

Abstract
Neuroinflammation and neurodegeneration often result from the aberrant deposition of aggregated host proteins, including amyloid‐β, α‐synuclein, and prions, that can activate inflammasomes. Inflammasomes function as intracellular sensors of both microbial pathogens and foreign as well as host‐derived danger signals. Upon activation, they induce an innate immune response by secreting the inflammatory cytokines interleukin (IL)‐1β and IL‐18, and additionally by inducing pyroptosis, a lytic cell death mode that releases additional inflammatory mediators. Microglia are the prominent innate immune cells in the brain for inflammasome activation. However, additional CNS‐resident cell types including astrocytes and neurons, as well as infiltrating myeloid cells from the periphery, express and activate inflammasomes. In this review, we will discuss current understanding of the role of inflammasomes in common degenerative diseases of the brain and highlight inflammasome‐targeted strategies that may potentially treat these diseases.
Keywords: disease, inflammasome, inflammation, microglia, neurodegeneration
Subject Categories: Immunology, Neuroscience
Glossary
- Blood–brain barrier (BBB)
a physiological barrier between the blood and the CNS parenchyma. The BBB is formed by endothelial cells that are joined by complex tight junctions and coated by a basement membrane and an additional membrane of astrocytic end‐feet, known as the glia limitans.
- Caspases
a family of cysteine‐dependent aspartate specific proteases that play a central role in inflammation and programmed cell death.
- Cerebrospinal fluid (CSF)
contained in the ventricles of the brain and the cranial and spinal subarachnoid spaces. It provides a mean for transporting different molecules, including cytokines, neurotransmitters, and hormones, but also resides innate immune cells.
- CNS macrophages
non‐parenchymal macrophages of the CNS that are located in meninges, choroid plexus, and perivascular spaces.
- Creutzfeldt–Jakob disease
fatal degenerative prion disease that can be sporadic, hereditary, or acquired. The acquired form of the disease is caused by exposure to the misfolded scrapie form of the PrP protein.
- Cuprizone‐induced demyelination
an experimental model to study local demyelination of the corpus callosum, which is induced by the administration of the copper chelator cuprizone (bis(cyclohexanone)oxaldihydrazone) to the food of mice during 5 weeks.
- Dopaminergic neurons
the main dopamine‐producing cells of the CNS and essential for controlling key functions of the brain, including voluntary movement, reward processing, mood, and working memory.
- Encephalomyelitis
general term to describe inflammation in the brain or spinal cord. Autoimmune encephalomyelitis is caused by an abnormal immune response to a self‐antigen.
- Experimental autoimmune encephalomyelitis (EAE)
the main rodent model of MS. EAE is actively induced by peripheral immunization with myelin‐specific proteins or peptides in combination with an adjuvant, or passively by transfer of encephalitogenic T cells.
- Familial Mediterranean Fever
the most common monogenic autoinflammatory disease and is characterized by periodic fevers with childhood onset, and frequently associated with serositis and joint pain. It predominantly affects Mediterranean populations.
- Grand‐mal seizures
or tonic‐clonic seizures: seizures that are characterized by the loss of consciousness and violent muscle contractions. This seizure comes in two phases: a tonic phase followed by a clonic phase. The brief tonic phase features loss of consciousness and muscle stiffening, while in the clonic phase the muscles go into rhythmic contractions.
- Induced pluripotent stem cells (iPSCs)
generated by genetic reprogramming of adult somatic cells. The advantage over other types of stem cells is that they are not derived from a human embryo.
- Ischemic infarct
an infarct caused by the interruption of the blood flow in the brain due to occlusion of a cerebral vessel.
- Middle cerebral artery occlusion
experimental stroke model that involves the permanent or transient occlusion of the middle cerebral artery. The middle cerebral artery is one of the three arteries that supply blood to the cerebrum. In the “filament model”, a suture filament is (transiently) introduced into the internal carotid artery and forwarded until the tip occludes the middle cerebral artery.
- Neonatal‐onset multisystem inflammatory disease (NOMID)
or chronic infantile neurologic cutaneous and articular (CINCA): the most severe cryopyrin‐associated periodic syndrome (CAPS). NOMID is characterized by neonatal‐onset skin lesions, chronic aseptic meningitis, and recurrent fever along with joint symptoms.
- Sepsis
a life‐threatening systemic inflammatory response syndrome (SIRS) caused by the body's response to an infection.
- Spatial memory
an important cognitive function that allows us to recall three‐dimensional objects or places.
- Steatohepatitis
a type of fatty liver disease and is characterized by the accumulation of lipids and the infiltration of inflammatory cells in the hepatic parenchyma.
- Striata
dopamine neurons from the substantia nigra pars compacta project to the caudate and the putamen of the basal ganglia, together referred to as the striatum.
- Substantia nigra pars compacta
structure in the brainstem that contains most of the dopamine‐producing neurons.
Introduction
The innate immune system is a rapid and coordinated cellular defense response that aims to eliminate the threat posed by both sterile and infectious insults. Recognition of these pathogenic agents is mediated by pattern recognition receptors (PRRs) that sense pathogen‐associated molecular patterns (PAMPs) and host‐ or environment‐derived danger‐associated molecular patterns (DAMPs). In the central nervous system (CNS), these PRRs are primarily expressed by microglia, astrocytes, and macrophages, but also oligodendrocytes, neurons, and endothelial cells express a repertoire of PRRs (Lampron et al, 2013; Walsh et al, 2014). PRRs can either be membrane‐bound, as is the case for the Toll‐like receptors (TLRs), to sense signals in the extracellular environment or in the endosome, or they can be intracellular, as with the nucleotide‐binding domain and leucine‐rich repeat‐containing receptors (NLRs) and AIM2‐like receptors (ALRs). An important subgroup of cytosolic PRRs that includes members of the NLR and ALR families as well as the tripartite motif (TRIM) family member pyrin critically contributes to the innate immune response by assembling so‐called “inflammasomes”.
General concepts of inflammasome biology
Inflammasomes are cytosolic multiprotein complexes which upon assembly activate the pro‐inflammatory caspase‐1 (see Glossary) that is responsible for the maturation and secretion of the inflammatory cytokines IL‐1β and IL‐18, and additionally induce pyroptosis (Lamkanfi & Dixit, 2014; Broz & Dixit, 2016). Briefly, inflammasome‐inducing stimuli trigger the oligomerization of PRR proteins and the recruitment of pre‐existing procaspase‐1 zymogens into the complex, leading to their proximity‐induced autoactivation to generate active caspase‐1. Consequently, caspase‐1 will cleave the biologically inactive pro‐peptides pro‐IL‐1β and pro‐IL‐18 into mature cytokines which are then secreted by the cell. Next to its role in the maturation of pro‐IL‐1β and pro‐IL‐18, caspase‐1 can also induce a pro‐inflammatory form of cell death, pyroptosis, that features early plasma membrane rupture, thereby releasing the soluble intracellular fraction that fuels the inflammatory response (Lamkanfi & Dixit, 2012, 2014). Central in this process is the executioner protein gasdermin D (GSDMD)—a substrate of murine caspase‐1 and caspase‐11, and human caspase‐1, caspase‐4, and caspase‐5—the amino‐terminal domain of which upon cleavage oligomerizes and perforates the plasma membrane to induce cell swelling and osmotic lysis (Shi et al, 2017) (Fig 1).
Figure 1. Inflammasome activation and signaling.
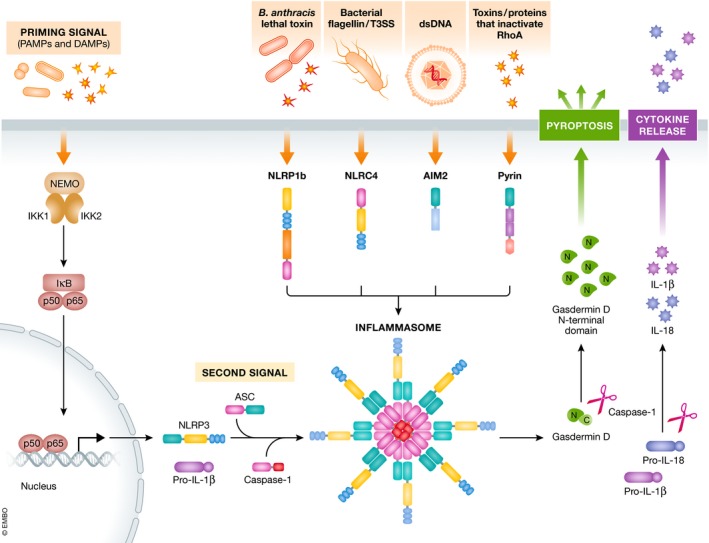
Inflammasomes assemble in a stimulus‐specific manner. Different DAMPs and PAMPs are able to induce NLRP3, while NLRP1b responds to Bacillus anthracis lethal toxin, NLRC4 recognizes bacterial flagellin and/or the type III secretion system of bacterial pathogens, AIM2 is specifically activated by dsDNA, and pyrin recognizes the inactivation of RhoA by toxins and effector proteins. Activation of the NLRP3 inflammasome involves a two‐step mechanism. The priming signal is detected by membrane‐bound PRRs, including TLRs and C‐type lectin receptors (CLRs) and induces NF‐κB‐dependent transcription of NLRP3 and pro‐IL‐1β precursor protein, and controls post‐translational modifications that license NLRP3 activation. The second activation signal is necessary for inflammasome formation, depending on the oligomerization and subsequent activation of procaspase‐1. Active caspase‐1 then cleaves pro‐IL‐1β and pro‐IL‐18 to their mature forms IL‐1β and IL‐18 which get secreted. In addition, caspase‐1 can cleave gasdermin D, releasing its N‐terminal fragment which translocates to the plasma membrane inducing pore formation and pyroptotic cell death. In contrast to NLRP3, other inflammasome receptors do not need this initial priming signal to induce inflammasome activation and cytokine release.
One way to classify inflammasome complexes is based on the receptor that initiates signaling (Lamkanfi & Dixit, 2012). The core inflammasome components consist of the cytosolic NLR, ALR, and pyrin receptors, the adaptor apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC) and procaspase‐1. ASC is composed of a pyrin domain (PYD) and a caspase recruitment domain (CARD) and functions as an adaptor that links the PYD of the NLR or pyrin and the CARD of procaspase‐1 (Fig 2). Some inflammasomes, such as NLRC4 and NLRP1b, do not require ASC and may directly recruit procaspase‐1 through their respective CARDs, although under wild‐type conditions ASC does contribute to optimal caspase‐1 activation and efficient secretion of IL‐1β and IL‐18 (Broz et al, 2010; Guey et al, 2014; Van Opdenbosch et al, 2014). Assembly and activation of inflammasomes requires detection of specific signals: Murine Nlrp1b responds to Bacillus anthracis lethal toxin (LeTx) (Boyden & Dietrich, 2006); NLRC4 detects intracellular flagellin and components of type III secretion systems (T3SS) of bacterial pathogens (Franchi et al, 2006; Miao et al, 2006, 2010); AIM2 physically binds cytosolic double‐stranded DNA (dsDNA) (Hornung et al, 2009); and pyrin indirectly responds to toxins that covalently inactivate the small GTPase RhoA (Xu et al, 2014). NLRP3, the most widely studied inflammasome, responds to a broad spectrum of activating stimuli that includes a suite of bacterial, fungal, and viral PAMPs, DAMPs such as ATP and uric acid crystals, and crystalline and aggregated substances such as asbestos, silica, and amyloid‐β fibrils (Lamkanfi & Dixit, 2012). NLRP3 activation is unique in the sense that it involves a two‐step process. A first signal, or priming signal, results in the NF‐κB‐dependent transcriptional upregulation of NLRP3 and pro‐IL‐1β, but also controls post‐translational modifications of NLRP3 as highlighted by the mapped ubiquitin and phosphorylation sites on NLRP3 (Yang et al, 2017). This is followed by a second “activation” signal that induces the oligomerization and activation of the NLRP3 inflammasome (Fig 1). Besides canonical NLRP3 inflammasome signaling, the non‐canonical NLRP3 inflammasome pathway involves activation of caspase‐11 (or its human orthologs caspase‐4 and caspase‐5) by cytosolic LPS, and the induction of pyroptosis through the cleavage of GSDMD and the release of high mobility group box 1 protein (HMGB1) and IL‐1α (Lamkanfi & Dixit, 2014; Shi et al, 2017). Apart from AIM2 and the NAIP/NLRC4 complexes, the common secondary messengers or physical ligands that bind and trigger inflammasome assembly await discovery.
Figure 2. Domain structure of inflammasomes.
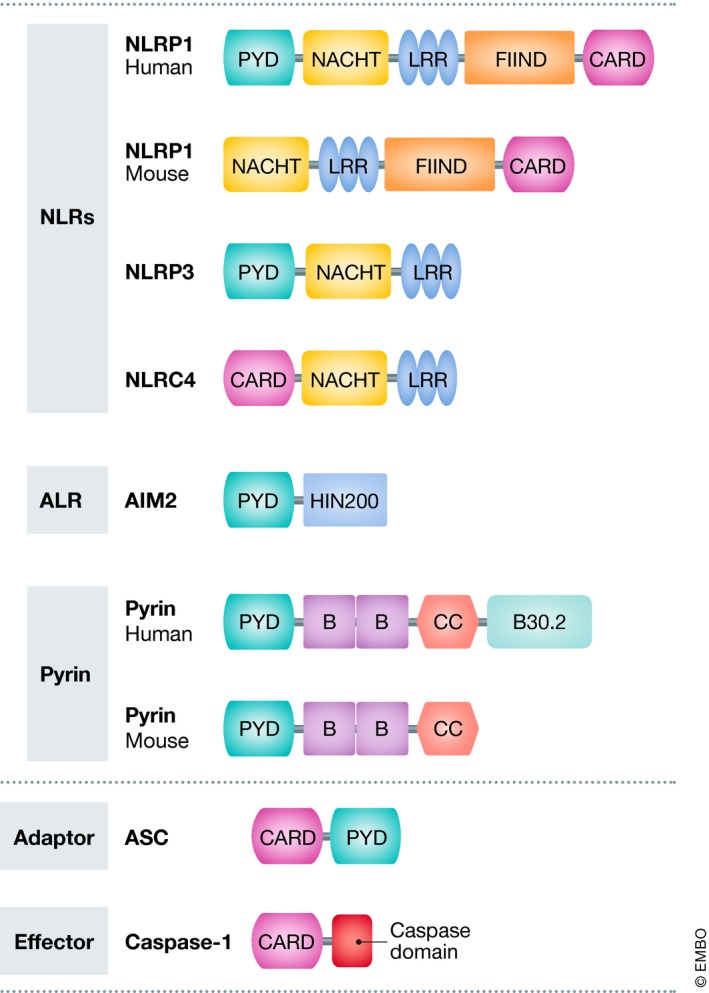
A subset of NLRs and ALRs can trigger the formation of inflammasomes. NLR family members have a nucleotide‐binding and oligomerization domain (NACHT/NBD), as well as leucine‐rich repeat (LRR) motifs, typically located in the center and carboxy terminus of the NLR proteins, respectively. The NACHT motif is usually flanked by an additional amino‐terminal domain, either CARD or PYD, and these domains are used for further sub‐classification of inflammasomes. These domains allow the recruitment of adaptor and effector proteins to the inflammasome signaling complex. The NLR gene family consists of 22 human members and 34 murine members, many of which the function is not always clear (Lamkanfi & Dixit, 2012; Broz & Dixit, 2016). In addition to the NLR‐containing inflammasomes, the ALR family member AIM2 can also assemble an inflammasome complex. AIM2 is characterized by an amino‐terminal PYD domain and one or two DNA‐binding HIN200 domains (Hornung et al, 2009). Pyrin, also known as TRIM20, features a PYD domain, two B‐boxes, and a coiled‐coil domain, whereas the human pyrin also has an additional C‐terminal B30.2 domain. ASC is the critical adaptor protein for many inflammasome complexes and is composed of CARD and PYD domains, the latter being necessary for homotypic interaction with a PYD‐containing inflammasome sensor (NLRP3, AIM2). Procaspase‐1 features a CARD domain, in addition to its caspase domain, and homotypic CARD interactions result in direct or indirect (via ASC) recruitment of procaspase‐1 to the inflammasome complex. Inflammasome activation involves ASC and procaspase‐1 recruitment, resulting in ASC oligomerization into a macromolecular aggregate, known as an ASC speck (Broz & Dixit, 2016).
Inflammasomes and the central nervous system
IL‐1β and IL‐18 have important functions in the CNS, and many cell types in the brain express their cognate receptors that initiate inflammatory signaling cascades that may contribute to neuronal injury and cell death (Allan et al, 2005; Alboni et al, 2010). Hence, increased levels of IL‐1β and IL‐18 are often observed upon CNS infection, brain injury, and neurodegenerative diseases (Heneka et al, 2014, 2018). IL‐1β and IL‐18 are also important for physiological functions in the CNS and have been shown to participate to processes of cognition, learning, and memory (Tsai, 2017). Pyroptosis additionally contributes to inflammasome‐driven pathology through the release of other inflammatory mediators and DAMPs. Apoptosis and necroptosis are additional cell death modes that have been shown to promote neuroinflammation and neuronal degeneration in several neurodegenerative pathologies, including multiple sclerosis, amyotrophic lateral sclerosis, Parkinson's disease, and Alzheimer's disease (Zhang et al, 2017; Yuan et al, 2019).
Although inflammasome signaling in the CNS is mainly attributed to microglia, the key innate immune cells of the brain, expression of inflammasome components has also been reported in other cell types of the CNS, including neurons (Kaushal et al, 2015), astrocytes (Freeman et al, 2017), perivascular CNS macrophages (Kawana et al, 2013), oligodendrocytes (Mckenzie et al, 2018), and endothelial cells (Gong et al, 2018) (Fig 3). However, current understanding of inflammasome activation in microglia and its role in CNS inflammation and disease is still fragmentary and primarily based on in vitro studies with primary microglia and microglial cell lines, and in vivo studies with transgenic knockout mice that lack expression of specific inflammasome components throughout the body. More appropriate research tools that allow CNS‐specific inflammasome targeting are needed to investigate the relative contribution of local inflammasome signaling in specific brain cell types to overall CNS pathology. Furthermore, evidence for inflammasome activation in patients with neurodegenerative disease often relies on detection of higher IL‐1β transcript levels and/or increased gene and protein expression of inflammasome components. However, increased expression levels of these NF‐κB responsive genes are largely indicative of an ongoing inflammatory response rather than direct support for inflammasome engagement. We here review the current knowledge on the relevance of inflammasome activation for the most common neurodegenerative pathologies (Fig 3 and Table 1) and highlight emerging strategies for the treatment of inflammasome‐driven diseases. Although inflammasome activation is also central to the pathology of many CNS infectious diseases, such as Zika, HIV, and West Nile virus, and bacterial infections that induce meningitis (Walsh et al, 2014; Mamik & Power, 2017; Heneka et al, 2018), these conditions are outside the scope of this review.
Figure 3. Inflammasome activation in neurodegenerative disease.
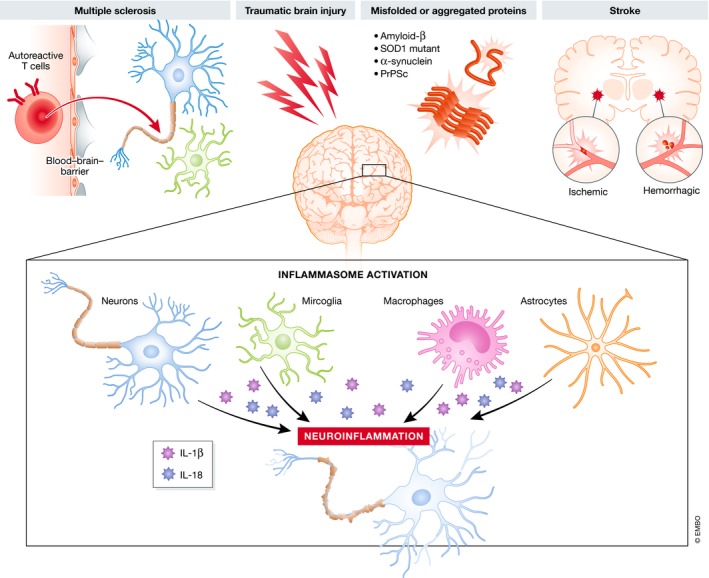
Inflammasomes can be activated in the CNS in response to acute injury (traumatic brain injury and stroke), autoimmune‐mediated injury (multiple sclerosis), and accumulation of misfolded or aggregated proteins in the brain (Alzheimer's disease, amyotrophic lateral sclerosis, Parkinson's disease, and prion disease). Inflammasome activation has been demonstrated in CNS‐resident cell types, including microglia, astrocytes, and neurons, but also in CNS‐infiltrating cells, such as in infiltrating macrophages. Although most research on neurodegenerative diseases has focused on the importance of the NLRP3 inflammasome, also other inflammasome types can be activated in the brain and have been demonstrated in neurodegenerative disorders. Overall, inflammasome activation results in caspase‐1‐mediated cleavage of pro‐IL‐1β and pro‐IL‐18, and the subsequent release of the mature cytokines. High levels of IL‐1β and IL‐18 can be detected in many neurodegenerative conditions and are considered to be crucial for the establishment of a chronic inflammatory environment, leading to neuronal dysfunction and eventually neurodegeneration.
Table 1.
Overview of the described neurodegenerative disease models
| Disease model | Description |
|---|---|
| Multiple sclerosis | |
| EAE | EAE is actively induced by peripheral immunization with myelin‐specific proteins or peptides in combination with an adjuvant, or passively by transfer of encephalitogenic T cells |
| Cuprizone | Administration of the copper chelator cuprizone will induce local demyelination of the corpus callosum |
| Stroke | |
| Permanent MCAO | Permanent occlusion of the middle cerebral artery is obtained using an intraluminal suture |
| Transient MCAO | Intraluminal suture MCAO utilizes a suture inserted into the middle cerebral artery to interrupt the blood flow for a specific duration and is afterward removed. Embolic stroke MCAO uses an autologous blood clot injected into the MCA to occlude the vessel |
| Intracerebral hemorrhage (ICH) | Stroke condition provoked by injection of autologous arterial blood into the basal ganglia |
| TBI and SCI | |
| Controlled cortical impact (CCI) | A mechanical model of traumatic brain injury. Following craniotomy, the CCI device mechanically transfers energy onto the dura mater damaging the cortex, and sometimes the subcortical structures |
| Impact acceleration model | The exposed skull is covered with a steel disk and a weight is dropped onto the steel disk |
| Contusion model of spinal cord injury | A transient force is applied by either an electromagnetic device or a weight‐drop to displace and damage the spinal cord |
| Alzheimer disease | |
| APP/PS1 | Mouse model expressing human APP695 with the Swedish double mutation (K670N/M671L) and a mutant human presenilin 1 (PS1‐dE9) |
| 3xTgAD | Mouse model expressing human APP695 with the Swedish double mutation (K670N/M671L), human PS1 with the M146V mutation, and human Tau with the P301L mutation |
| Tg2567 | Mouse model expressing human APP695 with the Swedish double mutation (K670N/M671L) at the β‐secretase cleavage site |
| 5xFAD | Mouse model expressing human APP and PSEN1 transgenes with a total of five AD‐linked mutations: the Swedish (K670N/M671L), Florida (I716V), and London (V717I) mutations in APP, and M146L and L286V mutations in PSEN1 |
| Amyotrophic lateral sclerosis | |
| TgSOD1‐G93A | Mouse model expressing a G93A mutant form of human SOD1 |
| hSOD1 G37R | Mouse model expressing a G37R mutant form of human SOD1 |
| hSOD1 G85R | Mouse model expressing a G85R mutant form of human SOD1 |
| Parkinson disease | |
| A53T | Mouse model expressing the mutant human A53T alpha‐synuclein. |
| LPS‐induced PD | Injection of LPS in the left substantia nigra pars compacta |
| 6‐hydroxydopamine‐induced PD | Injection of 6‐hydroxydopamine in the medial forebrain bundle |
| MPTP‐induced PD | Intraperitoneal injection of MPTP five times at 2‐h intervals |
| Prion disease | |
| Scrapie‐infected | Intracerebral infections with brain homogenate of scrapie strain 139A‐infected mice or intracerebral injection with RML6 (passage 6 of Rocky Mountain Laboratory strain mouse‐adapted scrapie prions) |
| Tg(CJD) | Mouse model expressing a misfolded mutant PrP (D177N/V128) |
| Huntington disease | |
| R6/2 | Transgenic mice expressing exon 1 of human huntingtin with expanded CAG/polyglutamine repeat |
Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis
Multiple sclerosis (MS) is the most common chronic inflammatory disease of the CNS. MS is characterized by a compromised blood–brain barrier (BBB) that leads to immune cell infiltration from the periphery and local microglia and astrocyte activation, which together promote inflammation, demyelination, and neurodegeneration (Baecher‐Allan et al, 2018). Experimental autoimmune encephalomyelitis (EAE) is a widely used rodent model of MS (Ransohoff, 2012), and both IL‐1β and IL‐18 were shown to critically contribute to EAE (Sutton et al, 2006; Gris et al, 2010; Lévesque et al, 2016). Most studies on the involvement of inflammasomes in MS have focused on the peripheral immune response that is shaped by lymphocytes and macrophages that enter the CNS during MS pathology. Caspase‐1, IL‐18, and IL‐1β are upregulated in peripheral blood mononuclear cells (PBMCs) and cerebrospinal fluid (CSF) of MS patients, as are levels of the NLRP3 inflammasome‐activating DAMPs ATP and uric acid (Inoue & Shinohara, 2013; Mamik & Power, 2017). Moreover, caspase‐1 expression was shown to be elevated in acute and chronic demyelinating lesions (Voet et al, 2018), and caspase‐1 and ASC have recently been proposed as candidate biomarkers for MS onset (Keane et al, 2018).
The role of the NLRP3 inflammasome in EAE‐associated T‐cell priming and trafficking into the CNS is supported by the analysis of NLRP3, caspase‐1, and ASC knockout mice (Furlan et al, 1999; Gris et al, 2010; Inoue et al, 2012a). Moreover, the pharmacological NLRP3 inflammasome inhibitor MCC950/CRID3 was shown to suppress IL‐1β production and attenuate EAE severity in wild‐type mice (Coll et al, 2015). Similarly, pharmacological blockade of caspase protease activity with Z‐Val‐Ala‐DL‐Asp‐fluoromethylketone or the inflammatory caspase prodrug VX‐765 reduced EAE symptoms in mice (Furlan et al, 1999; Mckenzie et al, 2018). Interestingly, IFNβ, the first line treatment for MS, is only effective in an NLRP3 inflammasome‐dependent EAE subtype, whereas NLRP3‐independent EAE could not be reversed by IFN‐β therapy (Inoue et al, 2012b, 2016). We recently provided direct genetic evidence for the relevance of inflammasome signaling in microglia and border‐associated macrophages during EAE (Voet et al, 2018). The anti‐inflammatory protein A20 has been shown to negatively regulate NLRP3 inflammasome activation (Vande Walle et al, 2014), and deletion of A20 in microglia and CNS macrophages exacerbated EAE in mice due to NLRP3 hyperactivation that resulted in increased IL‐1β secretion and CNS inflammation (Voet et al, 2018). CNS‐intrinsic inflammasome activation was further reported in another study that showed caspase‐1‐ and GSDMD‐mediated pyroptosis in microglia, as well as in myelin‐forming oligodendrocytes in the CNS of MS patients and EAE mice (Mckenzie et al, 2018).
The relevance of the NLRP3 inflammasome to MS has also been demonstrated in the model of cuprizone‐induced demyelination, in which NLRP3‐deficient mice presented delayed demyelination, oligodendrocyte loss, and neuroinflammation (Jha et al, 2010). Additional support was provided by similar observations in caspase‐1 and IL‐18 knockout mice, which unlike IL‐1β knockout mice showed protection (Jha et al, 2010). Moreover, IL‐1β and IL‐18 had a differential effect on the process of remyelination after cuprizone treatment, demonstrating a delay in remyelination in IL‐1β knockout mice, whereas accelerated remyelination was seen in IL‐18 knockouts (Jha et al, 2010).
Inflammasome activation in stroke
Neuroinflammation plays a crucial pathological role in stroke, and IL‐1β has been identified as a key cytokine in stroke pathology. So far, four distinct inflammasomes have been implicated in stroke, viz. NLRP1, NLRP3, NLRC4, and AIM2 (Barrington et al, 2017). Additionally, early studies implicated caspase‐11 in middle cerebral artery occlusion (MCAO), a mouse model of stroke (Kang et al, 2000). Expression levels of NLRP3 and NLRP1, as well as cleavage products of caspase‐1, IL‐1β, and IL‐18, were shown to be elevated in postmortem brain tissue of stroke patients (Fann et al, 2013). Studies with pharmacological inhibitors of inflammatory caspases and analysis of caspase‐1‐deficient mice—that were later shown to also lack caspase‐11 expression (Kayagaki et al, 2011)—showed protection in experimental models of stroke (Friedlander et al, 1997; Hara et al, 1997; Schielke et al, 1998; Rabuffetti et al, 2000; Ross et al, 2007). In agreement, mice deficient in both IL‐1α and IL‐1β exhibited dramatically reduced ischemic infarct volumes in transient MCAO (Boutin et al, 2001), whereas IL‐18‐deficient mice did not show protection in this model (Wheeler et al, 2003). NLRP3 deficiency in mice, treatment with the NLRP3 inflammasome inhibitor MCC950/CRID3, and intracerebroventricular injection of NLRP3 siRNAs all ameliorated clinical outcomes in experimental models of stroke (Ma et al, 2014; Yang et al, 2014; Yuan et al, 2015; Ismael et al, 2018b). Also, NLRC4‐ and AIM2‐deficient mice were shown to have significantly smaller infarct volumes compared to wild‐type mice subjected to tMCAO, with was associated with strongly reduced microglia cell activation and leukocyte recruitment to the infarct site (Denes et al, 2015). Finally, an expression analysis in the peri‐infarct zone of transient MCAO rats revealed elevated levels of the inflammasome components NLRP1, NLRP3, AIM2, NLRC4, and ASC (Lammerding et al, 2016), consistent with the potential activation of multiple inflammasomes in stroke.
Inflammasome activation in traumatic brain and spinal cord injury
Patients with traumatic brain injury (TBI) have significantly higher levels of inflammasome markers in their CSF, including ASC, caspase‐1, NLRP1, and NLRP3 (Adamczak et al, 2012; Wallisch et al, 2017). However, NLRP1b and ASC knockout mice did not show any improvement in motor recovery or lesion volume in conditions of TBI, compared to control mice, although their levels of IL‐1β were reduced (Brickler et al, 2016). NLRP3 expression localizing to neurons, microglia, and astrocytes was also detected in experimental TBI (Liu et al, 2013a), and NLRP3‐deficient mice as well as pharmacological blockade of NLRP3 activation were shown to improve recovery from TBI (Irrera et al, 2017). Treatment with IL‐1β neutralizing antibodies also was shown to improve the cognitive outcome following TBI (Clausen et al, 2009). In contrast to IL‐1β levels, which rapidly increase after TBI, IL‐18 levels gradually increase over several days, suggesting that IL‐1β may be involved in the early phase of TBI, while IL‐18 may contribute to pathogenesis at later phases of the disease. In agreement, IL‐18 inhibition resulted in improved neurologic recovery after 7 days with little effect on the early response being observed immediately after injury (Yatsiv et al, 2002).
Similar to TBI, spinal cord injury (SCI) induced higher levels of NLRP1, ASC, caspase‐1, IL‐1β, and IL‐18 in mice, and therapeutic neutralization of ASC was proposed to result in tissue sparing and functional improvement due to reduced inflammasome activation in neurons (de Rivero Vaccari et al, 2008). Furthermore, expression of heme oxygenase‐1 was shown to suppress NLRP1 inflammasome activation and neuronal cell death, and lead to improved functional recovery after SCI (Lin et al, 2016). Also, enhanced NLRP3 expression can be demonstrated in SCI, predominantly in neurons, but also in microglia and astrocytes (Zendedel et al, 2016).
Inflammasome activation in Alzheimer's disease
Alzheimer's disease (AD) is the most common age‐related neurodegenerative disorder and is characterized by progressive memory deficits and cognitive impairment (Sala Frigerio & De Strooper, 2016). AD pathology is dominantly explained by the amyloid hypothesis, which suggests that the increasing extracellular deposition of misfolded amyloid‐beta (Aβ) is the primary cause of the disease. Although there is overwhelming evidence for a pathogenic role for Aβ in AD, neuroinflammation is increasingly regarded as another key component that actively contributes to AD pathology (De Strooper & Karran, 2016; Sala Frigerio & De Strooper, 2016). Neuroinflammation in AD is primarily driven by the CNS‐resident microglia population (Sarlus & Heneka, 2017; Shi & Holtzman, 2018). Although it is recognized that microglia may exert benign and reparative activities in AD through the phagocytic removal of Aβ deposits, the accumulation of Aβ may also prime microglial cells and promote their activation and production of inflammatory mediators. Moreover, upon Aβ accumulation, microglial cells may become progressively impaired in their ability to phagocytose Aβ (Sarlus & Heneka, 2017; Shi & Holtzman, 2018). Furthermore, a microglial inflammatory state would also impact on other CNS‐resident cells and result in synaptic dysfunction and neuronal damage.
Many recent studies have implicated inflammasomes in the development of AD. Elevated expression of IL‐1β has been reported in microglia that surround Aβ plaques of AD patients (Griffin et al, 1989; Simard et al, 2006). Additionally, gene expression analysis of cultured PBMCs from AD patients revealed higher expression of NLRP3, ASC, caspase‐1, and caspase‐5 as well as the cytokines IL‐1β and IL‐18 (Saresella et al, 2016). The expression levels of NLRC4 and ASC were also found significantly elevated in brain samples of a subgroup of sporadic AD patients (Liu & Chan, 2014). Finally, genetic variants of NLRP1 have been associated with AD risk (Pontillo et al, 2012), and the amount of NLRP1 immunopositive neurons was strongly increased in AD brain (Kaushal et al, 2015).
In vitro studies have shown that fibrillar Aβ activates the NLRP3 inflammasome when phagocytosed by microglia, leading to activation of caspase‐1 and release of IL‐1β (Halle et al, 2008). NLRP3 inflammasome activation has also been documented in vivo in the transgenic APP/PS1 mouse model of AD, and deficiency in NLRP3 significantly ameliorated spatial memory deficits and hyperactive behavior in these mice, which was associated with reduced hippocampal and cortical Aβ deposition, smaller plaque volumes, decreased levels of pro‐inflammatory cytokines, and improved microglial phagocytic ability (Heneka et al, 2013). Similar results were obtained in APP/PS1 mice that were deficient in caspase‐1 (Heneka et al, 2013). Inhibition of the NLRP3 inflammasome by MCC950/CRID3 also promoted amyloid‐β clearance and ameliorated cognitive function in APP/PS1 mice (Dempsey et al, 2017). Indirect inhibition of NLRP3 inflammasome activation by clinically approved fenamate non‐steroidal anti‐inflammatory drugs that target cyclooxygenase enzymes and volume‐regulated anion channels (VRAC) also was shown to suppress microglia‐mediated neuroinflammation and memory loss in 3×TgAD mice (Daniels et al, 2016). Finally, pharmacological inhibition of caspase‐1 activity by VX‐765 reduced amyloid‐β accumulation, brain inflammation, and cognitive impairment (Flores et al, 2018). Collectively, these findings suggest that misfolded Aβ activates the microglial NLRP3 inflammasome, which triggers the release of pro‐inflammatory factors that perpetrate a chronic neuroinflammatory environment and promote AD pathology. In addition, NLRP3 inflammasome activity also results in the extracellular release of micrometer‐sized ASC particles that may function as danger signals and alert surrounding macrophages (Baroja‐Mazo et al, 2014). These ASC specks were shown to physically bind to Aβ to seed and spread Aβ pathology in a prion‐like manner by promoting misfolded protein aggregation and plaque formation in the APP/PS1 AD model (Venegas et al, 2017).
A pathogenic role for IL‐1β in AD has been demonstrated in mice with a deficiency in IL‐1 receptor antagonist (IL‐1ra), which resulted in increased vulnerability to intracerebroventricular injection with human oligomeric Aβ1–42 (Craft et al, 2005). Another study showed that IL‐1β injection in the cerebral hemisphere increases Aβ‐APP protein levels in wild‐type mice (Sheng et al, 1996). In contrast, sustained hippocampal IL‐1β overexpression in APP/PS1 mice was shown to reduce plaque pathology, an observation that might be explained by the increased phagocytic activity of microglia and macrophages (Shaftel et al, 2007). Similar observations were done in 3xTgAD mice, but tau phosphorylation was also increased in these mice, demonstrating the complex role of IL‐1 signaling in AD (Ghosh et al, 2013). IL‐1R1 deficiency in mice had no effect on Aβ deposition in Tg2567 AD mice (Das et al, 2006). Similarly, IL‐18 deletion did not confer a protective effect in APP/PS1 mice (Tzeng et al, 2018). However, these mice were shown to be highly susceptible to grand‐mal seizures and exhibited increased excitatory synaptic proteins and dendritic spine density, an imbalance which leads to a higher basal excitatory synaptic transmission (Tzeng et al, 2018). Therefore, IL‐18 may have an unexpected role in controlling neuronal activity.
Although the focus has been on the role of NLRP3 in AD, the role of other inflammasomes has also been characterized in the context of AD. The AIM2 inflammasome was shown to increase Aβ deposition, microglia activation, and cytokine production, but does not affect behavior or memory in transgenic 5xFAD mice (Wu et al, 2017). NLRP1 knockdown in APP/PS1 mice was shown to result in significantly reduced neuronal pyroptosis and to rescue cognitive impairments (Tan et al, 2014). Finally, the NLRC4 inflammasome was shown to enhance neuronal Aβ levels through activated pro‐inflammatory astrocytes (Liu & Chan, 2014).
Inflammasome activation in amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that is caused by the progressive loss of upper and lower motor neurons and leads to muscle weakness and wasting (Taylor et al, 2016). Twenty percent of familial ALS cases are caused by mutations in the gene that encodes superoxide dismutase 1 (SOD1), and many experimental models of ALS rely on expression of mutant SOD1. Deficiency in caspase‐1 and IL‐1β, as well as treatment with the recombinant IL‐1 receptor antagonist anakinra, all significantly extended the lifespan of mutant SOD1 transgenic animals (Meissner et al, 2010). Consistently, the expression of NLRP3, NLRC4, AIM2, and caspase‐1 activation has been demonstrated in neural tissue of mutant SOD1 transgenic animals (Pasinelli et al, 1998; Johann et al, 2015; Gugliandolo et al, 2018), and mutant SOD1 was shown to activate caspase‐1 and IL‐1β in microglia (Meissner et al, 2010; Lehmann et al, 2018). Increased caspase‐1 levels have also been detected in serum of ALS patients (Iłżecka et al, 2001), and analysis of postmortem spinal cord tissue showed increased NLRP3, ASC, caspase‐1, and IL‐18 expression levels, with spinal cord astrocytes identified as the main NLRP3 inflammasome‐expressing cell type (Johann et al, 2015). However, clinical studies with anakinra in ALS patients have not shown a significant reduction in disease progression (Maier et al, 2015), suggesting that inflammasome activation does not play a major role in ALS, or that the pathology is driven by IL‐18 or by DAMPs via pyroptosis.
Inflammasome activation in Parkinson's disease
Parkinson's disease (PD) is a progressive neurodegenerative disorder caused by the loss of dopaminergic neurons in the substantia nigra pars compacta. Lewy bodies are pathological hallmarks of the disease that predominantly consist of intra‐neuronal aggregates of fibrillar α‐synuclein (Przedborski, 2017). Analysis of the serum of PD patients revealed increased levels of IL‐1β and caspase‐1 (Zhou et al, 2016), and elevated IL‐1β levels were observed in the striata of PD patients (Mogi et al, 1994). The midbrain of A53T transgenic mice, which model PD based on the overexpression of mutant human A53T α‐synuclein, also contained increased IL‐1β concentrations (Zhou et al, 2016). Chronic expression of IL‐1β in the substantia nigra of rats by a recombinant adenovirus expressing IL‐1β was shown to induce progressive death of dopaminergic neurons and resulted in motor impairments (Ferrari et al, 2006). However, most of the evidence linking PD to inflammasome signaling comes from in vitro studies, and the importance of inflammasomes for the disease is not completely understood. α‐Synuclein was shown to trigger activation of the NLRP3 inflammasome in human monocytes and BV2 microglial cells (Codolo et al, 2013; Gustot et al, 2015; Zhou et al, 2016), but not in primary microglia (Gustin et al, 2015). In vivo, microinjection of the caspase‐1 inhibitor Ac‐YVAD‐CMK was shown to reduce the expression of NLRP3 inflammasome signaling proteins and improve the number of dopaminergic neurons in LPS‐induced and 6‐hydroxydopamine‐induced PD in rats (Mao et al, 2017). In agreement, NLRP3 knockout mice were resistant to the loss of nigral dopaminergic neurons induced by treatment with the neurotoxin 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP), which was associated with a reduction in caspase‐1 activation and IL‐1β and IL‐18 secretion (Yan et al, 2015). Recently, caspase‐1 was shown to cleave α‐synuclein in vitro, generating an aggregation‐prone protein that was toxic to cultured neurons (Wang et al, 2016). Finally, mutations in Parkin, PARK2, PARK6, and PINK1 have been identified in patients with autosomal recessive early‐onset PD, and microglia and macrophages from PARK2 and PINK1 knockout mice and patients with PARK2 mutations have been shown to display an exacerbated NLRP3 inflammasome response, possibly due to impaired expression of the anti‐inflammatory protein A20 that negatively regulates NLRP3 inflammasome activation (Mouton‐Liger et al, 2018).
Inflammasome activation in prion disease
Prion diseases are a group of rapidly progressive neurodegenerative disorders caused by misfolded aggregated infectious prion proteins (PrP) (Sigurdson et al, 2018). IL‐1β levels are increased in the CSF of patients with Creutzfeldt–Jakob disease (CJD) (Van Everbroeck et al, 2002), and IL‐1β and cleaved caspase‐1 levels were reported to be upregulated in the brains of scrapie‐infected mice (Schultz et al, 2004). Stimulation of microglia with an amyloidogenic PrP peptide induced NLRP3 activation and IL‐1β secretion in vitro (Shi et al, 2012). Stimulation of microglia with PrP fibrils was also shown to induce toxicity in neurons (Hafner‐Bratkovič et al, 2012). Mechanistically, NLRP3 inflammasome activation was suggested to negatively regulate autophagy in microglia, thereby contributing to neurodegeneration (Lai et al, 2018). Inhibition of IL‐1R signaling with anakinra was shown to reduce seizure susceptibility in a CJD mouse model (Bertani et al, 2017). However, genetic ablation of NLRP3 or ASC in mice did not affect prion pathogenesis, suggesting that the NLRP3 inflammasome does not play a significant role in prion disease (Nuvolone et al, 2015).
Inflammasome activation in Huntington's disease
Huntington's disease (HD) is an autosomal dominant progressive neurodegenerative disease that is caused by the expansion of a trinucleotide CAG repeat in the 5′ coding region of the huntingtin gene, leading to the expression of an abnormal protein that gradually damages cells in the brain (Caron et al, 2018). Caspase‐1 activation can be detected in the brains of HD patients and in mouse models of HD, and inhibition of caspase‐1 was shown to slow down disease progression in the R6/2 mouse model of HD (Ona et al, 1999). Mechanistically, caspase‐1 was shown to cleave mutant and wild‐type huntingtin in vitro (Wellington et al, 1998) and in vivo (Ona et al, 1999), potentially contributing to the neurodegeneration seen in HD. Similarly, treatment with the tetracycline derivative minocycline was shown to delay disease progression by inhibition of caspase‐1 and caspase‐3 expression (Chen et al, 2000).
Pharmacological targeting of inflammasomes
The central role of inflammasomes in neuroinflammatory responses, in particular NLRP3, makes it an attractive drug target for neurodegenerative diseases. Currently approved therapies in the clinic target the downstream effector cytokines released by inflammasome activation, such as anakinra (IL‐1 receptor antagonist), canakinumab (IL‐1β neutralizing antibody), and rilonacept (soluble decoy receptor for IL‐1β and IL‐1α) (Dinarello et al, 2012). While valuable in treating autoinflammatory diseases (Van Gorp et al, 2019), these IL‐1 inhibitors do not prevent pyroptosis and IL‐18‐driven immune activation. The need for subcutaneous administration of these biological agents represents another drawback of these biological agents (Moran et al, 2013; Rossi‐Semerano et al, 2015), especially in the case of anakinra, which requires daily injections due to a short half‐life of around 4–6 h (Granowitz et al, 1992). Moreover, constitutive neutralization of IL‐1β‐driven signaling increases the risk for infections, as observed in the recent CANTOS trial (Ridker et al, 2017). Finally, the concentration of anakinra in CSF of healthy non‐human primates did not surpass 0.2–0.3% of the peripheral serum concentration, demonstrating poor penetration of the BBB (Fox et al, 2010). These observations urge for the development of small molecule inflammasome antagonists with improved pharmacokinetic properties and efficacy, and that are able to cross the BBB.
Several small molecule inhibitors that target the inflammasome components caspase‐1, NLRP3, and GSDMD have been reported (Table 2). Since caspase‐1 activation is common to all inflammasome complexes, caspase‐1 inhibition would serve as a pan‐inflammasome inhibitory strategy, while compounds targeting NLRP3 have a narrower selectivity profile. Targeting the inflammasome adaptor ASC would have an intermediate selectivity profile; however, compounds that target ASC have not been reported to date (Mangan et al, 2018). A number of NLRP3 inhibitory compounds have been identified and validated in a suite of disease models (Table 2). However, for most agents it is not known whether they directly target NLRP3 or indirectly act on the pathway via other mechanisms, hampering clinical development of these molecules (Mangan et al, 2018). To date, only very few compounds targeting NLRP3 or caspase‐1 have entered clinical trials. RP‐1127, an intravenous formulation of the NLRP3 inflammasome inhibitor glyburide (Lamkanfi et al, 2009), is being tested in a clinical trial for stroke (EudraCT 2017‐004854‐41) after a positively evaluated pilot study (ClinicalTrials.gov: NCT01268683) (Sheth et al, 2014). RP‐1127 is also being tested in a clinical trial for TBI (ClinicalTrials.gov: NCT01454154). In addition, the caspase‐1 inhibitor VX‐765 entered a phase II clinical trial for use in epilepsy (ClinicalTrials.gov: NCT01048255) and psoriasis (ClinicalTrials.gov: NCT00205465), but further development has been discontinued (Mackenzie et al, 2010). The encouraging activity of small molecule NLRP3 inflammasome antagonists in preclinical studies warrants the development of inflammasome‐targeting therapies for clinical use.
Table 2.
Overview of pharmacological compounds targeting inflammasome signaling
| Compounds | Benefit (+)/detriment (−) | Neurodegenerative disease model |
|---|---|---|
| Caspase‐1 inhibitors | ||
| VX‐765 and VX‐740 | (−) No further development after phase II clinical trial for use in epilepsy and psoriasis (Mangan et al, 2018) |
Alzheimer disease (Flores et al, 2018) EAE (Mckenzie et al, 2018) |
| NLRP3 inflammasome inhibitors | ||
| Sulfonylurea‐based compounds | ||
| Glyburide |
(+) Specific for NLRP3 inflammasomes; significantly delays LPS‐induced mortality (Lamkanfi et al, 2009) (−) High dosage required (Marchetti et al, 2014); cardiovascular side effects (Riddle, 2003) |
TBI (Simard et al, 2012) Ischemic stroke (Simard et al, 2012) |
| CP‐412,245 and CP‐424,174 | (+) Oral administration of CP‐424,174 selectively blocks IL‐1 production in mice (Perregaux et al, 2001) | |
| CRID1 and CRID2 | (−) No in vivo evidence | |
| MCC950 (also known as CRID3 or CP‐456,773) |
(+) NLRP3 inflammasome specific (Coll et al, 2015); inhibits NLRP3 activation by all known stimuli (Mangan et al, 2018) (−) Precise molecular target unknown |
Alzheimer disease (Dempsey et al, 2017) EAE (Coll et al, 2015) TBI (Ismael et al, 2018a; Xu et al, 2018) Stroke (Ismael et al, 2018b; Ren et al, 2018) |
| 16673‐34‐0 | (+) Lacks cyclohexylurea group responsible for hypoglycemic activity; prevents NLRP‐mediated myocardial injury (Marchetti et al, 2014) | |
| Hybrid molecules (combining MCC950 and glyburide) | (−) Moderately effective at inhibiting NLRP3 compared to MCC950 (Hill et al, 2017) | |
| Fenamate classes of NSAIDs | ||
| Flufenamic and mefenamic acid |
(+) Inhibits NLRP3 by blocking VRACs (Daniels et al, 2016) (−) Lack of specificity, inhibits multiple inflammatory nodes (Daniels et al, 2016) |
Alzheimer disease (Daniels et al, 2016) |
| Michael acceptors | ||
| Parthenolide | (−) No suitable pharmacological properties (Baldwin et al, 2016); inhibits NF‐κB‐dependent signaling (Saadane et al, 2007) | Stroke (Dong et al, 2013) |
| BAY 11‐7082 | (−) Not specific; inhibits NF‐κB‐dependent signaling (Lee et al, 2012) | TBI (Irrera et al, 2017) |
| 3,4‐methylenedioxy‐β‐nitrostyrene (MNS) | (−) Modest potency (Baldwin et al, 2016) | |
| Acrylate and acrylamide derivatives (ex. IFN58, IFN39) | (+) Oral administration of IFN39 alleviates DNBS‐induced colitis in rats (Cocco et al, 2017) | |
| Novel boron compound series | ||
| NBC13 | (+) Significantly decreases LPS‐induced IL‐1β production in vivo (Baldwin et al, 2017) | |
| Other NLRP3 inhibitors | ||
| Fc11a‐2 | (+) In vivo efficacy in DSS‐induced colitis (Liu et al, 2013b) | |
| CY‐09 | (+) Therapeutic effect in mouse models of CAPS and type 2 diabetes (Jiang et al, 2017) | |
| JC‐171 | (+) Treatment effective in EAE in both prophylactic and therapeutic settings (Guo et al, 2017) | EAE (Guo et al, 2017) |
| OLT‐177 | (+) No adverse effects in preliminary clinical testing of healthy humans (Marchetti et al, 2018) | |
| β‐hydroxybutyrate (BHB) | (−) Not specific for NLRP3; can inhibit HDACs (Youm et al, 2015) | |
| GSDMD inhibitors | ||
| Antabuse | (+) Efficacious in sepsis models (preprint: Hu et al, 2018) | |
| Necrosulfonamide | (+) Efficacious in sepsis models (Rathkey et al, 2018) | |
Pyroptosis is emerging as another key mechanism that contributes to inflammasome‐driven pathology in metabolic, autoimmune, and neurodegenerative diseases. Cleavage of GSDMD by the inflammatory caspase‐1 and caspase‐11 (mouse), or caspase‐1, caspase‐4, and caspase‐5 (human) releases an amino‐terminal pore‐forming GSDMD domain that oligomerizes and inserts in the plasma membrane, inducing membrane rupture and leakage of the cytosolic contents (Vande Walle & Lamkanfi, 2016). Hence, GSDMD inhibition has been proposed as a novel therapeutic strategy to prevent inflammasome‐driven pathology in different diseases. Pyroptosis has recently been shown to be the critical mechanism of IL‐1β‐mediated systemic pathology in the autoinflammatory disease Familial Mediterranean Fever (FMF), which is caused by missense mutations in Mefv that activates the pyrin inflammasome (Kanneganti et al, 2018). Similarly, GSDMD was shown to control inflammasome‐driven pathology in neonatal‐onset multisystem inflammatory disease (NOMID), which is caused by activating mutations in the inflammasome sensor NLRP3 (Xiao et al, 2018). Indeed, FMF knockin mice and NOMID mice were shown to be fully protected from developing the respective autoinflammatory syndromes when crossed into a Gsdmd‐deficient background (Kanneganti et al, 2018; Xiao et al, 2018). GSDMD was also demonstrated to play a key role in the pathogenesis of steatohepatitis by controlling cytokine secretion, and Gsdmd knockout mice exhibit decreased severity of steatosis and inflammation in a high‐fat diet‐induced model of non‐alcoholic steatohepatitis (Xu et al, 2017). Pharmacologic inhibition of GSDMD by necrosulfonamide also was efficacious in sepsis models (Rathkey et al, 2018). Finally, antabuse (disulfiram), a drug used to treat alcohol addiction, was shown to inhibit GSDMD pore formation and IL‐1β secretion in human and mouse cells, and LPS‐induced septic death and IL‐1β secretion in mice (preprint: Hu et al, 2018). Together, these observations suggest that inhibition of GSDMD‐dependent pyroptosis may also hold promise for the treatment of inflammasome‐mediated neuroinflammatory pathology.
Concluding remarks and future perspectives
Although only discovered in 2002 (Martinon et al, 2002), inflammasomes are now widely regarded as central regulators of innate immunity playing an indispensable role in the host defense against stress conditions and pathogens that might be harmful to the host. However, inflammasome activation leading to excessive or prolonged inflammation can also contribute to a damaging and destructive environment resulting in the development of inflammatory diseases, including neurodegenerative diseases (Lamkanfi & Dixit, 2012).
Most research in the field has focused on the role of the NLRP3 inflammasome, largely neglecting the potential importance of other inflammasome complexes in CNS disorders. Future research will have to investigate and characterize less well‐known inflammasomes and expand the understanding of inflammasome signaling and its importance for neurodegeneration. The generation of new mouse models and reagents that allow selective targeting of the inflammatory mechanisms initiated by these inflammasomes will be crucial to this end. Also, although the importance of inflammasome activation has been demonstrated in many neurodegenerative diseases and experimental model systems, their specific role in the different CNS cell types is not always clear. Most research has focused on microglia, but other brain cell types also have functional inflammasomes which may contribute to disease pathology (Walsh et al, 2014). Since most studies published so far have investigated inflammasome activation using conventional “full body” knockout mice which do not allow to specify the relevant cell types involved, cell type‐specific inflammasome mutant mice will be needed to address the importance of inflammasome signaling in CNS‐specific cell types, assessing their importance for cytokine production, pyroptosis, and CNS inflammation. Also, new techniques including single‐cell RNA sequencing and mass cytometry, in combination with high‐resolution microscopy and in vivo cell imaging, should help to get a better view on the spectrum of inflammasomes that is expressed in the different CNS‐resident cell types in different (pathological) conditions.
Major progress needs to be made in translating findings from animal studies to humans. However, key differences exist between human and mouse inflammasomes. Humans have only a single NLRP1 gene, while mice have three isoforms of Nlrp1. Similarly, humans have two orthologues for caspase‐11, viz. caspase‐4 and caspase‐5 (Lamkanfi & Dixit, 2012). More research using human cell types, eventually based on iPS technology and patient‐derived material, will be needed to translate findings more effectively from rodents to the human conditions they model.
Finally, since inflammasome signaling is central to many neurodegenerative diseases, targeting their activation is regarded as a potential therapy to treat these diseases. Targeting the downstream IL‐1β and IL‐18 cytokines is probably not the best approach since these cytokines are needed for optimal immunity in the host. Moreover, cytokine‐targeting biologics have limited capacity to penetrate the blood–brain barrier. Therefore, modulating the activity of specific inflammasome sensors and cell type‐specific inflammasome targeting with small molecules will be vital in this context and may have better potential as therapies to treat patients that suffer from neurodegenerative diseases (Guo et al, 2015). Also, pyroptosis is now being identified as a critical mechanism driving inflammatory pathology, suggesting GSDMD inhibition as a potential anti‐inflammatory strategy (Kanneganti et al, 2018; Mckenzie et al, 2018; Xiao et al, 2018).
Conflict of interest
The authors declare that they have no conflict of interest.
Pending issues.
The cell type‐specific contribution of inflammasome signaling to neurodegenerative diseases, both peripheral and CNS resident.
Direct evidence for inflammasome activation and signaling in patients with neurodegenerative disease.
Characterization of the central activation mechanism and high‐resolution structure of inflammasome proteins.
Development of compounds able to cross the BBB and that selectively inhibits those inflammasomes that contribute to the pathogenicity.
Acknowledgements
S.V. is a predoctoral fellow with the Institute for the Promotion of Innovation by Science and Technology (IWT) and is supported by grants from the Charcot Foundation and the Queen Elisabeth Medical Foundation. S.S. is a predoctoral fellow with the FWO and is supported by a “Hope in Head” Rotary grant. Work in the G.v.L. laboratory is supported by research grants from the FWO, the Queen Elisabeth Medical Foundation, the CBC Banque Prize, the Charcot Foundation, the “Belgian Foundation Against Cancer”, and the “Concerted Research Actions” (GOA) of the Ghent University. M.L. is supported by European Research Council Grant 683144 (PyroPop).
EMBO Mol Med (2019) 11: e10248
See the Glossary for abbreviations used in this article.
Contributor Information
Mohamed Lamkanfi, Email: mlamkanf@its.jnj.com.
Geert van Loo, Email: geert.vanloo@irc.vib-ugent.be.
References
- Adamczak S, Dale G, de Rivero Vaccari JP, Bullock MR, Dietrich WD, Keane RW (2012) Inflammasome proteins in cerebrospinal fluid of brain‐injured patients as biomarkers of functional outcome. J Neurosurg 117: 1119–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alboni S, Cervia D, Sugama S, Conti B (2010) Interleukin 18 in the CNS. J Neuroinflammation 7: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan SM, Tyrrell PJ, Rothwell NJ (2005) Interleukin‐1 and neuronal injury. Nat Rev Immunol 5: 629–640 [DOI] [PubMed] [Google Scholar]
- Baecher‐Allan C, Kaskow BJ, Weiner HL (2018) Multiple sclerosis: mechanisms and immunotherapy. Neuron 97: 742–768 [DOI] [PubMed] [Google Scholar]
- Baldwin AG, Brough D, Freeman S (2016) Inhibiting the inflammasome: a chemical perspective. J Med Chem 59: 1691–1710 [DOI] [PubMed] [Google Scholar]
- Baldwin AG, Rivers‐Auty J, Daniels MJD, White CS, Schwalbe CH, Schilling T, Hammadi H, Jaiyong P, Spencer NG, England H et al (2017) Boron‐based inhibitors of the NLRP3 inflammasome. Cell Chem Biol 24: 1321–1335.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroja‐Mazo A, Martín‐Sánchez F, Gomez AI, Martínez CM, Amores‐Iniesta J, Compan V, Barberà‐Cremades M, Yagüe J, Ruiz‐Ortiz E, Antón J et al (2014) The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15: 738–748 [DOI] [PubMed] [Google Scholar]
- Barrington J, Lemarchand E, Allan SM (2017) A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol 27: 205–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertani I, Iori V, Trusel M, Maroso M, Foray C, Mantovani S, Tonini R, Vezzani A, Chiesa R (2017) Inhibition of IL‐1β signaling normalizes NMDA‐dependent neurotransmission and reduces seizure susceptibility in a mouse model of Creutzfeldt‐Jakob disease. J Neurosci 37: 10278–10289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ (2001) Role of IL‐1α and IL‐1β in ischemic brain damage. J Neurosci 21: 5528–5534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ED, Dietrich WF (2006) Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38: 240–244 [DOI] [PubMed] [Google Scholar]
- Brickler T, Gresham K, Meza A, Coutermarsh‐Ott S, Williams TM, Rothschild DE, Allen IC, Theus MH (2016) Nonessential role for the NLRP1 inflammasome complex in a murine model of traumatic brain injury. Mediators Inflamm 2016: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, von Moltke J, Jones JW, Vance RE, Monack DM (2010) Differential requirement for caspase‐1 autoproteolysis in pathogen‐induced cell death and cytokine processing. Cell Host Microbe 8: 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16: 407–420 [DOI] [PubMed] [Google Scholar]
- Caron NS, Dorsey ER, Hayden MR (2018) Therapeutic approaches to Huntington disease: from the bench to the clinic. Nat Rev Drug Discov 17: 729–750 [DOI] [PubMed] [Google Scholar]
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM et al (2000) Minocycline inhibits caspase‐1 and caspase‐3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6: 797–801 [DOI] [PubMed] [Google Scholar]
- Clausen F, Hånell A, Björk M, Hillered L, Mir AK, Gram H, Marklund N (2009) Neutralization of interleukin‐1β modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci 30: 385–396 [DOI] [PubMed] [Google Scholar]
- Cocco M, Pellegrini C, Martínez‐Banaclocha H, Giorgis M, Marini E, Costale A, Miglio G, Fornai M, Antonioli L, López‐Castejón G et al (2017) Development of an acrylate derivative targeting the NLRP3 inflammasome for the treatment of inflammatory bowel disease. J Med Chem 60: 3656–3671 [DOI] [PubMed] [Google Scholar]
- Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M (2013) Triggering of inflammasome by aggregated α‐synuclein, an inflammatory response in synucleinopathies. PLoS ONE 8: e55375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz‐Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A et al (2015) A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Hirsch E, Van Eldik LJ (2005) Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human β‐amyloid. J Neuroinflammation 2: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels MJD, Rivers‐Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, Booth SJ, White CS, Baldwin AG, Freeman S et al (2016) Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer's disease in rodent models. Nat Commun 7: 12504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P, Smithson LA, Price RW, Holloway VM, Levites Y, Chakrabarty P, Golde TE (2006) Interleukin‐1 receptor 1 knockout has no effect on amyloid deposition in Tg2576 mice and does not alter efficacy following Aβ immunotherapy. J Neuroinflammation 3: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Karran E (2016) The cellular phase of Alzheimer's disease. Cell 164: 603–615 [DOI] [PubMed] [Google Scholar]
- Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, Robertson AAB, Cooper MA, O'Neill LAJ, Lynch MA (2017) Inhibiting the NLRP3 inflammasome with MCC950 promotes non‐phlogistic clearance of amyloid‐β and cognitive function in APP/PS1 mice. Brain Behav Immun 61: 306–316 [DOI] [PubMed] [Google Scholar]
- Denes A, Coutts G, Lénárt N, Cruickshank SM, Pelegrin P, Skinner J, Rothwell N, Allan SM, Brough D (2015) AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc Natl Acad Sci USA 112: 4050–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, Simon A, van der Meer JWM (2012) Treating inflammation by blocking interleukin‐1 in a broad spectrum of diseases. Nat Rev Drug Discov 11: 633–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Qiao H, Zhang X, Zhang X, Wang C, Wang L, Cui L, Zhao J, Xing Y, Li Y et al (2013) Parthenolide is neuroprotective in rat experimental stroke model: downregulating NF‐κB, phospho‐p38MAPK, and caspase‐1 and ameliorating BBB permeability. Mediators Inflamm 2013: 370804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fann DY‐W, Lee S‐Y, Manzanero S, Tang S‐C, Gelderblom M, Chunduri P, Bernreuther C, Glatzel M, Cheng Y‐L, Thundyil J et al (2013) Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome‐mediated neuronal death in ischemic stroke. Cell Death Dis 4: e790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari CC, Pott Godoy MC, Tarelli R, Chertoff M, Depino AM, Pitossi FJ (2006) Progressive neurodegeneration and motor disabilities induced by chronic expression of IL‐1β in the substantia nigra. Neurobiol Dis 24: 183–193 [DOI] [PubMed] [Google Scholar]
- Flores J, Noël A, Foveau B, Lynham J, Lecrux C, LeBlanc AC (2018) Caspase‐1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer's disease mouse model. Nat Commun 9: 3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox E, Jayaprakash N, Pham T‐H, Rowley A, McCully CL, Pucino F, Goldbach‐Mansky R (2010) The serum and cerebrospinal fluid pharmacokinetics of anakinra after intravenous administration to non‐human primates. J Neuroimmunol 223: 138–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Amer A, Body‐Malapel M, Kanneganti T‐D, Özören N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A et al (2006) Cytosolic flagellin requires Ipaf for activation of caspase‐1 and interleukin 1β in salmonella‐infected macrophages. Nat Immunol 7: 576–582 [DOI] [PubMed] [Google Scholar]
- Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP‐Y (2017) NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med 214: 1351–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA, Yuan J (1997) Expression of a dominant negative mutant of interleukin‐1β converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med 185: 933–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, Desina G, Comi G, Flavell R, Su MS et al (1999) Caspase‐1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol 163: 2403–2409 [PubMed] [Google Scholar]
- Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O'Banion MK (2013) Sustained interleukin‐1 overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer's mouse model. J Neurosci 33: 5053–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Pan J, Shen Q, Li M, Peng Y (2018) Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J Neuroinflammation 15: 242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granowitz EV, Porat R, Mier JW, Pribble JP, Stiles DM, Bloedow DC, Catalano MA, Wolff SM, Dinarello CA (1992) Pharmacokinetics, safety and immunomodulatory effects of human recombinant interleukin‐1 receptor antagonist in healthy humans. Cytokine 4: 353–360 [DOI] [PubMed] [Google Scholar]
- Griffin WST, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, Araoz C (1989) Brain interleukin 1 and S‐100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86: 7611–7615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, Huang M, Schneider M, Miller SD, Ting JP‐Y (2010) NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol 185: 974–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guey B, Bodnar M, Manié SN, Tardivel A, Petrilli V (2014) Caspase‐1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc Natl Acad Sci USA 111: 17254–17259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gugliandolo A, Giacoppo S, Bramanti P, Mazzon E (2018) NLRP3 inflammasome activation in a transgenic amyotrophic lateral sclerosis model. Inflammation 41: 93–103 [DOI] [PubMed] [Google Scholar]
- Guo H, Callaway JB, Ting JP‐Y (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Fulp JW, Jiang Y, Li X, Chojnacki JE, Wu J, Wang X‐Y, Zhang S (2017) Development and characterization of a hydroxyl‐sulfonamide analogue, 5‐Chloro‐N‐[2‐(4‐hydroxysulfamoyl‐phenyl)‐ethyl]‐2‐methoxy‐benzamide, as a novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem Neurosci 8: 2194–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, Heurtaux T, Tardivel A, Heuschling P, Dostert C (2015) NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS ONE 10: e0130624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustot A, Gallea JI, Sarroukh R, Celej MS, Ruysschaert J‐M, Raussens V (2015) Amyloid fibrils are the molecular trigger of inflammation in Parkinson's disease. Biochem J 471: 323–333 [DOI] [PubMed] [Google Scholar]
- Hafner‐Bratkovič I, Benčina M, Fitzgerald KA, Golenbock D, Jerala R (2012) NLRP3 inflammasome activation in macrophage cell lines by prion protein fibrils as the source of IL‐1β and neuronal toxicity. Cell Mol Life Sci 69: 4215–4228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald K, Latz E, Moore K, Golenbock D (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid‐β. Nat Immunol 9: 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu‐Sasamata M, Yuan J, Moskowitz MA (1997) Inhibition of interleukin 1β converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci USA 94: 2007–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira‐Saecker A, Griep A, Axt D, Remus A, Tzeng T‐C et al (2013) NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493: 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14: 463–477 [DOI] [PubMed] [Google Scholar]
- Heneka MT, McManus RM, Latz E (2018) Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 19: 610–621 [DOI] [PubMed] [Google Scholar]
- Hill JR, Coll RC, Sue N, Reid JC, Dou J, Holley CL, Pelingon R, Dickinson JB, Biden TJ, Schroder K et al (2017) Sulfonylureas as concomitant insulin secretagogues and NLRP3 inflammasome inhibitors. ChemMedChem 12: 1449–1457 [DOI] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 458: 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JJ, Liu X, Zhao J, Xia S, Ruan J, Luo X, Kim J, Lieberman J, Wu H (2018) Identification of pyroptosis inhibitors that target a reactive cysteine in gasdermin D. bioRxiv 10.1101/365908 [PREPRINT] [DOI]
- Iłżecka J, Stelmasiak Z, Dobosz B (2001) Interleukin‐1β converting enzyme/Caspase‐1 (ICE/Caspase‐1) and soluble APO‐1/Fas/CD95 receptor in amyotrophic lateral sclerosis patients. Acta Neurol Scand 103: 255–258 [PubMed] [Google Scholar]
- Inoue M, Williams KL, Gunn MD, Shinohara ML (2012a) NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 109: 10480–10485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan J V, Miao EA, Shinohara ML (2012b) Interferon‐β therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal 5: ra38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Shinohara ML (2013) NLRP3 inflammasome and MS/EAE. Autoimmune Dis 2013: 859145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Chen P‐H, Siecinski S, Li Q‐J, Liu C, Steinman L, Gregory SG, Benner E, Shinohara ML (2016) An interferon‐β‐resistant and NLRP3 inflammasome‐independent subtype of EAE with neuronal damage. Nat Neurosci 19: 1599–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irrera N, Pizzino G, Calò M, Pallio G, Mannino F, Famà F, Arcoraci V, Fodale V, David A, Francesca C et al (2017) Lack of the Nlrp3 inflammasome improves mice recovery following traumatic brain injury. Front Pharmacol 8: 459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismael S, Nasoohi S, Ishrat T (2018a) MCC950, the selective inhibitor of nucleotide oligomerization domain‐like receptor protein‐3 inflammasome, protects mice against traumatic brain injury. J Neurotrauma 35: 1294–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismael S, Zhao L, Nasoohi S, Ishrat T (2018b) Inhibition of the NLRP3‐inflammasome as a potential approach for neuroprotection after stroke. Sci Rep 8: 5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, Chen VS, Gris D, Matsushima GK, Ting JP‐Y (2010) The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase‐1 and interleukin‐18. J Neurosci 30: 15811–15820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, Wang A, Tao J, Wang C, Liu Q et al (2017) Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med 214: 3219–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, Troost D, Beyer C (2015) NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63: 2260–2273 [DOI] [PubMed] [Google Scholar]
- Kang S‐J, Wang S, Hara H, Peterson EP, Namura S, Amin‐Hanjani S, Huang Z, Srinivasan A, Tomaselli KJ, Thornberry NA et al (2000) Dual role of caspase‐11 in mediating activation of caspase‐1 and caspase‐3 under pathological conditions. J Cell Biol 149: 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti A, Malireddi RKS, Saavedra PHV, Vande Walle L, Van Gorp H, Kambara H, Tillman H, Vogel P, Luo HR, Xavier RJ et al (2018) GSDMD is critical for autoinflammatory pathology in a mouse model of familial mediterranean fever. J Exp Med 215: 1519–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, Ghetti B, Koller BH, LeBlanc AC (2015) Neuronal NLRP1 inflammasome activation of Caspase‐1 coordinately regulates inflammatory interleukin‐1‐beta production and axonal degeneration‐associated Caspase‐6 activation. Cell Death Differ 22: 1676–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawana N, Yamamoto Y, Ishida T, Saito Y, Konno H, Arima K, Satoh JI (2013) Reactive astrocytes and perivascular macrophages express NLRP3 inflammasome in active demyelinating lesions of multiple sclerosis and necrotic lesions of neuromyelitis optica and cerebral infarction. Clin Exp Neuroimmunol 4: 296–304 [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S et al (2011) Non‐canonical inflammasome activation targets caspase‐11. Nature 479: 117–121 [DOI] [PubMed] [Google Scholar]
- Keane RW, Dietrich WD, de Rivero Vaccari JP (2018) Inflammasome proteins as biomarkers of multiple sclerosis. Front Neurol 9: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M, Yao H, Shah SZA, Wu W, Wang D, Zhao Y, Wang L, Zhou X, Zhao D, Yang L (2018) The NLRP3‐Caspase 1 inflammasome negatively regulates autophagy via TLR4‐TRIF in prion peptide‐infected microglia. Front Aging Neurosci 10: 116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM (2009) Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 187: 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM (2012) Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28: 137–161 [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM (2014) Mechanisms and functions of inflammasomes. Cell 157: 1013–1022 [DOI] [PubMed] [Google Scholar]
- Lammerding L, Slowik A, Johann S, Beyer C, Zendedel A (2016) Poststroke inflammasome expression and regulation in the peri‐infarct area by gonadal steroids after transient focal ischemia in the rat brain. Neuroendocrinology 103: 460–475 [DOI] [PubMed] [Google Scholar]
- Lampron A, ElAli A, Rivest S (2013) Innate immunity in the CNS: redefining the relationship between the CNS and its environment. Neuron 78: 214–232 [DOI] [PubMed] [Google Scholar]
- Lee J, Rhee MH, Kim E, Cho JY (2012) BAY 11‐7082 is a broad‐spectrum inhibitor with anti‐inflammatory activity against multiple targets. Mediators Inflamm 2012: 416036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann S, Esch E, Hartmann P, Goswami A, Nikolin S, Weis J, Beyer C, Johann S (2018) Expression profile of pattern recognition receptors in skeletal muscle of SOD1(G93A) amyotrophic lateral sclerosis (ALS) mice and sporadic ALS patients. Neuropathol Appl Neurobiol 44: 606–627 [DOI] [PubMed] [Google Scholar]
- Lévesque SA, Paré A, Mailhot B, Bellver‐Landete V, Kébir H, Lécuyer M‐A, Alvarez JI, Prat A, de Rivero Vaccari JP, Keane RW et al (2016) Myeloid cell transmigration across the CNS vasculature triggers IL‐1β–driven neuroinflammation during autoimmune encephalomyelitis in mice. J Exp Med 213: 929–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W‐P, Xiong G‐P, Lin Q, Chen X‐W, Zhang L‐Q, Shi J‐X, Ke Q‐F, Lin J‐H (2016) Heme oxygenase‐1 promotes neuron survival through down‐regulation of neuronal NLRP1 expression after spinal cord injury. J Neuroinflammation 13: 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H‐D, Li W, Chen Z‐R, Hu Y‐C, Zhang D‐D, Shen W, Zhou M‐L, Zhu L, Hang C‐H (2013a) Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res 38: 2072–2083 [DOI] [PubMed] [Google Scholar]
- Liu W, Guo W, Wu J, Luo Q, Tao F, Gu Y, Shen Y, Li J, Tan R, Xu Q et al (2013b) A novel benzo[d]imidazole derivate prevents the development of dextran sulfate sodium‐induced murine experimental colitis via inhibition of NLRP3 inflammasome. Biochem Pharmacol 85: 1504–1512 [DOI] [PubMed] [Google Scholar]
- Liu L, Chan C (2014) IPAF inflammasome is involved in interleukin‐1β production from astrocytes, induced by palmitate; implications for Alzheimer's disease. Neurobiol Aging 35: 309–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J (2014) NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol 75: 209–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie SH, Schipper JL, Clark AC (2010) The potential for caspases in drug discovery. Curr Opin Drug Discov Devel 13: 568–576 [PMC free article] [PubMed] [Google Scholar]
- Maier A, Deigendesch N, Müller K, Weishaupt JH, Krannich A, Röhle R, Meissner F, Molawi K, Münch C, Holm T et al (2015) Interleukin‐1 antagonist anakinra in amyotrophic lateral sclerosis‐A pilot study. PLoS ONE 10: e0139684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamik MK, Power C (2017) Inflammasomes in neurological diseases: emerging pathogenic and therapeutic concepts. Brain 140: 2273–2285 [DOI] [PubMed] [Google Scholar]
- Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E (2018) Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 17: 588–606 [DOI] [PubMed] [Google Scholar]
- Mao Z, Liu C, Ji S, Yang Q, Ye H, Han H, Xue Z (2017) The NLRP3 inflammasome is involved in the pathogenesis of Parkinson's disease in rats. Neurochem Res 42: 1104–1115 [DOI] [PubMed] [Google Scholar]
- Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, Federici M, Van Tassell BW, Zhang S, Abbate A (2014) A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury following ischemia‐reperfusion in the mouse. J Cardiovasc Pharmacol 63: 316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, Carta S, Tengesdal I, Nemkov T, D'Alessandro A et al (2018) OLT1177, a β‐sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci USA 115: E1530–E1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐β. Mol Cell 10: 417–426 [DOI] [PubMed] [Google Scholar]
- Mckenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, Lu J‐Q, Branton WG, Power C (2018) Caspase‐1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci USA 115: E6065–E6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner F, Molawi K, Zychlinsky A (2010) Mutant superoxide dismutase 1‐induced IL‐1β accelerates ALS pathogenesis. Proc Natl Acad Sci USA 107: 10346–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Alpuche‐Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A (2006) Cytoplasmic flagellin activates caspase‐1 and secretion of interleukin 1β via Ipaf. Nat Immunol 7: 569–575 [DOI] [PubMed] [Google Scholar]
- Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A (2010) Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107: 3076–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T (1994) Interleukin‐1β, interleukin‐6, epidermal growth factor and transforming growth factor‐α are elevated in the brain from parkinsonian patients. Neurosci Lett 180: 147–150 [DOI] [PubMed] [Google Scholar]
- Moran A, Bundy B, Becker DJ, Dimeglio LA, Gitelman SE, Goland R, Greenbaum CJ, Herold KC, Marks JB, Raskin P et al (2013) Interleukin‐1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double‐blind, placebo‐controlled trials. Lancet 381: 1905–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton‐Liger F, Rosazza T, Sepulveda‐Diaz J, Ieang A, Hassoun S‐M, Claire E, Mangone G, Brice A, Michel PP, Corvol J‐C et al (2018) Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20‐dependent negative feedback loop. Glia 66: 1736–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuvolone M, Sorce S, Schwarz P, Aguzzi A (2015) Prion pathogenesis in the absence of NLRP3/ASC inflammasomes. PLoS ONE 10: e0117208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li X‐J, Stieg PE et al (1999) Inhibition of caspase‐1 slows disease progression in a mouse model of Huntington's disease. Nature 399: 263–267 [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Borchelt DR, Houseweart MK, Cleveland DW, Brown RHJ (1998) Caspase‐1 is activated in neural cells and tissue with amyotrophic lateral sclerosis‐associated mutations in copper‐zinc superoxide dismutase. Proc Natl Acad Sci USA 95: 15763–15768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perregaux DG, Mcniff P, Laliberte R, Hawryluk N, Peurano H, Stam E, Eggler J, Griffiths R, Dombroski MA, Gabel CA (2001) Identification and characterization of a novel class of interleukin‐1 post‐translational processing inhibitors. J Pharmacol Exp Ther 299: 187–197 [PubMed] [Google Scholar]
- Pontillo A, Catamo E, Arosio B, Mari D, Crovella S (2012) NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis Assoc Disord 26: 277–281 [DOI] [PubMed] [Google Scholar]
- Przedborski S (2017) The two‐century journey of Parkinson disease research. Nat Rev Neurosci 18: 251–259 [DOI] [PubMed] [Google Scholar]
- Rabuffetti M, Sciorati C, Tarozzo G, Clementi E, Manfredi AA, Beltramo M (2000) Inhibition of caspase‐1‐like activity by Ac‐Tyr‐Val‐Ala‐Asp‐chloromethyl ketone induces long‐lasting neuroprotection in cerebral ischemia through apoptosis reduction and decrease of proinflammatory cytokines. J Neurosci 20: 4398–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM (2012) Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci 15: 1074–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR et al (2018) Chemical disruption of the pyroptotic pore‐forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol 3: eaat2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Kong Y, Liu Z, Zang D, Yang X, Wood K, Li M, Liu Q (2018) Selective NLRP3 (pyrin domain‐containing protein 3) inflammasome inhibitor reduces brain injury after intracerebral hemorrhage. Stroke 49: 184–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle M (2003) Editorial: sulfonylureas differ in effects on ischemic preconditioning‐is it time to retire glyburide? J Clin Endocrinol Metab 88: 528–530 [DOI] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD et al (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377: 1119–1131 [DOI] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW (2008) A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci 28: 3404–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross J, Brough D, Gibson RM, Loddick SA, Rothwell NJ (2007) A selective, non‐peptide caspase‐1 inhibitor, VRT‐018858, markedly reduces brain damage induced by transient ischemia in the rat. Neuropharmacology 53: 638–642 [DOI] [PubMed] [Google Scholar]
- Rossi‐Semerano L, Fautrel B, Wendling D, Hachulla E, Galeotti C, Semerano L, Touitou I, Koné‐Paut I & Inflammation) M (Maladies A et A‐I‐1) study G on the behalf of C Club R et (2015) Tolerance and efficacy of off‐label anti‐interleukin‐1 treatments in France: a nationwide survey. Orphanet J Rare Dis 10: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadane A, Masters S, Didonato J, Li J, Berger M (2007) Parthenolide inhibits IκB kinase, NF‐κB activation, and inflammatory response in cystic fibrosis cells and mice. Am J Respir Cell Mol Biol 36: 728–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala Frigerio C, De Strooper B (2016) Alzheimer's disease mechanisms and emerging roads to novel therapeutics. Annu Rev Neurosci 39: 57–79 [DOI] [PubMed] [Google Scholar]
- Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, Rainone V, Nemni R, Mancuso R, Clerici M (2016) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer's disease. Mol Neurodegener 11: 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarlus H, Heneka MT (2017) Microglia in Alzheimer's disease. J Clin Invest 127: 3240–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schielke GP, Yang GY, Shivers BD, Betz AL (1998) Reduced ischemic brain injury in interleukin‐1β converting enzyme‐deficient mice. J Cereb Blood Flow Metab 18: 180–185 [DOI] [PubMed] [Google Scholar]
- Schultz J, Schwarz A, Neidhold S, Burwinkel M, Riemer C, Simon D, Kopf M, Otto M, Baier M (2004) Role of interleukin‐1 in prion disease‐associated astrocyte activation. Am J Pathol 165: 671–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, Olschowka JA, Miller JH, Johnson RE, O'Banion MK (2007) Sustained hippocampal IL‐1β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest 117: 1595–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Ito K, Skinner RD, Mrak RE, Rovnaghi CR, Van Eldik LJ, Griffin WST (1996) In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin‐1 as a driving force in Alzheimer pathogenesis. Neurobiol Aging 17: 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth KN, Kimberly WT, Elm JJ, Kent TA, Mandava P, Yoo AJ, Thomalla G, Campbell B, Donnan GA, Davis SM et al (2014) Pilot study of intravenous glyburide in patients with a large ischemic stroke. Stroke 45: 281–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Yang L, Kouadir M, Yang Y, Wang J, Zhou X, Yin X, Zhao D (2012) The NALP3 inflammasome is involved in neurotoxic prion peptide‐induced microglial activation. J Neuroinflammation 9: 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Gao W, Shao F (2017) Pyroptosis: gasdermin‐mediated programmed necrotic cell death. Trends Biochem Sci 42: 245–254 [DOI] [PubMed] [Google Scholar]
- Shi Y, Holtzman DM (2018) Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol 18: 759–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdson CJ, Bartz JC, Glatzel M (2018) Cellular and molecular mechanisms of prion disease. Annu Rev Pathol 14: 497–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien J‐P, Rivest S (2006) Bone marrow‐derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49: 489–502 [DOI] [PubMed] [Google Scholar]
- Simard JM, Woo SK, Schwartzbauer GT, Gerzanich V (2012) Sulfonylurea receptor 1 in central nervous system injury: a focused review. J Cereb Blood Flow Metab 32: 1699–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton C, Brereton C, Keogh B, Mills KHG, Lavelle EC (2006) A crucial role for interleukin (IL)‐1 in the induction of IL‐17–producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 203: 1685–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M‐S, Tan L, Jiang T, Zhu X‐C, Wang H‐F, Jia C‐D, Yu J‐T (2014) Amyloid‐β induces NLRP1‐dependent neuronal pyroptosis in models of Alzheimer's disease. Cell Death Dis 5: e1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Brown RHJ, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539: 197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S‐J (2017) Effects of interleukin‐1beta polymorphisms on brain function and behavior in healthy and psychiatric disease conditions. Cytokine Growth Factor Rev 37: 89–97 [DOI] [PubMed] [Google Scholar]
- Tzeng T‐C, Hasegawa Y, Iguchi R, Cheung A, Caffrey DR, Thatcher EJ, Mao W, Germain G, Tamburro ND, Okabe S et al (2018) Inflammasome‐derived cytokine IL18 suppresses amyloid‐induced seizures in Alzheimer‐prone mice. Proc Natl Acad Sci USA 115: 9002–9007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Everbroeck B, Dewulf E, Pals P, Lübke U, Martin JJ, Cras P (2002) The role of cytokines, astrocytes, microglia and apoptosis in Creutzfeldt‐Jakob disease. Neurobiol Aging 23: 59–64 [DOI] [PubMed] [Google Scholar]
- Van Gorp H, Van Opdenbosch N, Lamkanfi M (2019) Inflammasome‐dependent cytokines at the crossroads of health and autoinflammatory disease. Cold Spring Harb Perspect Biol 11: pii: a028563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti T‐D, Lamkanfi M (2014) Activation of the NLRP1b inflammasome independently of ASC‐mediated caspase‐1 autoproteolysis and speck formation. Nat Commun 5: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, Beyaert R, Elewaut D, Kanneganti T‐D, van Loo G et al (2014) Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512: 69–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Walle L, Lamkanfi M (2016) Pyroptosis. Curr Biol 26: R568–R572 [DOI] [PubMed] [Google Scholar]
- Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira‐Saecker A, Schwartz S, Santarelli F, Kummer MP et al (2017) Microglia‐derived ASC specks cross‐seed amyloid‐β in Alzheimer's disease. Nature 552: 355–361 [DOI] [PubMed] [Google Scholar]
- Voet S, Mc Guire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P, Daems C, Staszewski O, Walle LV, Jordao MJC et al (2018) A20 critically controls microglia activation and inhibits inflammasome‐dependent neuroinflammation. Nat Commun 9: 2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallisch JS, Simon DW, Bayır H, Bell MJ, Kochanek PM, Clark RSB (2017) Cerebrospinal fluid NLRP3 is increased after severe traumatic brain injury in infants and children. Neurocrit Care 27: 44–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JG, Muruve DA, Power C (2014) Inflammasomes in the CNS. Nat Rev Neurosci 15: 84–97 [DOI] [PubMed] [Google Scholar]
- Wang W, Nguyen LTT, Burlak C, Chegini F, Guo F, Chataway T, Ju S, Fisher OS, Miller DW, Datta D et al (2016) Caspase‐1 causes truncation and aggregation of the Parkinson's disease‐associated protein α‐synuclein. Proc Natl Acad Sci USA 113: 9587–9592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M et al (1998) Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem 273: 9158–9167 [DOI] [PubMed] [Google Scholar]
- Wheeler RD, Boutin H, Touzani O, Luheshi GN, Takeda K, Rothwell NJ (2003) No role for interleukin‐18 in acute murine stroke‐induced brain injury. J Cereb Blood Flow Metab 23: 531–535 [DOI] [PubMed] [Google Scholar]
- Wu P‐J, Hung Y‐F, Liu H‐Y, Hsueh Y‐P (2017) Deletion of the inflammasome sensor Aim2 mitigates Aβ deposition and microglial activation but increases inflammatory cytokine expression in an alzheimer disease mouse model. NeuroImmunoModulation 24: 29–39 [DOI] [PubMed] [Google Scholar]
- Xiao J, Wang C, Yao J‐C, Alippe Y, Xu C, Kress D, Civitelli R, Abu‐Amer Y, Kanneganti T‐D, Link DC et al (2018) Gasdermin D mediates the pathogenesis of neonatal‐onset multisystem inflammatory disease in mice. PLoS Biol 16: e3000047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong Y‐N, Peng X, Xi JJ, Chen S et al (2014) Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513: 237–241 [DOI] [PubMed] [Google Scholar]
- Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, Zhang Z, Zhang D, Fan D, Nie Y et al (2017) Gasdermin D plays a key role as a pyroptosis executor of non‐alcoholic steatohepatitis in humans and mice. J Hepatol 68: 773–782 [DOI] [PubMed] [Google Scholar]
- Xu X, Yin D, Ren H, Gao W, Li F, Sun D, Wu Y, Zhou S, Lyu L, Yang M et al (2018) Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long‐term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis 117: 15–27 [DOI] [PubMed] [Google Scholar]
- Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, Zhou R (2015) Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160: 62–73 [DOI] [PubMed] [Google Scholar]
- Yang F, Wang Z, Wei X, Han H, Meng X, Zhang Y, Shi W, Li F, Xin T, Pang Q et al (2014) NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab 34: 660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu Z, Xiao TS (2017) Post‐translational regulation of inflammasomes. Cell Mol Immunol 14: 65–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsiv I, Morganti‐Kossmann MC, Perez D, Dinarello CA, Novick D, Rubinstein M, Otto VI, Rancan M, Kossmann T, Redaelli CA et al (2002) Elevated intracranial IL‐18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL‐18‐binding protein after experimental closed head injury. J Cereb Blood Flow Metab 22: 971–978 [DOI] [PubMed] [Google Scholar]
- Youm Y‐H, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D'Agostino D, Planavsky N, Lupfer C, Kanneganti TD et al (2015) The ketone metabolite β‐hydroxybutyrate blocks NLRP3 inflammasome‐mediated inflammatory disease. Nat Med 21: 263–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan B, Shen H, Lin L, Su T, Zhong S, Yang Z (2015) Recombinant adenovirus encoding NLRP3 RNAi attenuate inflammation and brain injury after intracerebral hemorrhage. J Neuroimmunol 287: 71–75 [DOI] [PubMed] [Google Scholar]
- Yuan J, Amin P, Ofengeim D (2019) Necroptosis and RIPK1‐mediated neuroinflammation in CNS diseases. Nat Rev Neurosci 20: 19–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zendedel A, Johann S, Mehrabi S, Joghataei M, Hassanzadeh G, Kipp M, Beyer C (2016) Activation and regulation of NLRP3 inflammasome by intrathecal application of SDF‐1a in a spinal cord injury model. Mol Neurobiol 53: 3063–3075 [DOI] [PubMed] [Google Scholar]
- Zhang S, Tang M, Luo H, Shi C, Xu Y (2017) Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Dis 8: e2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Lu M, Du R‐H, Qiao C, Jiang C‐Y, Zhang K‐Z, Ding J‐H, Hu G (2016) MicroRNA‐7 targets Nod‐like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson's disease. Mol Neurodegener 11: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]