

Abstract
Traumatic brain injury (TBI) is one of the leading causes of death and disability, particularly among the young and the elderly. Several therapeutic options have been investigated, including drug interventions or combinational therapies. Although many drugs have shown promising results in the preclinical stage, all have failed in large clinical trials. Targeting the dopamine system is a novel TBI approach that provides benefits to functional outcomes. TBI could damage the dopaminergic system. Alterations in dopamine levels can impact cellular dysfunction and central nervous system (CNS) inflammation. Experimental evidence suggests that dopamine should be considered a first-line treatment to protect cerebral autoregulation and promote cerebral outcomes in TBI. Furthermore, investigation of dopamine-related genetic factors in relation to injury severity could also be of great significance for promoting TBI treatment. Importantly, various clinical lines of evidence have indicated that many dopamine agonists are beneficial when administered following injury in TBI patients. However, side effects of dopamine treatment prevent their use in TBI treatment, and there is a need for ongoing large, prospective, double-blind randomized controlled trials (RCTs) with these medications by the use of standardized criteria and outcomes to fully understand their effectiveness in this patient group. Here, we review the roles of dopamine in TBI and discuss the role that dopaminergic therapies have in neuroprotective strategies.
Keywords: Dopamine, traumatic brain injury, dopamine transporter, neuroprotection
Introduction
Traumatic brain injury (TBI) is a major cause of death or disability worldwide. In the last 3 decades, various neuroprotective agents that could attenuate the downstream damaging events triggered by TBI have been explored. Although some drugs have demonstrated promising results in the preclinical stage, all have failed in large clinical trials [1]. Many of the neurological deficits that result from brain injury are caused by direct anatomical damage, but inhibition of chemical transmission also exerts important effects. These changes have been investigated and confirmed in epigenetic and behavioral studies [2,3]. Among the neurotransmitter systems, dopamine pathways seem especially vulnerable to brain injury due to the anatomic properties of the dopaminergic system. Animal models of TBI show both dopaminergic cell loss and biochemical disturbances of the dopaminergic system [4]. There is convincing evidence that dopamine dysfunction contributes to post-TBI deficits, while dopaminergic regulation could exert prominent neuroprotective effects.
Targeting dopamine signaling in TBI could be of great significance because dopamine impacts various brain regions, including the hippocampus [5,6], striatum [7], and frontal cortex (FC) [8,9]; damage in these regions has been associated with cognitive dysfunction in TBI. In addition, TBI has been associated with fluctuations in dopamine levels [10]. More importantly, the neuroprotective and therapeutic strategies that focus on the dopaminergic system have revealed great benefits, and the use of dopamine agonists has revealed benefits not only in preclinical experiments but also in clinical trials [11-13]. Thus, here we provide support for dopamine as a viable target in acute TBI, and this review focuses on the dopamine system related to brain injury.
Alterations in dopaminergic systems following TBI
Multiple brain regions are affected by TBI, including the FC [8,9], hippocampus [5,6], and striatum [7]. These three regions are particularly important because of their role in learning, memory, executive function and attention [14-18], and they could be impaired after TBI [19-23]. However, damage to brain tissue after TBI is not limited to discrete brain regions. Diffuse axonal injury in white matter tracts along with gray matter damage [24-27] further complicates the clinical presentation of TBI. The widespread disruption of neuronal projections has implications for all neurotransmitter systems, including dopamine. Whether depleted or at excessively high levels, dopamine can cause significant cellular dysfunction; thus, the changes in the levels of dopamine and related alterations in dopaminergic systems could have a great impact on functional outcomes (Figure 1). Potential strategies attenuating dopaminergic alterations could thus be of great significance for improving TBI treatment efficacy.
Figure 1.
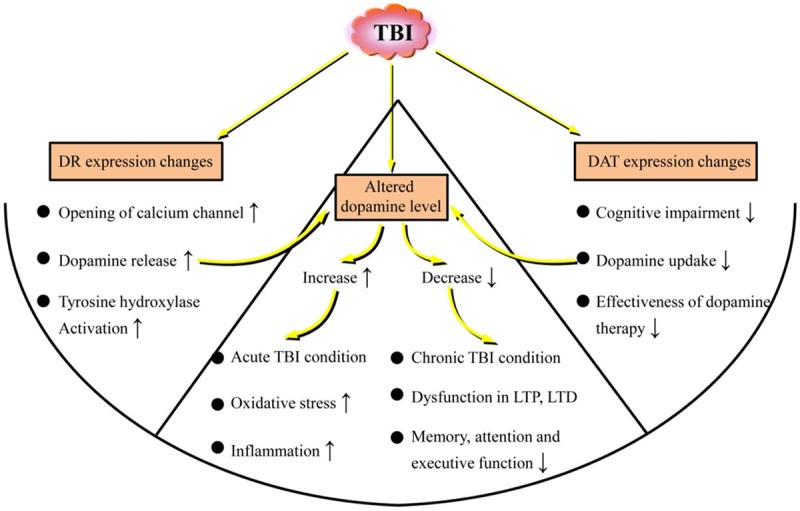
Changes in dopamine and related alterations in dopaminergic systems induced by traumatic brain injury (TBI). TBI could cause altered dopamine release, dopamine receptor (DR) expression changes, and changes in dopamine transporter (DAT) expression in the brain. Changes in levels and expression of dopamine and related alterations, including DR and DAT in dopaminergic systems, could have a great impact on functional outcomes.
Altered dopamine release following TBI
Dopamine is involved in various dysfunctional processes caused by TBI. There is evidence that dopamine release is affected by TBI [28,29], and this may contribute to various psychological disorders that are often observed [30,31]. Dopamine release is associated with cognitive behavior [32]. In addition, several mechanisms induced by TBI that could affect neurotransmission, especially dopamine transmission, have been elucidated; these mechanisms involve neuroinflammation that is induced after the initial insult in TBI [33,34] and play a critical role in secondary neurodegeneration [33]. The role that acutely released dopamine plays following TBI is complex (Figure 1). Whether the initial increase is neurotoxic or is an attempt to restore functional circuitry damaged by the mechanical insult is unclear [10]. A dramatic increase in dopamine within the central nervous system (CNS) has multiple consequences, including increased oxidative stress and the induction of inflammatory signals, while a decrease in dopamine, which often occurs in chronic TBI conditions, has other consequences, such as dysfunction in long-term depression (LTD) and long-term potentiation (LTP) processes, as well as deficits in memory, attention and executive functions [10].
A major site of dopamine activity is the nucleus accumbens (NAC), which includes an inner core and outer shell with very different functions [29]. If variations in dopamine release patterns are related to symptoms of TBI, such as cognitive dysfunction that is impacted by different parts of the NAC, differences in dopamine physiology between the core and shell are likely to have important implications for the relevant clinical manifestations. However, the effect of TBI on stimulus-related dopamine dynamics in different regions of the NAC still is in need of further research. Intriguingly, Chen et al. [29] reported that TBI was associated with major changes in dopamine release in both the core and shell of the NAC that correlated with the severity of injury, with the core being somewhat more vulnerable to TBI-associated changes. Furthermore, their research revealed that the most significant alterations in dopamine dynamics occurred at 1-2 weeks after injury, with some recovery over the longer term. These electrochemical findings in NAC could clarify the mechanisms related to post-TBI psychological syndromes and support the modulation of dopaminergic mechanisms as clinical therapeutic strategies for TBI.
Dopamine receptor expression changes in TBI
Dopamine can be synthesized at different places in the CNS and regulates cognition, emotion and various other physiological functions through binding to dopamine receptors [35]. Dopamine receptors can be generally divided into 5 subtypes, i.e., D1-D5 [36]. Based on the signal transduction mechanisms that are activated after binding of agonists to the different receptor subtypes, dopamine receptors have been classified as either D1-class receptors (D1 and D5), which are mainly distributed in the kidney, heart, and mesenteric tissue, or D2-class dopamine receptors (D2, D3, and D4), which are mainly distributed in the presynaptic adrenergic nerve endings and sympathetic ganglia [37-41]. D1 and D5 receptors activate the Gαs/olf family of G proteins to stimulate cAMP production by adenylate cyclase, while D2, D3, and D4 receptors couple to the Gαi/o family of G proteins and inhibit adenylate cyclase activity [36].
During TBI, transient mechanical violence acts on the nervous system, which leads to neuronal hyperexcitability and the opening of calcium channels as well as the activation of dopaminergic neurons followed by dopamine release [42-45]. Tyrosine hydroxylase (TH) activity in neurons is enhanced by the activation of calcium channels, and the activation of TH accelerates dopamine synthesis by dopaminergic neurons, resulting in the accumulation of a large amount of dopamine [46,47]. Thus, the activation of calcium channels could also impact dopamine levels. Interestingly, it has been confirmed that dopamine D1 receptor activity can exert modulatory effects on calcium channel currents [48]. Thus, activity of dopamine receptors themselves can also regulate the accumulation of dopamine. In addition, transient decreases in dopamine D1 receptor binding have been shown to occur immediately following injury [49] but do not persist chronically. The authors suggested that striatal dopamine D1 receptors were downregulated and then upregulated following isolated injury to the cerebral cortex. Overall, these data have indicated that in patients with TBI, the dopamine signaling pathways play an important role in brain injury and that dopamine receptors may also become novel targets for the treatment of brain injury [50].
Changes in dopamine transporter expression in the brain following TBI
Dopamine neurotransmission relies in part on dopamine transporter (DATs), which are membrane proteins that transport dopamine from the synapse to the cytosol [51]. Monoamine oxidase (MAO) metabolizes dopamine in the cytosol into 3,4-dihydroxyphenylacetic acid [52]. Dopamine that is not metabolized by this process is taken up into vesicles by the vesicular monoamine transporter 2 (VMT2) and is recycled to maintain dopamine homeostasis [53]. DATs are mainly expressed in the substantia nigra (SN), retrorubral field (RRF), and ventral tegmental area (VTA) of the ventral midbrain [54]. Significantly, decreased DAT levels in the rat FC [55] and striatum [56] after TBI have been found, and experimental studies have also demonstrated that TBI induces a loss of dopaminergic neurons in the SN in rats [4], suggesting that dopaminergic neurons in the SN are more vulnerable to TBI than neighboring neuronal populations. However, Shimada et al. [57] reported that there were no significant changes in DAT expression levels in the SN but found significantly decreased DAT expression in the RRF. Importantly, dopaminergic neurons are more important in the RRF than in the SN and VTA because the RRF cell group could be composed of the largest dopamine neurons [58].
Generally, various studies have demonstrated that DAT levels decreased continuously after TBI, and notably, that the rate at which DAT removes dopamine from the synapse could have a profound effect on dopamine levels in the cell. This is best evidenced by the severe cognitive deficits, hyperactivity and motor abnormalities exhibited by DAT knockout mice [59]. Therefore, the decreased levels of DAT expression after TBI could result in decreased dopamine neurotransmission in the brain. Since dopamine neurons in the RRF project to the amygdala, striatum, bed nucleus of the stria terminalis [60], and hippocampal formation [61], the decreased levels of expression of DAT in the RRF may also cause dysfunctions in these areas. Furthermore, decreased DAT expression levels may be related to the effectiveness of dopamine therapy in TBI patients, which is discussed further in the following text.
Interestingly, in a recent study, Jenkins et al. [62] demonstrated that striatal DAT abnormalities measured using 123I-ioflupane single-photon emission computed tomography (SPECT) are commonly observed following moderate/severe TBI. One-third of the SPECT scans were clinically reported as abnormal. These changes are a marker of reduced striatal dopamine. Their results suggested that these changes were due to the pattern of nigrostriatal tract damage produced by axonal injury. Compared with that in normal patients, cognitive impairment in patients with marked reductions in caudate DAT levels was greater, and similar cognitive impairments respond to dopaminergic treatments in other contexts. Therefore, DAT abnormalities following TBI may assist in treatment selection after TBI.
Cellular mechanisms regarding the neuroprotective effects of dopamine in TBI
Dopamine is a critical neurotransmitter for the normal function of the hippocampus, FC, and striatum [63-65]. The contribution of dopamine to intracellular signaling molecules makes dopaminergic regulation essentially important for multiple neuroprotection strategies for TBI (Figure 2).
Figure 2.
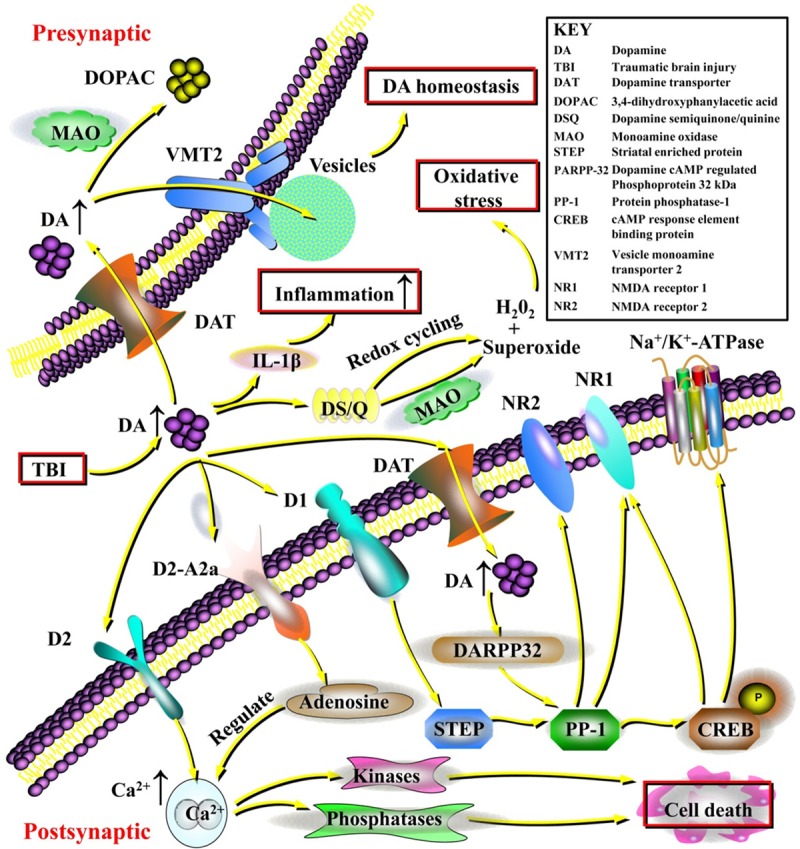
The contribution of dopamine to intracellular signaling and the essential neuroprotective effects of dopaminergic regulation following traumatic brain injury (TBI). Excessive dopamine could exert potent excitotoxic effects, inducing the generation of hydrogen peroxide (H2O2) and superoxide causing significant oxidative stress. In addition, dopamine can act as a potent inflammatory agent inducing inflammatory cytokines such as interleukin-1β (IL-1β), and dopaminergic regulation can potentially be important for reducing inflammation within the central nervous system (CNS). Excessive dopamine that is not metabolized by monoamine oxidase (MAO) could be recycled into vesicles by the vesicular monoamine transporter 2 (VMT2), maintaining dopamine homeostasis. Furthermore, the cellular mechanisms regarding the neuroprotective effects of dopamine in TBI have also been revealed. Dopamine exerts important effects on the regulation of Na+/K+-ATPase, Ca2+ release, and the N-methyl-D-aspartic acid receptor (NMDA) receptor through dopamine, cAMP-regulated phosphoprotein 32 kDa (DARPP-32) and protein phosphatase-1 (PP-1). Specifically, dopamine acting on D1 receptors can modify the activity of striatal enriched protein (STEP), which contributes to PP-1 activity. PP-1 also regulates nuclear transcription through cAMP response element-binding protein (CREB) phosphorylation and plays an important role in the phosphorylation of the NMDA NR1 subunit and the Na+/K+-ATPase. Dopamine signaling at the dopamine D2 receptor can induce increases in intracellular Ca2+ release and activation of calcium-dependent kinases and phosphatases, which is important for cell death signaling. Dopamine also forms a tight signaling relationship with adenosine via dopamine D2/adenosine A2a receptor interactions that can directly control intracellular Ca2+ release. Clarifying these dopaminergic signaling pathways could help identify promising therapies targeting dopamine and direct attention to this neurotransmitter for clinical applications.
Dopamine in cellular dysfunction
Dopamine, similar to glutamate, can also be a potent excitotoxic agent [66]. For example, high levels of dopamine in the synaptic cleft can be rapidly oxidized to form dopamine semiquinone/quinine [67]. In addition, oxidized dopamine via MAO activity [68] or redox cycling [69] can induce the generation of hydrogen peroxide and superoxide causing significant oxidative stress. Furthermore, dopamine signaling at the dopamine D2 receptor can induce increases in intracellular Ca2+ release and activation of calcium-dependent kinases and phosphatases, which is important for cell death signaling [70-72]. Animal models of TBI consistently result in widespread excitotoxic damage and increased amounts of oxidative stress in a number of different brain regions [73,74]. The initial increases in dopamine observed post TBI may promote excitotoxic disruption of and oxidative damage to the function of dopaminergic cells [56]. Interestingly, depleting dopaminergic projections into the striatum prior to the ischemic insult was found to be neuroprotective [75], indicating that dopamine could be neurotoxic.
Following TBI, there are known alterations in intracellular Ca2+ release [76,77], glutamatergic receptor function [78,79], and alterations in the function of Na+/K+-ATPase [80]. In addition, levels of excitatory amino acids and acetylcholine are markedly increased acutely in injured rats [81], and metabolic activity is also increased, resulting in adenosine triphosphate (ATP) depletion [82]. There are also decreases in both N-methyl-D-aspartic acid receptor (NMDA) NR1 and NR2 subunit expression a few hours after TBI [83]. Intriguingly, dopamine exerts an important effect on the regulation of Na+/K+-ATPase, calcium release, and the NMDA receptor through dopamine cAMP-regulated phosphoprotein 32 kDa (DARPP-32) and protein phosphatase-1 (PP-1) [84,85]. Dopaminergic signaling pathways can modify the phosphorylation of DARPP-32, which is followed by alterations in downstream PP-1 activity [85]. In hippocampal neurons, dopamine acting on D1 receptors can modify the activity of striatal enriched protein (STEP), which contributes to PP-1 activity [86,87]. PP-1 also regulates nuclear transcription through cAMP response element-binding protein (CREB) phosphorylation [88] and plays an important role in the phosphorylation of the NMDA NR1 subunit and Na+/K+-ATPase [87,89]. In addition to effects on PP-1, dopamine forms a tight signaling relationship with adenosine via dopamine D2/adenosine A2a receptor interactions that can directly control intracellular calcium release [90,91].
Dopamine in CNS inflammation
TBI causes a time-dependent upregulation of various genes during the postinjury period that include inflammatory cytokines such as interleukin 1 (IL-1), tumor necrosis factor (TNF), cyclooxygenase (COX) 1 and 2 and prostaglandin (PG) synthases. These may contribute to inflammation in the brain [92]. Thus, the neurodegenerative changes after TBI may be associated with inflammatory responses [93]. Moreover, significant decreases in TH-positive expression in the surviving dopaminergic neurons of the SN pars compacta (SNpc) and increased α-synuclein accumulation in the inflammation-infiltrated SN of rats exposed to chronic TBI were shown [34]. These phenomena may be one of the critical mechanisms by which dopamine transmission is impaired after TBI [33]. Strategies to reduce neuronal inflammation in TBI have provided benefits in functional outcomes [94-96]. However, the potential neuroprotective role of inflammatory cells cannot be ignored, and direct inhibition of inflammation may cause various side effects [97,98].
Dopamine can act as a potent inflammatory agent within the CNS. It is known that excessive dopamine levels could induce a proinflammatory environment [99], and inflammatory factors could be further augmented by dopamine supplementation, which can further increase interleukin-1β (IL-1β) production [100,101]. Intriguingly, in TBI, blocking IL-1β can be beneficial [94]. There is also the recognized vulnerability of dopaminergic neurons to the inflammatory cascade [102-104]. It has also been shown that drugs with dopaminergic action can reduce inflammation within the CNS [105]. This finding suggests that while endogenous dopamine can activate inflammatory pathways, activation of dopamine receptors with therapeutics may still provide reductions in inflammation. This dual nature of dopaminergic signaling also demonstrates the dopaminergic signaling complexities that need to be better understood as therapies targeting dopamine move towards clinical application.
Experimental evidence for the neuroprotective effects of dopamine against TBI impairments: beneficial outcomes
Multiple studies have demonstrated that cerebral autoregulation is absent or impaired in significant numbers of patients after TBI, even when values of cerebral perfusion pressure (CPP) and cerebral blood flow (CBF) were normal [106]. When autoregulation is impaired, decreases in CPP result in decreases in CBF; in moderate/severe TBI, such decreases in CBF may reach ischemic levels or lead to severe stroke, further augmenting various secondary injuries. Many retrospective studies have observed that impaired cerebral autoregulation could be associated with worsened cognitive outcomes (Glasgow Outcome Scale) [107-109]. In addition, previous research has indicated that phenylephrine (Phe), norepinephrine (NE), and epinephrine (EPI) all prevented reductions in CBF associated with fluid percussion injury (FPI) and limited neuronal cell necrosis in hippocampal areas CA1 and CA3 as a function of age and sex [110,111]. While cognition depends on more than the hippocampus, and cognitive testing was not performed in these studies, such results do suggest that support with vasoactive agents may affect cognitive outcomes after TBI.
Previously, it has been shown that dopamine equally protected cerebral autoregulation in male and female newborn pigs [112], indicating that dopamine improves outcomes irrespective of age and/or sex. However, other recent studies using different vasoactive agents have observed conflicting effects, building the argument for use of a precision medicine approach in the treatment of TBI. For example, Phe and NE have been shown to worsen cerebral autoregulation and histopathology in male but not female newborn piglets and male and female juvenile pigs after FPI [113,114]. In contrast, EPI prevents impairment of cerebral autoregulation and histopathology in male and female newborn and female juvenile pigs but not male juvenile pigs after FPI [111].
Since dopamine prevents impairments in cerebral autoregulation in both ages and sexes, dopamine should be recommended for the promotion of cognitive improvement after TBI independent of age and sex. A recent study [114] has indicated that dopamine protects autoregulation and prevents hippocampal neuronal cell necrosis via blocking ERK after brain injury in both male and female juvenile pigs. These results were novel in that they showed that dopamine was the only vasoactive agent studied thus far that improved outcome after TBI in both ages and sexes, and the authors suggested that dopamine should be explored as the vasoactive agent of choice in the treatment of pediatric TBI irrespective of age and sex for improving outcomes. However, a limitation was that histology was performed at an early time point; differences between treatment groups and sexes may disappear as more neurons die beyond the early time point. Therefore, additional studies will be needed to determine if the prevention of the loss of neurovascular unit integrity durably improves cerebral hemodynamics and cognitive function after pediatric TBI.
Influence of dopamine-related genes on neurobehavioral recovery after TBI
In clinical practice, it is common to observe different functional outcomes in individuals with similar degrees of TBI. Similarly, individual responses to medications can also vary enormously. These differences suggest that host factors may play an important role in the outcomes. Host genotype might be one such factor. Currently, although some important preliminary findings have been found [115,116], which will be discussed in the following text, our knowledge of the role of genetic factors in response to trauma and recovery from trauma, including reactivity to drugs, is limited and more research is needed.
Dopamine could be important for plasticity by enhancing synaptogenesis and neural sprouting [117], for recovery of motor function after TBI [118], and for cognitive reserve and the treatment of cognitive deficits after TBI [119]. Thus, polymorphisms in genes involved in the dopaminergic system could have great potential to influence behavioral outcomes after TBI. Candidate genes include those coding for dopamine receptor subtypes, dopamine reuptake (DAT), and dopamine metabolism (catechol-o-methyl transferase or COMT) [120]. Interestingly, positron emission tomography (PET) studies have shown changes in striatal D2 binding following working memory training [121], and lower dopamine D2 receptor densities have been associated with better motor sequence learning in healthy adults [122]. Recently, children and adolescents who were carriers of ANKK1 rs1800497 demonstrated greater improvements during working memory training than those who were not carriers [123]. Similarly, variation in SLC6A3 influenced improvements in working memory following cognitive training in preschool and school-age children [124,125]. Taken together, these studies suggest that the ANKK1 and SLC6A3 genes may provide higher cognitive [123] or neural [126] plasticity that may be especially relevant to recovery from injury to the brain.
The study of Treble-Barna et al. [127] was the first to provide preliminary evidence for the influence of variation in dopamine-related genes on neurobehavioral recovery following TBI in children. They examined the association of dopamine-related genes with short- and long-term neurobehavioral recovery in children who had sustained early childhood TBI. Genetic variation within the SLC6A3 (rs464049 and rs460000) gene was found to be associated with neurobehavioral recovery trajectories over time following TBI. In addition, genetic variation within ANKK1 (rs1800497 and rs2734849) and SLC6A3 (rs464049, rs460000, and rs1042098) was also associated with short- and long-term neurobehavioral recovery following TBI. The authors demonstrated that genetic variation in genes involved in D2 receptor (ANKK1) and DAT (SLC6A3) expression and density plays a role in neurobehavioral recovery following TBI.
The effect of genetic variation on treatment outcomes, including potential responses to drugs, is less advanced. However, it is encouraging that even with relatively small sample sizes, several groups have found genetic effects that influence cognitive outcomes. Larger sample sizes are needed to replicate these findings.
The clinical effectiveness and therapeutic significance of dopamine agonists for improving behavior after TBI
There has been longstanding evidence that dopaminergic therapy improves neuropsychiatric outcomes in TBI and non-TBI conditions. However, until recently, there has not been a rigorous evaluation of these findings in double-blind RCTs. To date, much of the evidence has come from case reports, case series or open-label trials. To date, only six studies were found to provide data on patients who received dopaminergic therapy [30,128-131], including the four drugs amantadine, levodopa, bromocriptine and rotigotine (Table 1). There is a need for larger prospective, double-blind RCTs with these medications using standardized criteria and outcomes to fully understand their effectiveness in this patient group.
Table 1.
Summary of clinical and experimental studies involving dopamine agonists for improving TBI outcomes

|
Amantadine is usually prescribed as an antiparkinsonian agent, as a treatment for neuroleptic-induced extrapyramidal symptoms, and as an antiviral agent. Amantadine appears to be a safe and effective means of reducing irritability and aggression among individuals who have had TBI for greater than 6 months’ duration and have sufficient creatinine clearance [30].
Levodopa has long been used for the treatment of Parkinson’s disease (PD). Lokk et al. [132] demonstrated that patients administered levodopa showed improvements in stroke severity over time. There were no side effects reported, and their findings will redirect attention to the clinical benefits of this type of drug treatment in rehabilitation. Future studies should address issues of optimal therapeutic windows and dosage of the medication as well as identify those patients with stroke who are likely to benefit from this treatment.
Bromocriptine, a direct dopamine agonist affecting primarily D2 receptors, also seems to have activity related to specific executive functions and attentional abilities in both animal and human studies. Whyte et al. [130] indicated that bromocriptine at a dose of 5 mg given twice a day to individuals with attentional complaints after TBI did not enhance attentional skills and may have been associated with an excess of adverse events. It is not clear whether intermittent dosing or lower doses might be beneficial.
Rotigotine has high affinity for the D1 receptor compared with many other available oral dopamine agonists. Gorgoraptis et al. [131] explored whether the dopamine agonist rotigotine would have a beneficial effect on hemispatial neglect in stroke patients in a double-blind, randomized, placebo-controlled experiment. The authors suggested a beneficial role of dopaminergic modulation on visual search and selective attention in patients with hemispatial neglect following stroke.
In addition, there could also be many dopamine agonists that have not been explored in patients. For example, methylphenidate (MPH) treatment in experimental models demonstrated cognitive benefit after cortical impact injuries [133-135]. Amphetamine (AMPH) use in experimental models of TBI and selective cortical injury models has also been shown to accelerate recovery [136-142]. These dopamine agonists could be of potential therapeutic effect for TBI patients, although no clinical data have been reported.
However, the side effects of dopamine-related treatment modalities in TBI should also be considered. Contrasting effects of dopamine therapy have been shown in TBI animal models. Worsening of the brain swelling processes subsequent to the effects of vasopressor therapy in a clinical setting needs to be more carefully evaluated [143]. There are various molecular changes in TBI with no clear-cut therapies available. Clarification of these molecular changes in the dopamine system in TBI may lead to novel therapeutic approaches in the near future.
Acknowledgements
This work is supported by grants from National Natural Science Foundation of China (Nos. 81372714, 81672480, 81872065, 81802506), Liaoning Provincial Natural Science Foundation of China (No. 201602244), Liaoning province innovation talents support program in Colleges and Universities (LR2016023), Distinguished Professor Project of Liaoning Province, Special Grant for Translational Medicine, Dalian Medical University (No. 2015002), Basic research projects in colleges and universities of Liaoning Province (No. LQ2017033).
Disclosure of conflict of interest
None.
Abbreviations
- TBI
traumatic brain injury
- FC
frontal cortex
- NAC
nucleus accumbens
- CNS
central nervous system
- MAO
monoamine oxidase
- DAT
dopamine transporter
- VMT2
vesicular monoamine transporter 2
- SN
substantia nigra
- RRF
retrorubral field
- VTA
ventral tegmental area
- DARPP-32
dopamine cAMP regulated phosphoprotein 32 kDa
- PP-1
protein phosphatase-1
- STEP
striatal enriched protein
- CREB
cAMP response element binding protein
- IL-1
interleukin 1
- TNF
tumor necrosis factor
- COX
cyclooxygenase
- PG
prostaglandin
- CPP
cerebral perfusion pressure
- CBF
cerebral blood flow
- Phe
phenylephrine
- NE
norepinephrine
- EPI
epinephrine
- FPI
fluid percussion injury
- PET
positron emission tomography
- MPH
methylphenidate
- AMPH
amphetamine
References
- 1.Carbonara M, Fossi F, Zoerle T, Ortolano F, Moro F, Pischiutta F, Zanier ER, Stocchetti N. Neuroprotection in traumatic brain injury: mesenchymal stromal cells can potentially overcome some limitations of previous clinical trials. Front Neurol. 2018;9:885. doi: 10.3389/fneur.2018.00885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong VS, Langley B. Epigenetic changes following traumatic brain injury and their implications for outcome, recovery and therapy. Neurosci Lett. 2016;625:26–33. doi: 10.1016/j.neulet.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaur P, Sharma S. Recent advances in pathophysiology of traumatic brain injury. Curr Neuropharmacol. 2018;16:1224–1238. doi: 10.2174/1570159X15666170613083606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen YH, Huang EY, Kuo TT, Miller J, Chiang YH, Hoffer BJ. Impact of traumatic brain injury on dopaminergic transmission. Cell Transplant. 2017;26:1156–1168. doi: 10.1177/0963689717714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kempadoo KA, Mosharov EV, Choi SJ, Sulzer D, Kandel ER. Dopamine release from the locus coeruleus to the dorsal hippocampus promotes spatial learning and memory. Proc Natl Acad Sci U S A. 2016;113:14835–14840. doi: 10.1073/pnas.1616515114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McNamara CG, Dupret D. Two sources of dopamine for the hippocampus. Trends Neurosci. 2017;40:383–384. doi: 10.1016/j.tins.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howe MW, Tierney PL, Sandberg SG, Phillips PE, Graybiel AM. Prolonged dopamine signalling in striatum signals proximity and value of distant rewards. Nature. 2013;500:575–9. doi: 10.1038/nature12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye Y, Mastwal S, Cao VY, Ren M, Liu Q, Zhang W, Elkahloun AG, Wang KH. Dopamine is required for activity-dependent amplification of Arc mRNA in developing postnatal frontal cortex. Cereb Cortex. 2017;27:3600–3608. doi: 10.1093/cercor/bhw181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trujillo P, van Wouwe NC, Lin YC, Stark AJ, Petersen KJ, Kang H, Zald DH, Donahue MJ, Claassen DO. Dopamine effects on frontal cortical blood flow and motor inhibition in Parkinson’s disease. Cortex. 2019;115:99–111. doi: 10.1016/j.cortex.2019.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bales JW, Kline AE, Wagner AK, Dixon CE. Targeting dopamine in acute traumatic brain injury. Open Drug Discov J. 2010;2:119–128. doi: 10.2174/1877381801002010119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frenette AJ, Kanji S, Rees L, Williamson DR, Perreault MM, Turgeon AF, Bernard F, Fergusson DA. Efficacy and safety of dopamine agonists in traumatic brain injury: a systematic review of randomized controlled trials. J Neurotrauma. 2012;29:1–18. doi: 10.1089/neu.2011.1812. [DOI] [PubMed] [Google Scholar]
- 12.Kraus MF, Smith GS, Butters M, Donnell AJ, Dixon E, Yilong C, Marion D. Effects of the dopaminergic agent and NMDA receptor antagonist amantadine on cognitive function, cerebral glucose metabolism and D2 receptor availability in chronic traumatic brain injury: a study using positron emission tomography (PET) Brain Inj. 2005;19:471–9. doi: 10.1080/02699050400025059. [DOI] [PubMed] [Google Scholar]
- 13.Sami MB, Faruqui R. The effectiveness of dopamine agonists for treatment of neuropsychiatric symptoms post brain injury and stroke. Acta Neuropsychiatr. 2015;27:317–26. doi: 10.1017/neu.2015.17. [DOI] [PubMed] [Google Scholar]
- 14.Daba Feyissa D, Sialana FJ, Keimpema E, Kalaba P, Paunkov A, Engidawork E, Höger H, Lubec G, Korz V. Dopamine type 1- and 2-like signaling in the modulation of spatial reference learning and memory. Behav Brain Res. 2019;362:173–180. doi: 10.1016/j.bbr.2019.01.028. [DOI] [PubMed] [Google Scholar]
- 15.Solari N, Hangya B. Cholinergic modulation of spatial learning, memory and navigation. Eur J Neurosci. 2018;48:2199–2230. doi: 10.1111/ejn.14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodman J, Packard MG. The role of the dorsal striatum in extinction: a memory systems perspective. Neurobiol Learn Mem. 2018;150:48–55. doi: 10.1016/j.nlm.2018.02.028. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y, Wang JZ. From structure to behavior in basolateral amygdala-hippocampus circuits. Front Neural Circuits. 2017;11:86. doi: 10.3389/fncir.2017.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raz A. Anatomy of attentional networks. Anat Rec B New Anat. 2004;281:21–36. doi: 10.1002/ar.b.20035. [DOI] [PubMed] [Google Scholar]
- 19.Kline AE, Leary JB, Radabaugh HL, Cheng JP, Bondi CO. Combination therapies for neurobehavioral and cognitive recovery after experimental traumatic brain injury: is more better? Prog Neurobiol. 2016;142:45–67. doi: 10.1016/j.pneurobio.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eme R. Neurobehavioral outcomes of mild traumatic brain injury: a mini review. Brain Sci. 2017;7 doi: 10.3390/brainsci7050046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fortress AM, Avcu P, Wagner AK, Dixon CE, Pang KCH. Experimental traumatic brain injury results in estrous cycle disruption, neurobehavioral deficits, and impaired GSK3β/β-catenin signaling in female rats. Exp Neurol. 2019;315:42–51. doi: 10.1016/j.expneurol.2019.01.017. [DOI] [PubMed] [Google Scholar]
- 22.Woytowicz EJ, Sours C, Gullapalli RP, Rosenberg J, Westlake KP. Modulation of working memory load distinguishes individuals with and without balance impairments following mild traumatic brain injury. Brain Inj. 2018;32:191–199. doi: 10.1080/02699052.2017.1403045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruet A, Bayen E, Jourdan C, Ghout I, Meaude L, Lalanne A, Pradat-Diehl P, Nelson G, Charanton J, Aegerter P, Vallat-Azouvi C, Azouvi P. A detailed overview of long-term outcomes in severe traumatic brain injury eight years post-injury. Front Neurol. 2019;10:120. doi: 10.3389/fneur.2019.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ojo JO, Mouzon B, Algamal M, Leary P, Lynch C, Abdullah L, Evans J, Mullan M, Bachmeier C, Stewart W, Crawford F. Chronic repetitive mild traumatic brain injury results in reduced cerebral blood flow, axonal injury, gliosis, and increased T-Tau and Tau oligomers. J Neuropathol Exp Neurol. 2016;75:636–55. doi: 10.1093/jnen/nlw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Filley CM, Kelly JP. White matter and cognition in traumatic brain injury. J Alzheimers Dis. 2018;65:345–362. doi: 10.3233/JAD-180287. [DOI] [PubMed] [Google Scholar]
- 26.Palacios EM, Sala-Llonch R, Junque C, Roig T, Tormos JM, Bargallo N, Vendrell P. White matter/gray matter contrast changes in chronic and diffuse traumatic brain injury. J Neurotrauma. 2013;30:1991–4. doi: 10.1089/neu.2012.2836. [DOI] [PubMed] [Google Scholar]
- 27.Kirov II, Tal A, Babb JS, Lui YW, Grossman RI, Gonen O. Diffuse axonal injury in mild traumatic brain injury: a 3D multivoxel proton MR spectroscopy study. J Neurol. 2013;260:242–52. doi: 10.1007/s00415-012-6626-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutson CB, Lazo CR, Mortazavi F, Giza CC, Hovda D, Chesselet MF. Traumatic brain injury in adult rats causes progressive nigrostriatal dopaminergic cell loss and enhanced vulnerability to the pesticide paraquat. J Neurotrauma. 2011;28:1783–801. doi: 10.1089/neu.2010.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen YH, Huang EY, Kuo TT, Ma HI, Hoffer BJ, Tsui PF, Tsai JJ, Chou YC, Chiang YH. Dopamine release impairment in striatum after different levels of cerebral cortical fluid percussion injury. Cell Transplant. 2015;24:2113–28. doi: 10.3727/096368914X683584. [DOI] [PubMed] [Google Scholar]
- 30.Hammond FM, Bickett AK, Norton JH, Pershad R. Effectiveness of amantadine hydrochloride in the reduction of chronic traumatic brain injury irritability and aggression. J Head Trauma Rehabil. 2014;29:391–9. doi: 10.1097/01.HTR.0000438116.56228.de. [DOI] [PubMed] [Google Scholar]
- 31.Huang EY, Tsui PF, Kuo TT, Tsai JJ, Chou YC, Ma HI, Chiang YH, Chen YH. Amantadine ameliorates dopamine-releasing deficits and behavioral deficits in rats after fluid percussion injury. PLoS One. 2014;9:e86354. doi: 10.1371/journal.pone.0086354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saddoris MP, Sugam JA, Cacciapaglia F, Carelli RM. Rapid dopamine dynamics in the accumbens core and shell: learning and action. Front Biosci (Elite Ed) 2013;5:273–88. doi: 10.2741/e615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat. 2015;11:97–106. doi: 10.2147/NDT.S65815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Acosta SA, Tajiri N, de la Pena I, Bastawrous M, Sanberg PR, Kaneko Y, Borlongan CV. Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson’s disease. J Cell Physiol. 2015;230:1024–32. doi: 10.1002/jcp.24830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Foll B, Gallo A, Le Strat Y, Lu L, Gorwood P. Genetics of dopamine receptors and drug addiction: a comprehensive review. Behav Pharmacol. 2009;20:1–17. doi: 10.1097/FBP.0b013e3283242f05. [DOI] [PubMed] [Google Scholar]
- 36.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi H, Takano H, Kodaka F, Arakawa R, Yamada M, Otsuka T, Hirano Y, Kikyo H, Okubo Y, Kato M, Obata T, Ito H, Suhara T. Contribution of dopamine D1 and D2 receptors to amygdala activity in human. J Neurosci. 2010;30:3043–7. doi: 10.1523/JNEUROSCI.5689-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernal S, Miner P, Abayev Y, Kandova E, Gerges M, Touzani K, Sclafani A, Bodnar RJ. Role of amygdala dopamine D1 and D2 receptors in the acquisition and expression of fructose-conditioned flavor preferences in rats. Behav Brain Res. 2009;205:183–90. doi: 10.1016/j.bbr.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinto A, Sesack SR. Ultrastructural analysis of prefrontal cortical inputs to the rat amygdala: spatial relationships to presumed dopamine axons and D1 and D2 receptors. Brain Struct Funct. 2008;213:159–75. doi: 10.1007/s00429-008-0180-6. [DOI] [PubMed] [Google Scholar]
- 40.Shinohara R, Taniguchi M, Ehrlich AT, Yokogawa K, Deguchi Y, Cherasse Y, Lazarus M, Urade Y, Ogawa A, Kitaoka S, Sawa A, Narumiya S, Furuyashiki T. Dopamine D1 receptor subtype mediates acute stress-induced dendritic growth in excitatory neurons of the medial prefrontal cortex and contributes to suppression of stress susceptibility in mice. Mol Psychiatry. 2018;23:1717–1730. doi: 10.1038/mp.2017.177. [DOI] [PubMed] [Google Scholar]
- 41.Rangel-Barajas C, Coronel I, Florán B. Dopamine receptors and neurodegeneration. Aging Dis. 2015;6:349–68. doi: 10.14336/AD.2015.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Louin G, Besson VC, Royo NC, Bonnefont-Rousselot D, Marchand-Verrecchia C, Plotkine M, Jafarian-Tehrani M. Cortical calcium increase following traumatic brain injury represents a pitfall in the evaluation of Ca2+-independent NOS activity. J Neurosci Methods. 2004;138:73–9. doi: 10.1016/j.jneumeth.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 43.Hiltebrand LB, Krejci V, Sigurdsson GH. Effects of dopamine, dobutamine, and dopexamine on microcirculatory blood flow in the gastrointestinal tract during sepsis and anesthesia. Anesthesiology. 2004;100:1188–97. doi: 10.1097/00000542-200405000-00022. [DOI] [PubMed] [Google Scholar]
- 44.Avila-Luna A, Gálvez-Rosas A, Alfaro-Rodríguez A, Reyes-Legorreta C, Garza-Montaño P, González-Piña R, Bueno-Nava A. Dopamine D1 receptor activation maintains motor coordination in injured rats but does not accelerate the recovery of the motor coordination deficit. Behav Brain Res. 2018;336:145–150. doi: 10.1016/j.bbr.2017.08.026. [DOI] [PubMed] [Google Scholar]
- 45.Guérin JP, Levraut J, Samat-Long C, Leverve X, Grimaud D, Ichai C. Effects of dopamine and norepinephrine on systemic and hepatosplanchnic hemodynamics, oxygen exchange, and energy balance in vasoplegic septic patients. Shock. 2005;23:18–24. doi: 10.1097/01.shk.0000150549.45338.6c. [DOI] [PubMed] [Google Scholar]
- 46.Jamal M, Tsukamoto I, Takata M, Ito A, Tanaka N, Miki T, Takakura A, Ameno K, Kubota Y, Konishi R, Kinoshita H. COA-Cl induces dopamine release and tyrosine hydroxylase phosphorylation: In vivo reverse microdialysis and in vitro analysis. Brain Res. 2019;1706:68–74. doi: 10.1016/j.brainres.2018.10.026. [DOI] [PubMed] [Google Scholar]
- 47.Hamanaka Y, Minoura R, Nishino H, Miura T, Mizunami M. Dopamine- and tyrosine hydroxylase-immunoreactive neurons in the brain of the American cockroach, Periplaneta Americana. PLoS One. 2016;11:e0160531. doi: 10.1371/journal.pone.0160531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu X, Grove JC, Hirano AA, Brecha NC, Barnes S. Dopamine D1 receptor modulation of calcium channel currents in horizontal cells of mouse retina. J Neurophysiol. 2016;116:686–97. doi: 10.1152/jn.00990.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karelina K, Gaier KR, Weil ZM. Traumatic brain injuries during development disrupt dopaminergic signaling. Exp Neurol. 2017;297:110–117. doi: 10.1016/j.expneurol.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bales JW, Yan HQ, Ma X, Li Y, Samarasinghe R, Dixon CE. The dopamine and cAMP regulated phosphoprotein, 32 kDa (DARPP-32) signaling pathway: a novel therapeutic target in traumatic brain injury. Exp Neurol. 2011;229:300–7. doi: 10.1016/j.expneurol.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foster JD, Vaughan RA. Phosphorylation mechanisms in dopamine transporter regulation. J Chem Neuroanat. 2017;83-84:10–18. doi: 10.1016/j.jchemneu.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naoi M, Maruyama W, Nagy GM. Dopamine-derived salsolinol derivatives as endogenous monoamine oxidase inhibitors: occurrence, metabolism and function in human brains. Neurotoxicology. 2004;25:193–204. doi: 10.1016/S0161-813X(03)00099-8. [DOI] [PubMed] [Google Scholar]
- 53.Patel J, Mooslehner KA, Chan PM, Emson PC, Stamford JA. Presynaptic control of striatal dopamine neurotransmission in adult vesicular monoamine transporter 2 (VMAT2) mutant mice. J Neurochem. 2003;85:898–910. doi: 10.1046/j.1471-4159.2003.01732.x. [DOI] [PubMed] [Google Scholar]
- 54.Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, Tomac AC. Characterization of a mouse strain expressing Cre recombinase from the 39 untranslated region of the dopamine transporter locus. Genesis. 2006;44:383–90. doi: 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- 55.Yan HQ, Kline AE, Ma X, Li Y, Dixon CE. Traumatic brain injury reduces dopamine transporter protein expression in the rat frontal cortex. Neuroreport. 2002;13:1899–901. doi: 10.1097/00001756-200210280-00013. [DOI] [PubMed] [Google Scholar]
- 56.Wagner AK, Sokoloski JE, Ren D, Chen X, Khan AS, Zafonte RD, Michael AC, Dixon CE. Controlled cortical impact injury affects dopaminergic transmission in the rat striatum. J Neurochem. 2005;95:457–65. doi: 10.1111/j.1471-4159.2005.03382.x. [DOI] [PubMed] [Google Scholar]
- 57.Shimada R, Abe K, Furutani R, Kibayashi K. Changes in dopamine transporter expression in the midbrain following traumatic brain injury: an immunohistochemical and in situ hybridization study in a mouse model. Neurol Res. 2014;36:239–46. doi: 10.1179/1743132813Y.0000000289. [DOI] [PubMed] [Google Scholar]
- 58.Fu Y, Yuan Y, Halliday G, Rusznák Z, Watson C, Paxinos G. A cytoarchitectonic and chemoarchitectonic analysis of the dopamine cell groups in the substantia nigra, ventral tegmental area, and retrorubral field in the mouse. Brain Struct Funct. 2012;217:591–612. doi: 10.1007/s00429-011-0349-2. [DOI] [PubMed] [Google Scholar]
- 59.Haleem DJ. Extending therapeutic use of psychostimulants: focus on serotonin-1A receptor. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:170–80. doi: 10.1016/j.pnpbp.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 60.Hu Z, Cooper M, Crockett DP, Zhou R. Differentiation of the midbrain dopaminergic pathways during mouse development. J Comp Neurol. 2004;476:301–11. doi: 10.1002/cne.20230. [DOI] [PubMed] [Google Scholar]
- 61.Hasue RH, Shammah-Lagnado SJ. Origin of the dopaminergic innervation of the central extended amygdala and accumbens shell: a combined retrograde tracing and immunohistochemical study in the rat. Comp Neurol. 2002;454:15–33. doi: 10.1002/cne.10420. [DOI] [PubMed] [Google Scholar]
- 62.Jenkins PO, De Simoni S, Bourke NJ, Fleminger J, Scott G, Towey DJ, Svensson W, Khan S, Patel M, Greenwood R, Cole JH, Sharp DJ. Dopaminergic abnormalities following traumatic brain injury. Brain. 2018;141:797–810. doi: 10.1093/brain/awx357. [DOI] [PubMed] [Google Scholar]
- 63.Kahnt T, Tobler PN. Dopamine modulates the functional organization of the orbitofrontal cortex. J Neurosci. 2017;37:1493–1504. doi: 10.1523/JNEUROSCI.2827-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kahnt T, Tobler PN. Dopamine regulates stimulus generalization in the human hippocampus. Elife. 2016;5:e12678. doi: 10.7554/eLife.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoshimi K, Kumada S, Weitemier A, Jo T, Inoue M. Reward-induced phasic dopamine release in the monkey ventral striatum and putamen. PLoS One. 2015;10:e0130443. doi: 10.1371/journal.pone.0130443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vaarmann A, Kovac S, Holmström KM, Gandhi S, Abramov AY. Dopamine protects neurons against glutamate-induced excitotoxicity. Cell Death Dis. 2013;4:e455. doi: 10.1038/cddis.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, Savas JN, Kiskinis E, Zhuang X, Krüger R, Surmeier DJ, Krainc D. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science. 2017;357:1255–1261. doi: 10.1126/science.aam9080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smythies J, Edelstein L. The desferrioxamine-prochlorperazine coma-clue to the role of dopamine-iron recycling in the synthesis of hydrogen peroxide in the brain. Front Mol Neurosci. 2014;7:74. doi: 10.3389/fnmol.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu M, Fritsch I. Application of electrochemical redox cycling: toward differentiation of dopamine and norepinephrine. Anal Chem. 2016;88:5574–8. doi: 10.1021/acs.analchem.6b00427. [DOI] [PubMed] [Google Scholar]
- 70.Azdad K, Gall D, Woods AS, Ledent C, Ferre S, Schiffmann SN. Dopamine D2 and adenosine A2A receptors regulate NMDA-mediated excitation in accumbens neurons through A2A-D2 receptor heteromerization. Neuropsychopharmacology. 2009;34:972–986. doi: 10.1038/npp.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta] 1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–8995. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.So CH, Verma V, Alijaniaram M, Cheng R, Rashid AJ, O’Dowd BF, George SR. Calcium signaling by dopamine D5 receptor and D5-D2 receptor hetero-oligomers occurs by a mechanism distinct from that for dopamine D1-D2 receptor heterooligomers. Mol Pharmacol. 2009;75:843–854. doi: 10.1124/mol.108.051805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 74.Yang T, Kong B, Gu JW, Kuang YQ, Cheng L, Yang WT, Xia X, Shu HF. Anti-apoptotic and anti-oxidative roles of quercetin after traumatic brain injury. Cell Mol Neurobiol. 2014;34:797–804. doi: 10.1007/s10571-014-0070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Momosaki S, Ito M, Yamato H, Iimori H, Sumiyoshi H, Morimoto K, Imamoto N, Watabe T, Shimosegawa E, Hatazawa J, Abe K. Longitudinal imaging of the availability of dopamine transporter and D2 receptor in rat striatum following mild ischemia. J Cereb Blood Flow Metab. 2017;37:605–613. doi: 10.1177/0271678X16635183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tehse J, Taghibiglou C. The overlooked aspect of excitotoxicity: glutamate-independent excitotoxicity in traumatic brain injuries. Eur J Neurosci. 2018 doi: 10.1111/ejn.14307. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 77.Kulbe JR, Hall ED. Chronic traumatic encephalopathy-integration of canonical traumatic brain injury secondary injury mechanisms with tau pathology. Prog Neurobiol. 2017;158:15–44. doi: 10.1016/j.pneurobio.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koola MM. Galantamine-memantine combination for cognitive impairments due to electroconvulsive therapy, traumatic brain injury, and neurologic and psychiatric disorders: kynurenic acid and mismatch negativity target engagement. Prim Care Companion CNS Disord. 2018;20 doi: 10.4088/PCC.17nr02235. [DOI] [PubMed] [Google Scholar]
- 79.Armstead WM. NMDA and age dependent cerebral hemodynamics after traumatic brain injury. Exp Toxicol Pathol. 2004;56:75–81. doi: 10.1016/j.etp.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 80.Ross ST, Soltesz I. Selective depolarization of interneurons in the early posttraumatic dentate gyrus: involvement od the Na(+)/K(+)-ATPase. J Neurophysiol. 2000;83:2916–930. doi: 10.1152/jn.2000.83.5.2916. [DOI] [PubMed] [Google Scholar]
- 81.Kelso ML, Oestreich JH. Traumatic brain injury: central and peripheral role of α7 nicotinic acetylcholine receptors. Curr Drug Targets. 2012;13:631–6. doi: 10.2174/138945012800398964. [DOI] [PubMed] [Google Scholar]
- 82.Prieto R, Tavazzi B, Taya K, Barrios L, Amorini AM, Di Pietro V, Pascual JM, Marmarou A, Marmarou CR. Brain energy depletion in a rodent model of diffuse traumatic brain injury is not prevented with administration of sodium lactate. Brain Res. 2011;1404:39–49. doi: 10.1016/j.brainres.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kumar A, Zou L, Yuan X, Long Y, Yang K. N-methyl-D-aspartate receptors: transient loss of NR1/NR2A/NR2B subunits after traumatic brain injury in a rodent model. J Neurosci Res. 2002;67:781–786. doi: 10.1002/jnr.10181. [DOI] [PubMed] [Google Scholar]
- 84.Nishi A, Bibb JA, Matsuyama S, Hamada M, Higashi H, Nairn AC, Greengard P. Regulation of DARPP-32 dephosphorylation at PKA- and Cdk5-sites by NMDA and AMPA receptors: distinct roles of calcineurin and protein phosphatase-2A. J Neurochem. 2002;81:832–41. doi: 10.1046/j.1471-4159.2002.00876.x. [DOI] [PubMed] [Google Scholar]
- 85.Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- 86.Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- 87.Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nishi A, Shuto T. Potential for targeting dopamine/DARPP-32 signaling in neuropsychiatric and neurodegenerative disorders. Expert Opin Ther Targets. 2017;21:259–272. doi: 10.1080/14728222.2017.1279149. [DOI] [PubMed] [Google Scholar]
- 89.Flores-Hernández J, Cepeda C, Hernández-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS. Dopamine enhancement of NMDA currents in dissociated mediumsized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol. 2002;88:3010–3020. doi: 10.1152/jn.00361.2002. [DOI] [PubMed] [Google Scholar]
- 90.Canals M, Marcellino D, Fanelli F, Ciruela F, de Benedetti P, Goldberg SR, Neve K, Fuxe K, Agnati LF, Woods AS, Ferré S, Lluis C, Bouvier M, Franco R. Adenosine A2A-dopamine D2 receptor-receptor heteromerization: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Biol Chem. 2003;278:46741–9. doi: 10.1074/jbc.M306451200. [DOI] [PubMed] [Google Scholar]
- 91.Fuxe K, Ferré S, Canals M, Torvinen M, Terasmaa A, Marcellino D, Goldberg SR, Staines W, Jacobsen KX, Lluis C, Woods AS, Agnati LF, Franco R. Adenosine A2A and dopamine D2 heteromeric receptor complexes and their function. J Mol Neurosci. 2005;26:209–20. doi: 10.1385/JMN:26:2-3:209. [DOI] [PubMed] [Google Scholar]
- 92.Shojo H, Borlongan CV, Mabuchi T. Genetic and histological alterations reveal key role of prostaglandin synthase and cyclooxygenase 1 and 2 in traumatic brain injury-induced neuroinflammation in the cerebral cortex of rats exposed to moderate fluid percussion injury. Cell Transplant. 2017;26:1301–13. doi: 10.1177/0963689717715169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shojo H, Kaneko Y, Mabuchi T, Kibayashi K, Adachi N, Borlongan CV. Genetic and histologic evidence implicates role of inflammation in traumatic brain injury-induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience. 2010;171:1273–82. doi: 10.1016/j.neuroscience.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 94.Clausen F, Hånell A, Björk M, Hillered L, Mir AK, Gram H, Marklund N. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2009;30:385–96. doi: 10.1111/j.1460-9568.2009.06820.x. [DOI] [PubMed] [Google Scholar]
- 95.Kelso ML, Scheff SW, Pauly JR, Loftin CD. Effects of genetic deficiency of cyclooxygenase-1 or cyclooxygenase-2 on functional and histological outcomes following traumatic brain injury in mice. BMC Neurosci. 2009;10:108. doi: 10.1186/1471-2202-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Khan M, Im YB, Shunmugavel A, Gilg AG, Dhindsa RK, Singh AK, Singh I. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation. 2009;6:32. doi: 10.1186/1742-2094-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience. 2009;158:1021–1029. doi: 10.1016/j.neuroscience.2008.06.052. [DOI] [PubMed] [Google Scholar]
- 98.Kriz J. Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol. 2006;18:145–157. doi: 10.1615/critrevneurobiol.v18.i1-2.150. [DOI] [PubMed] [Google Scholar]
- 99.Farber K, Kettenmann H. Physiology of microglial cells. Brain Res Rev. 2005;48:133–143. doi: 10.1016/j.brainresrev.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 100.Barnum CJ, Eskow KL, Dupre K, Blandino P Jr, Deak T, Bishop C. Exogenous corticosterone reduces L-dopa induced dyskinesia in the hemi-parkinsonian rat: role for interleukin-1β. Neuroscience. 2008;156:30–41. doi: 10.1016/j.neuroscience.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jiang B, Xu S, Hou X, Pimentel DR, Brecher P, Cohen RA. Temporal control of NF-κB activation by ERK differentially regulates interleukin-1beta-induced gene expression. J Biol Chem. 2004;279:1323–9. doi: 10.1074/jbc.M307521200. [DOI] [PubMed] [Google Scholar]
- 102.Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Exp Mol Med. 2006;38:333–347. doi: 10.1038/emm.2006.40. [DOI] [PubMed] [Google Scholar]
- 103.Teismann P, Schulz JB. Cellular pathology of Parkinson’s disease: astrocytes, microglia, and inflammation. Cell Tissue Res. 2004;318:149–161. doi: 10.1007/s00441-004-0944-0. [DOI] [PubMed] [Google Scholar]
- 104.Mastroeni D, Grover A, Leonard B, Joyce JN, Coleman PD, Kozik B, Bellinger DL, Rogers J. Microglia responses to dopamine in a cell culture model of Parkinson’s disease. Neurobiol Aging. 2009;30:1805–1817. doi: 10.1016/j.neurobiolaging.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brustolim D, Ribeiro-dos-Santos R, Kast RE, Altschuler EL, Soares MB. A new chapter opens in anti-inflammatory treatments: the antidepressant buproprion lowers production of tumor necrosis factor-alpha and interferon-gamma in mice. Int Immunopharmacol. 2006;6:903–907. doi: 10.1016/j.intimp.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 106.Rangel-Castilla L, Gasco J, Nauta HJ, Okonkwo DO, Robertson CS. Cerebral pressure autoregulation in traumatic brain injury. Neurosurg Focus. 2008;25:E7. doi: 10.3171/FOC.2008.25.10.E7. [DOI] [PubMed] [Google Scholar]
- 107.Freeman SS, Udomphorn Y, Armstead WM, Fisk DM, Vavilala MS. Young age as a risk factor for impaired cerebral autoregulation after moderatesevere pediatric brain injury. Anesthesiology. 2008;108:588–595. doi: 10.1097/ALN.0b013e31816725d7. [DOI] [PubMed] [Google Scholar]
- 108.Sorrentino E, Diedler J, Kasprowicz M, Budohoski KP, Haubrich C, Smielewski P, Outtrim JG, Manktelow A, Hutchinson PJ, Pickard JD, Menon DK, Czosnyka M. Critical thresholds for cerebrovascular reactivity after traumatic brain injury. Neurocrit Care. 2012;16:258–266. doi: 10.1007/s12028-011-9630-8. [DOI] [PubMed] [Google Scholar]
- 109.Czonsyka M, Miller C. Monitoring of cerebral autoregulation. Neurocrit Care. 2014;21:S95–S102. doi: 10.1007/s12028-014-0046-0. [DOI] [PubMed] [Google Scholar]
- 110.Armstead WM, Riley J, Vavilala MS. Norepinephrine protects autoregulation and reduces hippocampal necrosis after traumatic brain injury via block of ERK MAPK and IL-6 in juvenile pigs. J Neurotrauma. 2016;33:1761–1767. doi: 10.1089/neu.2015.4290. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 111.Armstead WM, Riley J, Vavilala MS. Sex and age differences in epinephrine mechanisms and outcomes after brain injury. J Neurotrauma. 2017;34:666–1675. doi: 10.1089/neu.2016.4770. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 112.Armstead WM, Riley J, Vavilala MS. Dopamine prevents impairment of autoregulation after TBI in the newborn pig through inhibition of upregulation of ET-1 and ERK MAPK. Ped Crit Care Med. 2013;14:e103–e111. doi: 10.1097/PCC.0b013e3182712b44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Armstead WM, Kiessling JW, Bdeir K, Kofke WA, Vavilala MS. Adrenomedullin prevents sex dependent impairment of cerebal autoregulation during hypotension after piglet brain injury through inhibition of ERK MAPK upregulation. J Neurotrauma. 2010;27:391–402. doi: 10.1089/neu.2009.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Curvello V, Hekierski H, Pastor P, Vavilala MS, Armstead WM. Dopamine protects cerebral autoregulation and prevents hippocampal necrosis after traumatic brain injury via block of ERK MAPK in juvenile pigs. Brain Res. 2017;1670:118–124. doi: 10.1016/j.brainres.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 115.Jordan BD. Genetic influences on outcome following traumatic brain injury. Neurochem Res. 2007;32:905–915. doi: 10.1007/s11064-006-9251-3. [DOI] [PubMed] [Google Scholar]
- 116.Diaz-Arrastia R, Baxter VK, Diaz-Arrastia R, Baxter VK. Genetic factors in outcome after traumatic brain injury: what the human genome project can teach us about brain trauma. J Head Trauma Rehabil. 2006;21:361–74. doi: 10.1097/00001199-200607000-00007. [DOI] [PubMed] [Google Scholar]
- 117.Stroemer RP, Kent TA, Hulsebosch CE. Enhanced neocortical neural sprouting, synaptogenesis, and behavioral recovery with D-amphetamine therapy after neocortical infarction in rats. Stroke. 1998;29:2381–2393. doi: 10.1161/01.str.29.11.2381. [DOI] [PubMed] [Google Scholar]
- 118.Martinsson L, Eksborg S. Drugs for stroke recovery: the example of amphetamines. Drugs Aging. 2004;21:67–79. doi: 10.2165/00002512-200421020-00001. [DOI] [PubMed] [Google Scholar]
- 119.Whyte J, Vaccaro M, Grieg-Neff P, Hart T. Psychostimulant use in the rehabilitation of individuals with traumatic brain injury. J Head Trauma Rehabil. 2002;17:284–299. doi: 10.1097/00001199-200208000-00003. [DOI] [PubMed] [Google Scholar]
- 120.McAllister TW. Polymorphisms in genes modulating the dopamine system: do they inf luence outcome and response to medication after traumatic brain injury? J Head Trauma Rehabil. 2009;24:65–8. doi: 10.1097/HTR.0b013e3181996e6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bäckman L, Nyberg L, Soveri A, Johansson J, Andersson M, Dahlin E, Neely AS, Virta J, Laine M, Rinne JO. Effects of working-memory training on striatal dopamine release. Science. 2011;333:718. doi: 10.1126/science.1204978. [DOI] [PubMed] [Google Scholar]
- 122.Karabanov A, Cervenka S, de Manzano O, Forssberg H, Farde L, Ullén F. Dopamine D2 receptor density in the limbic striatum is related to implicit but not explicit movement sequence learning. Proc Natl Acad Sci U S A. 2010;107:7574–9. doi: 10.1073/pnas.0911805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Söderqvist S, Matsson H, Peyrard-Janvid M, Kere J, Klingberg T. Polymorphisms in the dopamine receptor 2 gene region influence improvements during working memory training in children and adolescents. J Cogn Neurosci. 2014;26:54–62. doi: 10.1162/jocn_a_00478. [DOI] [PubMed] [Google Scholar]
- 124.Brehmer Y, Westerberg H, Bellander M, Fürth D, Karlsson S, Bäckman L. Working memory plasticity modulated by dopamine transporter genotype. Neurosci Lett. 2009;467:117–120. doi: 10.1016/j.neulet.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 125.Söderqvist S, Bergman Nutley S, Peyrard-Janvid M, Matsson H, Humphreys K, Kere J, Klingberg T. Dopamine, working memory, and training induced plasticity: implications for developmental research. Dev Psychol. 2012;48:836–843. doi: 10.1037/a0026179. [DOI] [PubMed] [Google Scholar]
- 126.Klingberg T. Childhood cognitive development as a skill. Trends Cogn Sci. 2014;18:573–579. doi: 10.1016/j.tics.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 127.Treble-Barna A, Wade SL, Martin LJ, Pilipenko V, Yeates KO, Taylor HG, Kurowski BG. Influence of dopamine-related genes on neurobehavioral recovery after traumatic brain injury during early childhood. J Neurotrauma. 2017;34:1919–1931. doi: 10.1089/neu.2016.4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schneider WN, Drew-Cates J, Wong TM, Dombovy ML. Cognitive and behavioural efficacy of amantadine in acute traumatic brain injury: an initial double-blind placebo-controlled study. Brain Inj. 1999;13:863–72. doi: 10.1080/026990599121061. [DOI] [PubMed] [Google Scholar]
- 129.Meythaler JM, Brunner RC, Johnson A, Novack TA. Amantadine to improve neurorecovery in traumatic brain injury-associated diffuse axonal injury: a pilot double-blind randomized trial. J Head Trauma Rehabil. 2002;17:300–13. doi: 10.1097/00001199-200208000-00004. [DOI] [PubMed] [Google Scholar]
- 130.Whyte J, Vaccaro M, Grieb-Neff P, Hart T, Polansky M, Coslett HB. The effects of bromocriptine on attention deficits after traumatic brain injury: a placebo-controlled pilot study. Am J Phys Med Rehabil. 2008;87:85–99. doi: 10.1097/PHM.0b013e3181619609. [DOI] [PubMed] [Google Scholar]
- 131.Gorgoraptis N, Mah YH, Machner B, Singh-Curry V, Malhotra P, Hadji-Michael M, Cohen D, Simister R, Nair A, Kulinskaya E, Ward N, Greenwood R, Husain M. The effects of the dopamine agonist rotigotine on hemispatial neglect following stroke. Brain. 2012;135:2478–91. doi: 10.1093/brain/aws154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lokk J, Salman Roghani R, Delbari A. Effect of methylphenidate and/or levodopa coupled with physiotherapy on functional and motor recovery after stroke--a randomized, double-blind, placebo-controlled trial. Acta Neurol Scand. 2011;123:266–73. doi: 10.1111/j.1600-0404.2010.01395.x. [DOI] [PubMed] [Google Scholar]
- 133.Kline AE, Chen MJ, Tso-Olivas DY, Feeney DM. Methylphenidate treatment following ablationinduced hemiplegia in rat: experience during drug action alters effects on recovery of function. Pharmacol Biochem Behav. 1994;48:773–779. doi: 10.1016/0091-3057(94)90345-x. [DOI] [PubMed] [Google Scholar]
- 134.Kline AE, Yan HQ, Bao J, Marion DW, Dixon CE. Chronic methylphenidate treatment enhances water maze performance following traumatic brain injury in rats. Neurosci Lett. 2000;280:163–166. doi: 10.1016/s0304-3940(00)00797-7. [DOI] [PubMed] [Google Scholar]
- 135.Wagner AK, Drewencki LL, Chen X, Santos FR, Khan AS, Harun R, Torres GE, Michael AC, Dixon CE. Chronic methylphenidate treatment enhances striatal dopamine neurotransmission after experimental traumatic brain injury. J Neurochem. 2009;108:986–997. doi: 10.1111/j.1471-4159.2008.05840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chudasama Y, Nathwani F, Robbins TW. D-Amphetamine remediates attentional performance in rats with dorsal prefrontal lesions. Behav Brain Res. 2005;158:97–107. doi: 10.1016/j.bbr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 137.Dhillon HS, Dose JM, Prasad RM. Amphetamine administration improves neurochemical outcome of lateral fluid percussion brain injury in the rat. Brain Res. 1998;804:231–237. doi: 10.1016/s0006-8993(98)00639-8. [DOI] [PubMed] [Google Scholar]
- 138.Feeney DM, Gonzales A, Law WA. Amphetamine restores locomotor function after motor cortex injury in the rat. Proc West Pharmacol Soc. 1981;24:15–17. [PubMed] [Google Scholar]
- 139.Hovda DA, Sutton RL, Feeney DM. Amphetamine-induced recovery of visual cliff performance after bilateral visual cortex ablation in cats: measurements of depth perception thresholds. Behav Neurosci. 1989;103:574–584. doi: 10.1037//0735-7044.103.3.574. [DOI] [PubMed] [Google Scholar]
- 140.M’Harzi M, Willig F, Costa JC, Delacour J. d-Amphetamine enhances memory performance in rats with damage to the fimbria. Physiol Behav. 1998;42:575–579. doi: 10.1016/0031-9384(88)90160-6. [DOI] [PubMed] [Google Scholar]
- 141.Queen SA, Chen MJ, Feeney DM. d-Amphetamine attenuates decreased cerebral glucose utilization after unilateral sensorimotor cortex contusion in rats. Brain Res. 1997;777:42–50. doi: 10.1016/s0006-8993(97)00717-8. [DOI] [PubMed] [Google Scholar]
- 142.Ramic M, Emerick AJ, Bollnow MR, O’Brien TE, Tsai SY, Kartje GL. Axonal plasticity is associated with motor recovery following amphetamine treatment combined with rehabilitation after brain injury in the adult rat. Brain Res. 2006;1111:176–186. doi: 10.1016/j.brainres.2006.06.063. [DOI] [PubMed] [Google Scholar]
- 143.Hylands M, Toma A, Beaudoin N, Frenette AJ, D’Aragon F, Belley-Côté É, Charbonney E, Møller MH, Laake JH, Vandvik PO, Siemieniuk RA, Rochwerg B, Lauzier F, Green RS, Ball I, Scales D, Murthy S, Kwong JSW, Guyatt G, Rizoli S, Asfar P, Lamontagne F. Early vasopressor use following traumatic injury: a systematic review. BMJ Open. 2017;7:e017559. doi: 10.1136/bmjopen-2017-017559. [DOI] [PMC free article] [PubMed] [Google Scholar]