

Abstract
Aims:
Patients with renovascular hypertension (RVH) exhibit elevated urinary mtDNA copy numbers, considered to constitute surrogate markers of renal mitochondrial injury. The modest success of percutaneous transluminal renal angioplasty (PTRA) in restoring renal function in RVH has been postulated to be partly attributable to acute reperfusion injury. We hypothesized that mitoprotection during revascularization would ameliorate PTRA-induced renal mitochondrial injury, reflected in elevated urinary mtDNA copy numbers, and improve blood pressure and functional outcomes 3 months later.
Methods:
We prospectively measured urinary copy number of the mtDNA genes COX3 and ND1 by qPCR in RVH patients before and 24hrs after PTRA, performed during IV infusion of vehicle (n=8) or the mitoprotective drug elamipretide (ELAM, 0.05mg/kg/hr, n=6). Five healthy volunteers (HV) served as controls. Urinary mtDNA levels were also assessed in RVH and normal pigs (n=7 each), in which renal mitochondrial structure and density were studied ex-vivo.
Results:
Baseline urinary mtDNA levels were elevated in all RVH patients versus HV, and directly correlated with serum creatinine levels. An increase in urinary mtDNA 24hrs after PTRA was blunted in PTRA+ELAM versus PTRA+Placebo. Furthermore, 3-months after PTRA, systolic blood pressure decreased and eGFR increased only in ELAM-treated subjects. In RVH pigs, mitochondrial damage was observed using electron microscopy in tubular cells and elevated urinary mtDNA levels correlated inversely with renal mitochondrial density.
Conclusions:
PTRA leads to an acute rise in urinary mtDNA, reflecting renal mitochondrial injury that in turn inhibits renal recovery. Mitoprotection might minimize PTRA-associated mitochondrial injury and improve renal outcomes after revascularization.
Keywords: Renovascular Hypertension, Mitochondria, mtDNA, PTRA, Revascularization
INTRODUCTION
Atherosclerotic occlusion of the renal arteries or their branches remains an important cause of renal dysfunction in the elderly population1. Reduced renal blood flow often induces renovascular hypertension (RVH), decreases glomerular filtration rate (GFR), and increases the risk of cardiovascular complications2.
Negative results from randomized clinical trials including the Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial3 have significantly lowered enthusiasm for renal revascularization4. Nevertheless, restoring blood flow with percutaneous transluminal renal angioplasty (PTRA) is helpful for some individuals, including those at risk of developing ischemic nephropathy or other high-risk manifestations of RVH5.
However, abrupt restoration of renal blood flow to a ischemic kidney may trigger inflammation, oxidative stress, and tissue injury considered to represent acute ischemia-reperfusion injury (IRI), which may be associated with sub-optimal outcomes after revascularization. For example, reperfusion by PTRA in swine RVH acutely increased systemic levels of the pro-inflammatory cytokine monocyte chemoattractant protein-1, associated with poorer PTRA outcomes6. Similarly, in patients with RVH systemic levels of the renal injury marker kidney injury molecule-1 transiently rise within 24 hours after stenting, accompained by transint renal hypoxia, implying reperfusion injury analagous to IRI7, 8. Elucidating the mechanisms implicated in PTRA-induced IRI in RVH may assist in developing novel adjunct therapies to improve renal outcomes after revascularization.
Experimental studies suggest that mitochondrial dysfunction might be an important contributor to IRI. Mitochondrial structural abnormalities and dysfunction were demonstrated in renal tubular cells of rats with IRI, associated with inflammation, oxidative stress, and apoptosis9–11. However, investigation of this injurious mechanism in human subjects has been limited by the need for renal biopsy, a procedure associated with potential risk12.
Fragments of mitochondrial DNA (mtDNA), which may break free from confinement of the double mitochondrial membrane and appear in the urine, have been touted as unique markers of renal mitochondrial injury. We have recently shown that urinary mtDNA copy number is elevated in RVH patients and correlates with markers of renal injury and dysfunction, implicating mitochondrial injury in chronic kidney damage in human RVH13. However, whether mitochondrial injury occurs acutely during PTRA remains unknown.
The mitochondria-targeted peptide elamipretide (ELAM), which concentrates in the inner mitochondrial membrane targeting the phospholipid cardiolipin, has shown potential to decrease renal damage in experimental6, 14 and clinical8 RVH. We therefore hypothesized that ELAM infusion during revascularization would ameliorate PTRA-induced renal mitochondrial injury, reflected in increasing urinary mtDNA copy numbers, and improve blood pressure and functional outcomes 3 months later. Furthermore, to validate urinary mtDNA copy numbers as surrogate markers of renal mitochondrial injury, we correlated urinary mtDNA levels with renal mitochondrial density and function in a swine model of RVH.
MATERIALS AND METHODS
Human RVH
This pilot study was carried out at Mayo Clinic with the approval of the Institutional Review Board and conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent before enrollment. Fourteen patients with identified RVH (renal artery Doppler ultrasound velocities across the stenosis >375cm/sec) participating in a previously described 3-day inpatient protocol study8 were enrolled. RVH patients were all maintained under controlled sodium intake (150mEq/d) and antihypertensive therapy (angiotensin-converting enzyme inhibitors or angiotensin receptor blockers) for 3 days during sample collection. Patients were selected for PTRA on clinical grounds (e.g. resistant hypertension, declining renal function) and randomized to a continuous infusion of either the mitochondrial-targeted peptide elamipretide (ELAM, Stealth Biotherapeutics. Inc. 0.05 mg/kg/hour) or placebo (n=6 and n=8, respectively) from 30 minutes before to 2.5 hours after PTRA. Most patients (8/14) had bilateral stenosis, and a total of 21 kidneys were stented (8 PTRA+ELAM and 13 PTRA+Placebo, p=0.64). Patients with serum creatinine >2.5mg/dL, renal disease requiring dialysis, significant medical conditions, or serum sodium <135mmol/L on the day of the PTRA were excluded. Blood pressure was measured by automated oscillometric recordings, and reported as the average of 3 values taken 3 times a day, as previously described8. Technically successful PTRA was confirmed by a completion angiogram after the end of the procedure.
The control group consisted of 5 age-, sex-, and body mass index- matched normotensive healthy volunteers (HV). In all patients, baseline clinical and laboratory parameters were collected via the electronic medical records, estimated GFR (eGFR) calculated using the chronic kidney disease epidemiology collaboration formula15, and urine samples collected to assess urine protein, albumin-creatinine-ratio, and urinary mtDNA copy numbers. In RVH patients, serum creatinine, the cell cycle arrest markers insulin-like growth factor-binding protein-(IGFBP)-7 and tissue inhibitor of metalloproteinases-(TIMP)-2, and urinary mtDNA copy numbers were measured again 24 hours after renal revascularization, and plasma mtDNA levels, plasma renin activity (PRA), eGFR, and blood pressure 3 months later.
Plasma and Urine mtDNA
Plasma and urinary copy numbers of the mitochondrial genes cytochrome-c oxidase-3 (COX3) and NADH dehydrogenase subunit-1 (ND1) levels were measured by quantitative real-time polymerase chain reaction (qPCR), as previously described13, 16. In brief, DNA was isolated from plasma (200µl) and urine samples (1.75ml) using a urine DNA isolation kit (Norgen Biotek, Ontario, Canada, Cat# 18100). Isolated DNA was then eluted in elution buffer (150µl), and DNA concentrations measured by a NanoDrop Spectrophotometer. DNA (7.2ng) from each sample was subsequently put into qPCR reactions for COX3 and ND1 primers. TaqMan copy number assays (Life Technology, Carlsbad, CA) were used to calculate the absolute copy numbers of DNA in each sample. COX3 and ND1 plasmid constructs were diluted and run together with the samples. qPCR was done on Applied Biosystems ViiA7 Real-Time qPCR systems. COX3 and ND1 levels were corrected to the nuclear control gene RNAse-P (Invitrogen, Cat# 4403326) using a human genomic DNA for the standard curve (Invitrogen, Cat# 360486) and were expressed as copies/µl. Urinary mtDNA levels were further adjusted by urinary creatinine (Arbor assays, Cat# K002-H1).
IGFBP-7 and TIMP-2 were measured by ELISA (R&D systems, Cat no. KIT DY1334–05 and Sigma-Aldrich, lot no. RAB0472, respectively) and their product (IGFBP-7 × TIMP-2) calculated. Changes in mtDNA and IGFBP-7 × TIMP-2 24 hours after renal revascularization were expressed as delta (24hr-baseline), and changes in eGFR 3 months after revascularization as delta (3months-baseline).
Swine RVH
Fourteen female domestic pigs were studied after 10 weeks of observation with the approval of the Mayo Clinic Institutional Animal Care and Use Committee, and in accordance with the NIH Guide for the Care and Use of Laboratory Animals. At baseline, under general anesthesia with intravenous ketamine (0.2mg/kg/min) and xylazine (0.03mg/kg/min), RVH was induced in 7 animals by placing an irritant coil in the main renal artery17, whereas normal pigs underwent a sham procedure. Ten weeks later, we evaluated the degree of stenosis using angiography and collected urine and plasma to assess copy numbers of the mitochondrial genes COX3 and ND1. Mean arterial pressure was assessed using an arterial catheter inserted in the left carotid artery, as previously described18. Animals were then euthanized with intravenous sodium pentobarbital (100 mg/kg, Fatal Plus, Vortech Pharmaceuticals, Dearborn, MI, USA). The kidneys were removed, dissected, and sections frozen in liquid nitrogen or preserved in formalin or Trump’s fixative for ex-vivo studies.
Renal tubular mitochondrial structure was examined by transmission electron microscopy (Philips CM10 Transmission Electron Microscope, Philips Electron Optics, Eindhoven, The Netherlands). Renal mitochondrial density was assessed under light microscopy by immunofluorescence staining with the mitochondrial outer membrane marker preprotein translocases of the outer membrane (TOM)-20 (Santa CruzBiotechnology, Dallas, TX, USA) and quantified in 15–20 random fields using a computer-aided image analysis program (ZENVR 2012 blue edition, Carl ZEISS SMT; Oberkochen, Germany).
Renal mitochondria were isolated using the MITO-ISO kit (Catalog #8268, ScienCell, Carlsbad, CA, USA)19. Mitochondrial cytochrome-c oxidase (COX)-IV activity and ATP/ADP levels were assessed by colorimetric/fluorometric methods (Abnova Cat# KA3950 and abcam, Cat# ab83355, respectively), as previously described20, 21. Stenotic kidney redox status was evaluated by the in situ production of superoxide anion, detected by fluorescence microscopy using dihydroethidium (DHE)14. Tubular injury was scored in stenotic kidney sections stained with periodic acid-Schiff and fibrosis in sections stained with Masson trichrome6.
Statistical analysis
Statistical analysis was performed with JMP version 13.0 (SAS Institute Inc. Cary, NC). Being a limited size pilot study, no power calculation was performed. The Shapiro-Wilk test was used to test data distribution. Normally distributed data were expressed as mean±SD and compared using ANOVA with Tukey’s post-hoc. Non-normally distributed data were expressed as median (interquartile range), and compared using non-parametric (Wilcoxon and Kruskal Wallis) tests with Steel-Dawss post-hoc. Longitudinal data were compared using parametric (paired t-test) and non-parametric (Wilcoxon Signed Rank) tests when appropriate. Regressions were calculated by the least-squares fit. A p value≤0.05 was considered statistically significant.
RESULTS
Table 1 shows the baseline clinical, laboratory, and demographic data of RVH patients and HV. Systolic blood pressure, mean arterial pressure, and PRA were similarly higher in both RVH groups compared to HV, whereas diastolic blood pressure did not differ among the groups. The number of antihypertensive and lipid-lowering drugs was similar between RVH+ELAM and RVH+Placebo groups. Serum creatinine levels were similarly higher and eGFR lower in RVH patients compared to HV. Urine protein and albumin-creatinine-ratio did not differ among the groups.
Table 1.
Clinical, laboratory, and demographic data of healthy volunteers (HV) and renovascular hypertensive (RVH) patients treated with percutaneous transluminal renal angioplasty (PTRA) and either vehicle or elamipretide (ELAM).
| Parameter | HV | RVH | |
|---|---|---|---|
| PTRA+ELAM | PTRA+Placebo | ||
| Demographics: | |||
| Number of patients | 5 | 6 | 8 |
| Age (years) | 65.0±8.3 | 65.3±7.1 | 69.5±9.7 |
| Gender (Female/Male) | 3/2 | 3/3 | 4/4 |
| Body Mass IndexϮ | 31.7 (27.9–32.3) | 29.2 (25.2–38.9) | 31.5 (25.2–39.6) |
| Related laboratory measures: | |||
| Systolic blood pressure (mmHg) | 114.2±16.7 | 149.5±16.6* | 150.6±21.2* |
| Diastolic blood pressure (mmHg) | 65.0±6.1 | 82.8±6.8 | 73.0±11.4 |
| Mean blood pressure (mm Hg) | 81.4±9.1 | 105.1±7.8* | 98.9±12.3* |
| PRA (ng/ml per hour) | 0.54±0.4 | 14.5±11.6* | 10.4±6.8* |
| Concomitant medication: | |||
| No. of Antihypertensive drugsϮ | 0 | 3.5 (2–5)* | 4 (2–6)* |
| Lipid-lowering drugs (No. of patients) | 0/5 | 5/6* | 5/8* |
| Renal function: | |||
| Serum creatinine (mg/dL) | 0.9±0.2 | 1.6±0.4* | 1.8±0.5* |
| eGFR-CDK-EPI (ml/min/1.73/m2) | 78.3±9.0 | 40.7±13.4* | 36.0±10.2* |
| Urine protein (mg/24hr) | 93.0 (72.0–93.0) | 134.0 (67.5–4910.5) | 79.0 (39.5–1038.5) |
| Albumin-creatinine-ratio (mg/g) | 48.5±43.6 | 118.0±154.3 | 131.2±171.5 |
eGFR-CKD-EPI: estimated glomerular filtration rate-chronic kidney disease epidemiology collaboration.
p≤0.05 vs. HV.
Median (interquartile range) reported because of skewed data.
Baseline mtDNA
Neither urinary nor plasma RNAse-P copy numbers differed among the groups (p=0.91 and p=0.94, respectively), whereas baseline urinary COX3 and ND1 levels were similarly elevated in both RVH groups (Figure 1A), and correlated directly with serum creatinine levels (Figure 1B). Statistically significant differences in urinary COX3 and ND1 copy number between the groups persisted after adjustment for urinary creatinine (all p<0.05). Contrarily, plasma COX3 and ND1 were similar among the groups (Figure S1A).
Figure 1. Urinary mitochondrial DNA (mtDNA) copy number is elevated in RVH.
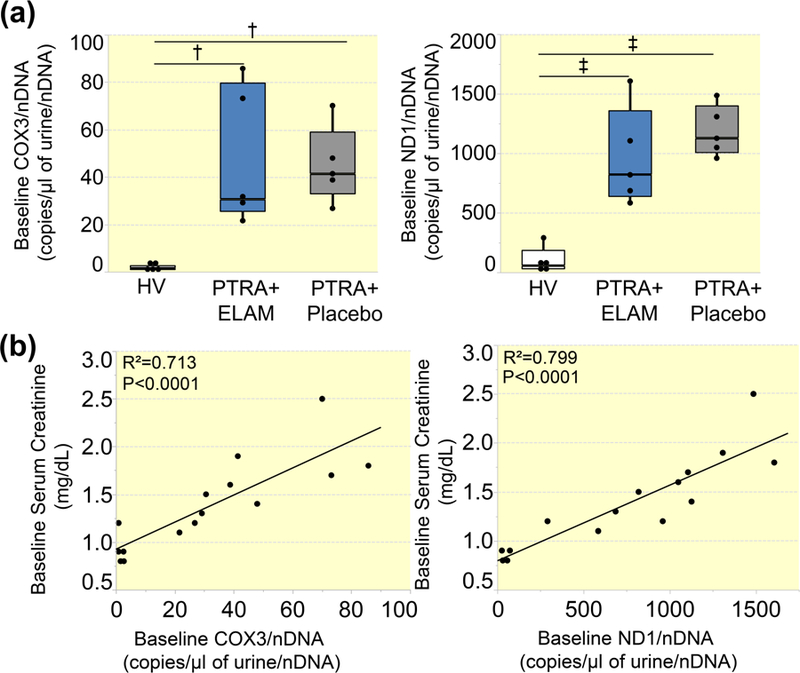
A: Urinary copy number of cytochrome-c oxidase-3 (COX3) and NADH dehydrogenase subunit-1 (ND1) were similarly elevated in RVH patients treated PTRA and either placebo or ELAM compared to healthy volunteers (HV). B: Urinary COX3 and ND1 copy number correlated directly with serum creatinine levels. †P<0.01, ‡P<0.001.
Changes after PTRA
Serum creatinine levels did not increase 24hrs after PTRA in either ELAM or Placebo groups (Figure 2A). Contrarily, circulating plasma IGFBP-7 × TIMP-2 increased only in ELAM-treated subjects within 24hrs of revascularization (Figure 2B). Urinary COX3 and ND1 increased in both groups 24hrs after PTRA (Figure 2C), but the increase was greater in RVH patients treated with PTRA+Placebo compared to those treated with PTRA+ELAM (Figure 2D). In contrast, plasma COX3 and ND1 levels remained unchanged in both groups 3 months after PTRA (Figure S1B), as were PRA levels (Figure S2).
Figure 2. Renal revascularization leads to an acute elevation in urinary mtDNA levels.
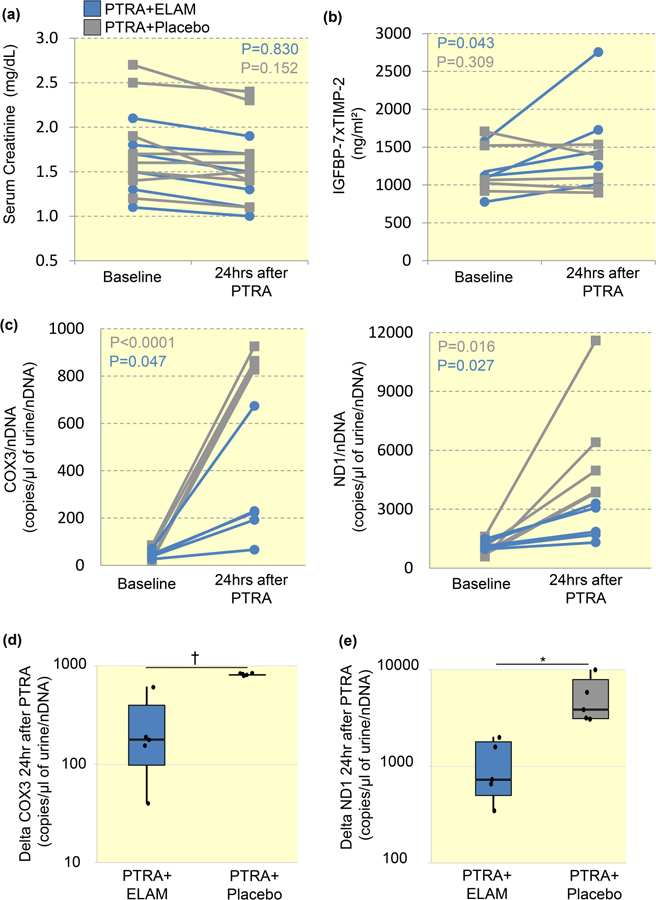
A: Serum creatinine levels did not increase in either ELAM or Placebo groups within 24hrs of PTRA. B: Circulating plasma levels of insulin-like growth factor-binding protein-(IGFBP)-7 × tissue inhibitor of metalloproteinases-(TIMP)-2 rose in ELAM-treated subjects at 24hrs after stenting. C: Urinary COX3 and ND1 increased in both groups within 24hrs of PTRA. D: Delta (24hrs after PTRA-baseline) change in urinary COX3 and ND1 were lower in RVH patients treated with PTRA+ELAM compared with those treated with PTRA+Placebo. *P<0.05, †P<0.01.
Adjunctive ELAM (but not Placebo) during PTRA was associated with an increase in eGFR and a decline in systolic blood pressure after 3 months (Figure 3A), whereas diastolic and mean arterial pressure similarly decreased in both PTRA-treated groups (Figure S3). There were no differences in the change of systolic, diastolic, mean arterial pressure, or eGFR between patients with unilateral and bilateral stenosis following PTRA (all p>0.05). Delta change in urinary COX3 and ND1 within 24hrs of PTRA correlated inversely with the change in IGFBP-7 × TIMP-2 levels of 24hrs (Figure 3C), but only delta change in urinary COX3 correlated with the change in eGFR of 3 months after PTRA (Figure 3D).
Figure 3. Adjunctive mitoprotection improved renal function in renovascular hypertension (RVH).
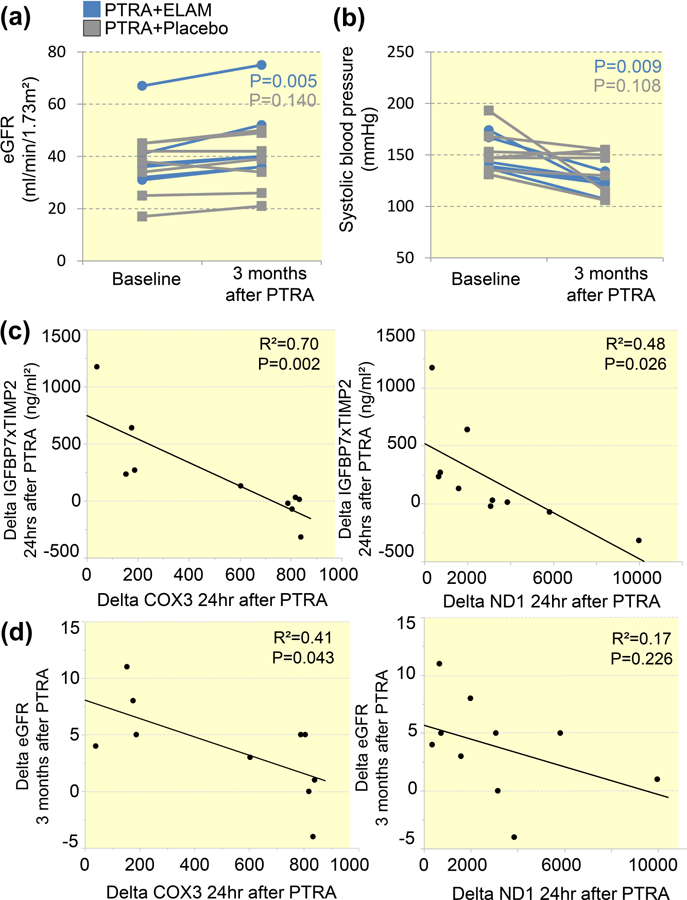
A: Over 3 months, estimated glomerular filtration rate (eGFR) increased only in RVH patients treated with percutaneous transluminal renal angioplasty (PTRA) and elamipretide (ELAM). B: Treatment with ELAM (but not placebo) during PTRA decreased systolic blood pressure 3 months after revascularization. C: Delta change in urinary COX3 and ND1 within 24hrs of PTRA correlated inversely with the change in IGFBP-7 × TIMP-2 levels of 24hrs. D: Only delta change in urinary COX3 correlated with the change in eGFR of 3 months after PTRA.
mtDNA in swine RVH
Ten weeks after induction of RVH, pigs achieved hemodynamically significant stenoses (77.1±8.6%), and mean arterial pressure was significantly elevated (127.3±10.1mmHg vs. 103.1±9.2mmHg, p=0.0005 vs. normal). Electron microscopy studies of stenotic-kidney segments revealed that tubular cells exhibit important mitochondrial structural abnormalities, including decreased matrix density and swelling (Figure S4A). Under light microscopy, TOM-20 immunoreactivity was significantly lower in the stenotic kidney of RVH compared to normal pigs, as were mitochondrial COX-IV activity and ATP generation (Figure S4B). Stenotic kidney oxidative stress, tubular injury, and tubulointerstitial fibrosis were higher in RVH compared to normal pigs (Figure S5).
Urinary COX3 and ND1 copy numbers were higher in RVH compared to normal pigs (Figure S6A) and correlated inversely with TOM-20 immunoreactivity (Figure S6B), COX-IV activity (Figure S6C), and ATP generation (Figure S6D). However, plasma COX3 and ND1 did not differ among the groups (Figure S7).
DISCUSSION
This study shows that in patients with RVH renal revascularization with PTRA leads to an acute rise in urinary mtDNA levels, likely reflecting renal mitochondrial injury, possibly secondary to subtle IRI. Interestingly, changes in COX3 and ND1 during revascularization correlated inversely with the change in cell cycle arrest biomarkers, suggesting that PTRA-associated mitochondrial injury may inhibit renal tubular cell repair. Changes in urinary mtDNA 24hrs after PTRA also correlated inversely with the change in eGFR at 3 months after PTRA, linking renal mitochondrial injury during reperfusion to ultimate renal recovery. Furthermore, adjunctive ELAM during PTRA improved blood pressure and renal function after revascularization, suggesting that mitoprotection to minimize PTRA-associated mitochondrial injury rescues the kidney. In pigs, RVH-induced renal mitochondrial injury, reflected by elevated urinary mtDNA levels, was associated with stenotic kidney oxidative stress, tubular injury, and fibrosis, linking mitochondrial injury and renal damage in experimental RVH. Lastly, porcine urinary mtDNA levels correlated inversely with renal mitochondrial density and function, establishing urinary mtDNA as a biomarker of renal mitochondrial injury.
Randomized prospective trials failed to establish additional clinical benefits for renal revascularization over optimized medical therapy regarding renal functional outcomes in patients with RVH3, 4. Yet, PTRA can be of benefit to recovering kidney function in selected patients whose kidneys are truly at risk from vascular occlusion5. However, stent revascularization in RVH is associated with transiently increased circulating pro-inflammatory cytokines and acute kidney injury markers, likely reflecting episodes of IRI6–8. Importantly, activation of inflammatory and injury signals may accelerate post-stenotic kidney injury, portending worse outcomes after PTRA22.
Mitochondrial dysfunction has been implicated in the pathogenesis of renal IRI and in kidneys subjected to a reduction in renal perfusion. On the other hand, rapid reperfusion of an ischemic kidney may trigger ROS generation and calcium overload, evoking mitochondrial membrane permeabilization, swelling, and release of mitochondrial ROS and pro-apoptotic proteins to the cytosol23. Studies in rats have shown that renal ischemia followed by reperfusion leads to mitochondrial structural abnormalities and dysfunction9–11. Likewise, defective mitochondrial homeostasis has been reported in the clipped rat24 and post-stenotic pig6 kidneys. We have previously shown that human RVH is associated with chronic increments in markers of mitochondrial injury, as measured by urinary mtDNA copy number, which correlate inversely with eGFR13. In line with this, the current study found that in RVH patients scheduled for renal revascularization, increased in urinary levels of fragments of the mitochondrial genome correlated directly with serum creatinine levels. The observation that circulating mtDNA levels were similar among the groups suggests that they were primarily renal-derived.
Moreover, this study extends our previous observations, and shows that urinary mtDNA levels further rise in patients with RVH within 24 hours of PTRA, implicating mitochondria in acute renal alterations occurring during PTRA. This renal injury is likely subtle, given that serum creatinine levels remained unchanged, yet was disclosed by a rise in urinary mtDNA. Hence, mitochondria might represent a novel therapeutic target to improve renal outcomes after revascularization.
ELAM is a novel aromatic-cationic peptide that targets and preserves cardiolipin, a phospholipid exclusively distributed in the inner mitochondrial membrane that plays a critical role in maintaining cristae structure and in electron transport chain assembly and function25. In addition, ELAM can bind cytochrome-c, favoring shuttling electrons among respiratory complexes26. In swine RVH, chronic daily treatment with ELAM preserves the stenotic-kidney microvasculature and improves function14. Continuous short-term intravenous administration of ELAM in conjunction with PTRA improved renal outcomes in pigs6 and humans8 with RVH. The current study underscores our previous observation that over 3 months, eGFR increases only in RVH patients treated with PTRA+ELAM8, suggesting a protective role of this compound in the face of PTRA-induced renal injury.
The mechanisms implicated in ELAM-induced renoprotection during PTRA may be related to tubular epithelial cell cycle arrest, an important mechanism that prevents division of damaged tubular cells exposed to acute insults27. IGFBP-7 and TIMP-2 are involved in G1 cell cycle arrest during cellular injury, and their product has been proposed as a prognostic marker in patients with established acute kidney injury28, 29. This study found that circulating plasma IGFBP-7 × TIMP-2 increased only in ELAM-treated subjects within 24hrs of revascularization. Notably, changes in urinary COX3 and ND1 during revascularization correlated inversely with the change in cell cycle arrest biomarkers, suggesting that PTRA-associated mitochondrial injury may blunt renal tubular cell repair, which ELAM contrarily enables. Moreover, the increase in urinary COX3, but not ND1 in the 24hrs after PTRA inversely correlated with the change in eGFR within 3 months later, suggesting that COX3 might be a better surrogate marker for PTRA-induced renal mitochondrial injury to predict functional changes after revascularization in patients with RVH. Overall, our findings implicate mitochondrial injury occurring during PTRA in renal functional ramification, and suggest that mitoprotection in conjunction with PTRA may curtail procedure-associated injury and improve renal outcomes following revascularization.
To establish the validity of urinary mtDNA copy numbers as surrogate markers of renal mitochondrial injury, we also measured plasma and urinary COX3 and ND1 levels in a swine model of RVH, in which kidney tissue was available for direct examination. Indeed, pigs with RVH exhibited important renal mitochondrial structural abnormalities detected by electron microscopy. This includes decreased matrix density and swelling, reflecting loss of cristae membranes and water influx in the mitochondrial matrix. These injury signals were also associated with decreased mitochondrial spatial density, reflected in lower immunoreactivity of TOM-20 compared to normal pigs. Furthermore, renal mitochondrial COX-IV activity and ATP/ADP ratio were lower in RVH compared to normal pigs, indicating impaired mitochondrial respiration and energy production. Urinary mtDNA levels were higher in RVH vs. normal pigs, congruent with our findings in RVH patients. Importantly, elevated urinary mtDNA was associated with stenotic kidney oxidative stress, tubular injury, and fibrosis, linking mitochondrial injury and renal damage in experimental RVH. Contrarily, plasma mtDNA levels were similar between RVH and normal pigs. These observations correlates with our finding in human RVH, but differ from our previous observations in pigs with atherosclerotic RVH30, suggesting that urinary mtDNA is a surrogate marker for mitochondrial damage in renal tubules, whereas atherosclerosis contributes to systemic mitochondrial injury in experimental RVH. Finally, urinary mtDNA correlated inversely with mitochondrial density and energy production in the post-stenotic swine kidney. This consistent link between structural and functional mitochondrial damage and increase in urinary mtDNA levels support its use as an index of renal mitochondrial injury in RVH.
Our pilot study is limited by the relatively small number of patients, but its longitudinal nature provided it with strength. While did not perform a power calculation for our pilot, our findings suggest that inclusion of 5 patients per group was sufficient to detect statistical significant changes in urinary mtDNA levels following renal revascularization. In addition, we do not have available urinary mtDNA levels 3 months after PTRA. While we do not have renal biopsies from patients with RVH to assess mitochondrial injury, our studies in RVH pigs clearly indicate that elevated urinary COX3 and ND1 levels correlated inversely with renal mitochondrial density, underscoring the utility of mtDNA levels as surrogate markers of renal mitochondrial injury. Additional prospective population studies are needed to confirm our findings, as well as define the degree to which levels of these novel markers are altered in patients with other forms of kidney disease.
In summary, we found that PTRA leads to an acute elevation in urinary mtDNA, likely reflecting renal mitochondrial injury due to reperfusion injury that in turn inhibits renal recovery. Furthermore, mitoprotection might minimize PTRA-associated mitochondrial injury and improve renal outcomes after revascularization. In RVH pigs, renal tubular mitochondria were damaged, and elevated urinary COX3 and ND1 levels correlated inversely with renal mitochondrial density. Therefore, our observations suggest that mitochondria are implicated in renal IRI during reperfusion and urinary mtDNA levels represent novel biomarkers of renal mitochondrial injury.
Supplementary Material
Acknowledgments
FUNDING
This study was partly supported by a grant from Stealth Biotherapeutics, Inc. and NIH grant numbers: DK100081, DK104273, HL123160, DK102325, DK118120, and DK106427.
Footnotes
CONFLICT OF INTEREST STATEMENT
Drs. S.C. Textor and L.O. Lerman hold a patent for the use of aromatic-cationic peptides in RVH.
CONFLICT OF INTEREST STATEMENT: The results presented in this paper have not been published previously in whole or part, except in abstract format.
REFERENCES
- 1.Hansen KJ, Edwards MS, Craven TE, Cherr GS, Jackson SA, Appel RG, Burke GL and Dean RH. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002;36:443–51. [DOI] [PubMed] [Google Scholar]
- 2.Zoccali C, Mallamaci F and Finocchiaro P. Atherosclerotic renal artery stenosis: epidemiology, cardiovascular outcomes, and clinical prediction rules. J Am Soc Nephrol 2002;13 Suppl 3:S179–83. [DOI] [PubMed] [Google Scholar]
- 3.Cooper CJ, Murphy TP, Cutlip DE, Jamerson K, Henrich W, Reid DM, Cohen DJ, Matsumoto AH, Steffes M, Jaff MR, Prince MR, Lewis EF, Tuttle KR, Shapiro JI, Rundback JH, Massaro JM, D’Agostino RB Sr., Dworkin LD and Investigators C. Stenting and medical therapy for atherosclerotic renal-artery stenosis. N Engl J Med 2014;370:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herrmann SM, Saad A and Textor SC. Management of atherosclerotic renovascular disease after Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL). Nephrol Dial Transplant 2015;30:366–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Textor SC, Misra S and Oderich GS. Percutaneous revascularization for ischemic nephropathy: the past, present, and future. Kidney Int 2013;83:28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eirin A, Li Z, Zhang X, Krier JD, Woollard JR, Zhu XY, Tang H, Herrmann SM, Lerman A, Textor SC and Lerman LO. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension 2012;60:1242–9. [DOI] [PubMed] [Google Scholar]
- 7.Wang W, Saad A, Herrmann SM, Eirin Massat A, McKusick MA, Misra S, Lerman LO and Textor SC. Changes in inflammatory biomarkers after renal revascularization in atherosclerotic renal artery stenosis. Nephrol Dial Transplant 2016;31:1437–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saad A, Herrmann SMS, Eirin A, Ferguson CM, Glockner JF, Bjarnason H, McKusick MA, Misra S, Lerman LO and Textor SC. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) During Stent Revascularization in Patients With Atherosclerotic Renal Artery Stenosis. Circ Cardiovasc Interv 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY and Seshan SV. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol 2011;22:1041–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh D, Chander V and Chopra K. Cyclosporine protects against ischemia/reperfusion injury in rat kidneys. Toxicology 2005;207:339–47. [DOI] [PubMed] [Google Scholar]
- 11.Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD and Szeto HH. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 2013;24:1250–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker PD. The renal biopsy. Arch Pathol Lab Med 2009;133:181–8. [DOI] [PubMed] [Google Scholar]
- 13.Eirin A, Saad A, Tang H, Herrmann SM, Woollard JR, Lerman A, Textor SC and Lerman LO. Urinary Mitochondrial DNA Copy Number Identifies Chronic Renal Injury in Hypertensive Patients. Hypertension 2016;68:401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eirin A, Ebrahimi B, Zhang X, Zhu XY, Woollard JR, He Q, Textor SC, Lerman A and Lerman LO. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc Res 2014;103:461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J and Ckd EPI. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150:604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eirin A, Saad A, Woollard JR, Juncos LA, Calhoun DA, Tang H, Lerman A, Textor SC and Lerman LO. Glomerular Hyperfiltration in Obese African American Hypertensive Patients Is Associated With Elevated Urinary Mitochondrial-DNA Copy Number. Am J Hypertens 2017;30:1112–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lerman LO, Schwartz RS, Grande JP, Sheedy PF and Romero JC. Noninvasive evaluation of a novel swine model of renal artery stenosis. J Am Soc Nephrol 1999;10:1455–65. [DOI] [PubMed] [Google Scholar]
- 18.Krier JD, Ritman EL, Bajzer Z, Romero JC, Lerman A and Lerman LO. Noninvasive measurement of concurrent single-kidney perfusion, glomerular filtration, and tubular function. Am J Physiol Renal Physiol 2001;281:F630–8. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Li ZL, Crane JA, Jordan KL, Pawar AS, Textor SC, Lerman A and Lerman LO. Valsartan regulates myocardial autophagy and mitochondrial turnover in experimental hypertension. Hypertension 2014;64:87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eirin A, Woollard JR, Ferguson CM, Jordan KL, Tang H, Textor SC, Lerman A and Lerman LO. The metabolic syndrome induces early changes in the swine renal medullary mitochondria. Transl Res 2017;184:45–56 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng Y, Eirin A, Zhu XY, Tang H, Chanana P, Lerman A, van Wijnen AJ and Lerman LO. Obesity-induced mitochondrial dysfunction in porcine adipose tissue-derived mesenchymal stem cells. J Cell Physiol 2018;233:5926–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eirin A, Ebrahimi B, Zhang X, Zhu XY, Tang H, Crane JA, Lerman A, Textor SC and Lerman LO. Changes in glomerular filtration rate after renal revascularization correlate with microvascular hemodynamics and inflammation in Swine renal artery stenosis. Circ Cardiovasc Interv 2012;5:720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orrenius S, Zhivotovsky B and Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 2003;4:552–65. [DOI] [PubMed] [Google Scholar]
- 24.Fedorova LV, Sodhi K, Gatto-Weis C, Puri N, Hinds TD Jr., Shapiro JI and Malhotra D. Peroxisome proliferator-activated receptor delta agonist, HPP593, prevents renal necrosis under chronic ischemia. PLoS One 2013;8:e64436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 2014;171:2029–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birk AV, Chao WM, Bracken C, Warren JD and Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol 2014;171:2017–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DiRocco DP, Bisi J, Roberts P, Strum J, Wong KK, Sharpless N and Humphreys BD. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am J Physiol Renal Physiol 2014;306:F379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kashani K, Al-Khafaji A, Ardiles T, Artigas A, Bagshaw SM, Bell M, Bihorac A, Birkhahn R, Cely CM, Chawla LS, Davison DL, Feldkamp T, Forni LG, Gong MN, Gunnerson KJ, Haase M, Hackett J, Honore PM, Hoste EA, Joannes-Boyau O, Joannidis M, Kim P, Koyner JL, Laskowitz DT, Lissauer ME, Marx G, McCullough PA, Mullaney S, Ostermann M, Rimmele T, Shapiro NI, Shaw AD, Shi J, Sprague AM, Vincent JL, Vinsonneau C, Wagner L, Walker MG, Wilkerson RG, Zacharowski K and Kellum JA. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care 2013;17:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bihorac A, Chawla LS, Shaw AD, Al-Khafaji A, Davison DL, Demuth GE, Fitzgerald R, Gong MN, Graham DD, Gunnerson K, Heung M, Jortani S, Kleerup E, Koyner JL, Krell K, Letourneau J, Lissauer M, Miner J, Nguyen HB, Ortega LM, Self WH, Sellman R, Shi J, Straseski J, Szalados JE, Wilber ST, Walker MG, Wilson J, Wunderink R, Zimmerman J and Kellum JA. Validation of cell-cycle arrest biomarkers for acute kidney injury using clinical adjudication. Am J Respir Crit Care Med 2014;189:932–9. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Li ZL, Eirin A, Ebrahimi B, Pawar AS, Zhu XY, Lerman A and Lerman LO. Cardiac metabolic alterations in hypertensive obese pigs. Hypertension 2015;66:430–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.