOI] [PubMed] [Google Scholar]
- 28. Newell-Litwa K., Seong E., Burmeister M., and Faundez V. (2007) Neuronal and non-neuronal functions of the AP-3 sorting machinery. J. Cell Sci. 120, 531–541 10.1242/jcs.03365 [DOI] [PubMed] [Google Scholar]
- 29. Hirokawa M., Morita H., Tajima T., Takahashi A., Ashikawa K., Miya F., Shigemizu D., Ozaki K., Sakata Y., Nakatani D., Suna S., Imai Y., Tanaka T., Tsunoda T., Matsuda K., Kadowaki T., et al. (2015) A genome-wide association study identifies PLCL2 and AP3D1–DOT1L–SF3A2 as new susceptibility loci for myocardial infarction in Japanese. Eur. J. Hum. Genet. 23, 374–380 10.1038/ejhg.2014.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bar-Peled L., and Sabatini D. M. (2014) Regulation of mTORC1 by amino acids. Trends Cell Biol. 24, 400–406 10.1016/j.tcb.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., and Sabatini D. M. (2010) Ragulator–Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 10.1016/j.cell.2010.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sancak Y., Peterson T. R., Shaul Y. D., Lindquist R. A., Thoreen C. C., Bar-Peled L., and Sabatini D. M. (2008) The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 10.1126/science.1157535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Partovian C., Ju R., Zhuang Z. W., Martin K. A., and Simons M. (2008) Syndecan-4 regulates subcellular localization of mTOR Complex 2 and Akt activation in a PKCα-dependent manner in endothelial cells. Mol. Cell 32, 140–149 10.1016/j.molcel.2008.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mattagajasingh S. N., Huang S.-C., Hartenstein J. S., and Benz E. J. Jr. (2000) Characterization of the interaction between protein 4.1R and ZO-2: a possible link between the tight junction and the actin cytoskeleton. J. Biol. Chem. 275, 30573–30585 10.1074/jbc.M004578200 [DOI] [PubMed] [Google Scholar]
- 35. Mezzano V., and Sheikh F. (2012) Cell–cell junction remodeling in the heart: possible role in cardiac conduction system function and arrhythmias? Life Sci. 90, 313–321 10.1016/j.lfs.2011.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stucke V. M., Timmerman E., Vandekerckhove J., Gevaert K., and Hall A. (2007) The MAGUK protein MPP7 binds to the polarity protein hDlg1 and facilitates epithelial tight junction formation. Mol. Biol. Cell 18, 1744–1755 10.1091/mbc.e06-11-0980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shahbazian D., Roux P. P., Mieulet V., Cohen M. S., Raught B., Taunton J., Hershey J. W., Blenis J., Pende M., and Sonenberg N. (2006) The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 25, 2781–2791 10.1038/sj.emboj.7601166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harding H. P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., and Ron D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 10.1016/S1097-2765(00)00108-8 [DOI] [PubMed] [Google Scholar]
- 39. Oh E. S., Woods A., and Couchman J. R. (1997) Syndecan-4 proteoglycan regulates the distribution and activity of protein kinase C. J. Biol. Chem. 272, 8133–8136 10.1074/jbc.272.13.8133 [DOI] [PubMed] [Google Scholar]
- 40. Oh E. S., Woods A., and Couchman J. R. (1997) Multimerization of the cytoplasmic domain of syndecan-4 is required for its ability to activate protein kinase C. J. Biol. Chem. 272, 11805–11811 10.1074/jbc.272.18.11805 [DOI] [PubMed] [Google Scholar]
- 41. Pandey P., Hawkes W., Hu J., Megone W. V., Gautrot J., Anilkumar N., Zhang M., Hirvonen L., Cox S., Ehler E., Hone J., Sheetz M., and Iskratsch T. (2018) Cardiomyocytes sense matrix rigidity through a combination of muscle and non-muscle myosin contractions. Dev. Cell 44, 326–336.e3 10.1016/j.devcel.2017.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murakami M., Horowitz A., Tang S., Ware J. A., and Simons M. (2002) Protein kinase C (PKC)δ regulates PKCα activity in a Syndecan-4-dependent manner. J. Biol. Chem. 277, 20367–20371 10.1074/jbc.M202501200 [DOI] [PubMed] [Google Scholar]
- 43. Riley K. N., Maldonado A. E., Tellier P., D'Souza-Schorey C., and Herman I. M. (2003) βcap73–ARF6 interactions modulate cell shape and motility after injury in vitro. Mol. Biol. Cell 14, 4155–4161 10.1091/mbc.e02-11-0726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gehmlich K., Geier C., Osterziel K. J., Van der Ven P. F., and Fürst D. O. (2004) Decreased interactions of mutant muscle LIM protein (MLP) with N-RAP and α-actinin and their implication for hypertrophic cardiomyopathy. Cell Tissue Res. 317, 129–136 10.1007/s00441-004-0873-y [DOI] [PubMed] [Google Scholar]
- 45. Schiller H. B., Friedel C. C., Boulegue C., and Fässler R. (2011) Quantitative proteomics of the integrin adhesome show a myosin II-dependent recruitment of LIM domain proteins. EMBO Rep. 12, 259–266 10.1038/embor.2011.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li A., Ponten F., and dos Remedios C. G. (2012) The interactome of LIM domain proteins: the contributions of LIM domain proteins to heart failure and heart development. Proteomics 12, 203–225 10.1002/pmic.201100492 [DOI] [PubMed] [Google Scholar]
- 47. Schallus T., Fehér K., Ulrich A. S., Stier G., and Muhle-Goll C. (2009) Structure and dynamics of the human muscle LIM protein. FEBS Lett. 583, 1017–1022 10.1016/j.febslet.2009.02.021 [DOI] [PubMed] [Google Scholar]
- 48. Hallhuber M., Burkard N., Wu R., Buch M. H., Engelhardt S., Hein L., Neyses L., Schuh K., and Ritter O. (2006) Inhibition of nuclear import of calcineurin prevents myocardial hypertrophy. Circ. Res. 99, 626–635 10.1161/01.RES.0000243208.59795.d8 [DOI] [PubMed] [Google Scholar]
- 49. Zong F., Fthenou E., Wolmer N., Hollósi P., Kovalszky I., Szilák L., Mogler C., Nilsonne G., Tzanakakis G., and Dobra K. (2009) Syndecan-1 and FGF-2, but not FGF receptor-1, share a common transport route and co-localize with heparanase in the nuclei of mesenchymal tumor cells. PLoS One 4, e7346 10.1371/journal.pone.0007346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Geier C., Gehmlich K., Ehler E., Hassfeld S., Perrot A., Hayess K., Cardim N., Wenzel K., Erdmann B., Krackhardt F., Posch M. G., Osterziel K. J., Bublak A., Nägele H., Scheffold T., et al. (2008) Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum. Mol. Genet. 17, 2753–2765 10.1093/hmg/ddn160 [DOI] [PubMed] [Google Scholar]
- 51. Frank R., and Overwin H. (1996) in Epitope Mapping Protocols (Morris G. E., ed) pp. 149–169, Humana Press, Totowa, NJ [Google Scholar]
- 52. Mlynarcik P., Bencurova E., Madar M., Mucha R., Pulzova L., Hresko S., and Bhide M. (2012) Development of simple and rapid elution methods for proteins from various affinity beads for their direct MALDI-TOF downstream application. J. Proteomics 75, 4529–4535 10.1016/j.jprot.2012.03.001 [DOI] [PubMed] [Google Scholar]
- 53. Echtermeyer F., Streit M., Wilcox-Adelman S., Saoncella S., Denhez F., Detmar M., and Goetinck P. (2001) Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J. Clin. Invest. 107, R9–R14 10.1172/JCI10559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 55. Keshava Prasad T. S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., Balakrishnan L., Marimuthu A., Banerjee S., Somanathan D. S., Sebastian A., et al. (2009) Human Protein Reference Database—2009 update. Nucleic Acids Res. 37, D767–D772 10.1093/nar/gkn892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Christensen G., Herum K. M., and Lunde I. G. (2018) Sweet, yet underappreciated: Proteoglycans and extracellular matrix remodeling in heart disease. Matrix Biol. 75–76, 286–299 10.1016/j.matbio.2018.01.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.