

ABSTRACT
Gestational Diabetes Mellitus (GDM) is the most common metabolic condition during pregnancy and may result in short- and long-term complications for both mother and offspring. The complexity of phenotypic outcomes seems influenced by genetic susceptibility, nutrient-gene interactions and lifestyle interacting with clinical factors. There is strong evidence that not only the adverse genetic background but also the epigenetic modifications in response to nutritional and environmental factors could influence the maternal hyperglycemia in pregnancy and the foetal metabolic programming. In this view, the correlation between epigenetic modifications and their transgenerational effects represents a very interesting field of study. The present review gives insight into the role of gene variants and their interactions with nutrients in GDM. In addition, we provide an overview of the epigenetic changes and their role in the maternal-foetal transmission of chronic diseases. Overall, the knowledge of epigenetic modifications induced by an adverse intrauterine and perinatal environment could shed light on the potential pathophysiological mechanisms of long-term disease development in the offspring and provide useful tools for their prevention.
KEYWORDS: Nutrigenetics, epigenetics, gestational diabetes, gene-nutrient interaction, hyperglycemia in pregnancy
Introduction
It is now widely accepted that environmental insults, including poor or unhealthy nutrition, lack of exercise, tobacco smoking, alcohol consumption, environmental pollutants, and psychological stress, increase an individual’s risk of metabolic diseases during the lifetime. As a consequence, many efforts are currently taken to gain knowledge about the mechanisms by which metabolic pathways are coordinated by acquired and genetic factors, in order to obtain novel insights into the treatment of these conditions [1–3].
As to genetic susceptibility, to date, several genetic loci correlated with metabolic disease risk have been identified by genome-wide association studies (GWAS) [4–7]. However, the gene variants, in form of single nucleotide polymorphisms (SNPs) or copy number variants (CNVs), explain only a small proportion of the individual risk.
The missing heritability component of the complexity of phenotypic outcomes may be revealed by epigenetic processes [8,9]. Epigenetics can be defined as the study of molecular mechanisms that establish and maintain mitotically stable patterns of gene expression yet do not alter DNA sequence [10]. These mechanisms can be affected by environmental factors such as diet, pollution, stress, smoke and others. As matter of fact, scientific literature has highlighted that the risk of developing diseases in later life can be also influenced by adverse condition exposures during early life [11,12]. This domain of research is solid, but the knowledge of the underlying mechanism is still in its infancy.
During specific periods (e.g. pre-conception, oocyte fertilization, gestation and the first few years of life), tissues and organs are particularly sensitive to several environmental insults and to lifestyle factors that condition the organism and shape susceptibility to disease later in life [13,14].
The analysis of epigenetic modifications occurring during pregnancy represents an interesting topic in the study of the environmental influence and foetal metabolic programming [15]. Several studies have showed how epigenetic changes induce life-long consequences in offspring exposed to unhealthy maternal nutrition and lifestyle, obesity, and Gestational Diabetes Mellitus (GDM) [16–19]. In this regard, the present review provides an overview on the role played by maternal genetic variants and epigenetic modifications in GDM and other metabolic conditions, as well on the maternal-foetal transmission of increased susceptibility to chronic diseases. We also examine future topics of research and the potential preventive interventions during early development to reduce the risk of metabolic diseases in both mothers and offspring.
Gestational diabetes and nutrigenetics
GDM can be defined as ‘diabetes diagnosed in the second or third trimester of pregnancy that was not clearly overt diabetes prior to gestation’ [20]. GDM shows a prevalence ranging between 1% and 28% worldwide, and generally regresses after delivery [21]. Consistent evidence has shown the relationships between GDM and subsequent type 2 diabetes (T2DM), hypertension, dyslipidaemia, vascular dysfunction, atherosclerosis and other markers of cardiovascular risk in the mother [22,23]. In addition, GDM can cause complications on the offspring, with short-term effects [24] including macrosomia, shoulder dystocia, birth injury, and prematurity as well as, in line with Freinkel hypothesis [25], long-term consequences upon body composition as well as anthropometric and metabolic functions [24,26].
The GDM prevalence has increased by more than 30% within one or two decades in a number of countries including the developing ones [27]. One of the possible causes of this increased prevalence could be ascribed to the advanced age of pregnancy, which in turn is related to the presence in pregnant women of risk factors, such as obesity and overweight, making them more susceptible to hyperglycemia during pregnancy [21,28].
However, some women developing GDM are not obese; suggesting that other factors, such as unhealthy nutrition and low physical activity before or during pregnancy may also represent risk factors of GDM [19,29].
In this scenario, as demonstrated by recent advances in molecular technology, a crucial role is played by genetic factors in the development, treatment response, and complications of diabetic pregnancy. In a systematic review, Zhang et al. [30] showed variants in seven genes significantly associated with GDM risk (ORs ranging from 1.15 to 1.46). Among these, six were related to insulin secretion (TCF7L2, GCK, KCNJ11, CDKAL1, IGF2BP2, MTNR1B) and one (IRS1) to insulin resistance, suggesting that inherited abnormalities of pancreatic islet b-cell function and/or b-cell mass may be implicated in the GDM aetiology. All these genes have been previously related to the T2DM risk [31,32].
Meta-analysis of candidate gene studies and GWAS have identified other T2DM-related common variants associated with GDM susceptibility [4,7], confirming an at least partly shared genetic basis between GDM and T2DM, given that insulin resistance and defects in insulin secretion play a central role in the pathogenesis of both these conditions.
A very important issue in the field of genetic susceptibility is represented by gene variants conferring individual differences in response to nutrition and diet-related chronic diseases [33]. Nutritional genomics, which encompasses nutrigenomics and nutrigenetics, studies the interaction-mechanisms of nutrients with DNA in human health. In this regard, nutrigenetics studies the effects of genetic variations on the nutritional response, while nutrigenomics investigates how nutrients and bioactive food compounds affect gene functions via epigenetic modifications [34].
Therefore, the nutrigenetics concept related to obesity, metabolic syndrome (MetS) and T2DM is largely based on the data associated with dietary fat, carbohydrate and fibres [35–37]. Through linkage analysis, candidate gene association studies and GWASs, polymorphisms in or near genes related to carbohydrate metabolism, lipid/lipoprotein metabolism, appetite control/food intake, energy expenditure and glucose homeostasis have been identified, suggesting the possible relationship among diet, gene expression and glucose homeostasis.
Nutrigenetic studies provide proof of how the inter-individual variability in response to dietary modifications is largely determined by genetic factors [38–47].
In this context, the nutrigenetics approach could be helpful to define genetic factors influencing maternal metabolism during GDM. Recently, the relationship between SNPs located in genes related to nutrients and metabolism, GDM risk and cardio-metabolic risk factors was identified [48,49], by evidencing a significant correlation between lipid parameters and variants in PPARγ, APOA5, MC4R, LDLR and FTO genes in GDM.
The presence of these gene variants and routinely assessed markers (such as lipid profile during pregnancy) could provide an opportunity to use genetic information in clinical practice to predict early cardiovascular disease (CVD) in previous GDM women as demonstrated by Franzago et al. in two studies carried out on 102 GDM cases versus 66 controls and 104 versus 124, respectively [48,49].
Recently, in light of these results, Franzago et al. [50] have also assessed the predictive role in CVD susceptibility of 3rd trimester lipid profile together with markers of subclinical atherosclerosis in a cohort of women three years after diagnosis of GDM, evidencing an association between 3rd trimester triglycerides and carotid artery intima-media thickness (cIMT). In addition, they found significant associations between APOA5 gene variant and cIMT as well as between CC APOA5/CC LDLR interaction and cIMT [50]. Although the results obtained in these studies need to be validated on a larger number of patients, these data highlight that GDM may represent a clinical window to identify ‘cardio-metabolic vulnerability’, therefore providing clinicians with an opportunity to plan early postpartum interventions [49].
Moreover, future studies are required to successfully implement innovative approaches in the field of Precision Nutrition through the analysis and monitoring of dietary behaviours, physical activity and phenotyping. Therefore, the identification of the nutrigenetic markers might be crucial in order to set up a strategy for the prevention, early diagnosis, and treatment of GDM.
However, the presence of constitutional genetic variants is not the only mechanism triggering the interaction between genes and diet-related disorders. In fact, due to the availability of novel high-throughput technologies, it has been possible to study not only genetic inheritance and its variations, but also genome stability, epigenome alterations, RNA and miRNA alterations, metabolite changes and their role in human metabolism, nutritional homeostasis and molecular events involved in nutrition-related diseases [51]. A clear example of such mechanisms is provided by recent epigenome-wide studies aimed to identify differentially methylated regions (DMRs) in the offspring, as a consequence of intrauterine exposure to maternal diabetes [52–54]. Del Rosario et al. [52] did not identify any specific differentially methylated promoter in human peripheral blood DNA from 28 nondiabetic Pima Indians, born from mothers with and without type 2 diabetes during pregnancy. However, the same authors on a larger series of 388 cases identified differentially methylated cytosine guanine dinucleotides (CpGs) in 39 genomic regions that achieved epigenome-wide significance in their association with exposure to a diabetic intrauterine environment [53].
These findings suggest that there is a need for more studies that are highly focussed on epigenetic mechanisms and their impact at any stage of life.
Epigenetic mechanisms
The main epigenetic mechanisms of gene expression regulation are represented by DNA methylation, histone modifications and small non-coding RNAs. These types of modifications play an important role in vast biological processes at the level of chromatin structure and organization [55]. Epigenetic changes can give rise to transgenerational inheritance, which can be carried through both male and female germline.
DNA methylation is a dynamic process and it is the best understood epigenetic system. It occurs at the 5′- position of cytosine residues, mainly within CpGs, 60–80% of which are methylated within the promoter regions of genes. In most instances, highly methylated DNA regions act to reduce gene expression [56]. Most DNA methylation states are stably maintained and inherited during cell division by the maintenance methyltransferases (DNMT1). These marks are critical for maintaining the physiological differentiated states of tissues and organs. Furthermore, DNMT3A, DNMT3B and co-factor DNMT3L are de novo DNA methyltransferases (DNMTs) which methylate DNA during embryogenesis and in differentiated cells.
Other mechanisms able to affect DNA methylation exist. In fact, the methyl group on the fifth carbon of the cytosine residue within the CpG can be oxidized by the ten-eleven translocation (TET) dioxygenase family, creating the ‘sixth base’ defined as 5-hydroxymethylcytosine (5hmC) [55]. High levels of 5hmC are generally found near the transcription start sites, making them essential for important regulatory functions [57].
The second mechanism of epigenetic regulation of gene activity is represented by modifications of histone tails. Histone marks are dynamic processes [58]; in fact, they can be easily induced and removed by many different enzymes. Histone modifications may increase the exposure of DNA to the transcription factors in the gene expression regulation [59].
Finally, microRNAs (miRNAs) are endogenous 18–22 nucleotides, small non-coding RNAs, that play an important role in the modulation of gene expression in many biological processes, including the development, differentiation, and regulation of cell cycle [60], and immune system homeostasis [61]. Additional evidence has shown that miRNAs are involved in multiple sides of beta-cell function and differentiation, contributing to the regulation of insulin secretion and beta cell identity and phenotype maintenance [60,62]. In spite of the presence of discordant data, lately, growing evidence indicates that circulating miRNAs may potentially represent new biomarkers of several diseases suggesting new pathogenic mechanisms [63,64].
Epigenetics and maternal nutrition
The current evidence
Nutrition is significant for the ‘metabolic memory’ [1], but it is not fully understood how nutrient signals during developmental stages influence metabolism and the associated lifestyle-related diseases in later life [65]. In any case, maternal nutritional disturbances are one of the most important foetal programming stimulus. In line with the ‘Barker hypothesis’ concept [66], intrauterine under- or over-nutrition program adaptations of the foetal metabolism to an adverse postnatal environment, deprived or enriched, respectively [67]. As extensively reported in the literature, dietary patterns, nutrients and bioactive compounds affect metabolic traits by epigenetic modifications, leading to changes in gene expression levels and genome stability.
Godfrey et al. [68] showed that in DNA extracted from umbilical cord tissue obtained at birth, methylation within the promoter of retinoid X receptor-a (RXRA), which encodes a transcription factor implicated in fat metabolism and insulin sensitivity, was correlated with body adiposity, as measured by imaging at age 6 or 9 years in two independent cohorts. Moreover, the methylation at this site was in turn strongly associated with maternal carbohydrate intake during early pregnancy.
Evidence from animal models
Novel biological insights evidenced that obesity predisposition and weight loss outcomes are correlated to changes in epigenetic patterns. Significantly, nutrients and related metabolites can directly modify elements of chromatin in different ways. For example, several findings in animal model studies, suggest that maternal high fat (HF) diet can alter foetal chromatin structure via covalent histone modifications [69–71] (Table 1). Gestational choline supply regulates the methylation of histone H3, the expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat foetal liver and brain [69]. On the other hand, Tosh et al. [70] observed that Igf1 mRNA expression modifications related to altered levels of demethylation of histone H3 at lysine residue 4 (H3K4Me2) during gestational food restriction in rats. Strakovsky et al. [71] investigated the HF diet in the gestational period, independent from maternal obesity and diabetes development. They showed, for the first time, an elevated amount of mRNA expression of several genes is associated with the hepatic gluconeogenic pathway in the liver of foetal offspring, corresponding to elevated glucose levels in the offspring at the time of delivery. Moreover, the authors also showed that HF diet during gestation was able to program phosphoenolpyruvate carboxykinase (Pck1) expression by histone modifications in offspring liver. Therefore, they suggested that an increase in hepatic glucose production will inevitably lead to altered glucose handling, with increased potential for the development of T2DM into adulthood [71].
Table 1.
Rodent studies related to maternal nutrition assessing the effects of epigenetic alterations and their consequences on offspring.
| Author Year [Reference] |
Animal Model | Maternal intervention | Offspring tissue | Method | Alterations in the offspring |
|---|---|---|---|---|---|
| Cannon 2004 [77] | C57BL/6J mouse | HFD LFD |
Liver | RRBS Gene Expression BeadChips |
No detectable DNA methylation differences Upregulation of genes involved in inflammation, cholesterol synthesis and RXR activation |
| Davison 2009 [69] | Sprague-Dawley rat | Choline-supplement/deficiency | Liver Frontal cortex |
MS-PCR RT-PCR Western blot |
Upregulation of DNA methylation of the G9a and Suv39h1 genes by choline-deficient diet Upregulation of H3K9Me2 and H3K27Me3 levels by choline supplementation |
| Tosh 2010 [70] | Sprague Dawley rat | FR | Liver Plasma |
ChIP Western blot qRT-PCR |
Decreased demethylation at H3K4 in the Igf1 region in the IUGR offspring Increased trimethylation of H3K4 in Igf1 region and increased of hepatic Igf1 mRNA expression in obese adult males offspring |
| Strakovsky 2011 [71] | Sprague-Dawley rat | HFD | Liver Plasma Serum |
RT-PCR ChIP |
Higher mRNA expression of gluconeogenic genes Increased plasma glucose levels Modifications of the Pck1 histone code in liver |
| Garbory 2012 [136] | C57BL/6J mouse | HFD | Placenta | Microarray qRT-PCR Western Blotting |
Dysregulation of 7 genes due to diet, sex or both, including the Y- and X-linked histone demethylase paralogues Kdm5c and Kdm5d |
| Borengasser 2013 [72] | Sprague Dawley rat | Over nutrition | WAT | RRBS RT-PCR |
Alterations in DNA methylation in developmentally important genes. Upregulation of lipogenic genes |
| Zhang 2015 [73] | Sprague-Dawley rat | HFD | Liver | MeDIP-seq MRE-seq |
Hypomethylation of 12,494 DMRs Hypermethylation of 6,404 DMRs Identification of DMGs involved in critical hepatic signaling networks |
| Petropoulos 2015 [102] | Cohen diabetes-sensitive rat |
HSD | Placenta Liver |
MeDIP | Different methylation of genes in the placenta and liver with a significant overlap |
| Wankhade 2017 [74] | C57BL6/J mouse | HFD | Liver | RNA-seq qRT-PCR RRBS |
Higher pro-fibrogenic genes expression Identification of 82 DMRs in O-MCD diet |
| Moody 2017 [75] | Sprague–Dawley rat | HFD | Liver | MeDIP-seq MRE-seq qRT-PCR |
Identification of DMGs clustered in the T2DM and the adipocytokine signaling pathways Alteration of several genes expression involved in lipid metabolism and inflammation |
| Keleher 2018 [76] | SM/J mouse | HFD | Liver Heart |
RNA-seq MeDIP-seq MRE-seq |
Identification of tens of thousands DMRs Alteration of several genes expression in liver and heart |
| Jiang 2018 [93] | ICR mouse | STZ | Placenta | qRT-PCR bisulfite genomic sequencing PCR |
Upregulation of 35 imprinted genes Down-regulation of 10 imprinted genes Down-regulation of Dlk1 and upregulation of Gtl2 due to their abnormal methylation status |
HFD, high fat diet; LFD, low fat diet; RRBS, reduced representation bisulfite sequencing; MS-PCR, methylation specific PCR; RT-PCR, reverse transcription-PCR; FR, food restriction; ChIP, Chromatin immunoprecipitation; qRT-PCR, quantitative real-time PCR; IUGR, intrauterine growth restricted; WAT, white adipose tissue; MeDIP-seq, methylated DNA immunoprecipitation sequencing; MRE-seq, methylation-sensitive restriction enzyme sequencing; DMRs, differentially methylated regions; DMGs, Differentially methylated genes; MeDIP, Methylated DNA immunoprecipitation arrays; HSD, high sucrose, low-copper diet; O-MCD diet, methionine choline deficient diet; STZ, streptozotocin.
Consistent with the premise that in utero programming leads to epigenetic changes, several studies have shown that maternal diet can influence metabolism in rat offspring also by affecting DNA methylation [72–77] (Table 1). Specifically, Borengasser [72] demonstrated that maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in male rat offspring. By using a combination of methyl-DNA immunoprecipitation (MeDIP) and methylation-sensitive restriction enzyme sequencing (MRE-seq), it has been demonstrated that HF diet also alters the DNA methylation of critical hepatic signaling genes [73]. Another study indicated that maternal obesity during gestation and lactation alters epigenetic and gut microbiome pathways to favour the development of fatty liver disease and inflammation in the offspring [74]. Moody et al [75] studied the relationship between DNA methylation and metabolic outcomes in response to a postnatal diet following a maternal HF diet. Although the maternal HF diet lays an epigenetic foundation, the authors showed that different postweaning diets result in a high degree of differential genome-wide DNA methylation in rat liver, especially within genes involved in metabolic pathways. These data provide evidence that DNA methylation responds to postnatal dietary changes, emphasizing the importance of dietary choices after birth and across the lifespan. Just recently, Keleher et al [76] identified dozens of differentially expressed genes due to maternal diet, along with tens of thousands of DMRs in the offspring. In the daughters, these epigenetic effects were accompanied by phenotypic changes relevant to obesity and diabetes. These data provided different conclusions as compared to previous investigations of Cannon et al. [77], who although highlighted the influence of maternal diet on adult tissue regulation, suggested that transcriptional changes were unlikely to be caused by DNA methylation differences in adult liver.
The Predictive Adaptive Response (PAR) theory highlighted that the foetus actively responds to its nutritional environment in preparation for its postnatal nutritional environment [78,79]. It should be noted that when the prenatal and postnatal environments do not match (e.g. prenatal undernutrition followed by postnatal nutritional abundance), the risk of metabolic disease increases, while when the prenatal and postnatal nutrition match, the offspring remains healthy. This theory is widely validated in animals, while the evidence in humans is controversial. The PAR hypothesis has received considerable support [80], but overall it has been criticized for some limitations. In fact, it has been derived from studies that relied on low birth-weight as an oversimplified marker of maternal nutrition and it does not adequately explain the increased disease risk of non-communicable disease (NCD) in offspring exposed to over-nutrition in both their prenatal and postnatal environments [19]. These studies allowed the development of the more integrative Developmental Origins of Health and Disease (DOHaD) theory, encompassing several key developmental periods as conception, gestation, infancy, and puberty, when specific exposures can protect or predispose individuals to chronic disease development. In particular, the DOHaD theory proposes that the origin of chronic diseases (e.g. obesity, diabetes, cardiovascular and neuropsychiatric diseases) is related to an early exposure to a suboptimal foetal environment [81].
Evidence from human studies
The 1944–1945 Dutch famine has provided us with a unique opportunity to study the effects on the offspring of a severe period of maternal undernutrition during different stages of gestation [82,83]. During this period, food rations decreased gradually from about 1800 calories (December 1943) to below 800 calories (April 1945) and the extra rations allowed for pregnant and lactating women and young children could not be provided. Studies carried out on the offspring of these women [82,83] demonstrated that chronic diseases in adult life were strongly related to the occurrence of the gestation during the exposure to the famine. In light of these insights, Heijmans [84] showed decreased methylation (likely related to a deficiency in methyl donors, such as the amino acid methionine) in the DMR of the maternally imprinted IGF2 gene in individuals exposed to the Dutch famine as compared with their unexposed, same-sex siblings, six decades later. During the critical period of development (i.e. gestation), maternal nutritional imbalance may influence the offspring health. Epidemiological and animal studies have shown the link between suboptimal early nutrition and poor growth in utero, with an increased risk of hypercholesterolemia, hypertension, T2DM and obesity in adulthood [85,86].
In their pilot study, Quilter et al. [87] examined the effects of various adverse intrauterine environments on DNA methylation at birth, by studying infants exposed to GDM or to prenatal growth restriction, as indicated by subsequent postnatal catch-up growth. The 14,000 genes analysis present on the methylation array revealed that many genes associated with significantly differentially methylated CpGs were common to both exposures, suggesting that these separate developmental trajectories to adult disease share common biological mechanisms. In addition, the majority of these differentially methylated genes were involved in metabolic disease, or growth and development, and indicate candidate mechanisms involved in the developmental programming of adult disease risk.
Epigenetics and gestational diabetes
The current evidence
Current research is increasingly focused on GDM and its foetal complications such as an increased risk of macrosomia (birth weight over 4 kg) or large-for-gestational-age (LGA; birth weight above the 90th centile for gestational age and gender) at birth [88]. In addition, there is a great interest in understanding the mechanistic impact of maternal obesity and hyperglycemia during pregnancy on the metabolic health of the next generation. Therefore, GDM represents a notable example of the Barker hypothesis [89] and fits well with the foetal metabolic programming and DOHaD hypotheses, since foetal exposure to diabetes and diabetes related metabolic derangements may alter the functional development of key organs and thus potentially increase children’s susceptibility to chronic diseases, as supported by several published reports [90,91]. Boney et al. [90] found that LGA children, exposed to an intrauterine environment of either diabetes or maternal obesity, are at increased risk of developing MetS in adult age. Subsequently, based on the data collected from the multi-ethnic (non-Hispanic white, African-American, and Hispanic) SEARCH Case-Control Study, Dabelea et al. [91] observed that intrauterine exposures to maternal diabetes and obesity accounted for 47% of cases of T2DM before 22 years of age in the offspring, likely as a consequence of intrauterine exposure to hyperglycemia. Despite this evidence to date, the literature displays a sizable knowledge gap in the field of epigenetics and GDM, since experimental data demonstrating that the increased risk of chronic diseases in the offspring of GDM mothers are associated to epigenetic mechanisms are still lacking. Nevertheless, the hypothesis that GDM may trigger these changes and that the differential epigenetic signatures could therefore serve as key biomarkers is taking off.
Epigenetic alterations in the placenta
In this view, a key role is likely to be played by the placenta, which is a critical protagonist in regulating foetal growth and development, by controlling maternal foetal nutrient exchanges via epigenetic mechanisms, which are mainly carried out by genomic imprinting. Adverse conditions in utero, such as GDM have been related with placental anatomy and physiology alterations, inducing perturbations in placental nutrient supply and, consequently, foetal growth and development. It is increasingly clear that proper epigenetic regulation is significant in placental development and function [92].
Evidence from animal models
Recently, Jiang et al. [93] used a GDM mouse model of intrauterine hyperglycemia, to demonstrate that the GDM intrauterine environment affects the placenta in both the first and the second filial generations. The authors revealed by microarray analysis of placental RNA, 35 upregulated and 10 down-regulated imprinted genes. In particular, Dlk1 was down-regulated and Gtl2 was up-regulated, as a consequence of their abnormal methylation status in the first and the second generation of mice. In detail, Dlk1 promotes the insulin/IGF-I signalling pathway activation and adipogenesis inhibition, while Gtl2 is a regulator of TGF-β and notch signalling pathway. In addition, these authors suggested that intrauterine hyperglycemia decreased placental weight in the first generation, transmitting it to the second generation through the paternal line
Evidence from human studies
Reichetzeder et al. [94] were the first to perform a robust quantitative assessment of placental global DNA methylation in over a thousand human placental samples, showing evidence that placental global DNA hypermethylation is associated with GDM, independently from the established risk factors.
Recently, a few studies carried out in humans have supported the epigenetic role in foetal metabolic programming of newborn exposed to maternal hyperglycemia during pregnancy, suggesting an important role of epigenetic alterations [78,95–103] (Table 2). Bouchard et al. [95,96] demonstrated that maternal hyperglycemia is associated with placental DNA methylation alterations at the leptin (LEP) and adiponectin (ADIPOQ) genes. The authors found a significant correlation between the 2-h glucose value and the degree of DNA methylation of the LEP gene in placenta on both foetal and maternal side in GDM women. Higher glucose values were correlated with lower degree of methylation on the foetal side, but with a higher degree of methylation on the maternal side [95]. Regard ADIPOQ, the authors reported that a high level of maternal insulin resistance in the second and third trimester was associated with lower DNA methylation of this gene on the maternal side. Because ADIPOQ and LEP are involved in energy metabolism and insulin sensitivity control, these epigenetic adaptations may have the potential to induce sustained glucose metabolism changes in the mother and the offspring later in life. The link between LEP and ADIPOQ epigenetic alterations and insulin sensitivity has been also confirmed by García-Cardona et al. [104], who determined the methylation levels of the promoters of these two genes in DNA from peripheral blood in one hundred and six adolescents. This study demonstrated that obese children with insulin resistance showed significantly decreased DNA methylation levels of ADIPOQ, associated with serum adiponectin levels. The authors supposed that the epigenetic modifications might underpin the development of obesity and other related metabolic disorders.
Table 2.
Human studies investigating epigenetic alterations in pregnant women with hyperglicemia and in their offspring.
| Author Year [Reference] |
Study Design | Sample size | Hyperglicemia criteria |
Tissue | Method | Main finding |
|---|---|---|---|---|---|---|
| Bouchard 2010 [95] | Case-control | 48 (23 IGT) | 2-h 75 g OGTT, IGT glucose ≥7.8 mmol/L at 2-h |
Placenta (foetal and maternal) UCB |
Bisulfite pyrosequencing |
Correlation between LEP DNA methylation and 2h glucose levels in IGT women |
| Bouchard 2012 [96] |
Cohort | 98 (31 IGT) | 2-h 75 g OGTT, IGT glucose ≥7.8 mmol/L at 2-h according to WHO |
Placenta (foetal and maternal) UCB MBS |
Bisulfite pyrosequencing |
Inverse correlation between ADIPOQ DNA methylation on the foetal side and 2h glucose levels in IGT women |
| Houde 2013 [97] | Cohort | 100 (26 IGT) |
2-h 75 g OGTT, IGT glucose ≥7.8 mmol/L at 2-h according to WHO |
Placenta (foetal and maternal) UCB MBS |
Bisulfite pyrosequencing qRT-PCR |
Positive correlation between ABCA1 DNA methylation on the maternal side and HDL-C and 2h glucose levels in IGT women correlation between DNA methylation on the fetal side and TGs in UCB Negative correlation between ABCA1 DNA methylation in UCB and 2h glucose levels |
| El Hajj 2013 [103] | Cohort | 251 offspring (88 OD-GDM 98 OI-GDM 65 O non-GDM) |
2-h 75 g OGTT, GDM glucose >180 mg/dL at 1 h and/or >155 mg/dL at 2 h | Placenta UCB |
Bisulfite pyrosequencing |
Decreased methylation of MEST, NR3C1, and ALUs in O-GDM |
| Ruchat 2013 [100] | Case-control | 44 offspring (30 O-GDM) |
2-h 75 g OGTT, GDM glucose ≥7.8 mmol/L at 2 h according to WHO |
Placenta (foetal) UCB |
Infinium HumanMethylation450 array | Number of genes potentially differentially methylated in the placenta and UCB in O-GDM |
| Quilter 2014 [87] | Cohort | C-HAPO cohort (n = 36) I-CBGS cohort [n = 96 (16 GDM)] |
WHO criteria | UCB | Human Methylation27 BeadChip |
Different methylation of some loci related to growth and diabetes |
| Houde 2014 [99] | Cohort | 126 (27 GDM) |
2-h 75 g OGTT, GDM glucose ≥7.8 mmol/L according to WHO criteria |
Placenta (foetal) | Bisulfite pyrosequencing qRT-PCR |
Lower LPL DNA methylation levels in GDM. Negative correlation between LPL DNA methylation levels in GDM and maternal 2h glucose levels/HDL-C |
| Desgagne 2014 [101] | Cohort | 140 (IGT 34) |
2-h 75 g OGTT, IGT glucose ≥7.8 mmol/L at 2 h according to WHO criteria |
Placenta (foetal) |
Bisulfite pyrosequencing qRT-PCR |
Lower IGF1R and IGFBP3 DNA methylation levels and correlation with maternal 2h glucose levels Association between IGF1R mRNA levels and newborns’ growth markers |
| Petropoulos 2015 [102] | Case control | 14 (7 mild hyperglicemia) |
GCT or a OGTT (1 week after GCT) |
Placenta | Infinium HumanMethylation450 array | Different methylation of some loci involved in endocrine function, metabolism, and insulin responses |
| Reichetzeder 2016 [94] | Cohort | 1030 (56 GDM) |
GDA and DGGG 2014 | Placenta (maternal) | LC-MS/MS | Increased global methylation in GDM |
| Côté 2016 [105] | Cohort | E-21 birth cohort (n = 133, 33 GDM) Gen3G birth cohort (n = 172, all controls) |
E-21: 2h OGTT, GDM glucose ≥7.8 mmol/L according to WHO criteria Gen3G: 2h OGTT according to IADPSG 2010 |
Placenta (foetal) | E-21: bisulfite pyrosequencing Gen3G: HumanMethylation450 array |
Inverse correlation between PRDM16, BMP7 and PPARGC1A DNA methylation levels and maternal glycemia at the 2nd and 3rd trimester |
| Gagné-Ouellet 2017 [17] | Prospective birth cohort | 66 offspring (24 O-GDM) |
2-h 75 g OGTT, GDM glucose ≥7.8 mmol/L according to WHO criteria |
Placenta (foetal) Offspring’s whole blood at 5 years |
Bisulfite pyrosequencing qRT-PCR |
Negative correlation between LPL DNA methylation and mRNA levels in placenta Positive correlation between LPL DNA methylation levels and anthropometric profile at 5 years of age |
| Chen 2017 [53] | Cohort | 388 Pima Indian offspring (187 O-T2DM, 201 O-BP) |
2-h 75 g OGTT, T2DM FBG≥7 mmol/L or 2-h glucose ≥11.1 mmol/L according to WHO criteria |
blood samples | Illumina HumanMethylation450 K | Different methylation at multiple genomic sites |
| Houshmand-Oeregaard 2017 [106] | Cohort | 206 adult offspring (82 O-GDM, 67 O-T1DM, 57 O-BP) |
Mother = 3-h 50g OGTT in women at risk with two consecutive FBG ≥4.1 mmol/l Offspring = 2-h 75g OGTT according to WHO 2006 criteria |
SAT plasma |
Bisulfite pyrosequencing qRT-PCR |
Increased ADIPOQ methylation levels and decreased ADIPOQ and RETN gene expression in SAT of O-GDM |
| Ott 2018 [107] | prospective observational cohort | 55 mother-child dyads (25 GDM) |
National Germany guidelines | SAT VAT UCB blood samples |
Bisulfite pyrosequencing qRT-PCR |
Alteration of ADIPOQ DNA methylation profiles in CB cells of O-GDM Reduction of mRNA adiponectin levels in SAT and VAT of GDM women |
| Ott 2019 [108] | Prospective observational | 55 mother-child dyads (25 GDM) |
National Germany guidelines | SAT VAT UCB blood samples |
Bisulfite pyrosequencing qRT-PCR |
Similar DNA methylation patterns across tissues Reduction of IR mRNA/protein expressions in SAT and VAT of GDM women |
IGT, Impaired Glucose Tolerance; OGTT, oral glucose tolerance test; UCB, umbilical cord blood; MBS, Maternal blood samples; qRT-PCR, quantitative Real-Time PCR; HDL-C, high-density lipoprotein cholesterol; TGs, triglycerides; OD-GDM, offspring of mother with dietetically treated gestational diabetes, OI-GDM offspring of mother with insulin-dependent GDM; C-HAPO, children from Hyperglicemia and Adverse Pregnancy Outcome; I-CBGS, infants from Cambridge Baby Growth Study; WHO, World Health Organization; GCT, Glucose Challenge Test; GDA, German Diabetes Association; DGGG, German Association for Gynaecology and Obstetrics; LC-MS/MS, Liquid Chromatography tandem Mass Spectrometry; E-21, ECOGENE-21; Gen3G, Genetics of Glucose regulation in Gestation and Growth; IADPSG, International Association of the Diabetes and Pregnancy Study Groups; O-GDM, offspring of women with GDM; O-T2DM, offspring of women with T2DM during pregnancy, O-BP, offspring of women from the background population; FBG, fasting blood glucose; O-T1DM, offspring of women with T1DM during pregnancy; SAT, subcutaneous adipose tissue; VAT, visceral adipose tissue;
Another study showed that DNA methylation levels at the maternally imprinted MEST gene were significantly lower in placenta and cord blood tissues exposed to GDM than in non-GDM women [103]. In addition, obese adults showed MEST hypomethylation compared with normal-weight controls (sex- and age-matched) in the blood. These findings support the hypothesis that epigenetic malprogramming of MEST in newborns of GDM mothers may contribute to obesity predisposition throughout life.
Houde et al. [97] assessed the associations between the maternal metabolic profile and ATP-binding cassette transporter A1 (ABCA1) DNA methylation levels in placenta and cord blood in GDM pregnancies. ABCA1 is a transporter of cholesterol from cells to apolipoproteins A1 and a contributor to high-density lipoprotein (HDL) formation. The authors reported that ABCA1 DNA methylation levels on the maternal side of the placenta were correlated with maternal HDL- cholesterol levels and glucose levels 2 h post-OGTT (oral glucose tolerance test). On the foetal side of the placenta, ABCA1 DNA methylation levels were associated with cord blood triglycerides levels. ABCA1 DNA methylation variability on both sides of the placenta were also associated with ABCA1 mRNA levels. By contrast, cord blood DNA methylation levels were negatively correlated with maternal glucose 2 h post-OGTT.
Houde et al. [98] reported for the first time associations between lipoprotein lipase (LPL) DNA methylation levels and changes in maternal glucose and lipid profiles in placenta samples exposed to GDM. In fact, the LPL DNA methylation in foetal placental tissue was lower in 27 GDM pregnancies as compared to 99 controls with a 1.6-fold higher expression of LPL as evidenced by mRNA analysis. Then, the same authors demonstrated that foetal placental DNA methylation levels at the LPL gene locus are positively associated with the anthropometric profile and body composition (fat mass, birth weight, mid-childhood weight) in children at 5 years of age. Overall, these results suggest the presence of GDM-induced placental LPL epivariations and support the evidence of foetal metabolic programming of childhood obesity through epigenetic alterations, underlining the harmful consequences of some in utero exposures [17].
Another relevant contribution has been provided by Côté et al. [105], who suggested that maternal glycemia is associated with foetal DNA methylation variations in placenta at PR domain-containing protein 16 (PRDM16), bone morphogenetic protein 7 (BMP7) and peroxisome proliferator-activated receptor-γ coactivator-1α (PPARGC1α) genes, involved in the regulation of newborns’ brown adipose tissue (BAT) and beige adipocytes (wBAT). Overall, the authors suggested that epigenetic programming at these loci is responsive to metabolic variations related to glucose homeostasis during pregnancy, which might affect BAT/wBAT activation and the development of obesity and T2DM later in life.
Using a cross-species approach in human and rat, Petropoulos et al. [102] evidenced that diabetes during pregnancy in rats and GDM in humans alter the methylome in the placenta of both the species, as well as in the liver of the rat offspring . These alterations involve similar functional processes (i.e. metabolic diseases and cardiovascular diseases) by affecting 27 overlapping genes in both species known to be associated with cytokine mediated signalling, immune processes, and metabolism. In particular, 12 of these genes displayed a methylation mark in the same direction in both the species, in which four were methylated and eight were demethylated. This study demonstrated that a genome-wide DNA methylation profile in the placenta significantly overlaps with the one in the offspring’s liver, supporting the use of the placenta in identifying biomarkers for predicting foetal outcomes. These data are consistent with a previous study by Ruchat et al. [100] that showed DNA methylation alterations in metabolic genes in cord blood and placenta of GDM offspring. In detail, 3,271 and 3,758 genes in placenta and cord blood, respectively were differentially methylated between samples exposed or not to GDM, with more than 25% (n = 1,029) being common to both tissues. Up to 115 of these genes (11%) were involved in the metabolic diseases pathway including diabetes mellitus.
Epigenetic alterations in other tissues
Placenta, however, does not appear to represent the only relevant tissue for the study of the epigenetic changes in GDM (Table 2). Interestingly, DNA methylation patterns can occur in a tissue-specific manner, but they can also be similar in other tissues. Some cross-tissue studies provided additional findings on alterations of DNA methylation patterns in hyperglycemic maternal-foetal conditions. For example, offspring born from GDM mothers who had been given dietary advice showed significantly increased ADIPOQ DNA methylation and decreased mRNA expression of ADIPOQ and RETN genes in subcutaneous adipose tissue (SAT); nevertheless, altered methylation and expression levels were not reflected in plasma protein levels [106]. This is an elegant human study proposing epigenetic, transcriptomic and proteomic data from a metabolically significant target tissue as SAT. It is worth noting that Ott et al. [107] analysed paired SAT and visceral adipose tissue (VAT) as well as blood samples of 25 GDM women vs 30 controls of mother-child dyads. GDM women were characterized by hypoadiponectinemia and presented significantly decreased mRNA levels in both SAT and VAT, independently of body mass index (BMI). Inverse relationships were observed between maternal adiponectin vs. glucose, C-peptide, insulin and homeostatic model assessment of insulin resistance (HOMA-IR). The altered maternal DNA methylation patterns appeared rather marginally involved, whereas they were variously altered in GDM offspring. In addition, plasma adiponectin levels were similar in offspring of both women with or without GDM. These studies emphasize the importance of investigating multiple tissues to understand the full scope of the effects of a maternal hyperglycemia in the offspring. In GDM, the investigations on molecular mechanisms of insulin resistance (IR) in VAT are lacking. Thereafter, the same authors [108] reported that, both in SAT and in VAT, insulin receptor (IR) mRNA/protein expressions were significantly reduced in GDM women, but the decrease was more pronounced in VAT and was independent of maternal BMI. In addition, VAT IR protein levels were inversely associated with maternal and neonatal anthropometric/metabolic parameters. Finally, DNA methylation patterns were similar in AT and blood cells, with small size modifications between groups in mothers and offspring [108].
miRNAs and GDM
DNA methylation is not the only epigenetic mechanism involved in GDM. More recently, also the miRNAs have been investigated as possible biomarkers of epigenetic modifications in GDM (Table 3). In fact, upregulation of miRNA miR-330-3p in the plasma of GDM patients has been recently demonstrated [109]. Previously, Zhao et al. [110] showed that miRNAs (miR-132, miR-29a, and miR-222) are differentially expressed between GDM women and controls in serum collected at 16th–19th gestational weeks. In contrast to Zhao et al. [110], Tagoma et al. [111] showed that miR-222 expression was higher in the plasma of GDM women compared to controls, as well as miR-195-5p evidenced the highest fold upregulation in GDM.
Table 3.
Studies investigating miRNAs in GDM and offspring.
| Author Year [Reference] |
Study design | Sample size |
GDM criteria | Tissue (GA wks) |
Method | Main finding |
|---|---|---|---|---|---|---|
| Zhao 2011 [110] |
Case-control | 48 (24 GDM) |
Two-step approach: 50 g GCT followed, if positive, by 3-h 75 g OGTT according to the ADA 2003 |
Maternal serum (16th–19th) |
TLDA chip qRT-PCR |
miR-132, miR-29a, miR-222 downregulation |
| Shi 2014 [113] |
Case-control | 26 (13 GDM) |
ADA 2006 | Maternal omental adipose tissue (cesarean delivery at 38th–39th) |
AFFX miRNA expression chips qRT-PCR |
miR-222 upregulation and negatively correlation with ERα and GLUT4 protein levels |
| Zhu 2015 [112] |
Case-control | 20 (10 GDM) |
Two-step approach: 50 g GCT followed, if positive, by 3-h 75 g OGTT according to the ADA 2011 |
Maternal plasma (16th–19th) |
High-throughput sequencing (Ion Torrent) qRT-PCR |
hsa-miR-16-5p, hsa-miR-17-5p, hsa-miR-19a-3p, hsa-miR-19b-3p, hsa-miR-20a-5p upregulation |
| Cao 2016 [114] |
Case-control | 395 (193 GDM) |
IADPSG 2010 | Placenta (37th-40th) |
qRT-PCR | miR-98 upregulation linked to the global DNA methylation via targeting MECP2 |
| Sebastiani 2017 [109] |
Case-control | 31 (21 GDM) |
A 2h 75 g OGTT according to the Italian guidelines (IADPSG 2010) |
Maternal plasma (24th–33rd) |
TaqMan array profiling analysis qRT- PCR |
miR-330-3p upregulation |
| Tagoma 2018 [111] |
Case-control | 22 (13 GDM) |
2-h 75 g OGTT according to the IADPSG 2010 | Maternal plasma (23rd-31st) |
RT-PCR | miR-195-5p upregulation |
| Houshmand-Oeregaard 2018 [115] |
observational follow-up | 206 offspring (82 O-GDM 67 O-T1D 57 O-BP) |
Mother = OGTT in women at risk with two consecutive FBG ≥4.1 mmol/l Offspring = 2h 75 g OGTT according to WHO 2006 criteria |
Skeletal muscle of adult offspring (26- to 35-year-old) |
Taqman miRNA assays | miR-15a, miR-15b upregulation in O-GDM and O-T1D |
GCT, glucose challenge test; OGTT, oral glucose tolerance test; ADA, American Diabetes Association; TLDA, TaqMan Low Density Array; qRT-PCR, quantitative reverse transcriptase polymerase chain reaction; IADPSG, International Association of the Diabetes and Pregnancy Study Groups; O-BP, offspring of women from the background population; OGDM, offspring of women with gestational diabetes; O-T1D, offspring of women with type 1 diabetes in pregnancy; FBG, fasting blood glucose; WHO, World Health Organization.
Zhu et al. [112] demonstrated that five miRNAs (hsa-miR-16-5p, hsa-miR-17-5p, hsa-miR- 19a-3p, hsa-miR-19b-3p, and hsa-miR-20a-5p) were upregulated in diabetic pregnant women with respect to controls. Shi et al. [113] determined the differential expression patterns of miRNAs in omental adipose tissues taken at the time of caesarean section from GDM patients and controls, suggesting miR-222 as a potential regulator of ER expression in estrogen-induced insulin resistance in GDM; and hence, it could be considered as a candidate biomarker and therapeutic target for GDM.
Cao et al. [114] examined the relationship between maternal GDM and miR-98. The authors found reduced expression of methyl-CpG-binding protein 2 (MECP2) and transient receptor potential cation channel subfamily C member 3 (TRPC3) in placental tissues from GDM patients, as a consequence of the increase of miR-98, especially for GDM patients over the age of 35 years. In addition, miR-98 over-expression was found to be associated with increased global DNA methylational level, which was reduced in miR-98 knockdown. Therefore, this study showed that miR-98 not only directly targets MECP2, but also indirectly regulates the target genes of MECP2. These findings imply that the expression of miR-98 may suggest a novel regulatory mechanism in GDM by the MECP2-TRPC3 pathway.
Noteworthily, the study by Houshmand-Oeregaard et al. [115] was the first to demonstrate that foetal exposure to maternal diabetes is associated with an increased expression of miR-15a and miR-15b inside the skeletal muscle cells in the offspring of 26- to 35-year-old.
It is also worth mentioning that obesity, diabetes, hypertension and CVD risk in offspring may originate from the altered epigenetic modifications in oocytes [116]. There are a few studies in humans about the effects of hyperglycemia on DNA methylation of oocytes. Wang et al. [117], using an in vitro maturation model, elucidated the effects of high-glucose concentration on DNA methylation of human oocytes. The authors suggested that in humans the high risk of chronic diseases in offspring from diabetic mothers may originate from abnormal DNA modifications in oocytes. This study presents several limitations, since it reports that the high-glucose concentrations altered the DNA methylation status of paternally expressed gene 3 (PEG3) and adiponectin in human IVM oocytes, without explaining whether this alteration is positive or negative for embryo development and offspring health. In addition, the number of oocytes used was limited and the effects of glucose levels on the whole process of oocyte maturation were not been elucidated.
Epigenetic modifications induced by lifestyle
In the last few years, animal and human studies have linked lifestyle factors to epigenetic changes, identifying the timing of early-life exposures as the factors for the different health outcomes in the offspring. In this regard, pregnant women are inevitably exposed to environmental insults of heterogeneous nature: not only nutrition, but also physical activity, tobacco smoking, alcohol consumption, environmental pollutants, psychological stress, and shift-work, all of which have been identified to modify epigenetic patterns [118–132] (Figure 1).
Figure 1.
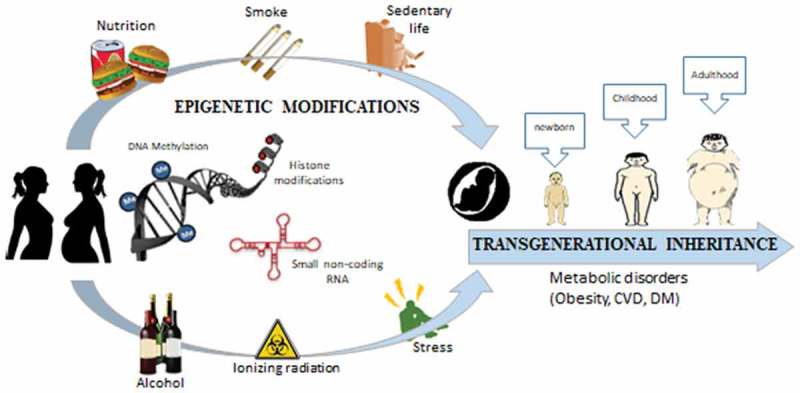
Epigenetic modifications induced by nutrition, hyperglycemia, smoking, radiation, psychological stress, alcohol consumption, etc. can lead to range of long-term metabolic disorders in offspring.
Sex-specific effects in the offspring
Evidence has shown that male and female offspring have different responses to the same early life exposure. For example, some rodent findings underscore the importance of including both males and females in diet studies [76,77,133,134]. The authors demonstrated that offspring’s sex affects the response to maternal diet; in fact, the daughters of high-fat-fed mothers had higher plasma leptin levels [135], higher blood pressure [77], and smaller livers than that of male counterpart [133], while the sons had a more marked difference in their transcriptomes [134].
Garbory et al. [136] observed sex-specific functional differences based on both epigenetic and transcriptomic analyses related to diet response in the mouse placenta. In detail, the authors reported that males and females diverged not only in terms of number and variation of the genes involved, but also more specifically in the functions and networks involved. In particular, the function and networks associated with sexually dimorphic genes for females were mainly related with cell signalling involving immune cells and the metabolism of aminoacids, whereas in males they were related with the development and function of the vascular system and metabolism of glucose and fatty acids. Remarkably, the pronounced sex-specificity of the offspring regarding nature and severity of the maternal diet effects should encourage us to consider the impact of the biological sex of the offspring also on GDM-induced epigenetic patterns in the offspring.
Regarding GDM, the sex-specificity effects are unclear and require further research. In humans, a meta-analysis of 20 studies showed an increased risk of GDM in women carrying a male foetus compared with women carrying a female one [137]. In addition, male foetus is correlated to β cell dysfunctions and higher postprandial glycemia suggesting a probable influence on the maternal glucose metabolism during pregnancy [138]; whereas the GDM development when carrying a female foetus predicted an overall future risk of early progression to T2DM [139]. Very recently, O’Neill [140] proposed that the sex-specific alterations in GDM maternal–foetal metabolism may clarify the sex-specific metabolic outcomes in offspring exposed to GDM in utero. In fact, the authors, characterizing the metabolome of 2nd trimester amniotic fluid (AF), identifying 44 and 58 metabolites altered by GDM exposure in male and female offspring, respectively. The significant changes in the metabolic pathways involved glucose, glutathione, fatty acid, sphingolipid, and bile acid metabolism, with specific changes identified based on offspring sex. These findings highlight the need to perform larger human studies that compare the GDM effects on the offspring of both the sexes.
Paternal influences
Finally, it must be stressed that, although literature is mainly focused on maternally mediated effects, the role of paternal contribute in modulating offspring’s health warrants attention, too [141]. In fact, several studies demonstrated a transmission of epigenetic alterations of sperm DNA related to paternal exposure to various contaminants, nutrition, and lifestyle-related conditions able to change the sperm epigenome [142,143]. The new and growing field of transgenerational epigenetics has introduced the Paternal Origins of Health and Disease (POHaD) paradigm [144].
Conclusions and future perspectives
Growing evidence has shown that epigenetic modifications mediated by maternal nutrition, gestational weight gain and metabolic perturbations during pregnancy can lead to a range of long-term metabolic disorders in the offspring (Figure1). The recent literature established the role of epigenetic marks as potential modulators and future predictors of human disease with a special focus on the very early stage of development. Furthermore, although there are gaps in the knowledge about the accurate mechanisms involved, recent suggestions have focused on peri-conceptional, intrauterine and postnatal periods as the most influential in foetal programming. The peri-conceptional period may represent the best window of opportunity to prevent foetal programming of NCDs. Yajnik et al. [145] defined gametogenesis, fertilisation, implantation, embryogenesis and placentation as periods of ‘primordial’ prevention. These authors suggested that not only conventional genetic inheritance shapes the future of the growing foetus, but also epigenetic influences, defined as ‘malleable’, determine its future [145]. In fact, epigenetic modifications are a modifiable component of the inter-generational transmission of phenotypic traits and thus can provide new exciting findings for susceptibility to obesity, diabetes, CVD, neuropsychiatric disorders and cancers. Recent epidemiological and experimental studies have demonstrated the importance and utility of possible future prognostic epigenetic analyses in healthcare [88].
In this regard, it has been demonstrated that GDM influences cellular and organ systems during the early life of the offspring and interacts with postnatal environmental and lifestyle factors. In fact, an increasing number of research studies have identified gene variants of susceptibility to GDM [9,48,49] as well as epigenetic alterations [100] that participate in the complexity of metabolic status of both GDM mothers and their offspring, inducing different levels of modifications bringing to hyperglycemia, impaired insulin sensitivity and correlated complications.
Over the last decade, the concept of ‘microbiome’ has come under increasing scrutiny and the knowledge about it is constantly expanding, suggesting potential future avenues of study on mechanisms linking maternal health to neonatal microbiota [146, 147]. Both maternal and neonatal microbiome could be influenced by GDM [147]; therefore, further studies are required to understand possible remodelling of the gut microbiome composition during the infant stage. Understanding maternal-foetal microbial vertical transmission effects and early-life colonisation could elucidate the long-term health impact of the offspring and develop intervention strategies in a timely manner.
Therefore, the knowledge of molecular mechanisms underlying health consequences of an altered in utero condition, such as in GDM, will help to both develop effective prenatal preventive strategies and limit the vicious cycle across generations. Overall, the primary goal is to shape the GDM impact on the epigenome-wide level by identifying genes and their pathways epigenetically involved. Additionally, it is also to establish how dietary patterns, nutrients, bioactive compounds and exercise affect the epigenome to trigger the development of the metabolic disturbances.
Understanding diabetes-related metabolic traits from an epigenetic perspective may offer new and optimal strategies to prevent or treat the occurrence of GDM complications in women and their children. To progress in this direction, it is clear that we should not only promote healthy nutrition and lifestyle during and after pregnancy in women of fertile age [23], but also assess the individual’s genetic predisposition and lifestyle [51]. Practising effective prevention by influencing the lifestyle of young girls and pregnant women in the relatively short period of peri-conceptional and gestational windows appears to be very attractive [145]. Therefore, a multi-sectoral approach combining all ‘omic’ levels (including nutrigenetic, epigenomic, and metagenomic data) will be required.
In light of the findings above discussed, further interventional and longitudinal research studies are required to widen the knowledge on this field. In this scenario, nutrigenetics and epigenetics in GDM can provide essential information and insights.
Authors’ contributions
MF and EV conceived this manuscript. MF, FF and EV carried out the search of literature about epigenetics, nutrigenetics and gestational diabetes. The manuscript was drafted by MF and EV. LS contributed to the editing the manuscript. All authors read and approved the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Hernández-Aguilera A, Fernández-Arroyo S, Cuyàs E, et al. Epigenetics and nutrition-related epidemics of metabolic diseases: current perspectives and challenges. Food Chem Toxicol. 2016;96:191–204. [DOI] [PubMed] [Google Scholar]
- [2].Katada S, Imhof A, Sassone-Corsi P.. Connecting threads: epigenetics and metabolism. Cell. 2012;148:24–28. [DOI] [PubMed] [Google Scholar]
- [3].van Dijk SJ, Tellam RL, Morrison JL, et al. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin Epigenetics. 2015;7:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lowe WL jr, Scholtens DM, Victoria S, et al. Genetics of gestational diabetes mellitus and maternal metabolism. Curr Diab Rep. 2016;16:15. [DOI] [PubMed] [Google Scholar]
- [5].Karaderi T, Drong AW, Lindgren CM. Insights into the genetic susceptibility to type 2 diabetes from genome-wide association studies of obesity-related traits. Curr Diab Rep. 2015;15:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang X, Strizich G, Hu Y, et al. Qi Q Genetic markers of type 2 diabetes: progress in genome-wide association studies and clinical application for risk prediction. J Diabetes. 2016;8:24–35. [DOI] [PubMed] [Google Scholar]
- [7].Ding M, Chavarro J, Olsen S, et al. Genetic variants of gestational diabetes mellitus: a study of 112 SNPs among 8722 women in two independent populations. Diabetologia. 2018;61:1758–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gluckman PD, Hanson MA, Beedle AS. Non-genomic transgenerational inheritance of disease risk. Bioessays. 2007;29:145–154. [DOI] [PubMed] [Google Scholar]
- [9].Schwenk RW, Vogel H, Schürmann A. Genetic and epigenetic control of metabolic health. Mol Metab. 2013;2:337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Christensen BC, Marsit CJ. Epigenomics in environmental health. Front Genet. 2011;2:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Boekelheide K, Blumberg B, Chapin RE, et al. Predicting later-life outcomes of early-life exposures. Environ Health Perspect. 2012;120:1353–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tobi EW, Slieker RC, Luijk R, et al,, . DNA methylation as a mediator of the association between prenatal adversity and risk factors for metabolic disease in adulthood. Sci Adv. 2018;4:eaao4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Blackmore HL, Ozanne SE. Maternal diet-induced obesity and offspring cardiovascular health. J Dev Orig Health Dis. 2013;4:338–347. [DOI] [PubMed] [Google Scholar]
- [14].Heindel JJ, Balbus J, Birnbaum L, et al. Developmental origins of health and disease: integrating environmental influences. Endocrinology. 2015;156:3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lance. 2011;378:169–181. [DOI] [PubMed] [Google Scholar]
- [16].Ma RC, Tutino GE, Lillycrop KA, et al. Maternal diabetes, gestational diabetes and the role of epigenetics in their long term effects on offspring. Prog Biophys Mol Biol. 2015;118:55–68. [DOI] [PubMed] [Google Scholar]
- [17].Gagné-Ouellet V, Houde AA, Guay SP, et al. Placental lipoprotein lipase DNA methylation alterations are associated with gestational diabetes and body composition at 5 years of age. Epigenetics. 2017;12:616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Godfrey KM, Reynolds RM, Prescott SL, et al. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017;5:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Agarwal P, Morriseau TS, Kereliuk SM, et al. Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Crit Rev Clin Lab Sci. 2018;55:71–101. [DOI] [PubMed] [Google Scholar]
- [20].American Diabetes Association (ADA) Standards of medical care in diabetes- 2018”. Diabetes Care. 2018;41(Suppl 1):S13–S27. [DOI] [PubMed] [Google Scholar]
- [21].Hod M, Kapur A, Sacks A, et al. The international Federation of Gynecology and Obstetrics (FIGO). Initiative on gestational diabetes mellitus: a pragmatic guide for diagnosis, management, and care. J Gynecol Obstet. 2015;131(Suppl 3):S173–211. [DOI] [PubMed] [Google Scholar]
- [22].Rayanagoudar G, Hashi AA, Zamora J, et al. Quantification of the type 2 diabetes risk in women with gestational diabetes: a systematic review and meta-analysis of 95,750 women. Diabetologia. 2016;59:1403–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Burlina S, Dalfrà MG, Lapolla A. Clinical and biochemical approach to predicting post-pregnancy metabolic decompensation. Diabetes Res Clin Pract. 2018;145:178–183. DOI: 10.1016/j.diabres.2018.02.035 [DOI] [PubMed] [Google Scholar]
- [24].Metzger BE, the HAPO Study Cooperative Research Group . Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study. Associations with neonatal anthropometrics. Diabetes. 2009;58:453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Freinkel N. Banting Lecture 1980: of pregnancy and progeny. Diabetes. 1980;29:1023–1035. [DOI] [PubMed] [Google Scholar]
- [26].Lowe WL Jr, Scholtens DM, Lowe LP, et al. Association of Gestational Diabetes With Maternal Disorders of Glucose Metabolism and Childhood Adiposity. JAMA. 2018;320(10):1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhu Y, Zhang C. Prevalence of gestational diabetes and risk of progression to type 2 diabetes: a global perspective. Curr Diab Rep. 2016;16(1):7 .Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Plows JF, Stanley JL, Baker PN, et al. The pathophysiology of gestational diabetes mellitus. Int J Mol Sci. 2018;19(11):pii: E3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang C, Ning Y. Effect of dietary and lifestyle factors on the risk of gestational diabetes: review of epidemiologic evidence. Am J Clin Nutr. 2011;94:1975S–1979S. Epub 2011 May 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang C, Bao W, Rong Y, et al. Genetic variants and the risk of gestational diabetes mellitus: a systematic review. Hum Reprod Update. 2013;19:376–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007;8:657–662. [DOI] [PubMed] [Google Scholar]
- [32].McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med. 2010;363:2339–2350. [DOI] [PubMed] [Google Scholar]
- [33].Stover PJ, James WPT, Krook A, et al. Emerging concepts on the role of epigenetics in the relationships between nutrition and health. J Intern Med. 2018;284(1):37–49. .Epub 2018 May 23. [DOI] [PubMed] [Google Scholar]
- [34].Simopoulos AP. Nutrigenetics/nutrigenomics. Annu Rev Public Health. 2010;31:53–68. [DOI] [PubMed] [Google Scholar]
- [35].Phillips CM, Tierney AC, Roche HM. Gene-nutrient interactions in the metabolic syndrome. J Nutrigenet Nutrigenomics. 2008;1:136–151. [DOI] [PubMed] [Google Scholar]
- [36].Roche HM, Phillips C, Gibney MJ. The metabolic syndrome: the crossroads of diet and genetics. Proc Nutr Soc. 2005;64:371–377. [DOI] [PubMed] [Google Scholar]
- [37].Phillips C, Lopez-Miranda J, Perez-Jimenez F, et al. Genetic and nutrient determinants of the metabolic syndrome. Curr Opin Cardiol. 2006;21:185–193. [DOI] [PubMed] [Google Scholar]
- [38].Moleres A, Ochoa MC, Rendo-Urteaga T, et al. Dietary fatty acid distribution modifies obesity risk linked to the rs9939609 polymorphism of the fat mass and obesity-associated gene in a Spanish case-control study of children. Br J Nutr. 2012;107:533–538. [DOI] [PubMed] [Google Scholar]
- [39].Ortega-Azorín C, Sorlí JV, Asensio EM, et al. Associations of the FTO rs9939609 and the MC4R rs17782313 polymorphisms with type 2 diabetes are modulated by diet, being higher when adherence to the Mediterranean diet pattern is low. Cardiovasc Diabetol. 2012;11:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Luan J, Browne PO, Harding AH, et al. Evidence for gene-nutrient interaction at the PPARgamma locus. Diabetes. 2001;50(3):686–689. [DOI] [PubMed] [Google Scholar]
- [41].Fisher E, Boeing H, Fritsche A, et al. Whole-grain consumption and transcription factor-7-like 2 (TCF7L2) rs7903146: gene-diet interaction in modulating type 2 diabetes risk. Br J Nutr. 2009;101:478–481. [DOI] [PubMed] [Google Scholar]
- [42].Hindy G, Sonestedt E, Ericson U, et al. Role of TCF7L2 risk variant and dietary fibre intake on incident type 2 diabetes. Diabetologia. 2012;55:2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wirstrom T, Hilding A, Gu HF, et al. Consumption of whole grain reduces risk of deteriorating glucose tolerance, including progression to prediabetes. Am J Clin Nutr. 2013;97:179–187. [DOI] [PubMed] [Google Scholar]
- [44].López-Ortiz MM, Garay-Sevilla ME, Tejero ME, et al. Analysis of the interaction between transcription factor 7-like 2 genetic variants with nopal and wholegrain fibre intake: effects on anthropometric and metabolic characteristics in type 2 diabetes patients. Br J Nutr. 2016;116(6):969–978. .Epub 2016 Aug 2. [DOI] [PubMed] [Google Scholar]
- [45].Lai CQ, Corella D, Demissie S, et al. Dietary intake of n-6 fatty acids modulates effect of apolipoprotein A5 gene on plasma fasting triglycerides, remnant lipoprotein concentrations, and lipoprotein particle size: the Framingham Heart Study. Circulation. 2006;113(17): 2062–2070. Epub 2006 Apr 24. [DOI] [PubMed] [Google Scholar]
- [46].Kang R, Kim M, Chae JS, et al. Consumption of whole grains and legumes modulates the genetic effect of the APOA5-1131C variant on changes in triglyceride and apolipoprotein A-V concentrations in patients with impaired fasting glucose or newly diagnosed type 2 diabetes. Trials. 2014;15:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Corella D, Lai CQ, Demissie S, et al. APOA5 gene variation modulates the effects of dietary fat intake on body mass index and obesity risk in the Framingham Heart Study. J Mol Med (Berl). 2007;85(2): 119–128. Epub 2007 Jan 9. [DOI] [PubMed] [Google Scholar]
- [48].Franzago M, Fraticelli F, Nicolucci A, et al. Molecular analysis of a genetic variants panel related to nutrients and metabolism: association with susceptibility to gestational diabetes and cardiometabolic risk in affected women. J Diabetes Res. 2017:ID 4612623DOI: 10.1155/2017/4612623Epub 2017 Jan 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Franzago M, Fraticelli F, Marchetti D, et al. Nutrigenetic variants and cardio-metabolic risk in women with or without gestational diabetes. Diabetes Res Clin Pract. 2018. 8;137:64–71. Epub 2018 Jan 8. [DOI] [PubMed] [Google Scholar]
- [50].Franzago M, Fraticelli F, Di Nicola M, et al. Early subclinical atherosclerosis in gestational diabetes: the predictive role of routine biomarkers and nutrigenetic variants. J Diabetes Res. 2018;2018:9242579 DOI: 10.1155/2018/9242579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Berná G, Oliveras-López MJ, Jurado-Ruíz E, et al. Nutrigenetics and nutrigenomics insights into diabetes etiopathogenesis. Nutrients. 2014;6:5338–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Del Rosario MC, Ossowski V, Knowler WC, et al. Potential epigenetic dysregulation of genes associated with MODY and type 2 diabetes in humans exposed to a diabetic intrauterine environment: an analysis of genome-wide DNA methylation. Metabolism. 2014;63(5):654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen P, Piaggi P, Traurig M, et al. Differential methylation of genes in individuals exposed to maternal diabetes in utero. Diabetologia. 2017;60(4):645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Houshmand-Oeregaard A, Hjort L, Kelstrup L, et al. DNA methylation and gene expression of TXNIP in adult offspring of women with diabetes in pregnancy. PLoS One. 2017;12(10):e0187038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Liyanage VR, Jarmasz JS, Murugeshan N, et al. DNA modifications: function and applications in normal and disease States. Biology (Basel). 2014;3:670–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Dolinoy DC, Weidman JR, Jirtle R. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol. 2007;23:297–307. [DOI] [PubMed] [Google Scholar]
- [57].Auclair G, Weber M. Mechanisms of DNA methylation and demethylation in mammals. Biochimie. 2012;94:2202–2211. [DOI] [PubMed] [Google Scholar]
- [58].Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. [DOI] [PubMed] [Google Scholar]
- [59].Lawrence M, Daujat S, Schneider R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet. 2016;32:42–56. [DOI] [PubMed] [Google Scholar]
- [60].Esguerra JLS, Mollet IG, Salunkhe VA, et al. Regulation of pancreatic beta cell stimulus-secretion coupling by microRNAs. Genes (Basel). 2014;5:1018–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wilczynska A, Bushell M. The complexity of miRNA-mediated repression. Cell Death Differ. 2015;22:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kaspi H, Pasvolsky R, Hornstein E. Could microRNAs contribute to the maintenance of b cell identity? Trends Endocrinol Metab. 2014;25:285–292. [DOI] [PubMed] [Google Scholar]
- [63].Ibarra A, Vega-Guedes B, Brito-Casillas Y, et al. Diabetes in pregnancy and MicroRNAs: promises and limitations in their clinical application. Noncoding RNA. 2018;4(4):pii: E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Guarino E, Delli Poggi C, Grieco GE, et al. Circulating MicroRNAs as biomarkers of gestational diabetes mellitus: updates and perspectives. Int J Endocrinol. 2018;2018:6380463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Mochizuki K, Hariya N, Honma K, et al. Relationship between epigenetic regulation, dietary habits, and the developmental origins of health and disease theory. Congenit Anom (Kyoto). 2017;57:184–190. [DOI] [PubMed] [Google Scholar]
- [66].Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35:595–601. [DOI] [PubMed] [Google Scholar]
- [67].El Hajj N, Schneider E, Lehnen H, et al. Epigenetics and life-long consequences of an adverse nutritional and diabetic intrauterine environment. Reproduction. 2014;148:R111–R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Godfrey KM, Sheppard A, Gluckman PD, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011;60:1528–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Davison JM, Mellott TJ, Kovacheva VP, et al. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J Biol Chem. 2009;284:1982–1989. Epub 2008 Nov 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tosh DN, Fu Q, Callaway CW, et al. Epigenetics of programmed obesity: alteration in IUGR rat hepatic IGF1 mRNA expression and histone structure in rapid vs. delayed postnatal catch-up growth. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1023–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Strakovsky RS, Zhang X, Zhou D, et al. Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. J Physiol. 2011;589:2707–2717. Epub 2011 Mar 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Borengasser SJ, Zhong Y, Kang P, et al. Maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in male rat offspring. Endocrinology. 2013;154(11):4113–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang Y, Wang H, Zhou D, et al. High-fat diet caused widespread epigenomic differences on hepatic methylome in rat. Physiol Genomics. 2015;47(10):514–523. [DOI] [PubMed] [Google Scholar]
- [74].Wankhade UD, Zhong Y, Kang P, et al. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS One. 2017;12(4):e0175675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Moody L, Chen H, Pan YX. Postnatal diet remodels hepatic DNA methylation in metabolic pathways established by a maternal high-fat diet. Epigenomics. 2017;9(11):1387–1402. [DOI] [PubMed] [Google Scholar]
- [76].Keleher MR, Zaidi R, Shah S, et al. Maternal high-fat diet associated with altered gene expression, DNA methylation, and obesity risk in mouse offspring. PLoS One. 2018;13(2):e0192606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Khan I, Dekou V, Hanson M, et al. Predictive adaptive responses to maternal high-fat diet prevent endothelial dysfunction but not hypertension in adult rat offspring. Circulation. 2004;110:1097–1102. [DOI] [PubMed] [Google Scholar]
- [78].Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305:1733–1736. [DOI] [PubMed] [Google Scholar]
- [79].Gluckman PD, Hanson MA, Low FM. The role of developmental plasticity and epigenetics in human health. Birth Defects Res C Embryo Today. 2011;93:12–18. [DOI] [PubMed] [Google Scholar]
- [80].Bateson P, Gluckman P, Hanson M. The biology of developmental plasticity and the predictive adaptive response hypothesis. J Physiol. 2014;592:2357–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gluckman PD, Hanson MA, Buklijas T. A conceptual framework for the developmental origins of health and disease. J Dev Orig Health Dis. 2010;1:6–18. [DOI] [PubMed] [Google Scholar]
- [82].Roseboom TJ, van der Meulen JH, Ravelli AC, et al. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol. 2001;185:93–98. [DOI] [PubMed] [Google Scholar]
- [83].Painter RC, Roseboom TJ, Bleker OP. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod Toxicol. 2005;20:345–352. [DOI] [PubMed] [Google Scholar]
- [84].Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Martin-Gronert MS, Ozanne SE. Mechanisms linking suboptimal early nutrition and increased risk of type 2 diabetes and obesity. J Nutr. 2010;140:662–666. [DOI] [PubMed] [Google Scholar]
- [86].Seki Y, Williams L, Vuguin PM, et al. Minireview: epigenetic programming of diabetes and obesity: animal models. Endocrinology. 2012;153:1031–1038. Epub 2012 Jan 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Quilter CR, Cooper WN, Cliffe KM, et al. Impact on offspring methylation patterns of maternal gestational diabetes mellitus and intrauterine growth restraint suggest common genes and pathways linked to subsequent type 2 diabetes risk. FASEB J. 2014;28:4868–4879. [DOI] [PubMed] [Google Scholar]
- [88].Wendland EM, Torloni MR, Falavigna M, et al. Gestational diabetes and pregnancy outcomes–a systematic review of the World Health Organization (WHO) and the International Association of Diabetes in Pregnancy Study Groups (IADPSG) diagnostic criteria. BMC Pregnancy Childbirth. 2012;12:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Block T, El-Osta A. Epigenetic programming, early life nutrition and the risk of metabolic disease. Atherosclerosis. 2017;266:31–40. [DOI] [PubMed] [Google Scholar]
- [90].Boney CM, Verma A, Tucker R, et al. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115:e290–e296. [DOI] [PubMed] [Google Scholar]
- [91].Dabelea D, Mayer-Davis EJ, Lamichhane AP, et al. Association of intrauterine exposure to maternal diabetes and obesity with type 2 diabetes in youth: the SEARCH case-control study. Diabetes Care. 2008;31:1422–1426. Epub 2008 Mar 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Nelissen EC, van Montfoort AP, Dumoulin JC, et al. Epigenetics and the placenta. Hum Reprod Update. 2011;17:397–417. [DOI] [PubMed] [Google Scholar]
- [93].Jiang Y, Yu YC, Ding GL, et al. Intrauterine hyperglycemia induces intergenerational Dlk1-Gtl2 methylation changes in mouse placenta. Oncotarget. 2018;9:22398–22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Reichetzeder C, Dwi Putra SE, Pfab T, et al. Increased global placental DNA methylation levels are associated with gestational diabetes. Clin Epigenetics. 2016;8: 82 eCollection 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Bouchard L, Thibault S, Guay SP, et al. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care. 2010;33:2436–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Bouchard L, Hivert MF, Guay SP, et al. Placental adiponectin gene DNA methylation levels are associated with mothers’ blood glucose concentration. Diabetes. 2012;61:1272–1280. Epub 2012 Mar 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Houde AA, Guay SP, Desgagné V, et al. Adaptations of placental and cord blood ABCA1 DNA methylation profile to maternal metabolic status. Epigenetics. 2013;8(12):1289–1302. .Epub 2013 Oct 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Houde AA, St-Pierre J, Hivert MF, et al. Placental lipoprotein lipase DNA methylation levels are associated with gestational diabetes mellitus and maternal and cord blood lipid profiles. J Dev Orig Health Dis. 2014;5:132–141. [DOI] [PubMed] [Google Scholar]
- [99].Houde AA, Ruchat SM, Allard C, et al. LRP1B, BRD2 and CACNA1D: new candidate genes in fetal metabolic programming of newborns exposed to maternal hyperglycemia. Epigenomics. 2015;7:1111–1122. Epub 2015 Nov 20. [DOI] [PubMed] [Google Scholar]
- [100].Ruchat SM, Houde AA, Voisin G, et al. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics. 2013;8:935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Desgagne V, Hivert MF, St-Pierre J, et al. Epigenetic dysregulation of the IGF system in placenta of newborns exposed to maternal impaired glucose tolerance. Epigenomics. 2014;6:193–207. [DOI] [PubMed] [Google Scholar]
- [102].Petropoulos S, Guillemin C, Ergaz Z, et al. Gestational diabetes alters offspring DNA methylation profiles in human and rat: identification of key pathways involved in endocrine system disorders, insulin signaling, diabetes signaling, and ILK signaling. Endocrinology. 2015;156:2222–2238. [DOI] [PubMed] [Google Scholar]
- [103].El Hajj N, Pliushch G, Schneider E, et al. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes. 2013;62:1320–1328. Epub 2012 Dec 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].García-Cardona MC, Huang F, García-Vivas JM, et al. DNA methylation of leptin and adiponectin promoters in children is reduced by the combined presence of obesity and insulin resistance. Int J Obes (Lond). 2014;38:1457–1465. [DOI] [PubMed] [Google Scholar]
- [105].Côté S, Gagné-Ouellet V, Guay SP, et al. PPARGC1α gene DNA methylation variations in human placenta mediate the link between maternal hyperglycemia and leptin levels in newborns. Clin Epigenetics. 2016;8:72 eCollection 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Houshmand-Oeregaard A, Hansen NS, Hjort L, et al. Differential adipokine DNA methylation and gene expression in subcutaneous adipose tissue from adult offspring of women with diabetes in pregnancy. Clin Epigenetics. 2017;9:37 eCollection 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ott R, Stupin JH, Melchior K, et al. Plagemann A Alterations of adiponectin gene expression and DNA methylation in adipose tissues and blood cells are associated with gestational diabetes and neonatal outcome. Clin Epigenetics. 2018;10(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Ott R, Melchior K, Stupin JH, et al. Plagemann A insulin receptor expression and altered DNA methylation in fat tissues and blood of women with GDM and offspring. J Clin Endocrinol Metab. 2019;104(1):137–149. [DOI] [PubMed] [Google Scholar]
- [109].Sebastiani G, Guarino E, Grieco GE, et al. Circulating microRNA (miRNA) expression profiling in plasma of patients with gestational diabetes mellitus reveals upregulation of miRNA miR-330-3p. Front Endocrinol (Lausanne). 2017;8:345 eCollection 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Zhao C, Dong J, Jiang T, et al. Early second-trimester serum miRNA profiling predicts gestational diabetes mellitus. PLoS One. 2011;6(8):e23925 .Epub 2011 Aug 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Tagoma A, Alnek K, Kirss A, et al. MicroRNA profiling of second trimester maternal plasma shows upregulation of miR-195-5p in patients with gestational diabetes. Gene. 2018;672:137–142. Epub 2018 Jun 4. [DOI] [PubMed] [Google Scholar]
- [112].Zhu Y, Tian F, Li H, et al. Profiling maternal plasma microRNA expression in early pregnancy to predict gestational diabetes mellitus. Int J Gynaecol Obstet. 2015;130:49–53. [DOI] [PubMed] [Google Scholar]
- [113].Shi Z, Zhao C, Guo X, et al. Differential expression of microRNAs in omental adipose tissue from gestational diabetes mellitus subjects reveals miR-222 as a regulator of ERα expression in estrogen-induced insulin resistance. Endocrinology. 2014;155:1982–1990. Epub 2014 Mar 6. [DOI] [PubMed] [Google Scholar]
- [114].Cao JL, Zhang L, Li J, et al. Up-regulation of miR-98 and unraveling regulatory mechanisms in gestational diabetes mellitus. Sci Rep. 2016;6:32268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Houshmand-Oeregaard A, Schrölkamp M, Kelstrup L, et al. Increased expression of microRNA-15a and microRNA-15b in skeletal muscle from adult offspring of women with diabetes in pregnancy. Hum Mol Genet. 2018;27(10):1763–1771. [DOI] [PubMed] [Google Scholar]
- [116].Ge ZJ, Luo SM, Lin F, et al. DNA methylation in oocytes and liver of female mice and their offspring: effects of high-fat-diet-induced obesity. Environ Health Perspect. 2014a;122:159–164. Epub 2013 Dec 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wang Q, Tang SB, Song XB, et al. High-glucose concentrations change DNA methylation levels in human IVM oocytes. Hum Reprod. 2018;25 DOI: 10.1093/humrep/dey006 [DOI] [PubMed] [Google Scholar]
- [118].Alegría-Torres JA, Baccarelli A, Bollati V. Epigenetics and lifestyle. Epigenomics. 2011;3:267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Palma-Gudiel H, Cirera F, Crispi F, et al. The impact of prenatal insults on the human placental epigenome: A systematic review. Neurotoxicol Teratol. 2018;66:80–93. [DOI] [PubMed] [Google Scholar]
- [120].Shah NR, Bracken MB. A systematic review and meta-analysis of prospective studies on the association between maternal cigarette smoking and preterm delivery. Am J Obstet Gynecol. 2000;182:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Breton C, Byun HM, Wenten M, et al. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;18:462–467. Epub 2009 Jun 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Fa S, Larsen TV, Bilde K, et al. Assessment of global DNA methylation in the first trimester fetal tissues exposed to maternal cigarette smoking. Clin Epigenetics. 2016;8:128 eCollection 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Maccani JZJ, Koestler DC, Houseman EA, et al. Placental DNA methylation alterations associated with maternal tobacco smoking at the RUNX3 gene are also associated with gestational age. Epigenomics. 2013;5:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Maccani MA, Avissar-Whiting M, Banister CE, et al. Maternal cigarette smoking during pregnancy is associated with downregulation of miR-16, miR-21, and miR-146a in the placenta. Epigenetics. 2010. 1;5:583–589. Epub 2010 Oct 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Zhou FC, Balaraman Y, Teng M, et al. Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol Clin Exp Res. 2011;35:735–746. Epub 2011 Jan 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Sharp GC, Arathimos R, Reese SE, et al,, . Maternal alcohol consumption and offspring DNA methylation: findings from six general population-based birth cohorts. Epigenomics. 2018;10:27–42. . Epub 2017 Nov 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Laker RC, Lillard TS, Okutsu M, et al. Exercise prevents maternal high-fat diet-induced hypermethylation of the Pgc- 1alpha gene and age-dependent metabolic dysfunction in the offspring. Diabetes. 2014;63:1605–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Chan JC, Nugent BM, Bale TL. Parental advisory: maternal and paternal stress can impact offspring neurodevelopment. Biol Psychiatry. 2018;83(10):886–894. .Epub 2017 Oct 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kertes DA, Kamin HS, Hughes DA, et al. Prenatal maternal stress predicts methylation of genes regulating the hypothalamicpituitary- adrenocortical system in mothers and newborns in the Democratic Republic of Congo. Child Dev. 2016;87:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Kertes DA, Bhatt SS, Kamin HS, et al. BNDF methylation in mothers and newborns is associated with maternal exposure to war trauma. Clin Clin Epigenet. 2017;9:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Monk C, Feng T, Lee S, et al. Distress during pregnancy: epigenetic regulation of placenta glucocorticoid-related genes and fetal neurobehavior. Am J Psychiatry. 2016;173:705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Palma-Gudiel H, Córdova-Palomera A, Eixarch E, et al. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: a meta-analysis. Epigenetics. 2015;10(10):893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Miller C, Krishna S, Zhoumeng L, et al. Early sex differences in hepatic metabolic signaling in offspring of obese female mice. FASEB. 2014;28(Supplement 1033.11). [Google Scholar]
- [134].Mischke M, Pruis MGM, Boekschoten MV, et al. Maternal western-style high fat diet induces sex-specific physiological and molecular changes in two-week-old mouse offspring. PLoS ONE. 2013;8:e78623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Bellisario V, Berry A, Capoccia S, et al. Gender-dependent resiliency to stressful and metabolic challenges following prenatal exposure to high-fat diet in the p66Shc −/− mouse. Front. Behav. Neurosci. 2014; 8: 285. PLoS ONE. 2013;8:e78623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Gabory A, Ferry L, Fajardy I, et al. Maternal diets trigger sex-specific divergent trajectories of gene expression and epigenetic systems in mouse placenta. PLoS One. 2012;7(11):e47986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Jaskolka D, Retnakaran R, Zinman B, et al. Sex of the baby and risk of gestational diabetes mellitus in the mother: a systematic review and meta-analysis. Diabetologia. 2015. November;58(11):2469–2475. . [DOI] [PubMed] [Google Scholar]
- [138].Retnakaran R, Kramer CK, Ye C, et al. Fetal sex and maternal risk of gestational diabetes mellitus: the impact of having a boy. Diabetes Care. 2015. May;38(5):844–851. . [DOI] [PubMed] [Google Scholar]
- [139].Retnakaran R, Shah BR. Sex of the baby and future maternal risk of Type 2 diabetes in women who had gestational diabetes. Diabet Med. 2016;33(7):956–960. [DOI] [PubMed] [Google Scholar]
- [140].O’Neill K, Alexander J, Azuma R, et al. Gestational diabetes alters the metabolomic profile in 2nd trimester amniotic fluid in a sex-specific manner. Int J Mol Sci. 2018;19(9):pii: E2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Stuppia L, Franzago M, Ballerini P, et al. Epigenetics and male reproduction: the consequences of paternal lifestyle on fertility, embryo development, and children lifetime health. Clin Epigenetics. 2015;11(7):120 .eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Champroux A, Cocquet J, Henry-Berger J, et al. A decade of exploring the mammalian sperm epigenome: paternal epigenetic and transgenerational inheritance. Front Cell Dev Biol. 2018;6:50 eCollection 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Soubry A, Hoyo C, Jirtle RL, et al. A paternal environmental legacy: evidence for epigenetic inheritance through the male germ line. Bioessays. 2014;36:359–371. Epub 2014 Jan 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Soubry A. POHaD: why we should study future fathers. Environ Epigenet. 2018;4:dvy007 eCollection 2018 Apr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Yajnik CS. Transmission of obesity-adiposity and related disorders from the mother to the baby. Ann Nutr Metab. 2014;64:8–17. Epub 2014 Jul 23. [DOI] [PubMed] [Google Scholar]
- [146].Cortez RV, Taddei CR, Sparvoli LG, et al. Microbiome and its relation to gestational diabetes. Endocrine. 2018. DOI: 10.1007/s12020-018-1813-z [DOI] [PubMed] [Google Scholar]
- [147].Wang J, Zheng J, Shi W, et al. Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus. Gut. 2018;67(9):1614–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]