

Abstract
Damage to genomic DNA leads to mutagenesis and disease. Repair of single base damage is initiated by DNA glycosylases, the first enzymes in the base excision repair pathway. Although eukaryotic packaging of chromosomal DNA in nucleosomes is known to decrease DNA glycosylase efficiency, the impact on individual glycosylases is unclear. Here, we present a model system in which we examine the repair of site-specific base damage in well-characterized nucleosome core particles by five different DNA glycosylases. We find that DNA glycosylase efficiency on nucleosome substrates depends not only on the geometric orientation of the damaged base, but also on its identity, as well as on the size, structure, and mechanism of the glycosylase. We show via molecular modeling that inhibition of glycosylase activity is largely due to steric obstruction by the nucleosome core.
Graphical Abstract
INTRODUCTION
The integrity of the genome is continually threatened by reactive radical species, toxins, and radiation.1 Single DNA base lesions resulting from such interactions are repaired enzymatically via the base excision repair (BER) pathway.2–6 BER is initiated by a DNA glycosylase, which catalyzes scission of the N-glycosidic bond connecting the aberrant base to the DNA backbone.7 Cleavage of this bond frees the lesion base and generates an abasic site on the DNA strand. The BER process is essential; persistence of damaged bases in the genome can cause DNA mutation, aging, cancer, and disease.1
DNA glycosylases can be categorized by preferred lesion or by protein architecture. To date, six structural superfamilies have been defined.8,9 The UDG and AAG superfamilies are characterized by compact, single-domain proteins with small DNA-interaction surfaces, while members of the helix-hairpin-helix (HhH) and helix-two turn-helix (H2TH) superfamilies contain two characteristic domains with the active site located at the inter-domain junction. The EndoV superfamily is exemplified by its titular member, T4 endonuclease V (also known as T4 pyrimidine dimer glycosylase, T4-Pdg), which catalyzes removal of the 5′ base in cyclobutane pyrimidine dimers (CPDs). Finally, the HEAT-like repeat (HLR) family proteins are built of an alpha-alpha superhelix fold comprised of six HEAT-like repeats.
Despite differences in structure and lesion specificity, all known DNA glycosylases (with the exception of HLR superfamily enzymes10) share common principles of action.11 First, glycosylase binding universally induces DNA bending, which facilitates “base flipping,” or extrusion of the lesion base from the DNA base stack and insertion into the active site pocket of the enzyme. The severity of structural distortion ranges from 22° to 70°, depending on the glycosylase. Second, recognition of the target lesion in the enzyme active site pocket is achieved through a combination of steric and hydrogen bonding interactions. Third, binding studies with transition state analogs and kinetic isotope experiments suggest that all DNA glycosylases operate by an SN1 (DN*AN) mechanism, although the catalytic details of the reaction divide glycosylases into two types.12 Monofunctional DNA glycosylases perform glycosidic bond cleavage only, while bifunctional glycosylases also catalyze cleavage of the sugar-phosphate backbone at the lesion site.
In eukaryotes, the BER process is challenged by the packaging of genomic DNA into chromatin. The fundamental unit of chromatin is the nucleosome, a structure comprised of 145–147 base pairs of DNA wrapped in 1.7 left-handed superhelical coils around a protein core (Figure 1).13 The protein core is made up of two copies each of the four histone proteins H2A, H2B, H3, and H4. When prepared in isolation, an individual nucleosome is called a nucleosome core particle (NCP). A pseudo-2-fold rotational axis of symmetry called the dyad axis runs through the center of the NCP, coinciding with the position of the central base pair of the bound DNA duplex. The position of each DNA base in the nucleosome is typically described relative to its displacement from this central base (translational positioning). In addition, the rotational orientation of each base can be described relative to the center of the histone core (rotational positioning). Bases with outward rotational positioning are accessible to solvent, while bases with inward rotational positioning face the histone core and are sequestered due to steric obstruction by the core.
Figure 1.

The architecture of a nucleosome core particle. Lesion incorporation sites OUT (red), MID (purple), and IN (blue) are highlighted. Image generated from PDB ID: 3LZ0.
In a recent review, Odell, et al. have identified several principles of glycosylase activity on NCPs.14 First, lesions that have outward rotational positioning are generally repaired more efficiently than lesions that have inward rotational positioning, although exceptions have been observed. Second, dynamic motion, including spontaneous, partial unwrapping of nucleosomal DNA, facilitates repair of lesions that are nominally sterically occluded. Third, lesions that are close to the dyad axis are repaired less efficiently than lesions that are far from it. These generalizations are reasonable considering the formidable steric block that is imposed by the histone core; however, questions regarding their universality remain. For example, these principles were developed from experimental observations of only a subset of known glycosylase/lesion pairs, including human uracil DNA glycosylase/uracil (hUNG/U),15–17 human endonuclease III/thymine glycol (hNTH/Tg),18 human endonuclease VIII (NEIL1)/Tg,19 human 8-oxoguanine DNA glycosylase/8-oxo-7,8-dihydroguanine (hOGG1/8-oxoG),20 and Escherichia coli (E. coli) uracil DNA glycosylase (UDG)/U.21,22 There are many glycosylase/lesion systems for which these principles have not been verified. Similarly, counterexamples to the correlation between solvent accessibility and glycosylase efficiency have been observed in some systems,15,17,21 calling into question the scope of applicability of the principles outlined above. Finally, differences in lesion position, DNA sequence, and reaction environment make it difficult to directly compare the results of these previous studies.
Here, we compare the ability of several glycosylases to remove their preferred lesions from homogeneous, well characterized NCPs under identical reaction conditions in order to determine how DNA packaging in eukaryotes affects glycosylase efficiency. Lesions were incorporated at sites with different rotational positioning, and solvent accessibility at each site was verified by hydroxyl radical footprinting (HRF). Glycosylase efficiency was measured as a function of rotational positioning for several glycosylase/lesion pairs. Specifically, we investigate the activity of UDG on U and 5-hydroxyuracil (5-OHU), of hOGG1 on 8-oxoG, of E. coli formamidopyrimidine DNA glycosylase (Fpg; also known as MutM) on 8-oxoG and 5-OHU, of human alkyladenine DNA glycosylase (hAAG; also known as MPG) on 1,N6-ethenoadenine (εA) and hypoxanthine (Hx), and of E. coli endonuclease III (EndoIII) on 5-OHU. We find that some glycosylases are able to remove solvent accessible lesions with high efficiency, while others are completely inhibited. Through kinetic and structural analysis, we show that a diversity of factors affects glycosylase activity on NCPs.
RESULTS AND DISCUSSION
Experimental Design and Rationale.
In order to determine the enzyme characteristics necessary for efficient initiation of BER in packaged DNA, we examined the reactivity of a subset of known glycosylases on a diverse set of base lesions (Scheme 1). Specifically, we report repair of lesions representing oxidation of G (8-oxoG), deamination of C and A (U and Hx, respectively), oxidative deamination of C (5-OHU) and alkylation of A (εA).3 Similarly, the glycosylases in this set represent four of the six structural superfamilies, both monofunctional and bifunctional mechanisms, and a wide range of molecular weights and DNA bending angles, and they target a variety of lesions (Table 1). We surmised that comparing the reactivity of glycosylases in this diverse set would enable us to isolate the characteristics necessary for efficient reactivity on NCPs. Although NCPs are only found in eukaryotes, we used prokaryotic DNA glycosylases in some cases because they are easier to obtain and better characterized than the human analogs (UDG and EndoIII), and they allow us to access structure/function relationships that do not exist in the collection of known human DNA glycosylases (Fpg). Nevertheless, the prokaryotic enzymes we use share strong structural homology with human enzymes, so interpretation of our results is expected to be valid no matter the biological source of our DNA glycosylases. Our studies did not include HLR superfamily enzymes because the weak glycosidic bonds of their substrate lesions are too unstable for our experiments. We also did not examine T4-Pdg because this enzyme flips an adenine opposite its target CPD lesion into its active site as opposed to the lesion itself.9
Scheme 1.

Structures of base lesions examined in this study.
Table 1.
Characteristics of DNA glycosylases used in this study.
| Glycosylase (Alias) | Organism | Preferred Lesionsa | MW (kDa) | Structural Superfamily8 | Mech.c,5 | DNA Bending Angle | Cuts ssDNA | Complement Specificity |
|---|---|---|---|---|---|---|---|---|
| UDG (Ung) |
E. coli | U, 5-OHU, 5-fU,52 | 25.6735 | UDG | M | 45°d, 11 | Yes11 | Moderate U·G > U·A35 |
| hOGG1 | H. sapiens | 8-oxoG, FapyG,2 8-oxoA, MeFapyG9 | 385 | HhH | B | 70°11 | No5 | Strong 8-oxoG·C, 8-oxoG·T ≫ 8-oxoG·G > 8-oxoG·A42 |
| Fpg (MutM) | E. coli | 8-oxoG, 8-oxoA, FapyG, FapyA, MeFapyG, Ug, 5-OHC, 5-OHU, Tg, DHT,32 Gh, Sp42 | 30.253 | H2TH | B | 66°11 | Yes34 | Moderate 8-oxoG·G, 8-oxoG·T, 8-oxoG·C > 8-oxoG·A42 |
| hAAG (MPG) | H. sapiens | 3mA, 7mG, 3mG, Hx, εA,2 Oxa44 | 24.3b | AAG | M | 22°11 | Yes Oxa, εA; no Hx, 7mG44 | Variable Hx·T ≫ Hx·C, > Hx·A, Hx·G; εA·A, εA·C > εA·G, εA·T;43 7mG·T > 7mG·C44 |
| EndoIII (Nth) |
E. coli | Tg, Ug, 5-OHC, 5-OHU, DHU, DHT, urea, other damaged pyrimidines32 | 23.537 | HhH | B | 55°39 | No (Fig. S1) | Moderate 5-OHC·G > 5-OHC·A; 5-OHU·G > 5-OHU·A54 |
Abbreviations: 3mA = 3-methyladenine; 3mG = 3-methylguanine; 5-fU = 5-fluorouracil; 5-OHC = 5-hydroxycytosine; 5-OHU = 5-hydroxyuracil; 7mG = 7-methylguanine; 8-oxoA = 8-oxo-7,8-dihydroadenine; 8-oxoG = 8-oxo-7,8-dihydroguanine; εA = 1-N6-ethenoadenine; DHT = 5,6-dihydrothymine; DHU = 5,6-dihydrouracil; FapyA = 4,6-diamino-5-formamidopyrimidine; FapyG = 2,6-diamino-4-oxo-5-formamidopyrimidine; Gh = guanidinohydantoin; Hx = hypoxanthine; MeFapyG = 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine; Oxa = oxanine; Tg = thymine glycol; Sp = spiroiminodihydantoin; Ug = uracil glycol.
The hAAG supplied by New England Biolabs and used in these experiments is the Δ79 N-terminal truncated version originally developed by the Ellenberger Laboratory. The molecular weight was calculated for the amino acid sequence reported by Lau, et al. (PDB ID: 1EWN).36
M = monofunctional; B = Bifunctional.
No crystal structure exists for E. coli UDG bound to DNA. The listed bending angle of 45° is for the human analog, hUNG. The two analogs share strong structural homology (see Supporting Information).
The substrates used in the experiments described here are based on the Widom 601 DNA sequence.23 This sequence is known as a strong positioning sequence due to its propensity to bind to the histone octamer in a unique orientation. Our DNA sequences differ from that isolated by the Widom group in two respects. First, our sequences retain the terminal EcoRV recognition sites incorporated by Vasudevan, et al. for their crystal structure determination of an NCP.24 Second, we modified the sequence at the lesion sites so that each lesion is paired with its physiologically relevant complement base: 8-oxoG is paired with C, U and 5-OHU are paired with G, and Hx and εA are paired with T. These minor sequence modifications are not expected to significantly perturb the properties of the resulting NCP. In choosing the Widom 601 sequence for our model system, two main advantages are conferred: first, the use of a strong positioning sequence ensures a homogeneous and predictable particle geometry; and second, the crystal structure can be used as a guide for the rational placement of lesions. The full sequences of all DNA duplexes used in this study are given in Scheme S1.
Guided by the 601 NCP crystal structure,24 we placed lesions in one of three positions near the center of the duplex: outward toward solution (OUT), approximately 90° away from solution (MID), or inward toward the histone core (IN) (Figure 1). These sites correspond to indices +2, –1, and –3 in DNA strand I of the crystal structure.24 Based on analysis of the crystal structure and on previous studies of BER in NCPs, we predicted that lesions placed at these positions would have decreasing solvent accessibility as OUT > MID > IN. Although the rotational positioning of the three lesion sites is expected to strongly influence glycosylase accessibility, all lesions were placed near the dyad axis, so the small differences in translational positioning were not expected to affect repair.
Hydroxyl Radical Footprinting Establishes Rotational Position of Lesions.
The solvent accessibility at each lesion site was verified by HRF.25 This analytical method utilizes hydroxyl radicals to cleave the DNA backbone at solvent accessible sites. The position of a bound protein, which shields the DNA backbone from damage, is revealed as a lack of DNA cleavage in the HRF damage profile. In NCPs, the HRF damage profile displays an oscillatory pattern as the DNA backbone precesses toward (weak cleavage) and away from (strong cleavage) the histone core with each turn of the helix.26
Upon hydroxyl radical treatment, the free duplex (i.e., not bound to the histone octamer) substrate 601 exhibits a largely unbiased cleavage profile (Figure 2). In contrast, an NCP substrate bearing Widom 601 DNA, NCP-601, shows an oscillatory pattern of cleavage, verifying the formation of stable, homogenous NCPs. In agreement with the crystal structure, the intensity of HRF cleavage decreases as OUT > MID > IN.
Figure 2.

Hydroxyl radical footprinting of DNA in a nucleosome core particle. The HRF damage profile was observed by PAGE. Lanes are as follows: Maxam-Gilbert sequencing ladder (A+G); negative control lane (NT, no treatment) in which no HRF reagents were added to a sample of 601·NCP; HRF reactivity on 601; and HRF reactivity on 601·NCP substrates. Base positions are indexed to the crystal structure of Vasudevan, et al.24 Lane profiles illustrate the relative reactivity as a function of base position, with the OUT, MID, and IN positions indicated by red, purple, and blue lines, respectively. The gel image has been straightened using SAFA software (see Supporting Information) in order to illustrate the relative migration of bands in neighboring lanes. (The unmodified gel image is shown in Figure S2.)
Footprinting analysis was also carried out on all lesion-containing NCPs (Figure 3, Figure S3). In every construct, an oscillatory pattern of damage is observed that matches the pattern exhibited by NCP-601. Even in NCPs containing εA, the bulkiest lesion we studied, lesion incorporation leads to only negligible differences in local solvent accessibility as compared to NCP-601. The lack of major structural perturbations allows us to discount variations in solvent accessibility when comparing repair efficiencies between glycosylases for a given rotational position. The results of the HRF assay verify stable NCP formation for all lesion-containing duplexes and allow us to accurately account for solvent accessibility when assessing the effect of rotational positioning on lesion repair.
Figure 3.

Histograms showing the integrated gel band area as a function of base position for NCP-εA substrates. The section of gel shown in the histograms is indicated by the yellow box in Figure 2. Notation and base positions are as in Figure 2.
General Considerations for Glycosylase Experiments.
Kinetics experiments were carried out to measure the effects that glycosylase identity, lesion identity, lesion specificity, DNA sequence context, NCP incorporation, and lesion positioning have on glycosylase reaction efficiency. Because reactions were carried out under single turnover conditions, the observed rate constant for each reaction, kobs, reflects the slowest step, up to and including the rate of chemistry.27
In order to accurately model glycosylase reactivity in eukaryotes, reactions were carried out in buffers that attempt to mimic conditions in the cell nucleus. Specifically, reactions were carried out at 37 °C in solutions buffered at pH 7.628 with Tris-HCl in the presence of 50 mM Na+ and 150 mM K+.29 Reaction solutions also included 200 μg/ml (~3 μM) bovine serum albumin (BSA) to act as a spectator protein and crowding agent, 1 mM EDTA to chelate free metal ions, and 1 mM DTT to maintain reducing conditions.30
Glycosylases Efficiently Repair Lesions in Free Duplexes.
All glycosylases examined show 80–99% product formation on free duplex substrates containing their preferred lesions (Figure 4, Figure S4). Incomplete glycosylase reactivity is often observed in these systems16,22,31 and may reflect partial degradation of the lesion to form a substrate that is poorly recognized by the glycosylase. For individual glycosylase/lesion pairs, kobs varies as a function of lesion position, and therefore as a function of sequence (Table 2). Similar sequence bias has been reported for glycosylase reactions. For example, depending on the sequence context of the lesion, the reported rate of excision of 8-oxoG by hOGG1 varies from 29.4 to 60 min–1.31 Similarly, efficiency of U excision by UDG can vary by as much as 10–15-fold, depending on the DNA sequence.22 Such variability can be attributed to differences in the chemical nature or local geometry of the bases surrounding the lesion.31,32
Figure 4.

Single turnover kinetics time courses. a) A schematic of the glycosylase reaction. b) A typical 8% denaturing PAGE gel showing the conversion of substrate (S) to product (P) with increasing reaction time. c) Reaction time courses for UDG/U, hOGG1/8oxoG, and hAAG/εA systems for free duplex (closed circles) or NCP (open circles) substrates. Data were fit using nonlinear least-squares regression. Error bars represent the standard deviation (n = 3). d) Maximum product yield observed in kinetics time courses for free duplex (FD; black) and NCP (gray) substrates.
Table 2.
Kinetic parameters for DNA glycosylase reactions.
| Enzyme | Substrate | kobs / min (% product)a | |
|---|---|---|---|
| Free Duplex | NCP | ||
| UDG | UOUT | 35 ± 2 (99%) | 5.8 ± 0.1 (99%) |
| UMID | 21.1 ± 0.6 (95%) | 16 ± 2 (11%) 0.22 ± 0.06 (8%) |
|
| UIN | 11.2 ± 0.6 (96%) | 9.5 ± 1.5 (11%) 0.3 ± 0.2 (4%) |
|
| hOGG1 | 8oxoGOUT | 5.5 ± 0.2 (93%) | 2.7 ± 0.5 (5%) |
| 8oxoGMID | 5.6 ± 0.2 (87%) | 6 ± 1 (10%) | |
| 8oxoGIN | 9.0 ± 0.3 (91%) | 7 ± 2 (8%) | |
| Fpg | 8oxoGOUT | 10.0 ± 0.6 (84%) | 4.6 ± 0.9 (9%) |
| 8oxoGMID | 10 ± 1 (80%) | 7 ± 2 (7%) | |
| 8oxoGIN | 12.2 ± 0.6 (91%) | 7 ± 2 (10%) | |
| hAAG | HxOUT | 0.222 ± 0.006 (95%) | 0.080 ± 0.003 (10%) |
| HxMID | 0.0747 ± 0.0009 (82%) | 0.082 ± 0.007 (6%) | |
| HxIN | 0.05 ± 0.01 (82%) | 0.07 ± 0.03 (5%) | |
| εAOUT | 0.0433 ± 0.0009 (91%) | 0.0111 ± 0.0002 (84%) | |
| εAMID | 0.0315 ± 0.0007 (90%) | 0.006 ± 0.001 (16%) | |
| εAIN | 0.0338 ± 0.0002 (88%) | N.R.b | |
| EndoIII | 5OHUOUT | 10 ± 1 (82%) | 1.6 ± 1.0 (5%) |
| Fpg | 5OHUOUT | 0.13 ± 0.02 (82%) | N.R.b |
| UDG | 5OHUOUT | 0.008 ± 0.001 (48%) | 0.007 ± 0.004 (3%) |
Error represents reported standard deviation from fitting by weighted nonlinear least-squares regression.
N.R.: No reaction.
The reaction rates reported here are generally slower than rates reported in other studies for the same glycosylase/lesion pair. For example, the rates we observe for the removal of 8-oxoG by hOGG1, 5.5–9.0 min–1, are an order of magnitude slower than the rates listed above for the same reaction. Such discrepancies could be due to nonspecific binding on our long DNA substrates11 or to differences in pH,33 ionic strength,34 or enzyme concentration (see Figure S5).17 Nevertheless, by conducting all of our experiments under identical reaction conditions, we maintain the ability to draw conclusions from differences in the reaction rates and product yields of our experimental systems.
Glycosylases Show Differential Reactivity on NCPs.
NCP incorporation affects each glycosylase/lesion system differently (Figure 4, Figure S4). For UDG/NCP-UOUT, product formation is quantitative and occurs at a rate of 5.8 min–1, which is less than an order of magnitude slower than the rate on the free duplex (35 min–1). As the solvent accessibility of the lesion decreases, the yield of product formation also decreases, and the kinetics become more complex. For example, for UDG/NCP-UMID, the reaction yield is only 19%, and two kinetic phases are resolved. For UDG/NCP-UIN, the reaction yield decreases further to 15%, and again, two kinetic phases are observed. Notably, in biphasic fits to NCP-UMID and NCP-UIN time courses, the rates of the fast phases are similar to the rates observed for the analogous free duplex substrates. Therefore, we attribute the fast phase in NCP samples to reaction of a small amount of free duplex contaminate carried over during NCP reconstitution. The slow phase then represents the rate of reaction on authentic NCP substrates, and may reflect the rate of large-scale dynamic modes that allow UDG to access otherwise occluded lesions.17
As in UDG/NCP-U systems, the degree of reactivity in hAAG/NCP-εA systems correlates with rotational positioning, and reaction rates are slower on NCP substrates than on free duplex substrates. Specifically, while efficient reactivity on NCP-εAOUT by hAAG results in the formation of 85% product, the product yield for NCP-εAMID is only 16%, and no product is observed for NCP-εAIN. In comparing kinetics, the rate of product formation on NCP-εAOUT is much slower than the rate observed for the analogous free duplex, εAOUT. Similarly, in NCP-εAMID, only a single kinetic phase can be resolved, and this rate is much slower than the rate of reaction on εAMID. Like UDG/NCP-U, it appears that decreasing solvent accessibility to the lesion results in corresponding decreases in both the rate of reaction and the product yield.
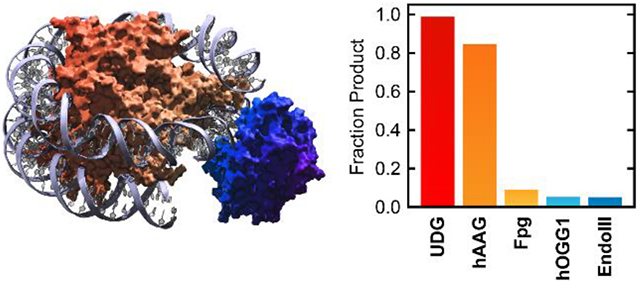
For all other systems studied here, including hOGG1/8-oxoG, Fpg/8-oxoG, hAAG/Hx, EndoIII/5-OHU, Fpg/5-OHU, and UDG/5-OHU, a product yield of 10% or less is observed for all NCP substrates, including those with outward-facing lesions. The small amount of product that is observed generally appears at a rate that is similar to the rate of product formation in the analogous free duplex substrate. Therefore, in these NCP systems, we attribute the observed product to reaction of residual free duplex and suggest that glycosylase activity is completely inhibited. Overall, incorporation into NCPs impairs the ability of glycosylases to process lesions, and the degree of impairment depends on the identities of the glycosylase and lesion, as well as on solvent accessibility.
In general, the results presented here agree with previous reports of glycosylase activity on NCPs. In particular, the dependence of glycosylase efficiency on rotational positioning has been observed in numerous experimental systems, including hUNG/U,16 hNTH/Tg,18 NEIL1/Tg,19 and UDG/U.21,22 Importantly, the ability for glycosylases to remove solvent accessible damage is by no means universal for all glycosylase/lesion pairs. Indeed, the lack of product formation we observe for hOGG1 activity on 8-oxoG-containing NCP substrates agrees with previous reports, which show that chromatin remodeling complexes must be added to allow removal of midway-facing 8-oxoG from NCPs.20
Enzyme Size, Structure, and Mechanism Affect Glycosylase Reactivity on NCPs.
The observation that not all DNA glycosylases react with similar efficiency on NCPs is intriguing; however, the cause of this discrepancy is not readily apparent. Here, we explore the roles of enzyme size, structure, and mechanism in dictating glycosylase efficiency on NCPs.
In seeking to identify which characteristics might prevent glycosylase reactivity on NCPs, enzyme structure is an obvious factor. After all, the histone core serves as a formidable steric block for any enzyme that interacts intimately with nucleosomal DNA. This principle is illustrated clearly by the relationship between glycosylase reactivity and the rotational positioning of the target lesion. It stands to reason that enzyme productivity should correlate inversely with enzyme size. Indeed, of the five glycosylases surveyed in this work, the two most active on NCPs, UDG and hAAG, are among the smallest (Table 1), with molecular weights of 25.635 and 24.336 kDa, respectively. Only EndoIII is smaller, with a molecular weight of 23.5 kDa.37
The shapes of the various glycosylases also impact their ability to interact intimately with nucleosomal DNA. Since no crystal structures of glycosylase-bound NCPs have been reported, we generated molecular models to assess the impact that steric obstruction by the histone core has on glycosylase binding. We built models of each glycosylase bound to its target OUT-facing lesion by merging a crystal structure of the DNA-bound glycosylase with a crystal structure of an NCP (Figure 5). In each model, the glycosylase is colored according to the distance between its surface and the histone octamer. Regions within 5 Å of the octamer are yellow, regions between 5 Å and 10 Å from the octamer are red, and regions beyond 10 Å from the octamer are blue/purple. Since there is no crystal structure of DNA-bound UDG, we used the crystal structure of DNA-bound hUNG instead.38 Notably, the truncation mutant of hUNG used in this analysis is similar in length and sequence to UDG (see Supporting Information), so structural analyses based on hUNG are expected to be valid for UDG as well. Importantly, all crystal structures used were solved at resolutions of 2.5 Å or less, so modeled interaction geometries are expected to be accurate to this length scale.
Figure 5.

Molecular models of glycosylase-bound NCPs. Front View: NCP binding surface of each glycosylase (NCP hidden for clarity). All models are shown at the same scale. Gray silhouettes indicating the size and position of NCP-bound hOGG1 are shown for comparison. Glycosylase surface models are colored according to the distance to the histone core. Residues of closest approach are labeled. Images generated from PDB IDs 3LZ0 (NCP), 1EMH (hUNG), 1EBM (hOGG1), 1K82 (Fpg), 1EWN (hAAG), and 1P59 (EndoIII).
Our analysis shows that each glycosylase interacts differently with the histone octamer. At one extreme, only two residues of bound hUNG, Y275 and R276, approach within 10 Å of the octamer. Similarly, the shortest interatomic distance between hAAG and the histone octamer is 7.8 Å, where M164 approaches H3. These large separation distances suggest that UDG and hAAG interact minimally with the histone octamer upon DNA binding. At the other extreme, K88 of Fpg lies within 2.0 Å of the C-terminus of histone H2A, and R31 of Fpg is only 4.2 Å away from H4. Such short separation distances indicate that the histone core may strongly interfere with Fpg binding at the lesion site. The distance between Q257 of Fpg and R42 of histone H3 is slightly longer, at 5.5 Å. However, the N-terminal tail of H3 (residues 1–38) is not resolved in the crystal structure, so steric interactions between Q257 and the H3 N-terminal tail could influence Fpg binding in solution. In hOGG1, a large surface patch including the side chains of residues R206, R197, and Y203, which stabilizes the highly compressed region of the DNA duplex opposite the extruded base, lies between 5.4 Å and 8.3 Å from the histone core. Like hOGG1, EndoIII displays a large surface patch that serves to sculpt or stabilize the complement DNA backbone near the lesion site. The closest of the surface patch residues, R78, R84, and R88, are only 3.1–4.7 Å from the octamer surface. In conjunction with the glycosylase reaction data, these molecular models suggests that efficient glycosylase reactivity on NCPs requires a low probability for steric interaction between the glycosylase and the histone core. Thus, high reaction efficiencies on NCPs are expected to extend beyond UDG and hAAG to other low molecular weight enzymes in the UDG and AAG structural superfamilies.
In addition to structural characteristics, DNA glycosylase efficiency on NCPs is also correlated with mechanistic details. For example, crystal structures suggest that hAAG and UDG, which show strong reactivity on NCPs, bend their DNA substrates at angles of 22° and 45°, while EndoIII, Fpg, and hOGG1, which show little or no reactivity on NCPs, bend their substrates more severely, at angles of 55°, 66°, and 70°, respectively.11,39 This correlation may be explained by the energetic cost associated with bending nucleosomal DNA. The greater the DNA bending angle, the more contacts between the DNA backbone and the histone core that must be disrupted. The severe distortions required for Fpg or hOGG1 reactivity may be prohibitively energetically expensive for these enzymes to react on NCPs.
The efficiency of lesion removal by DNA glycosylases on NCP substrates also depends on the lesion. For example, although UDG removes U from NCP-UOUT with high efficiency, UDG is inoperative on NCP-5OHUOUT, even though the enzyme can remove both U and 5-OHU from free duplex substrates.40 Similarly, hAAG excises εA, but not Hx, from NCPs. In the case of UDG, this discrepancy could be explained by the accumulation of confounding factors. Even on free duplex substrates, removal of 5-OHU by UDG is several orders of magnitude slower than removal of U. Analysis of the crystal structure of hUNG bound to pseudouracil-containing DNA suggests that this decrease in rate is likely due to poor accommodation of 5-OHU in the enzyme active site.38 Incorporation of 5-OHU into NCPs could decrease efficiency further by adding a steric challenge to glycosylase binding as well as a requirement to break DNA-histone contacts during DNA bending. In the case of hAAG, the observed differential reactivity between Hx and εA in NCPs suggests that histone binding impairs the ability of the glycosylase to recognize Hx, but not εA. This interpretation is consistent with a previous report showing that removal of Hx, but not εA, by murine 3-methyladenine glycosylase, the mouse homolog of hAAG, is impaired in DNA sequences with compressed minor grooves and enhanced base stacking interactions.41 Similarly, wrapping the DNA duplex around the histone octamer may distort the conformation of the DNA in the vicinity of the lesion in a way that affects the recognition of Hx, but not εA, by hAAG.
Recognition of the base opposite the lesion may also affect glycosylase efficiency on NCPs. For example, hOGG1 is specific for lesions paired with C or T42 and cannot react on NCPs (see Table 1). Like hOGG1, hAAG shows strong specificity for the base opposite Hx43 and cannot remove Hx from NCP substrates. If Hx is replaced with εA, the substrate specificity weakens considerably,43 and hAAG becomes reactive on NCPs. Similarly, hAAG and UDG can cleave single stranded DNA, while hOGG1 and EndoIII cannot.11,44 These correlations suggest that glycosylase reactivity may be inhibited in NCPs systems in which the glycosylase must interact with the base opposite the lesion in order to catalyze base excision. Steric interactions with the histone core could prevent such interactions from occurring.
Biological Implications of Sequestering DNA Damage.
In applying these results to biological systems, it should be noted that chromosomal DNA damage repair in the cell differs from the model system investigated in this study. First, the DNA sequence used here binds more strongly to the histone core than almost any other known sequence,23 and FRET studies have shown that nucleosomes undergo spontaneous dynamic motion that facilitates enzyme access to otherwise blocked DNA bases.45 The strongly binding Widom 601 sequence likely disfavors enzyme access that more weakly binding biological sequences might allow. Second, in the cell nucleus, additional factors that could affect glycosylase efficiency are not reflected in our system. For example, chromatin remodeling is necessary to expose sequestered DNA for replication, transcription, and to repair damage in nucleotide excision repair (NER) and double strand break (DSB) repair; however its role in BER remains undetermined.14 Our results suggest that in vivo, chromatin remodeling may be required for some glycosylases but not for others. Importantly, the fact that we observe robust glycosylase activity in some cases, despite our use of the Widom 601 DNA sequence and our lack of chromatin remodeling factors, underscores our conclusion that some glycosylases can operate in the challenging environment of packaged DNA.
Our observation that some DNA glycosylases react efficiently on NCP substrates while others do not raises an important question: could there be an evolutionary benefit to sequestering damaged bases from repair enzymes? Certainly, deficiencies in BER increase cancer risk. Nevertheless, halting BER in cases of acute stress could serve an evolutionary benefit by eliminating the risk of further genetic damage. For example, repair of clustered damage by bifunctional DNA glycosylases can lead to pernicious DSBs in DNA. In one study, E. coli strains in which the primary DNA glycosylases were knocked out did not form DSBs and were more viable than wild-type cells following exposure to ionizing radiation.46 Similarly, interruption of BER yields abandoned repair intermediates such as apurinic/apyrimidinic (AP) sites, which can subsequently form strand breaks, DNA-DNA interstrand cross-links, and protein adducts.47 In fact, recent work has shown that NCP binding catalyzes cleavage of DNA at AP sites.48 Since bifunctional glycosylases are ineffective on nucleosomes, sheltering base lesions in NCPs could protect DNA from DSBs and deleterious products derived from abandoned repair intermediates until the repair machinery is granted full access to the damage sites, potentially via currently unidentified chromatin remodeling factors.
CONCLUSION
In summary, our efforts have shown that DNA glycosylase activity on NCPs is highly variable. Factors affecting glycosylase efficiency include the solvent accessibility and identity of the damaged base as well as the size, structure, and mechanism of the glycosylase. Importantly, the sequestration of genomic DNA in nucleosomes affects many cellular processes besides BER. For instance, geometrical constraints imposed by the histone octamer are known to limit the efficiency of restriction endonucleases, RNA polymerases, transcription factors, double strand break repair and nucleotide excision repair enzymes,14 and CRISPR-Cas9 machinery.49–51 The experiments presented here therefore provide a basis for further studies into the effects of nucleosome incorporation on cellular DNA chemistry in general.
MATERIALS AND METHODS
Detailed methods are included in the Supporting Information.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by the National Institutes of Health (F32CA183573 to E. Olmon and R01ES019296 to S. Delaney). We thank J. Barton for providing EndoIII protein and T. Tsukiyama for developing histone expression plasmids. Molecular graphics were prepared in PyMOL. Molecular modeling was performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Footnotes
Supporting Information Available: This material is available free of charge via the Internet.
REFERENCES
- (1).Dizdaroglu M (2015) Oxidatively induced DNA damage and its repair in cancer. Mutat. Res. Rev. Mutat. Res 763, 212–245. [DOI] [PubMed] [Google Scholar]
- (2).Krokan HE, and Bjoras M (2013) Base excision repair Cold Spring Harb. Perspect. Biol (Friedberg EC, Elledge SJ, Lehmann AR, Lindahl T, and Muzi-Falconi M, Eds.) 5, a012583–a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Schermerhorn KM, and Delaney S (2015) A chemical and kinetic perspective on base excision repair of DNA. Acc. Chem. Res 47, 1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kim Y-J, and Wilson DMI (2012) Overview of base excision repair biochemistry. Curr. Mol. Pharmacol 5, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hegde ML, Hazra TK, and Mitra S (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 18, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).David SS, O’Shea VL, and Kundu S (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).David SS, and Williams SD (1998) Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev 98, 1221–1262. [DOI] [PubMed] [Google Scholar]
- (8).Brooks SC, Adhikary S, Rubinson EH, and Eichman BF (2013) Recent advances in the structural mechanisms of DNA glycosylases. Biochim. Biophys. Acta 1834, 247–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dalhus B, Laerdahl JK, Backe PH, and Bjørås M (2009) DNA base repair - recognition and initiation of catalysis. FEMS Microbiol. Rev 33, 1044–1078. [DOI] [PubMed] [Google Scholar]
- (10).Mullins EA, Shi R, Parsons ZD, Yuen PK, David SS, Igarashi Y, and Eichman BF (2015) The DNA glycosylase AlkD uses a non-base-flipping mechanism to excise bulky lesions. Nature 527, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Stivers JT, and Jiang YL (2003) A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev 103, 2729–2759. [DOI] [PubMed] [Google Scholar]
- (12).Drohat AC, and Maiti A (2014) Mechanisms for enzymatic cleavage of the N-glycosidic bond in DNA. Org. Biomol. Chem 12, 8367–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).McGinty RK, and Tan S (2015) Nucleosome structure and function. Chem. Rev 115, 2255–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Odell ID, Wallace SS, and Pederson DS (2013) Rules of engagement for base excision repair in chromatin. J. Cell. Physiol 228, 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nilsen H, Lindahl T, and Verreault A (2002) DNA base excision repair of uracil residues in reconstituted nucleosome core particles. EMBO J. 21, 5943–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hinz JM, Rodriguez Y, and Smerdon MJ (2010) Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc. Natl. Acad. Sci. USA 107, 4646–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ye Y, Stahley MR, Xu J, Friedman JI, Sun Y, McKnight JN, Gray JJ, Bowman GD, and Stivers JT (2012) Enzymatic excision of uracil residues in nucleosomes depends on the local DNA structure and dynamics. Biochemistry 51, 6028–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Odell ID, Barbour J-E, Murphy DL, Della-Maria JA, Sweasy JB, Tomkinson AE, Wallace SS, and Pederson DS (2011) Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Molec. Cell. Biol 31, 4623–4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Odell ID, Newick K, Heintz NH, Wallace SS, and Pederson DS (2010) Non-specific DNA binding interferes with the efficient excision of oxidative lesions from chromatin by the human DNA glycosylase, NEIL1. DNA Repair 9, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Menoni H, Gasparutto D, Hamiche A, Cadet J, Dimitrov S, Bouvet P, and Angelov D (2007) ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A.Bbd nucleosomes. Mol. Cell. Biol 27, 5949–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cole HA, Tabor-Godwin JM, and Hayes JJ (2010) Uracil DNA glycosylase activity on nucleosomal DNA depends on rotational orientation of targets. J. Biol. Chem 285, 2876–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Rodriguez Y, and Smerdon MJ (2013) The structural location of DNA lesions in nucleosome core particles determines accessibility by base excision repair enzymes. J. Biol. Chem 288, 13863–13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chua EYD, Vasudevan D, Davey GE, Wu B, and Davey CA (2012) The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 40, 6338–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Vasudevan D, Chua EYD, and Davey CA (2010) Crystal structures of nucleosome core particles containing the “601” strong positioning sequence. J. Mol. Biol 403, 1–10. [DOI] [PubMed] [Google Scholar]
- (25).Jain SS, and Tullius TD (2008) Footprinting protein–DNA complexes using the hydroxyl radical. Nat. Protoc 3, 1092–1100. [DOI] [PubMed] [Google Scholar]
- (26).Bashkin J, Hayes JJ, Tullius TD, and Wolffe AP (1993) Structure of DNA in a nucleosome core at high salt concentration and at high temperature. Biochemistry 32, 1895–1898. [DOI] [PubMed] [Google Scholar]
- (27).Jarem DA, Wilson NR, Schermerhorn KM, and Delaney S (2011) Incidence and persistence of 8-oxo-7,8-dihydroguanine within a hairpin intermediate exacerbates a toxic oxidation cycle associated with trinucleotide repeat expansion. DNA Repair 10, 887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Seksek O, and Bolard J (1996) Nuclear pH gradient in mammalian cells revealed by laser microspectrofluorimetry. J. Cell Sci 109, 257–262. [DOI] [PubMed] [Google Scholar]
- (29).Palmer LG, and Civan MM (1977) Distribution of Na+ , K+ and Cl– between nucleus and cytoplasm in chironomus salivary gland cells. J. Membr. Biol 33, 41–61. [DOI] [PubMed] [Google Scholar]
- (30).Go Y-M, and Jones DP (2010) Redox control systems in the nucleus: mechanisms and functions. Antioxid. Redox Signal 13, 489–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sassa A, Beard WA, Prasad R, and Wilson SH (2012) DNA sequence context effects on the glycosylase activity of human 8-oxoguanine DNA glycosylase. J. Biol. Chem 287, 36702–36710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ide H (2001) DNA substrates containing defined oxidative base lesions and their application to study substrate specificities of base excision repair enzymes. Prog. Nucleic Acid Res. Mol. Biol 68, 207–221. [DOI] [PubMed] [Google Scholar]
- (33).O’Brien PJ, and Ellenberger TE (2003) Human alkyladenine DNA glycosylase uses acid-base catalysis for selective excision of damaged purines. Biochemistry 42, 12418–12429. [DOI] [PubMed] [Google Scholar]
- (34).Ishchenko AA, Bulychev NV, Maksakova GA, Johnson F, and Nevinsky GA (1999) Single-stranded oligodeoxyribonucleotides are substrates of Fpg protein from Escherichia Coli. IUBMB Life 48, 613–618. [DOI] [PubMed] [Google Scholar]
- (35).Bennett SE, Sanderson RJ, and Mosbaugh DW (1995) Processivity of Escherichia coli and rat liver mitochondrial uracil-DNA glycosylase is affected by NaCl concentration. Biochemistry 34, 6109–6119. [DOI] [PubMed] [Google Scholar]
- (36).Lau AY, Wyatt MD, Glassner BJ, Samson LD, and Ellenberger TE (2000) Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc. Natl. Acad. Sci. USA 97, 13573–13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Asahara H, Wistort PM, Bank JF, Bakerian RH, and Cunningham RP (1989) Purification and characterization of Escherichia coli endonuclease III from the cloned nth gene. Biochemistry 28, 4444–4449. [DOI] [PubMed] [Google Scholar]
- (38).Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, and Tainer JA (2000) Uracil-DNA glycosylase-DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc. Natl. Acad. Sci. USA 97, 5083–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Fromme JC, and Verdine GL (2003) Structure of a trapped endonuclease III-DNA covalent intermediate. EMBO J. 22, 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hatahet Z, Kow YW, Purmal AA, Cunningham RP, and Wallace SS (1994) New substrates for old enzymes. J. Biol. Chem 18814–18820. [PubMed] [Google Scholar]
- (41).Wyatt MD, and Samson LD (2000) Influence of DNA structure on hypoxanthine and 1,N6-ethenoadenine removal by murine 3-methyladenine DNA glycosylase. Carcinogenesis 21, 901–908. [DOI] [PubMed] [Google Scholar]
- (42).Krishnamurthy N, Haraguchi K, Greenberg MM, and David SS (2008) Efficient removal of formamidopyrimidines by 8-oxoguanine glycosylases. Biochemistry 47, 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).O’Brien PJ, and Ellenberger TE (2004) Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J. Biol. Chem 279, 9750–9757. [DOI] [PubMed] [Google Scholar]
- (44).Hitchcock TM, Dong L, Connor EE, Meira LB, Samson LD, Wyatt MD, and Cao W (2004) Oxanine DNA glycosylase activity from mammalian alkyladenine glycosylase. J. Biol. Chem 279, 38177–38183. [DOI] [PubMed] [Google Scholar]
- (45).Ngo TTM, and Ha T (2015) Nucleosomes undergo slow spontaneous gaping. Nucleic Acids Res. 43, 3964–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Blaisdell JO, and Wallace SS (2001) Abortive base-excision repair of radiation-induced clustered DNA lesions in Escherichia coli. Proc. Natl. Acad. Sci. USA 98, 7426–7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Greenberg MM (2014) Abasic and oxidized abasic site reactivity in DNA: Enzyme inhibition, cross-linking, and nucleosome catalyzed reactions. Acc. Chem. Res 47, 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zhou C, Sczepanski JT, and Greenberg MM (2012) Mechanistic studies on histone catalyzed cleavage of apyrimidinic/apurinic sites in nucleosome core particles. J. Am. Chem. Soc 134, 16734–16741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hinz JM, Laughery MF, and Wyrick JJ (2015) Nucleosomes inhibit Cas9 endonuclease activity in vitro. Biochemistry 54, 7063–7066. [DOI] [PubMed] [Google Scholar]
- (50).Isaac RS, Jiang F, Doudna JA, Lim WA, Narlikar GJ, and Almeida RA (2016) Nucleosome breathing and remodeling constrain CRISPR-Cas9 function. Elife 5, e13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Horlbeck MA, Witkowsky LB, Guglielmi B, Replogle JM, Gilbert LA, Villalta JE, Torigoe SE, Tijan R, and Weissman JS (2016) Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife 5, e12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kow YW (2002) Repair of deaminated bases in DNA. Free Rad. Biol. Med 33, 886–893. [DOI] [PubMed] [Google Scholar]
- (53).Boiteux S, O’Connor TR, Lederer F, Gouyette A, and Laval J (1990) Homogeneous Escherichia coli FPG Protein. J. Biol. Chem 265, 3916–3922. [PubMed] [Google Scholar]
- (54).Jiang D, Hatahet Z, Robert J, Kow YW, Susan S, Melamede RJ, and Wallace SS (1997) Characterization of Escherichia coli endonuclease VIII. J. Biol. Chem 272, 32230–32239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.