

Abstract
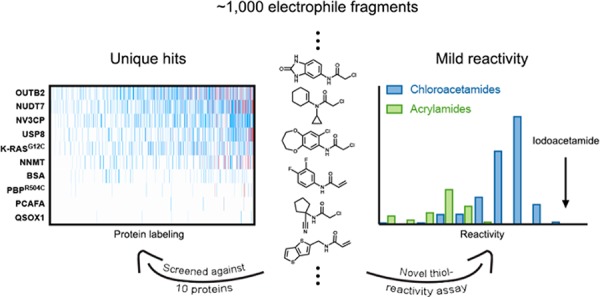
Covalent probes can display unmatched potency, selectivity, and duration of action; however, their discovery is challenging. In principle, fragments that can irreversibly bind their target can overcome the low affinity that limits reversible fragment screening, but such electrophilic fragments were considered nonselective and were rarely screened. We hypothesized that mild electrophiles might overcome the selectivity challenge and constructed a library of 993 mildly electrophilic fragments. We characterized this library by a new high-throughput thiol-reactivity assay and screened them against 10 cysteine-containing proteins. Highly reactive and promiscuous fragments were rare and could be easily eliminated. In contrast, we found hits for most targets. Combining our approach with high-throughput crystallography allowed rapid progression to potent and selective probes for two enzymes, the deubiquitinase OTUB2 and the pyrophosphatase NUDT7. No inhibitors were previously known for either. This study highlights the potential of electrophile-fragment screening as a practical and efficient tool for covalent-ligand discovery.
Introduction
Targeted covalent inhibitors have many advantages as chemical probes and drug candidates:1,2 in particular, prolonged duration of action,3 improved potency, and exquisite selectivity when nonconserved protein nucleophiles are targeted.4 There has, therefore, been increasing interest in covalently acting compounds, in both academia5 and the pharmaceutical industry.5−8 This trend is underlined by the recent FDA approval of the rationally designed covalent drugs ibrutinib, afatinib, osimertinib, and neratinib.
Discovering new covalent inhibitors, however, remains challenging. Historically, the most widespread approach to designing such inhibitors has relied on the incorporation of an electrophile into an already optimized reversible recognition element,4,9−11 most notably in kinase inhibitors.4,12−16 More recently, large-scale covalent virtual screens have emerged as a method for the discovery of covalent binders.17−23 While successful, in silico docking still has its limitations: it is limited to targets for which a crystal structure (or a high-quality model) is available, it cannot efficiently address protein flexibility, and it cannot predict the intrinsic reactivity of electrophiles and thus may result in highly reactive compounds. Empirical high-throughput screening (HTS) for covalent binders is typically avoided,24 owing to concerns about promiscuous activity.25−27 A major risk in screening large covalent libraries is that hits will be dominated by overly reactive compounds rather than by specific recognition.28
Fragment-based screening, which focuses on very low molecular weight compounds, is a successful hit discovery approach for reversible inhibitors29,30 that has led to several drugs and chemical probes.30,31 In comparison to traditional HTS, fragment-based screening offers better coverage of chemical space and higher probability of binding due to lower molecular complexity.32,33 The major limitation in fragment-based screening is the weak binding affinity of fragment hits, which not only necessitates very sensitive biophysical detection methods, coupled with elaborate validation cascades, to eliminate attendant artifacts but also makes progressing hits to potency difficult and expensive. In particular, it requires a large compound series with typically ambiguous structure–activity relationships, because no method to date can reliably rationalize which are the dominant interactions of the original fragment. Screening covalent fragments addresses these limitations. This is because covalent binders are easy to detect by mass spectrometry, because the dominant interaction is unambiguous (namely, the covalent bond), which simplifies the design of follow-up series, and because the primary hits are already potent.
A prominent covalent-fragment screening approach is disulfide tethering,34,35 which entails incubating a library of disulfide-containing fragments with the target. Disulfide exchange with the target cysteine selects for fragments that are reversibly stabilized in its vicinity. Disulfide tethering was successfully applied to a variety of targets containing both native and introduced cysteine residues.36 Recently, it led to the discovery of a promising K-RasG12C inhibitor.37 Disulfides are not, however, suitable as cellular probes, and replacing them with a suitable electrophile is, in general, no less challenging than starting from a reversible ligand.
A potential solution is to directly screen mild electrophile fragments. Electrophile fragment screens were recently performed on a small scale, with libraries of up to ∼100 compounds in vitro against a recombinant target38−43 or in a cellular phenotypic context.44−46 Small-scale screens were also performed with reversible covalent fragments.47,48 We hypothesized that significantly increasing the library size and screening it against a diverse panel of targets will allow robust discovery of covalent ligands. An additional advantage of irreversible binding fragments is the relative ease of cocrystal determination in comparison to reversible fragments with low residence time in the binding site.40,49,50
Here, we report a holistic covalent fragment screening approach. We have screened approximately 1000 electrophiles against 10 different proteins. In combination with a newly reported high-throughput thiol-reactivity assay, our approach circumvents problems ascribed to irreversible binding: by robustly evaluating reactivity and screening several proteins, we could detect, and thus avoid, promiscuous hitters. We could exploit the advantages of covalent screening: namely, sensitive detection of binding at relatively low concentrations, which yielded potent and unique primary hits in the majority (7 out of 10) of cases. Moreover, we demonstrate that, by combining the approach with high-throughput crystallography, quality leads can be rapidly developed, as shown for OTUB2 and NUDT7, two targets that have thus far lacked probes.
Results
Assembling an Electrophile-Fragment Library
We constructed our electrophile-fragment library by focusing on two mild electrophilic “warheads”: acrylamides and chloroacetamides. Acrylamides are represented in many rationally designed covalent drugs, such as ibrutinib and osimertinib. Although chloroacetamides are more reactive,51 they still show selectivity in chemical-proteomic screens.44 Not many fragments containing these electrophiles were available for purchase from commercial vendors, likely due to the longstanding bias against covalent modifiers.25,26 Nevertheless, we limited our screen to commercially available compounds to enable the broadest future use of the library. Additional considerations were to ensure an overall low molecular weight and to enrich the library with related analogues in order to allow preliminary structure–activity relationships to be deduced directly from a primary screen. The final library (Figure 1) contains 993 compounds, 76% of which are chloroacetamides (n = 752) and 24% of which are acrylamides (n = 241), with 92% exhibiting a molecular weight below 300 Da, including, in the case of chloroacetamides, the chlorine leaving group (36 Da). Thus, the molecular-weight distribution of the reversible recognition elements is shifted to even lower masses (see Figure S1 in the Supporting Information for the molecular-weight distribution of the adducts). If the electrophilic moiety is included, 95% of the compounds have fewer than 20 heavy atoms. The library also adheres to the so-called “rule of three”,52 with almost all compounds containing fewer than three hydrogen-bond donors, acceptors, rotatable bonds, and c log P < 3 (in the unreacted form; Figure 1). A substructure analysis of the library (Figure S2 in the Supporting Information) shows that there are relatively few electrophile-containing substructures that are represented by more than 10 fragments in the library. Similarly, clustering identified only 10 clusters with more than 10 members, indicating a high degree of diversity in the library (Figure S2E in the Supporting Information).
Figure 1.

Electrophile-fragment library adheres to the “rule-of-three”. Distribution of (A) chloroacetamides and acrylamides in the library (B) molecular weights of the fragments, including the electrophile moiety, (C) number of heavy atoms, (D) c log P values, (E) number of rotatable bonds, (F) number of hydrogen-bond acceptors, and (G) number of hydrogen-bond donors. The library largely adheres to the “rule of three” for fragment libraries.
Only a Few Fragments Are Highly Reactive
To address a major concern in covalent-molecule screening, namely, that high reactivity could lead to a high proportion of irrelevant hits, we developed a high-throughput thiol-reactivity assay that assess the reactivity of all the fragments in our library. This entails incubating fragments with reduced DTNB (Ellman’s reagent; 5,5-dithio-bis-2-nitrobenzoic acid) and following the absorbance of TNB2– at the 412 nm wavelength for up to 7 h. By fitting the data to a second-order reaction rate equation, we were able to extrapolate the kinetic constant for the alkylation (see example in Figure 2 and see Data set 1 and Figure S3 in the Supporting Information). Since no specific recognition is expected between TNB2– and the electrophile, this rate constant reflects the intrinsic reactivity of the electrophile toward thiols. The majority of the compounds showed an excellent fit to the kinetic model (63.5% had R2 > 0.9; 71% had R2 > 0.8; Figure S4A in the Supporting Information). The poorest-fitting data were obtained for the least reactive compounds (Figure S4B in the Supporting Information), for which the reaction rate was below the dynamic range of the assay. We verified that the reduction of DTNB with TCEP does not influence the measured rates (Figure S3 in the Supporting Information). For a subset of compounds, we showed that the rates agree well with GSH consumption t1/2 (Table S1 in the Supporting Information).
Figure 2.

High-throughput thiol-reactivity measurements across electrophile fragments. (A) Schematic description of the high-throughput thiol-reactivity assay. In the presence of TCEP, DTNB is reduced to TNB2–, which has a strong absorbance at 412 nm and is yellow under natural light. Alkylation of TNB2– by an electrophile fragment reduces the observed absorbance. (B) Example of the reactivity measurement for PCM-0102854. (C) Example of a second-order kinetic rate calculation. The data are fitted to a second-order reaction. [A] is the concentration of the electrophile, and [B] is the concentration of TNB2–. The rate is determined by a linear regression of the data across 4 h of measurement. (D) Distribution of the rates of all the electrophile fragments in the library. (E) Thiol-reactivity-rate distributions for the fragments containing prevalent substructures. (F) Chemical structures of chloroacetamide substructures in the library that are represented by at least 10 fragments (see Figure S2 in the Supporting Information for the same analysis for acrylamides).
Several results arise from the analysis of the kinetic data from the entire library. First, the most reactive fragments were labeled only ∼100-fold faster than the least reactive fragments; the majority of library fragments (78%) falls within only a 30-fold difference. Second, the chloroacetamides are clearly more reactive (show faster kinetics) than the acrylamides (p = 4.6 × 10–38; two-sided t test), in accordance with previous experiments for a handful of fragments.51 Finally, for reference, we measured the reactivity of iodoacetamide, a generally used, nonselective thiol alkylator, and found it to be 21-fold more reactive than the average chloroacetamide and 26-fold more reactive than the median choloroacetamide (Data set 1 in the Supporting Information).
A comparison of the reactivities of various common substructures (i.e., at least 10 instances) adjacent to the electrophile reveals additional trends (Figure 2E). For instance, the four most reactive substructures are piperazine-based chloroacetamides. Two of the least reactive substructures are 2-chloro-N-cyclohexylacetamides and chloroacethydrazides. In general, chloroacetamides that are based on primary amines/anilines are significantly (p < 0.0001) less reactive than those that are based on secondary amines/anilines (Figure S5A in the Supporting Information). We found 29 matched pairs of fragments in the library, which differ by a single methyl on the chloroacetamide. N-Methylated chloroacetamides are significantly more reactive than their nonmethylated counterparts (p = 0.0002; Figure S5B in the Supporting Information). The median increase in the reactivity by such N-methylation is 1.7-fold. We should note that the variability in reactivities of acrylamides as a function of the substructure near the electrophile is much lower (Figure S2C in the Supporting Information).
Overall, while there is variability in the intrinsic reactivity of the fragments toward thiols, it is sufficiently small to allow the identification of quality hits in a screening campaign. Moreover, these data indicate that the two selected electrophiles are indeed sufficiently mild to ensure that the main driver of protein labeling is recognition rather than reactivity.
Intact-Protein Mass Spectrometry Identifies Hits against a Diverse Panel of Proteins
We screened our library against a diverse panel of 10 cysteine-containing proteins (Table 1). The targets were selected on the basis of their therapeutic potential, and most of them lacked any validated covalent inhibitor or probe, or indeed any known chemical probe. A screen of bovine serum albumin (BSA) served as control. Four of the proteins in the panel contained a solvent-exposed catalytic cysteine, while the other six did not.
Table 1. Panel of Protein Targets for Screening.
| proteina | MWb | hitsc | nonpromiscuous hitsd | catalytic Cyse | Cys residuesf | possible therapeutic indicationsg |
|---|---|---|---|---|---|---|
| QSOX1 | 58084 | 0/993 | 0 | noh | 12 | cancer53,54 |
| PCAF | 19452 | 0/993 | 0 | no | 3 | HIV,55 cancer56,57 |
| PBPR504C | 57903 | 2/983 | 2 | no | 1 | antibiotic resistance58 |
| K-RasG12C | 19245 | 10/968 | 7 | no | 3 | cancer37,59 |
| USP8 | 44429 | 20/923 | 7 | yes | 12 | cancer,60 Cushing’s disease61 |
| NNMT | 31248 | 30/299 | 22 | no | 8 | cancer,62 diet-induced obesity63 |
| OTUB2 | 27312 | 47/938 | 42 | yes | 4 | viral infection,64 diabetes,65 ALS66 |
| NUDT7 | 26672 | 36/973 | 24 | yes | 4 | diabetes67 |
| NV3CP | 19284 | 10/824 | 9 | yes | 5 | viral infection68 |
| BSA | 66464 | 0/981 | 0 | no | 23 | negative control |
Acronyms are as follows: quiescin sulfhydryl oxidase 1 (QSOX1); penicillin-binding protein 3 (PBP); ubiquitin-specific peptidase 8 (USP8); nicotinamide N-methyltransferase (NNMT); norovirus 3C protease (NV3CP); bovine serum albumin (BSA).
Molecular weight of construct in Da.
Number of fragments with >50% labeling/compounds for which labeling could be evaluated.
Number of hits that did not label any other protein to >50%.
Indicates whether a catalytic cysteine is present in the protein.
Number of cysteine residues in protein.
Indicates possible therapeutic rationale to inhibit this target.
QSOX1 contains a catalytic disulfide.
Each protein was incubated with the electrophilic library in pools of five compounds per well. The pool’s composition maximized the difference in mass between fragments in the same well. Incubations were performed with each fragment at 200 μM, for 24 h at 4 °C, to allow the screening of proteins that are not stable for long time periods at higher temperatures. Following incubation, we used intact protein liquid chromatography/mass spectrometry (LC/MS) to identify the fragments and quantify their labeling (Figure 3A). We confirmed that screening the compounds in pools of five does not significantly affect hit identification. There was good agreement between the percent of labeling of NUDT7 by 100 random individual compounds and that obtained in pools of five (R2=0.78; Figure S6 and Data set 2 in the Supporting Information).
Figure 3.

Intact protein LC/MS screen identifies hits for most targets (A) (top) Outline of our screening pipeline. Compounds are pooled, five in each well, incubated with a target protein for 24 h and read via LC/MS. (bottom) Example of the MS deconvoluted spectrum for NUDT7 with no compound (blue) and after 24 h incubation with five compounds (green). Note that the shift in the mass of the protein corresponds to 100% labeling of PCM-0102951. (B) Summary of the quantified labeling of 10 proteins by the electrophile library. Blue represents 100% binding and white no labeling or data not available (see labeling assignment in Methods).
Overall, while the hit rate varied greatly between different proteins (Figure 3B), we were able to find hits for almost all of the screened proteins, except for PCAF, QSOX1, and the negative control BSA (see Data set 3 in the Supporting Information for the labeling quantification and Figure S7 in the Supporting Information for the structures of nonpromiscuous hits).
Promiscuity Does Not Correlate with Reactivity
We define promiscuous compounds to be those that label two or more proteins by more than 50% or three or more proteins by more than 30%. Despite this stringent definition, only 27 of the electrophilic fragments are promiscuous (Figure S8 and Data set 3 in the Supporting Information). Under an even stricter definition, i.e., more than 30% labeling of any two proteins, only an additional 36 compounds become promiscuous. We should note here that by promiscuity we only refer to nonspecific irreversible binding and not to the inherent reversible binding promiscuity of low-molecular-weight fragments, which is a favorable property in screening for protein binders. Some compounds labeled the protein twice in the primary screen (Data set 4 in the Supporting Information). Our comparison of the reactivity distribution of these compounds with that of the rest of the set shows that such “double labeling” compounds are not significantly more reactive on average (Figure S9 in the Supporting Information; p = 0.09).
Unexpectedly, promiscuous labeling does not correlate well with thiol-reactivity (Figure S10 in the Supporting Information; R2 = 0.09). For instance, compound PCM-0102496 (Figure S8 in the Supporting Information) labeled NNMT, USP8, and NUDT7 by more than 50%, although its alkylation rate is in the lowest quartile of reactivity (1.09 × 10–8 M–1 s–1). On the other hand, some of the most reactive compounds, such as PCM-0102140, PCM-0102859, and PCM-0102150 (Figure S10A,C in the Supporting Information), did not significantly label any of the proteins assayed.
We evaluated the possibility that promiscuous compounds label amino acids other than cysteines. We incubated five promiscuous compounds with NUDT7 and USP8, followed by trypsin digestion and LC/MS/MS analysis, to identify modification sites. Despite rare lysine and histidine modifications, the compounds preferably reacted with cysteines (Data set 5 and Figure S11 in the Supporting Information), thus largely ruling out this hypothesis.
The degradation of compounds between protein screening and reactivity measurement can also explain the discrepancy between promiscuity and reactivity. To control for this, we resourced 14 compounds—10 promiscuous compounds and 4 random compounds that did not label any protein. We reevaluated the reactivity of these fresh compounds and found, with one exception (PCM-0102982), that the rates agreed well with the previous measurements (Table S1 in the Supporting Information). These results suggest that degradation is not the source of the discrepancy.
Many of the promiscuous binders contain similar chemical motifs. For instance, a large family of aminothiazole chloroacetamides (Figures S10B and S12 in the Supporting Information) are frequent hitters in our screens. Of note, these compounds are not significantly more reactive than other chloroacetamides in our thiol-reactivity assays (p = 0.183 in a one-sided t test).
Fragment Growing Identifies Novel OTUB2 Inhibitors
OTUB2 is a deubiquitinase (DUB) from the ovarian tumor domain (OTU) DUB superfamily.69 OTUB2, initially identified in HeLa cells,70 preferentially cleaves Lys63-linked polyUb chains and can also cleave Lys11- and Lys48-linked chains.71 OTUB2 is important for the choice between the homologous-recombination and the nonhomologous-end-joining DNA repair pathways.72 OTUB2 has also been found to function as a negative regulator of virus-triggered type I IFN induction64 and has been linked to the inhibition of NF-κB signaling and the regulation of beta cell survival in human pancreatic islets.65 Finally, OTUB2 has been identified as a potential biomarker for sporadic amyotrophic lateral sclerosis (ALS).66 Thus, OTUB2 plays an important role in several biological pathways, suggesting that the development of OTUB2-specific inhibitors can have therapeutic potential.
The primary screening against OTUB2 produced 47 fragments with >50% labeling, of which 42 were nonpromiscuous and 37 strictly nonpromiscuous (Figure S13 in the Supporting Information). We evaluated the protein labeling of 26 of these compounds at 200 and 100 μM (24 h; 4 °C; Table 2 in the Supporting Information); 9 compounds showed >50% labeling even at the lower concentration. In order to prioritize fragments for optimization, we turned to high-throughput crystallography.
We were able to determine the cocrystal structures of 15 OTUB2/fragment complexes by streamlined parallel cocrystallization involving 24 h of preincubating a protein with a fragment and seeding with apo-protein crystals. In 11 of these complexes, the fragments formed a covalent bond with the catalytic cysteine 51, located in the enzyme’s active site (Figure 4A and Table S3 in the Supporting Information).
Figure 4.

Discovery of a selective OTUB2 inhibitor by fragment growing. (A) Cocrystal structures of OTUB2 in complex with (from top left) PCM-0102998, PCM-0103080, PCM-0102660, PCM-0103011, PCM-0103007, PCM-0102954, PCM-0103050, PCM-0102153, PCM-0102305, PCM-0102821, and PCM-0102500 (see Figure S13 in the Supporting Information). Structures with compounds containing the chloroacethydrazide motif are boxed. (B) Percent covalent labeling of OTUB2 with compounds containing the identified motif, PCM-0102300, PCM-0103009, PCM-0102142, PCM-0102998, PCM-0102954, and PCM-0102355 (Figure S13 in the Supporting Information), at 200 μM (blue) and 100 μM (green). Compounds boxed in (A) are marked with asterisks. (C) Percent covalent labeling of OTUB2 with selected next-generation compounds at 100 μM (see Figure S16 in the Supporting Information for all analogues and Table S4 in the Supporting Information for percent labeling). (D) Dose–response measurement of percent labeling by next-generation OTUB2 binders. All labeling in (B)–(D) was measured via LC/MS after 24 h incubation at 4 °C. (E) Inhibition of OTUB2 in an enzymatic assay (2.5 h preincubation in the presence of 2 mM free cysteine). (F) Gel-based ABPP selectivity assessment using a fluorescent activity-based DUB probe73 in HEK293 cells overexpressing OTUB2 (see Figure S21 in the Supporting Information for the full gel and additional controls). Cells were incubated with DMSO or increasing concentrations of OTUB2-COV-1 and then lysed, and the DUBs were fluorescently labeled with an alkyne ABPP probe: the higher the level of labeling by an inhibitor, the lower the level of labeling by the ABPP probe. The only bands that are reduced by OTUB2-COV-1 up to 30 μM correspond to OTUB2-GFP (at 65 kDa) and a degradation product (∼55 kDa). This demonstrates both OUTB2’s cellular engagement with OTUB2-COV-1 in cells and its high selectivity over other DUBs (see Figure S20 in the Supporting Information for the corresponding experiment in lysates). (G) Chemical structures of selected next-generation OTUB2 binders. The chloroacethydrazide motif is highlighted in red.
The carbonyls of all chloroacetamides occupied the oxyanion hole formed by the amide backbones of D48, G49, N50, and C51. To progress selected fragments, the analysis of labeling results and crystal structures led us to focus on a series of fragments that share a common chloroacethydrazide motif (Figure 4B), two of which were seen in crystal structures. In both cases, the shared moiety participates in an extensive hydrogen-bonding network with the protein active site (Figure S14 in the Supporting Information). The side chain of E174 switched its rotamer (in comparison to all other apo and fragment-bound cocrystal structures) in order to mediate one such hydrogen bond to the hydrazide motif (Figure S15 in the Supporting Information). In both structures, the hydrophobic moiety connected to the hydrazide is pointing toward the solvent, making few, obviously productive, contacts with the protein. We concluded that we might be able to optimize compound binding by changing this moiety.
We purchased 21 analogues, all containing the chloroacethydrazide motif (Figure S16 in the Supporting Information). On incubation with OTUB2 at 100 μM (24 h; 4 °C), six showed >50% labeling, representing the reversible recognition stemming from the shared motif (Figure 4C and Table S4 in the Supporting Information). Two particularly promising analogues, OTUB2-COV-1 and OTUB2-COV-17, showed 100% labeling (Figure 4C). We sourced additional analogues of OTUB2-COV-1 with various para substitutions of the phenyl ring and assessed their labeling efficiency at various concentrations. Compounds OTUB2-COV-1, OTUB2-COV-22, and OTUB2-COV-23 showed 46%, 55%, and 49% labeling, respectively, at 5 μM (24 h; 4 °C; Figure 4D).
We next assessed their inhibition of the enzymatic activity of OTUB2 (Figure 4E). The best inhibitor, OTUB2-COV-1, showed an IC50 value of 31.5 μM at a relatively short incubation time of 30 min in the presence of 2 mM cysteine. The IC50 improved to 15.4 μM with a longer incubation period of 2.5 h, supporting the irreversible binding mechanism. A jump dilution experiment (Figure S17 in the Supporting Information) further validated irreversible binding. There was a very good correlation between the labeling efficiency of these compounds and their inhibitory effect. We attempted to determine the cocrystal structure of OTUB2 labeled with the lead compound, OTUB2-COV-1. While the chloroacethydrazide motif adopted the same conformation as the original library hits, we could not detect density for the cyclopropyl-phenyl moiety (Figure S18 in the Supporting Information).
The improvement in the analogues appears to stem from better recognition rather than reactivity: the primary chloroacethydrazide hits gave little labeling of other proteins in the panel (Data set 3 in the Supporting Information), and their mild reactivity in the thiol-reactivity assay (average rate k = 3.9 × 10–8 M–1 s–1) was comparable to that of some of the new analogues in the same assay (OTUB2-COV-23, k = 2.74 × 10–8 M–1 s–1; OTUB2-COV-22, k = 3.69 × 10–8 M–1 s–1; Data set 1 in the Supporting Information). We determined the kinact/KI value of OTUB2-COV-1 to be 3.75 M–1 s–1 (Figure S19 in the Supporting Information), which is similar to that of recently reported covalent binders.49,50
To assert the compounds’ selectivity against other DUBs, we used cell and lysate gel-based activity based protein profiling (ABPP). We used our previously developed alkyne activity-based DUB probe73 to fluorescently label all cysteine DUBs. OTUB2 is endogenously expressed at very low levels and so is not visible in comparison to other highly expressed DUBs (Figures S20 and S21 in the Supporting Information). To overcome this impediment, we have used either HEK293 cells overexpressing an OTUB2-GFP construct (Figure 4F and Figure S21 in the Supporting Information) or native HEK293 lysates spiked with recombinant OTUB2 (Figure S20 in the Supporting Information).
The experiment in cells proved the cellular engagement of OTUB2-COV-1 with OTUB2 with a dose-dependent reduction of DUB probe labeling (Figure 4F). Strikingly, even at concentrations as high as 30 μM, OTUB2 appears to be the only DUB target of OTUB2-COV-1, with no detectable differences between the compound-treated cells/lysate and the DMSO control (Figure 4F and Figure S20A,C in the Supporting Information; compare lanes 1 and 16). These results indicate exquisite selectivity across all DUBs detected by the probe.
Finally, we performed a competitive isoTOP-ABPP experiment74 in order to profile the proteomic targets of OTUB2-COV-1 in HEK293T cells (10 μM compound; 2 h incubation; Data set 6 and Figure S22A in the Supporting Information). We were unable to detect OTUB2 itself in this experiment, likely due to low endogenous expression (see Figures S20 and S21 in the Supporting Information). OTUB2-COV-1 significantly labeled (Heavy/Light ratio >4) only 26 of the 2998 cysteines detected by the experiment (<1%). Moreover, none of the cysteines with a Heavy/Light ratio >2 belonged to a DUB. This both asserts the proteomic selectivity of the probe and confirms its selectivity within the DUB family.
Fragment Merging Leads to Potent NUDT7 Inhibitors
NUDT7, a peroxisomal CoA pyrophosphohydrolase, belongs to a protein family characterized by a 23-amino-acid motif referred to as the “NUDIX box”. These proteins have been reported to hydrolyze a diverse range of substrates, including (d)NTPs, nucleotide sugars, diadenosine polyphosphates, and capped RNA.75 The NUDT7 gene contains a CoA-binding motif and a C-terminal peroxisomal targeting signal (PTS).76,77 Expression of NUDT7 is highest in liver, with NUDT19 likely acting as the complementary CoA and CoA ester hydrolase in kidney.78 Leptin double-knockout mice, which display alterations in CoA homeostasis and exhibit a diabetic phenotype, have been reported to express reduced levels of NUDT7 with a concomitant increase in pantothenate kinase activity.67 To the best of our knowledge, no small-molecule inhibitors or probes have been reported for NUDT7 so far.
The primary screening against NUDT7 produced 36 fragments with >50% labeling, of which 24 were nonpromiscuous and 20 strictly nonpromiscuous (Figure S23 in the Supporting Information). A series of similar fragments sharing a common 2-phenylpyrrolidine motif stood out, with four compounds labeling 100% (Figure 5). We validated compound binding via differential scanning fluorimetry (DSF), in which 26 of the 30 compounds that showed covalent labeling also stabilized NUDT7 by 4.5–13.4 °C (Figure S24 in the Supporting Information). Specifically, compounds PCM-0102298, PCM-0102938, PCM-0102558, PCM-0102951, PCM-0102716, and PCM-0102512 stabilized NUDT7 by 4.5–8 °C (Figure 5A,B).
Figure 5.

Discovery of a potent NUDT7 inhibitor by fragment merging. (A) Chemical structures of similar hit compounds that exhibited 68% (PCM-0102716), 88% (PCM-0102512), and 100% (PCM-0102558, PCM-0102298, PCM-0102951, and PCM-0102938) labeling of NUDT7 in the primary screen. Compound NUDT7-REV-1 is a noncovalent fragment (purple) that was identified as a NUDT7 binder in a crystallography soaking screen (see (E)). NUDT7-COV-1 (blue) is a merged compound based on PCM-0102716 (magenta) and NUDT7-REV-1. (B) The six hits identified in the primary screen stabilize NUDT7 by 4.5–8.1 °C in a Tm shift assay. (C) Labeling percentage of compounds PCM-0102558, PCM-0102951, PCM-0102298, PCM-0102716, PCM-0102512, and PCM-0102938 at 5–200 μM, 24 h, 4 °C. (D) Cocrystal structures of NUDT7 with compounds PCM-0102951, PCM-0102558, and PCM-0102716. (E) Overlay of the crystal structures of NUDT7 with compound PCM-0102716 (pink) and with the noncovalent fragment NUDT7-REV-1 (purple). (F) The cocrystal structure of NUDT7 with the merged compound NUDT7-COV-1 adopts the exact same binding mode as the two separate fragments. (G) Enzymatic inhibition of NUDT7 by NUDT7-COV-1 and NUDT7-REV-1. The data shown include results with and without a 30 min protein preincubation in the presence of the compounds. (H) Intracellular target engagement is demonstrated by thermal stabilization of FLAG-NUDT7 by NUDT7-COV-1 in intact HEK293 cells. After transfection, cells were treated with 20 μM NUDT7-COV-1 or DMSO for 30 min before being heated to the indicated temperatures.
In order to optimize this series, we determined the cocrystal structures of compounds PCM-0102951, PCM-0102558, and PCM-0102716 bound to NUDT7 (Table S5 in the Supporting Information) using a cocrystallization protocol similar to that for OTUB2. The structures show that all compounds form a covalent bond with the catalytic cysteine 73. Surprisingly, however, despite their chemical similarity, the three compounds adopt different binding poses (Figure 5D). An evaluation by dose–response labeling of the six compounds sharing the 2-phenylpyrrolidine motif showed that, after incubation (4 °C, 24 h), all six compounds label 100% up to 25 μM. At 5 μM concentration, the compounds label less (60–80%), except for PCM-0102558, which still shows 100% labeling (Figure 5C).
We have previously completed a crystallographic fragment screen with noncovalent fragments at the XChem facility at Diamond Light Source and have identified 18 fragments bound to the putative substrate binding region of NUDT7 (https://doi.org/10.5281/zenodo.1244111). We have synthesized, on the basis of one of the initial hits, a series of diphenylacetamide analogues and have soaked them into NUDT7 crystals, which yielded a structure in complex with compound NUDT7-REV-1 (Figure 5A), A comparison of this structure with the covalent NUDT7/PCM-0102716 structure reveals an almost perfect overlap of one of the phenyl rings (Figure 5E), suggesting a clear strategy for fragment merging.
The merged compound, NUDT7-COV-1 (see chemical synthesis in File S1 in the Supporting Information), combines the key features of both fragments and has much improved properties. In the cocrystal structure, the merged compound adopts exactly the predicted pose (root-mean-square deviation of 0.4 Å over the shared atoms; Figure 5F), and at 5 μM, NUDT7-COV-1 labels NUDT7 to 95% in 15 min (2 μM protein). This improvement is likely due to enhanced recognition, since NUDT7-COV-1 is less than 3-fold more reactive than its parent compound, PCM-0102716 (k = 4.22 × 10–7 M–1 s–1 vs. k = 1.63 × 10–7 M–1 s–1, respectively). We should note that both the parent PCM-0102716 and NUDT7-COV-1 contain a pyrrolidine stereocenter and are racemic mixtures. Their cocrystal structures reveal that in both the S enantiomer is the active species.
In a NUDT7 enzymatic activity assay, without preincubation, the merged compound has an IC50 value of 2.7 μM. After a 30 min preincubation with the compound, the IC50 value improves to 1.1 μM. Notably, none of the noncovalent hits from crystallographic fragment screening show detectable activity in the enzymatic assay, including the parent noncovalent fragment we used for merging (Figure 5G and Figures S25 and S26 in the Supporting Information). We determined the kinact/KI value of NUDT7-COV-1 to be 757 M–1 s–1 (kinact = 0.01 s–1, KI = 13.21 μM; Figure S27 in the Supporting Information), in comparison to kinact/KI = 233.1 M–1 s–1 for the best primary hit from the screen, PCM-0102558, for which we were not able to determine the kinact and KI values separately.
We evaluated NUDT7 cellular target engagement by NUDT7-COV-1 via a cellular thermal shift assay (CETSA) in intact HEK293 cells. Indeed, NUDT7-COV-1 showed significant stabilization of FLAG-tagged NUDT7 in comparison to DMSO, confirming that the compound binds the target in living cells (Figure 5H). Finally, we performed an isoTOP-ABPP experiment, in which we profiled the cellular targets of NUDT7-COV-1 (10 μM; 2 h incubation; HEK293T cells; Data set 7 and Figure S22B in the Supporting Information). NUDT7 itself could not be detected, similarly to OTUB2, likely due to low endogenous expression levels. Only 37 of the 2025 detected cysteines were labeled by NUDT7-COV-1 with a Heavy/Light ratio >4. None of the cysteines with a ratio of Heavy/Light >2 belonged to a NUDIX-hydrolase domain family member.
Electrophile Fragments Can Be Suitable for Cellular Screens
Many previous studies established a correlation between intrinsic reactivity and cellular toxicity for a range of electrophiles.79 To see if such a correlation exists for our electrophilic fragments, we performed a cellular viability assay for each of our compounds with three different model cell lines, HEK293, HB2, and CCD841 (Data set 8 in the Supporting Information).
At a concentration of 10 μM and after 48 h of incubation, 47%, 58%, and 60% of the compounds negligibly effect the viability of HEK293, HB2, and CCD841 cells, respectively (>75% viable; Figure S28A–C in the Supporting Information). There is good correlation between the toxicity of the compounds across the three cell lines (Figure S28D–F in the Supporting Information). We observed a switch-like toxicity effect, in which compounds with reaction rates of k = 1 × 10–7 M–1 s–1 or less hardly affect viability; only 30%, 19%, and 18% of these compounds affect by more than 25% the viability of HEK293, HB2, and CCD841 cells, respectively. Whereas compounds with rates higher than k = 1 × 10–7 M–1 s–1 show a sharp decline in viability as a function of reactivity (Figure S28A–C in the Supporting Information), 89%, 80%, and 75% of these compounds reduce cellular viability by more than 25%. These results indicate that this library can be suitable for cellular phenotypic screening in addition to in vitro screening against purified proteins.
Discussion
Discovering selective covalent acting compounds is challenging. We approach this problem by significantly increasing the chemical space of recognition elements, through the use of mild electrophiles, while carefully accounting for reactivity and promiscuity. We describe the screening of 993 commercially available electrophile fragments against 10 different proteins. Previous electrophile screening campaigns were limited in scope: Nonoo et al.39 assayed only 10 acrylamides against 3 proteins, Jost et al.41 tested 6 diverse electrophiles against 11 proteins, Kathman et al.38 and Johansson et al.49 screened 100 methyl acrylates against 4 proteins and 1 protein (HOIP), respectively, and, recently, Craven et al.40 screened 138 electrophiles against the kinase CDK2. While most of these studies were restricted to well-studied targets with known inhibitors, they pioneered electrophilic fragment screening and served as a proof of concept that the method is viable. By significantly expanding the library and screening a diverse array of targets, we demonstrate the broad applicability of this approach by producing valuable hits for “orphan” targets.
We observe, moreover, that the combination of screening carbon electrophiles alongside exploiting high-throughput crystallography results in an effective method for progressing hits to leads and designing probes: the covalent bond in the hit provides a clear, dominant chemical rule for designing follow-ups and provides sufficient potency such that only a few analogues are necessary for a potent and selective probe to pass the threshold. Thus, we needed to purchase or synthesize fewer than 50 compounds in order to achieve selective chemical probes against two targets.
Another key point of screening carbon electrophiles, especially in comparison to the well-established disulfide tethering approach, is that it directly optimizes both recognition element binding and electrophile orientation toward the target cysteine. Disulfide hits from a tethering screen typically cannot be used as cellular probes due to the reducing environment, and the transition from an active disulfide fragment to a similarly active carbon electrophile can be demanding and require the synthesis of many test compounds. For example, Ostrem et al.37 identified a tethering hit against K-RasG12C but had to synthesize nearly 100 carbon electrophiles to reach suitable labeling efficacy. Instead, our screen immediately identified compound PCM-0102818 (Figure S7 in the Supporting Information), which is highly similar to the previously optimized compounds, and labeled 63% of K-RasG12C, highlighting the efficiency of directly screening carbon electrophiles. The expanded library size does, however, appear to be crucial to providing sufficient coverage of the chemical space: our previous screen of K-RasG12C with a much smaller number of electrophiles18 (only 62 acrylamide fragments) failed to yield plausible hits.
A main novelty of our study is the development of a high-throughput thiol-reactivity assay that allowed us to comprehensively characterize the intrinsic reactivity of the entire library. There are a few reports of the use of TNB2– as an indicator of the formation of the reactive species benzoquinones or chloramines,80,81 but to the best of our knowledge, it has never been used systematically to quantitate intrinsic reactivity toward thiols. We show that there is good agreement between GSH-based reactivity evaluation and our method (Table S1 in the Supporting Information). However, since GSH binding measurements cannot be parallelized, they take, on average, 5 h per compound, whereas our assay can measure hundereds of compounds over the course of 4 h. Another advantage is that the assay can be performed on a plate reader, which is typically more accessible to users than the NMR or LC/MS required to assay direct GSH binding. One caveat of this method is that it cannot account for different kinetic rates in compounds that contain more than one electrophilic center, but this limitation holds also for other methods, such as GSH consumption, and only very few compounds in our library contain more than one electrophile.
On the basis of this assay, a major result of this study is the relatively narrow range of reactivity displayed by the wide majority of the electrophilic fragments, an observation that is robust due to the large number of compounds we could screen with the new high-throughput reactivity assay. Previous studies that have characterized the thiol reactivity of various electrophiles which are commonly explored as warheads for chemical probes38,51 were limited in terms of the number of evaluated compounds, likely due to the low-throughput assays used to determine thiol reactivity. For example, when they examined acrylamides, Kathman et al.38 determined the pseudo-first-order reaction rate for only three model compounds, and because one showed a significantly higher reaction rate, it was suggested that acrylamides as a class of electrophiles have too variable a reactivity for screening. Instead, we could assess the reactivity of close to 250 acrylamides. We, too, observed outliers with high reactivity, but the vast majority displayed low reactivity and narrow variability. Indeed, under the current conditions, the rates of 90 acrylamides fall below the dynamic range of our assay, showing rates similar to that of the background blank reaction. Acrylamide hits were also rarer in the 10 protein screens, overall suggesting that, as a class of electrophiles, they are suitable for screening.
We compared the reactivity of fragments that were screened in the same well with some of the validated OTUB2 chloroacethydrazide binders and found that we could detect hits even in the presence of more reactive fragments: PCM-0103009 labeled to 100% in the presence of PCM-0102797, which is 4.3-fold more reactive. PCM-0102142 also labeled to 100% in the presence of PCM-0102715, which is 9.4-fold more reactive. Finally, PCM-0102426 labeled 60% in the presence of PCM-0102273, which is 14.1-fold more reactive but did not label at all. A consequence of this result is that we could screen the library in pools of 5—saving considerable time and protein—without compromising hit identification, as we demonstrated for 100 random compounds (Figure S6 in the Supporting Information). We should note here that fragments that label at 100% may hinder another hit from the same well; this can be circumvented by expanding such wells and retesting only those fragments as single agents. Still, for future screens, it will likely be beneficial to pool compounds on the basis of similar intrinsic reactivity in addition to maximum mass difference.
A related observation is that promiscuity is, in fact, not a function of reactivity, which has historically been the main reason covalent compounds are assumed to be problematic. While promiscuous compounds were observed (Data set 3 in the Supporting Information) and will be removed from future screens, the most promiscuous compounds did not necessarily have high intrinsic reactivity. For instance, PCM-0102957 (Figure S8 in the Supporting Information), one of the most promiscuous compounds in the library, has an intrinsic reactivity similar to that of iodoacetamide (1.9 × 10–6 M–1 s–1). In contrast, another promiscuous compound, PCM-0102496 (Figure S8 in the Supporting Information), displays very low reactivity. A previous study that tried to correlate the intrinsic reactivity of electrophiles to in vitro covalent binding found similar discrepancies.82 We discounted two possible explanations for this observation: that these unexpected compounds do not label cysteine but rather other amino acids, such as lysine (Data set 5 and Figure S11 in the Supporting Information), and that the low reactivities are an artifact of compound degradation (Table S1 in the Supporting Information). Other explanations, such as photoreactivity, redox cycling, and other confounding mechanisms, might still be at play. This remains an area of active research, and future screens might shed more light on this phenomenon.
Nevertheless, the data allow us to recommend a threshold reactivity of k = 1 × 10–7 M–1 s–1, below which electrophiles are likely to be useful starting points for generating hits suitable for further optimization and progression to cell active probes. This proposed threshold is based on the loose correlation between reactivity and promiscuity (Figure S10 in the Supporting Information) and the correlation between reactivity and cellular toxicity (Figure S28A–C in the Supporting Information). An exception to this rule of thumb would be structural motifs we identify as promiscuous, such as the aminothiazole series (Figure S12 in the Supporting Information), which showed up as frequent hitters, even in noncovalent screening,83 and are considered PAINS compounds.25 An important criterion for selecting hits for follow-up optimization is the existence of shared substructures among hits. The shared chloroacethydrazide motif for OTUB2 and the chemical similarity of the NUDT7 hits increased their confidence as true positives for follow-up. Additional shared motifs were found for other targets; for instance, there were five NNMT hits that shared an N-carbamoyl-2-chloroacetamide motif (Figure S7 in the Supporting Information) and three low-reactivity aminothiazoles hitting USP8 (with the caveat mentioned above for aminothiazoles in general).
The hit rates obtained with this library are similar to or slightly higher than those observed in screens with noncovalent fragments:84,85 2–4% for NNMT, OTUB2, and NUDT7 and 0.2–0.9% for other proteins. These would be attenuated by screening at different concentrations. Here, all primary screens were performed at 200 μM, but on the basis of our results, we can now recommend a concentration of 100 μM when catalytic cysteines are targeted, while at 200 μM should be used for less nucleophilic target cysteines. Other factors that can affect the hit rate are the incubation time and temperature. Our relatively long incubation time of 24 h may help in the identification of weak binders, while running the reaction at 4 °C may eliminate some nonspecific, reactivity-driven binding.
A major application of our screening approach is the ability to evaluate the potential ligandability of target cysteine residues. It is clear by looking at the overall labeling statistics (Figure 3B) that different target cysteines show different potentials for electrophilic labeling. Proteomic approaches for the identification of functional or reactive cysteines44,46,86−88 can identify potential target cysteines on a much larger scale than ever before. However, the throughput of the proteomics pipeline does not allow assessing the covalent ligandability. In a recent proteomic screen of 50 electrophilic fragments, for example, Backus et al.44 identified several cysteine residues available for labeling in NNMT but did not deem it to be a probe target since only a single fragment was able to significantly label it. In contrast, in our library we found 30 compounds that label NNMT by more than 50%, and in an enzymatic assay with a much shorter incubation time (2 h), 10 of these were able to inhibit it (>20% inhibition; Data set 9 in the Supporting Information). Indeed, a recently published report demonstrated that a selective probe could be developed against NNMT in lysates but not in whole cells.89
Our approach, of course, also has some limitations. First, the requirement for purified protein means it will be difficult when protein production itself is challenging. Second, the current library targets only cysteine residues, which are relatively rare; this limitation might be overcome in future libraries by screening different chemistries targeting alternative amino acids, which would require additional developments and calibration. Finally, the identity of the labeled cysteine cannot be determined directly from the primary screen. This is a minor caveat, since one can either counterscreen a mutant lacking the target cysteine and compare the results or, as we have done for two proteins in this study, follow up with LC/MS/MS to verify the identity of the target residue (Figure S11 in the Supporting Information).
Overall, we conclude that our approach can be widely adopted: the screening does not require specialized equipment or algorithms, the compounds of the library are commercially available from a single vendor, and high-throughput crystallography is now widely supported at synchrotrons worldwide. Our method should, therefore, become a powerful tool for the fast and robust development of covalent ligands against many different proteins, as demonstrated by the development of two new covalent probes for targets that have lacked inhibitors to date.
Methods
Library Acquisition and Handling
A total of 993 compounds were acquired from Enamine (https://www.enamine.net/) as 20 mM DMSO stocks in 96-deep-well plates. A working copy was formatted to 384-well plates and was kept at room temperature under nitrogen. The rest of the library was aliquoted to 384-well plates and frozen in −20 °C. Echo 550 liquid handler (Labcyte Inc.) was used to make screening plates with appropriate volumes of compound. Chemical descriptors of the library were calculated using Pipeline-Pilot (Biovia).
Thiol Reactivity Assay
A 50 μM sample of DTNB was incubated with 200 μM TCEP in 20 mM sodium phosphate buffer pH 7.4 and 150 mM NaCl for 5 min at room temperature, in order to obtain TNB2–. Portions of the compounds (200 μM) were subsequently added to the TNB2–, followed by immediate UV absorbance measurement at 412 nm at 37 °C. The absorbances were acquired every 15 min for 7 h. The assay was performed in a 384-well plate using a Tecan Spark10 M plate reader. The background absorbance of compounds was subtracted by measuring the absorbance at 412 nm of each compound under the same conditions without DTNB. Compounds were measured in triplicate.
The data were fitted to a second-order reaction equation such that the rate constant k is the slope of ln([A][B0]/[B][A0]), where [A0] and [B0] are the initial concentrations of the compound (200 μM) and TNB2– (100 μM), respectively, and [A] and [B] are the remaining concentrations as a function of time as deduced from the spectrometric measurement. Linear regression using Prism was performed to fit the rate against the first 4 h of measurements.
GSH Reactivity Assay
A 200 μM sample of the electrophile was incubated with 1 mM GSH and 40 μM rhodamine B as internal standard in 100 mM potassium phosphate buffer pH 7.4 and ACN at a ratio of 9:1, respectively. All solvents were bubbled with argon. Reaction mixtures were kept at 37 °C with shaking under argon. Every 1 h, for up to 6 h, 20 μL from the reaction mixture was mixed with 80 μL of water and immediately injected into the LC/MS. LC/MS runs were performed on a Waters ACUITY UPLC BEH instrument in positive ion mode using electrospray ionization. UPLC separation used a C18 column (2.1 mm i.d., 50 mm length). The column was held at 40 °C and the autosampler at 10 °C. Mobile phase A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in acetonitrile. The run flow was 0.4 mL/min. The gradient used was 100% A for 2 min, increasing linearly to 90% B for 5 min, holding at 90% B for 1 min, changing to 0% B in 0.1 min, and holding at 0% for 1.9 min. The reaction was followed by the peak area of the electrophile normalized by the area of the rhodamine B. Natural logarithms of the results were fitted to a linear regression, and t1/2 was calculated as t1/2 = ln 2/–slope.
Electrophile Library Screen
Plates for electrophile library screens were prepared by combining 0.5 μL of 20 mM stock solution of 4 or 5 compounds into 1 well in a 384-well plate. Compound pooling was based on maximum difference in molecular weight. The whole library was sorted by the molecular weight of the adduct and then split into five groups. Each pool is composed of five fragments with the same corresponding position in each of the groups.
Incubations were performed at 200 μM for each compound and 2 μM of protein (10 μM for BSA) for 24 h at 4 °C with moderate shaking. Incubation buffers varied among proteins (PBS pH 7.4 for QSOX1 and BSA; 10 mM HEPES pH 7.5, 0.3 M NaCl, 0.5 mM TCEP for PCAF, UPS8, and NUDT7; 10 mM Tris pH 8.2 500 mM NaCl for PBP3R504C; 10 mM Na2HPO4 pH 7.5 100 mM NaCl 5 mM β-mercaptoethanol for NV3CP; 20 mM Na phosphate pH 7.5 for NNMT; 50 mM NaCl, 20 mM Tris pH 8.0 for K-RasG12C). The reaction was stopped by quenching with formic acid, 0.4% final concentration.
The LC/MS runs were performed on a Waters ACUITY UPLC class H instrument, in positive ion mode using electrospray ionization. UPLC separation used a C4 column (300 Å, 1.7 μm, 21 mm × 100 mm). The column was held at 40 °C and the autosampler at 10 °C. Mobile solution A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in acetonitrile. The run flow was 0.4 mL/min. The gradient used for BSA was 20% B for 2 min, increasing linearly to 60% B for 4 min, holding at 60% B for 2 min, changing to 0% B in 0.1 min, and holding at 0% for 1.9 min. The gradient for the other proteins was 20% B for 2 min, increasing linearly to 60% B for 3 min, holding at 60% B for 1.5 min, changing to 0% B in 0.1 min, and holding at 0% for 1.4 min. The mass data were collected on a Waters SQD2 detector with an m/z range of 2–3071.98 at a range of 750–1550 m/z for NV3CP, K-RasG12C, and NUDT7, 700–1300 m/z for QSOX1, PBP3R504C, USP8, and PCAF and 1000–2000 m/z for BSA. The desolvation temperature was 500 °C with a flow rate of 1000 L/h. The voltages used were 0.69 kV for the capillary and 46 V for the cone. Raw data were processed using openLYNX and deconvoluted using MaxEnt.
Labeling Assignment
For each measured well, processed peaks were searched to match the unlabeled protein, common small adducts of the unlabeled protein (that could not be the results of fragment labeling and were seen in the control sample), or labeled protein. The labeling percentage for a compound was determined as the labeling of a specific compound (alone or together with other compounds) divided by the overall detected protein species. Peaks whose mass could not be assigned were discarded from the overall labeling calculation. Wells were flagged if there was no peak of unlabeled protein, an undefined peak of over 30%, or double labeling of a compound. Flagged wells were manually inspected, and the labeling assignment was modified if needed. Wells were regarded as “bad wells” if their LC and MS spectra appeared to be of a degraded protein (low intensity and deformed peak shape) or if after deconvolution there were no clear peaks (high noise levels). All of the compounds from bad wells were assigned as no available data in Data set 3 in the Supporting Information.
Quantitative Processing of Mass Spectrometry Data
A Python script for processing the MaxEnt deconvoluted spectra is supplied as File 2 in the Supporting Information. Below we briefly outline the logic of the processing.
We first identify the 10 highest peaks for each well and discard the rest. The peaks are then normalized from ion counts to percentages, where the highest peak is defined as 100%. The unlabeled protein mass is deduced from a reference well that contains just the protein. Up to four noncompound “adducts” are also assigned from that well (buffer adducts, protein oxidations, etc.); these often keep their proportion when a compound labels the protein.
We discard peaks lower than 10% of the maximum peak. For compound labeling assignment (i.e., not the reference well) we also discard peaks that are less than 100 Da heavier than the unlabeled protein (could not be compound). We iterate the remaining peaks, first assigning single modification, prioritizing “adducts”. In a second pass we try to assign double labeling (adduct + compound or two compounds). In identifying compound peaks, we allowed a “noise” level of ±4 Da; we note that in rare cases we did identify peaks not corresponding to any compound slightly above this noise level, but the automatic processing disregards these. If there are more than 4 peaks exceeding 45%, the well is flagged as a “Bad Well”.
For three of the proteins we identified a second major species: K-RasG12C ∼+48 Da in 25/200 wells, USP8 ∼+48 Da in 50/200 wells, and NV3CP ∼+78 Da in 116/200 wells. In these wells we manually assigned compound identity, taking into account both main species of the unmodified protein and compound labeling against other proteins in the benchmark.
Substructure and Clustering Analysis
We defined the “electrophilic” scaffold of each compound as the naked ring system of each compound decorated only by the electrophile. For each such electrophilic scaffold we ran a substructure search to identify class members. For chloroacetamides with a free NH we forced the NH to remain unsubstituted in the substructure search. Clustering (Figure S2E in the Supporting Information) was performed in Pipeline-Pilot (9.1.0.13) using ECFP_4 fingerprints and a 0.5 maximum Tanimoto coefficient cutoff.
Protein Sources, Expression, and Purification
BSA was purchased from MP biomedicals (cat. 160069). QSOX1 (mouse aa 36–550) was a generous gift from Prof. Deborah Fass (Weizmann Institute) and was produced as described in Grossman et al.90 NV3CP was produced following the procedure described in Hussey et al.68 K-RasG12C (1–169) was expressed and purified as described in Nnadi et al.18
PBP3R504C
The soluble 50–579 aa fragment lacking the N-terminal transmembrane helix of the ftsi gene from Pseudomonas aeruginosa PAO1 encoding for the penicillin-binding protein 3 (PaPBP3) was amplified by PCR and subcloned into pET47b using restriction enzymes BamHI and HindIII. The clinical mutation arginine 504 to cysteine was introduced by the Qiaquick protocol. The R504C mutant was expressed and purified as follows: transformed BL21 (DE3) cells were grown in LB media and induced with 1 mM IPTG; protein overexpression was carried out at 18 °C for 16 h; purification was achieved by reversed Ni2+ affinity chromatography using the N-terminal His6 tag followed by tag cleavage using recombinant HRV 3C protease; the protein was then injected onto a 16/60 HiLoad Superdex 200 column (GE Healthcare) and eluted in 20 mM Tris-HCl (pH 8) and 400 mM NaCl.
NUDT7
Human NUDT7 (residues 14–235) was cloned into pNIC28-Bsa4 with a TEV-cleavable N-terminal hexahistidine tag. After transformation into E. coli (BL21(DE3)-R3), expression was performed in TB auto induction medium (FroMedium), supplemented with 20 g/L glycerol, 50 μg/mL kanamycin, and 34 μg/mL chloramphenicol. Cultures were grown for 4 h at 37 °C, and then the temperature was decreased to 20 °C and the cultures were grown for another 20 h. Cells were spun at 5000 rpm for 10 min and then resuspended in 0.5 mg/mL lysozyme, 1 μg/mL benzonase, and 20 mM imidazole and stirred for 2 h at room temperature. Triton X-100 (1%) was added, and the cells were frozen at −80 °C. On thawing, cells were centrifuged for 1 h at 4000g and the supernatant applied to a His GraviTrap column (GE Healthcare) equilibrated with binding buffer (10 mM HEPES, 5% glycerol, 500 mM NaCl, 0.5 mM TCEP, pH 7.5). After washing with binding buffer supplemented to 20 mM imidazole, NUDT7 was eluted with buffer supplemented to 500 mM imidazole. The eluted protein was applied to a PD-10 desalting column (GE Healthcare) and eluted with binding buffer supplemented to 20 mM imidazole. The N-terminal affinity tag was removed by TEV cleavage overnight, and uncleaved protein was removed by applying it again to a His GraviTrap column. The flow-through was concentrated and purified further by size exclusion chromatography using a YARRA SEC-2000 PREP column (Phenomenex) equilibrated with binding buffer. Fractions containing protein were pooled, concentrated, and stored at −80 °C.
PCAF (aa 23–190) and USP8 (aa 705–1081)
These compounds were produced using the same procedure as for NUDT7.
NNMT
Wild type hNNMT was cloned into pET-28-TEVH vector harboring an N-terminal His 6-tag. The vector was transformed into E. coli BL21(DE3) cells. Following induction with 1 mM IPTG the culture grew overnight at 25 °C. The cells were suspended in lysis buffer (50 mM Tris pH 8, 0.5 M NaCl, 5 mM imidazole, 2 mM DTT, 5% glycerol supplemented with protease inhibitor cocktail (Calbiochem), 1 mM PMSF, 0.2 mg/mL lysozyme, and 20 μgr/mL DNase). The cells were lysed using a cooled cell disrupter. The clarified lysate was loaded onto a HisTrap_FF_crude column (GE Healthcare) equilibrated with binding buffer (50 mM Tris pH 8, 0.5 M NaCl, 25 mM Imidazole, 5% glycerol). The enzyme was eluted with the same buffer containing 0.25 M imidazole and injected immediately to a size exclusion column (HiLoad_26/60_Superdex_75). hNNMT eluted in a single peak. The protein was flash frozen using liquid nitrogen in aliquots and kept at −80 °C.
OTUB2A
pET20b vector containing human OTUB2A (residues 1–234) was transformed into the E. coli expression strain BL21(DE3)-R3. Expression was performed in TB medium, supplemented with 0.4% glucose, 50 μg/mL ampicillin, and 34 μg/mL chloramphenicol. Cultures were grown at 37 °C until an absorbance at 600 nm of 1.2. The temperature was then decreased to 18 °C, and the cultures were grown for a further 90 min. Benzyl alcohol (10 mM) was added, and the cells were grown for a further 20 min before inducing with 0.5 mM IPTG and grown overnight. The following day, cells were centrifuged at 6000 rpm for 30 min and the pellet was resuspended in the lysis buffer (50 mM Tris pH 7.5, 500 mM NaCl, 5% glycerol).
Cells were lysed by sonication for 3 min (20 s on, 50 s off). The lysate was centrifuged at 17000 rpm for 45 min. The supernatant was bound to Ni-NTA agarose beads that were pre-equilibrated with binding buffer (50 mM Tris pH 8, 500 mM NaCl, 20 mM imidazole), for 1 h at 4 °C. The beads were washed with binding buffer to pack the column.
Protein was eluted with elution buffer (50 mM Tris pH 8, 500 mM NaCl, 200 mM imidazole) supplemented with 0.5 mM DTT. The His tag was removed by TEV cleavage while OTUB2A was dialyzed overnight in a buffer containing 50 mM Tris pH 8, 500 mM NaCl, 0.5 mM DTT. Dialyzed protein was incubated again with Ni-NTA agarose beads for 1 h. The untagged protein was collected as the flow through, concentrated, and purified further by size exclusion chromatography using a HiLoad 26/600 Superdex 75 pg apparatus (GE Healthcare Lifesciences) equilibrated with gel filtration buffer (20 mM Tris pH 8, 50 mM NaCl, 5 mM DTT). Fractions containing the protein were pooled, concentrated to 25 mg/mL, and stored at −80 °C.
OTUB2A Crystallization
Microcrystals of OTUB2A were by obtained by mixing 50 nL of OTUB2A (25 mg/mL) with 100 nL of reservoir solution (16% PEG4K, 0.1 M HEPES pH 7.0, 8% 2-propanol, 5 mM DTT) in a sitting drop plate at 20 °C. These microcrystals were used for making a seed stock.
In order to first attempt the soaking strategy, 20 nL of the seed stock was used to grow large prism-shaped crystals in less than 24 h. An ECHO 550 acoustic liquid handler (Labcyte) was used to transfer individual fragments from the covalent fragment library to drops containing crystals. Briefly, the compound solution was added to each crystallization drop, resulting in a final compound concentration of 4 mM with 20% DMSO, calculated on the basis of the initial drop volume. Crystals were incubated for 2 and 24 h at room temperature. All structures except the compound PCM-0103080 were obtained by cocrystallization.
For cocrystallization of OTUB2A with the compounds of the covalent fragment library, 100 μL of the crystallization cocktail (16% PEG4K, 0.1 M HEPES pH 7.0, 8% 2-propanol, 5 mM DTT) was dispensed in the reservoir of a sitting drop plate. An ECHO 550 acoustic liquid handler was then used to dispense 75 nL of protein and 1–4 mM of the compound on top of the protein drop. The mix was incubated at 20 °C overnight. The next day, 75 nL of the reservoir solution was added on top of the drop along with 20 nL of the seed stock. The plate was incubated at 20 °C, and crystals were obtained within 24 h.
All crystals were harvested with 20% ethylene glycol as cryoprotection and flash-cooled in liquid nitrogen. All X-ray diffraction data were collected on the beamline I04-1 at the Diamond Light Source (Harwell, U.K.) unless stated otherwise.
OTUB2 Structure Determination
Diffraction data were automatically processed by software pipelines at the Diamond Light Source.91 Initial refinement and map calculation was carried out with DIMPLE.92 PanDDA93 was used for hit identification. Further refinement and model building was performed with REFMAC94 and COOT,95 respectively. Coordinates and structure factors for all data sets are deposited in the RCSB Protein Data Bank under PDB IDs 5QIO, 5QIP, 5QIQ, 5QIR, 5QIS, 5QIT, 5QIU, 5QIV, 5QIW, 5QIX, 5QIY, and 5QIZ. Data collection and refinement statistics are available from the PDB pages.
OTUB2 Inhibition Assays
The assays were performed in “non-binding surface flat bottom low flange” black 384-well plates (Corning) at room temperature in a buffer containing 50 mM Tris·HCl, 100 mM NaCl, pH 7.6, 2.0 mM cysteine, 1 mg/mL 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonic acid (CHAPS), and 0.5 mg/mL γ-globulins from bovine blood (BGG). Each well had a final volume of 20.4 μL. The compounds were dissolved in 10 mM DMSO stocks, and appropriate volumes were transferred to the empty plates using a Labcyte Echo acoustic dispenser. A DMSO back-fill was performed to obtain equal volumes of DMSO (400 μL) in each well. N-Ethylmaleimide (NEM, 10 mM) was used a positive control (100% inhibition) and DMSO as negative control (0% inhibition). A 10 μL portion of buffer was added, and the plate was vigorously shaken for 20 s. Next, 5 μL of OTUB2 (full length) was added to a final concentration of 25 nM followed by incubation for 30 or 150 min. A 5 μL portion of the substrate (Ub-Rho) was added (final concentration 400 nM), and the increase in fluorescence over time was recorded using a BMG Labtech Clariostar plate reader (excitation 487 nm, emission 535 nm). The initial enzyme velocities were calculated from the slopes, normalized to the positive and negative controls, and plotted using GraphPad Prism 7 to obtain the IC50 values.
DUB ABPP Assays
All assays were performed in a buffer containing 50 mM Tris, pH 7.4, 5 mM MgCl2, 250 mM sucrose, 5 mM DTT, and 2 mM ATP. Purified recombinant OTUB2, HEK293T cell lysate (2.5 mg/mL), and HEK293T cell lysate spiked with purified recombinant OTUB2 (0.05 and 0.1 μg/μL) were incubated with 50 μM of the inhibitors for 3 h at 37 °C. Iodoacetamide (10 mM) was used as a positive control. Next, a Rho-Ub-PRG probe (10 μM) was added and the samples were incubated for another 30 min at 37 °C. Proteins were resolved on a 4–12% NuPage Novex Bis-Tris gel using MOPS running buffer. Rho-Ub-PRG bound DUBs were visualized by fluorescence scanning of the gel on a GE Typhoon GoldSeal FLA9500 scanner (excitation 473 nm), and protein loading was checked by Expedeon InstantBlue staining.
DUB inhibition of NUDT7 inhibitor NUDT7-COV-1 was assessed using a similar method. HEK293T cell lysate was incubated with a concentration series of 0.1–100 μM of the compound, and N-ethylmaleimide (15 mM) was used as positive control.
OTUB2 Overexpression and In-Cell Inhibition Assay
HEK293 cells were transfected with plasmids overexpressing GFP, GFP-OTUB2 wild type (GFP-OTUB2 wt), and GFP-OTUB2 catalytic cysteine-to-serine mutant (GFP-OTUB2 CS)73 separately in 6-well plates using polyethylenimine (PEI, Polysciences, lnc.) according to the manufacturer’s instructions. At 24 h following transfection, cells were incubated with COV-1 compound at the indicated final concentrations at 37 °C for 4 h. Cells were harvested and resuspended in HR buffer (50 mM Tris, 5 mM MgCl2, 250 mM sucrose, 1 mM DTT, pH 7.4). Cell lysis was achieved by sonication (Bioruptor, Diagenode, high intensity for 10 min with an ON/OFF cycle of 30 s) at 4 °C. After a centrifugation step (14000 rpm for 15 min) to remove cell debris, the protein concentration of the supernatant was determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific) by measuring the absorbance at 280 nm. A 40 μg portion of each protein sample was treated with rhodamine–ubiquitin–propargylamide probe (final concentration 1 μM) at 37 °C for 1 h. Reactions were stopped by the addition of LDS (lithium dodecyl sulfate) sample buffer (Invitrogen Life Technologies, Carlsbad, CA, USA) containing 2.5% β-mercaptoethanol, followed by boiling for 7 min. A 20 μg portion of each sample was resolved on 4–12% Bis-Tris NuPAGE Gels (Invitrogen) using MOPS buffer (Invitrogen Life Technologies, Carlsbad, CA, USA). Labeled enzymes were visualized by in-gel fluorescence using a Typhoon FLA 9500 imaging system (GE Healthcare Life Sciences) (rhodamine channel for probe, Cy5 channel for protein marker). The gel was then transferred to nitrocellulose membranes and immunoblotted using rabbit anti-GFP serum96 and mouse anti-β-actin (Sigma-Aldrich). Immunoblots were visualized using a LICOR Odyssey system. The following fluorescent secondary antibodies purchased from LICOR were used: antimouse-680, antirabbit-800.
Jump Dilution Assay
All assays were performed in triplicate. The assay was performed in a buffer containing 50 mM Tris-HCl, 100 mM NaCl, pH 7.6, 2.0 mM cysteine, 1 mg/mL 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonic acid (CHAPS), and 0.5 mg/mL γ-globulins from bovine blood (BGG). The final concentrations used were 25 nM OTUB2, 400 nM Ub-Rho, 100 μM or 1 μM or a jump dilution of 100 μM to 1 μM inhibitor. Samples containing 20 μL of a 5 μM OTUB2 solution were mixed with 20 μL of 200 μM inhibitor in buffer (2% DMSO), 2% DMSO in buffer, or 20 mM N-ethylmaleimide (NEM) in buffer (2% DMSO). These three samples were incubated for 150 min at room temperature. A 5 μL portion of each sample was then diluted into a 500 μL solution containing 400 nM Ub-Rho. After a brief mixing 20 μL of each of these solutions was quickly transferred to a “non-binding surface flat bottom low flange” black 384-well plate (Corning) and the increase in fluorescence over time was recorded using a BMG Labtech Clariostar plate reader (excitation 487 nm, emission 535 nm). As a control, samples were taken along in which 40 μL of a 200 μM and 2 μM inhibitor solution in buffer (2% DMSO) or 2% DMSO in buffer were added to 20 μL of a 100 nM OTUB2 solution. After 150 min incubation 20 μL of a 1.6 μM Ub-Rho solution was added, after which 20 μL of each solution was transferred to the same 384-well plate mentioned above and the increase in fluorescent intensity was measured concomitantly. Fluorescent intensities were plotted against time using GraphPad Prism 7.
OTUB2-COV-1 kinact/KI Determination
The assay was performed in duplicate in a “non-binding surface flat bottom low flange” black 384-well plate (Corning) in a buffer containing 50 mM Tris-HCl, 100 mM NaCl, pH 7.6, 2.0 mM cysteine, 1 mg/mL 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonic acid (CHAPS), and 0.5 mg/mL γ-globulins from bovine blood (BGG). A serial dilution of the inhibitor (200–150–100–50–25–12.5–5–1.25–0 μM final concentration) was made by transferring different amounts (400–0 nL) of a 10 mM stock solution in DMSO into an empty plate using a Labcyte Echo550 acoustic dispenser. All volumes were equalized to 400 nL by means of a DMSO back-fill. A 15 μL portion of a 533 nM Ub-Rho solution was added to each well using a Biotek Multiflow FX liquid dispenser, and the plate was shaken vigorously for 10 s and then centrifuged for 1 min at 1000 rpm. Next, 5 μL of a 100 nM OTUB2 solution was added to each well, the plate was quickly loaded into a BMG Clariostar plate reader, and the increase in fluorescence (excitation 487 nm, emission 535 nm) over time was recorded immediately. All data fitting and calculations were done using GraphPad Prism 7 software. The fluorescence intensities were plotted against time (in seconds) after a baseline correction using the DMSO control for each inhibitor concentration. The data were fitted to the equation
NUDT7 Crystallization
NUDT7 crystals were obtained by mixing 100 nL of 30 mg/mL protein in 10 mM Na-HEPES pH 7.5, 500 mM NaCl, 5% glycerol with 50 nL of reservoir solution containing 0.1 M BisTris pH 5.5, 0.1 M ammonium acetate, and 6% (w/v) PEG 10000. Compact, hexagon-shaped crystals with typical dimensions between 50 and 100 μm appeared within several days from sitting drop plates at 20 °C. Cocrystals of NUDT7 in complex with NUDT7-REV-1 and NUDT7-COV-1 were obtained by soaking NUDT7 crystals with a mixture containing 600 nL of 100 mM of the respective compound in DMSO with 1200 nL of reservoir solution. Crystals were incubated overnight at room temperature and then harvested (without further cryoprotection) and flash-cooled in liquid nitrogen. Crystals of NUDT7 with covalent fragments were grown by mixing 100 nL of 30 mg/mL protein in 10 mM Na-HEPES pH 7.5, 500 mM NaCl, and 5% glycerol with 30 nL of 20 mM compound in DMSO in sitting-drop crystallization plates containing 0.1 M BisTris pH 5.5, 0.1 M ammonium acetate, and 4–16% (w/v) PEG 10000 in the reservoir at 20 °C. After overnight incubation of protein and compound, 100 nL of reservoir solution and 30 nL of a crystal seed solution obtained from a previous crystallization experiment were added to the drop. Hexagon-shaped crystals appeared within several days. Prior to data collection, all crystals were transferred to a solution consisting of the precipitation buffer supplemented with 25% ethylene glycol and subsequently flash-cooled in liquid nitrogen. All X-ray diffraction data were collected on beamline I04-1 and beamline I03 at the Diamond Light Source (Harwell, U.K.).
NUDT7 Structure Determination
Diffraction data were automatically processed by software pipelines at the Diamond Light Source.91 Initial refinement and map calculation was carried out with DIMPLE.92 PanDDA93 was used for hit identification, and further refinement and model building was performed with REFMAC94 and COOT,95 respectively. All structure determination steps were performed within the XChemExplorer97 data management and workflow tool.
Coordinates and structure factors for all data sets are deposited in the Protein Data Bank under group deposition ID G_1002045. Data collection and refinement statistics are summarized in Table 5 in the Supporting Information. The complete PanDDA analysis and all processed data from the NUDT7 fragment campaign (including information about soaked compounds) can be accessed via the ZENODO data repository under DOI 10.5281/zenodo.1244111.
NUDT7 Activity Assay
Mass spectrometry assays monitoring acetyl-CoA hydrolysis by NUDT7 were performed on a Agilent 6530 RapidFire QTOF mass spectrometer in a 384-well plate format using polypropylene plates (Greiner, code 781280) and an assay buffer containing 20 mM HEPES pH 7.5, 200 mM NaCl, and 5 mM MgCl2. All bulk liquid handling steps were performed using a multidrop combi reagent dispenser (Thermo Scientific, Code 5840300) equipped with a small tube plastic tip dispensing cassette (Thermo Scientific, Code 24073290). For inhibitor IC50 determinations an 11-point and 2-fold serial dilution was prepared from a 50 mM stock solution in DMSO, which was transferred to give four replicates using an ECHO 550 acoustic dispenser (Labcyte). The transferred volume was 400 nL, giving a final DMSO concentration of 0.4%. In addition, a DMSO control (400 nL) was transferred into alternate wells in columns 12 and 24 and 50 mM EDTA (NUDT7 inhibitor) was dispensed into alternate wells of column 24 as the background control. An 80 μL portion of assay buffer was added to all wells, and NUDT7 was prepared to 500 nM (10× final concentration in assay buffer), and acetyl-CoA was prepared to 200 μM (10× final concentration in assay buffer). A 10 μL portion of NUDT7 was dispensed into half of the assay plate (for two of the compound replicates), and the plate was incubated at room temperature for 30 min. A 10 μL portion of NUDT7 was then dispensed into the remaining half of the assay plate (for the remaining two compound replicates). A 10 μL portion of acetyl-CoA was immediately dispensed into all wells of the assay plate to initiate the reaction, and the enzyme reaction was allowed to proceed for 15 min. The enzyme reaction was stopped by addition of 10 μL of 50 mM EDTA, and the plate was transferred to a RapidFire RF360 high-throughput sampling robot. Samples were aspirated under vacuum and loaded onto a C4 solid-phase extraction (SPE) cartridge equilibrated and washed for 5.5 s with 1 mM octylammonium acetate in LCMS grade water to remove nonvolatile buffer components. After the aqueous wash, analytes of interest were eluted from the C4 SPE onto an Agilent 6530 accurate mass Q-TOF in an organic elution step (85% acetonitrile in LC-MS grade water). Ion data for the acetyl-CoA and hydrolyzed product were extracted and peak area data integrated using RapidFire integrator software (Agilent). Percent conversion of substrate to product was calculated in Excel, and IC50 curves were generated using Graphpad prism version 7.0. The assay had a Z score of 0.79 with the 30 min preincubation and 0.75 without preincubation. For kinact/KI determination, product conversion was followed as a function of time in the presence of various concentrations of inhibitor. The time window was restricted to the linear conversion domain for DMSO and buffer. Kobs vs [inhibitor] plots were fitted either to Y = kinactX/(KI + X) to separately determine kinact and KI or to a linear line for which the slope is directly kinact/KI.
NUDT7 Thermal Shift Assay
A 5 μM portion of NUDT7 was incubated prior to the measurements with 200 μM of compound in 10 mM HEPES pH 7.5, 0.3 M NaCl, 0.5 mM TCEP for 24 h at 4 °C. A 1 μL portion of 5x SYPRO Orange (Sigma) was added to 19 μM of incubated protein in a MicroAmp Fast Optical 96-Well Reaction Plate sealed with MicroAmp Optical Adhesive Film. Measurements were performed using StepOnePlus rtPCR from 25 to 95 °C with 0.3 °C steps. Tm was determined using StepOne Software v2.3. The reported Tm was calculated as the average of three triplicates for each compound.
NUDT7 Cellular Thermal Shift Assay
HEK293 cells were cultured at 37 °C in a humidified 5% CO2 atmosphere in DMEM supplemented with GlutaMAX and 10% FBS. Cells were grown in T175 flasks until around 70% confluence and transfected with Flag-NUDT7 using Lipofectamine2000 transfection reagent. Twenty-four hours post-transfection, cells were detached, and 13 × 106 cells were seeded in T75 flasks for treatment and control samples, respectively. After 24 h cells were treated with either DMSO or 20 μM NUDT7-COV-1 for 30 min at 37 °C. Cells were harvested, washed with PBS, and aliquoted into PCR tubes. PBS was removed by centrifugation (300g, 3 min, room temperature). Cell pellets were heated to temperatures ranging from 37 to 73 °C with 4 °C increments for 3 min (UNO96, VWR), cooled to room temperature for 3 min, and then transferred onto ice. Lysis was performed in lysis buffer (50 mM Tris pH 7.5, 0.8% v/v NP-40, 5% v/v glycerol, 1.5 mM MgCl2, 100 mM NaCl, 25 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 1 mM DTT, 10 μg/mL TLCK, 1 μg/mL leupeptin, 1 μg/mL aprotinin, 1 μg/mL soy bean trypsin) by three freeze–thaw cycles in liquid nitrogen. The resulting lysates were centrifuged at 21000g for 20 min at 4 °C to remove aggregated proteins. Protein concentration was determined for the soluble fraction followed by by SDS-PAGE analysis followed and Western blotting. After transfer the nitrocellulose membrane was blocked with blocking buffer (5% (m/v) BLOT-QuickBlocker (Merck) in PBST (phosphate-buffered saline with 0.05% (v/v) Tween 20)) and probed with primary antibody (mouse anti-FLAG (Merck, F3165) 1:1500 in blocking buffer) overnight at 4 °C and secondary antibody (goat antimouse Alexa Fluor 750 (Life Technologies, A-21037) 1:10000 in blocking buffer) for 1 h at room temperature. Blots were imaged on an Odyssey CLx imager (LI-COR).
Cell Viability Assay
HEK293, HB2, or CCD841 cells grew in either RPMI or DMEM mediums supplemented with 10% FCS, 1% PS, and 1% l-glutamine (all from Biological Industries). Exclusion of Mycoplasma contamination was monitored and conducted by test with a Mycoalert kit (LONZA). Cells were trypsnized and counted, and 1000 cells/well were plated in 50 μL of growth medium into 384-well white TC plates (Greiner) using a MultiDrop 384 (Thermo Scientific) Washer Dispenser II. The number of viable cells was monitored using a CellTiter-Glo Luminescent kit (Promega) in accordance with the manufacturer’s protocol. Luminescence was measured using the luminescence module of a PheraStar FS plate reader (BMG Labtech). Data analysis was performed using GeneData 12 analytic software.
NNMT Activity Assay
Compounds were transferred into black microplates (Greiner 784900) using Labcyte Echo acoustic dispensing. Assay ready plates were then sealed with heat seals. If they were not used immediately, plates were frozen at −20 °C and held in polypropylene boxes with silica gel desiccant.
Reagents were obtained as follows: nicotinamide (Sigma 47865-U), SAM (Sigma A7007), SAH-FITC (Axis Shield, RPBB350), Anti-SAH (Axis Shield, RPBB278). All liquid handling was done with a GNF Washer/dispenser II. All reagents were prepared in 20 mM phosphate buffer, pH = 7.6. A 6 μL portion of 3X NNMT (120 nM) was added to assay plates and incubated for 10 min at room temperature. A 6 μL portion of 3X substrate mixture (120 mM Nicotinamide, 6 μM SAM) was added and incubated for 1 h at 30 °C. A 6 μL portion of 3X detection mixture (150 ng/mL SAH-FITC and 30 μg/mL anti-SAH antibody) was prepared and incubated for 30 min at room temperature before adding to reaction mixtures. Plates were further incubated for another 2 h at 30 °C, protected from light. Fluorescence polarization reactions were read with a BMG Pherastar FS instrument using a 485/520/520 nm module. Data were normalized to DMSO (100%) and no enzyme (0%) controls using Genedata Screener software.
Profiling of OTUB2 and NUDT7 Reactive Cysteines by Competitive isoTOP-ABPP
The profiling was performed according to a slightly modified competitive isoTOP-ABPP protocol.74 Briefly, HEK-293T cells were cultured at 37 °C under 5% CO2 atmosphere in DMEM culture medium supplemented with 10% FBS and 1% PS. Cells were grown to 80% confluence and incubated with DMSO or 10 μM compound (OTUB2 or NUDT7) for 2 h in serum-free media. Cells were harvested, lysed by sonication in ice-cold PBS containing 0.1% Triton X-100, and centrifuged at 20000g for 30 min to remove cell debris. Protein concentrations were determined by a BCA protein assay, and proteomes were normalized to 2 mg/mL in 1 mL for each sample. Each of the DMSO- and compound-incubated proteomes was treated with 100 μM IAyne for 1 h at room temperature. The proteomes were then reacted with 1 mM CuSO4, 100 μM TBTA ligand, 100 μM either light or heavy variants of the azido-TEV-biotin tags, and 1 mM TCEP for 1 h. After the click reaction, the proteomes were centrifuged at 8000g for 5 min and the precipitated proteins were washed two times using cold methanol. The two samples were mixed, resuspended in 1.2% SDS/PBS, and diluted to 0.2% SDS/PBS for streptavidin enrichment and subsequent trypsin and TEV digestion. The resulting TEV digests were analyzed by LC-MS/MS on a Q-Exactive Orbitrap instrument, and the MS data were searched and quantified by the Open-pfind algorithm.98
Acknowledgments
N.L. is the incumbent of the Alan and Laraine Fischer Career Development Chair. N.L. wishes to acknowledge funding from the Israel Science Foundation (Grant No. 1097/16), The Rising Tide Foundation, The Israel Cancer Research Foundation, and the Israeli Ministry of Science and Technology (Grant No. 3-14763). This work was supported by the Dutch Organization for Scientific Research NWO (VICI grant 724.013.002 to H.O.) We thank Yves Leestemaker for assistance with the DUB ABPP assays. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, the Canada Foundation for Innovation, the Eshelman Institute 22 for Innovation, Genome Canada through Ontario Genomics Institute (OGI-055)], the Innovative Medicines Initiative (EU/EFPIA) (ULTRA-DD Grant No. 115766), Janssen, Merck KGaA (Darmstadt, Germany), MSD, Novartis Pharma AG, the Ontario Ministry of Research, Innovation and Science (MRIS), Pfizer, Sao Paulo Research Foundation-FAPESP, Takeda, and Wellcome (106169/Z/14/Z). Supported by a research grant from the Nancy and Stephen Grand Israel National Center for Personalized Medicine. We thank Ilana Berlin for providing the GFP-OTUB2 plasmids. We thank Prof. Deborah Fass for the generous gift of QSOX1 protein.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b02822.
Additional information on the distribution of the electrophile-fragment library properties including MW distribution of the adduct form and diversity of substructures, experimental data for thiol-reactivity assay in different TCEP concentrations, reactivity rates for resourced compounds and comparison with GSH t1/2, correlations between reactivity rates and goodness of fit to the second-order reaction equation, between reactivity and occurrences of double labeling, between reactivity and overall labeling, between reactivity and cellular toxicity, and between reactivity of unsubstituted and methyl-substituted chloroacetamides, comparison of labeling with single compound vs compounds in pools of five, labeling of OTUB2 with selected compounds, identification of cysteine modification sites for sample compounds for USP8 and NUDT7, structures of unique hits, promiscuous compounds, compounds containing aminothiazoles, and second-generation OTUB2 binders, cocrystal structures of OTUB2 with two compounds containing the acethydrazide motif and with OTUB2-COV-1 and crystallographic statistics, experimental data of jump dilution experiment for OTUB2 with OTUB2-COV-1, gel-based ABPP selectivity of OTUB2 inhibitors in lysates and in cells, stabilization of NUDT7 by covalent fragments, and enzymatic inhibition of NUDT7, Kinact/Ki determination and competitive isoTOP-ABPP for both OTUB2-COV-1 and NUDT7-COV-1 (PDF)
Data for thiol-reactivity assay, LC/MS/MS of NUDT7 and USP8 with a selection of compounds, isoTOP-ABPP ratios for all peptides, cellular viability and NNMT enzymatic inhibition, labeling data for all fragments in the electrophile library against 10 proteins, double-labeling occurrence, and comparison between labeling of one compound vs pool of five compounds (XLSX)
Detailed synthetic procedures and 1H and 13C NMR spectra and LC/MS for synthesized compounds (PDF)
Labeling quantification script from raw LC/MS data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Noe M. C.; Gilbert A. M.. Targeted Covalent Enzyme Inhibitors. In Annual Reports in Medicinal Chemistry; Elsevier: 2012; Vol. 47, pp 413–439. [Google Scholar]
- Johnson D. S.; Weerapana E.; Cravatt B. F. Strategies for Discovering and Derisking Covalent, Irreversible Enzyme Inhibitors. Future Med. Chem. 2010, 2 (6), 949–964. 10.4155/fmc.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw J. M.; McFarland J. M.; Paavilainen V. O.; Bisconte A.; Tam D.; Phan V. T.; Romanov S.; Finkle D.; Shu J.; Patel V.; Ton T.; Li X.; Loughhead D. G.; Nunn P. A.; Karr D. E.; Gerritsen M. E.; Funk J. O.; Owens T. D.; Verner E.; Brameld K. A.; Hill R. J.; Goldstein D. M.; Taunton J. Prolonged and Tunable Residence Time Using Reversible Covalent Kinase Inhibitors. Nat. Chem. Biol. 2015, 11 (7), 525–531. 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. S.; Zhang C.; Shokat K. M.; Taunton J. Structural Bioinformatics-Based Design of Selective, Irreversible Kinase Inhibitors. Science 2005, 308 (5726), 1318–1321. 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer R. A. Covalent Inhibitors in Drug Discovery: From Accidental Discoveries to Avoided Liabilities and Designed Therapies. Drug Discovery Today 2015, 20 (9), 1061–1073. 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Discovery 2011, 10 (4), 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Mah R.; Thomas J. R.; Shafer C. M. Drug Discovery Considerations in the Development of Covalent Inhibitors. Bioorg. Med. Chem. Lett. 2014, 24 (1), 33–39. 10.1016/j.bmcl.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Baillie T. A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem., Int. Ed. 2016, 55 (43), 13408–13421. 10.1002/anie.201601091. [DOI] [PubMed] [Google Scholar]
- Matthews D. A.; Dragovich P. S.; Webber S. E.; Fuhrman S. A.; Patick A. K.; Zalman L. S.; Hendrickson T. F.; Love R. A.; Prins T. J.; Marakovits J. T.; Zhou R.; Tikhe J.; Ford C. E.; Meador J. W.; Ferre R. A.; Brown E. L.; Binford S. L.; Brothers M. A.; DeLisle D. M.; Worland S. T. Structure-Assisted Design of Mechanism-Based Irreversible Inhibitors of Human Rhinovirus 3C Protease with Potent Antiviral Activity against Multiple Rhinovirus Serotypes. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (20), 11000–11007. 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z.; Scheerens H.; Li S.-J.; Schultz B. E.; Sprengeler P. A.; Burrill L. C.; Mendonca R. V.; Sweeney M. D.; Scott K. C. K.; Grothaus P. G.; Jeffery D. A.; Spoerke J. M.; Honigberg L. A.; Young P. R.; Dalrymple S. A.; Palmer J. T. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2 (1), 58–61. 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Huhn A. J.; Guerra R. M.; Harvey E. P.; Bird G. H.; Walensky L. D. Selective Covalent Targeting of Anti-Apoptotic BFL-1 by Cysteine-Reactive Stapled Peptide Inhibitors. Cell Chem. Biol. 2016, 23 (9), 1123–1134. 10.1016/j.chembiol.2016.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage S. J.; Jones L. H.; Gray N. S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20 (2), 146–159. 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A.; Koch P.; Laufer S. A.; Knapp S. The Cysteinome of Protein Kinases as a Target in Drug Development. Angew. Chem., Int. Ed. 2018, 57 (16), 4372–4385. 10.1002/anie.201707875. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Bourne P. E. Progress with Covalent Small-Molecule Kinase Inhibitors. Drug Discovery Today 2018, 23 (3), 727–735. 10.1016/j.drudis.2018.01.035. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Inesta-Vaquera F.; Niepel M.; Zhang J.; Ficarro S. B.; Machleidt T.; Xie T.; Marto J. A.; Kim N.; Sim T.; Laughlin J. D.; Park H.; LoGrasso P. V.; Patricelli M.; Nomanbhoy T. K.; Sorger P. K.; Alessi D. R.; Gray N. S. Discovery of Potent and Selective Covalent Inhibitors of JNK. Chem. Biol. 2012, 19 (1), 140–154. 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Hur W.; McDermott U.; Dutt A.; Xian W.; Ficarro S. B.; Zhang J.; Sharma S. V.; Brugge J.; Meyerson M.; Settleman J.; Gray N. S. A Structure-Guided Approach to Creating Covalent FGFR Inhibitors. Chem. Biol. 2010, 17 (3), 285–295. 10.1016/j.chembiol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N.; Miller R. M.; Krishnan S.; Uchida K.; Irwin J. J.; Eidam O.; Gibold L.; Cimermančič P.; Bonnet R.; Shoichet B. K.; Taunton J. Covalent Docking of Large Libraries for the Discovery of Chemical Probes. Nat. Chem. Biol. 2014, 10 (12), 1066–1072. 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nnadi C. I.; Jenkins M. L.; Gentile D. R.; Bateman L. A.; Zaidman D.; Balius T. E.; Nomura D. K.; Burke J. E.; Shokat K. M.; London N. Novel K-Ras G12C Switch-II Covalent Binders Destabilize Ras and Accelerate Nucleotide Exchange. J. Chem. Inf. Model. 2018, 58 (2), 464–471. 10.1021/acs.jcim.7b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo Warshaviak D.; Golan G.; Borrelli K. W.; Zhu K.; Kalid O. Structure-Based Virtual Screening Approach for Discovery of Covalently Bound Ligands. J. Chem. Inf. Model. 2014, 54 (7), 1941–1950. 10.1021/ci500175r. [DOI] [PubMed] [Google Scholar]
- Ouyang X.; Zhou S.; Su C. T. T.; Ge Z.; Li R.; Kwoh C. K. CovalentDock: Automated Covalent Docking with Parameterized Covalent Linkage Energy Estimation and Molecular Geometry Constraints. J. Comput. Chem. 2013, 34 (4), 326–336. 10.1002/jcc.23136. [DOI] [PubMed] [Google Scholar]
- Scholz C.; Knorr S.; Hamacher K.; Schmidt B. DOCKTITE— A Highly Versatile Step-by-Step Workflow for Covalent Docking and Virtual Screening in the Molecular Operating Environment. J. Chem. Inf. Model. 2015, 55 (2), 398–406. 10.1021/ci500681r. [DOI] [PubMed] [Google Scholar]
- Zhu K.; Borrelli K. W.; Greenwood J. R.; Day T.; Abel R.; Farid R. S.; Harder E. Docking Covalent Inhibitors: A Parameter Free Approach to Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54 (7), 1932–1940. 10.1021/ci500118s. [DOI] [PubMed] [Google Scholar]
- Shraga A.; Olshvang E.; Davidzohn N.; Khoshkenar P.; Germain N.; Shurrush K.; Carvalho S.; Avram L.; Albeck S.; Unger T.; Lefker B.; Subramanyam C.; Hudkins R. L.; Mitchell A.; Shulman Z.; Kinoshita T.; London N. Covalent Docking Identifies a Potent and Selective MKK7 Inhibitor. Cell Chemical Biology. 2019, 26 (1), 98–108.e5. 10.1016/j.chembiol.2018.10.011. [DOI] [PubMed] [Google Scholar]
- Sirois S.; Hatzakis G.; Wei D.; Du Q.; Chou K.-C. Assessment of Chemical Libraries for Their Druggability. Comput. Biol. Chem. 2005, 29 (1), 55–67. 10.1016/j.compbiolchem.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53 (7), 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Baell J.; Walters M. A. Chemistry: Chemical Con Artists Foil Drug Discovery. Nature 2014, 513 (7519), 481–483. 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- Huth J. R.; Mendoza R.; Olejniczak E. T.; Johnson R. W.; Cothron D. A.; Liu Y.; Lerner C. G.; Chen J.; Hajduk P. J. ALARM NMR: A Rapid and Robust Experimental Method to Detect Reactive False Positives in Biochemical Screens. J. Am. Chem. Soc. 2005, 127 (1), 217–224. 10.1021/ja0455547. [DOI] [PubMed] [Google Scholar]
- Metz J. T.; Huth J. R.; Hajduk P. J. Enhancement of Chemical Rules for Predicting Compound Reactivity towards Protein Thiol Groups. J. Comput.-Aided Mol. Des. 2007, 21 (1–3), 139–144. 10.1007/s10822-007-9109-z. [DOI] [PubMed] [Google Scholar]
- Leach A. R.; Hann M. M.; Burrows J. N.; Griffen E. J. Fragment Screening: An Introduction. Mol. Biosyst. 2006, 2 (9), 429–446. 10.1039/b610069b. [DOI] [PubMed] [Google Scholar]
- Baker M. Fragment-Based Lead Discovery Grows up. Nat. Rev. Drug Discovery 2013, 12 (1), 5–7. 10.1038/nrd3926. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. Fragment-Based Approaches in Drug Discovery and Chemical Biology. Biochemistry 2012, 51 (25), 4990–5003. 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- Hall R. J.; Mortenson P. N.; Murray C. W. Efficient Exploration of Chemical Space by Fragment-Based Screening. Prog. Biophys. Mol. Biol. 2014, 116 (2–3), 82–91. 10.1016/j.pbiomolbio.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Hann M. M.; Leach A. R.; Harper G. Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery. J. Chem. Inf. Comput. Sci. 2001, 41 (3), 856–864. 10.1021/ci000403i. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Braisted A. C.; Raphael D. R.; Randal M.; Stroud R. M.; Gordon E. M.; Wells J. A. Site-Directed Ligand Discovery. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (17), 9367–9372. 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson D. A.; Wells J. A.; Braisted A. C. Tethering: Fragment-Based Drug Discovery. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 199–223. 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- Wilson C. G.; Arkin M. R. Probing Structural Adaptivity at PPI Interfaces with Small Molecules. Drug Discovery Today Technol. 2013, 10 (4), e501–e508. 10.1016/j.ddtec.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503 (7477), 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathman S. G.; Xu Z.; Statsyuk A. V. A Fragment-Based Method to Discover Irreversible Covalent Inhibitors of Cysteine Proteases. J. Med. Chem. 2014, 57 (11), 4969–4974. 10.1021/jm500345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonoo R. H.; Armstrong A.; Mann D. J. Kinetic Template-Guided Tethering of Fragments. ChemMedChem 2012, 7 (12), 2082–2086. 10.1002/cmdc.201200404. [DOI] [PubMed] [Google Scholar]
- Craven G. B.; Affron D. P.; Allen C. E.; Matthies S.; Greener J. G.; Morgan R. M. L.; Tate E. W.; Armstrong A.; Mann D. J. High-Throughput Kinetic Analysis for Target-Directed Covalent Ligand Discovery. Angew. Chem., Int. Ed. 2018, 57 (19), 5257–5261. 10.1002/anie.201711825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jöst C.; Nitsche C.; Scholz T.; Roux L.; Klein C. D. Promiscuity and Selectivity in Covalent Enzyme Inhibition: A Systematic Study of Electrophilic Fragments. J. Med. Chem. 2014, 57 (18), 7590–7599. 10.1021/jm5006918. [DOI] [PubMed] [Google Scholar]
- Cardoso R.; Love R.; Nilsson C. L.; Bergqvist S.; Nowlin D.; Yan J.; Liu K. K.-C.; Zhu J.; Chen P.; Deng Y.-L.; Dyson H. J.; Greig M. J.; Brooun A. Identification of Cys255 in HIF-1α as a Novel Site for Development of Covalent Inhibitors of HIF-1α/ARNT PasB Domain Protein-Protein Interaction. Protein Sci. 2012, 21 (12), 1885–1896. 10.1002/pro.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeley A.; Ábrányi-Balogh P.; Keserü G. M. Design and Characterization of a Heterocyclic Electrophilic Fragment Library for the Discovery of Cysteine-Targeted Covalent Inhibitors. MedChemComm 2019, 10 (2), 263–267. 10.1039/C8MD00327K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus K. M.; Correia B. E.; Lum K. M.; Forli S.; Horning B. D.; González-Páez G. E.; Chatterjee S.; Lanning B. R.; Teijaro J. R.; Olson A. J.; Wolan D. W.; Cravatt B. F. Proteome-Wide Covalent Ligand Discovery in Native Biological Systems. Nature 2016, 534, 570. 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A. M.; Miyamoto D. K.; Huffman T. R.; Bateman L. A.; Ives A. N.; Akopian D.; Heslin M. J.; Contreras C. M.; Rape M.; Skibola C. F.; Nomura D. K. Chemoproteomic Screening of Covalent Ligands Reveals UBA5 As a Novel Pancreatic Cancer Target. ACS Chem. Biol. 2017, 12 (4), 899–904. 10.1021/acschembio.7b00020. [DOI] [PubMed] [Google Scholar]
- Backus K. M. Applications of Reactive Cysteine Profiling. Curr. Top. Microbiol. Immunol. 2018, 420, 375–417. 10.1007/82_2018_120. [DOI] [PubMed] [Google Scholar]
- Miller R. M.; Paavilainen V. O.; Krishnan S.; Serafimova I. M.; Taunton J. Electrophilic Fragment-Based Design of Reversible Covalent Kinase Inhibitors. J. Am. Chem. Soc. 2013, 135 (14), 5298–5301. 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S.; Miller R. M.; Tian B.; Mullins R. D.; Jacobson M. P.; Taunton J. Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis. J. Am. Chem. Soc. 2014, 136 (36), 12624–12630. 10.1021/ja505194w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson H.; Isabella Tsai Y.-C.; Fantom K.; Chung C.-W.; Kümper S.; Martino L.; Thomas D. A.; Eberl H. C.; Muelbaier M.; House D.; Rittinger K. Fragment-Based Covalent Ligand Screening Enables Rapid Discovery of Inhibitors for the RBR E3 Ubiquitin Ligase HOIP. J. Am. Chem. Soc. 2019, 141 (6), 2703–2712. 10.1021/jacs.8b13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathman S. G.; Span I.; Smith A. T.; Xu Z.; Zhan J.; Rosenzweig A. C.; Statsyuk A. V. A Small Molecule That Switches a Ubiquitin Ligase From a Processive to a Distributive Enzymatic Mechanism. J. Am. Chem. Soc. 2015, 137 (39), 12442–12445. 10.1021/jacs.5b06839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan M. E.; Abramite J. A.; Anderson D. P.; Aulabaugh A.; Dahal U. P.; Gilbert A. M.; Li C.; Montgomery J.; Oppenheimer S. R.; Ryder T. Others. Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J. Med. Chem. 2014, 57 (23), 10072–10079. 10.1021/jm501412a. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Carr R.; Murray C.; Jhoti H. A “rule of Three”for Fragment-Based Lead Discovery?. Drug Discovery Today 2003, 8 (19), 876–877. 10.1016/S1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- Ilani T.; Alon A.; Grossman I.; Horowitz B.; Kartvelishvily E.; Cohen S. R.; Fass D. A Secreted Disulfide Catalyst Controls Extracellular Matrix Composition and Function. Science 2013, 341 (6141), 74–76. 10.1126/science.1238279. [DOI] [PubMed] [Google Scholar]
- Katchman B. A.; Ocal I. T.; Cunliffe H. E.; Chang Y.-H.; Hostetter G.; Watanabe A.; LoBello J.; Lake D. F. Expression of Quiescin Sulfhydryl Oxidase 1 Is Associated with a Highly Invasive Phenotype and Correlates with a Poor Prognosis in Luminal B Breast Cancer. Breast Cancer Res. 2013, 15 (2), R28. 10.1186/bcr3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.; Li J.; Muller M.; Yan S.; Mujtaba S.; Pan C.; Wang Z.; Zhou M.-M. Selective Small Molecules Blocking HIV-1 Tat and Coactivator PCAF Association. J. Am. Chem. Soc. 2005, 127 (8), 2376–2377. 10.1021/ja044885g. [DOI] [PubMed] [Google Scholar]
- Chaikuad A.; Lang S.; Brennan P. E.; Temperini C.; Fedorov O.; Hollander J.; Nachane R.; Abell C.; Müller S.; Siegal G.; Knapp S. Structure-Based Identification of Inhibitory Fragments Targeting the p300/CBP-Associated Factor Bromodomain. J. Med. Chem. 2016, 59 (4), 1648–1653. 10.1021/acs.jmedchem.5b01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stimson L.; Rowlands M. G.; Newbatt Y. M.; Smith N. F.; Raynaud F. I.; Rogers P.; Bavetsias V.; Gorsuch S.; Jarman M.; Bannister A.; Kouzarides T.; McDonald E.; Workman P.; Aherne G. W. Isothiazolones as Inhibitors of PCAF and p300 Histone Acetyltransferase Activity. Mol. Cancer Ther. 2005, 4 (10), 1521–1532. 10.1158/1535-7163.MCT-05-0135. [DOI] [PubMed] [Google Scholar]
- Kos V. N.; McLaughlin R. E.; Gardner H. A. Elucidation of Mechanisms of Ceftazidime Resistance among Clinical Isolates of Pseudomonas Aeruginosa by Using Genomic Data. Antimicrob. Agents Chemother. 2016, 60 (6), 3856–3861. 10.1128/AAC.03113-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes M. R.; Zhang J.; Li L.-S.; Hansen R.; Peters U.; Guo X.; Chen Y.; Babbar A.; Firdaus S. J.; Darjania L.; Feng J.; Chen J. H.; Li S.; Li S.; Long Y. O.; Thach C.; Liu Y.; Zarieh A.; Ely T.; Kucharski J. M.; Kessler L. V.; Wu T.; Yu K.; Wang Y.; Yao Y.; Deng X.; Zarrinkar P. P.; Brehmer D.; Dhanak D.; Lorenzi M. V.; Hu-Lowe D.; Patricelli M. P.; Ren P.; Liu Y. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell. 2018, 172 (3), 578. 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Byun S.; Lee S.-Y.; Lee J.; Jeong C.-H.; Farrand L.; Lim S.; Reddy K.; Kim J. Y.; Lee M.-H.; Lee H. J.; Bode A. M.; Won Lee K.; Dong Z. USP8 Is a Novel Target for Overcoming Gefitinib Resistance in Lung Cancer. Clin. Cancer Res. 2013, 19 (14), 3894–3904. 10.1158/1078-0432.CCR-12-3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian F.; Cao Y.; Bian L.; Sun Q. USP8: A Novel Therapeutic Target for Cushing’s Disease. Endocrine 2015, 50 (2), 292–296. 10.1007/s12020-015-0682-y. [DOI] [PubMed] [Google Scholar]
- Ulanovskaya O. A.; Zuhl A. M.; Cravatt B. F. NNMT Promotes Epigenetic Remodeling in Cancer by Creating a Metabolic Methylation Sink. Nat. Chem. Biol. 2013, 9 (5), 300–306. 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus D.; Yang Q.; Kong D.; Banks A. S.; Zhang L.; Rodgers J. T.; Pirinen E.; Pulinilkunnil T. C.; Gong F.; Wang Y.-C.; Cen Y.; Sauve A. A.; Asara J. M.; Peroni O. D.; Monia B. P.; Bhanot S.; Alhonen L.; Puigserver P.; Kahn B. B. Nicotinamide N-Methyltransferase Knockdown Protects against Diet-Induced Obesity. Nature 2014, 508 (7495), 258–262. 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Zheng H.; Mao A.-P.; Zhong B.; Li Y.; Liu Y.; Gao Y.; Ran Y.; Tien P.; Shu H.-B. Regulation of Virus-Triggered Signaling by OTUB1- and OTUB2-Mediated Deubiquitination of TRAF3 and TRAF6. J. Biol. Chem. 2010, 285 (7), 4291–4297. 10.1074/jbc.M109.074971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A.; Vinik Y.; Shatz-Azoulay H.; Isaac R.; Streim S.; Jona G.; Boura-Halfon S.; Zick Y. Otubain 2 Is a Novel Promoter of Beta Cell Survival as Revealed by siRNA High-Throughput Screens of Human Pancreatic Islets. Diabetologia 2013, 56 (6), 1317–1326. 10.1007/s00125-013-2889-x. [DOI] [PubMed] [Google Scholar]
- Kudo L. C.; Parfenova L.; Vi N.; Lau K.; Pomakian J.; Valdmanis P.; Rouleau G. A.; Vinters H. V.; Wiedau-Pazos M.; Karsten S. L. Integrative Gene-tissue Microarray-Based Approach for Identification of Human Disease Biomarkers: Application to Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2010, 19 (16), 3233–3253. 10.1093/hmg/ddq232. [DOI] [PubMed] [Google Scholar]
- Jackowski S.; Leonardi R. Deregulated Coenzyme A, Loss of Metabolic Flexibility and Diabetes. Biochem. Soc. Trans. 2014, 42 (4), 1118–1122. 10.1042/BST20140156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussey R. J.; Coates L.; Gill R. S.; Erskine P. T.; Coker S.-F.; Mitchell E.; Cooper J. B.; Wood S.; Broadbridge R.; Clarke I. N.; Lambden P. R.; Shoolingin-Jordan P. M. A Structural Study of Norovirus 3C Protease Specificity: Binding of a Designed Active Site-Directed Peptide Inhibitor. Biochemistry 2011, 50 (2), 240–249. 10.1021/bi1008497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky A.; Ovaa H.; Kolli N.; Gan-Erdene T.; Wilkinson K. D.; Ploegh H. L.; Kessler B. M. Chemistry-Based Functional Proteomics Reveals Novel Members of the Deubiquitinating Enzyme Family. Chem. Biol. 2002, 9 (10), 1149–1159. 10.1016/S1074-5521(02)00248-X. [DOI] [PubMed] [Google Scholar]
- Balakirev M. Y.; Tcherniuk S. O.; Jaquinod M.; Chroboczek J. Otubains: A New Family of Cysteine Proteases in the Ubiquitin Pathway. EMBO Rep. 2003, 4 (5), 517–522. 10.1038/sj.embor.embor824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen T. E. T.; Hospenthal M. K.; Geurink P. P.; Elliott P. R.; Akutsu M.; Arnaudo N.; Ekkebus R.; Kulathu Y.; Wauer T.; El Oualid F.; Freund S. M. V.; Ovaa H.; Komander D. OTU Deubiquitinases Reveal Mechanisms of Linkage Specificity and Enable Ubiquitin Chain Restriction Analysis. Cell 2013, 154 (1), 169–184. 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K.; Nakajima K.; Ui A.; Muto-Terao Y.; Ogiwara H.; Nakada S. Fine-Tuning of DNA Damage-Dependent Ubiquitination by OTUB2 Supports the DNA Repair Pathway Choice. Mol. Cell 2014, 53 (4), 617–630. 10.1016/j.molcel.2014.01.030. [DOI] [PubMed] [Google Scholar]
- Ekkebus R.; Van Kasteren S. I.; Kulathu Y.; Scholten A.; Berlin I.; Geurink P. P.; De Jong A.; Goerdayal S.; Neefjes J.; Heck A. J. R.; Komander D.; Ovaa H. On Terminal Alkynes That Can React with Active-Site Cysteine Nucleophiles in Proteases. J. Am. Chem. Soc. 2013, 135 (8), 2867–2870. 10.1021/ja309802n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Weerapana E.; Blewett M. M.; Cravatt B. F. A Chemoproteomic Platform to Quantitatively Map Targets of Lipid-Derived Electrophiles. Nat. Methods 2014, 11 (1), 79–85. 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan A. G. The Nudix Hydrolase Superfamily. Cell. Mol. Life Sci. 2006, 63 (2), 123–143. 10.1007/s00018-005-5386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly S.-J.; Tillander V.; Ofman R.; Alexson S. E. H.; Hunt M. C. The Nudix Hydrolase 7 Is an Acyl-CoA Diphosphatase Involved in Regulating Peroxisomal Coenzyme A Homeostasis. J. Biochem. 2008, 144 (5), 655–663. 10.1093/jb/mvn114. [DOI] [PubMed] [Google Scholar]
- Gasmi L.; McLennan A. G. The Mouse Nudt7 Gene Encodes a Peroxisomal Nudix Hydrolase Specific for Coenzyme A and Its Derivatives. Biochem. J. 2001, 357 (1), 33–38. 10.1042/0264-6021:3570033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumar S. A.; Kerr E. W.; Geldenhuys W. J.; Montgomery G. E.; Fagone P.; Thirawatananond P.; Saavedra H.; Gabelli S. B.; Leonardi R. Nudt19 Is a Renal CoA Diphosphohydrolase with Biochemical and Regulatory Properties That Are Distinct from the Hepatic Nudt7 Isoform. J. Biol. Chem. 2018, 293 (11), 4134–4148. 10.1074/jbc.RA117.001358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwöbel J. A. H.; Koleva Y. K.; Enoch S. J.; Bajot F.; Hewitt M.; Madden J. C.; Roberts D. W.; Schultz T. W.; Cronin M. T. D. Measurement and Estimation of Electrophilic Reactivity for Predictive Toxicology. Chem. Rev. 2011, 111 (4), 2562–2596. 10.1021/cr100098n. [DOI] [PubMed] [Google Scholar]
- Esterbauer H.; Schwarzl E.; Hayn M. A Rapid Assay for Catechol Oxidase and Lactase Using 2-Nitro-5-Thiobenzoic Acid. Anal. Biochem. 1977, 77 (2), 486–494. 10.1016/0003-2697(77)90262-7. [DOI] [PubMed] [Google Scholar]
- Dypbukt J. M.; Bishop C.; Brooks W. M.; Thong B.; Eriksson H.; Kettle A. J. A Sensitive and Selective Assay for Chloramine Production by Myeloperoxidase. Free Radical Biol. Med. 2005, 39 (11), 1468–1477. 10.1016/j.freeradbiomed.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Dahal U. P.; Obach R. S.; Gilbert A. M. Benchmarking in Vitro Covalent Binding Burden as a Tool to Assess Potential Toxicity Caused by Nonspecific Covalent Binding of Covalent Drugs. Chem. Res. Toxicol. 2013, 26 (11), 1739–1745. 10.1021/tx400301q. [DOI] [PubMed] [Google Scholar]
- Devine S. M.; Mulcair M. D.; Debono C. O.; Leung E. W. W.; Nissink J. W. M.; Lim S. S.; Chandrashekaran I. R.; Vazirani M.; Mohanty B.; Simpson J. S.; Baell J. B.; Scammells P. J.; Norton R. S.; Scanlon M. J. Promiscuous 2-Aminothiazoles (PrATs): A Frequent Hitting Scaffold. J. Med. Chem. 2015, 58 (3), 1205–1214. 10.1021/jm501402x. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Greer J. A Decade of Fragment-Based Drug Design: Strategic Advances and Lessons Learned. Nat. Rev. Drug Discovery 2007, 6 (3), 211–219. 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Huth J. R.; Fesik S. W. Druggability Indices for Protein Targets Derived from NMR-Based Screening Data. J. Med. Chem. 2005, 48 (7), 2518–2525. 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]
- Shannon D. A.; Banerjee R.; Webster E. R.; Bak D. W.; Wang C.; Weerapana E. Investigating the Proteome Reactivity and Selectivity of Aryl Halides. J. Am. Chem. Soc. 2014, 136 (9), 3330–3333. 10.1021/ja4116204. [DOI] [PubMed] [Google Scholar]
- Weerapana E.; Wang C.; Simon G. M.; Richter F.; Khare S.; Dillon M. B. D.; Bachovchin D. A.; Mowen K.; Baker D.; Cravatt B. F. Quantitative Reactivity Profiling Predicts Functional Cysteines in Proteomes. Nature 2010, 468 (7325), 790–795. 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery D. A.; Bogyo M. Chemical Proteomics and Its Application to Drug Discovery. Drug Discovery Today. 2004, 9, S19–S26. [PubMed] [Google Scholar]
- Lee H.-Y.; Suciu R. M.; Horning B. D.; Vinogradova E. V.; Ulanovskaya O. A.; Cravatt B. F. Covalent Inhibitors of Nicotinamide N-Methyltransferase (NNMT) Provide Evidence for Target Engagement Challenges in Situ. Bioorg. Med. Chem. Lett. 2018, 28 (16), 2682–2687. 10.1016/j.bmcl.2018.04.017. [DOI] [PubMed] [Google Scholar]
- Grossman I.; Ilani T.; Fleishman S. J.; Fass D. Overcoming a Species-Specificity Barrier in Development of an Inhibitory Antibody Targeting a Modulator of Tumor Stroma. Protein Eng., Des. Sel. 2016, 29 (4), 135–147. 10.1093/protein/gzv067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G.; McAuley K. E. Automated Data Collection for Macromolecular Crystallography. Methods 2011, 55 (1), 81–93. 10.1016/j.ymeth.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Wojdyr M.; Keegan R.; Winter G.; Ashton A. W. DIMPLE - a Pipeline for the Rapid Generation of Difference Maps from Protein Crystals with Putatively Bound Ligands. Acta Crystallogr. A 2013, 69 (a1), s299 10.1107/S0108767313097419. [DOI] [Google Scholar]
- Pearce N. M.; Krojer T.; Bradley A. R.; Collins P.; Nowak R. P.; Talon R.; Marsden B. D.; Kelm S.; Shi J.; Deane C. M.; Von Delft F. A Multi-Crystal Method for Extracting Obscured Crystallographic States from Conventionally Uninterpretable Electron Density. Nat. Commun. 2017, 8, 15123. 10.1038/ncomms15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 (4), 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 (4), 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Kant R.; Fish A.; Janssen L.; Janssen H.; Krom S.; Ho N.; Brummelkamp T.; Carette J.; Rocha N.; Neefjes J. Late Endosomal Transport and Tethering Are Coupled Processes Controlled by RILP and the Cholesterol Sensor ORP1L. J. Cell Sci. 2013, 126 (15), 3462–3474. 10.1242/jcs.129270. [DOI] [PubMed] [Google Scholar]
- Krojer T.; Talon R.; Pearce N.; Collins P.; Douangamath A.; Brandao-Neto J.; Dias A.; Marsden B.; Von Delft F.F. The XChemExplorer Graphical Workflow Tool for Routine or Large-Scale Protein--Ligand Structure Determination. Acta Crystallogr. D Struct. Biol. 2017, 73 (3), 267–278. 10.1107/S2059798316020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H. Comprehensive Identification of Peptides in Tandem Mass Spectra Using an Efficient Search Engine. Nat. Biotechnol. 2018, 10.1038/nbt.4236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.